

Numerous studies have reported links from smoking and e-cigarette use to adverse cardiovascular and pulmonary outcomes and pathologies.1–4 However, the molecular mechanisms are incompletely understood.
We previously showed that the receptor for advanced glycation end products (RAGE) is a potential upstream mediator of e-cigarette-induced endothelial barrier dysfunction, in that sera of e-cigarette users increased permeability of cultured endothelial cells and this was attenuated by the RAGE inhibitor, FPS-ZM1 (RAGEi).1 Here, we studied the effect of acute and subacute e-cigarette exposure on endothelial function in rats measured as flow-mediated dilation (FMD), which we have shown is impaired by a wide range of inhalational exposures.2,3
For acute exposure, anesthetized, 10-week-old Sprague-Dawley rats (n=4M+4F/group) were exposed via nose cones to undiluted aerosol from tank-style e-cigarettes containing flavorless propylene glycol/vegetable glycerin (PG/VG) and freebase nicotine (12 mg/mL), aerosol from PG/VG alone, smoke from Marlboro Red cigarettes, or air, in a single session of 10 cycles of pulsatile 5s exposure over 5 minutes using a Gram Research analytical vaping machine drawing 55 ml. Smoke generated under identical conditions yielded 30.24±7.75 mg/m3 PM2.5 in the nose cone as measured gravimetrically (4 replicates), and e-cig aerosol yielded 4036±1439 mg/m3 based on mass of e-liquid liberated per puff (10 replicates). For each exposure condition, groups received either 1 mg/kg of RAGEi or DMSO (vehicle) i.p. 1h pre-exposure. FMD was measured pre-exposure and 10 minutes after end of exposure as we have described.2 All procedures were approved by the UCSF IACUC. Exposure to e-cigarette aerosol with or without nicotine or to cigarette smoke, but not to air, significantly impaired FMD in vehicle groups; but impairment was prevented by RAGEi (Figure 1A).
Figure 1. Inhibition of RAGE prevents FMD impairment by cigarette smoke and e- cigarette aerosol.
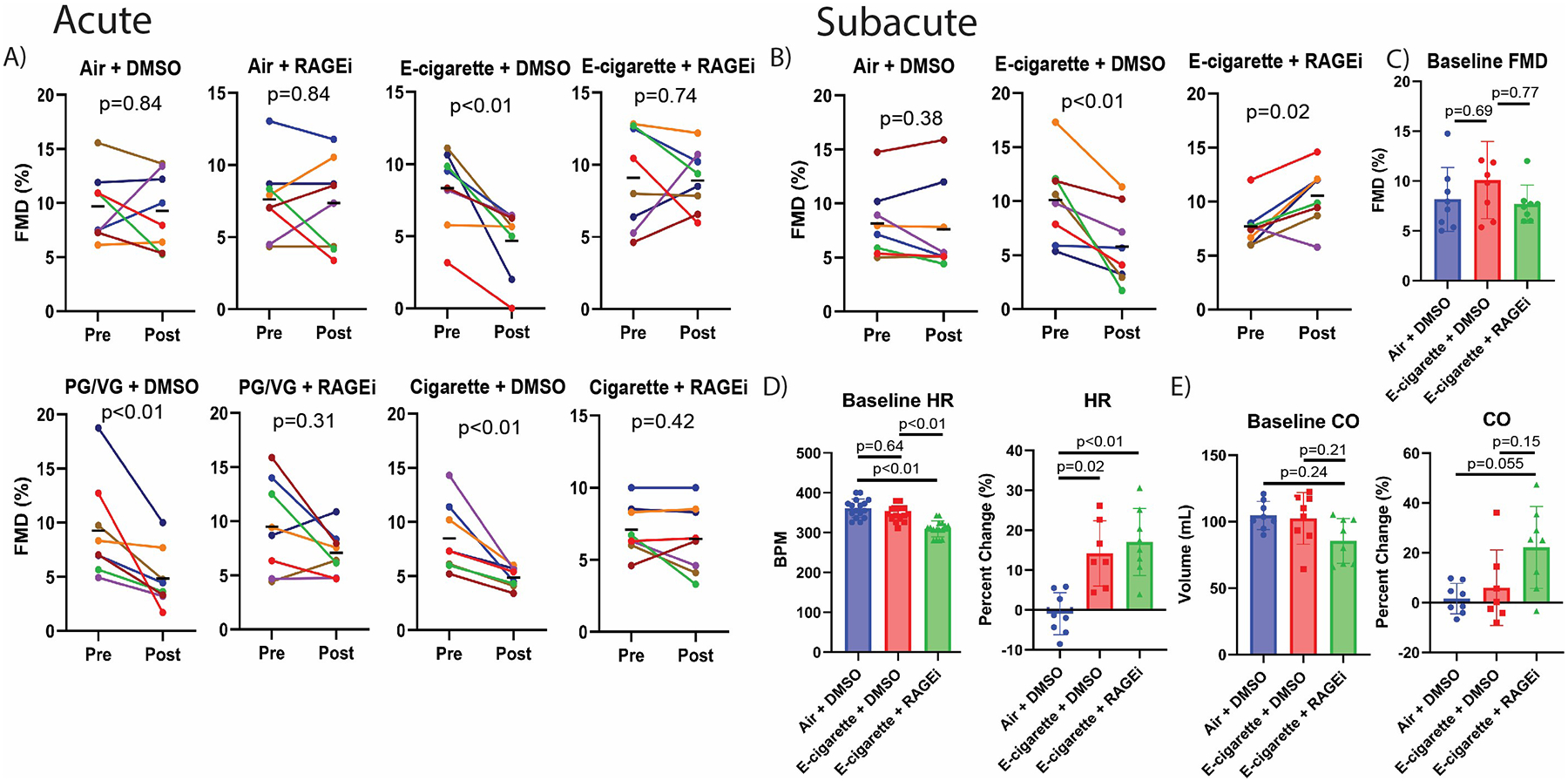
(A) Changes in FMD within each group after acute exposure. Each colored line shows FMD of an individual rat measured pre- and post-exposure; bars indicate group means. (B) Changes in FMD within each group on the final day of subacute exposure. C) Baseline FMD before final subacute exposure. (D, E) Baseline heart rate (HR) and cardiac output (CO) before final subacute exposure and percent change of HR and CO from pre- to post-final exposure. p values from Wilcoxon matched-pairs signed rank test for A-B, and Kruskal-Wallis with Dunn’s correction for C-E. p≤0.05 was considered significant. Error bars=SD.
