

Introduction
The AAA proteins, members of the larger AAA+ superfamily, are a family of enzymatic machines involved in diverse cellular functions ranging from DNA repair and replication to organelle biogenesis, membrane trafficking, transcriptional regulation, and protein quality control. AAA proteins were defined as ATPases associated with various cellular activities, but this definition disguises the breadth and importance of their functions and the common structures and mechanisms of action that underlie their activities. At the International meeting on the cellular functions of AAA proteins in Kyoto, Japan, the organizers asked participants to come up with ‘a common molecular basis’ for understanding these proteins. The response was an impressive display that establishes the AAA proteins, alongside G proteins, as mediators of assembly and disassembly of macromolecular complexes.
Structure—a motor within a machine
AAA proteins do a lot of heavy work in the cell: erecting or disassembling complexes, unfolding or unwinding macromolecules, and transporting cellular cargo. This mechanical work requires a chemo-mechanical converter, the AAA module, which derives its energy from ATP hydrolysis. AAA modules are composed of 200–250 amino acids predicted to have conserved secondary structures (Neuwald et al., 1999). The motor module is attached to other domains that act as tool heads, the latter interacting with substrates either directly or through ‘adaptor’ molecules. The connectors between the motor and the tool heads thus act as coupling devices, akin to cams, converting movements of the motor into the specialized motion required for a particular activity.
Several new crystal structures discussed at the meeting, together with previously resolved structures of the D2 domain of N-ethyl maleimide-sensitive factor (NSF) (Lenzen et al., 1998) and the δ′ subunit of the DNA clamp loader (Guenther et al., 1997), confirmed the remarkably well conserved structure and mechanism of interaction with nucleotides. The conserved features of AAA modules imply a common mechanism for the operation of the motor, but differences in nucleotide-sensing residues and in the machinery assembled around the motor all determine the direction and timing of the forces that are exerted. The resulting variations produce a bewildering array of biological activities.
AAA proteins have either one (D1) or two non-identical (D1 and D2) AAA modules (Neuwald et al., 1999), each of which consists of a core with an α–β–α nucleotide binding fold and a C-terminal domain with at least three α-helices (Figure 1). AAA modules tend to oligomerize into hexameric rings, with all subunits oriented in the same direction (Lenzen et al., 1998; Zhang et al., 2000). The helical domain helps to make subunit contacts in the ring and is important for communicating conformational changes in response to nucleotide.
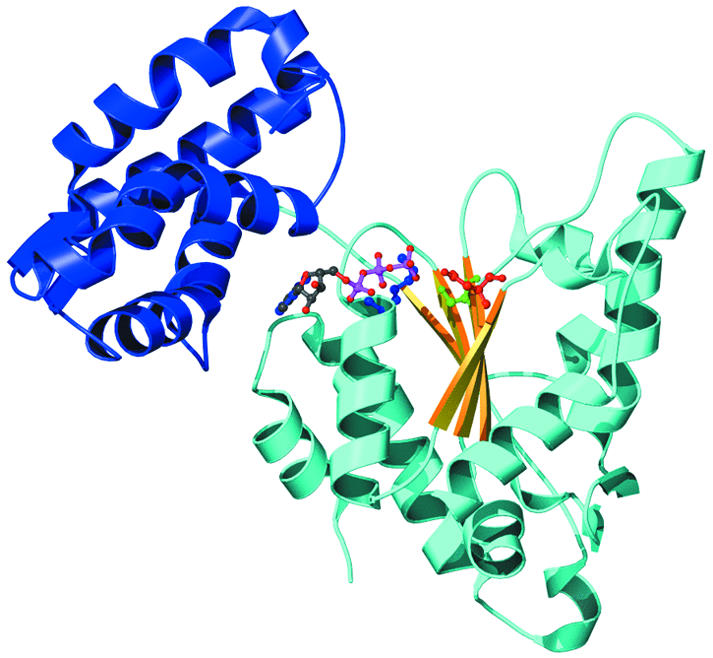
Fig. 1. Structure of the AAA module based on the crystal structure data of HslU from Bochtler et al. (1999). The N-terminal α–β–α nucleotide subdomain is in cyan (α helices) and yellow (β sheet), and the C-terminal helical domain is in blue. ADP (green and magenta) binds between the subdomains, with the Walker motif residues Lys63 (blue) and Asp256 (red) and the Sensor 1 residue Ser307 nearby.
