

SUMMARY
The zebrafish is amenable to variety of genetic approaches. However, lack of conditional deletion alleles limits stage- or cell-specific gene knockout. Here, we applied an existing protocol to establish a floxed allele for gata2a, but failed to do so due to off-target integration and incomplete knock-in. To address these problems, we applied simultaneous co-targeting with Cas12a to insert loxP sites in cis, together with transgenic counter-screening and comprehensive molecular analysis to identify off-target insertions and confirm targeted knock-ins. We subsequently used our approach to establish endogenously floxed alleles of foxc1a, rasa1a, and ruvbl1, each in a single generation. We demonstrate the utility of these alleles by verifying Cre-dependent deletion, which yielded expected phenotypes in each case. Finally, we used the floxed gata2a allele to demonstrate an endothelial autonomous requirement in lymphatic valve development. Together, our results provide a framework for routine generation and application of endogenously floxed alleles in zebrafish.
eTOC
The zebrafish is amenable to genetic approaches, but conditional gene knockout has been limited due the lack of floxed deletion alleles. Shin et al. describe reliable methods for generating, validating, and applying zebrafish bearing endogenously floxed alleles, providing a foundation for widespread application of conditional knockout approaches in zebrafish.
INTRODUCTION
The zebrafish has become a widely accepted model system for studying developmental biology based on several notable benefits of its life cycle. Zebrafish embryos are transparent and develop externally, allowing visualization of morphogenesis in vivo1. Embryogenesis is highly synchronous and rapid, with organ systems functioning by one to two days post fertilization (dpf)2. Coupled with these benefits is the genetic amenability of the zebrafish. Adults are easy to maintain and exhibit high fecundity, with a single pair often yielding hundreds of eggs on a given day. Zebrafish researchers leveraged these characteristics to perform large-scale forward genetic screens for a host of embryonic phenotypes3–5. Subsequent development of sequence-specific nucleases, such as those that target clustered regularly interspaced short palindromic repeats (CRISPR) in bacteria, has allowed introduction of targeted germline deletions in the zebrafish genome for reverse genetic approaches6,7. Thus, a combination of genetic accessibility and unique embryonic characteristics make the zebrafish an ideal model for interrogating gene function during development.
Nearly all zebrafish mutants perturb the function of a given gene from the earliest point at which it is expressed during development, making it a challenge to study direct roles in subsequent processes. While mosaic analysis through cell transplantation can address issues of cell autonomy in these mutants, ascribing primary functional effects at later stages remains challenging. Early efforts to address this issue relied on identification of temperature sensitive alleles, but these were limited8. More recently, conditional transgenic systems have been developed, but these require the use of dominant activating or inhibitory transgenes that may interfere with unrelated pathways9. Thus, a reliable and definitive conditional genetics platform is lacking in zebrafish, limiting analysis at postembryonic and adult stages.
The mouse became an established genetic model through development of techniques for making germline deletions using embryonic stem cells10–12. Subsequently, mouse researchers leveraged this platform for conditional genetics using the Cre/lox system13. Cre is a bacteriophage recombinase that stimulates recombination between loxP sequences14. When loxP sites are arranged directly in cis, recombination deletes the intervening sequence. By modifying endogenous mouse loci to flank coding exons with loxP sites (referred to as “floxed”), together with transgenic expression of a cell-specific Cre, it is possible to achieve tissue-specific gene knockout13. Timing of the knockout can be controlled using Cre fused to a modified estrogen ligand binding domain that binds to tamoxifen (CreERT15). To date, there are over 3000 published floxed alleles for approximately 2300 mouse genes, along with more than 300 transgenic lines expressing CreERT, allowing comprehensive genetic analysis in any biological context.
DESIGN
In zebrafish, Cre/lox has been applied for lineage tracing and conditional transgene expression9. However, its application for conditional gene deletion in zebrafish has been limited by the difficulty in generating endogenously floxed alleles. Indeed, only two previous studies have successfully made such alleles in zebrafish. Burg et al. used single-stranded oligodeoxynucleotides (ssODNs) with short homology arm sequences and a loxP site as a template for homology-directed repair (HDR), together with generating a double-stranded break (DSB) the target site using Cas9 nuclease, to generate a floxed tbx20 allele16. However, this approach inserted each loxP site sequentially, requiring two generations to establish a floxed allele. By contrast, Hoshijima et al. relied on plasmid template with 1kb homology arms to replace an endogenous exon of potassium voltage-gated channel, subfamily H, member 6a (kcnh6a) with a floxed version in a single generation17. In both cases, Cre-mediated excision of the floxed allele resulted in expected cardiac defects16,17. Here, we present our experience applying the Hoshijima et al. technique to generate a floxed gata2a allele, with a practical emphasis on pitfalls and points for improvement. We leveraged our experience to develop an improved protocol, which we applied to generate floxed alleles for foxc1a, rasa1a, and ruvbl1. Finally, we used the gata2a floxed allele with a tissue-specific inducible CreERT transgene to demonstrate an endothelial autonomous requirement during lymphatic valve development.
RESULTS
Generation of a gata2a conditional knock-in allele
To generate a floxed allele we applied the Hoshijima protocol17 to target gata2a, which encodes a zinc finger transcription factor expressed in blood, endothelial cells, and neurons18–20. We constructed a targeting plasmid with homology arms 1 kb up- and downstream of exon 5, which was flanked by loxP sites (Figure 1A, B). Homology arms matched the haplotype sequence in adults used to produce embryos for injection, as recommended17. Immediately downstream of exon 5 we placed a crystallin, alpha A (cryaa) promoter and enhanced green fluorescent (EGFP) cassette (Figure 1B; Supplementary File S1) to identify transgenic individuals by green lens fluorescence21. This cassette is flanked by FRT sites for removal if it interferes with normal gene function. The targeting cassette was flanked by I-SceI sites to release a linear HDR template17. Exon 5 deletion yields an in-frame exon junction that removes the C-terminal zinc finger (Supplementary Figure S1A, B), which is essential for DNA binding22. This allele is structurally the same as the widely used mouse Gata2tm1Sac allele23 (Supplementary Figure S1C).
Figure 1. Generation of a gata2a conditional deletion allele.
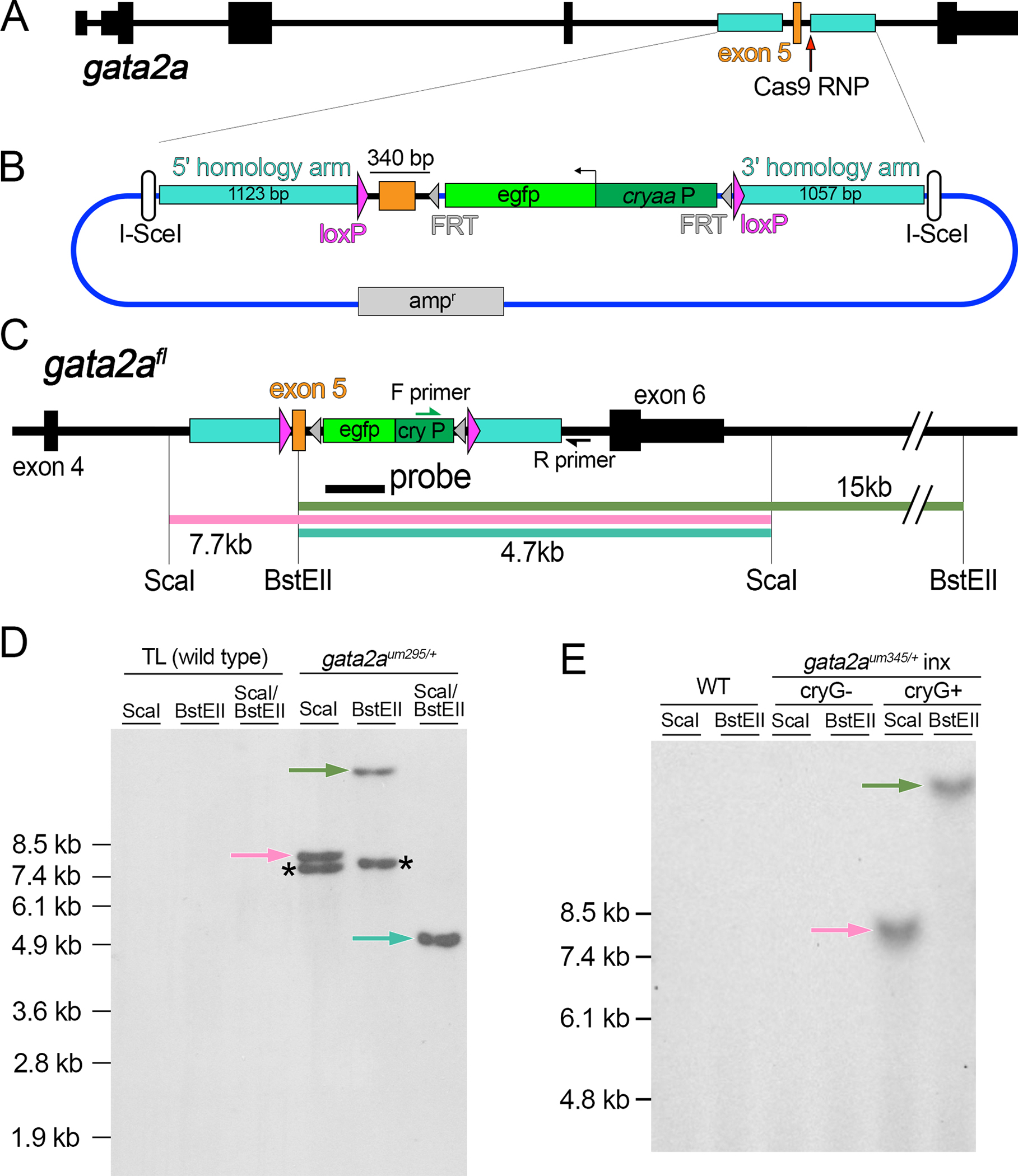
(A) Zebrafish gata2a locus. (B) Targeting plasmid for floxed gata2a allele. egfp – enhanced green fluorescent protein, cryaa:P – cryaa promoter element, ampr – ampicillin resistance cassette. (C) Restriction map of gata2afl. PCR primers and probe are shown. (D, E) Southern analysis of embryos of indicated genotype. Blots hybridized with a DIG-labeled egfp probe detected by chemiluminescence. Arrows denote fragments expected from precise integration, asterisked fragments from random integration.
To stimulate HDR, we used Cas9/sgRNA ribonucleoprotein complex (RNP) to introduce a DSB downstream of gata2a exon 5 (Figure 1A). In the targeting construct, we mutated the Cas9 spacer and split it from the PAM sequence with the 3’ loxP site to prevent cleavage (Supplementary File 1). We co-injected 1-cell stage embryos with Cas9 RNP, targeting plasmid, and I-SceI. At 3 days post fertilization (dpf), PCR across the 3’ homology arm junction revealed 6 out of 12 individual embryos exhibited a fragment of correct size and junction sequence in cryaa:egfp-positive embryos (Figure S2A–C). We repeated injections, grew cryaa:egfp-positive embryos to adulthood, and identified 12 founders out of 115 that transmitted cryaa:egfp to progeny embryos. Of these, only one (gata2aum295) gave PCR-positive embryos with the primers above, suggesting that most founders carried off-target insertions.
Subsequent PCR analysis of gata2aum295 revealed that only the 3’ loxP site and cryaa:egfp cassette inserted at the target (Supplementary Figure S2D–F), while Southern analysis revealed an off-target integration. Precise knock-in would yield a 7.7kb or 15kb fragment following digest of genomic DNA with ScaI or BstEII, respectively, and 4.7kb with a double digest followed by Southern hybridization using an Egfp probe (Figure 1C, D). However, additional fragments were also apparent indicating random insertion of the vector (Figure 1D). Unfortunately, this off-target integration co-segregated with gata2aum295 demonstrating linkage, although we did not detect Cre-mediated recombination between the off-target and single inserted loxP sites in gata2aum295 embryos (Supplementary Figure S3A, B). Therefore, we introduced a 5’ loxP site upstream of the targeted exon 5 by first eliminating the off-target cryaa:egfp cassette (Supplementary Figure S3B, C; referred to as gata2aum329, which retains some targeting construct sequence and a single loxP at the off-target site). We then injected homozygous gata2aum329 embryos with a Cas12a RNP targeted to a sequence upstream of exon 5 and a double-stranded (ds) ODN template with 30 bp flanking homology arms and a loxP site in direct orientation relative to the integrated 3’ loxP site (Supplementary Figure S3C, D). We identified a founder with a precise 5’ loxP integration (1 out of 15 fish screened; Supplementary Figure S3E) and the resulting floxed allele (gata2aum345) is referred to as gata2afl (Figure 1C, E).
To test gata2afl functionality, we crossed gata2a+/fl carriers to gata2a+/um27 adults, which bear a frameshift deletion upstream of the Gata2a zinc finger domains24. As expected, one-half of progeny embryos showed cryaa:egfp expression and retain exon 5 in the absence of Cre (Figure 2A, B; Supplementary Table S1). By contrast, cre mRNA injection caused loss of cryaa:egfp and exon 5 (Figure 2B, Supplementary Table S1). As expected24, we also observed fully penetrant loss of swim bladder inflation in heterozygous gata2aum27 embryos with Cre-mediated exon 5 deletion (gata2aum27/Δ), while siblings were normal (Figure 2C, Supplementary Table S1). Uninjected trans-heterozygous gata2aum27/fl larvae inflated their swim bladders at 5 dpf and appeared indistinguishable from gata2a+/fl siblings (Figure 2D). Consistently, gata2afl/fl adults were viable and fertile. Uninjected gata2afl/fl larvae from crosses between gata2afl/fl and gata2a+/fl adults also expressed cryaa:egfp and displayed normal swim bladder inflation at 6 dpf (Figure 2E, Supplementary Table S2). By contrast, gata2afl/fl larvae injected with a zebrafish codon-optimized cre mRNA (zfcre) at 1-cell stage showed loss of cryaa:egfp and no swim bladder inflation (Figure 2F, Supplementary Table S2).
Figure 2. Functional validation of a floxed gata2a allele.