Sub-acute exposure involved rats as described above, but conscious in restraint cones, exposed to PG/VG/nicotine aerosol or air in a single session of 120 cycles of pulsatile 5s exposure over 1h/day for 2 weeks. RAGEi or DMSO was administered i.p. 1h pre-exposure daily during acclimation and exposure (6 groups, 3 weeks total). Echocardiography was conducted one day before end of experiment, measuring parameters pre- and post-exposure. On the last day, FMD was assessed before and after one session of 10 cycles of pulsatile 5s exposure over 5 minutes. Rats receiving vehicle exhibited impairment of FMD after the final e-cigarette exposure, while surprisingly, rats treated with RAGEi exhibited improved FMD after e-cigarette exposure (Figure 1B). There was no difference in pre-exposure (baseline) FMD between groups on the last day (Figure 1C). Pre-exposure baseline heart rate in the RAGEi group was lower (Figure 1D). However, the final exposure increased heart rate in both e-cigarette groups, while cardiac output increased in rats treated with RAGEi compared with both vehicle groups (Figure 1D,E), without changes in ejection fraction and ventricular chamber size (not shown).
Our results indicate that RAGE is an important mediator of vascular dysfunction induced by both smoking and vaping, which is consistent with our prior report that chronic e-cigarette users had higher circulating levels of the RAGE ligands HMGB1 and S100A8, whereas chronic smokers had higher levels of RAGE itself.1 Our findings are also concordant with a recent report connecting endothelial dysfunction to pulmonary irritation,4 and our own report that acute impairment of FMD by exposure to smoke requires the vagus nerve.2 Although depressed FMD is largely a function of endothelial dysfunction due to less bioavailable NO, the enhanced FMD shown in Fig. 1B (E-cigarette+RAGEi) may have been caused by increased NO production, improved smooth muscle sensitivity, or a combination of both effects as observed by Jin et al.4 in isolated aorta of mice exposed to acetaldehyde. It will be important to elucidate the connection between airway irritation and the RAGE pathway.
Acknowledgments
Research reported in this publication was supported by grant U54 HL147127 from the National Heart, Lung, and Blood Institute at the National Institutes of Health (NIH/NHLBI) and the US Food and Drug Administration Center for Tobacco Products, by grant T32PT6041 from the California Tobacco-Related Disease Research Program, and by a generous donation from the Elfenworks Foundation in memory of Deb O’Keefe. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the FDA.
Footnotes
No disclosures
References
- 1.Mohammadi L, Han DD, Xu F, Huang A, Derakhshandeh R, Rao P, Whitlatch A, Cheng J, Keith RJ, Hamburg NM, et al. Chronic E-Cigarette Use Impairs Endothelial Function on the Physiological and Cellular Levels. Arterioscler Thromb Vasc Biol. 2022;42(11):1333–1350. doi: 10.1161/ATVBAHA.121.317749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nabavizadeh P, Liu J, Rao P, Ibrahim S, Han DD, Derakhshandeh R, Qiu H, Wang X, Glantz SA, Schick SF, et al. Impairment of Endothelial Function by Cigarette Smoke Is Not Caused by a Specific Smoke Constituent, but by Vagal Input From the Airway. Arterioscler Thromb Vasc Biol. 2022;42(11):1324–1332. doi: 10.1161/ATVBAHA.122.318051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rao P, Han DD, Tan K, Mohammadi L, Derakhshandeh R, Navabzadeh M, Goyal N, Springer ML. Comparable Impairment of Vascular Endothelial Function by a Wide Range of Electronic Nicotine Delivery Devices. Nicotine Tob Res. 2022;24:1055–1062. doi: 10.1093/ntr/ntac019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin L, Lynch J, Richardson A, Lorkiewicz P, Srivastava S, Theis W, Shirk G, Hand A, Bhatnagar A, Srivastava S, et al. Electronic cigarette solvents, pulmonary irritation, and endothelial dysfunction: role of acetaldehyde and formaldehyde. Am J Physiol Heart Circ Physiol. 2021;320(4):H1510–H1525. doi: 10.1152/ajpheart.00878.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]