

Abstract
Genetic variation at the transmembrane protein 106B gene (TMEM106B) has been linked to risk of frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) through an unknown mechanism. We found that presence of the TMEM106B rs3173615 protective genotype was associated with longer survival after symptom onset in a postmortem FTLD-TDP cohort, suggesting a slower disease course. The seminal discovery that filaments derived from TMEM106B is a common feature in aging and, across a range of neurodegenerative disorders, suggests that genetic variants in TMEM106B could modulate disease risk and progression through modulating TMEM106B aggregation. To explore this possibility and assess the pathological relevance of TMEM106B accumulation, we generated a new antibody targeting the TMEM106B filament core sequence. Analysis of postmortem samples revealed that the TMEM106B rs3173615 risk allele was associated with higher TMEM106B core accumulation in patients with FTLD-TDP. In contrast, minimal TMEM106B core deposition was detected in carriers of the protective allele. Although the abundance of monomeric full-length TMEM106B was unchanged, carriers of the protective genotype exhibited an increase in dimeric full-length TMEM106B. Increased TMEM106B core deposition was also associated with enhanced TDP-43 dysfunction, and interactome data suggested a role for TMEM106B core filaments in impaired RNA transport, local translation, and endolysosomal function in FTLD-TDP. Overall, these findings suggest that prevention of TMEM106B core accumulation is central to the mechanism by which the TMEM106B protective haplotype reduces disease risk and slows progression.
Editor’s summary
Variants in the gene for TMEM106B, a transmembrane protein that regulates lysosomal functions, modulate the risk for frontotemporal lobar degeneration (FTLD). Lysosomal dysfunctions may contribute to FTLD, but recent discoveries of TMEM106b filaments suggest that protein aggregations could also be implicated. Here, Marks and colleagues developed an antibody to detect the TMEM106B filament core and used biochemical extractions from postmortem brain tissue to show that TMEM106B core deposition was increased in carriers of a FTLD-risk SNP, correlated with TDP-43 dysfunction and associated with TDP-43 pathology. Although further mechanistic studies are needed, these findings indicate that the presence of TMEM106B core depositions might modulate disease risk. —Daniela Neuhofer
INTRODUCTION
Frontotemporal dementia (FTD) is the second most common cause of dementia in patients under 65 after Alzheimer’s disease (AD) (1) and encompasses a spectrum of related neurodegenerative disorders that are defined clinically by changes in personality, behavior, and language. Pathologically, FTD is caused by frontotemporal lobar degeneration (FTLD) of the brain, with ~50% of all FTLD cases characterized by proteinaceous inclusions of transactive response (TAR) DNA binding protein of 43 kDa (TDP-43) (FTLD-TDP) (2, 3). Genetic variation at the transmembrane protein 106B (TMEM106B) locus has been linked to risk of FTLD-TDP (4–8), including the coding variant rs3173615 [the major/risk allele (C) encodes a threonine at residue 185, whereas the minor/protective allele (G) encodes serine], which is in strong linkage disequilibrium (LD) with other single-nucleotide polymorphisms (SNPs) (9). The protective allele at the TMEM106B locus was also associated with slowed progression of TDP-43 pathology in individuals without FTLD in two community-based studies of aging, the Religious Orders Study and the Rush Memory and Aging Project (10). In particular, after the exclusion of cases with FTLD or related pathology, the rs1990622 protective allele [in high LD with rs3173615 (9)] was linked to decreased propagation of hyperphosphorylated TDP-43 (pTDP-43) pathology from the amygdala to the hippocampus/entorhinal cortex and ultimately neocortical regions (10). Similarly, in patients with FTLD-TDP, the rs1990622 protective allele was associated with reduced pTDP-43 burden in the frontal cortex (11). Collectively, these studies suggest that the genetic link between TMEM106B and disease risk is driven by TDP-43 pathology, but the mechanism by which TMEM106B variants affect TDP-43 deposition remains unclear.