In different AAA proteins, additional functional domains are found attached at the N- or C-terminal end or inserted within loops connecting secondary structural elements. In some cases, an independently folding N-terminal domain (N-domain) forms part of the substrate-binding surface of D1. In variants with no N-domain, other domains are attached to the analogous surface. For example, the I-domain of HslU (Bochtler et al., 1997) or the zinc-binding loop of the δ′ subunit of Escherichia coli DNA clamp-loader complex (Guenther et al., 1997). The N-domains or inserted domains interact directly or indirectly with substrates.
Other conserved motifs within the AAA module, Sensor 1 and Sensor 2, are important because they interact with the bound metal/nucleotide complex, transducing changes in the nucleotide state into precisely timed conformational changes and domain movements (Ogura and Wilkinson, 2001). W. Weis (Stanford, CA) reported that mutating Sensor 1 residues of NSF eliminates its ability to disassemble complexes without affecting its ATPase activity, emphasizing the importance of Sensor 1 in coupling ATP hydrolysis to changes in the substrate interaction domains.
A new twist to an old story
NSF and p97 (also called Cdc48p or VCP) are components of membrane fusion pathways that are important in processes such as the delivery of neurosecretory vesicles to the cell membrane and reconstitution of the Golgi system following cell division. Membrane fusion requires the formation of complexes between SNARE proteins in the vesicle and the corresponding target membrane. A critical step in the process is the NSF- or p97-catalyzed disassembly of the SNARE complexes following the fusion event, allowing SNAREs to be recycled (May et al., 2001). Interaction of the AAA proteins with SNAREs is mediated by adaptor proteins, αSNAP or p47, which bind to the N-domain of NSF or p97, respectively.
The mechanism of disassembly catalyzed by NSF or p97 was the focus of structure-derived models from two groups. Electron microscopic imaging of NSF indicates that N-domains can either be packed close to the sides of the hexamer or extend radially outwards depending on the nucleotide state. W. Weis (Stanford, CA) pointed out that it is possible to model a rigid body motion of the N-domain about the connection to the D1 domain, allowing the N-domains to rotate from the perimeter to a position nearer the central axis. Evidence that the N-domains of NSF can form a transient trimer led to the proposal of periodic three-in, three-out movements, which would allow them to interact symmetrically with the ternary αSNAP adaptor complex. Rotation of the N-domains could serve to either pull apart or unwind the helical bundles holding SNARE complexes together (Figure 2).

Fig. 2. Proposed mechanisms for AAA proteins. Blue represents substrate and green AAA proteins. (A) Twisting motions between D1 and D2 domains or between N-domains and D1 could lead to unwinding of helical substrate bundles, as is proposed for SNARE disassembly. (B) A pulling apart mechanism, proposed for microtubule disassembly, requires that the substrate components form stable interactions with separate AAA subunits and consequently dissociate from each other, pulled along as the AAA subunits separate. (C) Prying apart of subunits could occur upon partial ring opening, as seems to be the case for DNA clamp loaders. Nucleotide-dependent conformational changes could introduce tension in the ring, causing subunits to separate and forcing bound substrate subunits apart. (D) Threading occurs by unraveling of the protein substrate from one end, as in vectorial translocation of proteins by Clp ATPases. Movement of the polypeptide chain could occur by repeated cycles of substrate binding and release within the channel.
A different model emphasizing relative movements between the two AAA domains was proposed for p97 by P. Freemont (London, UK). When the crystal structure of p97 N-D1 and NSF D2 were aligned with the high resolution EM models of the intact p97, the two AAA domains fit best when arranged tail-to-tail. Such orientation would result in ATP induced conformational changes occurring in opposite directions in each half of p97, allowing a reciprocating rotational motion between the two domains during successive cycles of ATP hydrolysis. In the crystal, the N-domains of p97 are found on the perimeter of the D1 hexamer, which is also the site at which the adaptor, p47, has been visualized by electron microscopy (F. Indig, Bethesda, MD). The reciprocal motion of the domains combined with synchronous changes in binding would produce a ratcheting effect that could have the effect of unwinding the coiled domains of SNARE complexes (Figure 2).
