

Summary
Background:
In sepsis and acute respiratory distress syndrome (ARDS) heterogeneity has contributed to difficulty identifying effective pharmacotherapies. In ARDS, two molecular phenotypes (“Hypoinflammatory” and “Hyperinflammatory”) have consistently been identified, with divergent outcomes and treatment responses. In this study, we sought to derive molecular phenotypes in critically ill adult sepsis patients, determine their overlap with prior ARDS phenotypes, and whether they respond differently to treatment in completed sepsis trials.
Methods:
We used clinical data and plasma biomarkers from two prospective sepsis cohorts [VALID (N=1140) and EARLI (N=818)] in latent class analysis (LCA) to identify the optimal number of classes in each cohort independently. We used validated models trained to classify ARDS phenotypes to evaluate concordance of sepsis and ARDS phenotypes. We applied these models to the PROWESS-SHOCK and VASST trials to assign phenotypes and evaluate heterogeneity of treatment.
Findings:
A two-class model best fit both VALID and EARLI (p<0.0001). In VALID, 804/1140 (71%) were classified as Hypoinflammatory, and 336/1140 (29%) as Hyperinflammatory; in EARLI, the proportions were 530/818 (65%) and 288/818 (35%) respectively. Comparatively, we observed higher plasma pro-inflammatory cytokines, more vasopressor-use, more bacteraemia, lower Protein C, and higher mortality in the Hyperinflammatory phenotype (p<0.0001 for all). Classifier models indicated strong concordance between sepsis phenotypes and previously identified ARDS phenotypes (area under the curve 0.86-0.96, depending on model). Findings were similar excluding participants with both sepsis and ARDS. In PROWESS-SHOCK, 1142/1680 (68%) were Hypoinflammatory and 538/1680 (32%) Hyperinflammatory, and response to activated protein C differed by phenotype (p=0.0043). In VASST, phenotype proportions were similar to other cohorts; however, no treatment interaction with the type of vasopressor was observed (p=0.72).
Interpretation:
Molecular phenotypes previously identified in ARDS are also identifiable in multiple sepsis cohorts and respond differently to activated protein C. Molecular phenotypes may represent a treatable trait in critical illness beyond the patient’s syndromic diagnosis.
INTRODUCTION
Sepsis and acute respiratory distress syndrome (ARDS) have for decades been referred to as “graveyards for pharmacotherapy” on account of the countless pharmaceutical agents that showed promise in pre-clinical and early-phase clinical studies, only to fail in larger randomised controlled trials (RCTs).(1, 2, 3) One reason for these repeated failures may be the considerable biological heterogeneity that characterizes sepsis and ARDS, where both syndromes are defined entirely by non-specific features, rather than specific pathologic entities.(2, 3) Taking a cue from oncology,(4, 5, 6) critical care has begun to make progress towards identifying molecular phenotypes; in some cases, these phenotypes appear to respond differently to therapies, suggesting that a precision medicine approach may hold promise for ending the therapeutic drought.(7, 8)
To that end, latent class analysis (LCA) of clinical and protein biomarker data has consistently identified two distinct molecular phenotypes of ARDS in multiple cohorts, including one cohort that enrolled only sepsis-associated ARDS patients.(9, 10, 11, 12, 13, 14, 15, 16) These phenotypes, termed “Hyperinflammatory” and “Hypoinflammatory” based on patterns of inflammatory plasma cytokines observed in each group, have widely divergent clinical features, including significantly different clinical outcomes, and appear to respond differently to therapies including mechanical ventilation,(9, 17) fluid therapy,(10) simvastatin,(11) and corticosteroids(13). If molecular phenotypes are consistently identifiable across critical illness syndromes, this finding could lay the foundation for a new taxonomy, opening the door to targeted clinical trials and therapies. However, it remains unknown whether these ARDS phenotypes are also observed in sepsis and whether they respond differently to therapies applied in sepsis.
To address these knowledge gaps, we analysed clinical and biological data from two observational cohorts and two RCTs. We hypothesized that in sepsis, LCA would identify phenotypes similar to the molecular phenotypes previously identified in ARDS. Since plasma protein C levels are consistently lower in the Hyperinflammatory phenotype, we hypothesized that this phenotype would preferentially benefit from activated Protein C. Finally, we hypothesized that the Hypoinflammatory phenotype would benefit from vasopressin use compared to norepinephrine, based on prior data suggesting a benefit among less severe septic patients.(18)
METHODS (Overview Figure 1)
Figure 1: Summary of the Analysis Plan.
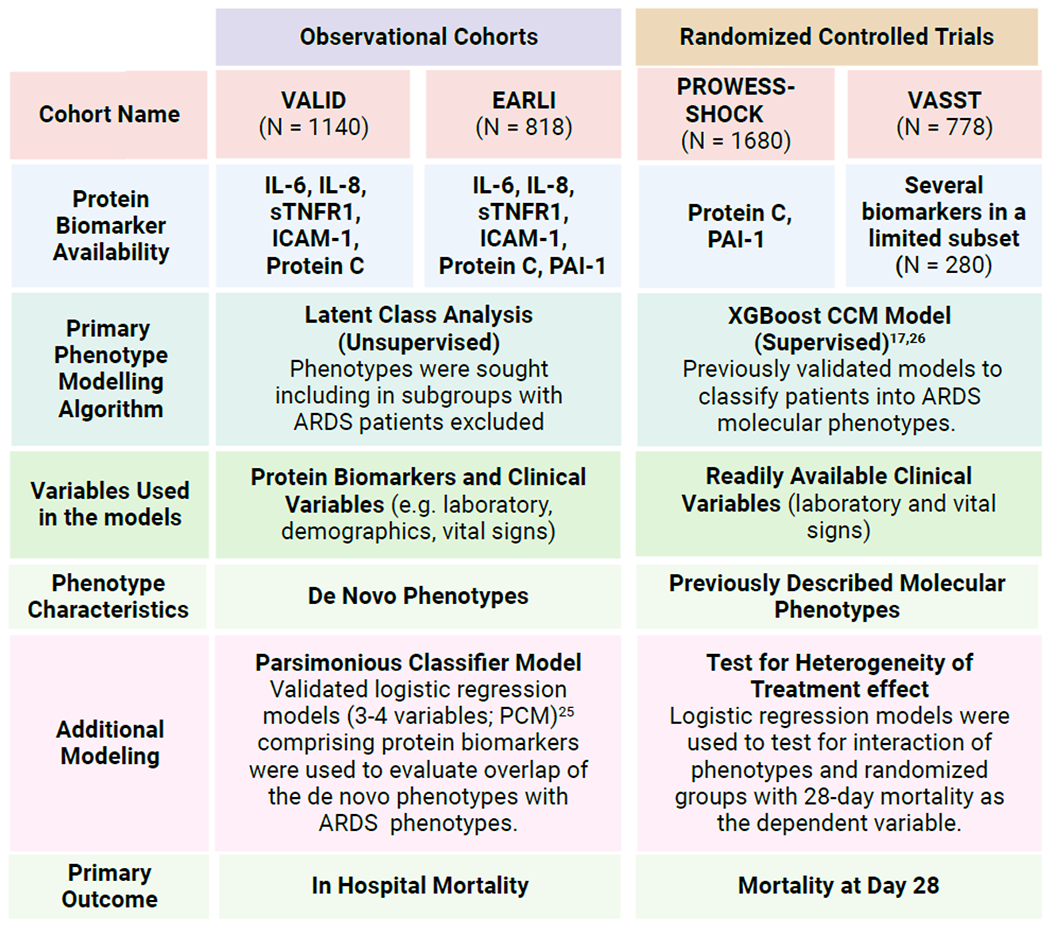
Latent class analysis (LCA) was used in VALID and EARLI because sufficient number of biomarkers were available across the entire cohort to recapitulate prior work used for discovering molecular phenotypes. In comparison, in PROWESS-SHOCK and VASST protein biomarker availability was insufficient for performing LCA as per our prior procedures. VALID: Validating Acute Lung Injury markers for Diagnosis; EARLI: Early Assessment of Renal and Lung Injury; PROWESS-SHOCK: Prospective Recombinant Human Activated Protein C Worldwide Evaluation in Severe Sepsis and Septic Shock trial; VASST: Vasopressin and Septic Shock trial; ICAM-1: intercellular adhesion molecule-1, IL-6: interleukin 6; IL-8: interleukin 8; PAI-1: plasminogen activator inhibitor-1; sTNFR-1: soluble tumour necrosis factor receptor-1; CCM = clinical classifier model; PCM = Parsimonious classifier model .