Recently, four independent groups made the seminal discovery that filaments derived from the C terminus of TMEM106B (corresponding to residues 120 to 254) are a common feature in aging and across a range of neurodegenerative disorders (12–15). Although the functional relevance of this observation has yet to be determined, the localization of the rs3173615 coding SNP (C > G, p.T185S) within the TMEM106B filament core raises the possibility that TMEM106B genetic variation could modulate TMEM106B aggregation. However, the lack of commercially available specific and sensitive antibodies to detect the TMEM106B filament core has hindered efforts to explore this idea. To address this need, we generated a polyclonal antibody targeting the TMEM106B filament core sequence, which we validated using sequence-verified expression constructs and postmortem brain tissue from patients with FTLD-TDP with confirmed TMEM106B filament pathology (13). After authentication, the new TMEM106B core antibody was used to examine the relationship between TMEM106B genetic variation and aggregation in a large cohort of well-characterized and pathologically confirmed FTLD-TDP cases (16, 17). This analysis revealed that accumulation of the TMEM106B filament core in the frontal cortex was strongly associated with the rs3173615 risk allele in a gene dosage–dependent manner. We also observed a reduction in dimeric full-length TMEM106B in homozygous carriers of the risk genotype. Moreover, the abundance of insoluble TMEM106B core fragments was associated with both pTDP-43 burden and TDP-43 loss of function, supporting the idea that TMEM106B genetic variants may influence TDP-43 dysfunction through TMEM106B core deposition. Accordingly, we found that TDP-43 targets were enriched in the network of proteins that associated with aggregated TMEM106B as identified via mass spectrometry (MS). The interactome of insoluble TMEM106B also included a number of ribosomal subunits and proteins involved in endosomal transport, providing mechanistic insight into the consequences of TMEM106B filament deposition in the brain. Together, our data delineate a model in which genetic variants in TMEM106B alter aggregation of the TMEM106B core and its downstream effects on protein translation and intracellular trafficking, ultimately influencing TDP-43 dysfunction and disease progression.
RESULTS
Extended survival after symptom onset is associated with the TMEM106B protective haplotype
Given previous evidence linking genetic variation at the TMEM106B locus to risk of FTLD-TDP (4–8), we wanted to determine whether this locus might also affect disease trajectory. To do so, we assessed rs3173615 genotype to identify carriers of the TMEM106B protective haplotype (Table 1) and subsequently evaluated the relationship with survival after symptom onset. A longer survival after symptom onset was associated with the rs3173615 protective genotype in FTLD-TDP [median survival of 12 years in individuals homozygous for the minor allele (GG/SS185) compared with median survival of 8 years in carriers of the major allele (CC/TT185 and CG/TS185); P = 0.009] (Table 2), suggesting that the presence of the TMEM106B protective haplotype slows disease progression.
Table 1. Patient characteristics in cohort with TMEM106B rs3173615 SNP status.
The sample median (minimum, maximum) is given for continuous variables, and number of cases (percentages) is provided for categorical variables. Information was unavailable regarding age at onset and survival after symptom onset (n = 22).
| Characteristic | FTLD-TDP (N = 228) |
|---|---|
| Sex (female) | 106 (46.5%) |
| Age at onset (years) | 64.72 (39.98, 86.49) |
| Survival after onset (years) | 8.00 (1.00, 25.00) |
| Age at death (years) | 74.16 (49.98, 99.56) |
| TMEM106B rs3173615 | |
| CC/TT185 | 106 (46.5%) |
| CG/TS185 | 104 (45.5%) |
| GG/SS185 | 18 (7.9%) |
Table 2. Association between survival after symptom onset and TMEM106B rs3173615 status in FTLD-TDP.