The models of NSF and p97 action contrast with a proposal from F. McNally (Davis, CA) on the mechanism of action of katanin, a microtubule severing AAA protein. Katanin subunits form hexamers that are only transiently stable in the presence of ATP. Katanin assembles on microtubules and, in response to ATP hydrolysis, rapidly dissociates taking the associated microtubule subunits with it (Figure 2), hence the reference to this activity as a ‘pull-apartase’. Since most AAA proteins do not appear to dissociate during their catalytic cycles, the pulling-apart activity of katanin may be an extreme case of the prying-apart mechanism, requiring domain movements or partial ring opening, but not dissociation, to impart severing forces on the subunits of target complexes (Figure 2).
Learning to adapt: multiple cellular functions of p97
In addition to its role in membrane fusion, p97 has been implicated in a surprising number of other cellular processes, including apoptosis, cell cycle regulation and ubiquitin-dependent protein degradation. Not surprisingly, p97 is essential for viability. C. Ruhrberg (London, UK) reported that targeted deletion of p97 leads to lethality in day 10.5 mouse embryos and in cultured cells. New biochemical and genetic evidence is emerging to define the divergent functions of p97 and to implicate specific adaptor proteins in mediating these activities.
H. Meyer (New Haven, CT) described an adaptor that may direct p97 to the ubiquitin-dependent degradation pathway. Using p97 as bait, two proteins, Ufd1 (required for ubiquitin-fusion protein degradation) and Npl4 (required for nuclear transport), were pulled down from rat liver cytosol. The Ufd1-Npl4 binary complex binds p97 and regulates nuclear envelope processing by targeting an unknown factor to the ubiquitin pathway. The functional interaction of p97 and Ufd1-Npl4 was clarified in yeast by S. Jentsch (Martinsried, Germany), who showed that formation of the complex leads to the activation of a membrane-bound transcription factor, SPT23. SPT23 is a relative of mammalian NF-κB and controls unsaturated fatty acid levels. It is synthesized as a latent ER/nuclear membrane-localized precursor, and is activated by limited ubiquitin-proteasome degradation, similar to that seen with NF-κB p105 processing.
Binding of p47 and Ufd1-Npl4 to p97 is mutually exclusive, suggesting that adaptors compete for binding to p97. The ability of one adaptor to sequester p97 away from other pathways can be used to identify new adaptors and perhaps identify new functions for p97. Using a yeast two-hybrid system, M. Tagaya (Tokyo, Japan) identified SVIP (small VCP interacting protein) as a novel adaptor protein for p97. Although SVIP function is as yet unknown, its overexpression in cells caused extensive vacuolation and deformation of ER and microtubules.
A novel link between p97 and the ubiquitin-proteasome degradation pathway was presented by Chou-Chi Li (Frederick, MD), who identified p97 as a multiubiquitin chain targeting factor. Loss of p97 functions results in decreased degradation of proteasome substrates and accumulation of multi-ubiquitylated proteins. Ubiquitin chains bind directly to p97 through its N-domain, and deletion of the N-domain correlates with a defect in degradation of a model proteasome substrate in vitro.
Dungeons and drag-ins
The AAA family includes many protein quality control enzymes: the Clp/Hsp100 family of molecular chaperones, many of which act as regulatory components of ATP-dependent proteases; the regulatory subunits of the 26S proteasome; and the bifunctional ATP-dependent proteases, FtsH and Lon, each of which encode an AAA domain and a protease domain within a single polypeptide.
The proteolytic sites of Clp proteases and the proteasome are sequestered in a chamber in the center of two rings and are accessible only through their axial channels (Horwich et al., 1999). Proteins must first be unwound into an extended conformation in order to pass through the channels. The ATPase rings are axially aligned on both sides of the protease. In EM images shown by A. Steven (Bethesda, MD), substrates initially bind to the distal ring surface of intact complexes of ClpAP and ClpXP, and upon ATP addition, disappear from the surface and appear within the central chamber of ClpP. When the proteolytic chamber of ClpAP was partially blocked, a segmental movement of the protein through the complex could be seen.
