

Abstract
Chronic infection with the gammaherpesvirus Epstein Barr virus (EBV) is a risk factor for several autoimmune diseases, and poor control of EBV viral load and enhanced anti-EBV responses elevate this risk further. However, the role of host genetic variation in the regulation of immune responses to chronic gammaherpesvirus infection and control of viral replication remains unclear. To address this question, we infected C57BL/6J (B6) and genetically divergent wild-derived inbred PWD/PhJ (PWD) mice with murine gammaherpesvirus-68 (MHV-68), a gammaherpesvirus similar to EBV, and determined the effect of latent gammaherpesvirus infection on the CD4 T cell transcriptome. Chronic MHV-68 infection of B6 mice resulted in a dramatic upregulation of genes characteristic of a cytotoxic T helper cell (ThCTL) phenotype, including Gzmb, Cx3cr1, Klrg1, and Nkg7, a response that was highly muted in PWD mice. Flow cytometric analyses revealed an expansion of CX3CR1+KLRG1+ ThCTL-like cells in B6 but not PWD mice. Analysis of MHV-68 replication demonstrated that in spite of muted adaptive responses, PWD mice had superior control of viral load in lymphoid tissue, despite an absence of a defect in MHV-68 in vitro replication in PWD macrophages. Depletion of NK cells in PWD mice, but not B6 mice, resulted in elevated viral load, suggesting genotype-dependent NK cell involvement in MHV-68 control. Taken together, our findings demonstrate that host genetic variation can regulate control of gammaherpesvirus replication through disparate immunological mechanisms resulting in divergent long-term immunological sequelae during chronic infection.
Introduction
Epstein Barr virus (EBV) is a ubiquitous gammaherpesvirus that infects >90% of the population in high income nations. Infection typically occurs during childhood and is usually asymptomatic. The virus is transmitted via the oral route, initiated by lytic replication in the oral epithelium, followed by lytic replication in B cells in the tonsils and systemic dissemination, with eventual control of the lytic replication by the host immune system. EBV then establishes a life-long latent infection in memory B cells, with periodic reactivation and lytic replication, resulting in virus shedding. While asymptomatic in young children, primary EBV infection of adolescents and adults results in infectious mononucleosis, a prolonged syndrome caused by a massive immune response to EBV, which eventually resolves and results in viral latency and chronic infection.
Chronic infection with EBV has been shown to be a risk factor in several types of cancers, as well as several autoimmune diseases, including systemic lupus erythematosus (SLE)(1, 2), rheumatoid arthritis (RA)(3), and multiple sclerosis (MS)(4, 5). Numerous studies have examined the possible underlying mechanisms behind the associated increased risk of autoimmunity and chronic EBV infection, with two major non-mutually exclusive hypotheses emerging: 1) molecular mimicry of autoantigens by EBV antigens, and 2) skewing of the immune system by EBV infection and latency, resulting in a state favorable to development of autoimmunity(6). The association between EBV and antigenically unrelated autoimmune diseases lends further support for the latter hypothesis. Intriguingly, among EBV seropositive individuals, higher antibody titers against EBV antigens and higher EBV viral loads were shown to be HLA genotype-dependent and strongly associated with higher risk for MS(7, 8), suggesting that host poor control of EBV replication is genotype-dependent, and can in turn enhance the risk of autoimmune diseases. Together with the emerging findings that control of EBV replication may be host genotype dependent(9), these data strongly support the existence of yet-to-be-defined genetic factors controlling EBV replication and subsequent immune dysregulation.
A barrier to studying the host response to EBV is the absence of a robust small animal model of EBV infection, owing to the narrow host tropism typical of herpesviruses(10). However, the identification of murine gammaherpesvirus-68 (MHV-68), a structurally similar and related gammaherpesvirus to EBV, which readily establishes a latent life-long infection in wild and laboratory mice(10), allows for modeling of gammaherpesvirus infection and its consequences in a mammalian host. MHV-68 is a well-accepted model of gammaherpesvirus pathogenesis and chronic infection(11). Like EBV, MHV-68 undergoes initial lytic replication, followed by control of infection via adaptive immune responses, and establishment of a latent infection primarily in B lymphocytes in lymphoid organs(12). Many foundational studies have used MHV-68 to reveal the critical host immunological requirements for control of gammaherpesvirus replication, including indispensable roles for type I interferon, CD4, and CD8 T cells (13). However, the vast majority of these studies have used the classic laboratory inbred strain, C57BL/6 (B6), due to the wide availability of genetic tools on this background.
In this study, we leveraged conventional laboratory inbred B6 and genetically divergent wild-derived inbred PWD/PhJ (PWD) mice to determine the role of host genotype in MHV-68 infection. PWD mice introduce a much greater level of genetic diversity compared with standard laboratory strains(14). Importantly, wild mice, which recently served as founders for wild-derived inbred strains, evolved under relevant selective pressures, including those from infectious diseases(15). Previous studies from our laboratory demonstrated that PWD mice, compared with B6 mice, have a unique immune cell transcriptome and lower susceptibility to CNS autoimmunity, as modeled through EAE(16). Furthermore, studies utilizing B6.ChrPWD chromosome substitution (consomic) strains, in which chromosomes from the PWD strain were incorporated into the B6 background, demonstrated genetic control over CNS autoimmunity(17, 18) and environmental risk factors(19), as well as resistance to experimental inflammatory bowel disease(20) and acute infection with influenza A virus(21). In this study, we analyzed the susceptibility of B6 and PWD mice to infection with MHV-68, as well as the subsequent immune responses required for control of this virus. We found that chronic MHV-68 infection of B6 mice resulted in a dramatic reprogramming of gene expression in T helper cells, with an expansion of cytotoxic T helper cells (ThCTL) expressing KLRG1 and CX3CR1, a response that was unexpectedly and profoundly muted in PWD mice. Analysis of MHV-68 replication demonstrated that in spite of muted T cell responses, PWD mice had superior control of viral load. In vitro viral replication studies demonstrated a lack of a replication defect in PWD macrophages. However, depletion of NK cells resulted in enhanced MHV-68 replication in PWD but not B6 mice. Taken together, our findings suggest that host genotype determines not only susceptibility to gammaherpesvirus infection, but also the immune mechanisms by which it is controlled and subsequent long term immunological sequelae.
Materials and Methods
Animals and MHV-68 Infection
C57BL/6J (B6) and PWD/PhJ (PWD) mice were purchased from Jackson Laboratories (Bar Harbor, Maine, USA), then bred and housed in a single room within the vivarium at the University of Vermont for 2 or more generations prior to experimentation. MHV-68 (clone G2.4) was provided by Edward J. Usherwood, PhD (Geisel School of Medicine Dartmouth, Hanover, NH, USA). Virus was propagated by infection of 3T3 cells at a MOI of 0.1, as previously described(22, 23). Infectious virus was measured via plaque titration by infecting confluent 3T3 cells using 1:10 serial dilutions of viral inoculate in DMEM (Gibco) plus 10% (v/v) fetal bovine serum (FBS; Gibco) at 37° C for one hour, after which cells were overlaid with 0.75% w/v solution of carboxymethylcellulose (CMC; Sigma Aldrich, USA) in DMEM plus 10% (v/v) FBS and Pen-Strep (P/S; Gibco) and incubated for 7 days. Post incubation cells were fixed with methanol and stained with crystal violet (Fisher Scientific, USA), and then the plaques were enumerated microscopically to determine titer (PFU/ml).
B6 and PWD littermate mice were randomized to MHV-68 infection or saline control injection at an average of 9 weeks of age (range: 6–14 weeks). MHV-68 infection was administered via i.p. injection of 104 PFU of MHV-68 in total volume of 100 μl PBS, to allow for viral replication and latency in the spleen that is fully established by ~1 month post-infection(24). Mice were euthanized at 5- 9- 16- or 35-days post infection and spleen was collected and processed as described for each assay. To address the physiological mucosal route of infection, in some experiments MHV-68 infection was administered via intranasal inoculation (i.n.) of 104 PFU of MHV-68 in total volume of 50 μl PBS under light anesthesia with isoflurane. Mice were euthanized at 7- and 35- days post infection to assess acute and latent timepoint respectively. Spleen and lung tissue were collected and processed as described for each assay. The experimental procedures used in this study were approved by the Institutional Animal Care and Use Committee of the University of Vermont.
Cell Sorting and RNA Isolation
B6 and PWD mice were subjected to MHV-68 infection or control treatment as described above. At 35 days post injection, viral latency, mice were euthanized and spleens were collected for cell sorting and RNA isolation as previously described(19). Briefly, spleens were digested and depleted for B cells using Spleen Dissociation Medium and EasySep B cell positive selection respectively (STEMCELL Technologies, Inc., Canada). The remaining cells were purified by fluorescence-activated cell sorting (FACS) using fluorophore-conjugated antibodies (BioLegend, USA) against cell surface markers on CD4 T cells (CD19−TCRβ+ CD4+ CD8− CD25−). Dead cells were excluded using the Far-Red Live-Dead staining kit (ThermoFisher, USA). RNA was isolated from the isolated CD4 T cells using the Qiagen RNeasy Mini or Micro Kits, and samples were selected for downstream analysis based on RNA integrity number (typically 6–9). Four biological replicates (individual mice) for each strain, sex, and treatment combination were selected.
Transcriptional Profiling and Statistical Analysis of Microarray Data
Transcriptional profiling was conducted via microarray analysis performed at the University of Vermont Cancer Center Genomics Facility using the Mouse Affymetrix Clariom D Genechip and the GeneChip™ WT Pico Target Preparation reagent kit (ThermoFisher 9026220) as previously described(19). Completed arrays were stained using the Affymetrix® GeneChip® staining reagents and scanned with the 7G Affymetrix® GeneChip® Scanner 3000. Resulting raw intensity CEL files were imported into Expression Console software (Affymetrix, USA), and CHP files were generated for gene level analysis. CHP files were imported into Transcriptome Analysis Console (TAC) software v4.0.2.15 (Affymetrix), and gene level expression analysis was performed using the default ANOVA settings (e-Bayesian method). The following comparison variables were used: genotype (B6 and PWD), sex (male and female), and treatment (MHV-68 and PBS). To detect differentially expressed genes as a function of MHV-68 viral latency, pairwise comparisons were done between MHV-68 infected and PBS controls for the following sample groupings: all mice together, all B6 mice, all PWD mice, B6 females only, B6 males only, PWD females only, and PWD males only. Differentially expressed genes (DEGs) were identified using a differential expression threshold of FDR <0.2 and FC > |2|, and DEG lists for all of the different comparisons are provided in Supplemental Table 1.
Bioinformatic Analysis
Pathway enrichment analysis of upregulated DEGs was conducted using two parallel approaches – Ingenuity Pathway Analysis™ (IPA; Qiagen, Inc, USA) and Enrichr(25–27) gene set enrichment analysis tool. For pathway enrichment analysis using IPA, the gene expression datasets with the applied differential expression threshold of FDR <0.2 and FC > |2| were exported from TAC software and uploaded to IPA. The IPA Core Analysis function, Comparison Analysis function, and the Canonical Pathway function were used to identify the top canonical pathways (p < 0.05, Z score > |2|) and compare the effect of MHV-68 latency across the four genotype-sex combinations (B6 females, B6 males, PWD females, and PWD male), following methodology used in our previous studies(16, 18, 19, 28). For Enrichr-based analysis, the list of shared upregulated DEGs between B6 males and females was uploaded to the Enrichr gene set enrichment tool and analyzed for pathway enrichment using the Elsevier Pathway Collection (p < 0.01).
To identify immune cell subset-specific pattern of expression of the upregulated DEGs ImmGen database(29) and MyGeneSet tool was utilized. Gene lists of the top 20 upregulated genes by fold change the four genotype-sex combinations as well as all B6 and all PWD mice, were exported from TAC software and inputted into the MyGeneSet tool for analysis. Enrichment analysis was conducted using the ImmGen ULI RNA-Seq data set for all available cell populations of interest and displayed as a median normalized heatmap.
To address the potential involvement of pro-viral and anti-viral CD4 T cell subsets on the observed phenotypes given MHV-68 infection, the DEG lists were probed for genes associated with T follicular helper (Tfh)(30–32), Th17(33, 34), and Th1(35) cells, as identified through an in-depth literature search. Resulting fold change values of gene expression in B6 females, B6 males, PWD females, and PWD males were reported and displayed as a heatmap.
Flow Cytometry
At 5-, 7- (for i.n. infection) 9-, 16-, and 35-days post infection mice were euthanized and spleens and/or lungs, in the case of i.n. infections, were collected and processed for staining for flow cytometry as previously described(36). Briefly, spleens were mechanically dissociated by a syringe plunger between two pieces of mesh, until a single cell suspension was created. Lung samples were processed via mechanical dissociated by a syringe plunger, digested for 40 minutes at 37°C under rocking agitation, and then filtered until a single cell suspension was created. Resulting cell suspensions for both spleen and lung tissue underwent red blood cell lysis by incubation in 0.8% ammonium chloride solution (STEMCELL Technologies) and subsequent cell processing was dependent on the objective of flow cytometry analysis (intracellular cytokines, surface staining with permeabilization, Tfh and Th17 cell assessment, or NK cell assessment).
For intracellular cytokine analysis, cells were stimulated with 5 ng/mL PMA, 250 ng/mL ionomycin, and brefeldin A (Golgi Plug reagent; BD Bioscience, USA) for 4 hours prior to staining for flow cytometry analysis. For flow cytometry analysis of intracellular cytokines and surface staining with permeabilization, cells were stained with UV-Blue LIVE/DEAD fixable stain (Invitrogen, USA) and then surface stained for different combinations of the following markers: CD45, CD19, CD11b, CD25, TCRβ, CD4, CD8, CX3CR1 and KLRG1 (BioLegend). Cells were then fixed and permeabilized depending on analysis objective. Cells for intracellular cytokine analysis were fixed with 1% paraformaldehyde (Sigma Aldrich) and 0.05% saponin permeabilization buffer and labeled with antibodies against IFNγ and TNFα (BioLegend). For T-bet and GzmB staining, cells were surface labeled on ice, then fixed and permeabilized using BioLegend’s True-Nuclear™ Transcription Factor Buffer Set and labeled for antibodies against T-bet and GZMB.
To assess the presence of Tfh and Th17 cells, cells underwent both intercellular cytokine analysis and surface staining with permeabilization. For intracellular cytokine analysis cells were stimulated for 4 hours as described above. In both intercellular cytokine analysis and surface staining with permeabilization, cells were labeled with UV-Blue LIVE/DEAD fixable stain (Invitrogen, USA), surface stained using BD Biosciences BV buffer for different combinations of the following markers: CD45, CD11b, CD19, TCRβ, CD4, CD8, CX3CR1, KLRG1, PD-1, CD27 and CXCR5 (BioLegend and BD Biosciences), and then fixed with 1% paraformaldehyde (Sigma Aldrich) and permeabilized using 0.05% saponin permeabilization buffer. Cells were then stained depending on analysis objective. For intercellular cytokine analysis subsequent cells were labeled with antibodies against IFNγ and IL-17A (BioLegend), while cells that underwent surface staining with permeabilization were labeled with GzmB (BioLegend).