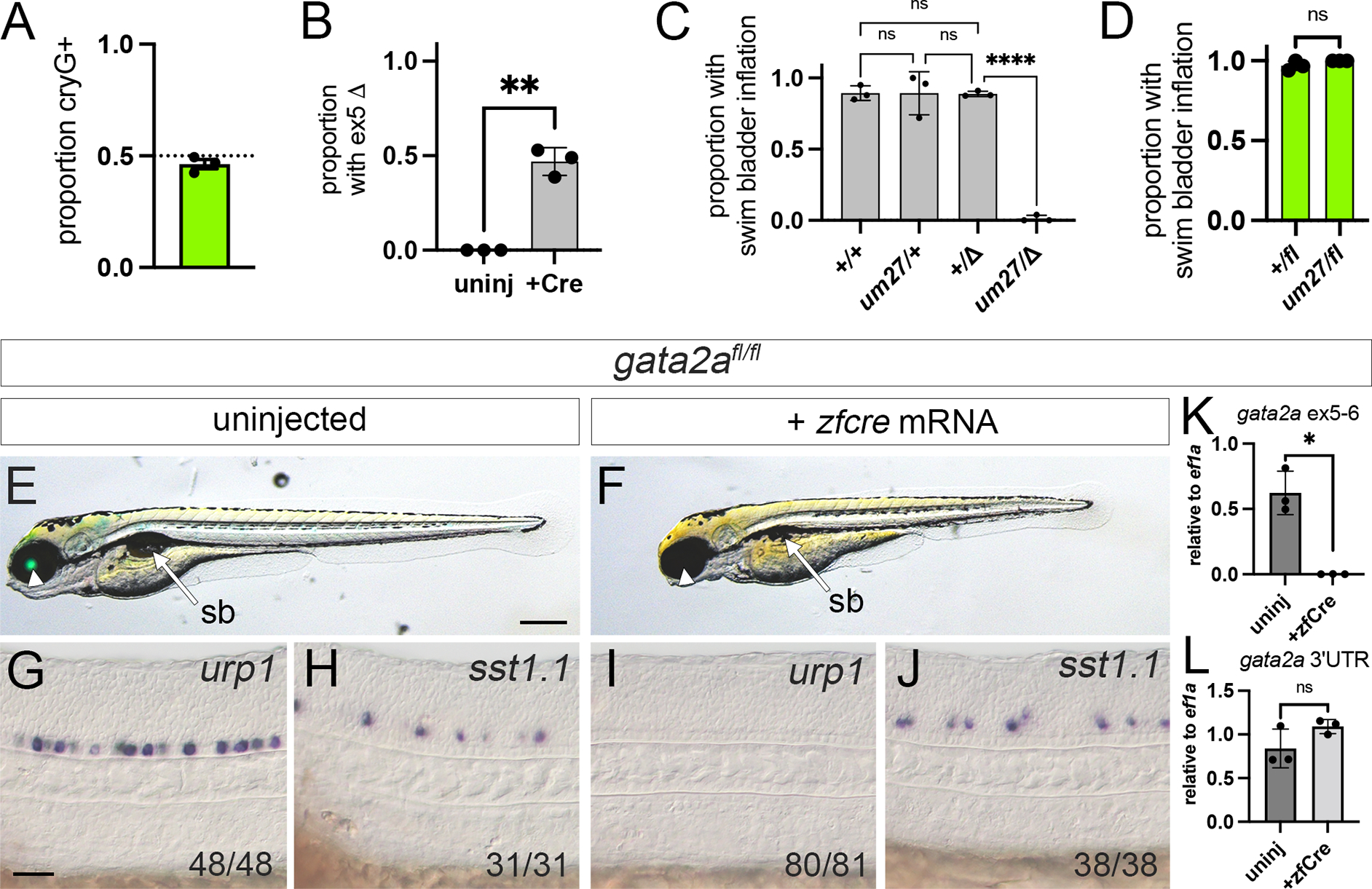
(A-D) Embryos from crosses between gata2a+/um27 and gata2a+/fl individuals (n = 3 clutches). (A) Proportion of embryos expressing lens Egfp (cryG+) at 2 dpf. (B) Proportion of uninjected or cre mRNA-injected embryos with exon 5 deletion. ** p<0.01, paired t-test. (C) Proportion of embryos of indicated genotype with inflated swim bladder (sb) following cre mRNA injection. “Δ” denotes exon 5 deletion confirmed by PCR. Analysis of variance, p<0.0001. **** p<0.0001, Tukey’s multiple comparison test. (D) Proportion of uninjected cryaa:egfp-positive embryos of indicated genotype with inflated swim bladder at 5 dpf. ns – not significant, Wilcoxon test. (E-J) Homozygous gata2afl/fl (E, F) larvae at 5 dpf or (G-J) embryos at 24 hpf. Lateral views, anterior to the left, dorsal is up. (E, G, H) Uninjected or (F, I, J) injected with zfcre mRNA at 1-cell stage. (E, F) Overlay of transmitted light and green fluorescence at 5 dpf. Arrowhead denotes lens Egfp expression, or absence thereof, arrow indicates sb. Scale bar is 250 μm. (G-J) Differential interference contrast (DIC) images of 24 hpf embryos following whole mount in situ hybridization with antisense riboprobes against indicated transcript. Scale bar is 50 μm. (K, L) Quantitative RT-PCR of uninjected or zfcre mRNA-injected gata2afl/fl embryos at 24 hpf for gata2a transcript with (K) exon 5 and 6, or (L) 3’ UTR. Paired t-test, * - p<0.05, ns – not statistically significant.
We next assessed spinal neuron and circulatory defects in gata2afl/fl embryos 18,19,24. Uninjected gata2afl/fl embryos express urotensin related peptide 1 (urp1) in the ventral-most Kolmer-Agduhr (KA) neurons in the floor plate and somatostatin 1, tandem duplicate 1 (sst1.1) in more dorsally-located KA neurons at 24 hours post fertilization (hpf; Figure 2G, H). By contrast, urp1-positive cells were absent in gata2afl/fl embryos injected with zfcre mRNA, while sst1.1-expressing KA neurons were present (Figure 2I, J), similar to gata2aum27 mutant embryos19. Gata2a+/fl sibling embryos injected with zfcre mRNA exhibited a normal heartbeat and circulation throughout aortic arch blood vessels, with venous circulatory return from cranial vessels through the posterior cerebral vein and primary head sinus (Supplementary Movie S1). Trunk circulation through the dorsal aorta (DA) and posterior cardinal vein (PCV) was also normal (Supplementary Movie S1). In zfCre-injected gata2afl/fl embryos, we observed a heartbeat and blood cell circulation through aortic arch blood vessels and cranial veins, albeit weaker than in gata2a+/fl siblings (Supplementary Movie S2). However, trunk circulation was absent (Supplementary Movie S2), similar to gata2aum27 mutants24.
Analysis of gata2a transcript in gata2afl/fl embryos by qRT-PCR using primers in exon 5 and 6 indicated expression in uninjected embryos, but not those injected with zfcre mRNA (Figure 2K). By contrast, gata2a transcript detected using 3’ UTR primers was not reduced by exon 5 deletion, consistent with an in-frame exon 4/6 fusion (Figure 2L, Supplementary Figure S1B). Together, these observations indicate that gata2afl provides wild type gata2a function and Cre-dependent deletion of exon 5 in gata2afl/fl phenocopies gata2aum27 mutants. Thus, gata2afl behaves as a conditional loss-of-function deletion allele.
Simultaneous dual CRISPR targeting can improve complete knock-in frequency.
In generating gata2afl, we failed to achieve complete knock-in, which we hypothesized was due to the location of the DSB. Our targeting strategy, which mirrored previous work17, placed the Cas9 spacer at the 3’ loxP insertion site, 340 downstream of the 5’ loxP site (Figure 1A, B). This stretch of homology could allow HDR-mediated insertion of only the cryaa transgene and 3’ loxP site. We reasoned that simultaneous DSBs at both loxP insertion sites would force HDR using the homology arms outside of the loxP sites, thereby improving complete knock-in. Alternatively, only targeting the site of 5’ loxP insertion could better facilitate full knock-in since embryos screened for cryaa:egfp expression will likely also harbor the adjacent 3’ loxP site. To test these possibilities, we identified CRISPR targets flanking foxc1a, which encodes a Forkhead transcription factor25. Since foxc1a is a single exon gene, we targeted immediately downstream of the promoter identified by DANIO-CODE26, and 150 bp downstream of the annotated 3’ UTR (Figure 3A, B; Supplementary File S2). The loxP sites flank a 2.2 kb fragment comprising foxc1a coding and UTR sequences, and haplotype-matched homology arms extend 400 bp up- and 1kb downstream of these sites (Figure 3B; Supplementary File S2).
Figure 3. Dual CRISPR targeting improves complete knock-in of a floxed cassette.
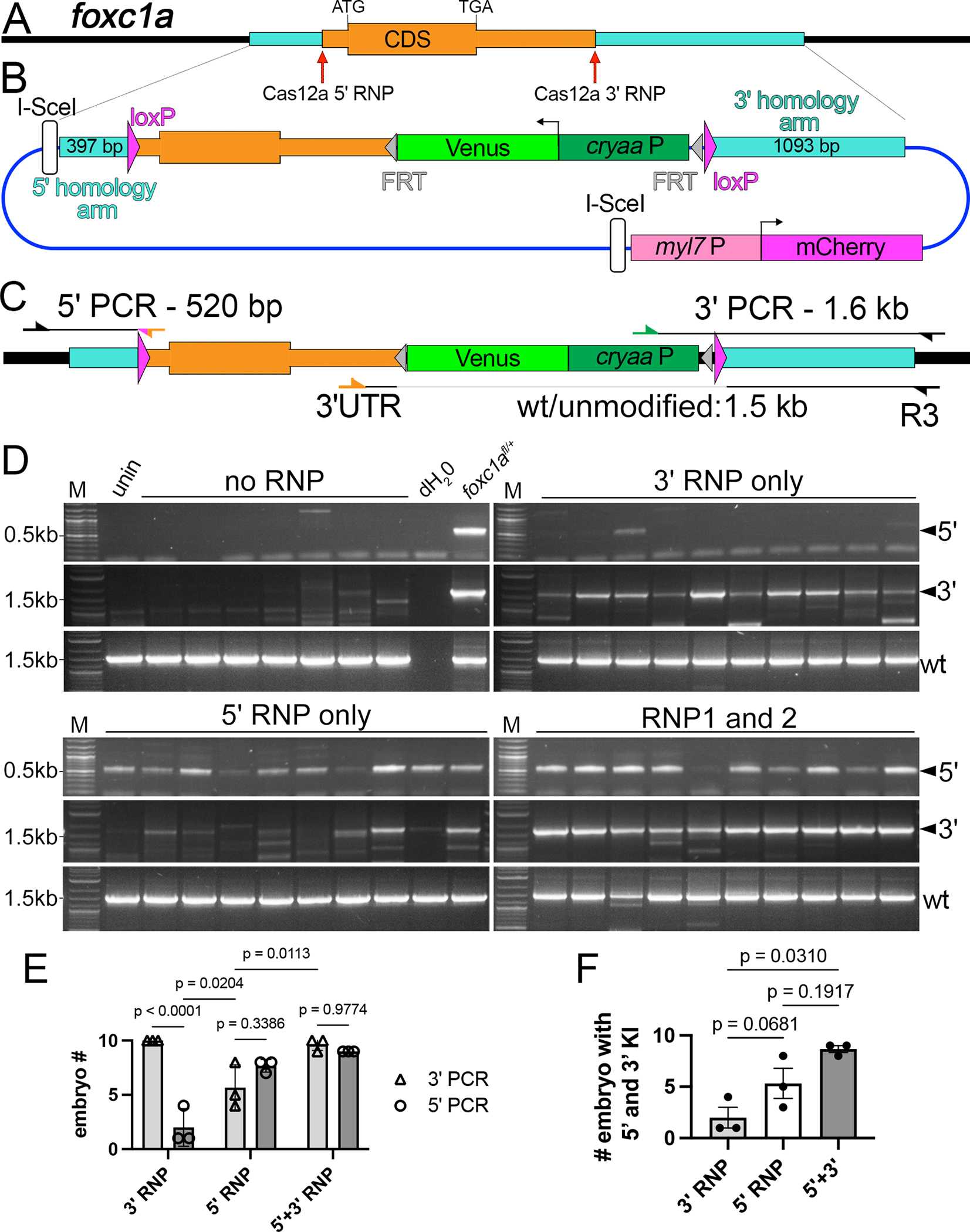
(A) Zebrafish foxc1a locus; Homology arms (turquoise) and Cas12a ribonucleoprotein (RNP) targets indicated, as are coding sequence (CDS), start (ATG) and stop codons (TGA). (B) Targeting plasmid for foxc1a. cryaaP – cryaa promoter. Not to scale as myl7:mcherry cassette is immediately adjacent to the 3’ homology arm; see Supplementary File S2. (C) Fox1afl locus with PCR primer locations indicated. (D) 5’ and 3’ junction PCRs from 2 dpf Venus/mCherry-positive individual embryos injected with indicated RNPs. (E) Number of embryos injected with indicated RNP showing either 5’ and 3’ knock-in. One-way ANOVA, p=0.0114; Holm-Sidak multiple comparison test, p-values indicated. (F) Number of embryos injected with indicated RNP(s) with both 5’ and 3’ knock-in. Tukey’s multiple comparison test, p-values shown (E,F) Each point is a replicate from 10 normal embryos.
We used Cas12a as a nuclease since it can stimulate higher HDR rates than Cas9 and is amenable to targeting AT-rich non-coding sequence due to its 5’-TTTN PAM27,28. To compare effects of DSB location, we injected individual Cas12a RNPs targeting either loxP insertion site, or co-injected both RNPs, along with targeting plasmid and I-SceI. We assessed knock-in by PCR across 5’ and 3’ junctions in 10 individual cryaa:venus-positive embryos at 72 hpf in three replicate injections (Figure 3C; reverse primer for 5’ product is anchored in the loxP site and does not detect endogenous foxc1a). Embryos injected with plasmid and I-SceI, but without RNP, do not exhibit knock-in but showed amplification with control primers to detect the endogenous locus (Figure 3D). Injections with only 3’ RNP showed robust knock-in at the 3’ junction, but not on the 5’ side (Figure 3D, E). Conversely, 5’ RNP induced robust 5’ knock-in as well as 3’ knock-in, though amplification appeared inconsistent and less robust on the 3’ side (Figure 3D, E). Simultaneous injection of both RNPs further increased 3’ knock-in while maintaining high rates of 5’ knock-in (Figure 3D, E). Accordingly, the proportion of individuals exhibiting both 5’ and 3’ knock-in was significantly higher when comparing dual targeting to 3’ RNP alone (Figure 3D, F). We also observed a benefit from targeting only the 5’ insertion site, but this was less consistent (Figure 3F). Together, these results suggest that simultaneous dual CRISPR targeting is useful to stimulate complete knock-in of a floxed targeting cassette.
Transgenic counter-screening can identify germline carriers of off-target insertions.
Our foxc1a targeting construct included a cardiomyocyte-specific myosin, light chain 7 (myl7) promoter:mcherry cassette outside of the homology arms to detect off-target insertions17,29 (Figure 3B, Supplementary File S2). A plasmid bearing this cassette was previously described, but never applied17. To test the utility of this marker, we repeated dual RNP injections and separated 2 dpf embryos by venus and mcherry expression, followed by PCR across homology junctions (Figure 3C). One-third of injected embryos co-expressed venus and mcherry (19 out of 52) or only venus (19 out of 52). A smaller proportion exhibited only mcherry or no expression (7 out of 52 for each class). Surprisingly, only embryos co-expressing both transgenes consistently showed evidence of complete knock-in (Figure 4A), suggesting that myl7:mcherry does not distinguish on- and off-target integration at this stage.
Figure 4. Generation of foxc1afl using transgenic counter-screening.