Substrates for unfolding and degradation can interact directly with a domain in the AAA component, but, just as with membrane-fusion AAA proteins, substrate selection may also be modulated by adaptor proteins. A unique adaptor for ClpA, named ClpS, was described by B. Bukau (Freiburg, Germany). ClpS interacts with ClpA and acts as a specificity factor by inhibiting degradation of some soluble substrates, while activating degradation of aggregated proteins.
New studies now establish the molecular surface involved in interactions with substrates and with the protease component. Cryo-electron microscopy of HslUV (ClpYQ) by A. Steven and a new X-ray crystal structure by J. Wang (New Haven, CT) both showed the I-domains pointing away from the protease and the opposite ring face making firm contact with the protease. M. Bochtler (Martinsried, Germany) showed that removing the I-domain of HslU blocks the ability to degrade some but not all substrates. J. Wang’s analysis of HslU crystals in several nucleotide states, suggests that the domains undergo nucleotide-dependent movements. One movement closes or opens a central pore and could govern substrate movement through the channel. Another movement twists portions of the I-domains, possibly providing a basis for a nucleotide-dependent substrate binding-release cycle.
The ATPase–protease interfaces in ClpXP and ClpAP are analogous to those in HslUV. M. Maurizi (Bethesda, MD) showed that both ClpA and ClpX have surface loops required for activation of ClpP. The crystal structure of ClpA shows this surface to be analogous to the surface of HslU that interacts with HslV. The N-domains of ClpX and ClpA, like the I-domain of HslU, are on the surface distal to the protease and affect their activity against specific substrates. This surface is comparable to the surface on which the N-domains of NSF and p97 are located, suggesting that AAA proteins share a common mechanism for communicating ATP-dependent conformational changes to substrates or adaptor proteins bound to the N-domains.
Unwinding the winding path
Progress in understanding the mechanism by which ATP-dependent proteases unfold proteins and deliver them to the protease has been an exciting development. Recognition motifs located near the N- or C-terminus are needed to target proteins for complete degradation by ClpAP or ClpXP. M. Maurizi pointed out that, once a substrate is bound, the rate-limiting step in degradation is unfolding of the protein. However, some protein domains are resistant to unfolding, and A. Matouschek (Evanston, IL) was able to exploit that property to study the direction of unfolding and translocation by ClpAP and ClpXP. An unstable domain immediately adjacent to the degradation motif was degraded, but, if a stable domain was placed between the motif and the unstable domain, the latter was spared. Thus, translocation into the protease proceeds vectorially from the end bearing the degradation motif. Moreover, when circularly permuted protein substrates were compared, degradation rates inversely correlated with the stability of the secondary structural element adjacent to the degradation motif rather than with the global thermodynamic stability of the protein. Thus, ClpA and ClpX unfold proteins by breaking local secondary structures in a sequential manner (unraveling) during translocation.
Directional translocation of substrate into ClpAP was followed in real time by B. Reid (New Haven, CT) using fluorescence energy transfer measurements. A fluorescent probe located near the C-terminal tag entered ClpP without a lag, whereas probes located several hundred amino acids away were translocated after a delay, confirming that the C-terminus itself enters ClpP during the initial phase of translocation. Since degradation is highly processive with the release of few, if any, intermediates, these results imply that disruption of the initial secondary structural element is rate-limiting.
Sequential unraveling of a protein from an accessible end bearing a recognition motif can explain how ClpA and ClpX, acting as autonomous chaperones, are able to disassemble stable protein or protein–DNA complexes (Wickner et al., 1994; Levchenko et al., 1995). Unraveling would destabilize the tertiary structure of the target protein, thereby disrupting its quaternary interactions and destabilizing any assembly of which it is part. This mechanism might serve as a paradigm for disassembly reactions catalyzed by other AAA proteins as well (Figure 2).
Proteasomes and substrate gating
The Clp mechanism of protein unfolding and directional translocation probably applies to all ATP-dependent proteases. The regulatory particle of the 26S proteasome is composed of a loosely connected lid and base (Glickman et al., 1998). The base is comprised of eight subunits, six of which are AAA proteins, and exhibits an ATP-dependent chaperone-like activity in vitro. This activity may promote protein unfolding as shown by T. Kishimoto (Yokohama, Japan), who devised an assay based on disassembly of the cyclin B/Cdc2 complex. Inactivation of cyclin B was independent of proteolysis but required the regulatory particle and its constituent AAA subunits. These results imply that, subsequent to their capture by the 26S proteasome, ubiquitylated proteins are unfolded before they are transferred to the core particle for degradation.