For flow cytometry staining to assess NK cells, cells were labeled with UV-Blue LIVE/DEAD fixable stain (Invitrogen, USA), surface stained with antibodies against CD45, CD19, TCRβ, CD4, CD8, CX3CR1, KLRG1, ASGM1, CD49b, and NK1.1 (BioLegend), and fixed with 1% paraformaldehyde (Sigma Aldrich).
All stained cells were analyzed on an LSRII cytometer (BD Biosciences) or a Cytek Aurora (Cytek Biosciences, USA). Flow cytometry data analysis was performed using FlowJo Software versions 10.7.1 – 10.8.1 (BD Biosciences).
Viral Load Assessment by Infectious Virus Titer and Viral DNA Load
Infectious viral titers in the spleen were determined by plaque assay following a modified procedure to the one described above for initial infectious viral titer and as previously described(37). Briefly, spleens were frozen at −80°C in media utilized for plaque assays (DMEM+10%v/v FBS+ P/S) prior to the assay. Samples were thawed and virus was released via sonication and clarified via centrifugation. Serial dilutions of the resulting sample viral suspensions were added in 0.5ml volumes to 3T3 cell monolayers and incubated at 37°C for an hour to allow the virus to absorb, before overlaying with 0.75% w/v solution of carboxymethylcellulose. After 7 days of incubation at 37°C cells were fixed with methanol and stained with crystal violet (Fisher Scientific, USA), and then the plaques were enumerated microscopically to calculate PFU/spleen. For assessment of infectious viral titers from BMDM cultures, supernatant was collected from infected BMDMs at 0-, 24-, 48-, and 72-hours post infection and frozen at −80°C prior to use. Completely thawed supernatant was serially diluted and added in 0.5ml volumes to 3T3 cell monolayers, subsequent processing of samples followed the process described above for spleen derived samples.
Viral DNA was quantified via quantitative PCR (qPCR) using a modified approach from what was previously described(38). DNA was extracted from spleen tissue or BMDMs using Qiagen DNeasy Blood and Tissue Kit and concentration was determined via Nanodrop (Thermo Scientific NanoDrop 2000 Spectrophotometer). qPCR was preformed according to the manufacturer’s instructions using the using DyNAmo ColorFlash SYBR Green kit (ThermoFisher) and 100nm primers complementary to the sequence of the MHV-68 ORF 50 gene(38, 39) and samples were run on Quant Studio 3 Real-Time PCR System (Applied Biosystems, USA). MHV-68 ORF 50 gene abundance was normalized by the abundance of the cellular DNA reference gene Mapk14 and calculated by a comparative cycle threshold (Ct) method equation 2−(deltaCt) and multiplied by a factor of 10,000 for ease of visualization, i.e., Relative Abundance = 2−(MHV-68 ORF 50 Ct – Mapk14 Ct) × 10,000. For all analysis of viral DNA load with the exception of NK cell depletion experiments, samples were not normalized against a reference control. For analysis of viral DNA load post NK cell depletion, samples were further normalized against the B6 IgG control population deltaCt (i.e., δ-B6 IgG Control deltaCt) to control for baseline variability to allow for pooling of multiple experiments.
Differentiation and Infection of Bone Marrow Derived Macrophages
Bone marrow derived macrophages (BMDMs) were generated as previously described(40) from age- and sex- matched male and female B6 or PWD mice. Differentiated BMDMs were replated in DMEM plus 10% (v/v) FBS, P/S and L-glutamine (Gibco) in 12-well plates at 6×105 cells/well and allowed to adhere overnight at 37°C. BMDM infection protocol was derived and modified from previously published methods(41, 42). In brief, BMDMs were infected with MHV-68 at a multiplicity of infection (MOI) of 0.4 PFU/cell for an hour at 37°C to allow for adsorption, and then washed twice with PBS prior to media replenishment (DMEM plus 10% (v/v) FBS). Supernatants and cell lysates were collected at 24-, 48-, and 72-hours post infection for later assessment of viral load.
In vivo NK Cell Depletion
On the day prior to MHV-68 infection (D −1), B6 mice were i.p. injected with 0.1 mg anti-NK1.1 (clone PK136; BioLegend) or mouse IgG2a, κ isotype control (BioLegend) and PWD mice were injected with 0.05 ml ASGM1 (clone Poly21460; BioLegend) or 0.1 mg of rabbit IgG isotype control (InVivoMAb polyclonal rabbit IgG; Bio X Cell, USA) respectively, delivered in a total volume of 100 μl PBS. NK Cell depletion was verified by flow cytometry analysis of TCRβ−CD19−CD4−CD8−CD49b+ NK1.1+/ASGM1+ cell populations.
Statistical Analysis
Statistical analyses not pertaining to microarray data were carried out using GraphPad Prism software, versions 8.3 – 9. Details of the analyses are provided in the figure legends; including the specific tests used to assess the significance of the observed differences and indication of adjustments for multiple comparisons when appropriate. Comparisons were assessed for effects between genotypes (B6 and PWD) or within genotype sex effects as indicated. All center values represent the mean, and error bars represent the standard error of the mean. A P value <0.05 was considered significant. Comparisons are indicated by the brackets and P values are reported using asterisks where significant (* P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, **** P ≤ 0.0001). A lack of an indicated asterisk p value therefore signifies a lack of a significant difference.
Results
Genotype- and sex-dependent regulation of the CD4 T cell transcriptome during chronic gammaherpesvirus infection
While EBV and MHV-68 target B cells as the latent reservoir, robust virus-specific and bystander CD4 T cell responses are elicited and sustained during chronic infection, which are necessary to control primary infection and to prevent virus reactivation(43, 44). In order to determine the role of genotype and sex in the regulation of the host immune response to chronic gammaherpesvirus infection, we performed transcriptional profiling of CD4 T cells isolated from male and female B6 and PWD mice latently infected with MHV-68, compared with uninfected controls. 6–8 week old littermate mice were randomized to infection or control PBS injections. Infection with 104 PFU of MHV-68 was performed via the i.p. route, which allows for robust replication and latency in the spleen, with latency fully established by ~1 month post-infection(24). At 35 days post-infection, conventional CD4 T cells were isolated from the spleen by FACS (TCRβ+CD4+CD8−CD25−CD11b−CD11c−), followed by RNA extraction and bulk transcriptional profiling essentially as in our previously studies(45) (see Materials and Methods).
Principal component analysis demonstrated robust sample clustering based on genotype (B6 versus PWD), with secondary clustering by treatment (PBS or MHV-68) amongst samples from B6 mice, which was much less evident in PWD (Fig. 1A). This was further confirmed via proportion of variance analysis, which revealed that host genotype demonstrated the largest impact on the CD4 T cell transcriptome, followed by MHV-68 infection and sex (Fig. 1B). Differential expression analysis, comparing cells from uninfected controls to those from MHV-68 infected mice, revealed that latent MHV-68 infection resulted in divergent outcomes by genotype and sex. Latent infection resulted predominantly in the upregulation of gene expression, but with large differences in the number of significant differentially expressed genes (DEGs) across genotype-sex combinations, with B6 females > B6 males >> PWD females > PWD males (Fig. 1C and Supplemental Table 1). Comparison of upregulated DEGs that passed the differential expression threshold between B6 females, B6 males, and PWD females revealed a large overlap in upregulated DEGs and identified a subset of 23 upregulated DEGs shared between the three groups (Fig. 1D), with varying magnitudes of upregulation for the same DEGs depending on host genotype and sex (Fig. 1E). Similarly, comparison of the top 20 (by fold change) upregulated DEGs between B6 females and B6 males revealed an overlap in upregulated DEGs between the two sexes with the magnitude of upregulation being greater in females then males (Fig. 1F). These results suggest that the observed difference in DEG number is primarily in the magnitude of the response, rather than overtly distinct transcriptional responses across genotype and sex. Taken together, these results demonstrate that latent MHV-68 infection drives pronounced changes in CD4 T gene expression in B6 mice, with a moderate female bias, and that these changes are surprisingly muted in PWD mice.
Figure 1. Host genotype drives CD4 T Cell gene expression in MHV-68 latently infected mice.
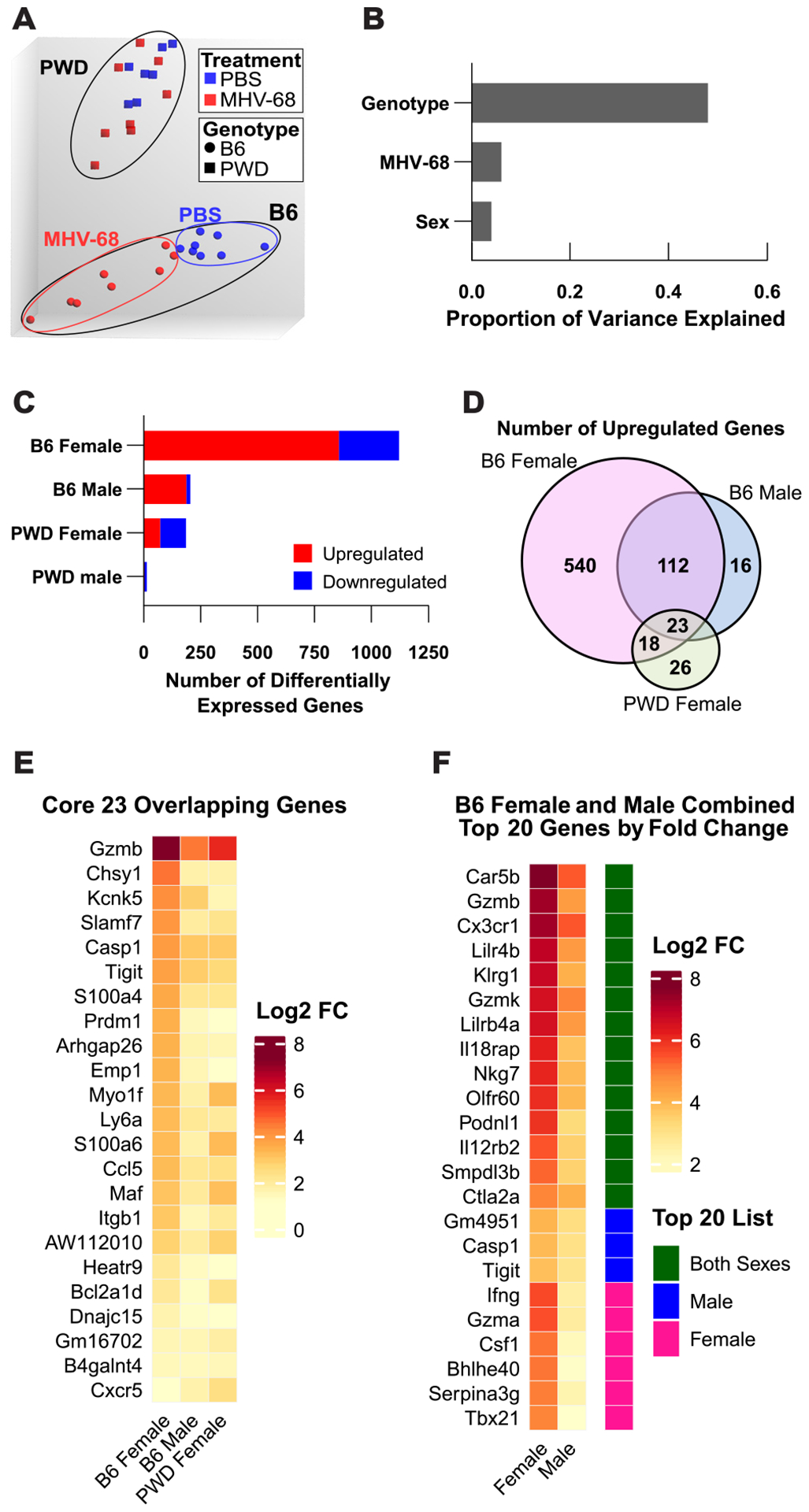
6–8 week old female and male B6 (8F + 8M) and PWD (7F + 8M) littermate mice were randomized to infection with 104 PFU of MHV-68 (4F+4M B6 and 4F+4M PWD) or control PBS (4F+4M B6 and 3F+4M PWD) i.p. injections. At 35 days post-infection, CD4 T cells were isolated from the spleen by FACS (TCRβ+CD4+CD8−CD25−CD11b−CD11c−), RNA was isolated, and bulk transcriptional profiling was performed using Affymetrix microarrays and Applied Biosystems TAC software as described in Materials and Methods. (A) Principal component analysis demonstrating clustering by genotype (shape) and treatment (color), (B) and corresponding proportion of variance in gene expression explained by MHV-68 infection, sex, and genotype. (C) Number of genes passing the differential expression threshold (FDR <0.2 and FC |2|) as a function of latent MHV-68 infection for both genotypes and sexes. (D) Comparison of shared upregulated genes passing the differential expression threshold between B6 females, B6 males, and PWD females, (E) and corresponding heatmap demonstrating the log2 fold change of the 23 shared upregulated genes between B6 females, B6 males, and PWD females. (F) Heatmap demonstrating log2 fold change and gene expression overlap for compiled gene lists of the top 20 upregulated genes (by fold change) for B6 females or B6 males, with green denoting genes present in both sexes top 20 upregulated gene list and blue (male) or pink (female) denoting genes that were specific to the top 20 upregulated gene list of one sex.
Chronic gammaherpesvirus infection results in a prominent cytotoxic signature in CD4 T cells in B6 mice
To understand the functional significance of MHV-68-driven changes in CD4 T cell gene expression, we undertook bioinformatic analyses focused on genes upregulated during latent infection, given their numeric preponderance (Fig. 1C) and their magnitude of change (Fig. 2A). Pathway enrichment analysis of upregulated DEGs using two parallel approaches (Ingenuity software and Enrichr) revealed an upregulation in the activation of several cell-mediated cytotoxic pathways in B6 but not PWD mice, including interferon signaling and natural killer cell signaling and activation, as well as canonical pathways associated with autoimmune diseases such as Th1 and Th17 activation, and disease-specific pathways like SLE and MS (Fig. 2B and C). A more immune-focused analysis of the pattern of expression of the top 20 upregulated genes (by fold change) using the ImmGen database, revealed a strong enrichment of genes canonically associated with cytotoxic function in NKT cells, NK cells, and activated CD8 T cells (the latter specifically in the context of LCMV infection) (Fig. 2D). These genes included (among many others) granzyme genes (Gzma, Gzmb, and Gzmk), T effector subset markers Klrg1, Cx3cr1, and Ccl5, and transcription factor genes Pdrm1 and Tbx21 (encoding BLIMP-1 and T-bet, respectively), with many of these genes upregulated 10–200-fold during latent MHV-68 infection (Fig. 2A). This pattern of gene expression and enrichment is reminiscent of the so-called T helper cytotoxic (ThCTL) phenotype described in cases of influenza A virus (46), EBV, and MHV-68 infection(43, 44, 47).