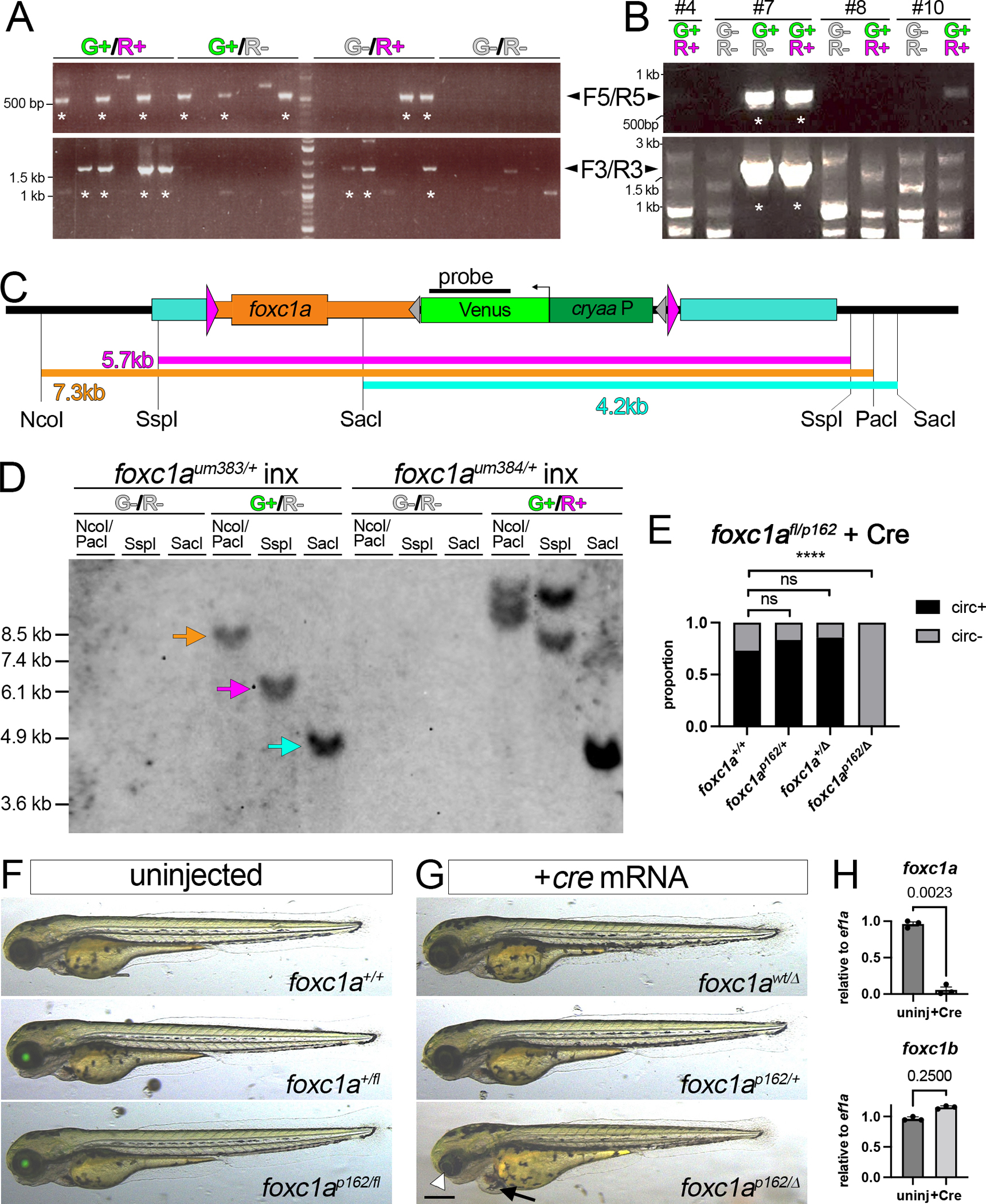
(A, B) PCR of 3 dpf embryos scored for Venus (G+) and mCherry (R+) expression with indicated primer pairs (F5/R5 or F3/R3, see Figure 3C). Asterisk denotes lanes with expected size. (A) Individual embryos injected with Cas12a RNPs, targeting construct, and I-SceI. (B) Pooled F1 embryos from indicated founder. (C) Foxc1afl restriction map. (D) Southern analysis of F2 embryos of indicated genotype screened for Venus (“G”) or mCherry (“R”) expression. Blot hybridized with DIG-labeled venus probe detected by chemiluminescence. (E) Proportion of 3 dpf larvae of indicated genotype with or without circulation. Total embryos: foxc1a+/+ - 26, foxc1p162/+ - 12, foxc1a+/Δ - 14, foxc1ap162/Δ - 16. Fisher’s exact test, ****p<0.0001. (F, G) Overlay of transmitted light and green fluorescence at 3 dpf. Larvae from cross between foxc1a+/fl and foxc1a+/p162 carriers. (F) Uninjected embryos of indicated genotype with normal morphology. (G) Embryos injected with cre mRNA. Arrowhead denotes small eye and arrow indicates cardiac edema (bottom). (F, G) Scale bar is 250 μm. (H) Quantitative RT-PCR for foxc1a and foxc1b in foxc1afl/fl embryos. Embryos left uninjected or injected with zfcre mRNA at 1-cell stage. Paired t-test, p values are shown.
We repeated injections, grew embryos that co-expressed cryaa:venus and myl7:mcherry to adulthood and identified founders. From 45 P0 fish, 7 gave progeny that were only double-positive for cryaa:venus and myl7:mcherry, with all but one being negative for 5’ and 3’ junction PCRs (for examples see embryo pools #4, 8, and 10 in Figure 4B, Supplementary Table S3). Two founders transmitted a mix of cryaa:venus;myl7:mcherry double-positive and cryaa:venus single-positive sibling embryos, one of which gave embryos with evidence of complete knock-in (Figure 4B, founder 7; Supplementary Table 3). We separately grew cryaa:venus-positive;myl7:mcherry-negative (foxc1aum383) and cryaa:venus;myl7:mcherry double-positive (foxc1aum384) embryos from founder 7. We then crossed individual foxc1a+/um383 or foxc1a+/um384 carriers and separated progeny based on transgene expression, followed by Southern blot analysis. In cryaa:venus-positive embryos from foxc1aum383 parents, we observed fragment sizes consistent with precise knock-in at the target, while genomic DNA from cryaa:venus-negative embryos did not hybridize to the Venus probe (Figure 4C, D; foxc1aum383 is hereafter referred to as foxc1afl). By contrast, we observed multiple fragments, none of the expected size, from double-positive cryaa:venus;myl7:mcherry foxc1aum384 embryos (Figure 4D), indicating multiple off-target insertions. These observations confirm that multiple germline insertions independently occurred in P0 founder 7 and demonstrate that transgenic counter-screening can distinguish F1 progeny bearing on- or off-target insertions.
We next crossed foxc1afl carriers to heterozygous foxc1ap162 adults, which bear a nonsense mutation in the Forkhead domain30. All resulting embryos appeared normal regardless of genotype indicating that the cryaa:venus cassette does not affect foxc1a function (Figure 4F). All sibling embryos injected with cre mRNA lack cryaa:venus expression, while foxc1ap162/fl embryos showed deletion of the floxed allele (foxc1ap162/Δ) and exhibited small eyes and cardiac edema (Figure 4E, G), as expected31. We also did not observe circulation in any foxc1ap162/Δ embryos compared to relatively normal heterozygous and homozygous wild type siblings (Figure 4E), consistent with our previous findings18. Occasional circulatory defects in wild type embryos suggests mild toxicity from Cre itself in these experiments (Figure 4E). Foxc1afl/fl homozygous adults were normal and fertile, while foxc1afl/fl embryos injected with zfcre mRNA exhibited loss of aortic arch and trunk circulation (Supplementary Movies S3, S4). Accordingly, we observe loss of foxc1a expression following injection of foxc1afl/fl larvae with zfcre mRNA and expression of foxc1b is unchanged (Figure 4H). Together, these observations demonstrate that foxc1afl behaves as a conditional loss-of-function allele following Cre-mediated deletion.
Generation of additional floxed alleles.
To further test our knock-in approach, we generated floxed alleles for rasa1a and ruvbl1, the latter of which was targeted by co-authors (H. M. Y., C. E. B., C. G. B.) in an independent lab. Ruvbl1 (also known as pontin) encodes an ATPase required for chromatin modification and cilia assembly32,33. Since the conditional Ruvbl1tm1a allele in mouse is a floxed exon 3, we generated a targeting construct to insert loxP sites flanking zebrafish ruvbl1 exon 3 (Figure 5A, B). In this case, we used a single Cas12a RNP targeting the 5’ loxP insertion site, based on our findings above (see Figure 3D–F). Targeting strategy and follow-up analyses were otherwise the same. From injected embryos, we screened eleven P0 adults via group in-crosses from which we observed embryos expressing cryaa:venus and myl7:mcherry or cryaa:venus only with evidence of both 5’ and 3’ insertions (Figure 5D). Subsequent individual outcrosses identified three founders that gave progeny expressing both cryaa:venus and myl7:mcherry. One P0 adult gave a mix of progeny expressing both transgenes or only cryaa:venus, the latter of which were positive for PCR across both 5’ and 3’ junctions and were grown to adulthood as ruvbl1chb10 (Figure 5C, D; hereafter referred to as ruvbl1fl). We verified precise insertion and lack of off-target integration by Southern analysis of F2 cryaa:venus-positive progeny (Figure 5C, E). Homozygous ruvbl1fl/fl larvae appeared normal (Figure 5F, G) and adults were viable and fertile. By contrast, ruvbl1fl/fl larvae injected with zfcre mRNA exhibit trunk curvature and cardiac edema (Figure 5F, G) as previously observed in ruvbl1hi1055b mutant larvae33, and loss of ruvbl1 transcript containing exon 3 and an overall reduction of transcript (Figure 5H). These findings indicate that ruvbl1fl functions as a conditional floxed allele.
Figure 5. A floxed ruvbl1 allele (ruvbl1fl).
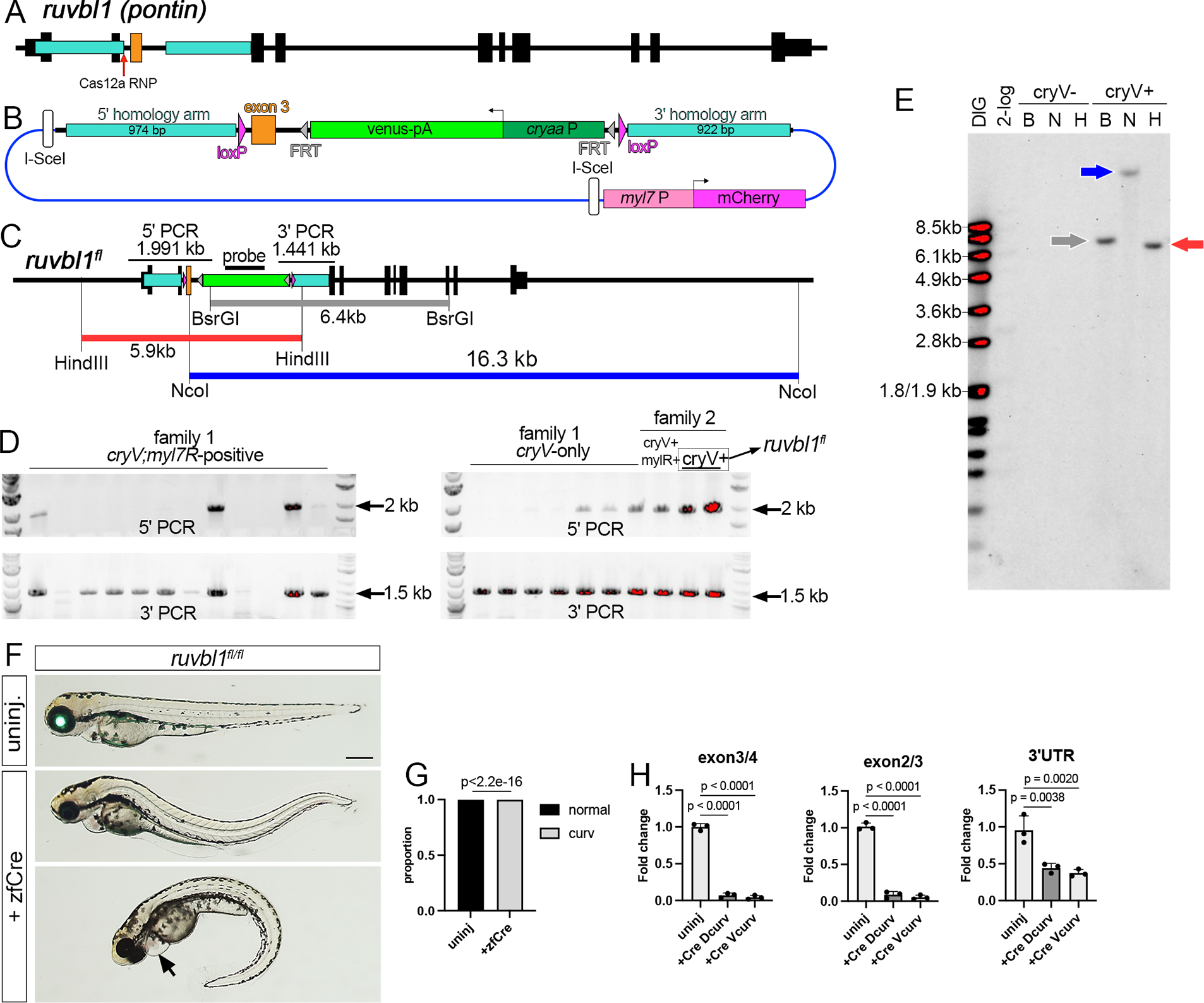
(A) Ruvbl1 locus. (B) Ruvbl1fl targeting construct. (C) Ruvbl1fl restriction map. 5’ and 3’ PCR screening fragments indicated. (D) PCR of individual F1 embryos from group family P0 incrosses. cryV denotes Venus expression, myl7R is mCherry expression. Family 2 founder transmitting cryV-only embryos (boxed) was identified by individual outcross. (E) Southern analysis of F2 cryV-positive and -negative siblings from outcross of F1 ruvbl1fl/+ carriers (B – BsrGI, N – NcoI, H – HindIII). Blot hybridized with DIG-labeled venus probe detected using chemiluminescence. (F) Ruvbl1fl/fl larvae at 3 dpf; Lateral views, anterior to the left, dorsal is up. Overlays of epifluorescent and transmitted light images. Top panel, uninjected embryo expressing cryaa:venus. Bottom panels, embryos injected with zfcre mRNA showing dorsal (middle) or ventral (bottom) trunk curvature, cardiac edema (indicated by arrow), and loss of cryaa:venus. Scale bar is 250 μm. (G) Trunk curvature penetrance in uninjected (n=113) or zfcre-injected (n=379) ruvbl1fl/fl larvae. Fishers exact test. (H) qRTPCR across indicated exon junctions or in 3’ UTR. Dunnett’s multiple comparison test, p-values shown.