Observational Patient Cohorts
To test the hypothesis that in sepsis we would identify similar molecular phenotypes as in ARDS, we performed LCA in two prospective observational cohorts. The first was the Validating Acute Lung Injury biomarkers for Diagnosis (VALID) study, an ongoing prospective cohort of critically ill patients enrolled on the morning after admission to the medical, surgical, trauma or cardiovascular ICUs at Vanderbilt University Medical Center. VALID participants were included for this analysis if they met diagnostic criteria for sepsis on ICU day 1 or 2. Although in VALID the number of patients excluded from the study due to unavailability of protein biomarkers was larger, the demographics and baseline characteristics of these patients were similar to the studied population (Table S1). The second cohort was the Early Assessment of Renal and Lung Injury (EARLI) study, an ongoing prospective cohort of critically ill patients enrolled on or before ICU day 1 at UCSF Medical Center and Zuckerberg San Francisco General Hospital. Patients are identified in the Emergency Department and eligible for the study if an ICU admission has been requested for the patient. EARLI participants were included for this analysis if they met criteria for sepsis on ICU day 1 or 2.
Figure S1A and S1B show the screening and selection of patients in the two cohorts. Details of both study protocols and inclusion/exclusion criteria have been previously published.(14) Studies were approved by their respective Institutional Review Boards, and informed consent was obtained from subjects or their surrogates, or in some cases waived, as previously described.(19, 20)
In both studies, diagnosis and source of sepsis and/or ARDS were determined by at least two board-certified physicians independently reviewing all clinical data from participants’ hospitalizations, blinded to biological data, and prior to LCA. Sepsis was defined using Sepsis-2 criteria, since both cohorts began enrolling before publication of Sepsis-3 in 2016,(21) and ARDS was defined using the American-European Consensus Conference definition.(22)
Assay Procedures
Biological samples were collected on ICU day 2 in VALID, and either in the emergency department or ICU day 1 in EARLI. Blood samples were processed and plasma stored at −80°C until batch quantification. Plasma protein biomarkers, selected on the basis of contributing meaningfully to prior LCAs in ARDS,(9, 11, 12) were measured using multiplex bead-based assays or enzyme-linked immunoassay (ELISA)(details in online supplement) and included interleukin (IL)-8, IL-6, Protein C, soluble tumour necrosis factor receptor (sTNFR)-1, intracellular adhesion molecule-1, and plasminogen activator inhibitor-1 (EARLI only).
Latent Class Analysis
We performed LCA in each cohort independently using the same procedures as prior studies.(14, 23) Clinical and protein biomarker data from the time of study enrolment were used as class-defining variables in the modelling (Table S2). Outcome data and severity scores (e.g. APACHE and SOFA scores) were not included in the modelling. We built five models, consisting of one-five classes respectively. We determined the best-fitting model using the Bayesian Information Criteria (BIC), Vuong-Lo-Mendell-Rubin likelihood ratio (VLMR) test, entropy, and the number of observations in the smallest class.(23) Once the optimal model was identified, we assigned class membership based on the highest probability. Details of data handling and model development are provided in the online supplement. We compared differences between classes in clinical characteristics and outcomes, which included ICU-free days (censored at day 28) and in-hospital mortality.
In order to determine whether findings were being driven by sepsis patients with ARDS, we repeated the LCA in each cohort excluding patients that developed ARDS on either Day 1 or 2 of study enrolment.
Classifier Models
Biomarker-based parsimonious classifier models (PCMs) have been developed and validated to accurately classify ARDS phenotypes.(24) PCMs rely on research plasma biomarkers such as IL-8, Protein C, and/or sTNFR-1, serum bicarbonate, and vasopressor-use and were trained in a combined cohort of three ARDS RCTs and validated in a fourth RCT. To determine the concordance of the LCA-derived sepsis phenotypes in this analysis with previously identified ARDS phenotypes, we classified phenotypes using several PCMs in EARLI and VALID, using the coefficients that were generated in ARDS cohorts. PCM classification performance was compared to the gold standard of LCA-derived class assignment using the area under receiver-operating characteristic curves (AUC).
Since plasma biomarkers are not routinely available in clinical practice, we have also previously developed and validated a classifier model which uses routinely available clinical data to identify LCA-derived phenotypes. This clinical-data-only classifier model (CCM), which was derived using machine learning algorithms (XGBoost), also performs with high accuracy.(17, 25) Briefly, the CCMs were trained in a combined cohort of three prior ARDS RCTs to predict the Hyperinflammatory phenotype using baseline vital signs and laboratory values. The model was validated in a holdout RCT of ARDS. We further tested the concordance of the sepsis phenotypes identified in this analysis with those previously identified in ARDS by applying the CCM in EARLI and VALID. For both the PCM and CCM, we used a probability cut-off of ≥ 0.5 to assign the Hyperinflammatory phenotype. Further details of these analyses are in the online supplement.
Testing for Heterogeneity of Treatment Effect
To determine whether the molecular phenotypes respond differently to randomly allocated treatments for sepsis, we undertook secondary analyses of two completed RCTs. PROWESS-SHOCK was a double-blind placebo-controlled RCT testing recombinant human activated Protein C (drotrecogin alfa; APC) for the treatment of septic shock in 1680 patients.(26) APC did not significantly reduce mortality at 28 days. VASST was a double-blind RCT comparing norepinephrine to early vasopressin for treatment of catecholamine-dependent septic shock in 778 patients.(18) Vasopressin did not significantly reduce mortality compared to norepinephrine. Since plasma samples or sufficient biomarker data were unavailable for LCA in these trials, molecular phenotypes were identified using the CCM described above. In a subset of VASST, a limited set of protein biomarker data were available for comparison between the molecular phenotypes. We tested for heterogeneity of treatment effect (HTE) using logistic regression models with phenotype classification, treatment allocation, and their product as predictors, and the primary study outcome in both studies, 28-day mortality, as the dependent variable. As the CCM generates a probability of belonging to the Hyperinflammatory phenotype, we repeated the above analysis using probability of Hyperinflammatory classification, treatment allocation, and their product as the predictors in the model.
Other statistical considerations
Descriptive data are presented as mean (± standard deviation; SD) for normally distributed data, median (interquartile range; IQR) for non-normally / skewed data, or count (%), and we tested differences between groups using the Student’s t-test, Wilcoxon rank test, or χ2 test, respectively. Additionally, when comparing counts between two groups, we generated odds ratios (OR) with 95% confidence intervals (95% CI). We plotted Kaplan-Meier survival curves censored at day 28 from study enrolment day to compare survival between phenotypes stratified by treatment groups. LCA was performed using Mplus (V.8.1). All other analyses were performed using R-software on RStudio V.1.0.143.
Role of the funding source
The funders of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. The corresponding author had full access to all the data and final responsibility to submit the manuscript for publication.