β, regression coefficient; CI, confidence interval; min, minimum; max, maximum. β values, 95% CIs, and P values result from unadjusted linear regression models or linear regression models adjusted for age at death and sex. β values are interpreted as the difference in survival after symptom onset. P < 0.025 are considered statistically significant after correcting for multiple testing.
| Unadjusted analysis | Adjusted analyses | ||||
|---|---|---|---|---|---|
|
|
|||||
| FTLD (N = 206) | Median survival [years (min, max)] | β (95% CI) | P value | β (95% CI) | P value |
|
| |||||
| CC/TT185 | 8 (2, 25) | 0.00 (reference) | NA | 0.00 (reference) | NA |
|
| |||||
| CG/TS185 | 8 (1, 25) | 0.523 (−0.802–1.847) | 0.4373 | 0.725 (−0.539–1.989) | 0.2594 |
|
| |||||
| GG/SS185 | 12 (3, 16) | 3.036 (0.638–5.434) | 0.0133 | 3.095 (0.781–5.409) | 0.0090 |
The TMEM106B core antibody detects TMEM106B filaments
After the discovery that filaments derived from TMEM106B are visible by cryo–electron microscopy (cryo-EM) and accumulate with aging and across a range of neurodegenerative disorders (12–15), we hypothesized that TMEM106B aggregation may explain the genetic link between TMEM106B haplotype and disease. To investigate this idea, we first used the cryo-EM structural map of TMEM106B filaments to identify an immunogenic sequence present on the surface of filaments (to facilitate immunolabeling) and subsequently generated an antibody against residues 191 to 206 in the TMEM106B filament core sequence (Fig. 1A, epitope colored in blue). To validate our TMEM106B core antibody, we generated a full-length TMEM106B expression construct, which we confirmed is both expressed and glycosylated to a similar extent as endogenous TMEM106B using a commercially available antibody recognizing the N terminus of the protein and deglycosylation by peptide N-glycosidase F (PNGase) (fig. S1A) (18). We also used site-directed mutagenesis to generate a TMEM106B (p.T185) construct that mimics the coding change introduced by the rs3173615 risk genotype (residue marked in red in Fig. 1A) and used both variants in addition to a myc/His-tagged plasmid to examine antibody specificity. As expected, immunoblotting verified that our antibody detects all TMEM106B constructs (fig. S1B), including both full-length and higher–molecular weight TMEM106B-positive protein complexes, and that immunoreactivity was unaffected by the p.T185S coding change. Last, we generated a TMEM106B deletion construct lacking the protein sequence used as an immunogen for our antibody (Δ191 to 206). Confirming antibody specificity, we demonstrated by immunoblotting that our TMEM106B core antibody recognizes the full-length protein but not that encoded by the Δ191 to 206 deletion construct (fig. S1C, left), whereas a commercially available antibody generated against the N terminus of TMEM106B recognized both constructs (fig. S1C, right).
Fig. 1. TMEM106B core antibody detects TMEM106B filaments and a glycosylated 29-kDa species.