Figure 2. Latent MHV-68 infection generates a prominent cytotoxic signature in CD4 T cells in B6 mice.
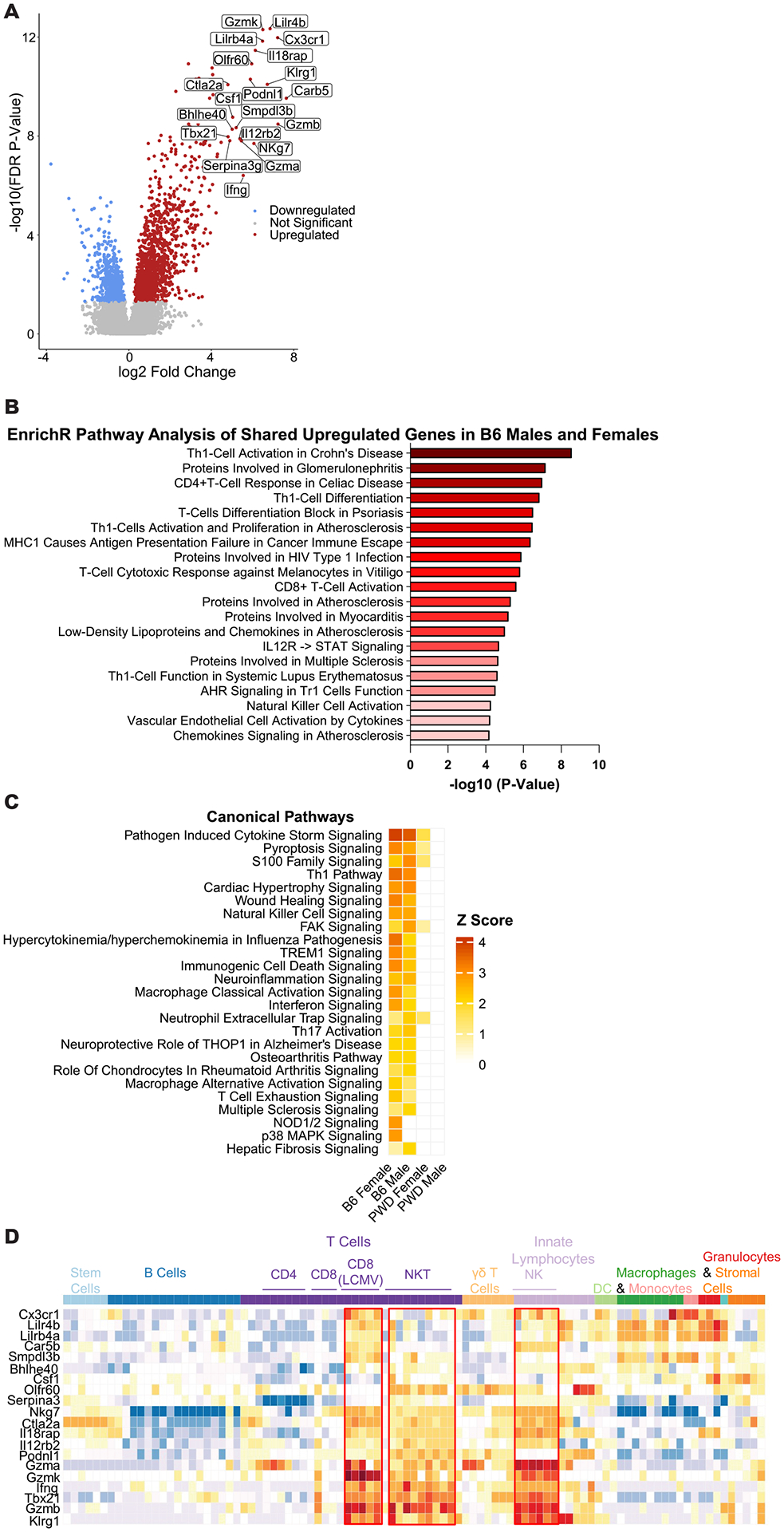
Analysis of upregulated differentially expressed genes in CD4 T cells from B6 MHV-68 latently infected mice, as determined by transcriptional analysis described in Figure 1. (A) Volcano plot demonstrating upregulated genes passing the differential expression threshold (FDR <0.2 and FC |2|) in B6 females as a representative population. The top 20 upregulated genes by fold change are labeled. (B) Pathway analysis for all upregulated genes shared between B6 females and males, top 20 enriched pathways by P value as determined using Enrichr’s Elsevier Pathway Collection are shown (see Materials and Methods). (C) Pathway enrichment analysis of all differentially expressed genes to compare the effect of MHV-68 latency across the four genotype-sex combinations (B6 females, B6 males, PWD females, and PWD male) was conducted utilizing Ingenuity software (see Materials and Methods), top 25 enriched canonical pathways by Z score are shown. (D) Heatmap demonstrating the pattern of expression of the top 20 upregulated genes by fold change in B6 females as a representative population, across selected immune cell types of interest generated utilizing the ImmGen database and MyGeneSet tool (see Materials and Methods).
To further assess the expansion of the ThCTL phenotype during latent MHV-68 infection in B6 mice as suggested by transcriptomic analysis, flow cytometry was utilized. Splenic leukocytes were isolated from B6 and PWD mice latently infected with MHV-68 via the i.p. route, as well as naïve controls. Cell subset markers to assess ThCTL abundance were selected using the upregulated DEG’s identified during transcriptomic analysis, with CX3CR1 and KLRG1 as surrogate markers for the ThCTL phenotype. This analysis revealed a substantial expansion of a CD4+CX3CR1+KLRG1+ population during latent infection in B6 mice that was almost completely absent in infected PWD mice and, as expected, in naïve mice of either genotype (Fig. 3A–B and E). Further analysis also demonstrated a significant increase in the frequency of single positive CX3CR1+ (Fig. 3C and F), and KLRG1+ (Fig. 3D and G) CD4+ subsets during latent infection in B6 mice compared with PWD. Furthermore, B6 mice demonstrated a significantly higher frequency of CD4 T cells expressing IFNγ (Fig. 3H and I) and T-bet (Fig. 3J and K) as compared with PWD mice, also in line with the DEG analysis. Since KLRG1 and CX3CR1 expression typically marks activated effector CD8 T cells(48–50), which are also critical for control of MHV-68(22, 51), we also examined effector CD8 T cell subsets. As expected, B6 mice had a large expansion of a CX3CR1+KLRG1+ CD8 T cell subset, which was greatly reduced, albeit not completely absent, in PWD mice (Fig. 3L and O). The same pattern was seen for expression of IFNγ (Fig. 3M and P) and T-bet (Fig. 3N and Q) in CD8 T cells. Given the differences between B6 females and males in the CD4 T cell transcriptional profiles, with a stronger response in females (Fig. 1 and Fig. 2), flow cytometric data were analyzed splitting the sexes. We observed a trend in increased frequency of CX3CR1+KLRG1+ and IFNγ+ CD4 T cells in female versus male B6 mice, and a significant increase in B6 females compared to B6 males in T-bet expression in CD4 and CD8 T cells, and IFNγ+ in CD8 T cells (Fig. 3E, I, K, Q, and P respectively). Together with the transcriptional profiling results, these data demonstrate that latent MHV-68 infection generates a robust expansion of a discrete population of ThCTL-like effector CX3CR1+KLRG1+ CD4 T cells, which is somewhat more pronounced in B6 female vs. B6 male mice, and completely absent in PWD mice of either sex.
Figure 3. ThCTL cell expansion in latent MHV-68 is restricted to B6 mice.
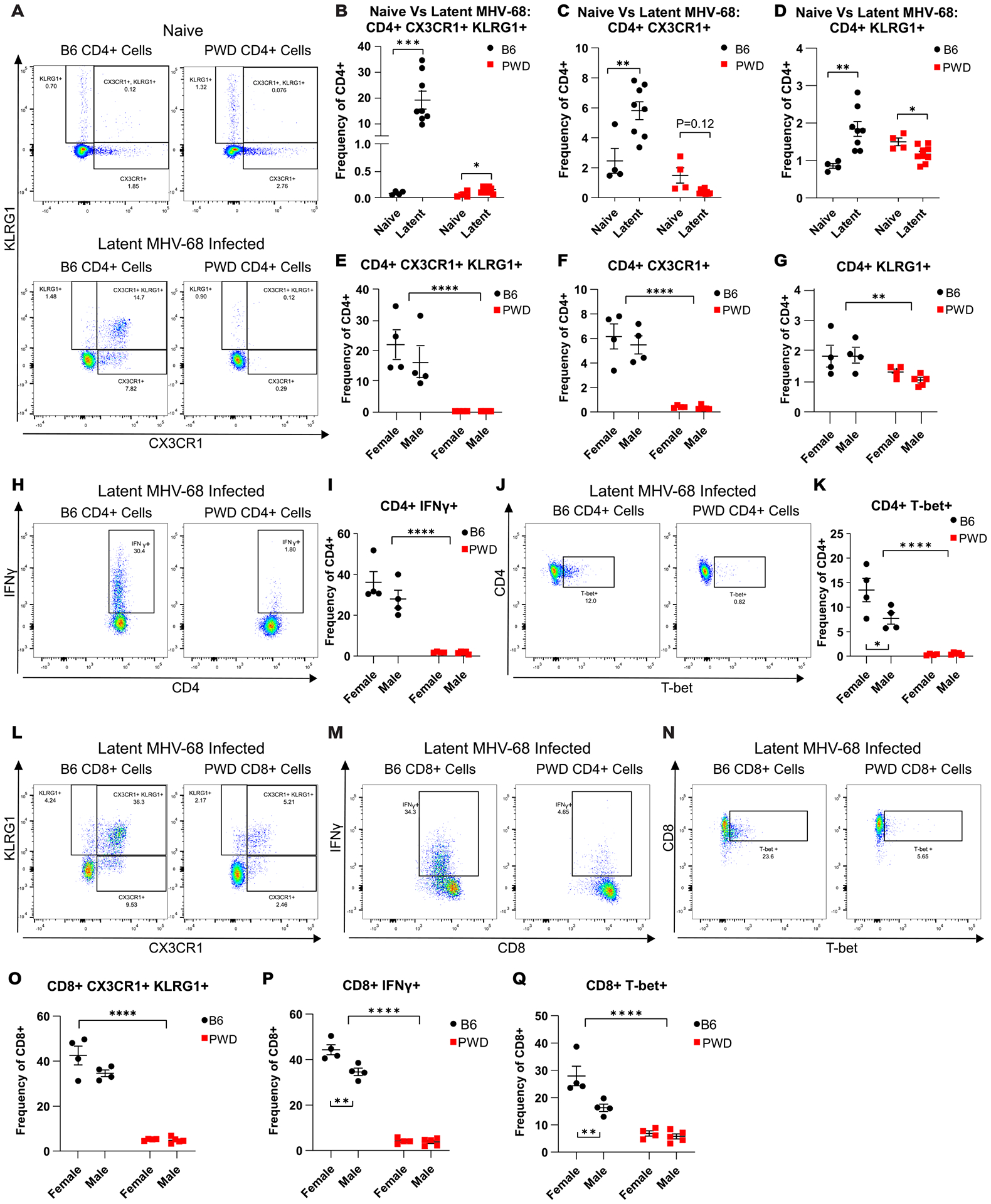
6–8 week old female and male B6 (4F + 4M) and PWD (4F + 5M) mice were i.p. infected with MHV-68 as described in Figure 1. At 35 days post infection splenic leukocytes were isolated and flow cytometry was preformed to assess ThCTL abundance, using CX3CR1 and KLRG1 as surrogate markers (see Materials and Methods). (A-D) Representative CD4 T cell (CD4+TCRβ+CD11b−CD19−) staining profiles and corresponding summary scatterplots comparing CD4+ CX3CR1+KLRG1+ cell subsets from naïve (4 B6 and 4 PWD across two experiments) and latent MHV-68 infected B6 and PWD mice (B6 4F and 4M and PWD 4F and 4M). Significance of differences between naïve and latent timepoints within genotype was determined by unpaired T-tests, or Welch’s T-test in cases where the variance between groups was significantly different as calculated by an F test. Representative staining profiles and corresponding summary scatterplots of (E – G) CD4+ CX3CR1+KLRG1+, (H and I) CD4+IFNγ+ and (J and K) CD4+T-bet+ cell subsets from latent MHV-68 infected B6 and PWD mice. Representative staining profiles and corresponding scatterplots of CD8 T cell (CD8+TCRβ+CD11b−CD19−) subsets from latently MHV-68 infected B6 and PWD mice for CD8+CX3CR1+KLRG1+, CD8+IFNγ+, and CD8+T-bet+ (L-Q). Significance of differences between B6 and PWD genotypes, as well as differences between males and females within genotype, were determined by two-way ANOVA with Šídák’s multiple comparisons test. Comparisons are indicated by the brackets where significant (p<0.05).
To determine the kinetics of expansion of ThCTL’s during MHV-68 infection, splenocytes were harvested from B6 and PWD mice at 5-, 9-, 16- and 35-days post i.p. infection in order to capture the full spectrum of infection from acute phase to latency, and flow cytometric analysis was performed. Analysis of CX3CR1 and KLRG1 expression in CD4 T cells over the course of infection in B6 mice revealed a time-dependent increase in single-positive CX3CR1+ and KLRG1+ cell populations during acute infection and early latency (D9–15), which shifted to a double positive CX3CR1+KLRG1+ population during established viral latency (day 35) (Fig. 4A). Examination of the effect of genotype on cell subsets over the course of infection demonstrated a muted ThCTL (CD4+CX3CR1+KLRG1+) response across infection timepoints in PWD mice compared with B6 mice (Fig. 4B). This blunted response in PWD mice was also consistent in the single positive CX3CR1+ and KLRG1+ subsets across infection timepoints (Fig. 4C and D), except for the KLRG1+ cell subset at 5- and 9-days post infection. At these early time points, PWD mice demonstrated a higher frequency of KLRG1+ cells compared with B6 mice, although this preceded the expansion of KLRG1+ cells seen on D16, when this population was greatly increased in B6 relative to PWD (Fig. 4C), and thus likely indicates a baseline difference. Expression of IFNγ and granzyme B (GzmB) in CD4 T cells followed a similar pattern, with greatly reduced expression in PWD mice (Fig. 4E and F). Taken together, these results demonstrate a robust expansion of ThCTL-like cells in B6 mice, which is initiated during acute infection and sustained during latency, and almost completely absent in PWD mice.
Figure 4. ThCTL cell expansion develops over the course of infection in B6 mice but remains muted across time in PWD mice.