RESULTS (see Figure 1 for overview)
Study population: VALID (N = 1140) and EARLI (N =818)
Baseline characteristics of the populations are described in Table 1. Patients in VALID were younger (56; SD 16 years) compared to EARLI (65; SD 16 years), and proportionately more patients were white (VALID 85% vs 48% EARLI). Vasopressor-use at enrolment was lower in VALID (48%) compared to EARLI (56%), although Acute Physiology and Chronic Health Evaluation (APACHE) II scores were similar. Prevalence of bacteraemia, as determined by positive blood cultures within seven days of study enrolment, was similar in the two cohorts (VALID 20% and EARLI 18%). In-hospital mortality was higher in EARLI (29%) compared to VALID (24%).
Table 1. Baseline Characteristics of VALID and EARLI.
Data are presented as n (%), mean (± Standard deviation), or median (interquartile range).
| VALID (n=1140) | EARLI (n=818) | |
|---|---|---|
| Age (years) | 56 ± 16 | 65 ± 16 |
|
| ||
| Sex (% female) | 490 (43%) | 358 (44%) |
|
| ||
| Race (%) | ||
|
| ||
| White | 971 (85%) | 394 (48%) |
|
| ||
| Black | 150 (13%) | 107 (13%) |
|
| ||
| Asian | -- | 213 (26%) |
|
| ||
| Others | 19 (2%) | 104 (13%) |
|
| ||
| APACHE II Score | 27 (22 – 33) | 26 (20 – 33) |
|
| ||
| Vasopressor use (%) | 548 (48%) | 459 (56%) |
|
| ||
| Temperature (C) | 37.8 ± 1.1 | 37·8 ± 1.4 |
|
| ||
| Systolic Blood Pressure (mmHg) | 87 ± 16 | 88 ± 21 |
|
| ||
| Heart Rate (bpm) | 120 ± 22 | 125 ± 26 |
|
| ||
| Respiratory Rate (breaths/min) | 29 (24 – 35) | 34 (29 – 40) |
|
| ||
| Hypoxia category | ||
| None | 250 (23%) | 196 (27%) |
| Mild | 231 (21%) | 145 (20%) |
| Moderate | 344 (32%) | 225 (31%) |
| Severe | 261 (24%) | 168 (23%) |
|
| ||
| Hematocrit (%) | 30 ± 7 | 30 ± 7 |
|
| ||
| WBC Count (106/mL) | 14 (9 – 21) | 13 (9 – 19) |
|
| ||
| Platelet (109/L) | 168 (97 – 250) | 158 (93 – 230) |
|
| ||
| Sodium (mEq/dL) | 136 ± 5 | 135 ± 6 |
|
| ||
| Creatinine (mg/dL) | 1.5 (1.0 – 2.8) | 1.4 (0.9 – 2.4) |
|
| ||
| Bicarbonate (mmol/L) | 21 ± 5 | 20 ± 6 |
|
| ||
| Albumin (g/dL) | 2·7 ± 0·6 | 2.4 ± 0·7 |
|
| ||
| Bilirubin (mg/dL) | 1.1 (0.7 – 2.3) | 1.0 (0.6 – 1.6) |
|
| ||
| Interleukin-6 (pg/mL) | 61 (19-248) | 99 (23 – 688) |
|
| ||
| Interleukin-8 (pg/mL) | 21 (9 – 73) | 19 (8 – 81) |
|
| ||
| Soluble TNF Receptor-1 (pg/mL) | 3602 (2058 – 6773) | 4159 (1976 – 9319) |
|
| ||
| Protein C (% Control) | 60 ± 33 | 83 ± 58 |
|
| ||
| ICAM-1 (ng/ml) | 674 (455 – 1004) | 623 (359 – 1118) |
|
| ||
| PAI1 (ng/ml) | -- | 6 (3-20) |
|
| ||
| Blood Culture Positive | 233 (20%) | 144 (18%) |
|
| ||
| Mechanical ventilation (%) | 716 (63%) | 393 (48%) |
|
| ||
| ICU-free days* | 20 (0-24) | 22 (0 – 25) |
|
| ||
| In-hospital mortality | 276 (24%) | 236 (29%) |
Hypoxia categories were created based on the PaO2/FiO2 or (SPO2/FiO2 converted to PaO2/FiO2) where a value > 300 mmHg was assigned none, 300 - ≥ 200 mmHg was mild, 200 - ≥ 100 mmHg was moderate, and < 100 mmHg was severe.
Intensive care unit (ICU) free days were censored at Day 28, such that patients that died before Day 28 were assigned zero ICU-free days.
APACHE = Acute Physiology and Chronic Health Evaluation, BP = Blood Pressure, TNF = tumor-necrosis factor, PAI-1: plasminogen activator inhibitor-1, ICAM-1: intercellular adhesion molecule-1, ICU = Intensive care unit.
Latent Class Analysis in VALID and EARLI
In VALID, as more classes were added to the model, the BIC decreased. The greatest decrease, however, was observed when going from a 1-class to a 2-class solution. Based on VLMR, the 2-class model was a better fit than a 1-class model (p<0.0001; Table S3), and models with more classes did not show significantly improved fit. Therefore, we determined the 2-class model as the optimal fitting model for this cohort. Based on highest probability of class membership, 804 participants (71%) were classified to Class 1 and 336 (29%) to Class 2.The median probability of class membership was high, with a median of 0.99 in both class 1 (IQR 0.99–1.0) and Class 2 (IQR 0.91–1.0).
In EARLI, similar patters were observed with the BIC as VALID. Based on VLMR, the 2-class model was a better fit than a 1-class model (p<0.0001; Table S3), and models comprising more classes did not fit significantly better. 530 (65%) participants were assigned to Class 1 and 288 (35%) were assigned to Class 2. The median probability for class membership in EARLI was 0.99 in both class 1 (IQR 0.96–1.0) and Class 2 (IQR 0.89–1.0).
Comparison of Class Features in VALID and EARLI
Class 1 and Class 2 had divergent baseline characteristics, with similar patterns across both cohorts (Tables S4–5). Demographics were largely similar between the two classes. Class 2 was defined by higher levels of proinflammatory cytokines such as IL-8, IL-6, sTNFR-1 and markers of organ dysfunction such as creatinine and bilirubin (Figures 2A–B). Conversely, platelets, serum bicarbonate, and protein C levels were lower in Class 2. Vasopressor-use was more common in Class 2 than Class 1 (VALID: 69% vs 39%; EARLI: 85% vs 41%; p<0.0001 for both). In both cohorts, bacteraemia was more common in Class 2 than Class 1 (VALID 31% vs 16%; OR 2.3, 95% CI: 1.7 – 3.2; EARLI 31% vs 10%, OR 3.9, 95% CI: 2.7 – 5.6 ). Species level breakdown for positive blood cultures in each phenotype is presented in Figures S2A–B.
Figure 2: Mean standardized values for continuous class-defining variables used in the latent class analysis models.