RASA1 is a GTPase activating protein (GAP) that stimulates conversion of Ras-bound GTP to GDP, leading to Ras inactivation34. In humans, loss-of-function RASA1 mutations lead to capillary and arteriovenous malformations35, while mouse knockouts show embryonic vascular and lymphatic defects36,37. The zebrafish genome encodes two RASA1 orthologs, rasa1a and rasa1b, for which mutants have not been described. Therefore, we first generated deletion alleles in each gene. Zebrafish larvae bearing a homozygous 5-bp deletion in the Rasa1a GAP domain leading to a frameshift (rasa1aum255) show a low penetrance of ocular, gut and cardiac edema, and loss of circulation by 4 dpf. Phenotypic penetrance is significantly increased on a rasa1bum290 homozygous mutant background, with all double homozygous embryos displaying these defects (Figure S4A–C), while larvae only homozygous for rasa1bum290 are normal and viable. Based on these phenotypes, we inserted loxP sites flanking rasa1a exons 20 and 21, which encode part of the GAP domain and, when deleted, would lead to a frameshift similar to rasa1aum255 (Figures 6A, B, and S4A; Supplementary File 4). Following injection, we identified 4 out of 64 adults that transmitted cryaa:Venus, but not myl7:mcherry, one of which showed PCR amplification across both 5’ and 3’ junctions (Figure S4D, E; founder 5, referred to as rasa1aum414). Southern analysis of rasa1aum414 progeny supported knock-in at the target, although a MscI fragment appeared larger than expected (Figure 6D). PCR across the 5’ junction in rasa1aum414 embryos using conditions that favored long amplification yielded a 4.5 kb fragment that included the polyadenylation signal from the myl7:mcherry cassette, ampicillin resistance and lacZ genes, and intact I-SceI sites (Figure S4F, Supplementary File 5). This suggests that rasa1aum414 arose from HDR mediated repair on the 3’ end and non-homologous end joining-mediated insertion of the 5’ homology arm and vector sequence. The initial positive 5’ PCR (see Figure S4D) likely resulted from spurious internal priming between tandem duplicate 5’ homology arm sequences, similar to previous observations38,39. Despite this insertion, rasa1afl;rasa1bum290 double homozygous individuals exhibited normal embryonic development (Figure 6E, G) and were viable and fertile as adults, while exhibiting fully penetrant edema and loss of circulation following injection of mRNA encoding zfCre at 1-cell stage (Figure 6E–G). Quantitative RT-PCR showed complete Cre-dependent loss of rasa1a mRNA containing exon 21, with reduction of total rasa1a, but not rasa1b transcript in rasa1afl homozygous mutants (Figure 6H). These observations demonstrate that the exogenous vector sequence inserted upstream of exon 20 does not affect rasa1a function and that rasa1afl acts as conditional deletion allele.
Figure 6. A floxed rasa1a allele (rasa1afl).
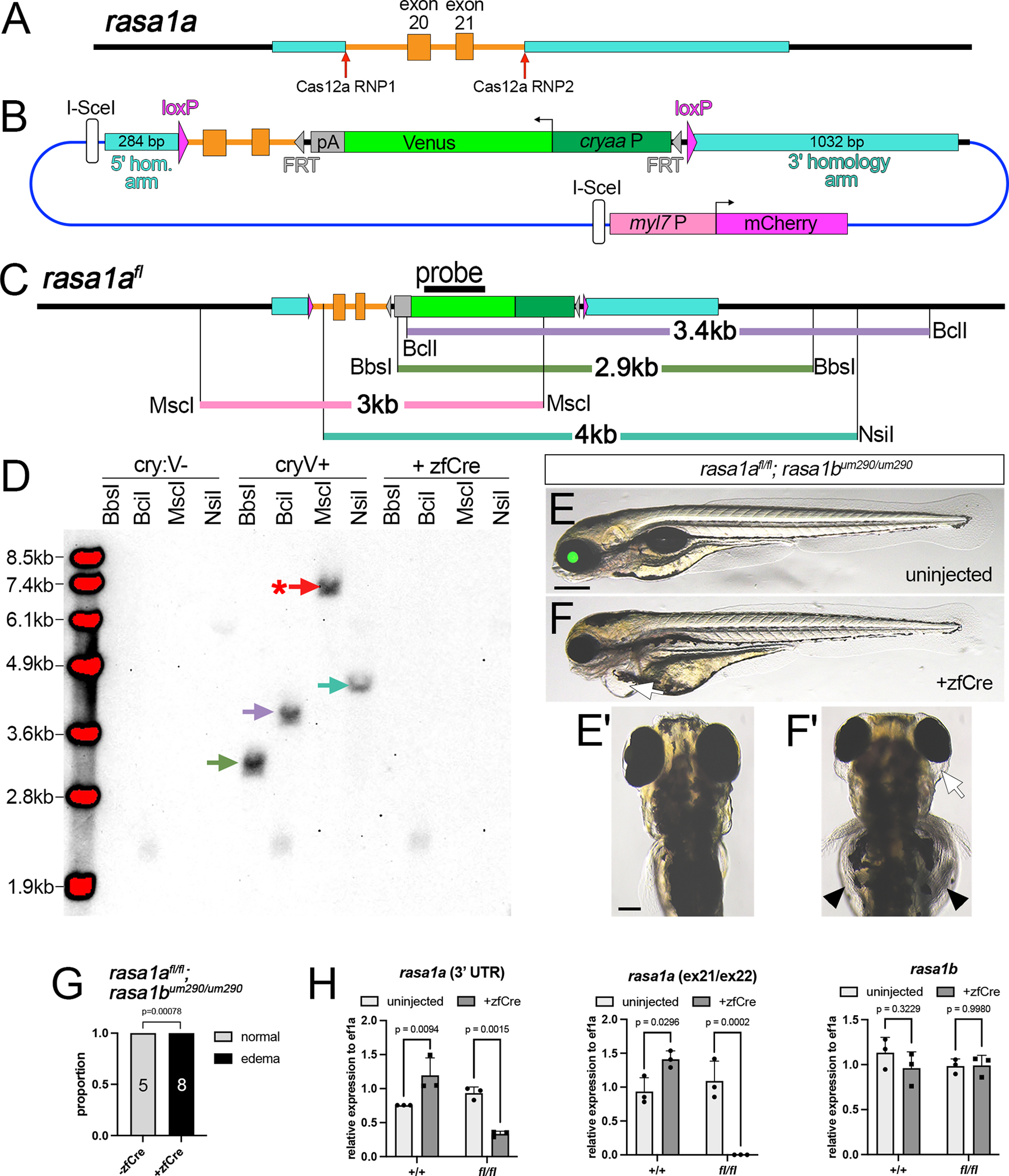
(A) Rasa1a exons 20 and 21. (B) Rasa1fl targeting construct. (C) Rasa1afl restriction map. (D) Southern analysis of F1 cryaa:venus-positive and -negative siblings, or those injected with zfCre mRNA. Blot hybridized with DIG-labeled venus probe detected using chemiluminescence. Asterisk denotes larger fragment than expected. (E, F) rasa1afl/fl;rasa1bum290/um290 larvae at 4 dpf. Scale bar, 250 μm. (E, E’) Uninjected larva; lateral and dorsal views, respectively. (F) Lateral view of larva injected with zfcre mRNA showing cardiac edema (arrow). (F’) Dorsal view of larva in (F) showing ocular (arrow) and gut edema (arrowhead). (E’, F’) Scale bar, 100 μm. (G) Penetrance of edema in 4 dpf larvae of indicated genotype. Fisher’s exact test, p-values shown. (H) qRT-PCR for indicated transcript in uninjected or zfcre-injected embryos of indicated genotype. Sidak’s multiple comparison test, p-values shown.
Endothelial cell-specific gata2a knockout causes defective lymphatic valve development
To demonstrate the application of a floxed allele for cell and stage-specific knockout in zebrafish, we used gata2afl to determine the endothelial autonomous requirement for gata2a. For this purpose, we developed a transgenic line where CreERT2 was driven by gata2aECE, an intronic gata2a enhancer element that expresses in endothelial cells18 (Figure 7A). In this case, we incorporated cryaa:mcherry as a marker to identify transgenic carriers. We identified a founder from which progeny embryos displayed endothelial-specific recombination (Tg(gata2aECE:CreERT2;cryaa:mcherry)um337; referred to as gata2aECE:iCre) and bred this line out to obtain a single copy insertion (Figure 7B). To confirm inducible endothelial-specific Cre activity, we crossed gata2aECE:iCre to fish bearing Tg(ubb:loxP-cerulean-loxP;h2b-cherry)jh63, in which cells expressing Cre will switch to expressing a nuclear-localized form of mCherry40 (referred to as ubb:SwitchRed). Gata2aECE:iCre;ubb:SwitchRed embryos show endothelial-specific H2B-mCherry expression at 6 dpf when exposed to 5 μM 4-hydroxytamoxifen (4OHT) from 1 to 3 dpf (Figure 7C, D). We observed H2B-mCherry in endothelial cells lining the facial lymphatic vessels, as well as those in aortic arch and brain blood vessels (Figure 7C). In trunk vessels, H2B-mCherry was restricted to endothelial cells within the posterior cardinal vein and the thoracic duct, with mosaic expression in intersegmental vessels and very few cells apparent in the dorsal aorta (Figure 7D).
Figure 7. Endothelial gata2a is required for lymphatic valve function and formation.
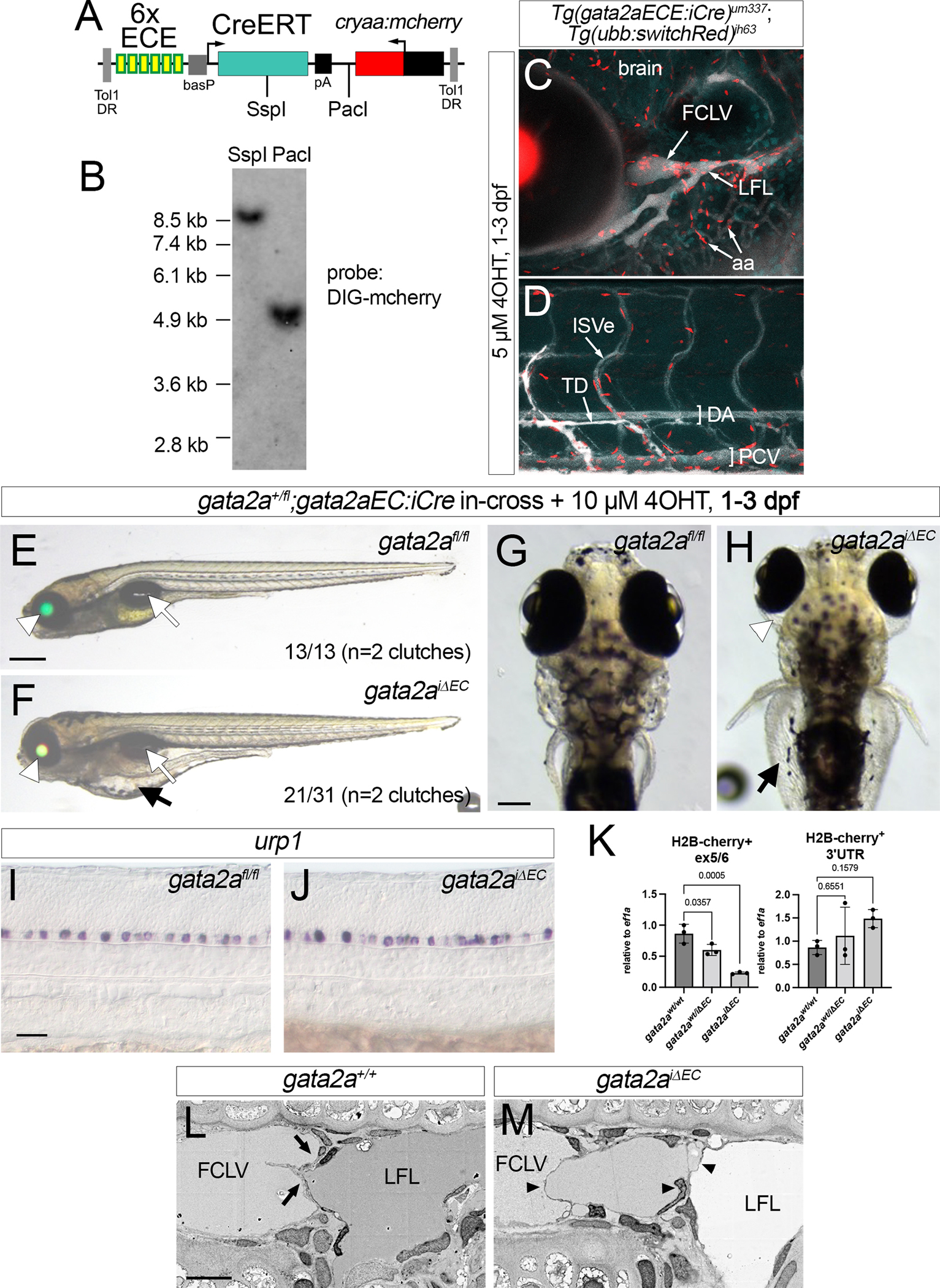
(A) Restriction map of Tol1 gata2aECE:iCre transgene. (B) Southern analysis of Tg(gata2aECE:iCre)um337 embryos. Blot hybridized with a DIG-labeled mcherry probe detected by chemiluminescence. (C, D) Confocal micrograph overlays of Tg(gata2aECE:iCre)um337; (ubb:switchRed)jh63 7 dpf larvae showing expression of H2B-Cherry (red), cerulean (blue) and Qtracker705 introduced via lymphangiography (white). Lateral views, anterior to the left, dorsal is up. (C) Cranial vessels; (D) trunk vessels. FCLV – facial collecting lymphatic vessel, LFL – lateral facial lymphatic vessel, aa – aortic arch blood vessels, ISVe – intersomitic vein, TD – thoracic duct, DA – dorsal aorta, PCV – posterior cardinal vein. (E, F) 6 dpf and (G, H) 7 dpf larvae from indicated cross with 10μM 4OHT treatment from 1 dpf to 3 dpf. (G, H) Scale bar, 100 μm. (E, G) gata2afl/fl larvae without iCre. (F, H) gata2afl/fl; Tg(gata2aEC:iCre)um337 larvae, referred to as gata2aiΔEC. (E, F) Transmitted light, green and red fluorescence overlay. White arrowhead denotes lens, white arrow inflated swim bladder. Scale bar, 250 μm. (F, H) Black arrows indicate gut edema. (H) White arrowhead indicates ocular edema. (I, J) Urp1 expression by whole mount in situ hybridization in 24 hpf embryos of indicated genotype treated with 5 μM 4OHT from 6 hpf. Scale bar, 50 μm. (K) Quantitative RT-PCR for gata2a exon 5/6 or 3’ UTR from H2B-mcherry-positive cells from gata2aiΔEC;ubi:switchRed at 5 dpf and 6 dpf treated with 5 μM 4OHT from 6 hpf to 4 dpf. Exon 5/6, analysis of variance, p<0.005; for 3’, not significant. Pairwise comparisons performed using Dunnett’s multiple comparison test, p values indicated. (L, M) Scanning electron micrographs of lymphatic valves in 7 dpf larvae of indicated genotype following 5 μM 4OHT from 1 dpf to 3 dpf. Lateral views, dorsal is up, anterior to the left. Normal bicuspid valve leaflets indicated (arrows). Multiple abnormal leaflets (black arrowheads) appear in gata2aiΔEC larvae. FCLV – facial collecting lymphatic vessel. LFL – lateral facial lymphatic vessel. Scale bar, 5 μm.