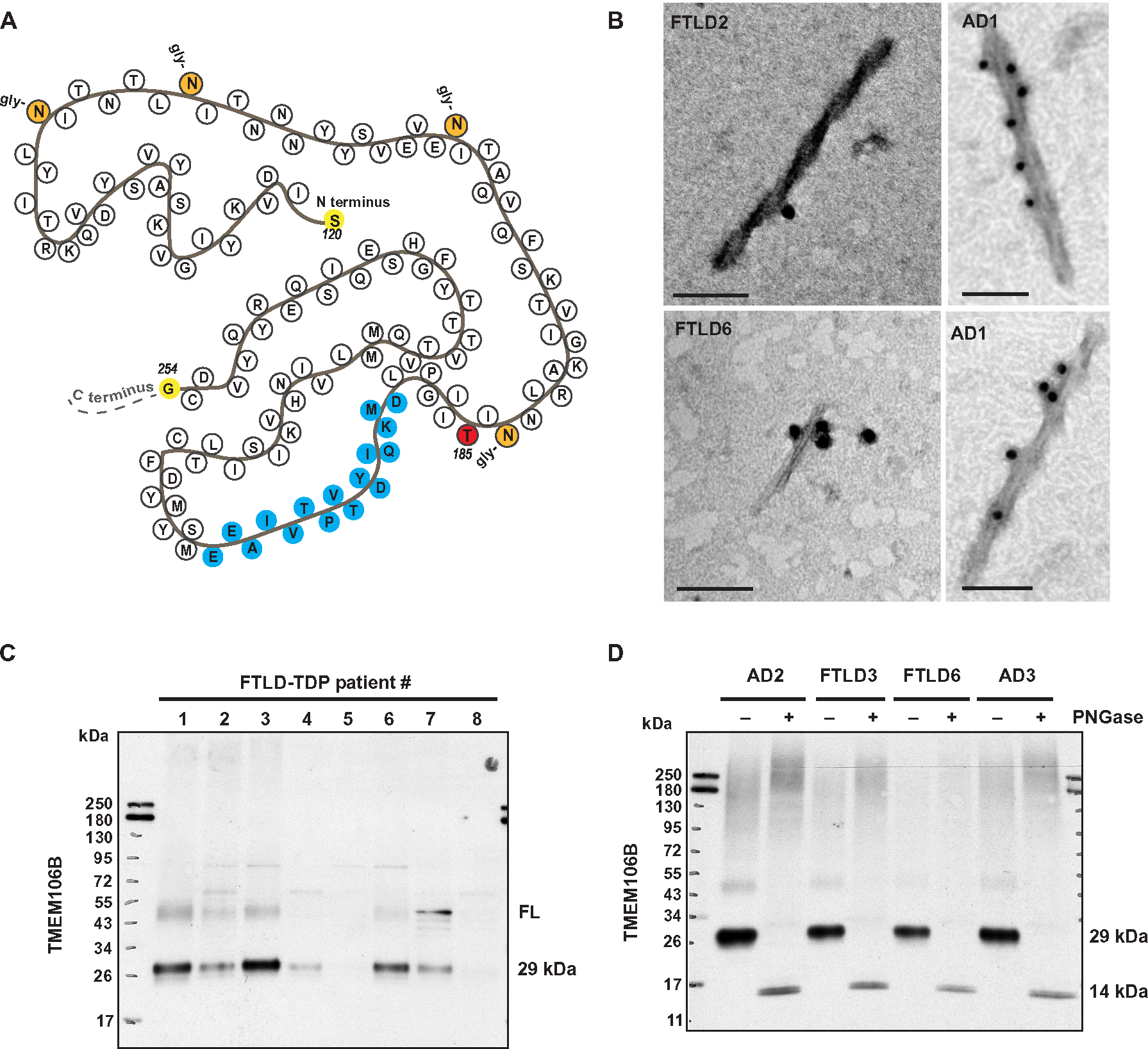
(A) Schematic illustration of the TMEM106B filament structure solved by cryo-EM, with the epitope for the TMEM106B core antibody we developed colored in blue. The rs3173615 coding variant, with p.T185 as the reference sequence, is colored in red. Experimentally detected glycosylation (gly-) sites are also noted, with N and C termini (at residues 120 and 254, respectively) colored in yellow. (B) TMEM106B-positive filaments were detected in the sarkosyl-insoluble fraction from patients with FTLD-TDP and AD by iEM. Scale bars, 100 nm. (C) Sarkosyl-insoluble fractions from FTLD-TDP cases (n = 8) with cryo-EM confirmed TMEM106B filaments were evaluated by SDS– polyacrylamide gel electrophoresis (PAGE). Immunoblotting with the TMEM106B core antibody detected a 29-kDa species and full-length TMEM106B (FL) in some cases. (D) Sarkosyl-insoluble fractions from FTLD-TDP [n = 2, both also shown in (B) and (C)] and AD (n = 2) were incubated in the presence or absence of PNGase (peptide N-glycosidase F) to induce deglycosylation, followed by immunoblotting for TMEM106B to demonstrate that the 29-kDa TMEM106B species is glycosylated.