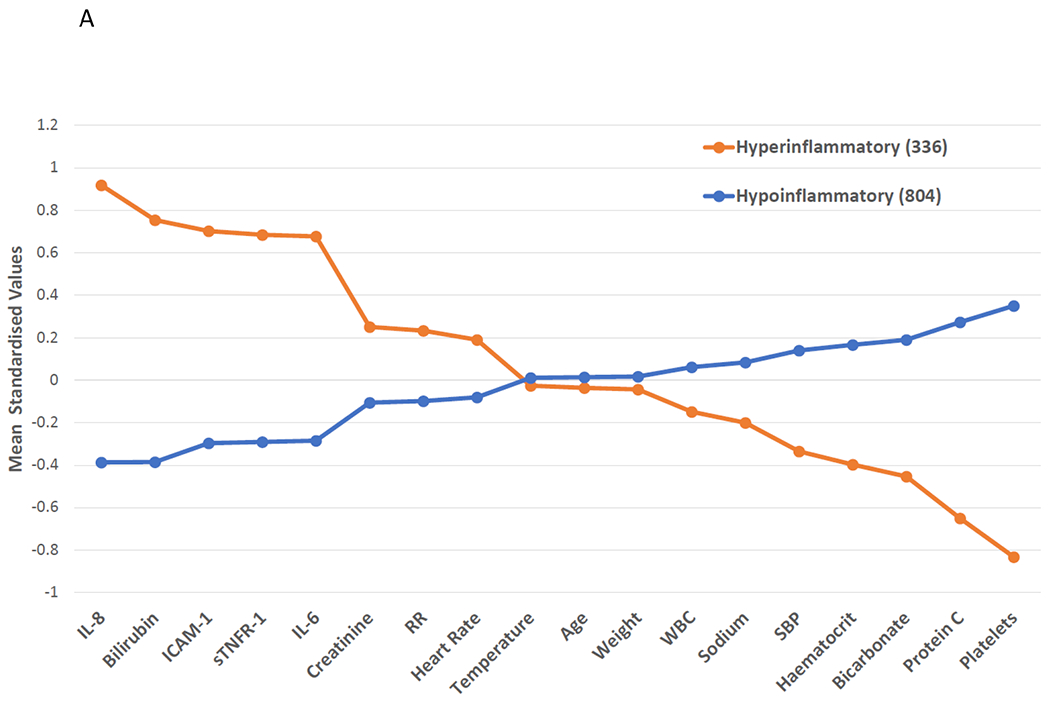
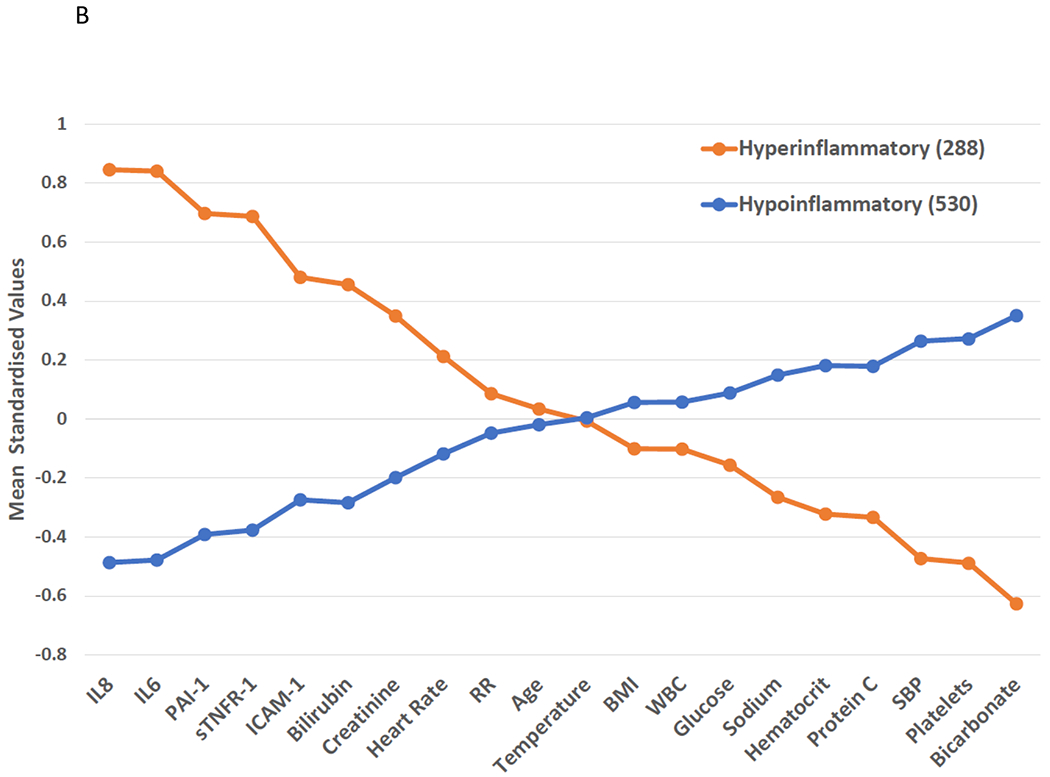
The variables are sorted from left to right in descending order for the highest values in the Hyperinflammatory phenotype. Standardized values were calculated by assigning the mean of the variables as 0 and standard deviation as 1. All variables were collected on the day of study enrolment. Panel A: VALID cohort. Panel B: EARLI Cohort. BMI: body mass index, SBP: systolic blood pressure, ICAM-1: intercellular adhesion molecule-1, IL-6: interleukin 6, IL-8: interleukin 8, PAI-1: plasminogen activator inhibitor-1, sTNFR-1: soluble tumour necrosis factor receptor-1, WBC: white blood cell count, RR = Respiratory Rate, HR = Heart Rate.
Outcomes were significantly worse in Class 2 compared to Class 1 (Table 2). In VALID, in-hospital mortality was 43% in Class 2 compared to 17% in Class 1 (OR 2.5, 95 CI: 2.1 – 3.1); in EARLI, these proportions were 45% and 20% respectively (OR 2.3, 95% CI: 1.8 – 2.9).
Table 2. Clinical outcomes in the two molecular phenotypes.
Differences in outcome between the Hypoinflammatory and Hyperinflammatory phenotypes across four sepsis cohorts.
| Hypoinflammatory | Hyperinflammatory | P-value | ||||||
|---|---|---|---|---|---|---|---|---|
| N | ICU free days | Mortality | N | ICU free days | Mortality | ICU free days | Mortality | |
| VALID * | 804 | 21 (11 -24) | 133 (17%) | 336 | 7 (0 - 22) | 143 (43%) | < 0.0001 | < 0.0001 |
| EARLI * | 530 | 23 (17 - 25) | 107 (20%) | 288 | 12 (0 - 23) | 129 (45%) | < 0.0001 | < 0.0001 |
| VALID* ARDS excluded | 532 | 23 (17 - 25) | 70 (13%) | 236 | 16 (0 - 23) | 80 (34%) | < 0.0001 | < 0.0001 |
| EARLI* ARDS excluded | 346 | 24 (21 - 25) | 48 (14%) | 226 | 22 (0 - 24) | 77 (34%) | < 0.0001 | < 0.0001 |
| PROWESS-SHOCK ** | 1142 | 13 (0 - 20) | 233 (20%) | 538 | 0 (0 - 14) | 192 (36%) | < 0.0001 | <0.0001 |
| VASST ** | 455 | 11 (0 - 20) | 127 (28%) | 323 | 0 (0 - 12) | 163 (51%) | < 0.0001 | <0.0001 |
In-hospital mortality;
Mortality at Day 28.
Intensive care unit (ICU) free days were censored at Day 28, such that patients that died before Day 28 were assigned zero ICU-free days. P-values for ICU free days were generated using the Wilcoxon-rank test and for mortality using the Chi-square test.
Concordance of Sepsis Classes with Previously Identified ARDS Phenotypes
When comparing phenotype classification using PCMs against LCA-derived classes, we observed high performance metrics for the PCMs in VALID (AUC’s 0.90-0.92; Figure S3A) and EARLI (AUC’s 0.93-0.96; Figure S3B), indicating high concordance between the sepsis classes and prior ARDS phenotypes (i.e. Class 1 corresponded to Hypoinflammatory and Class 2 to Hyperinflammatory phenotype). Details of individual model performance in each cohort, including confidence intervals of the AUCs and sensitivity and specificity using a probability cut-off of ≥ 0.5 to assign phenotype are presented in Table S6.
The previously validated CCM also indicated high concordance, with AUC’s in VALID and EARLI of 0.87 (95% CI: 0.85 – 0.89) and 0.90 (95% CI: 0.88 – 0.92) respectively. Hereafter, we refer to the sepsis classes as Hypoinflammatory (Class 1) and Hyperinflammatory (Class 2) phenotype for both cohorts.