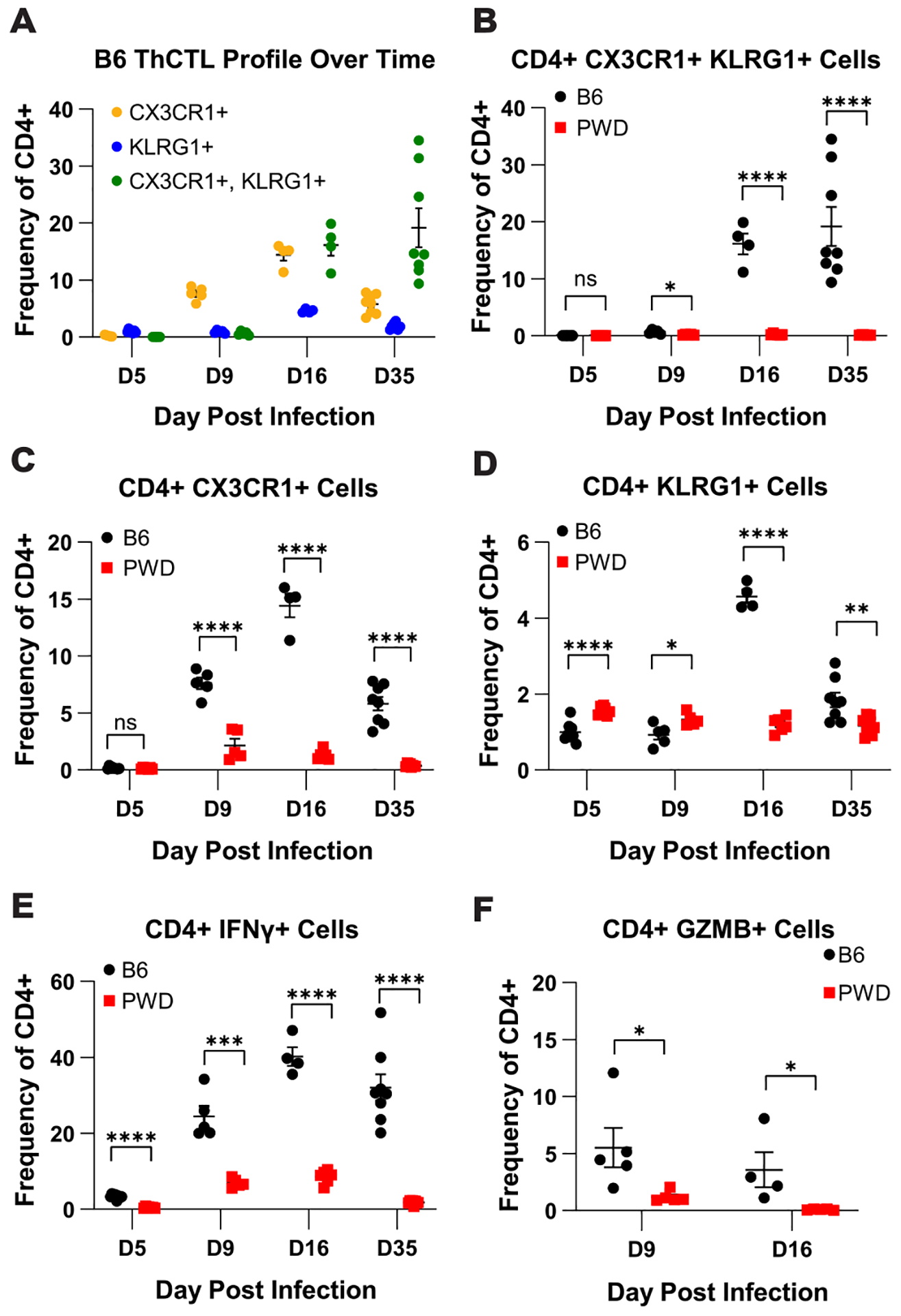
B6 and PWD mice were i.p. infected MHV-68 as described in Figure 1. At 5 (D5) (4F + 4M B6 and 4F + 3M PWD), 9 (D9) (3F + 2M B6 and 3F + 2M PWD), 16 (D16) (2F + 2M B6 and 4F + 2M PWD), or 35 (D35) (4F + 4M B6 and 4F + 4M PWD) days post infection splenic leukocytes were isolated and flow cytometry was preformed to determine ThCTL kinetics during MHV-68 Infection (see Materials and Methods). Male and female data were pooled by genotype. (A) Kinetics of the CX3CR1+KLRG1+ cell subset frequency in CD4 T cells (TCRβ+CD4+CD11b−CD19−) during MHV-68 infection in B6 mice. Comparison of (B) Cx3CR1+KLRG1+, (C) CX3CR1+, (D) KLRG1+, (E) IFNγ+, and (F) GZMB+ cell populations as frequency of total CD4 T cells (TCRβ+CD4+CD11b−CD19−) in B6 and PWD mice over the course of MHV-68 infection. Significance of differences between B6 and PWD mice at each time point was determined via multiple unpaired T-tests and are indicated by the brackets where significant (p<0.05).
PWD mice demonstrate superior control of MHV-68 viral load
To determine if the expansion of the ThCTL phenotype in B6 but not PWD mice was associated with differential control of viral replication, we assessed viral load by measuring relative abundance of MHV-68 ORF50 DNA via qPCR. Analysis at viral latency (day 35 post i.p. infection) – the same timepoint as transcriptome analysis (Fig. 1 and 2) – revealed a ~20-fold reduction in the relative abundance of viral DNA in PWD mice as compared to B6 mice and no significant sex difference in either PWD or B6 mice (Fig. 5A). To further validate these findings, plaque assays for infectious titers were conducted at both day 5 (acute phase) and day 35 (latency) post i.p. infection and demonstrated similar results, with a reduction in infectious virus at day 5, and a lack of infectious virus during latency (Fig. 5B), as expected. To assess if this genetic control of viral replication existed throughout the course of infection, we assessed viral load over the course of i.p. infection via qPCR. PWD mice had a significantly lower viral load as compared to B6 mice, ranging from a ~5 – 1000-fold reduction depending on timepoint, especially during acute infection on day 9 (Fig. 5C). These results demonstrate superior control of viral load in PWD mice, which is achieved during acute infection and maintained throughout latency, and develops in the absence of a major apparent expansion of CD4 and CD8 T cell effector responses (Fig. 1 – 4).
Figure 5. PWD mice demonstrate lower viral load across MHV-68 infection.
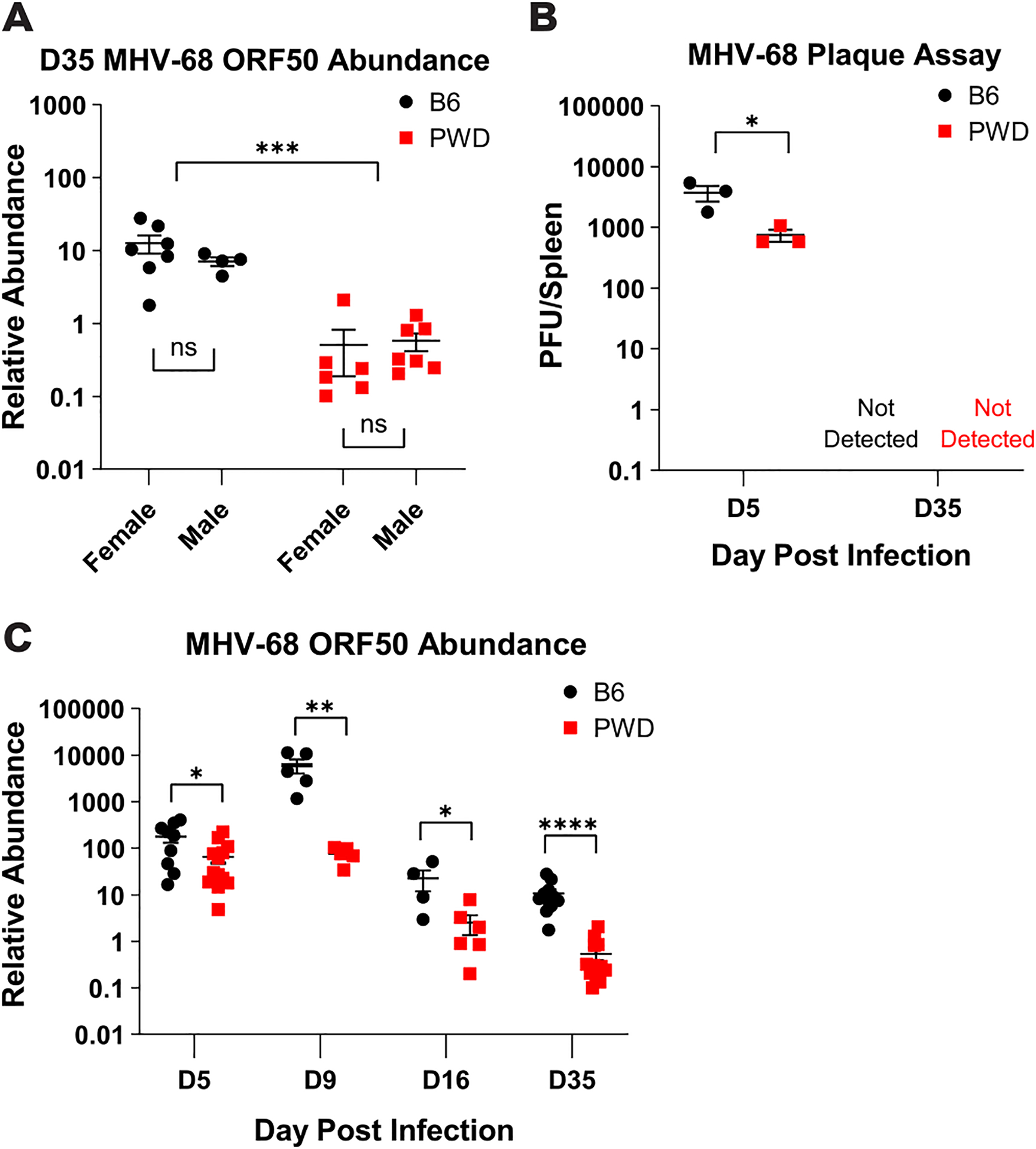
Female and male 6–8 week old B6 and PWD mice were i.p. infected with MHV-68 as described in Figure 1. At 5- (D5) (9F B6 and 6F and 7M PWD, pooled across two experiments), 9- (D9) (3F + 2M B6 and 3F + 2M PWD), 16- (D16) (2F + 2M B6 and 4F + 2M PWD), or 35- (D35) (7F + 4M B6 and 6F + 7M PWD pooled across two experiments) days post infection splenic tissue was harvested and processed for DNA isolation and quantification of viral load by MHV-68 ORF50 qPCR, or determination of infectious viral titers by plaque assay (see Materials and Methods). (A) MHV-68 ORF50 relative abundance (normalized by host DNA abundance) in B6 and PWD mice at the timepoint of transcriptome analysis (D35). Significance of differences between B6 and PWD genotypes, as well as differences between males and females within genotype, were determined by two-way ANOVA with Šídák’s multiple comparisons test. (B) Comparison of plaque forming units (PFU) per spleen in infected B6 and PWD mice, sexes pooled, at D5 and D35. (C) Analysis of viral load in B6 and PWD mice, sexes pooled, across the course of MHV-68 infection via MHV-68 ORF50 DNA relative abundance. Significance of differences between B6 and PWD genotypes at each infection time point were determined by multiple unpaired T-tests with a P value threshold of 0.05. Comparisons are indicated by the brackets where significant (p<0.05).
All of the experiments described above were carried out using the i.p. route of infection to achieve maximal replication in lymphoid tissue and latency. However, for EBV and MHV-68, the natural route of infection is mucosal, followed by spread to lymphoid tissues. To validate our findings using a more physiological relevant mucosal route of infection, mice were infected with 104 PFU of MHV-68 via intranasal inoculation (i.n.). At both 7- (acute) and 35- (latent) days post infection, spleen and lung tissues were harvested for assessment of viral load via qPCR and flow cytometric profiling of CD4 and CD8 T cell phenotypes. Analysis of viral load in the spleen recapitulated our previous findings, with a ~10-fold (P = 0.235) and ~100-fold (P ≤ 0.01) reduction in the relative abundance of viral DNA in PWD mice compared with B6 at 7- and 35 days post infection, respectively (Fig. 6A). Surprisingly, assessment of lung viral load revealed no significant difference between B6 and PWD mice (Fig. 6B), suggesting that superior control of viral load in PWD mice is limited to lymphoid tissue and not acute replication at mucosal sites. Determination of CD4 and CD8 T cell phenotypes in the spleen during latency confirmed the previously noted B6-specific expansion of CD4 and CD8 T cell effector responses, which was greatly reduced in PWD mice. This was demonstrated by significant increases in the frequency of CX3CR1+KLRG1+ ThCTL and CXCR1+KLRG1- Th cells (Fig. 6C–E), IFNγ expressing CD4 T cells (Fig. 6F and G), CD8+CX3CR1+KLRG1+ cells (Fig. 6H and I) and IFNγ expressing CD8 T cells (Fig. 6J and K) in B6 mice compared to PWD mice. Taken together these data demonstrate superior control of MHV-68 replication in PWD mice, which occurs in the absence of ThCTL expansion, and is independent of the route of infection. They also suggest that this restriction of viral replication occurs specifically in lymphoid tissue and not at the mucosal site of initial infection.
Figure 6. PWD mice demonstrate lower splenic viral load and a lack of ThCTL expansion after intranasal MHV-68 infection.