To provide additional validation of our antibody, we obtained postmortem brain tissue from the frontal cortices of FTLD-TDP cases confirmed to contain TMEM106B filaments by cryo-EM (table S1) (13). After extraction of the sarkosyl-insoluble fraction, we evaluated immunoreactivity for the TMEM106B core antibody by immuno–electron microscopy (iEM) and Western blot. As expected, TMEM106B-positive filaments were detected by iEM in the sarkosyl-insoluble fraction from patients with FTLD-TDP (Fig. 1B, left), as well as from patients with pathologically confirmed AD (Fig. 1B, right, and table S1). In addition, consistent with a previous study (14), Western blotting with the TMEM106B core antibody revealed that accumulation of a 29-kDa core fragment in most FTLD-TDP cases with cryo-EM confirmed TMEM106B fibrils (Fig. 1C). Because the predicted molecular weight of the TMEM106B filament core (corresponding to residues 120 to 254) is ~14 kDa, it has been proposed that glycosylation contributes to the higher-than-expected molecular weight of the insoluble TMEM106B core fragment. To test this idea, we incubated the sarkosyl-insoluble fraction with PNGase to promote deglycosylation of TMEM106B as described (18) and subsequently evaluated the reactions by Western blot. In agreement with the idea that glycosylation increases the molecular weight of the TMEM106B core to 29 kDa, we observed a shift in the molecular weight of the TMEM106B-positive fragment to around 14 kDa upon deglycosylation (Fig. 1D). Together, these results indicate that our TMEM106B core antibody is specific for TMEM106B, immunoreactivity is independent of rs3173615 genotype, and the 29-kDa TMEM106B fragment is both glycosylated and filamentous.
TMEM106B core accumulation is associated with TMEM106B risk haplotype
To investigate the possibility that TMEM106B aggregation may explain the relationship between rs3173615 genotype and disease risk in FTLD-TDP, we isolated the sarkosyl-insoluble fraction from a larger cohort of pathologically confirmed FTLD-TDP cases (n = 292; see Table 3 for patient characteristics) and examined TMEM106B core accumulation by immunoblotting. As shown in the representative Western blot in Fig. 2A (top), robust deposition of the 29-kDa TMEM106B core was observed in homozygous carriers of the rs3173615 risk SNP (CC/TT185), whereas carriers of the homozygous protective genotype (GG/SS185) exhibited minimal to undetectable accumulation of TMEM106B in the sarkosyl-insoluble fraction (Fig. 2A, top). Quantitation of the sarkosyl-insoluble 29-kDa TMEM106B core fragment revealed that deposition occurs in a gene dosage-dependent manner in FTLD-TDP, with the highest accumulation detected in CC/TT185 carriers, intermediate accumulation observed in heterozygous CG/TS185 carriers, and the lowest TMEM106B core burden observed in GG/SS185 carriers (Fig. 2B and data file S1). Statistically significant differences in TMEM106B core accumulation between genotypes were detected both in unadjusted analyses (all P < 0.0001; Fig. 2B and table S2) and analyses adjusted for age at death and sex (all P < 0.0001; table S2). To exclude the possibility that TMEM106B core burden is influenced by changes in the abundance of full-length TMEM106B, we examined total TMEM106B in soluble brain lysates. Because Chen-Plotkin and colleagues (19) previously reported that full-length TMEM106B can be detected in both dimeric and monomeric form, although the dimer is only detectable under cold conditions, we used a similar approach to assessing whether rs3173615 genotype would be associated with alterations in either monomeric or dimeric TMEM106B (Fig. 2A, bottom). In our efforts to detect both species for quantification, we tested multiple commercial antibodies and chose an antibody directed against the N terminus of TMEM106B for further experiments (fig. S2, A and B). Although the abundance of monomeric TMEM106B was unchanged (Fig. 2C, right), the amount of dimeric TMEM106B was significantly (P < 0.01) lower in carriers of the risk genotype (CC/TT185) compared with the protective genotype (GG/SS185) (Fig. 2C, left). This finding may suggest that dimeric TMEM106B protects against either the formation or deposition of the core fragment, which is not a simple consequence of higher full-length TMEM106B protein expression. Consistent with this, we did not observe differences in TMEM106B expression at the RNA level (fig. S2C). Last, we evaluated TMEM106B core burden in the sarkosyl-insoluble fraction from control cases (cognitively normal), which revealed that accumulation of a 29-kDa TMEM106B fragment similarly occurs in a rs3173615 genotype–dependent manner (fig. S3, A and B), consistent with cryo-EM data showing that TMEM106B core deposition is not exclusive to FTLD-TDP.