Analyses Excluding ARDS
We repeated the LCA in both cohorts excluding participants diagnosed with ARDS on study days 1 or 2, leaving 768 participants in VALID and 572 in EARLI. Findings were very similar to the main analyses, with the 2-class model determined to be the best fitting for both cohorts (Table S3), with similarly divergent baseline characteristics and worse outcomes in the Hyperinflammatory phenotype (Table S7–8). The AUCs of the parsimonious ARDS classifier models in this subset ranged from 0.87-0.95 (Table S6), again indicating concordance of these sepsis-only phenotypes with previously identified ARDS phenotypes.
Molecular Phenotypes in PROWESS-SHOCK and VASST
Next, using the CCM in PROWESS-SHOCK, 1142 (68%) participants were classified to the Hypoinflammatory and 538 (32%) to the Hyperinflammatory phenotypes. We observed divergent baseline characteristics between the phenotypes (Table S9). Bacteraemia was more common in the Hyperinflammatory phenotype (37% vs 26%; OR 1.7, 95% CI: 1.4 – 2.2; Figure S2C). We observed significantly lower protein C and higher PAI-1 in Hyperinflammatory compared to Hypoinflammatory participants (p<0.0001 for both; Figure S4A), consistent with findings in EARLI (Table S5), and prior findings in ARDS molecular phenotypes.(9, 10, 11) 28-day mortality was significantly higher in the Hyperinflammatory compared to the Hypoinflammatory phenotype (36% vs 20%; OR 2.2, 95% CI: 1.7 – 2.7; Table 2). Notably, we observed a significant treatment interaction between APC randomization groups and the molecular phenotypes for the primary outcome (p=0.0043 for the interaction term), where APC treatment associated with higher mortality in the Hypoinflammatory phenotype (APC 23% vs Placebo 17%) and lower mortality in the Hyperinflammatory phenotype compared to placebo (APC 32% vs Placebo 39%). The survival plot for phenotypes stratified by treatment groups are presented in Figure S5A. Next, we evaluated the interaction of the continuous probabilities generated by the CCM for belonging to the Hyperinflammatory phenotype with the randomisation allocation group. Using this continuous scale, we observed similar findings, with a significant coefficient for the interaction term of probability and phenotype to predict mortality (p = 0.0048), with increased mortality associated with APC therapy in patients with lower probability of the Hyperinflammatory phenotype, and decreased mortality with APC therapy in patients with higher probability for the Hyperinflammatory phenotype (Figure 3).
Figure 3: Differential treatment response to activated protein C (APC) according to probability of belonging to the Hyperinflammatory phenotype.
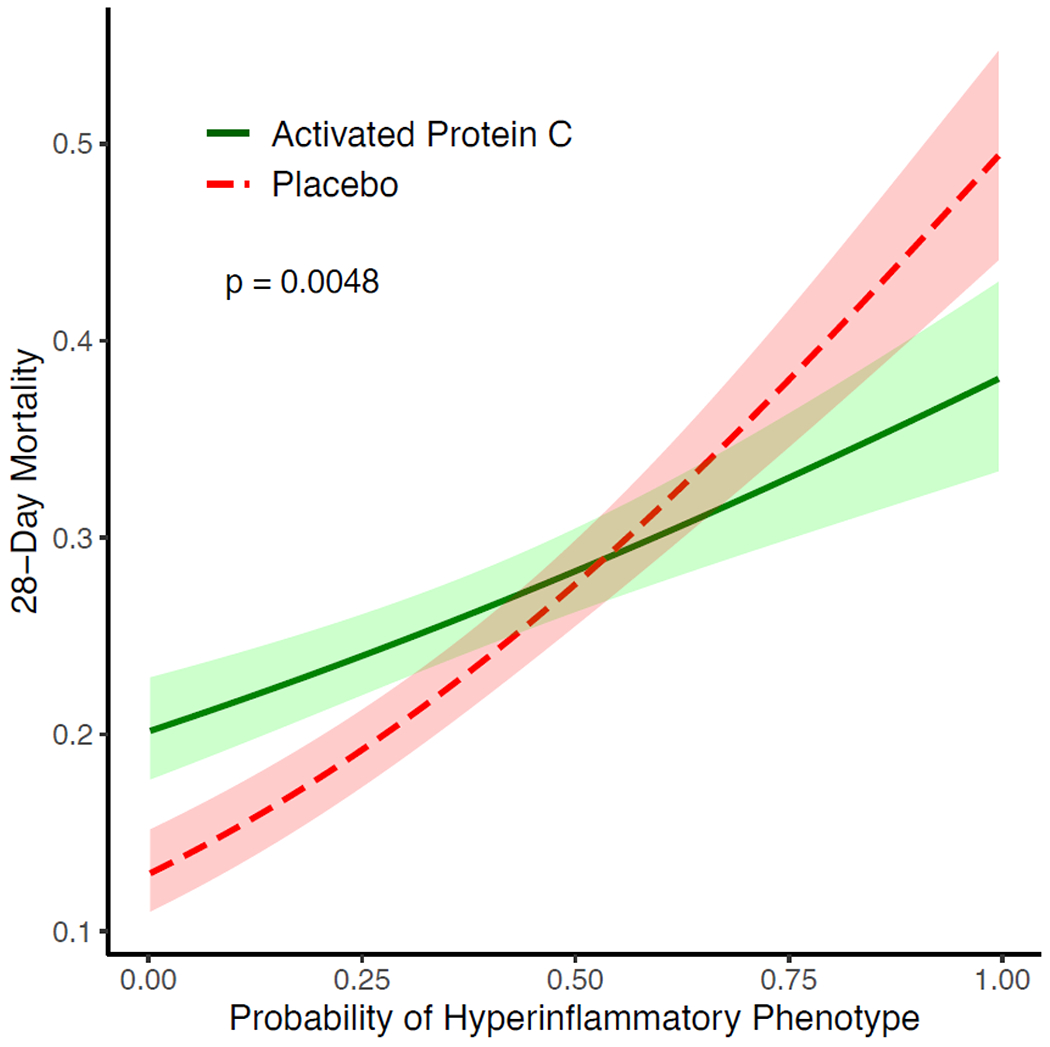
A logistic regression model was fit to predict mortality at Day 28 in the PROWESS-SHOCK trial, with the probability of belonging to the Hyperinflammatory phenotype (x-axis), treatment allocation, and their interaction term as predictor variables. The lines plot the estimated mortality in either placebo (red) or activated protein C (green) with 95% confidence intervals over a range of probabilities. P-value was generated using Wald test for the interaction term of probability and treatment allocation in the logistic regression model.
In VASST, using the CCM, 455 (58%) of participants were Hypoinflammatory and 323 (42%) were Hyperinflammatory. Differences in baseline clinical characteristics between the phenotypes are similar to all three other cohorts and described in Table S10. In a subset of patients with available biomarker data (n=285), pro-inflammatory biomarkers such as IL-6, IL-8, and TNF-α were significantly higher in the Hyperinflammatory phenotype (Figure S4B), consistent with the LCA-derived phenotypes in EARLI and VALID. 28-day mortality was significantly higher in the Hyperinflammatory phenotype compared to the Hypoinflammatory phenotype (51% vs 28%, OR 2.6, 95% CI: 2.0 – 3.6); Table 2). We did not observe a significant treatment interaction with randomised vasopressor intervention and the phenotypes for the primary outcome (p=0.72 for the interaction term; survival plot Figure S5B).
DISCUSSION
For the first time to our knowledge, we report that previously described molecular phenotypes of ARDS are consistently identifiable in multiple cohorts of critically ill sepsis patients. Moreover, even after excluding patients with ARDS from these analyses, the identified sepsis phenotypes were highly concordant with the Hypoinflammatory and Hyperinflammatory ARDS phenotypes. Including our prior studies in ARDS, we have now evaluated these phenotypes in over 10,000 critically-ill patients. We also report a higher prevalence of bacteraemia in the Hyperinflammatory phenotype, which has not been described previously. Finally, in retrospective analysis of a completed RCT, we observed differential responses to APC between the phenotypes, with survival benefit for APC in the Hyperinflammatory and harm in the Hypoinflammatory phenotype.