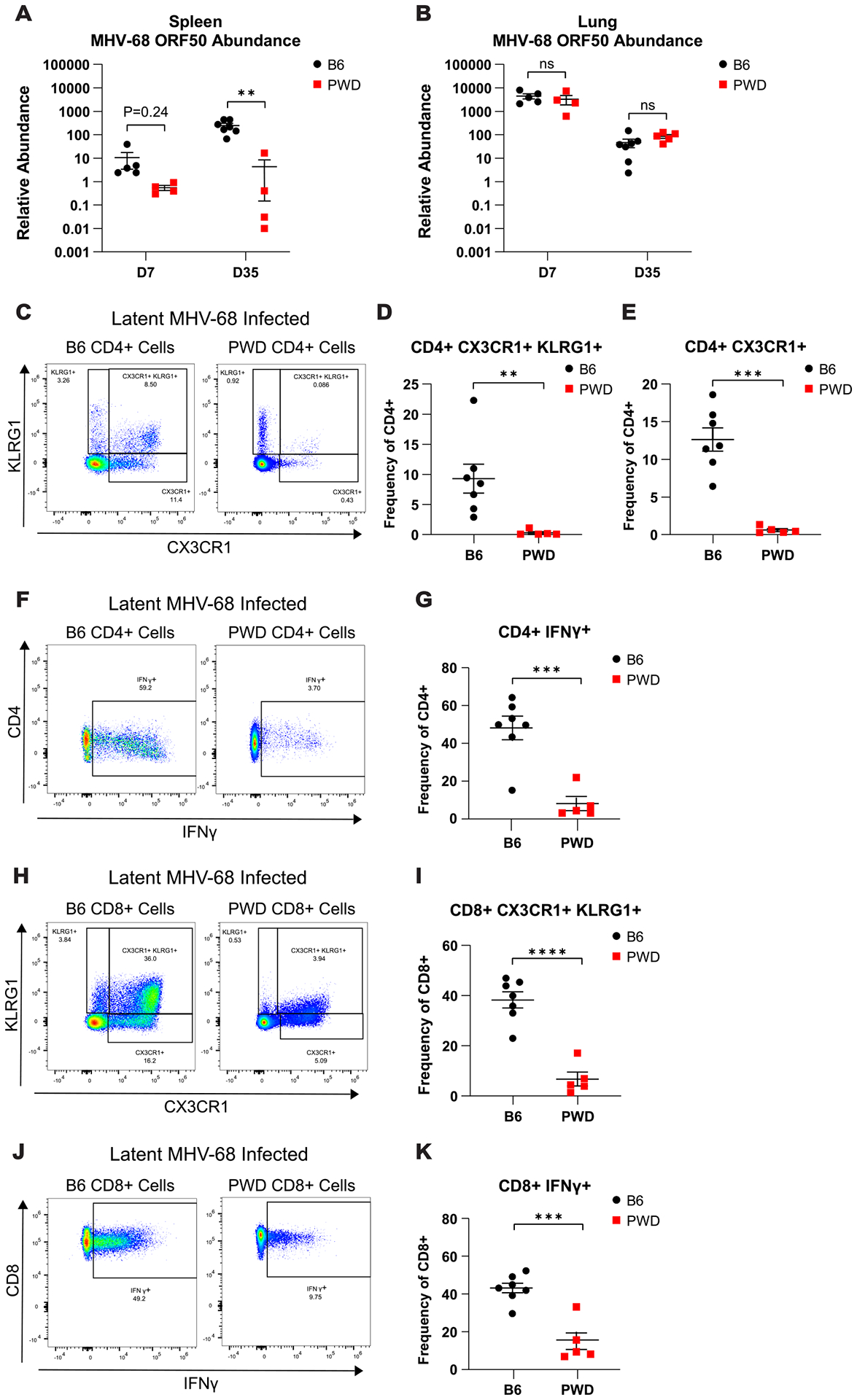
B6 and PWD mice were infected with 104 PFU of MHV-68 via intranasal inoculation (i.n.). At 7- (D7) (5M B6 and 4M PWD) and 35- (D35) (7M B6 and 5M PWD) days post infection spleen and lung tissues were harvested and processed for DNA isolation and quantification of viral load by MHV-68 ORF50 qPCR, or determination of ThCTL abundance in the spleen at D35 via flow cytometry (see Materials and Methods). Assessment of MHV-68 ORF50 relative abundance of B6 and PWD mice at both D7 and D35 in the (A) spleen and (B) lung. Representative splenic CD4 T cell (CD4+TCRβ+CD11b−CD19−) staining profiles and corresponding summary scatterplots comparing (C and D) CD4+ CX3CR1+KLRG1+, (C and E) CD4+ CX3CR1+, and (F and G) CD4+IFNγ+ from latent (D35) i.n. MHV-68 infected B6 and PWD mice. Representative splenic CD8 T cell (CD8+TCRβ+CD11b−CD19−) staining profiles and corresponding summary scatterplots comparing (H and I) CD8+CX3CR1+KLRG1+ and (J and K) CD8+IFNγ+ from latent (D35) MHV-68 i.p. infected B6 and PWD mice. Significant differences between B6 and PWD mice, sexes pooled, at each time point were determined via unpaired T-tests, or Welch’s T-test in cases where the variance between groups was significantly different as calculated by an F test, and are indicated by the brackets where significant (p<0.05). Data represents one independent experiment.
Wild-derived mice are resistant to infection by several viruses, including influenza A virus and West Nile virus, due to cell-intrinsic genetic factors that restrict viral replication, such as Mx1(52) and Oasl1(53). In order to investigate whether the reduction in viral load in PWD mice was due to a similar cell-intrinsic resistance to MHV-68 infection, in vitro replication assays using either PWD or B6 bone marrow derived macrophages (BMDMs) – a cell type permissive to MHV-68 infection – were conducted. BMDMs were cultured from naive PWD and B6 mice and infected with MHV-68 at a multiplicity of infection (MOI) of 0.4, as was done in previous studies(41, 42). Cell supernatants, for viral plaque assay, and cell lysates, for qPCR for viral DNA, were collected at 24-, 48-, and 72-hours post infection in order to assess acute viral replication. Analysis of cell supernatants demonstrated that at 48- and 72-hours post infection PWD BMDMs surprisingly had produced higher titers of infectious virus compared with B6 mice (Fig. 7A). On the other hand, cell-associated MHV-68 DNA abundance was lower in PWD BMDMs at 48-hours post infection, but comparable at other time points (Fig. 7B). This indicates that macrophages from PWD mice, if anything, can support more efficient viral replication and release, as indicated by greater infectious virus titers released into the supernatant. Taken together, these results demonstrate that while PWD mice exhibit superior control over viral replication in vivo, this is not driven by cell-intrinsic resistance to viral replication in macrophages, suggesting that the immune system may be required, and/or other target cell types may be involved.
Figure 7. Efficient replication of MHV-68 in PWD macrophages suggests a lack of cell intrinsic resistance.
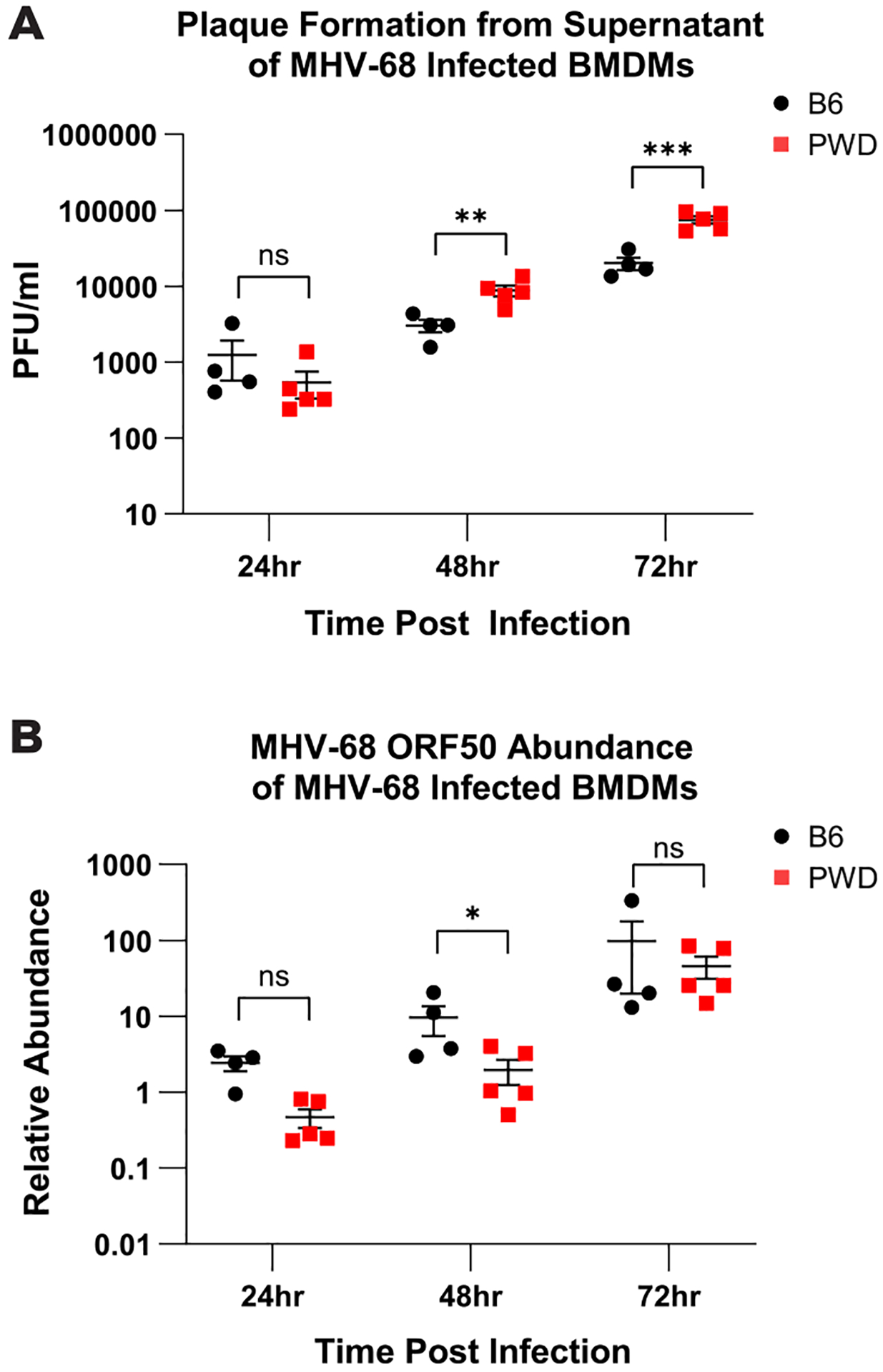
Bone marrow derived macrophages (BMDMs) were cultured from naive B6 (3F +1M) and PWD (3F and 2M) mice and infected with MHV-68 at a multiplicity of infection (MOI) of 0.4. Cell supernatants, for viral plaque assay, and cell lysates, for qPCR for viral DNA abundance, were collected at 24- 48- and 72-hours post infection (see Materials and Methods). (A) Comparison of plaque forming units (PFU) from the supernatant of B6 and PWD infected BMDMs at 24-, 48-, and 72-hours post infection. (B) Assessment of MHV-68 ORF50 relative abundance of B6 and PWD BMDMs at 24-, 48-, and 72-hours post infection. Significant differences between B6 and PWD mice, sexes pooled, at each time point were determined via unpaired T-tests, or Welch’s T-test in cases where the variance between groups was significantly different as calculated by an F test, and are indicated by the brackets where significant (p<0.05).
Pro-viral and anti-viral Th subsets do not segregate with differential control of MHV-68 replication in B6 and PWD mice
Given the almost complete lack of ThCTL response in spite of efficient viral control in PWD mice, we assessed the potential involvement of previously described pro-viral and anti-viral CD4 T cell subsets (namely, Th1, Th17, T follicular helper (Tfh), and T regulatory (Treg) cells) in the control of MHV-68 infection (13, 30–35, 54), using a combinatorial transcriptomic and flow cytometric approach. First, we probed our DEG lists from CD4 T cells isolated from chronically MHV-68 i.p. infected vs. naïve mice (Fig. 1–2) for genes that are known to be associated with T follicular helper (Tfh), Th17, and Th1 lineages (see Materials and Methods). A number of Tfh signature genes, including Tigit, Maf, Il21, Icos, Cxcr5, and Pdcd1 were upregulated by MHV-68 infection in B6 mice, and surprisingly, and unlike what was seen for ThCTL genes, this response was mostly conserved in PWD mice (Fig. 8A). In contrast, other Tfh-associated genes (many of which are reciprocally regulated between Tfh versus ThCTL or Th1 development) including Tcf7, Prdm1, and Runx2 (55–57), were differentially expressed in B6 females only, and in a direction opposing Tfh signatures and favoring Th1 or ThCTL development (Fig. 8A). These findings were also supported by strong upregulation of Th1 signature genes in female and male B6 mice, which was absent in PWD mice (Fig. 8A), corroborating our previously pathway enrichment findings (Fig. 2). Lastly, we saw no evidence of Th17 gene expression in either genotype or sex (Fig. 8A).
Figure 8. Pro-viral and anti-viral Th subsets do not segregate with differential control of MHV-68 replication in B6 and PWD mice.
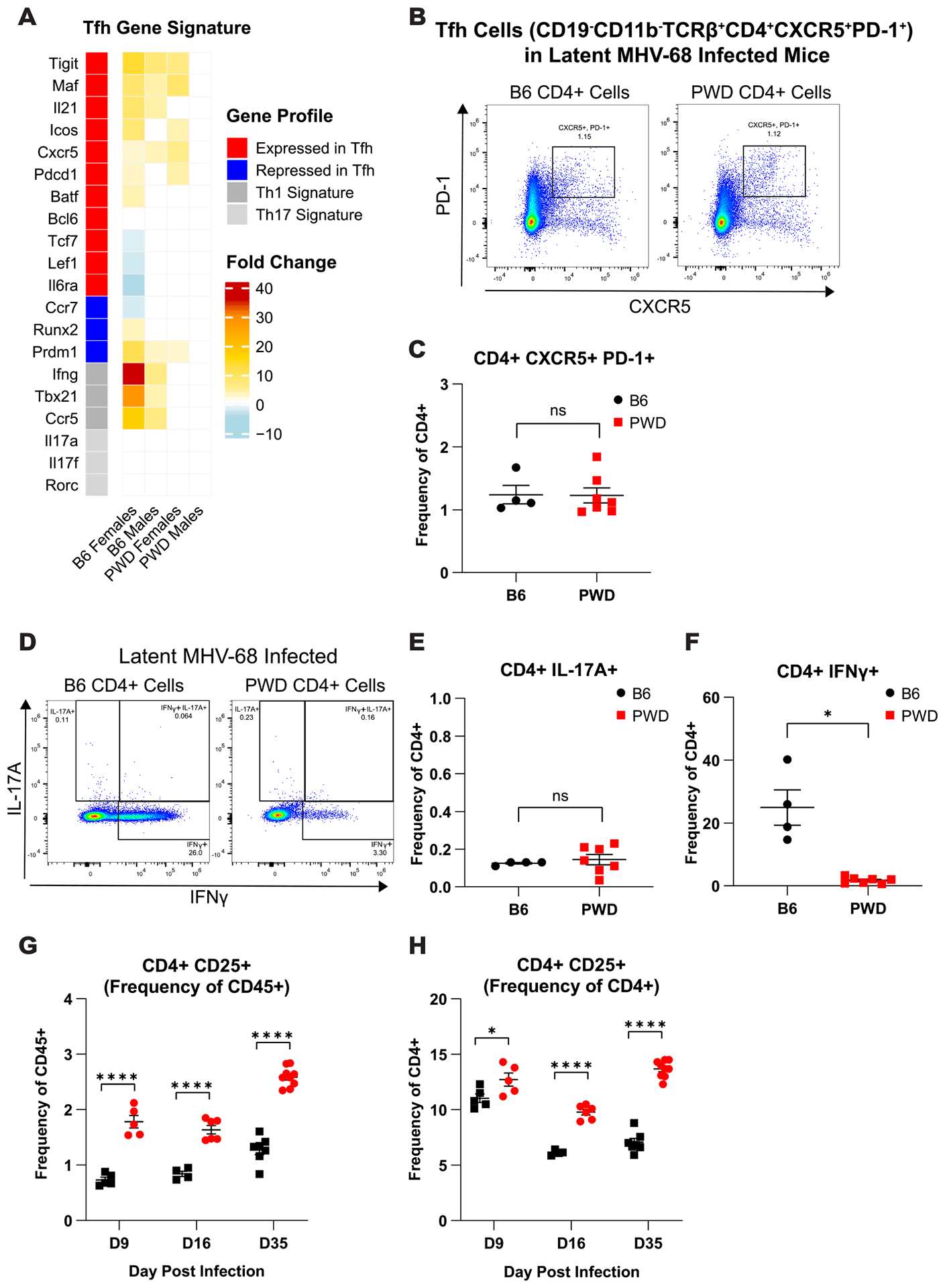
(A) Heatmap of DEGs demonstrating log fold change induced by MHV-68 infection, annotated by Th subset as associated with T follicular helper (Tfh), Th17, and Th1 cells from B6 and PWD MHV-68 infected mice 35 days (D35) post i.p. infection, as determined by CD4 transcriptional analysis described in Figure 1 and Materials and Methods. (B–H) Flow cytometric analysis of anti-viral and pro-viral Th subsets in latent (D35) MHV-68 i.p. infected female and male B6 (2F + 2M) and PWD (5F + 2M) mice. Representative CD4 T cell (CD4+TCRβ+CD11b−CD19−) staining profiles and corresponding summary scatterplots comparing (B and C) Tfh (CD4+CXCR5+PD-1+), (D and E) Th17 (CD4+IL-17A+) and (D and F) IFNγ-expressing Th1 (CD4+IFNγ+) cells. Significant differences between B6 and PWD mice, sexes pooled, were determined via unpaired T-tests, or Welch’s T-test in cases where the variance between groups was significantly different as calculated by an F test, and are indicated by the brackets where significant (p<0.05). Assessment of (CD4+TCRβ+CD11b−CD19−) CD25+ CD4 T cells as a surrogate marker for T regulatory cells in B6 and PWD mice at 9- (3F + 2M B6 and 3F + 2M PWD), 16- (2F + 2M B6 and 4F + 2M PWD), and 35- (4F and 3M B6 and 4F and 5M PWD)(data from two independent experiments), shown as (G) frequency of total CD45+ splenocytes and (H) frequency of CD4+ cells. Significance of differences between B6 and PWD mice at each time point was determined via unpaired T-tests or Welch’s T-test in cases where the variance between groups was significantly different as calculated by an F test, and are indicated by the brackets where significant (p<0.05).