We incrossed gata2a+/fl;gata2aECE:iCre adults and treated resulting progeny with DMSO or 4OHT from 1 to 3 dpf. Homozygous gata2afl/fl larvae without iCre, identified by cryaa:egfp but not cryaa:mcherry and confirmed with genotyping, exhibited inflated swim bladders and normal circulation (Figure 7E, G). By contrast, 4OHT-treated gata2afl/fl;gata2aECE:iCre larvae (referred to hereafter as gata2aiΔEC) displayed edema around the eyes and gut at a penetrance similar to that of whole body gata2a knockout larvae18 (Figure 7F, H). In these experiments, we initially identified gata2aiΔEC larvae by lens expression of mCherry and Egfp, followed by genotyping for gata2afl/fl. In contrast to cre mRNA injection (see Figure 2F), gata2aiΔEC larvae retain cryaa:egfp due to the specificity of CreERT2 expression (Figure 7F). Despite lymphedema, gata2aiΔEC larvae were otherwise normal, with inflated swim bladders and active trunk circulation (Figure 7E, F), unlike gata2aum27 mutants18. Furthermore, gata2aiΔEC embryos treated from 6 hpf to 24 hpf with 4OHT exhibited normal urp1 expression in ventral KA neurons (Figure 7I, J), unlike gata2afl/fl embryos injected with zfcre mRNA (Figure 2I). Quantification of gata2a transcript in H2B-mcherry-positive cells isolated by fluorescence activated cell sorting (FACS) from gata2aiΔEC larvae bearing ubb:SwitchRed (i.e. cells having exhibited Cre-mediated recombination) showed significant reduction in transcript containing exon 5, but not the 3’ UTR, compared to wild type or gata2a+/iΔEC larvae (Figure 7K). In contrast to gata2afl/fl embryos injected with zfcre mRNA, wild type transcript was still detectable in gata2aiΔEC larvae at low levels suggesting that recombination was robust, but incomplete when using gata2aECE:iCre (compare with Figure 2K, L).
Edema in gata2aum27 mutant embryos coincides with defects in lymphatic valve formation18. Therefore, we performed electron microscopy to investigate lymphatic valve morphology. In wild type larvae at 7 dpf, we observed flattened bicuspid valve leaflets separating the facial collecting lymphatic vessel (FCLV) and lateral facial lymphatic vessel (LFL), as previously described18 (Figure 7L). By contrast, lymphatic valve leaflets appeared disorganized in gata2aiΔEC larvae that had been treated with 4OHT from 1 to 3 dpf. We observed multiple leaflets spanning the lumen between the FCLV and LFL in a disorganized manner (Figure 7M), similar to what we have previously observed in gata2aum27 mutant larvae18.
DISCUSSION
In this work we describe our efforts to generate and apply floxed deletion alleles in zebrafish. Our initial application of a previous knock-in protocol for this purpose led us to identify several important caveats and points for improvement. We subsequently leveraged our experience to develop a robust pipeline that we applied to establish floxed alleles for three loci, each in a single generation. Importantly, one of these alleles was generated in an independent lab, underscoring the utility of our approach within the zebrafish community. Together, our efforts here provide a blueprint for more widespread generation and application of floxed alleles in zebrafish.
The starting point for generating a floxed allele is target exon considerations. For gata2a and ruvbl1 we implemented targeting strategies based on mouse alleles. Zebrafish and mouse genes often have conserved organization and functional domains making this a reliable approach. Indeed, both gata2afl and ruvbl1fl behave as expected for conditional deletion alleles. Alternatively, genotype and phenotype characteristics of existing zebrafish mutants should be considered, as we did for rasa1a. Once a target is identified, sequencing the region in wild type adults is essential to match homologous arm sequences with the haplotype of injected embryos17. This also ensures proper design of PCR primers and CRISPR targets. While we did not directly test the effects of using non-homologous sequence, there is considerable variability between and within wild type zebrafish strains41,42 and minor sequence variants in mouse significantly reduce knock-in rates43.
An important targeting consideration in generating floxed alleles is location of the CRISPR-induced DSB relative to loxP sites and homology arms. At the gata2a locus a DSB at the 3’ loxP insertion site only achieved partial knock-in, likely due to sufficient homologous sequence for HDR downstream of the 5’ loxP site within the targeting construct. Moving the DSB to the 5’ loxP slightly improved knock-in in P0 individuals at the foxc1a locus and we used this approach to successfully establish the ruvbl1fl allele. Simultaneous dual CRISPR targeting was even more robust for foxc1a, where there was a much larger span between the loxP sites. Therefore, for shorter targets (e.g. a single exon less than 500 bp), we suggest targeting a DSB to the loxP insertion site (the 5’ loxP target in our designs) opposite of that adjacent to the cryaa marker. While it is still likely that partial knock-in events will occur when targeting only the 5’ site, screening for cryaa:venus expression should reduce the occurrence of these types of insertions. To introduce DSBs, we employed Cas12a instead of Cas9. Unlike Cas9, which yields blunt ends, Cas12a leaves overhangs, while its PAM sequence lies opposite its cleavage site and the spacer can be up to 24 nucleotides44. Thus, initial indels created by Cas12a-stimulated cleavage can often be re-targeted. Together, these characteristics likely contribute to the ability of Cas12a to stimulate higher HDR rates than Cas9 in both zebrafish and mammalian cells27,28. The TTTN PAM used by Cas12a also favors its application for targeting AT-rich non-coding sequences used for intronic loxP insertion sites in floxed alleles.
An issue with targeted knock-in in multiple models is that non-homologous endjoining is favored over HDR as a repair event, leading to robust random insertion of the targeting construct. Indeed, the first protocols for generating targeted knockouts in mouse ES cells incorporated a counter selection approach to reduce off-target insertions12. Accordingly, random integration occurred in most P0 founders across all targeted loci in our study. To reliably identify these events and confirm targeted knock-in, we successfully employed a combination of transgenic screening and molecular approaches. Transgenic counter-screening allowed us to clearly distinguish F1 individuals with on- and off-target insertions. However, this approach was not effective in injected embryos, where predominance of random insertions expressing both cryaa:venus and myl7:mcherry over rare HDR-mediated events likely masks identification of the latter. We also demonstrate that molecular validation by Southern analysis is essential to assess confirm precise integration and further rule out random integration. While considered a dated technical approach, it provides the simplest and most definitive means to identify off-target insertions in an unbiased manner. Our results with rasa1afl further underscore the importance for Southern analysis as a final molecular validation step since initial PCR screening of F1 progeny proved to be spurious. Several previous studies have demonstrated similar cases where false-positive PCR can confound identification of bona fide knock-in founders38,39, making orthogonal validation approaches essential. Indeed, given the spectrum of different insertion events that we observed in our work, we strongly recommend a combination of PCR analysis coupled with transgenic counter screening and subsequent validation by Southern analysis to definitively validate endogenously floxed alleles.
Our results using gata2afl/fl demonstrates the utility of floxed alleles for genetic analysis at zebrafish larval stages. We previously relied on the gata2aum27 allele, which causes numerous embryonic defects that can confound analysis at larval stages18,24, a common issue with most zebrafish mutants. Notably, gata2aum27 mutant embryos have circulatory defects that could indirectly affect lymphatic valve development, which is sensitive to mechanosensory input in other models45. By contrast, gata2aiΔEC larvae displayed normal circulation and neuronal specification, yet defects in lymphatic valve function and morphogenesis with a severity and penetrance similar to gata2aum27 mutants18. This observation is consistent with the known endothelial autonomous role for mouse Gata2 in lymphatic valve formation46. Of note, normal circulatory function in gata2aiΔEC embryos was somewhat surprising given the severity of circulation defects in gata2aum27 embryos and endothelial expression of gata2a in embryos18,24. It is possible that gata2a acts at a stage prior to the 4OHT treatment used in our experiments. Alternatively, gata2a acts in endothelial subtypes not targeted by gata2aECE:iCre. For example, gata2aECE:iCre did not induce recombination in arterial endothelial cells, raising the possibility that only this cell type in the trunk blood vessels is affected by loss of gata2a. The possibility also remains that circulatory defects in gata2aum27 embryos are not endothelial autonomous. In any case, the availability of the gata2afl allele, together with other Cre lines, now allows us to better investigate the cellular nature of the early circulatory defects.
The development of efficient CRISPR nucleases now enables zebrafish researchers to engineer knock-in alleles. Initial efforts in this regard introduced short epitope tags or reporter transgenes using a single DSB to insert exogenous sequence6,17,47. More recent studies have used a single DSB with a plasmid targeting vector to insert a conditional splice trap cassette into an intron via HDR48–50. These alleles also incorporate reporter transgenes to observe endogenous expression and to assess Cre-mediated recombination. However, splice traps may not always lead to loss-of-function and reliable reporter expression is limited to strongly expressed genes. Moreover, splice traps will not work for small genes comprising only one or two exons. In general, a floxed deletion allele is more likely to be definitively loss-of-function. Although these alleles can be more challenging to make, the improvements we have identified should aid researchers in this regard. The mouse community currently benefits from thousands of floxed alleles and hundreds of Cre lines allowing genetic analysis in any setting. These tools have enabled the mouse to become the preeminent genetic model for studying vertebrate development. The zebrafish is currently constrained by the lack of such tools. We hope that our efforts here can serve as an initial framework to establish more floxed lines that allow conditional gene manipulation in zebrafish.
Limitations of the study
Our protocol for targeted gene replacement in a single generation remains labor- and time-intensive and rates of precise knock-in can still be variable. Our approach also requires techniques that are no longer commonplace in most labs and considered dated (e.g. Southern analysis), but nonetheless proved to be essential. Further advances can likely be made to improve knock-in rates, including optimization of template topology51 and other parameters that govern HDR. Despite these limitations, our work underscores that it is feasible to routinely generate floxed zebrafish lines and that careful validation is essential to successful implementation of our approach.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Nathan Lawson (nathan.lawson@umassmed.edu)
Materials availability
Plasmids generated in this study are available by request from the lead contact. Transgenic and knock-in zebrafish lines are available by request from the lead contact.
Data and code availability
All original data and images are available from the Lead Contact upon request. This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animals
All zebrafish used in this study were handled according to approved University of Massachusetts Chan Medical School and Boston Children’s Hospital Institutional Animal Care and Use Committee protocols. Establishment and characterization of gata2aum27, foxc1ap162, and Tg(ubi:CtoH2b-cherry)jh63 lines has been described elsewhere24,30,40. Generation of all other alleles is described below. All adult zebrafish were maintained at 28°C in a recirculating system. All analysis was performed on embryos or larvae at indicated stages. All stages under investigation in this study are prior to specification of sex in zebrafish, therefore this was not considered as a variable.
METHOD DETAILS
Target locus sequencing
We sequenced target loci in genomic DNA isolated from finclips from 12 individual TL wild type fish (6 males and 6 female). For gata2a, we amplified by PCR using superPfx (Cowin Biotech) using primers: 5’-GGGGAATTGCGCTGTACGCCTATAAA-3’ and 5’-AATATGGACCTTTGAGGTACCACCCC-3’ to obtain genomic sequence flanking exon 5 (GRCz11, chr11:3,849,652–3,852,157). For foxc1a, we used primers: 5’-CTTAACCTCGCTGTATGGCTGTAG-3’ and 5’-CCCACAAGGCAGTGGCTAGCAGAACTCATG-3’ to amplify a spanning genomic sequence (chr2:686,529–689,296) of upstream including 5’ homologous arm, promoter, coding exon and 3’UTR, and 5’- CGGGAATAACAGCTGTCAAATGTC-3’ and 5’- AACCGCTGTAATCACTACCAACCGCTCATG-3’ to amplify 3’ homologous sequence (chr2:685,341–687,455). Amplicons were cloned into pBluescript linearized with SmaI. For rasa1a, we used 5’-GCGTGTACGTCCATCTTGCGAATGTAC-3’ and 5’- CAACTGCCTCATATCAACTATTCAGGCTG-3’ to obtain genomic sequence flanking exon20 and exon21 (chr5:47,953,001–47,954,255) subcloned into pCR Blunt II TOPO vector using Zero Blunt TOPO PCR cloning kit (ThermoFisher). We sequenced individual clones from each ligation and aligned them to reference genomic sequence (GRCz11). For embryo generation for injection, we used a male and a female bearing the same haplotype for each locus that also matched the homology arms in the respective targeting construct (see Supplementary Files 1–4).
Plasmid construction
For generation of a gata2a targeting vector, 5’ and 3’ homologous arms corresponding to GRCz11 chr11:3,851,035–3,852,157 and chr11:3,849,652–3,850,708, respectively, were PCR-amplified from sequence validated pBluescript clones described above. After digestion of pBS-ISceI using ClaI and SacII in MCSs, these PCR products were combined with a gBlock DNA fragment (IDT) containing gata2a exon 5 genomic sequence (chr11:3,850,717–3,851,034), a 5’ loxP sequence, FRT-flanked cryaa:egfp, and a 3’ loxP site in a HiFi Assembly reaction (NEB) with pBS-ISceI to give pBSIce-gata2aKI (Supplementary File 1).To construct a foxc1a targeting vector, 5’ loxP sequence and point mutations (5’-TTTC-3’ to 5’-CCCT-3’) on the Cas12a RNP1 PAM sequence were added in the upstream of the promoter sequence (chr2:689,007) in pBluescript vector described above. DNA fragments of modified foxc1a locus with 5’ homologous arm (chr2:689,008–689,404) and loxP, and 3’ homologous arm (chr2:685,632–686,723) were separately PCR-amplified from the vectors. After digestion of pKHR8 (Addgene#74625; Hoshijima et al. 2016) using BamHI and SalI in MCSs, the DNA fragments were assembled using NEbuilder HiFi DNA assembly kit to give pKHR8-foxc1aKI (Supplementary File 2). To construct a rasa1a targeting vector, gBlock encoding rasa1a genomic sequence (chr5:47,953,030–47,953,972) encompassing the 5’ homology arm (chr5:47,953,030–47,953,311), exons 20 and 21 with a BamHI-5’ loxP-NotI sequence to disrupt the Cas12a RNP1 target was synthesized (IDT) while 3’ homologous arm (chr5:47,953,973–47,954,995) was PCR-amplified from sequence-validated plasmid described above. After digestion of pKHR8 using BamHI and SalI, the gBlock and PCR amplicons were assembled using NEbuilder HiFi DNA assembly kit to give pKHR8-rasa1aKI (Supplementary File 4). To construct a ruvbl1 targeting vector, the 5’ homology arm (chr6:40,832,688–40,841,106) was PCR-amplified with 5’- GCGGGTCTCTTGTGTTTGATTTTAACG-3’ and 5’- CCTTACCTTGCCTGTTCCAG -3’, the 5’-loxp and mutated CRISPR target site were introduced by NEbuilder HiFi DNA assembly kit with 5’- TATAACTTCGTATAGCATACATTATACGAAGTTATAGTTTCAGAGCTGTTGTTTAAGATAGTATGTATAGTCTGCCCATTTGATTTCTATCCA-3’ and 5’- GACTGGTTTTATTTACACTGCACTCTATATTTATAATGGTCAATATAATATAACTTCGTATAGCATACATTATACGAAGTTATAG-3’. The 3’ homology arm (chr6:40,834,179–40,845,039) was PCR-amplified with 5’- TCAACTTGCCGTGGTAAACAAATATGGG-3’ and 5’- TTGATACGCAGACCTGTTCG-3’. All the fragments were sequentially assembled into pKHR8 vector by NEbuilder HiFi DNA assembly kit. A Cre Gateway middle entry clone was generated by PCR using 5’- GGGGACAAGTTTGTACAAAAAAGCAGGCTTGACCaTGCCCAAGAAGAAGAGG-3’ and 5’-GGGGACCACTTTGTACAAGAAAGCTGGGTAatcgccatcttccagcaggcg-3’, followed by BP cloning into pDONR221 (ThermoFisher), to give pME-CreNS. pCS-CreNS was constructed via a multisite LR reaction using pCSDest2, pME-CreNS and p3E-MCS1 52,53. Alternatively, we used a Cre with zebrafish optimized codon usage (zfCre). For this purpose, we first constructed pME-zfCreERT2 by HiFi assembly using two gBlock DNA fragments (IDT) and pME-MCS1 vector 54 linearized by inverse PCR using primers (5’- GTGAGTCGTATTACATGGTCATAGCTG -3’ and 5’- GGCCGTCGTTTTACAACGTCGTGACTG -3’). To generate pME-zfCre, pME-zfCreERT2 was digested using XhoI to remove zfERT2 and self-ligated. We constructed pCS-zfCre using pCSDest2, pmE-zfCre and p3E-MCS1 in a multisite reaction using LR Clonase II (Thermofisher). For transgenic expression of CreERT2, we first modified the Tol1 backbone vector, pToneDest (Addgene#67969)55, by adding a cryaa:mcherry cassette to give pToneDestCryR. We then performed a multisite reaction using LR Clonase II with pToneDestCryR, p5E-6xgata2aECEbas 18, pENTR/D-CreERT2 (Addgene#27231)56, and p3E-MCS1 to give pTol1-gata2aECE:CreERT2;cryaa:mcherry.