These findings have several important implications. First, the finding of molecular phenotypes across ARDS and sepsis suggests that this phenotyping schema captures a well conserved and relatively uniform response to critical illness that is consistently detectable by quantifiable measurements. Our findings imply that the Hyperinflammatory phenotype may represent a consistent clinical-biological “trait” present in many critically ill patients irrespective of syndromic diagnoses. The identification of these molecular phenotypes in patients with acute hypoxaemic respiratory failure without ARDS further substantiates this hypothesis.(27) Further, the finding that these phenotypes respond differently to numerous interventions,(9, 10, 11, 13, 17) now including activated protein C, suggests that this “trait” may be treatable.
In both sepsis and ARDS, current syndromic definitions rely exclusively on clinical data without consistent links to pathology or biology. The molecular phenotyping schema presented in our study support consideration of a shift in that paradigm.(28) The phenotypes described in these analyses are defined by inherent clinical and biological features, which are combined in multivariate models. The reduction of dimensionality, both in terms of biology and complexity, offered by these phenotypes may be more likely to reveal biological pathways that are amenable to interventions. To that end, the finding of heterogeneity of treatment effect with APC adds to a list of interventions such as simvastatin, corticosteroids, fluid management and positive end-expiratory pressure which appear to have phenotype-specific effects in retrospective analysis of previous studies, further supporting the need for prospective testing of phenotype-specific responses in future trials.(9, 10, 11, 13)
Molecular phenotypes offer a potential pathway to prognostic and/or predictive enrichment of future trials. However, it is unclear whether the observed potential benefit of APC in the Hyperinflammatory phenotype is due to prognostic or predictive enrichment or both. Given APC is purported to have antithrombotic and anti-inflammatory effects in sepsis, we speculate that the observed treatment benefit in the phenotype characterized by higher levels of endothelial injury markers and more severe dysregulation of coagulation and fibrinolysis (i.e. the hyperinflammatory phenotype) may be due to predictive enrichment of the population. This hypothesis is further reinforced by the finding that the effect size of benefit was more evident as probabilities of belonging to Hyperinflammatory phenotype increased. In the original trial,(26) no differential treatment effect was observed when the population was stratified by either plasma protein C levels or sepsis severity, suggesting that the phenotypes capture information that is not readily captured by simpler approaches. Furthermore, a previous LCA performed on PROWESS SHOCK, using only clinical data (no protein biomarkers) as class-defining variables, identified six phenotypes with no significant HTE.(29) This finding is consistent with other secondary analyses of ARDS RCTs that have shown the importance of including protein biomarkers for identifying clusters where significant HTE was observed with the randomized interventions.(30) Our approach contrasts with the prior LCA of PROWESS-SHOCK because we first used biological data to identify the phenotypes, and subsequently trained models using only clinical data to identify these phenotypes, as opposed to using only clinical data to identify de novo phenotypes. Nevertheless, the findings of HTE in the phenotypes in PROWESS-SHOCK should be interpreted with caution because these are secondary analyses, and further uncertainty is introduced because the CCM is a simplified classifier of the LCA-derived phenotype.
Although this phenotyping schema appears to be robust, reproducible, and generalisable, several important knowledge gaps remain. First, it should be unambiguous that the routine clinical identification of the phenotypes is currently unwarranted. The main purpose of the phenotypes at this stage is to provide investigators with a consistent biologically and clinically meaningful schema that reduces the observed heterogeneity in critical illness syndromes, which have historically been treated as uniform entities in prior RCTs. Specifically, this phenotyping schema provides an intuitive approach to testing interventions in groups with divergent biology and trajectories; these groups have otherwise been treated uniformly in prior RCTs. Currently, there is an international collaboration that proposes to evaluate interventions in a phase II clinical trial in patients stratified according to these molecular phenotypes (NIHR154493).
Second, the optimal approach to identifying the phenotypes at the bedside remains uncertain. While our classifier models (PCM and CCM) are both promising in their respective performance metrics, each has their own limitations. The PCMs we have previously described are advantageous as they include several of the protein biomarkers (IL-8, IL-6, sTNFR-1, and protein C) that define the phenotypes. However, point-of-care quantification for most of these biomarkers remains experimental and is currently not widely available. In a small study of patients with COVID-19 associated ARDS, we have reported feasibility of point-of-care quantification of IL-6 and sTNFR-1 and real time, prospective, phenotype classification using PCMs.(31) These findings require validation in larger studies, and the ongoing PHIND study is currently enrolling patients to evaluate feasibility and validity of this approach in a multicentre observational study (NCT04009330).
While the CCM does not rely on quantification of protein biomarkers, its implementation as a real time classification tool in the clinical setting is limited by two factors. First, the XGBoost algorithm is a “black box”, meaning its inner workings are not visible to the operator. This characteristic may limit confidence of the bedside clinician to prescribe an intervention based on the CCM and/or use it for clinical decision-making. Second, the time period during which the CCM is effective beyond the initial phase of critical illness remains unknown. This information will be critical, as several interventions that patients receive early in their care seek to normalise physiology (e.g. lower heart rates, increase blood pressure, etc), which may impact model performance. Although the latent classes appear to remain stable over the first three days of critical illness,(32) data evaluating the biological trajectories of the phenotypes and clinical implications thereof remains underdeveloped and represents a third important unaddressed knowledge gap of our phenotyping approach. A fourth knowledge gap that warrants further investigation is whether including anti-inflammatory biomarkers and/or biomarkers of endothelial recovery in LCA models may point towards additional subclasses and/or phenotypes. We would anticipate that the addition of a single such biomarker is unlikely to dramatically alter the findings of the presented LCA given that the phenotypes, by definition, are identified through multivariate modelling algorithms. To that end, the ARDS phenotyping schema described by Bos and colleagues only identified two phenotypes (which were biologically aligned with the Hypoinflammatory and Hyperinflammatory phenotypes), despite including IL-10 (an anti-inflammatory protein),(33) reinforcing the hypothesis that several biomarkers of alternative pathways will likely be needed to discover additional novel classes.
Our study has several strengths. LCAs were performed in two large independent observational cohorts, and the phenotypes’ clinical value evaluated in two RCTs. The identification of phenotypes using the CCM suggests that the phenotypes may be clinically implementable in the near future. This study also has some important limitations. We present only secondary analyses of RCTs conducted retrospectively. Another important limitation of this study, and more broadly the proposed phenotyping schema, is that it is likely too simplistic and fails to fully capture the complexity of these population. For example, other groups have described between three to four subgroups in critically ill patients with sepsis, based on transcriptional(34, 35, 36, 37) or clinical data(38). Specifically, in this schema, the Hypoinflammatory phenotype is the majority class yet remains biologically undifferentiated and heterogeneous. Taken together, these findings suggest that there are probably other meaningful subgroups subsumed within the Hypoinflammatory phenotype. More importantly, the evaluation of how these other phenotyping schema overlap with the Hypoinflammatory and Hyperinflammatory phenotypes, and whether these phenotypes can be cumulatively informative for patient care, are exciting future questions for the field. We anticipate that incorporating additional high resolution biological and clinical data will add additional complexity to these phenotypes, with ongoing iteration, refinement, and improvement needed. Further, the use of corticosteroids and other immunomodulatory drugs prior to biomarker quantification may have influenced the phenotype classification. This data was not systematically recorded in EARLI and VALID. Finally, although the CCM showed good face validity, its performance may further be improved by including variables that are known to be important in prognostication of sepsis and the inflammatory state, such as lactate and ferritin,(39, 40, 41) which were unavailable in our training data sets.