Table 3. Patient characteristics in the FTLD-TDP postmortem cohort.
RIN, RNA integrity number in the frontal cortex; tSTMN2 RNA, truncated STMN2 RNA concentration in the frontal cortex; pTDP-43 protein, phosphorylated TDP-43 protein concentration in the frontal cortex. The sample median (minimum, maximum) is given for continuous variables, and number of cases (percentages) is provided for categorical variables. information was unavailable regarding age at onset (n = 21), survival after symptom onset (n = 21), age at death (n = 1), sex (n = 1), RIN (n = 69), pTDP-43 protein (n = 39), tSTMN2 RNA (n = 55), and TDP-43 subtype (n = 1).
| Characteristic | FTLD-TDP (N = 292) |
|---|---|
| Sex (female) | 139 (47.60%) |
| Age at onset (years) | 65 (39.98, 89.62) |
| Survival after onset (years) | 7.00 (0.61, 25.0) |
| Age at death (years) | 72.57 (44.47, 99.56) |
| Motor neuron disease co-occurence | 76 (26%) |
| TMEM106B rs3173615 | |
| CC/TT185 | 129 (44.2%) |
| CG/TS185 | 136 (46.6%) |
| GG/SS185 | 27 (9.2%) |
| TDP-43 subtype | |
| A | 150 (51.4%) |
| B | 84 (28.8%) |
| C | 57 (19.5%) |
| RIN | 9.50 (6.10, 10) |
| pTDP-43 protein | 4451 (173.5, 37109) |
| 29-kDa TMEM106B protein | 18.59 (0.0, 470.6) |
| tSTMN2 RNA | 560.4 (8.18, 5210) |
Fig. 2. TMEM106B core deposition associates with risk haplotype.
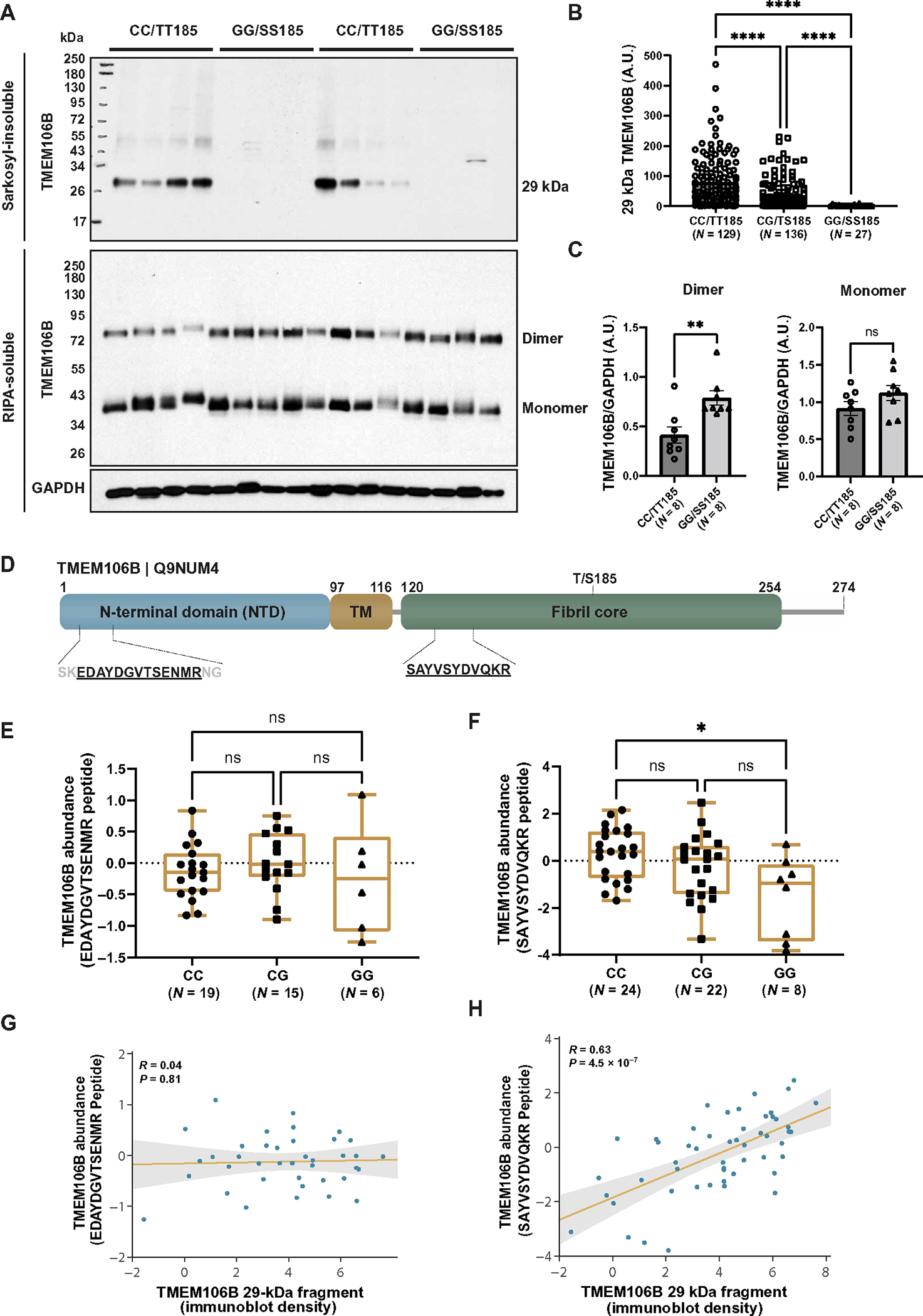
(A) The sarkosyl-insoluble fraction was extracted from the frontal cortices of patients with FTLD-TDP (n = 292 total: 129 CC, 136 CG, and 27 GG) and separated by SDS-PAGE (top). In addition, the radioimmunoprecipitation assay (RIPA)–soluble fraction from the frontal cortices of FTLD-TDP patients (n = 16 total: 8 CC and 8 GG) was separated by