Translocation of proteins through narrow channels could also be regulated by gating mechanisms. Two aspects of gating in the proteasome have been characterized by D. Finley (Boston, MA). The first one entails the core particle, where in the latent state, N-terminal extensions of the α-ring subunits form a gate blocking entry into the channel. The α3 subunit acts as the lock holding the other α-subunit N-terminal peptides in place. Crystallographic analysis shows that deleting the α3 N-terminus causes the other N-termini to retract and opens the channel. The gating mechanism may be mediated by one of the proteasomal AAA proteins, Rpt2 (S4). A mutation in the nucleotide-binding Walker A motif of Rpt2 results in a severe peptide hydrolysis defect, which can be suppressed by the α-subunit open-channel mutation, implying that the rpt2 peptide hydrolysis defect results from failure to open the channel.
FtsH and other AAA proteases
FtsH is a bifunctional ATP-dependent protease, in which a metallo-protease domain is fused at the C-terminus of an AAA module. FtsH-like proteases are widespread from bacterial cells to mammalian mitochondria (Langer, 2000). The N-terminal domain of FtsH has two membrane spanning segments, but the catalytic domains remain within a soluble compartment. Homology modeling and mutagenesis were used by T. Ogura (Kumamoto, Japan) to provide evidence that E. coli FtsH is similar to other ATP-dependent proteases in compartmentalizing its unfolding and proteolytic activities. A hexamer model of FtsH, based on alignment with the ATP-bound form of NSF D2, contains a central channel that undergoes a conformational change in converting from the ATP- to the ADP-bound state. Subunit association brings two conserved arginine residues into position to play a critical role in ATP hydrolysis, which may explain why FtsH is functional only in the oligomeric state.
Evidence in favor of a translocation mechanism consistent with the above model of FtsH was obtained by Y. Akiyama and K. Ito (Kyoto, Japan). Degradation of membrane-associated substrates by E. coli FtsH requires an accessible cytosolic tail of at least 20 amino acids. Data obtained from studies in which tail length was manipulated are consistent with the presence of a tethering site within the translocation channel and the accessibility of this site only to extensions of sufficient length. Both N- and C-terminal extensions could be degraded effectively, and effectiveness did not depend on a specific sequence. A provocative model in which the transmembrane segments of FtsH also mediate specific substrate interaction was discussed. Soluble substrates are degraded by FtsH lacking the transmembrane segments, but the latter are required for degradation of membrane proteins. These data suggest that both transmembrane segments and cytosolic tails of substrates may act as determinants for recognition by FtsH.
Ringing in the nu(cleic acid)
Processivity is an essential feature of DNA replication. D. Jeruzalmi (New York) described loading of the β processivity clamp onto circular DNA in E. coli, which is accomplished by the clamp loader complex, composed of γ, δ, δ′, χ and ψ subunits. The major function of the AAA ATPase in the clamp loader complex is to partially dissociate a stable β dimer. γ and δ′ are both AAA proteins, but only γ is an active ATPase. Upon binding of ATP to γ, changes within the complex cause δ to bind β, a bi-lobed dimer, and pry it partially open. DNA slips into the open dimer and, upon ATP hydrolysis, the complex dissociates and β closes, thus encircling the DNA.
The major features of bacterial DNA replication are recapitulated in mammals. In eukaryotes, DNA polymerase is stabilized on the DNA by an accessory sliding clamp, PCNA (proliferating cell nuclear antigen). PCNA is loaded on the DNA by a clamp loader, RFC (replication factor C). RFC is a pentameric ring structure consisting of five AAA family members. T. Tsurimoto (Kyoto, Japan) used electron microscopy and atomic force microscopy to study the interaction between RFC and PCNA and their structural changes during repeated cycles of ATP hydrolysis. RFC has a closed two-finger structure in the absence of nucleotides but can be converted into an open structure in the presence of ATP. The PCNA clamp is held between the two fingers of RFC. Opening and closing of RFC in response to binding and release of ATP allows the attached PCNA to clamp onto DNA.