In summary, molecular phenotypes, which were previously described in ARDS, are also identifiable in sepsis and have now been evaluated in over 10,000 critically ill patients. The Hyperinflammatory phenotype is consistently and reproducibly associated with higher pro-inflammatory biomarkers, lower platelets and protein C, higher incidence of shock and bacteraemia, and increased mortality. These phenotypes appear to respond differently to activated protein C, adding to evidence accumulated from studies of simvastatin, fluid management, and mechanical ventilation that the hyper-inflammatory phenotype may represent a treatable trait. Further studies prospectively testing this hypothesis are needed to definitively determine whether the current paradigm of syndromic classification of critical illness should be amended to incorporate biological phenotypes.
Supplementary Material
Research in context.
Evidence before this study
We searched PubMed and Google Scholar for either sepsis or ARDS, using the additional search terms “molecular phenotypes”, “phenotypes”, “heterogeneity”, and “heterogeneity of treatment effect” for research published between January 2010 and January 2023, with no language restrictions. Additionally, we considered work by co-authors and colleagues on the subject of critical care phenotyping. Heterogeneity is increasingly being recognised as a principal factor leading to the multitude of negative clinical trials in critical illness syndromes such as sepsis and acute respiratory distress syndrome (ARDS). To mitigate this heterogeneity in ARDS, investigators have consistently described two phenotypes of ARDS, called the Hypoinflammatory and Hyperinflammatory phenotypes, with distinct clinical characteristics and outcomes, and they appear to respond differently to randomised interventions in secondary analyses of randomised control trials. It is not known whether these phenotypes are specific to ARDS or whether they also exist in other critical illness syndromes such as sepsis.
Added value of this study
In this study, for the first time, we report that the Hypoinflammatory and Hyperinflammatory phenotypes were also identifiable in two observational cohorts of critically ill patients with sepsis. The Hyperinflammatory phenotype was associated with higher proinflammatory cytokines and organ failure. Using machine-learning models, we classified the phenotypes in two previously-conducted trials of sepsis and observed similar characteristics. In all four cohorts, mortality was significantly higher in the Hyperinflammatory phenotype, and these patients had longer ICU stays. In the PROWESS-SHOCK trial, which originally showed no treatment benefit, we observed differential treatment response to activated protein C, with apparent treatment benefit in the Hyperinflammatory phenotype and harm in Hypoinflammatory phenotype.
Implications of all the available evidence
Including this study, we have now identified the Hypoinflammatory and Hyperinflammatory phenotypes in over 10 000 critically ill patients with sepsis and/or ARDS, including paediatric populations. These findings suggest that the Hyperinflammatory phenotype may represent a treatable trait that goes beyond syndromic diagnosis. These phenotypes may enhance future clinical trials by providing investigators with a biologically and clinically consistent schema that enriches critical care populations, which historically have been treated as uniform in prior RCTs. Such prospectively designed trials will be required prior to use of these phenotypes for clinical decision-making.
Acknowledgements
This study was funded by the U.S. National Institute of Health (GM142992 to PS, HL140026 to CSC, HL164937, HL158906, and HL164937-01 to LBW. We would like to acknowledge the contributions and generosity of all the participants, clinical providers, and investigators of the observational cohorts (VALID and EARLI) and randomized controlled trials (PROWESS SHOCK and VASST) used in our study. This presentation is based on research using data from Eli Lilly that has been made available through Vivli, Inc. Vivli has not contributed to or approved, and is not in any way responsible for, the contents of this publication. Eli Lilly did not contribute to the study design, analysis, or production of the publication.
Declaration of interest
PS reports funding from the National Institute of Health (NIH), National Institute of General Medical Sciences (NIGMS), and consulting fees from AstraZeneca. LBW reports funding from the NIH, research funding from the Department of Defense, Genentech, Boehringer Ingelheim CSL, and Behring, receives consulting fees from Akebia, Santhera, Global Blood Therapeutics, and Boehringer Ingelheim, and has stock options in Virtuoso Surgical. CSC reports funding from the NIH, research grants from Roche Genentech and Quantum Leap Healthcare Collaborative, consulting fees from Vasomune, Gen1e Life Sciences, NGM Bio, Cellenkos, and Janssen, and holds a patent on metagenomic sequencing for sepsis diagnosis (co-recipient). MAM reports research funding from Roche-Genentech, Quantum Therapeutics, NIH/NHLBI/NAIAD, Department of Defense (DoD), and California Institute of Regenerative Medicine, and consulting fees from Johnson and Johnson, Gilead Therapeutics, and Novartis Therapeutics. MMC reports funding from the NIH, research funding from the NIH and DoD, intellectual property royalties from an issued patent (#11,410,777). JAR reports has received an investigator-initiated grant from Grifols that was provided to and administered by UBC, Canadian Institutes of Health Research (CIHR), and 3 from the St. Paul’s Foundation (SPF). reports receiving consulting fees in the last 3 years from SIB Therapeutics LLC, Ferring Pharmaceuticals, PAR Pharma, and was a funded member of the Data and Safety Monitoring Board (DSMB) of an NIH-sponsored trial of plasma in COVID-19 (PASS-IT-ON). JAR also reports patents owned by the University of British Columbia (UBC) that are related to the use of PCSK9 inhibitor(s) in sepsis, the use of vasopressin in septic shock, and a patent owned by Ferring for use of selepressin in septic shock, and was a founder, Director and shareholder in Cyon Therapeutics Inc. (now closed) and is a shareholder in Molecular You Corp. KDL reports grant funding from National Institute of Health: NIDDK, and consulting fees from Biomerieux, UptoDate, SeaStar Medical, AM Pharma, and Baxter, and owns stock or stock options in Amgen.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Sharing
Data from these studies can be provided to others upon reasonable request on approval of a written request to Dr Pratik Sinha, Prof Carolyn Calfee, Prof Lorraine Ware, Prof James Russell, and Prof Keith Walley. Data for PROWESS-SHOCK were publically available and accessed via a data sharing platform after a scientific review process though Vivli Inc.