Next, we assessed the frequency of the above-mentioned Th subsets in latent MHV-68 i.p. infected B6 and PWD mice using flow cytometry. We found no significant differences in Tfh (CD4+CXCR5+PD-1+) (Fig. 8B and C) and Th17 (CD4+Il17+) (Fig. 8D and E) cells between B6 or PWD mice, while the frequency of IFNγ expressing Th1 cells (CD4+IFNγ+) were greatly increased in B6 mice compared with PWD (Fig. 8D and F). Using CD25 as a surrogate marker for Treg cells (58–60), we found that the frequency of CD4+CD25+ T cells, shown either as a frequency of total CD45+ (Fig. 8G) or CD4+ T cells (Fig. 8H), was significantly greater in PWD mice compared with B6 mice. These data demonstrate that superior control of viral replication in PWD mice compared with B6 occurs in the presence of: 1) comparable frequency of pro-viral Tfh and Th17 cells, 2) higher frequency of pro-viral Tregs, and 3) in the absence of a strong anti-viral Th1 response. Taken together, these results suggest that differential abundance of anti-viral and pro-viral T helper subsets does not account for differences in viral replication between PWD and B6 mice, and that ThCTL and Th1 expansion during chronic infection in B6 likely represents a downstream consequence of inferior viral control.
NK cell mediated mechanisms contribute to superior viral load control in PWD mice
Given the early control of viral replication observed in vivo in PWD mice (Fig. 5), which occurred in the absence of a cell-intrinsic barrier to virus replication, at least in macrophages (Fig. 7), with minimal apparent engagement of effector T cell responses (Fig. 4), we hypothesized that innate immune anti-viral responses could play a role in the ability of PWD mice to control MHV-68 replication. As NK cells are critical in early response to viruses, specifically the clearance of virally infected cells(61), we investigated the contribution of NK cells to the control of MHV-68 viral load in PWD and B6 mice using antibody-based depletion. NK cell depletion in B6 mice can be effectively achieved using antibodies to the NK1.1 marker, which unfortunately represents a strain-specific allelic variant which we confirmed is not expressed in PWD mice (Fig. 9A). An alternative depletion involves antibodies to the marker asialo-ganglioside-monosialic acid (ASGM1)(62–64). We found that ASGM1 expression in PWD and B6 mice marks a non-T/B cell population that co-expresses CD49b (Fig. 9A and B), another widely used NK marker(65, 66), suggesting that ASGM1 expression is conserved on PWD NK cells. Therefore, to determine the contribution of NK cells to control of viral load, NK cells were depleted in PWD and B6 mice by anti-ASGM1 or anti-NK1.1 treatment, respectively, on day −1, and NK-depleted or isotype control-treated mice were then infected with MHV-68 via i.p. injection and followed to peak viral replication at 9 days post infection. Successful NK cell depletion was verified via flow cytometric analysis (Fig. 9A and C). Analysis of splenic infectious virus titers demonstrated that NK depletion in PWD mice increased viral titer to a level that was no longer significantly different from either IgG-control or NK-depleted B6 mice (Fig. 9D). Analysis of viral DNA load demonstrated that NK depletion in PWD mice significantly elevated viral load compared with IgG-treated controls, but not quite to the level seen in control or NK-depleted B6 mice (Fig. 9E). NK depletion in B6 mice did not impact viral load as assessed by infectious virus titer or viral DNA load, recapitulating previously published findings demonstrating a lack of role for NK cells in controlling MHV-68 infection in B6 mice(37).
Figure 9. NK cells contribute to superior viral load control in PWD mice.
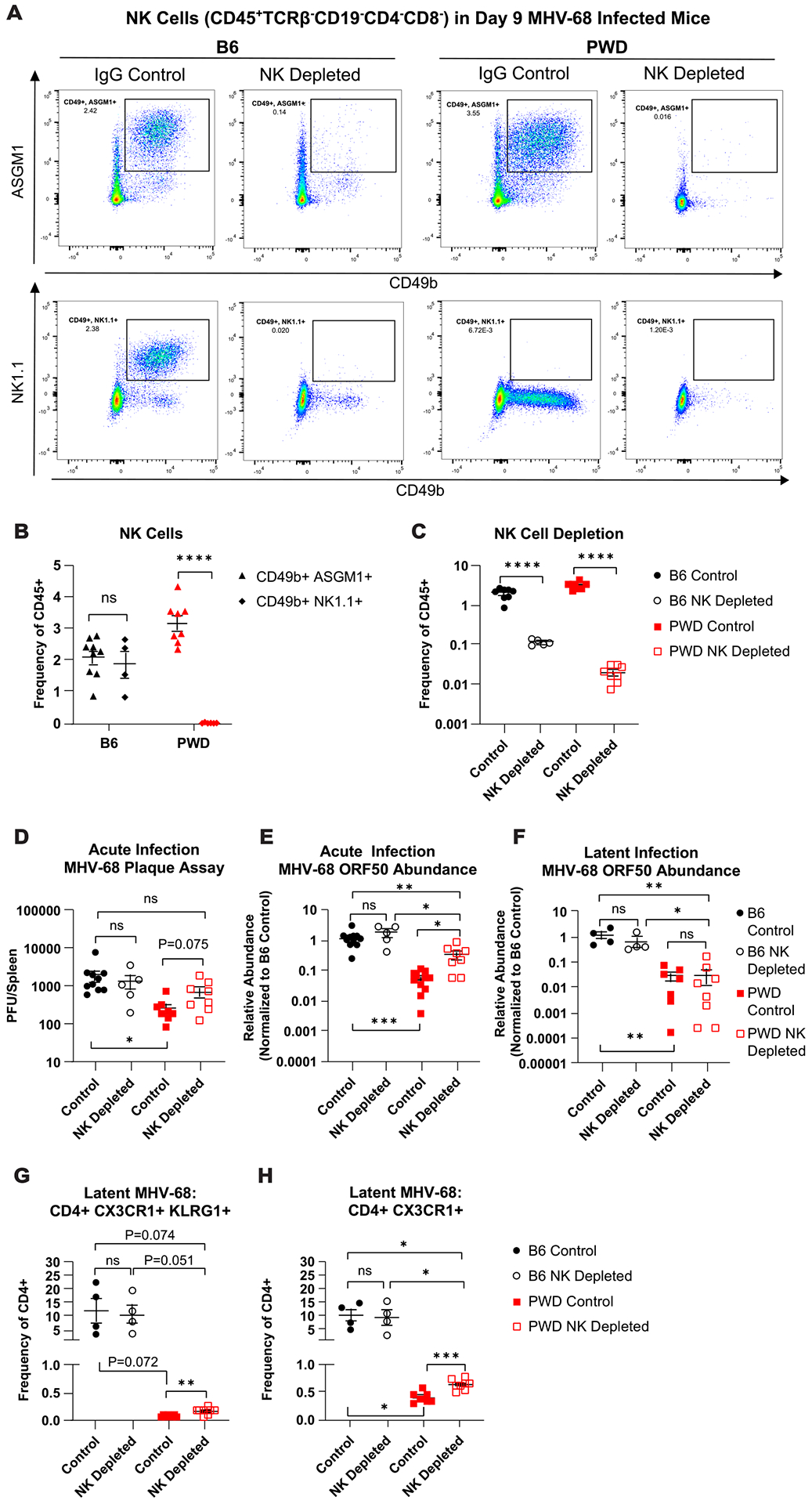
Female and male B6 and PWD mice were treated with anti-NK1.1 (acute n = 2F + 3M B6, latent n = 2F + 2M B6) or anti-ASGM-1 antibodies (acute n = 2F + 6M PWD, latent n = 6F + 2M PWD), respectively (NK depleted) or IgG isotype control antibodies (control) (acute n = 8F + 3M B6 and 4F + 8M PWD, latent n = 2F + 2M B6 and 5F + 2M PWD) and one day later infected with 104 PFU of MHV-68. Data shown are pooled from three independent experiments. At 9- (D9) and 35- (D35) days post infection, spleens were collected and processed for flow cytometry, qPCR, or infectious viral titration (see Materials and Methods). (A) Representative staining profiles of NK cell markers CD49b, NK1.1, and ASGM1 on CD45+TCRβ− CD19− CD4− CD8− cell populations in NK depleted or IgG control-treated D9 B6 and PWD mice. (B) Assessment of CD49b+ASGM1+ and CD49b+NK1.1+ NK cell populations in D9 MHV-68 infected B6 and PWD mice, represented as frequency of total CD45+ splenocytes. (C) NK cell populations (TCRβ− CD19− CD4− CD8− CD49b+ASGM1+) as frequency of CD45+ cells in D9 B6 and PWD NK depleted and IgG control mice. (D) Comparison of plaque forming units (PFU) between D9 B6 and PWD IgG control and NK depleted mice. Relative abundance of MHV-68 ORF DNA normalized to B6 IgG control, in B6 and PWD IgG control and NK depleted mice at (E) D9 and (F) D35, as assessed by qPCR. Assessment of ThCTL cells – (CD4+TCRβ+CD11b−CD19−) (G) CD4+CX3CR1+KLRG1+ and (H) CD4+CX3CR1+ at D35 in B6 and PWD IgG control and NK depleted mice. Significant differences between or within B6 and PWD mice, sexes pooled, given IgG control or NK depletion conditions were determined via unpaired T-tests, or Welch’s T-test in cases where the variance between groups was significantly different as calculated by an F test, and are indicated by the brackets where significant (p<0.05).
To characterize the impact of acute NK cell depletion on subsequent chronic levels of gammaherpesvirus persistence, NK cells were depleted in B6 and PWD mice one day prior to viral infection as described above, infected with MHV-68 via i.p. injection, and followed to latency at 35 days post infection. Assessment of splenic viral DNA levels revealed a lack of significant difference in viral load between NK depleted and IgG control PWD and, as expected, B6 mice (Fig. 9F), suggesting that NK mediated control of acute viral replication does not impart a sustained effect on the level of latent virus. Subsequent assessment of ThCTL cell frequencies via flow cytometry revealed a slight but significant increase in CD4+CX3CR1+KLRG1+ and CD4+CX3CR1+ in NK depleted PWD mice compared with IgG controls, although far below the level of B6 mice, which remained unchanged by NK depletion (Fig. 9G and H). Taken together, these data indicate that NK cells in PWD mice contribute to the control of acute gammaherpesvirus replication and suggest host genetic variation can regulate gammaherpesvirus replication through disparate immunological mechanisms.
Discussion
It has been well documented that chronic infection with EBV is a risk factor for several autoimmune diseases(1, 3–5), however the underlying mechanisms and role of host genetics is still unclear, although immune dysregulation driven by chronic EBV infection is a likely contributor. Animal models represent a tool to bridge the gap between observation and causation/mechanism through the control of genetic and environmental risk factors in a regulated setting(67). Due to the restricted host tropism of EBV, MHV-68, a structurally similar and related gammaherpesvirus to EBV, has been well-accepted and utilized as a small animal model of gammaherpesvirus infection(10–12). In this study, we leveraged conventional laboratory inbred B6 and genetically divergent wild-derived PWD mice to determine the role of host genotype in MHV-68 infection, as well as the subsequent immune responses required for control of this virus. This approach revealed several unique phenotypes in PWD mice compared with B6 mice, revealing intriguing blind spots that had resulted from previous studies of host determinants of gammaherpesvirus immunity in a narrow set of host genotypes. First, while chronic MHV-68 infection in B6 mice resulted in a massive expansion of T helper cells with cytotoxic characteristics (ThCTL), this phenotype was almost completely absent in PWD mice. In spite of this apparent lack of adaptive immune activation, PWD mice controlled viral replication in lymphoid tissue better than B6 mice. This was at least in part mediated by NK cells, rather than a cell-intrinsic barrier to infection, at least as supported by enhanced in vitro replication of MHV-68 in PWD macrophages. Taken together, our results suggest that genetically-determined efficient control of gammaherpesvirus replication may minimize subsequent adaptive immune dysregulation, such as ThCTL expansion.
Related to the role of EBV-induced immune dysregulation as a risk factor for autoimmune disease, ThCTL cells have been found to be expanded in individuals with SLE(68), RA(69), and MS(70, 71). In a recent study, using single cell transcriptomics, Schafflick et al., identified a ThCTL population in the cerebral spinal fluid (CSF) that represented the mostly highly expanded T cell subset in patients with MS compared with healthy controls. The group identified several marker genes expressed by this ThCTL population, most of which corresponded with the upregulated differentially expressed genes of MHV-68 latently infected B6 mice identified in our study (Supplemental Table 1); including GZMB, CX3CR1, CCL5, NKG7, EOMES, and TBX21 among others(72). Another recent study identified an expansion of Eomes- and GzmB-expressing ThCTL in the blood and CNS lesions of secondary progressive MS patients(71). Additional studies have implicated the involvement and/or expansion of ThCTL’s in other autoimmune diseases where chronic EBV infection has been implicated as a risk factor(68, 69). The expansion of ThCTL’s in these autoimmune diseases implicates a plausible mechanism by which chronic EBV infection could augment autoimmune disease risk through skewing of the immune system. By extension, given our present findings together with the emerging findings that control of EBV replication could be host genotype dependent(9) and the identification of HLA genotype-dependent responses to EBV infection and subsequent elevated autoimmune disease risk(7, 8), it can be suggested that genotype-dependent inefficient control of gammaherpesvirus infection can result in immune deviation and a predisposition towards an autoimmune state.