DNA replication normally occurs once per cell division. Timing is regulated in part by the AAA protein, DnaA. DnaA is the DNA replication initiation protein and is active in the ATP-bound state but not in the ADP-bound state. T. Katayama (Tokyo, Japan) found that the sliding clamp of DNA polymerase III, in a reaction mediated by another protein, IdaB, accelerates the rate of ATP hydrolysis by DnaA. DnaA assembles on DNA as part of the initiation complex, and loading of the sliding clamp promotes hydrolysis of bound ATP, converting DnaA to the inactive state and blocking further initiation.
AAA proteins are also part of the replication complex required to complete DNA synthesis. This includes the MCM (minichromosome maintenance) complex, composed of the six AAA proteins MCM2-7. Y. Ishimi (Tokyo, Japan) reported on the helicase activity of a subcomplex of the MCM complex, a hexameric ring with two molecules each of MCM4, 6 and 7. DNA unwinding occurs upon passage of single-stranded DNA through the ring. Not all MCM subunits are catalytic, suggesting that the different proteins play distinct roles in helicase activity. Catalytic and non-catalytic subunits are also found in other AAA proteins, such as the clamp-loader complex and dynein. Heterologous composition may allow ring opening or other asymmetric structural changes needed for these enzymes to conduct their activities.
Ishimi and H. Takisawa (Toyonaka, Japan) reported that MCM proteins are phosphorylated by two types of protein kinases, cyclin-dependent Cdk kinase and Cdc7-Dbf4 kinase, which contributes to cell cycle-dependent regulation of DNA synthesis. Interestingly, certain MCM proteins are also regulated by ubiquitylation. I. Cheng and B. Tye (Ithaca, NY) showed that during M phase a small fraction of MCM3 protein is targeted for ubiquitylation which appears to be an essential step for the initiation of DNA replication in S. cerevisiae.
Recombination: Holliday junction branch migration
DNA translocation by AAA protein rings is dramatically illustrated by E. coli RuvB, a hexameric AAA protein that encircles double-stranded DNA and promotes branch migration of Holliday junctions during homologous recombination. H. Iwasaki (Osaka, Japan) showed that RuvB, together with its partner RuvA, translocates along the DNA extending the region encompassed by the heteroduplex. An interesting issue is how RuvB loads onto the DNA. RuvB is a heptamer in solution, whereas it is a hexamer when bound to DNA, and we can speculate that the ring opens by loss of a subunit and then closes to encircle the DNA that has passed through the gap. RuvB-like proteins also have a role in transcription. K. Kohno (Nara, Japan), reported on a distant RuvB homolog in S. cerevisiae, Tih2p/Rvb2p, which interacts with the TATA binding protein and RNA polymerase II. Tih2p has helicase activity and is required for transcription of G1 cyclin and of ribosomal proteins.
Escherichia coli restriction endonuclease McrBC also has ring-shaped structure and functions similar to AAA proteins but, interestingly, McrBC is a GTPase rather than an ATPase. According to D. Panne (Cambridge, MA), single rings of McrB move along the DNA until they meet another oligomer. This interaction activates the endonuclease activity of McrBC. McrBC may represent a link between AAA proteins and G proteins, both of which use the energy of ATP or GTP hydrolysis to drive conformational changes and other protein domain movements.
It takes two to untangle
ClpB/Hsp104 can dissolve protein aggregates but additional molecular chaperones are required to promote complete refolding of the solubilized protein (Glover and Lindquist, 1998). M. Yoshida (Tokyo, Japan) showed that in Thermus thermophilus both Hsp70 and GroEL chaperone systems are needed in addition to ClpB to obtain maximum yields of resolubilized aggregates. Combined with the finding that E. coli ClpS can facilitate degradation of aggregated proteins by ClpAP, these studies reveal that cells have mechanisms not only for avoiding protein aggregation but also for dealing with aggregates once they are formed.
As no mammalian ClpB/Hsp104 proteins have been found, it seems likely that another AAA protein family serves the function of Hsp104 in mammalian cells. New AAA proteins with molecular chaperone activity towards protein aggregates would be of great interest. One candidate is PIP-1, a polyglutamine-binding AAA protein described by A. Kakizuka (Osaka, Japan). PIP-1 is expressed in nuclear inclusions in Huntington’s disease patients brain, Lewy bodies, and expanded polyglutamine-expressing transgenic mice. Interestingly, expression of a mutant PIP-1 defective in ATPase activity produced cytoplasmic vacuolization, another condition associated with polyglutamine diseases. Perhaps PIP-1 or some other aggregate-binding AAA protein may be found to reverse protein aggregation or to dissolve the protein tangles associated with human amyloid diseases.