References
- 1.Riedemann NC, Guo RF, Ward PA. The enigma of sepsis. J Clin Invest. 2003;112(4):460–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marshall JC. Why have clinical trials in sepsis failed? Trends Mol Med. 2014;20(4):195–203. [DOI] [PubMed] [Google Scholar]
- 3.Matthay MA, McAuley DF, Ware LB. Clinical trials in acute respiratory distress syndrome: challenges and opportunities. Lancet Respir Med. 2017;5(6):524–34. [DOI] [PubMed] [Google Scholar]
- 4.Corren J, Lemanske RF, Hanania NA, Korenblat PE, Parsey MV, Arron JR, et al. Lebrikizumab treatment in adults with asthma. N Engl J Med. 2011;365(12):1088–98. [DOI] [PubMed] [Google Scholar]
- 5.Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE Jr., Davidson NE, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353(16):1673–84. [DOI] [PubMed] [Google Scholar]
- 6.Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180(5):388–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinha P, Calfee CS. Phenotypes in acute respiratory distress syndrome: moving towards precision medicine. Curr Opin Crit Care. 2019;25(1):12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reddy K, Sinha P, O’Kane CM, Gordon AC, Calfee CS, McAuley DF. Subphenotypes in critical care: translation into clinical practice. Lancet Respir Med. 2020;8(6):631–43. [DOI] [PubMed] [Google Scholar]
- 9.Calfee CS, Delucchi K, Parsons PE, Thompson BT, Ware LB, Matthay MA, et al. Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med. 2014;2(8):611–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Famous KR, Delucchi K, Ware LB, Kangelaris KN, Liu KD, Thompson BT, et al. Acute Respiratory Distress Syndrome Subphenotypes Respond Differently to Randomized Fluid Management Strategy. Am J Respir Crit Care Med. 2017;195(3):331–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calfee CS, Delucchi KL, Sinha P, Matthay MA, Hackett J, Shankar-Hari M, et al. Acute respiratory distress syndrome subphenotypes and differential response to simvastatin: secondary analysis of a randomised controlled trial. Lancet Respir Med. 2018;6(9):691–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sinha P, Delucchi KL, Thompson BT, McAuley DF, Matthay MA, Calfee CS, et al. Latent class analysis of ARDS subphenotypes: a secondary analysis of the statins for acutely injured lungs from sepsis (SAILS) study. Intensive Care Med. 2018;44(11):1859–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sinha P, Furfaro D, Cummings MJ, Abrams D, Delucchi K, Maddali MV, et al. Latent Class Analysis Reveals COVID-19-related Acute Respiratory Distress Syndrome Subgroups with Differential Responses to Corticosteroids. Am J Respir Crit Care Med. 2021;204(11):1274–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sinha P, Delucchi KL, Chen Y, Zhuo H, Abbott J, Wang C, et al. Latent class analysis-derived subphenotypes are generalisable to observational cohorts of acute respiratory distress syndrome: a prospective study. Thorax. 2022;77(1):13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dahmer MK, Yang G, Zhang M, Quasney MW, Sapru A, Weeks HM, et al. Identification of phenotypes in paediatric patients with acute respiratory distress syndrome: a latent class analysis. Lancet Respir Med. 2022;10(3):289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitsios GD, Yang L, Manatakis DV, Nouraie M, Evankovich J, Bain W, et al. Host-Response Subphenotypes Offer Prognostic Enrichment in Patients With or at Risk for Acute Respiratory Distress Syndrome. Crit Care Med. 2019;47(12):1724–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maddali MV, Churpek M, Pham T, Rezoagli E, Zhuo H, Zhao W, et al. Validation and utility of ARDS subphenotypes identified by machine-learning models using clinical data: an observational, multicohort, retrospective analysis. Lancet Respir Med. 2022;10(4):367–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Russell JA, Walley KR, Singer J, Gordon AC, Hebert PC, Cooper DJ, et al. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med. 2008;358(9):877–87. [DOI] [PubMed] [Google Scholar]
- 19.Kerchberger VE, Brown RM, Semler MW, Zhao Z, Koyama T, Janz DR, et al. Impact of Clinician Recognition of Acute Respiratory Distress Syndrome on Evidenced-Based Interventions in the Medical ICU. Crit Care Explor. 2021;3(7):e0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwede M, Lee RY, Zhuo H, Kangelaris KN, Jauregui A, Vessel K, et al. Clinician Recognition of the Acute Respiratory Distress Syndrome: Risk Factors for Under-Recognition and Trends Over Time. Crit Care Med. 2020;48(6):830–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149(3 Pt 1):818–24. [DOI] [PubMed] [Google Scholar]
- 23.Sinha P, Calfee CS, Delucchi KL. Practitioner’s Guide to Latent Class Analysis: Methodological Considerations and Common Pitfalls. Crit Care Med. 2020;(In press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sinha P, Delucchi KL, McAuley DF, O’Kane CM, Matthay MA, Calfee CS. Development and validation of parsimonious algorithms to classify acute respiratory distress syndrome phenotypes: a secondary analysis of randomised controlled trials. Lancet Respir Med. 2020;8(3):247–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sinha P, Churpek MM, Calfee CS. Machine Learning Classifier Models Can Identify Acute Respiratory Distress Syndrome Phenotypes Using Readily Available Clinical Data. Am J Respir Crit Care Med. 2020;202(7):996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, Finfer S, et al. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med. 2012;366(22):2055–64. [DOI] [PubMed] [Google Scholar]
- 27.Heijnen NFL, Hagens LA, Smit MR, Cremer OL, Ong DSY, van der Poll T, et al. Biological Subphenotypes of Acute Respiratory Distress Syndrome Show Prognostic Enrichment in Mechanically Ventilated Patients without Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med. 2021;203(12):1503–11. [DOI] [PubMed] [Google Scholar]
- 28.Maslove DM, Tang B, Shankar-Hari M, Lawler PR, Angus DC, Baillie JK, et al. Redefining critical illness. Nat Med. 2022;28(6):1141–8. [DOI] [PubMed] [Google Scholar]
- 29.Gardlund B, Dmitrieva NO, Pieper CF, Finfer S, Marshall JC, Taylor Thompson B. Six subphenotypes in septic shock: Latent class analysis of the PROWESS Shock study. J Crit Care. 2018;47:70–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sinha P, Spicer A, Delucchi KL, McAuley DF, Calfee CS, Churpek MM. Comparison of machine learning clustering algorithms for detecting heterogeneity of treatment effect in acute respiratory distress syndrome: A secondary analysis of three randomised controlled trials. EBioMedicine. 2021;74:103697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sinha P, Calfee CS, Cherian S, Brealey D, Cutler S, King C, et al. Prevalence of phenotypes of acute respiratory distress syndrome in critically ill patients with COVID-19: a prospective observational study. Lancet Respir Med. 2020;8(12):1209–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delucchi K, Famous KR, Ware LB, Parsons PE, Thompson BT, Calfee CS, et al. Stability of ARDS subphenotypes over time in two randomised controlled trials. Thorax. 2018;73(5):439–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bos LD, Schouten LR, van Vught LA, Wiewel MA, Ong DSY, Cremer O, et al. Identification and validation of distinct biological phenotypes in patients with acute respiratory distress syndrome by cluster analysis. Thorax. 2017;72(10):876–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong HR, Cvijanovich N, Lin R, Allen GL, Thomas NJ, Willson DF, et al. Identification of pediatric septic shock subclasses based on genome-wide expression profiling. BMC Med. 2009;7:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davenport EE, Burnham KL, Radhakrishnan J, Humburg P, Hutton P, Mills TC, et al. Genomic landscape of the individual host response and outcomes in sepsis: a prospective cohort study. Lancet Respir Med. 2016;4(4):259–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scicluna BP, van Vught LA, Zwinderman AH, Wiewel MA, Davenport EE, Burnham KL, et al. Classification of patients with sepsis according to blood genomic endotype: a prospective cohort study. Lancet Respir Med. 2017;5(10):816–26. [DOI] [PubMed] [Google Scholar]
- 37.Sweeney TE, Azad TD, Donato M, Haynes WA, Perumal TM, Henao R, et al. Unsupervised Analysis of Transcriptomics in Bacterial Sepsis Across Multiple Datasets Reveals Three Robust Clusters. Crit Care Med. 2018;46(6):915–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seymour CW, Kennedy JN, Wang S, Chang CH, Elliott CF, Xu Z, et al. Derivation, Validation, and Potential Treatment Implications of Novel Clinical Phenotypes for Sepsis. JAMA. 2019;321(20):2003–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leventogiannis K, Kyriazopoulou E, Antonakos N, Kotsaki A, Tsangaris I, Markopoulou D, et al. Toward personalized immunotherapy in sepsis: The PROVIDE randomized clinical trial. Cell Rep Med. 2022;3(11):100817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Posma RA, Froslev T, Jespersen B, van der Horst ICC, Touw DJ, Thomsen RW, et al. Prognostic impact of elevated lactate levels on mortality in critically ill patients with and without preadmission metformin treatment: a Danish registry-based cohort study. Ann Intensive Care. 2020;10(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Nooijer AH, Kotsaki A, Kranidioti E, Kox M, Pickkers P, Toonen EJM, et al. Complement activation in severely ill patients with sepsis: no relationship with inflammation and disease severity. Crit Care. 2023;27(1):63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data from these studies can be provided to others upon reasonable request on approval of a written request to Dr Pratik Sinha, Prof Carolyn Calfee, Prof Lorraine Ware, Prof James Russell, and Prof Keith Walley. Data for PROWESS-SHOCK were publically available and accessed via a data sharing platform after a scientific review process though Vivli Inc.