In vitro transcription
To synthesize cre and zfcre mRNA, we performed in vitro transcription using a mMESSAGEmMACHINE SP6 kit (ThermoFisher) with respective pCS vectors linearized with NotI (NEB). To synthesize Cas9 sgRNA, we annealed the following oligonucleotides: 5’-tagGAGAGGGACGAGCGAGGCC, 5’- aaaCGGCCTCGCTCGTCCCTCT and cloned them into pDR274 digested with BsaI to give pDR274-gata2a_3pex5sgRNA1. The resulting plasmids was linearized with HindIII (NEB) and used to synthesize sgRNA using the T7 MEGAscript kit (ThermoFisher). Preparation of DNA templates and generation of crRNAs targeting gata2a (5’ site for loxP insertion RNP spacer; 5’-ACATGACCATGGGGTTGTTCCTT-3’), foxc1a (5’RNP target: 5’-CGATGCGCGCTCCGAGAGAAAGAG-3’; 3’RNP spacer: 5’CTGCGGCACACTTGAACGATCGTC-3’), rasa1a (5’ RNP spacer: 5’- TGAAAATGTATTATTAATGCCTTG -3’; 3’ RNP spacer: 5’- GTAAAATGAAAATAACATATTATA-3’), and ruvbl1 (RNP spacer: 5’- ATGTCCATTTTAATTATACAGTG-3’) with a full length direct-repeat for Lachnospiraceae bacterium Cas12a were performed as described previously 57, using bottom strand oligos (5’-TGACCATGGGGTTGTTCCTTATA ATCTACACTTAGTAG-3’, 5’-CTCTTTCTCTCGGAGCGCGCATCG ATCTACACTTAGTAG-3’ and 5’- GACGATCGTTCAAGTGTGCCGCAG ATCTACACTTAGTAG-3’, 5’CAAGGCATTAATAATACATTTTCA ATCTACACTTAGTAG-3’ and 5’- TATAATATGTTATTTTCATTTTAC ATCTACACTTAGTAG-3’, respectively). Tol1 transposase mRNA was synthesized from linearized pToneTP as described elsewhere 55.
SpCas9 and LbCas12a Protein purification
3xNLS-SpCas9 protein expression and purification was performed as previously described58. LbCas12a-2xNLS protein expression and purification was performed as previously described57.
Line generation
For targeting gata2a, 50pg of pBSIce-gata2aKI, 60pg of sgRNA, 0.8pg of 3xNLS-SpCas9 protein and 1mU of I-SceI (NEB) was injected per embryo at early 1-cell stage. At 2 dpf, embryos were separated based on expression of EGFP in the lens. Genomic DNA from individual embryos was purified by adding 50μL of 0.05M NaOH, incubating at 98°C for 10 minute, and then adding 10μL of 0.5M Tris pH7.5 (HCl). Precise knock-in of 3’ homologous arm was evaluated by PCR using KAPA2G Fast HotStart ReadyMix (KAPA Biosystems) with a primer set (5’-CCACTAGTTCTAGAGCGGC-3’ and 5’-GAACTGTTGGCTACTAACACTAATACTG-3’) across the junction with endogenous sequence using genomic DNAs of individual embryos as templates. Subsequently, injected embryos with EGFP lens expression were grown to adulthood. Founders were screened by individual outcross with wild type adults followed by screening for lens expression of EGFP in progeny. The same primer set as above was used for PCR screening of genomic DNA from pooled embryos with lens EGFP expression. To delete the off-target loxP-flanked exon5 sequence, we injected 10 pg of cre mRNA into embryos from an incross of gata2aum295/um295 parents and grew these to adulthood to give rise to gata2aum329. To insert a loxP site 5’ of the targeted gata2a exon 5 in gata2aum329 fish, we designed oligodeoxynucleotides (ODN) to include loxP sequence flanked by XhoI and EcoRI restriction sites and 30bp of homology (IDT). ODNs were annealed in 50mM NaCl solution to generate double stranded ODN template (dsODN) by incubating at 98oC for 10 minutes followed by 95°C, 90°C, 85°C, 80°C, 75°C, 70°C, 65°C and 63°C each for 20 seconds before 62°C. We co-injected 45pg of dsODN and 57 fmol of LbCas12a-2xNLS RNP (see above) per embryo at early 1-cell stage. Embryos were derived from an incross of homozygous gata2aum329/um329 adults. After injection, integration was evaluated by PCR using 5 PRIME HotMasterTaq DNA polymerase (Quantabio; primers : 5’-TTAGAAGAACCGGTCTTCAGTATG-3’ and 5’CTGTAGGCAAGAAAGCAGAGTATTGTCA-3’) on genomic DNA of pooled or individual embryos followed by digest with XhoI (NEB). Following validation of knock-in, injections were repeated and embryos grown to adulthood. Founders were identified using the same PCR primers and XhoI digestion as above. For generation of foxc1aum383, 40 pg of pKHR8-foxc1aKI, 9.5 fmol of Cas12a 5’ RNP, 9.5 fmol of Cas12a 3’ RNP and 1mU I-SceI were co-injected into wild type TL embryos at early 1-cell stage. Injected embryos were sorted at 2 dpf for expression of Venus in the lens and/or mCherry in the heart. Integration was evaluated by PCR using KAPA2G Fast HotStart ReadyMix on genomic DNA from individual embryos with primer sets that span 5’ and 3’ junctions between homology arms and endogenous sequence (5’ junction primers; 5’- CAGATTTTACCTCTGGGTATTATACGA-3’ and 5’- TACTGGCCACCTCTTATAACTTC-3’, 3’ junction primers; 5’- GATGAAGCTACATGGCTGTAGAACGTCAG-3’ and 5’- AACCGCTGTAATCACTACCAACCGCTCATG-3’). After initial analysis, injections were repeated and only embryos expressing both Venus and mCherry were grown to adulthood. Founders were screened by individual outcross to wild type followed by screening for Venus and mCherry expression at 3 dpf. Embryos were separated based on Venus and mCherry expression, then pooled and used to isolate genomic DNA. We performed PCR across 5’ and 3’ junction sequences as above. For generation of rasa1aum414, 18pg of pKHR8-rasa1aKI, 1.25 fmol of Cas12a RNP1, 1.25 fmol of Cas12a RNP2 and 1mU I-SceI were co-injected into wild type TL embryos at 1-cell stage. Similarly, somatic integration of embryos at 2 dpf after sorting for expression of GFP in lens and mCherry in heart was evaluated by PCR on individual genomic DNAs using 5’ and 3’ junction primer sets (5’ junction primers; 5’-GCGTGTACGTCCATCTTGCGAATGTAC-3’and 5’- TACATTTTCAGCGGCCGCATAACTTC-3’, 3’ junction primers; 5’- GATGAAGCTACATGGCTGTAGAACGTCAG-3’and 5’- CAACTGCCTCATATCAACTATTCAGGCTG-3’) (Figure S7C). For founder screening, embryos were sorted for Venus and mCherry expression at 3 dpf and pooled to isolate genomic DNA for PCR evaluation using 5’ and 3’ junction primer sets described above. After isolating rasa1aum414 allele and western blot assay to confirm single insertion, we noticed a nonspecific insertion of targeting vector sequence upstream of 5’ homologues arm and PCR-amplified the insertion using flanking primers (5’-CAGATCAGACAGAGATCTACATGCATC-3 and 5’-TACATTTTCAGCGGCCGCATAACTTC-3’) followed by whole PCR product sequence (Plasmidsaurus). For generation of ruvbl1fl, 50 pg of pKHR8-ruvbl1, 24 fmol of Cas12a RNP, and 1mU I-SceI were co-injected into wild type TL embryos at early 1-cell stage. The integration was evaluated by PCR with primer sets that span 5’ and 3’ junctions between homology arms and endogenous sequence. (5’ junction primer: 5’-CCCACAGCGTAGATCCTTGT-3’ and 5’-GGGAGGATTGGGAAGACAAT-3’, 3’ junction primer: 5’-GCACTGAATGGCTCAGACAA-3’ and 5’-TGCCTGCACTTCCTTACCTT-3’).
To generate an endothelial-specific inducible Cre line, we co-injected 25 pg of pTol1-gata2aECE:CreERT2 and 50 pg of Tol1 transposase mRNA into wild type 1-cell stage embryos. Only embryos expressing mCherry in the lens were grown to adulthood. Founders were identified by individual outcross and screening of progeny for mCherry lens expression at 3 dpf. Founders were subsequently validated by crossing to an adult bearing Tg(ubi:CtoH2b-cherry)jh63 followed by tamoxifen treatment. A founder was identified that gave robust endothelial recombination and designated as Tg(gata2aECE:CreERT2;cryaa:mcherry)um337. Following successive outcrosses with wild type, Tg(gata2aECE:CreERT2;cryaa:mcherry)um337 a single copy insertion of the transgene was confirmed by Southern analysis (see below).
mRNA Injections
To delete loxP-flanked sequences we injected 10 to 25 pg of cre mRNA, or 6.25pg of zfcre mRNA into incrosses of fish that were heterozygous or homozygous for gata2aum345 or foxc1aum383 as indicated in the text.
Tamoxifen administration
A 10 mM 4-hydroxytamoxifen (4OHT; MilliporeSigma) stock solution was made by dissolving in DMSO, aliquoted, and stored at −80°C. For treatment, embryos were treated for the indicated timepoints by dissolving 10 mM stock to a final of 5 or 10 μM in standard egg water. In cases where larvae were to be subjected to confocal imaging, embryos were also treated with 0.003% phenylthiourea (PTU). Embryos were placed in egg water with fresh 4OHT every 24 hours. After the indicated treatment period, embryos were washed several times and maintained in PTU until 6dpf for imaging. For scanning electron microscopy (SEM), embryos were treated with tamoxifen at 5 μM between 1 dpf and 3 dpf grown until 7 dpf. Larvae were then analyzed by SEM and genotyped as described previously18.
Fluorescent-activating cell sort (FACS)
FACS was performed at room temperature using cells dissociated from embryos carrying the gata2aum345/um345, gata2a+/um345 or gata2a+/+, carrying transgene of the Tg(gata2aECE:CreERT2;cryaa:mcherry)um337 and Tg(ubi:CtoH2b-cherry)jh63 at 5 dpf or 6 dpf with 4OHT administration between 6 hpf and 4 dpf under PTU treatment. Briefly, embryos (n=300 to 500) in 1x TrypLE (ThermoFIsher) at 28.5°C were minced with razor blade, dissociated within 15 minutes using P1000 pipetman (Gilson), and strained using 40μm pore-sized cell strainer (Falcon). Trypsin was inactivated by adding fetal bovine serum and cells rinsed with Leibovitz’s L-15 media without phenol red (ThermoFisher). Cells were spun down at 300g for 5 minutes at room temperature and resuspended with collection solution (L-15 media/10% embryo extract) for FACS. mCherry-positive and CFP-negative cells (150 × 103 to 350 ×103 cells) were sorted by Flow Cytometry Core Lab at UMass Chan Medical School.