Conclusions
AAA modules are at the heart of molecular machines catalyzing simple mechanical movements that make or break contacts between macromolecules. This results in local or global protein unfolding, assembly or disassembly of complexes, or transport of macromolecules relative to each other. These activities underlie processes critical to DNA replication and recombination, secretion, membrane sorting, cellular reorganization and organelle biogenesis, protein disaggregation and degradation, and intracellular transport. A remaining challenge is to describe the molecular details of how nucleotide binding and hydrolysis contribute to the varied domain motions involved in protein unfolding, disassembly and translocation. Another challenge will be to define how substrates are recognized by AAA proteins either directly or through adaptor proteins, and how binding and release of a substrate is coordinated with the domain movements within the AAA protein to bring about structural alteration of the substrate.

The ‘International Meeting on Cellular Functions of AAA Proteins’ organized by Koreaki Ito and Teru Ogura took place in Kyoto, Japan, March 13–16, 2001.

The authors* and organizers of the meeting (from left to right): Koreaki Ito, Teru Ogura, Mike Maurizi* and Chou Chi Li*
References
- Bochtler M., Ditzel, L., Groll, M. and Huber, R. (1997) Crystal structure of heat shock locus V (HslV) from Escherichia coli. Proc. Natl Acad. Sci. USA, 94, 6070–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman M.H., Rubin, D.M., Coux, O., Wefes, I., Pfeifer, G., Cjeka, Z., Baumeister, W., Fried, V.A. and Finley, D. (1998) A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell, 94, 615–623. [DOI] [PubMed] [Google Scholar]
- Glover J.R. and Lindquist, S. (1998) Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell, 94, 73–82. [DOI] [PubMed] [Google Scholar]
- Guenther B., Onrust, R., Sali, A., O’Donnell, M. and Kuriyan, J. (1997) Crystal structure of the δ′ subunit of the clamp-loader complex of E. coli DNA polymerase III. Cell, 91, 335–345. [DOI] [PubMed] [Google Scholar]
- Horwich A.L., Weber-Ban, E.U. and Finley, D. (1999) Chaperone rings in protein folding and degradation. Proc. Natl Acad. Sci. USA, 96, 11033–11040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer T. (2000) AAA proteases: cellular machines for degrading membrane proteins. Trends Biochem. Sci., 25, 247–251. [DOI] [PubMed] [Google Scholar]
- Lenzen C.U., Steinmann, D., Whiteheart, S.W. and Weis, W.I. (1998) Crystal structure of the hexamerization domain of N-ethylmaleimide-sensitive fusion protein. Cell, 94, 525–536. [DOI] [PubMed] [Google Scholar]
- Levchenko I., Luo, L. and Baker, T.A. (1995) Disassembly of the Mu transposase tetramer by the ClpX chaperone. Genes Dev., 9, 2399–2408. [DOI] [PubMed] [Google Scholar]
- May A.P., Whiteheart, S.W. and Weis, W.I. (2001) Unraveling the mechanism of the vesicle transport ATPase NSF, the N-ethylmaleimide-sensitive factor. J. Biol. Chem., 276, 21991–21994. [DOI] [PubMed] [Google Scholar]
- Neuwald A.F., Aravind, L., Spouge, J.L. and Koonin, E.V. (1999) AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res., 9, 27–43. [PubMed] [Google Scholar]
- Ogura T. and Wilkinson, A.J. (2001) AAA+ superfamily ATPases: common structure-diverse function. Genes Cells, 6, 575–597. [DOI] [PubMed] [Google Scholar]
- Wickner S., Gottesman, S., Skowyra, D., Hoskins, J., McKenney, K. and Maurizi, M.R. (1994) A molecular chaperone, ClpA, functions like DnaK and DnaJ. Proc. Natl Acad. Sci. USA, 91, 12218–12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. et al. (2000) Structure of the AAA ATPase p97. Mol. Cell., 6, 1473–1484. [DOI] [PubMed] [Google Scholar]