The role of NK cells in gammaherpesvirus infection has been a topic of considerable interest in viral immunity. While there is considerable evidence that NK cells contribute to control of EBV replication in humans(73), multiple studies in mice have suggested a lack of a major role for these innate immune cells. Utilizing B6 WT control and NK cell deficient mice, as well as NK cell depletion Usherwood and colleagues examined the impact of NK cells during MHV-68 infection. The group demonstrated a lack of significant difference in viral load across several infection time points in the examined NK depleted or NK deficient mice compared with controls, suggesting that NK cells have little impact on control of MHV-68 infection(37). These findings were further supported through an investigation into the contribution of NK cells on the innate immune responses to MHV-68 infection by Thomson and colleagues. The authors demonstrated a lack of significant difference in viral load between NK cell depleted and control B6 mice across viral infection in several infectious compartments, despite the identification of an expanded NK cell population in B6 mice 2 days post infection and in absence of evidence of MHV-68 inhibition on NK cell function(74). The conclusions of these studies, indicating a lack of role of NK cells in MHV-68 infection, are in line with our findings following NK cell depletion in B6 mice but are in contrast to those of NK cell depletion in PWD mice – which suggest that NK cell mediated control of acute viral load is dependent on genotype, and caution against overinterpretation of the results of an experimental animal model based on a single genotype (i.e. B6); a notion that is reinforced by our recent findings that PWD myeloid cells behave more similarly to human myeloid cells(75). It should be noted that in order to achieve NK cell depletion in PWD mice, experimental design dictated the use of anti-ASGM1 treatment, as opposed to anti-NK1.1 in B6 mice, as to our knowledge an NK-specific depletion antibody has yet to be identified for PWD mice. While depletion with ASGM1 has been previously supported in the literature for NK cell depletion in various strains(62–64), ASGM1 is not as exclusive to NK cells as NK1.1, adding the caveat of potential off target cell depletion. This experimental constraint notwithstanding, it is plausible that this difference in the role of NK cells during MHV-68 infection could be related to the genetic control of PWD NK cells during infection. Lastly, it is worth noting that studies of experimental MHV-68 infection in a wild-derived wood mouse host (Apodemus sylvaticus) compared with standard inbred BALB/c mice demonstrated marked differences in pathogenesis, including reduced viral loads and splenomegaly, and altered anti-viral immune responses(76), consistent with our findings in PWD mice.
Our discovery that NK cell-mediated control of gammaherpesvirus infection is host genotype-dependent is supported by the extensive literature demonstrating the importance of NK cells in the control of alpha- and betaherpesviruses, such as cytomegalovirus (CMV)(77, 78). In this regard, several studies have identified major host immunogenetic determinants of murine cytomegalovirus (MCMV) susceptibility. This includes the identification of the Cmv1 locus located within the NK gene complex of chromosome 6, which has been shown to control survival and resistance of MCMV infection, and identification of Ly49H, an NK cell activation receptor located within the Cmv1 locus, that has been associated with NK-mediated resistance to MCMV in B6 mice(77). Furthermore, additional studies demonstrated the existence of strain-specific variants in genes encoding Ly49 family NK activating receptors in NOD/LtJ, PWK/Pas, and BALB/c mice, which recognize MCMV-infected cells in a H2 haplotype-dependent manner(79). These studies also suggest that these strain-specific variants can exhibit differential control of MCMV proliferation(79) and utilize divergent mechanisms to mediate MCMV resistance(80, 81). Considering the evidence of genetically differential control of herpesvirus replication via NK cells, our presented data on the role of NK cells in MHV-68 infected PWD mice align more closely to that of the NK mediated control seen in MCMV infection as opposed to the previously published lack of NK control of MHV-68 in B6 mice. Taken together, these results suggest that host genotype plays a determinant role in the mechanism associated with control of gammaherpesvirus viral replication and the subsequent immune responses, which could impact the baseline immunological state and propensity towards autoimmunity. In this light, studies have demonstrated a link between CMV-driven expansion of NK cell populations and delayed MS onset(82), which is further strengthened by evidence for reduced MS risk associated with CMV seroconversion(5), and possible protective roles of NK cell subsets in MS(83, 84) and its models(85).
A surprising finding from our intranasal infection studies was that superior control of MHV-68 replication in PWD mice was seen only in the spleen and not in the lung (Fig. 6A and B). This suggests several possibilities. First, acute replication in this model occurs in lung epithelial cells and alveolar macrophages, followed by spread to lymphoid organs and replication predominantly in B cells, followed by latency in B cells (12). It is thus possible that there is a B cell-specific restriction of MHV-68 replication and/or latency establishment in PWD mice, which is absent in epithelial cells and macrophages, with the latter consistent with our in vitro replication results in macrophages (Fig. 7). Alternatively, there may be a PWD-specific restriction of viral spread from the lung (and the peritoneal cavity during i.p. infection) to the spleen and lymphoid organs. Lastly, there may be specific immunological factors favoring elimination of infected cells specifically in the spleen in PWD mice, such as for example, more effective lymphoid tissue-specific NK responses, as described above. Future mechanistic studies can help differentiate between these intriguing possibilities.
It has been shown in a recent study that host genetics has a strong impact on NK cell abundance and phenotype as assessed utilizing a group of genetically diverse mice called the Collaborative Cross (CC) mice(86), and that these traits are associated with specific loci, as determined by quantitative trait loci (QTL) mapping(87). Given this, our future studies will focus on identification of novel variants in specific host genes driving immunological control of gammaherpesvirus infection. To this end, the B6.ChrPWD chromosome substitution model used in our previous studies(17, 18), or the CC model, which carries alleles from PWK/PhJ mice that are closely related to PWD, can be employed. These studies will aim to leverage distinct immunological responses to MHV-68 infection, such as those demonstrated by B6 and PWD mice, to identify loci associated with these unique phenotypes. Additionally, these models can be used to examine the impact of chronic gammaherpesvirus infection in the context of murine autoimmune models, which so far have been restricted predominantly to the B6 models(24, 88, 89). Together, these studies will help elucidate the complex mechanisms underlying balanced host responses to these ubiquitous viruses, which have co-evolved so successfully with their mammalian hosts, potentially revealing prophylactic interventions aimed at lowering the risk of autoimmune sequelae during chronic infection.
Supplementary Material
Key Findings.
Host genotype alters immunological mechanisms of gammaherpesvirus infection control
C57BL/6J mice expand ThCTL cell populations in response to chronic MHV-68 infection
Superior control of MHV-68 viral load in PWD/PhJ mice is dependent on NK cells
Acknowledgments
The authors would like to acknowledge the Vermont Integrative Genomics Resource and the Flow Cytometry and Cell Sorting facility at the University of Vermont Larner College of Medicine for the use of their facilities and resources.
References
- 1.Jog NR, Young KA, Munroe ME, Harmon MT, Guthridge JM, Kelly JA, Kamen DL, Gilkeson GS, Weisman MH, Karp DR, Gaffney PM, Harley JB, Wallace DJ, Norris JM, and James JA. 2019. Association of Epstein-Barr virus serological reactivation with transitioning to systemic lupus erythematosus in at-risk individuals. Ann Rheum Dis 78: 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jog NR, and James JA. 2021. Epstein Barr Virus and Autoimmune Responses in Systemic Lupus Erythematosus. Frontiers in Immunology 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Houen G, and Trier NH. 2020. Epstein-Barr Virus and Systemic Autoimmune Diseases. Front Immunol 11: 587380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ascherio A, and Munger KL. 2007. Environmental risk factors for multiple sclerosis. Part I: The role of infection. Annals of Neurology 61: 288–299. [DOI] [PubMed] [Google Scholar]
- 5.Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, Elledge SJ, Niebuhr DW, Scher AI, Munger KL, and Ascherio A. 2022. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 375: 296–301. [DOI] [PubMed] [Google Scholar]
- 6.Bar-Or A, Pender MP, Khanna R, Steinman L, Hartung HP, Maniar T, Croze E, Aftab BT, Giovannoni G, and Joshi MA. 2020. Epstein-Barr Virus in Multiple Sclerosis: Theory and Emerging Immunotherapies. Trends Mol Med 26: 296–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agostini S, Mancuso R, Guerini FR, D’Alfonso S, Agliardi C, Hernis A, Zanzottera M, Barizzone N, Leone MA, Caputo D, Rovaris M, and Clerici M. 2018. HLA alleles modulate EBV viral load in multiple sclerosis. J Transl Med 16: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hedstrom AK, Huang J, Michel A, Butt J, Brenner N, Hillert J, Waterboer T, Kockum I, Olsson T, and Alfredsson L. 2019. High Levels of Epstein-Barr Virus Nuclear Antigen-1-Specific Antibodies and Infectious Mononucleosis Act Both Independently and Synergistically to Increase Multiple Sclerosis Risk. Front Neurol 10: 1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Houldcroft CJ, and Kellam P. 2015. Host genetics of Epstein-Barr virus infection, latency and disease. Rev Med Virol 25: 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barton E, Mandal P, and Speck SH. 2011. Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu. Rev. Immunol 29: 351–397. [DOI] [PubMed] [Google Scholar]
- 11.Olivadoti M, Toth LA, Weinberg J, and Opp MR. 2007. Murine gammaherpesvirus 68: a model for the study of Epstein-Barr virus infections and related diseases. Comp Med 57: 44–50. [PubMed] [Google Scholar]
- 12.Flano E, Woodland DL, and Blackman MA. 2002. A mouse model for infectious mononucleosis. Immunologic Research 25: 201–217. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Tibbetts SA, and Krug LT. 2021. Conquering the Host: Determinants of Pathogenesis Learned from Murine Gammaherpesvirus 68. Annual Review of Virology 8: 349–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gregorova S, and Forejt J. 2000. PWD/Ph and PWK/Ph inbred mouse strains of Mus m. musculus subspecies--a valuable resource of phenotypic variations and genomic polymorphisms. Folia biologica 46: 31–41. [PubMed] [Google Scholar]
- 15.Viney M, Lazarou L, and Abolins S. 2015. The laboratory mouse and wild immunology. Parasite Immunol 37: 267–273. [DOI] [PubMed] [Google Scholar]
- 16.Bearoff F, del Rio R, Case LK, Dragon JA, Nguyen-Vu T, Lin C-Y, Blankenhorn EP, Teuscher C, and Krementsov DN. 2016. Natural genetic variation profoundly regulates gene expression in immune cells and dictates susceptibility to CNS autoimmunity. Genes Immun 17: 386–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montgomery TL, Künstner A, Kennedy JJ, Fang Q, Asarian L, Culp-Hill R, D’Alessandro A, Teuscher C, Busch H, and Krementsov DN. 2020. Interactions between host genetics and gut microbiota determine susceptibility to CNS autoimmunity. Proceedings of the National Academy of Sciences 117: 27516–27527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bearoff F, Case LK, Krementsov DN, Wall EH, Saligrama N, Blankenhorn EP, and Teuscher C. 2015. Identification of Genetic Determinants of the Sexual Dimorphism in CNS Autoimmunity. PLoS ONE 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krementsov DN, Asarian L, Fang Q, McGill MM, and Teuscher C. 2018. Sex-Specific Gene-by-Vitamin D Interactions Regulate Susceptibility to Central Nervous System Autoimmunity. Frontiers in Immunology 9: 1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lahue KG, Lara MK, Linton AA, Lavoie B, Fang Q, McGill MM, Crothers JW, Teuscher C, Mawe GM, Tyler AL, Mahoney JM, and Krementsov DN. 2020. Identification of novel loci controlling inflammatory bowel disease susceptibility utilizing the genetic diversity of wild-derived mice. Genes & Immunity 21: 311–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sabikunnahar B, Lahue KG, Asarian L, Fang Q, McGill MM, Haynes L, Teuscher C, and Krementsov DN. 2022. Sex differences in susceptibility to influenza A virus infection depend on host genotype. PLoS One 17: e0273050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Obar JJ, Crist SG, Gondek DC, and Usherwood EJ. 2004. Different Functional Capacities of Latent and Lytic Antigen-Specific CD8 T Cells in Murine Gammaherpesvirus Infection. The Journal of Immunology 172: 1213–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sunil-Chandra NP, Efstathiou S, Arno J, and Nash AA. 1992. Virological and pathological features of mice infected with murine gammaherpesvirus 68. Journal of General Virology 73: 2347–2356. [DOI] [PubMed] [Google Scholar]
- 24.Casiraghi C, Shanina I, Cho S, Freeman ML, Blackman MA, and Horwitz MS. 2012. Gammaherpesvirus latency accentuates EAE pathogenesis: relevance to Epstein-Barr virus and multiple sclerosis. PLoS Pathog 8: e1002715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’Ayan A. 2013. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, and Ma’ayan A. 2016. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, Jeon M, and Ma’ayan A. 2021. Gene Set Knowledge Discovery with Enrichr. Current Protocols 1: e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blankenhorn EP, Butterfield R, Case LK, Wall EH, del Rio R, Diehl SA, Krementsov DN, Saligrama N, and Teuscher C. 2011. Genetics of experimental allergic encephalomyelitis supports the role of T helper cells in multiple sclerosis pathogenesis. Annals of Neurology 70: 887–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heng TSP, Painter MW, Elpek K, Lukacs-Kornek V, Mauermann N, Turley SJ, Koller D, Kim FS, Wagers AJ, Asinovski N, Davis S, Fassett M, Feuerer M, Gray DHD, Haxhinasto S, Hill JA, Hyatt G, Laplace C, Leatherbee K, Mathis D, Benoist C, Jianu R, Laidlaw DH, Best JA, Knell J, Goldrath AW, Jarjoura J, Sun JC, Zhu Y, Lanier LL, Ergun A, Li Z, Collins JJ, Shinton SA, Hardy RR, Friedline R, Sylvia K, and Kang J. 2008. The Immunological Genome Project: networks of gene expression in immune cells. Nature Immunology 9: 1091–1094. [DOI] [PubMed] [Google Scholar]