PCR and genotyping
Larvae from crosses between gata2a+/um27 and gata2a+/um345, with or without cre mRNA injection, individual embryos were separated by cryaa:egfp expression and subsequently genotyped for gata2aum27 using a KASP assay as previously (Biosearch Technologies)18. Excision of floxed exon 5 was determined by PCR using the following primers: 5’-TTAGAAGAACCGGTCTTCAGTATG-3’ and 5’-GATCGCAGCCAAGCTTAACATTAAA-3’. Gata2aiΔEC larvae and embryos were identified using a KASP probe. For embryos from foxc1ap162/um383 complementation cross, we first sorted embryos cryaa:venus expression at 3 dpf. Individual embryos were then genotyped for foxc1ap162 by KASP assay, as previously18 and for deletion of foxc1a using PCR. Genotyping of foxc1afl was done by KASP assay. . Larvae from incrosses between rasa1a+/um414; rasa1b+/um290, with or without cre mRNA injection, individual embryos were separated by edema and/or cryaa:egfp expression at 4 dpf and subsequently genotyped for rasa1bum290 using KASP assays and deletion of exon 20 and exon 21 by PCR. Genotyping of rasa1aum414 or rasa1aum255 was done by KASP assay. All primer and KASP sequences can be found in Key Resources.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Digoxigenin-AP, Fab fragments | MilliporeSigma | 11093274910 |
| Chemicals, peptides, and recombinant proteins | ||
| 2×Super Pfx MasterMix | Cowin Biotech | CW2965F |
| Gateway™ LR Clonase™ II Enzyme mix | ThermoFisher Scientific | 11791020 |
| KAPA2G Fast HotStart ReadyMix | KAPA Biosystems | 7961235001 |
| AccuStart II DNA polymerase | Quantabio | 95141 |
| PowerSYBR Green PCR Master Mix | ThermoFisher Scientific | 4367659 |
| SuperScript™ III First-Strand Synthesis SuperMix | ThermoFisher Scientific | 18080400 |
| TURBO™ DNase | ThermoFisher Scientific | AM2238 |
| PACE 2x Genotyping Master Mix High ROX 500nM | 3Cr Biosciences | 001-0010 |
| ISceI | New England Biolabs | R0694 |
| ClaI | New England Biolabs | R0197 |
| SacII | New England Biolabs | R0157 |
| XhoI | New England Biolabs | R0146 |
| NotI-HF | New England Biolabs | R3189 |
| HindIII-HF | New England Biolabs | R3104 |
| EcoRI-HF | New England Biolabs | R3101 |
| ScaI-HF | New England Biolabs | R3122 |
| BstEII-HF | New England Biolabs | R3162 |
| NcoI-HF | New England Biolabs | R3193 |
| PacI | New England Biolabs | R0547 |
| SspI-HF | New England Biolabs | R0132 |
| BbsI-HF | New England Biolabs | R3539 |
| BclI-HF | New England Biolabs | R3160 |
| MscI | New England Biolabs | R0534 |
| NsiI-HF | New England Biolabs | R3127 |
| BsrGI-HF | New England Biolabs | R3575 |
| BamHI-HF | New England Biolabs | R3136 |
| SalI-HF | New England Biolabs | R3138 |
| (Z)-4-Hydroxytamoxifen | MilliporeSigma | H7904 |
| DIG-labeled DNA marker VII | MilliporeSigma | 11669940910 |
| DIG Easy Hyb™ Granules | MilliporeSigma | 11796895001 |
| DIG DNA Labeling Mix | MilliporeSigma | 11277065910 |
| Casein Blocking Buffer 10x | MilliporeSigma | B6429 |
| CDP-Star®, ready-to-use | MilliporeSigma | 12041677001 |
| Qtracker 705 vascular labels | ThermoFisher Scientific | Q21061MP |
| TRIzol™ Reagent | ThermoFisher Scientific | 15596026 |
| SpCas9 | Wolfe Lab | N/A |
| LbCas12a-2xNLS | Wolfe Lab | N/A |
| Critical commercial assays | ||
| pGEM®-T Vector Systems | Promega | A3600 |
| NEBuilder® HiFi DNA Assembly Cloning Kit | New England Biolabs | E5520S |
| mMESSAGE mMACHINE™ SP6 Transcription Kit | ThermoFisher Scientific | AM1340 |
| MEGAscript™ T7 Transcription Kit | ThermoFisher Scientific | AM1334 |
| Blood & Cell culture DNA Midi kit | QIAGEN | 13343 |
| NucleoSpin Gel and PCR Clean-up kit | Takara Bio | 740609 |
| MultiSite Gateway Three Fragment Vector Construction kit | ThermoFisher Scientific | 12537-023 |
| AllPrep DNA/RNA Micro Kit | QIAGEN | 80284 |
| CloneJET PCR Cloning Kit | ThermoFisher Scientific | K1231 |
| Zero Blunt™ TOPO™ PCR Cloning Kit | ThermoFisher Scientific | 451245 |
| Experimental models: Organisms/strains | ||
| Zebrafish: gata2aum27 | Zhu et al.24 | ZDB-ALT-111201-1 |
| Zebrafish: foxc1ap162 | Banerjee et al.30 | ZDB-ALT-140721-2 |
| Zebrafish: Tg(ubi:CtoH2b-cherry)jh63 | Zhang et al.40 | ZDB-ALT-151007-31 |
| Zebrafish: gata2aum345 (aka gata2afl) | this study | um345 |
| Zebrafish: foxc1aum383 (aka foxc1afl) | this study | um383 |
| Zebrafish: rasa1aum414 (aka rasa1afl) | this study | um414 |
| Zebrafish: rasa1aum255 | this study | um255 |
| Zebrafish: rasa1aum290 | this study | um290 |
| Zebrafish: ruvbl1chb10 (aka ruvbl1fl) | this study | chb10 |
| Zebrafish: Tg(gata2aECE:iCre)um337 | this study | um337 |
| Oligonucleotides | ||
| See Supplementary Table 4 for all oligonucleotide sequences | ||
| Recombinant DNA | ||
| Plasmid: pKHR8 | Hoshijima et al.17 | RRID:Addgene_74625 |
| Plasmid: p3E-MCS1 | Moore et al.52 | RRID:Addgene_49004 |
| Plasmid: pmE-CreERT2 | Mosimann et al.56 | RRID:Addgene_27321 |
| Plasmid: pToneDest | Shin et al.55 | RRID:Addgene_67691 |
| Plasmid: pToneTP | Shin et al.55 | RRID:Addgene_67692 |
| Plasmid: p5E-6xgata2aECEbas | Shin et al.55 | RRID:Addgene_132968 |
| Plasmid: pCSDest2 | Villefranc et al.53 | RRID:Addgene_22424 |
| Plasmid: pBSIce-gata2aKI | this study | N/A |
| Plasmid: pBSIsce-KI | this study | N/A |
| Plasmid: pCS-CreNS | this study | N/A |
| Plasmid: pCS-zfCre | this study | N/A |
| Plasmid: pDR274-gata2a_3pex5sgRNA1 | this study | N/A |
| Plasmid: pJET-mCherry | this study | N/A |
| Plasmid: pJET-mVenus | this study | N/A |
| Plasmid: pKHR8-foxc1aKI | this study | N/A |
| Plasmid: pKHR8-rasa1aKI | this study | N/A |
| Plasmid: pmE-CreNS | this study | N/A |
| Plasmid: pmE-MCS1 | this study | N/A |
| Plasmid: pmE-zfCre | this study | N/A |
| Plasmid: pmE-zfCreERT2 | this study | N/A |
| Plasmid: pTol1-gata2aECe:CreERT2;cryaa:mcherry | this study | N/A |
| Software and algorithms | ||
| ZEN2010 | Zeiss | RRID:SCR_014340 |
| MultiTime Series PLUS Macro | Zeiss | RRID:SCR_014340 |
| AxioVision SE64 Rel 4.9.1 software | Zeiss | RRID:SCR_002677 |
| Adobe Photoshop CC | Adobe | RRID:SCR_014199 |
| Adobe Premiere CC | Adobe | RRID:SCR_021315 |
| Adobe Illustrator CC | Adobe | RRID:SCR_010279 |
| ImageJ2 version 2.3.0/1.53q with FIJI | NIH | RRID:SCR_002285 |
| IC Capture 2.5 software | IMAGINGSOURCE | https://www.theimagingsource.com/enus/support/download/iccapture-2.5.1525.3931/ |
| Prism | GraphPad Software | RRID:SCR_002798 |
| R Package for Statistical Computing | www.r-project.org | RRID:SCR_001905 |
| Other | ||
| Scanning Electron microscope | Hitachi High-Technology Corp | SU8010 |
| 710NLO confocal/two-photon microscope | Zeiss | RRID:SCR_017377 |
| Leica Fluorescence Dissection microscope | Leica | M165FC |
| Digital camera, AxioCam | Zeiss | MRc5 |
| Axioskop2 Plus | Zeiss | N/A |
| Imaging source USB CCD B&W 2.0 camera | IMAGINGSOURCE | DMK21AU04 |
| Spectrolinker™ UV Crosslinker | Spectronics Corp | XL-1000 |
| hybridization incubator | Robbins Scientific | Model 400 |
| ChemiDoc | BIO-RAD | 12003153 |
| StepOnePlus Real-Time PCR Systems | ThermoFisher Scientific | RRID:SCR_015805 |
| Nylon Membrane, positively charged | MilliporeSigma | 11209299001 |
| Hybridization Bags | Cosmo Bio Co | SE-S-1002-EX |
RT-qPCR
Total RNA was purified from Cre-injected or uninjected embryos (5 to 10 per sample) with TRIzol reagent (ThermoFisher). For gata2a, foxc1a, and ruvbl1, homozygous carriers of the floxed allele were incrossed, respectively, and used for RNA isolation. For embryos from an incross of rasa1a+/um414 adults, we separated heads and tails of individuals at 3 dpf in Trizol and DNA lysis buffer, respectively. Individual DNAs were isolated and genotyped. After genotyping, individual RNAs from 5 embryos of matched genotype (rasa1a+/+ or rasa1aum414/um414) were pooled and subjected to qRT-PCR analysis. For FACS-isolated mcherry+ cells (1.50–3.5×105) we used the Allprep DNA/RNA Micro kit (QIAGEN). cDNA was synthesized using 10ng of total RNA with SuperScript III First-Strand Synthesis SuperMix (ThermoFisher). For foxc1a, we removed residual genomic DNA before the reverse transcription reaction by treating with TURBO DNase (ThermoFisher). qPCR was performed with PowerSYBR Green PCR Master Mix (ThermoFisher) using ΔΔCT method on StepOne Plus Real-Time PCR machine (ThermoFisher). We used eef1l1 for normalization. All primers for qRT-PCR are listed in the Key Resources Table.
Southern analysis
We purified genomic DNA from pooled embryos using the Blood & Cell Culture DNA Midi kit (QIAGEN). 5 or 10 μg of DNA was fully digested overnight with indicated restriction enzyme at the appropriate temperature. Digested samples were loaded onto a 0.7% agarose gel made with Tris acetate EDTA buffer alongside 100–200 ng of digoxigenin (DIG)-labeled DNA marker VII (MilliporeSigma) and run at 25V for 18 hours. After electrophoresis, the gel was sequentially incubated in 250 mM HCl for 25 minutes, 0.5M NaOH/1.5M NaCl for 45 minutes, and then 0.5M Tris pH7.5-HCl/1.5M NaCl for 30 minutes. Genomic DNA was then transferred from the gel to positively charged nylon membrane (MilliporeSigma) by standard capillary transfer overnight. Membrane was denatured on filter paper soaked in 0.4M NaOH for 5 minutes and UV-crosslinked at 420 × 102 μj/cm2 in an XL-1000 UV CrossLinker (Spectronics Corp). Membranes were then prehybridized with DIG Easy Hyb solution (MilliporeSigma) in a HybriBag (Cosmo bio) at 42°C for 30 minutes with agitation. Prehybridization solution was replaced with fresh DIG Easy Hyb solution containing DIG labeled DNA probe at a final concentration between 50–100 ng/ml and incubated overnight at 42°C with agitation in a hybridization incubator (Robbins Scientific). An EGFP probe was PCR-amplified (5’-ATGGTGAGCAAGGGCGAGGAGCTG-3’ and 5’-ACTTGTACAGCTCGTCCATGCCG-3’) with DIG-labeled dNTP mix (MilliporeSigma) and AccuStart II DNA polymerase (Quantabio) using pBSIce-gata2aKI as a template followed by gel purification. Venus and mCherry fragments were PCR-amplified (venus:5’-ATGGTGAGCAAGGGCGAGGAGCTG-3’ and 5’-GGCGGCGGTCACGAACTCCAG-3’; mcherry: 5’-ATGGTGAGCAAGGGCGAGGAG-3’ and 5’-CTTACTTGTACAGCTCGTCCATG-3’) from pKHR8-foxc1aKI and separately subcloned into pJET2.1 (ThermoFisher) to give pJET-mVenus and -mCherry, respectively. DIG-labeled venus and mcherry DNA probes were then generated as above. Following hybridization, membranes were washed with 2x SSC/0.1% SDS at room temperature for 5 minutes, 0.5x SSC/0.1% SDS at 65°C for 20 minutes and 0.25x SSC/0.1% SDS at 65°C for 20 minutes, with agitation. After rinsing with maleic buffer (0.1M Maleic acid/150mM NaCl/0.3% Tween20-pH7.5), membrane was blocked in 1x Casein blocking buffer (MilliporeSigma) diluted in maleic buffer at room temperature for 30 minutes and incubated with anti-DIG antibody conjugated with alkaline phosphatase (1:50,000 dilution; MilliporeSigma) in blocking buffer at room temperature for 30 minutes. Membranes were washed with maleic buffer at room temperature for 15 minutes twice, preactivated with 0.1M Tris-pH9.5/0.1M NaCl/50mM MgCl2 then reacted with CDP-star ready-to-use substrate (MilliporeSigma). Chemiluminescence was detected by exposing membrane to film using CL-XPosure (ThermoFisher) or direct imaging on a ChemiDoc (Bio-rad).
In situ hybridization
DNA templates for urp1 or sst1.1. were generated by PCR as described elsewhere 19. Following PCR, fragments were gel purified and then used as templates to generate DIG-labeled RNA probes using T3 polymerase. In situ hybridization was performed as previously described59.
Imaging
Confocal imaging was performed using a LMS 710NLO (Zeiss) with ZEN10. Prior to imaging, larvae were subjected to lymphangiography using Qtracker 705 quantum dots as described previously 18. Bright field and fluorescent images ware taken on M165FC (Leica) equipped with AxioCamMRc (Zeiss) using AxioVision SE64 Rel 4.9.1 software (Zeiss). Videos of blood circulation were acquired using differential interference contrast imaging on an Axioskop2 Plus (Zeiss) equipped with DMK21AU04 (IMAGINGSOURCE) using IC capture 2.5 software (IMAGINGSOURCE). Video files were saved as .mp4 and imported into Adobe Premiere Pro for labeling (Adobe). SEM sample preparation and image acquisition were performed as previously described18.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical tests were performed using Prism (GraphPad Software LLC), except Fishers Exact test, which was performed using the R software package. In all pairwise comparison, we tested for normal distribution using the Shapiro-Wilk test. All statistical tests applied in this study are noted in Figure legends. All error bars are mean with standard deviation.
Supplementary Material
File S1. Genbank-formatted file with annotated sequence of gata2a targeted knock-in plasmid, related to Figure 1.
File S2. Genbank-formatted file with annotated sequence of foxc1a targeted knock-in plasmid, related to Figure 3.
File S3. Genbank-formatted file with annotated sequence of ruvbl1 targeted knock-in plasmid, related to Figure 5.
File S4. Genbank-formatted file with annotated sequence of rasa1a targeted knock-in plasmid, related to Figure 6.
File S5. Genbank-formatted file with annotated sequence of vector insert upstream of rasa1a exon 20 in rasa1afl, related to Figure 6.
Movie S1. Heartbeat and circulation in a gata2a+/fl embryo at 2 dpf injected with zfcre mRNA, related to Figure 2.
Movie S2. Heartbeat and circulation in a gata2afl/fl embryo at 2 dpf injected with zfcre mRNA, related to Figure 2.
Movie S3. Heartbeat and circulation in a foxc1a+/fl embryo at 2 dpf injected with zfcre mRNA, related to Figure 4.
Movie S4. Heartbeat and lack of circulation in a foxc1afl/fl embryo at 2 dpf injected with zfcre mRNA, related to Figure 4.
Highlights.
Improved methods for generating conditional deletion alleles in zebrafish
Generation of four zebrafish lines bearing endogenously floxed alleles
Blueprint for characterization and validation of floxed alleles in zebrafish
Use of a floxed allele to define cell-specific gene function in zebrafish
ACKNOWLEDGEMENTS
This work was supported by NIH grants R35HL140017 and R21OD030004 to N. D. L. K.L. and S.A.W. were supported by NIH grants R01GM115911, R01HL150669, and 5F31HL147482. M. J. P. was supported by R01DK080730. We thank Sarah Oikemus for critical reading of the manuscript. The Burns Laboratory was supported by NIH grants R01HL139806 (C.G.B.) and R35HL135831 (C.E.B.).