SDS-PAGE using cold conditions to visualize both dimeric and monomeric forms of TMEM106B (bottom). Immunoblotting was performed using TMEM106B and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (RIPA-soluble fraction only) antibodies. (B) Quantification was performed on the 29-kDa TMEM106B-positive fragment (A, top). Data presented as means ± SEM, with statistical analysis performed using a one-way ANOVA with Dunn’s multiple comparisons (****P < 0.0001). (C) Quantification was performed on full-length TMEM106B dimer (left) and monomer (right), using GAPDH to control for protein loading [immunoblots presented in (A, bottom)]. Data presented as means ± SEM, with statistical analysis performed using a Student’s t test (**P < 0.01). ns, not significant; A.U., arbitrary units. (D) Schematic diagram of the TMEM106B protein illustrating the N-terminal domain, transmembrane domain (TM), and C-terminal fibril core. Location of peptide sequences identified by mass spectrometry are depicted. (E and F) Quantification of N-terminal (E) or C-terminal (F) peptides mapped to the TMEM106B protein demonstrate abundance in the sarkosyl-insoluble fraction of FTLD-TDP cases stratified by rs3173615 genotype. Fifty-four cases were processed for mass spectrometry. The N-terminal peptide was detected in 19 CC, 15 CG, and 6 GG cases and was not detected in 14 cases. The C-terminal core domain peptide was detected in all cases (n = 24 CC, 22 CG, and 8 GG). Statistical analysis performed using the Kruskal-Wallis test with Dunn’s multiple comparisons (*P < 0.05). (G and H) Regression analysis of N-terminal (G) or C-terminal (H) peptides with 29-kDa TMEM106B fragment quantification by immunoblot.