- 30.Gauld SB, De Santis JL, Kulinski JM, McGraw JA, Leonardo SM, Ruder EA, Maier W, and Tarakanova VL. 2013. Modulation of B-cell tolerance by murine gammaherpesvirus 68 infection: requirement for Orf73 viral gene expression and follicular helper T cells. Immunology 139: 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collins CM, and Speck SH. 2015. Interleukin 21 signaling in B cells is required for efficient establishment of murine gammaherpesvirus latency. PLoS Pathog 11: e1004831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collins CM, and Speck SH. 2014. Expansion of murine gammaherpesvirus latently infected B cells requires T follicular help. PLoS Pathog 10: e1004106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jondle CN, Johnson KE, Aurubin C, Sylvester P, Xin G, Cui W, Huppler AR, and Tarakanova VL. 2021. Gammaherpesvirus Usurps Host IL-17 Signaling To Support the Establishment of Chronic Infection. mBio 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jondle CN, and Tarakanova VL. 2022. T Cell-Intrinsic Interleukin 17 Receptor A Signaling Supports the Establishment of Chronic Murine Gammaherpesvirus 68 Infection. J Virol 96: e0063922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jondle CN, Johnson KE, Mboko WP, and Tarakanova VL. 2021. T Cell-Intrinsic Interferon Regulatory Factor 1 Expression Suppresses Differentiation of CD4(+) T Cell Populations That Support Chronic Gammaherpesvirus Infection. J Virol 95: e0072621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sabikunnahar B, Caldwell S, Varnum S, Hogan T, Cooper A, Lahue KG, Bivona JJ III, Cousens PM, Symeonides M, Ballif BA, Poynter ME, and Krementsov DN. 2023. Long Noncoding RNA U90926 Is Induced in Activated Macrophages, Is Protective in Endotoxic Shock, and Encodes a Novel Secreted Protein. The Journal of Immunology 210: 807–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Usherwood EJ, Meadows SK, Crist SG, Bellfy SC, and Sentman CL. 2005. Control of murine gammaherpesvirus infection is independent of NK cells. Eur J Immunol 35: 2956–2961. [DOI] [PubMed] [Google Scholar]
- 38.Usherwood EJ, Ward KA, Blackman MA, Stewart JP, and Woodland DL. 2001. Latent Antigen Vaccination in a Model Gammaherpesvirus Infection. Journal of Virology 75: 8283–8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Virgin H. W. t., Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, and Speck SH. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol 71: 5894–5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raza A, Crothers JW, McGill MM, Mawe GM, Teuscher C, and Krementsov DN. 2017. Anti-inflammatory roles of p38α MAPK in macrophages are context dependent and require IL-10. Journal of Leukocyte Biology 102: 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wood BM, Mboko WP, Mounce BC, and Tarakanova VL. 2013. Mouse gammaherpesvirus-68 infection acts as a rheostat to set the level of type I interferon signaling in primary macrophages. Virology 443: 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tarakanova VL, Leung-Pineda V, Hwang S, Yang C-W, Matatall K, Basson M, Sun R, Piwnica-Worms H, Sleckman BP, and Virgin HW. 2007. γ-Herpesvirus Kinase Actively Initiates a DNA Damage Response by Inducing Phosphorylation of H2AX to Foster Viral Replication. Cell Host & Microbe 1: 275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stuller KA, and Flano E. 2009. CD4 T cells mediate killing during persistent gammaherpesvirus 68 infection. J Virol 83: 4700–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meckiff BJ, Ladell K, McLaren JE, Ryan GB, Leese AM, James EA, Price DA, and Long HM. 2019. Primary EBV Infection Induces an Acute Wave of Activated Antigen-Specific Cytotoxic CD4(+) T Cells. J Immunol 203: 1276–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krementsov DN, Asarian L, Fang Q, McGill MM, and Teuscher C. 2018. Sex-Specific Gene-by-Vitamin D Interactions Regulate Susceptibility to Central Nervous System Autoimmunity. Front Immunol 9: 1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marshall NB, Vong AM, Devarajan P, Brauner MD, Kuang Y, Nayar R, Schutten EA, Castonguay CH, Berg LJ, Nutt SL, and Swain SL. 2017. NKG2C/E Marks the Unique Cytotoxic CD4 T Cell Subset, ThCTL, Generated by Influenza Infection. J Immunol 198: 1142–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu Z, Blackman MA, Kaye KM, and Usherwood EJ. 2015. Functional heterogeneity in the CD4+ T cell response to murine gamma-herpesvirus 68. J Immunol 194: 2746–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Voehringer D, Blaser C, Brawand P, Raulet DH, Hanke T, and Pircher H. 2001. Viral Infections Induce Abundant Numbers of Senescent CD8 T Cells1. The Journal of Immunology 167: 4838–4843. [DOI] [PubMed] [Google Scholar]
- 49.Thimme R, Appay V, Koschella M, Panther E, Roth E, Hislop AD, Rickinson AB, Rowland-Jones SL, Blum HE, and Pircher H. 2005. Increased expression of the NK cell receptor KLRG1 by virus-specific CD8 T cells during persistent antigen stimulation. J Virol 79: 12112–12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, Garg R, de la Torre JC, and von Andrian UH. 2016. The Chemokine Receptor CX3CR1 Defines Three Antigen-Experienced CD8 T Cell Subsets with Distinct Roles in Immune Surveillance and Homeostasis. Immunity 45: 1270–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gredmark-Russ S, Cheung EJ, Isaacson MK, Ploegh HL, and Grotenbreg GM. 2008. The CD8 T-cell response against murine gammaherpesvirus 68 is directed toward a broad repertoire of epitopes from both early and late antigens. J Virol 82: 12205–12212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haller O, Frese M, and Kochs G. 1998. Mx proteins: mediators of innate resistance to RNA viruses. Rev Sci Tech 17: 220–230. [DOI] [PubMed] [Google Scholar]
- 53.Mashimo T, Lucas M, Simon-Chazottes D, Frenkiel MP, Montagutelli X, Ceccaldi PE, Deubel V, Guenet JL, and Despres P. 2002. A nonsense mutation in the gene encoding 2’−5’-oligoadenylate synthetase/L1 isoform is associated with West Nile virus susceptibility in laboratory mice. Proc Natl Acad Sci U S A 99: 11311–11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu Z, Blackman MA, Kaye KM, and Usherwood EJ. 2015. Functional heterogeneity in the CD4+ T cell response to murine γ-herpesvirus 68. J Immunol 194: 2746–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Strutt TM, McKinstry KK, Marshall NB, Vong AM, Dutton RW, and Swain SL. 2013. Multipronged CD4+ T-cell effector and memory responses cooperate to provide potent immunity against respiratory virus. Immunological Reviews 255: 149–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marshall NB, Vong AM, Devarajan P, Brauner MD, Kuang Y, Nayar R, Schutten EA, Castonguay CH, Berg LJ, Nutt SL, and Swain SL. 2017. NKG2C/E Marks the Unique Cytotoxic CD4 T Cell Subset, ThCTL, Generated by Influenza Infection. The Journal of Immunology 198: 1142–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sheikh AA, and Groom JR. 2021. Transcription tipping points for T follicular helper cell and T-helper 1 cell fate commitment. Cellular & Molecular Immunology 18: 528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, and Byrne MC. 2002. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity 16: 311–323. [DOI] [PubMed] [Google Scholar]
- 59.Chen X, and Oppenheim JJ. 2011. Resolving the identity myth: key markers of functional CD4+FoxP3+ regulatory T cells. Int Immunopharmacol 11: 1489–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sakaguchi S 2004. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annual review of immunology 22: 531–562. [DOI] [PubMed] [Google Scholar]
- 61.Van Erp E, Van Kampen M, Van Kasteren P, and De Wit J. 2019. Viral Infection of Human Natural Killer Cells. Viruses 11: 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou G, Juang SW, and Kane KP. 2013. NK cells exacerbate the pathology of influenza virus infection in mice. Eur J Immunol 43: 929–938. [DOI] [PubMed] [Google Scholar]
- 63.Monnier J, and Zabel BA. 2014. Anti-Asialo GM1 NK Cell Depleting Antibody Does Not Alter the Development of Bleomycin Induced Pulmonary Fibrosis. PLOS ONE 9: e99350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Manangeeswaran M, Lewkowicz AP, Israely T, Ireland DDC, and Verthelyi D. 2020. CpG Oligonucleotides Protect Mice From Alphavirus Encephalitis: Role of NK Cells, Interferons, and TNF. Frontiers in immunology 11: 237–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abel AM, Yang C, Thakar MS, and Malarkannan S. 2018. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Frontiers in Immunology 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Arase H, Saito T, Phillips JH, and Lanier LL. 2001. Cutting Edge: The Mouse NK Cell-Associated Antigen Recognized by DX5 Moncoclonal Antibody is CD49b (α2 Integrin, Very Late Antigen-2)1. The Journal of Immunology 167: 1141–1144. [DOI] [PubMed] [Google Scholar]
- 67.Krementsov DN, and Teuscher C. 2013. Environmental factors acting during development to influence MS risk: insights from animal studies. Multiple Sclerosis Journal 19: 1684–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kozłowska A, Hrycaj P, Łącki JK, and Jagodziński PP. 2010. Perforin level in CD4+ T cells from patients with systemic lupus erythematosus. Rheumatology International 30: 1627–1633. [DOI] [PubMed] [Google Scholar]
- 69.van de Berg PJ, van Leeuwen EM, ten Berge IJ, and van Lier R. 2008. Cytotoxic human CD4+ T cells. Current Opinion in Immunology 20: 339–343. [DOI] [PubMed] [Google Scholar]
- 70.Schafflick D, Xu CA, Hartlehnert M, Cole M, Schulte-Mecklenbeck A, Lautwein T, Wolbert J, Heming M, Meuth SG, Kuhlmann T, Gross CC, Wiendl H, Yosef N, and Meyer Zu Horste G. 2020. Integrated single cell analysis of blood and cerebrospinal fluid leukocytes in multiple sclerosis. Nat Commun 11: 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Raveney BJE, Sato W, Takewaki D, Zhang C, Kanazawa T, Lin Y, Okamoto T, Araki M, Kimura Y, Sato N, Sano T, Saito Y, Oki S, and Yamamura T. 2021. Involvement of cytotoxic Eomes-expressing CD4(+) T cells in secondary progressive multiple sclerosis. Proc Natl Acad Sci U S A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schafflick D, Xu CA, Hartlehnert M, Cole M, Schulte-Mecklenbeck A, Lautwein T, Wolbert J, Heming M, Meuth SG, Kuhlmann T, Gross CC, Wiendl H, Yosef N, and Meyer zu Horste G. 2020. Integrated single cell analysis of blood and cerebrospinal fluid leukocytes in multiple sclerosis. Nature Communications 11: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chijioke O, Landtwing V, and Münz C. 2016. NK Cell Influence on the Outcome of Primary Epstein-Barr Virus Infection. Front Immunol 7: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thomson RC, Petrik J, Nash AA, and Dutia BM. 2008. Expansion and Activation of NK Cell Populations in a Gammaherpesvirus Infection. Scandinavian Journal of Immunology 67: 489–495. [DOI] [PubMed] [Google Scholar]
- 75.Snyder JP, Gullickson SK, del Rio-Guerra R, Sweezy A, Vagher B, Hogan TC, Lahue KG, Reisz JA, D’Alessandro A, Krementsov DN, and Amiel E. 2022. Divergent Genetic Regulation of Nitric Oxide Production between C57BL/6J and Wild-Derived PWD/PhJ Mice Controls Postactivation Mitochondrial Metabolism, Cell Survival, and Bacterial Resistance in Dendritic Cells. The Journal of Immunology 208: 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hughes DJ, Kipar A, Sample JT, and Stewart JP. 2010. Pathogenesis of a model gammaherpesvirus in a natural host. J Virol 84: 3949–3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dokun AO, Kim S, Smith HRC, Kang H-SP, Chu DT, and Yokoyama WM. 2001. Specific and nonspecific NK cell activation during virus infection. Nature Immunology 2: 951–956. [DOI] [PubMed] [Google Scholar]
- 78.Yokoyama WM, Matsumoto K, Scalzo AA, and Brown MG. 1997. Molecular genetics of the natural killer gene complex and innate immunity. Biochemical Society Transactions 25: 691–695. [DOI] [PubMed] [Google Scholar]
- 79.Pyzik M, Charbonneau B, Gendron-Pontbriand EM, Babić M, Krmpotić A, Jonjić S, and Vidal SM. 2011. Distinct MHC class I-dependent NK cell-activating receptors control cytomegalovirus infection in different mouse strains. J Exp Med 208: 1105–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Desrosiers MP, Kielczewska A, Loredo-Osti JC, Adam SG, Makrigiannis AP, Lemieux S, Pham T, Lodoen MB, Morgan K, Lanier LL, and Vidal SM. 2005. Epistasis between mouse Klra and major histocompatibility complex class I loci is associated with a new mechanism of natural killer cell-mediated innate resistance to cytomegalovirus infection. Nat Genet 37: 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kielczewska A, Pyzik M, Sun T, Krmpotic A, Lodoen MB, Munks MW, Babic M, Hill AB, Koszinowski UH, Jonjic S, Lanier LL, and Vidal SM. 2009. Ly49P recognition of cytomegalovirus-infected cells expressing H2-Dk and CMV-encoded m04 correlates with the NK cell antiviral response. J Exp Med 206: 515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martínez-Rodríguez JE, Cobo-Calvo A, Villar LM, Munteis E, Blanco Y, Rasal R, Vera A, Muntasell A, Alvarez-Lafuente R, Saiz A, Alvarez-Cermeño JC, Martínez-Yélamos S, Roquer J, and López-Botet M. 2016. Adaptive natural killer cell response to cytomegalovirus and disability progression in multiple sclerosis. Multiple Sclerosis Journal 22: 741–752. [DOI] [PubMed] [Google Scholar]
- 83.Infante-Duarte C, Weber A, Krätzschmar J, Prozorovski T, Pikol S, Hamann I, Bellmann-Strobl J, Aktas O, Dörr J, Wuerfel J, Stürzebecher CS, and Zipp F. 2005. Frequency of blood CX3CR1-positive natural killer cells correlates with disease activity in multiple sclerosis patients. Faseb j 19: 1902–1904. [DOI] [PubMed] [Google Scholar]
- 84.Beliën J, Goris A, and Matthys P. 2022. Natural Killer Cells in Multiple Sclerosis: Entering the Stage. Front Immunol 13: 869447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hertwig L, Hamann I, Romero-Suarez S, Millward JM, Pietrek R, Chanvillard C, Stuis H, Pollok K, Ransohoff RM, Cardona AE, and Infante-Duarte C. 2016. CX3CR1-dependent recruitment of mature NK cells into the central nervous system contributes to control autoimmune neuroinflammation. Eur J Immunol 46: 1984–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Noll KE, Ferris MT, and Heise MT. 2019. The Collaborative Cross: A Systems Genetics Resource for Studying Host-Pathogen Interactions. Cell Host & Microbe 25: 484–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dupont MSJ, Guillemot V, Campagne P, Serafini N, Marie S, Montagutelli X, Di Santo JP, and Vosshenrich CAJ. 2021. Host genetic control of natural killer cell diversity revealed in the Collaborative Cross. Proceedings of the National Academy of Sciences 118: e2018834118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Marquez AC, Shanina I, and Horwitz MS. 2020. Multiple Sclerosis-Like Symptoms in Mice Are Driven by Latent gammaHerpesvirus-68 Infected B Cells. Front Immunol 11: 584297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Casiraghi C, Marquez AC, Shanina I, and Horwitz MS. 2015. Latent virus infection upregulates CD40 expression facilitating enhanced autoimmunity in a model of multiple sclerosis. Sci Rep 5: 13995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.