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Beis D, and Stainier DY (2006). In vivo cell biology: following the zebrafish trend. Trends Cell Biol 16, 105–112. 10.1016/j.tcb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, and Schilling TF (1995). Stages of embryonic development of the zebrafish. Dev Dyn 203, 253–310. 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 3.Mullins MC, Navajas Acedo J, Priya R, Solnica-Krezel L, and Wilson SW (2021). The zebrafish issue: 25 years on. Development 148. 10.1242/dev.200343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haffter P, Granato M, Brand M, Mullins MC, Hammerschmidt M, Kane DA, Odenthal J, van Eeden FJ, Jiang YJ, Heisenberg CP, et al. (1996). The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development 123, 1–36. 10.1242/dev.123.1.1. [DOI] [PubMed] [Google Scholar]
- 5.Driever W, Solnica-Krezel L, Schier AF, Neuhauss SC, Malicki J, Stemple DL, Stainier DY, Zwartkruis F, Abdelilah S, Rangini Z, et al. (1996). A genetic screen for mutations affecting embryogenesis in zebrafish. Development 123, 37–46. 10.1242/dev.123.1.37. [DOI] [PubMed] [Google Scholar]
- 6.Hruscha A, Krawitz P, Rechenberg A, Heinrich V, Hecht J, Haass C, and Schmid B (2013). Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 140, 4982–4987. 10.1242/dev.099085. [DOI] [PubMed] [Google Scholar]
- 7.Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JR, and Joung JK (2013). Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol 31, 227–229. 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson SL, and Weston JA (1995). Temperature-sensitive mutations that cause stage-specific defects in Zebrafish fin regeneration. Genetics 141, 1583–1595. 10.1093/genetics/141.4.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carney TJ, and Mosimann C (2018). Switch and Trace: Recombinase Genetics in Zebrafish. Trends Genet 34, 362–378. 10.1016/j.tig.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Doetschman T, Gregg RG, Maeda N, Hooper ML, Melton DW, Thompson S, and Smithies O (1987). Targetted correction of a mutant HPRT gene in mouse embryonic stem cells. Nature 330, 576–578. 10.1038/330576a0. [DOI] [PubMed] [Google Scholar]
- 11.Thomas KR, and Capecchi MR (1987). Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 51, 503–512. 10.1016/0092-8674(87)90646-5. [DOI] [PubMed] [Google Scholar]
- 12.Mansour SL, Thomas KR, and Capecchi MR (1988). Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature 336, 348–352. 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- 13.Gu H, Marth JD, Orban PC, Mossmann H, and Rajewsky K (1994). Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science 265, 103–106. 10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- 14.Nagy A (2000). Cre recombinase: the universal reagent for genome tailoring. Genesis 26, 99–109. [PubMed] [Google Scholar]
- 15.Feil R, Brocard J, Mascrez B, LeMeur M, Metzger D, and Chambon P (1996). Ligand-activated site-specific recombination in mice. Proc Natl Acad Sci U S A 93, 10887–10890. 10.1073/pnas.93.20.10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burg L, Palmer N, Kikhi K, Miroshnik ES, Rueckert H, Gaddy E, MacPherson Cunningham C, Mattonet K, Lai SL, Marin-Juez R, et al. (2018). Conditional mutagenesis by oligonucleotide-mediated integration of loxP sites in zebrafish. PLoS Genet 14, e1007754. 10.1371/journal.pgen.1007754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoshijima K, Jurynec MJ, and Grunwald DJ (2016). Precise Editing of the Zebrafish Genome Made Simple and Efficient. Dev Cell 36, 654–667. 10.1016/j.devcel.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shin M, Nozaki T, Idrizi F, Isogai S, Ogasawara K, Ishida K, Yuge S, Roscoe B, Wolfe SA, Fukuhara S, et al. (2019). Valves Are a Conserved Feature of the Zebrafish Lymphatic System. Dev Cell. 10.1016/j.devcel.2019.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrzejczuk LA, Banerjee S, England SJ, Voufo C, Kamara K, and Lewis KE (2018). Tal1, Gata2a, and Gata3 Have Distinct Functions in the Development of V2b and Cerebrospinal Fluid-Contacting KA Spinal Neurons. Front Neurosci 12, 170. 10.3389/fnins.2018.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pase L, Layton JE, Kloosterman WP, Carradice D, Waterhouse PM, and Lieschke GJ (2009). miR-451 regulates zebrafish erythroid maturation in vivo via its target gata2. Blood 113, 1794–1804. 10.1182/blood-2008-05-155812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurita R, Sagara H, Aoki Y, Link BA, Arai K, and Watanabe S (2003). Suppression of lens growth by alphaA-crystallin promoter-driven expression of diphtheria toxin results in disruption of retinal cell organization in zebrafish. Dev Biol 255, 113–127. 10.1016/s0012-1606(02)00079-9. [DOI] [PubMed] [Google Scholar]
- 22.Martin DI, and Orkin SH (1990). Transcriptional activation and DNA binding by the erythroid factor GF-1/NF-E1/Eryf 1. Genes & development 4, 1886–1898. 10.1101/gad.4.11.1886. [DOI] [PubMed] [Google Scholar]
- 23.Charles MA, Saunders TL, Wood WM, Owens K, Parlow AF, Camper SA, Ridgway EC, and Gordon DF (2006). Pituitary-specific Gata2 knockout: effects on gonadotrope and thyrotrope function. Mol Endocrinol 20, 1366–1377. 10.1210/me.2005-0378. [DOI] [PubMed] [Google Scholar]
- 24.Zhu C, Smith T, McNulty J, Rayla AL, Lakshmanan A, Siekmann AF, Buffardi M, Meng X, Shin J, Padmanabhan A, et al. (2011). Evaluation and application of modularly assembled zinc-finger nucleases in zebrafish. Development 138, 4555–4564. 10.1242/dev.066779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Topczewska JM, Topczewski J, Solnica-Krezel L, and Hogan BL (2001). Sequence and expression of zebrafish foxc1a and foxc1b, encoding conserved forkhead/winged helix transcription factors. Mech Dev 100, 343–347. 10.1016/s0925-4773(00)00534-7. [DOI] [PubMed] [Google Scholar]
- 26.Baranasic D, Hortenhuber M, Balwierz PJ, Zehnder T, Mukarram AK, Nepal C, Varnai C, Hadzhiev Y, Jimenez-Gonzalez A, Li N, et al. (2022). Multiomic atlas with functional stratification and developmental dynamics of zebrafish cis-regulatory elements. Nat Genet 54, 1037–1050. 10.1038/s41588-022-01089-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shahbazi R, Sghia-Hughes G, Reid JL, Kubek S, Haworth KG, Humbert O, Kiem HP, and Adair JE (2019). Targeted homology-directed repair in blood stem and progenitor cells with CRISPR nanoformulations. Nat Mater 18, 1124–1132. 10.1038/s41563-019-0385-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moreno-Mateos MA, Fernandez JP, Rouet R, Vejnar CE, Lane MA, Mis E, Khokha MK, Doudna JA, and Giraldez AJ (2017). CRISPR-Cpf1 mediates efficient homology-directed repair and temperature-controlled genome editing. Nat Commun 8, 2024. 10.1038/s41467-017-01836-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang CJ, Tu CT, Hsiao CD, Hsieh FJ, and Tsai HJ (2003). Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Dev Dyn 228, 30–40. 10.1002/dvdy.10356. [DOI] [PubMed] [Google Scholar]
- 30.Banerjee S, Hayer K, Hogenesch JB, and Granato M (2015). Zebrafish foxc1a drives appendage-specific neural circuit development. Development 142, 753–762. 10.1242/dev.115816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferre-Fernandez JJ, Sorokina EA, Thompson S, Collery RF, Nordquist E, Lincoln J, and Semina EV (2020). Disruption of foxc1 genes in zebrafish results in dosage-dependent phenotypes overlapping Axenfeld-Rieger syndrome. Hum Mol Genet 29, 2723–2735. 10.1093/hmg/ddaa163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mao YQ, and Houry WA (2017). The Role of Pontin and Reptin in Cellular Physiology and Cancer Etiology. Front Mol Biosci 4, 58. 10.3389/fmolb.2017.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Zhao L, Yuan S, Zhang J, and Sun Z (2017). Axonemal dynein assembly requires the R2TP complex component Pontin. Development 144, 4684–4693. 10.1242/dev.152314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trahey M, and McCormick F (1987). A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science 238, 542–545. 10.1126/science.2821624. [DOI] [PubMed] [Google Scholar]
- 35.Eerola I, Boon LM, Mulliken JB, Burrows PE, Dompmartin A, Watanabe S, Vanwijck R, and Vikkula M (2003). Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet 73, 1240–1249. 10.1086/379793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Henkemeyer M, Rossi DJ, Holmyard DP, Puri MC, Mbamalu G, Harpal K, Shih TS, Jacks T, and Pawson T (1995). Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature 377, 695–701. 10.1038/377695a0. [DOI] [PubMed] [Google Scholar]
- 37.Lapinski PE, Kwon S, Lubeck BA, Wilkinson JE, Srinivasan RS, Sevick-Muraca E, and King PD (2012). RASA1 maintains the lymphatic vasculature in a quiescent functional state in mice. J Clin Invest 122, 733–747. 10.1172/JCI46116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meyerhans A, Vartanian JP, and Wain-Hobson S (1990). DNA recombination during PCR. Nucleic Acids Res 18, 1687–1691. 10.1093/nar/18.7.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Won M, and Dawid IB (2017). PCR artifact in testing for homologous recombination in genomic editing in zebrafish. PLoS One 12, e0172802. 10.1371/journal.pone.0172802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang D, Gates KP, Barske L, Wang G, Lancman JJ, Zeng XI, Groff M, Wang K, Parsons MJ, Crump JG, and Dong PDS (2017). Endoderm Jagged induces liver and pancreas duct lineage in zebrafish. Nat Commun 8, 769. 10.1038/s41467-017-00666-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Balik-Meisner M, Truong L, Scholl EH, Tanguay RL, and Reif DM (2018). Population genetic diversity in zebrafish lines. Mamm Genome 29, 90–100. 10.1007/s00335-018-9735-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Butler MG, Iben JR, Marsden KC, Epstein JA, Granato M, and Weinstein BM (2015). SNPfisher: tools for probing genetic variation in laboratory-reared zebrafish. Development 142, 1542–1552. 10.1242/dev.118786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng C, and Capecchi MR (1992). Reexamination of gene targeting frequency as a function of the extent of homology between the targeting vector and the target locus. Mol Cell Biol 12, 3365–3371. 10.1128/mcb.12.8.3365-3371.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, et al. (2015). Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771. 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sabine A, Agalarov Y, Maby-El Hajjami H, Jaquet M, Hagerling R, Pollmann C, Bebber D, Pfenniger A, Miura N, Dormond O, et al. (2012). Mechanotransduction, PROX1, and FOXC2 cooperate to control connexin37 and calcineurin during lymphatic-valve formation. Dev Cell 22, 430–445. 10.1016/j.devcel.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 46.Kazenwadel J, Betterman KL, Chong CE, Stokes PH, Lee YK, Secker GA, Agalarov Y, Demir CS, Lawrence DM, Sutton DL, et al. (2015). GATA2 is required for lymphatic vessel valve development and maintenance. J Clin Invest 125, 2979–2994. 10.1172/JCI78888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wierson WA, Welker JM, Almeida MP, Mann CM, Webster DA, Torrie ME, Weiss TJ, Kambakam S, Vollbrecht MK, Lan M, et al. (2020). Efficient targeted integration directed by short homology in zebrafish and mammalian cells. Elife 9. 10.7554/eLife.53968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sugimoto K, Hui SP, Sheng DZ, and Kikuchi K (2017). Dissection of zebrafish shha function using site-specific targeting with a Cre-dependent genetic switch. Elife 6. 10.7554/eLife.24635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li W, Zhang Y, Han B, Li L, Li M, Lu X, Chen C, Lu M, Zhang Y, Jia X, et al. (2019). One-step efficient generation of dual-function conditional knockout and geno-tagging alleles in zebrafish. Elife 8. 10.7554/eLife.48081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu F, Kambakam S, Almeida MP, Ming Z, Welker JM, Wierson WA, SchultzRogers LE, Ekker SC, Clark KJ, Essner JJ, and McGrail M (2022). Cre/lox regulated conditional rescue and inactivation with zebrafish UFlip alleles generated by CRISPR-Cas9 targeted integration. Elife 11. 10.7554/eLife.71478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghanta KS, Chen Z, Mir A, Dokshin GA, Krishnamurthy PM, Yoon Y, Gallant J, Xu P, Zhang XO, Ozturk A, et al. (2021). 5′ modifications improve potency and efficacy of dna donors for precision genome editing. eLife 10, e72216. 10.7554/eLife.72216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moore JC, Sheppard-Tindell S, Shestopalov IA, Yamazoe S, Chen JK, and Lawson ND (2013). Post-transcriptional mechanisms contribute to Etv2 repression during vascular development. Dev Biol 384, 128–140. 10.1016/j.ydbio.2013.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Villefranc JA, Amigo J, and Lawson ND (2007). Gateway compatible vectors for analysis of gene function in the zebrafish. Dev Dyn 236, 3077–3087. 10.1002/dvdy.21354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, Parant JM, Yost HJ, Kanki JP, and Chien CB (2007). The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn 236, 3088–3099. 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]
- 55.Shin M, Male I, Beane TJ, Villefranc JA, Kok FO, Zhu LJ, and Lawson ND (2016). Vegfc acts through ERK to induce sprouting and differentiation of trunk lymphatic progenitors. Development 143, 3785–3795. 10.1242/dev.137901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mosimann C, Kaufman CK, Li P, Pugach EK, Tamplin OJ, and Zon LI (2011). Ubiquitous transgene expression and Cre-based recombination driven by the ubiquitin promoter in zebrafish. Development 138, 169–177. 10.1242/dev.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu P, Luk K, Shin M, Idrizi F, Kwok S, Roscoe B, Mintzer E, Suresh S, Morrison K, Frazao JB, et al. (2019). Enhanced Cas12a editing in mammalian cells and zebrafish. Nucleic Acids Res 47, 4169–4180. 10.1093/nar/gkz184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu Y, Zeng J, Roscoe BP, Liu P, Yao Q, Lazzarotto CR, Clement K, Cole MA, Luk K, Baricordi C, et al. (2019). Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med 25, 776–783. 10.1038/s41591-019-0401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hauptmann G, and Gerster T (1994). Two-color whole-mount in situ hybridization to vertebrate and Drosophila embryos. Trends Genet 10, 266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
File S1. Genbank-formatted file with annotated sequence of gata2a targeted knock-in plasmid, related to Figure 1.
File S2. Genbank-formatted file with annotated sequence of foxc1a targeted knock-in plasmid, related to Figure 3.
File S3. Genbank-formatted file with annotated sequence of ruvbl1 targeted knock-in plasmid, related to Figure 5.
File S4. Genbank-formatted file with annotated sequence of rasa1a targeted knock-in plasmid, related to Figure 6.
File S5. Genbank-formatted file with annotated sequence of vector insert upstream of rasa1a exon 20 in rasa1afl, related to Figure 6.
Movie S1. Heartbeat and circulation in a gata2a+/fl embryo at 2 dpf injected with zfcre mRNA, related to Figure 2.
Movie S2. Heartbeat and circulation in a gata2afl/fl embryo at 2 dpf injected with zfcre mRNA, related to Figure 2.
Movie S3. Heartbeat and circulation in a foxc1a+/fl embryo at 2 dpf injected with zfcre mRNA, related to Figure 4.
Movie S4. Heartbeat and lack of circulation in a foxc1afl/fl embryo at 2 dpf injected with zfcre mRNA, related to Figure 4.
Data Availability Statement
All original data and images are available from the Lead Contact upon request. This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.