As an additional strategy to validate the specificity of our antibody in an unbiased manner, we used MS to evaluate the sarkosyl-insoluble fraction from 54 FTLD-TDP cases grouped by rs3173615 genotype (including 24 CC/TT185, 22 CG/TS185, and 8 GG/SS185). Similar to prior methods developed for deep proteomic profiling of the AD brain (20), we used a high-throughput strategy coupling tandem mass tag–based multiplex proteomics and MS with off-line fractionation to reliably quantify approximately 8500 proteins, including TMEM106B itself (Fig. 2D). Although one N-terminal TMEM106B peptide was detected within the sarkosyl-insoluble fraction in a subset of FTLD-TDP cases, there was no relationship between the abundance of this peptide and rs3173615 genotype (Fig. 2, D and E). In contrast, a peptide located within the TMEM106B filament core was detected in the sarkosyl-insoluble fraction from all FTLD-TDP cases and exhibited a relationship between peptide abundance and rs3173615 genotype (Fig. 2, D and F). Moreover, although there was no correlation (P = 0.81) between the N-terminal peptide abundance and 29-kDa fragment accumulation measured by immunoblot (Fig. 2G), 29-kDa fragment deposition was significantly correlated (P < 0.0001) with abundance of the peptide located within the TMEM106B filament core (Fig. 2H). These findings corroborate results in our larger FTLD-TDP postmortem series (Fig. 2, A and B), validating both the generated TMEM106B core antibody and the observation that TMEM106B core deposition occurs in a rs3173615 genotype–dependent manner.
Having firmly established that TMEM106B core accumulation associates with risk haplotype using both immunoblotting and MS approaches, we next sought to examine the impact of haplotype on the cellular and subcellular localization of TMEM106B in the FTLD-TDP brain. We first performed a series of control experiments to validate the use of our antibody for immunohistochemistry (IHC) on formalin-fixed paraffin–embedded tissue sections from human frontal cortex. Using serum collected from rabbits before (prebleed) or after immunization with the TMEM106B peptide, we confirmed that positive immunoreactivity was only observed with serum collected from immunized rabbits (fig. S4A). We next confirmed antibody specificity by preincubation with the peptide used as an immunogen before staining, which eliminated the 3,3′-diaminobenzidine (DAB)–positive signal (fig. S4B). Several commercial antibodies were also purchased to compare with our TMEM106B core antibody, although positive immunolabeling under identical staining conditions was only detected with a commercial antibody from Sigma-Aldrich (fig. S4C, top right), which exhibited a similar staining morphology to our TMEM106B core antibody (fig. S4C, top left).
After antibody validation, we next assessed cell type distribution of TMEM106B in the FTLD-TDP frontal cortex (fig. S5). Robust TMEM106B immunoreactivity was detected in several cell types, including large perinuclear deposits in neurons (fig. S5A, insets 1 and 2) and strong immunoreactivity in glia (fig. S5A, insets 3 and 4). Immunofluorescence (IF) with cell type–specific markers confirmed this apparent cellular distribution, given that we observed strong (but not exclusive) colocalization of TMEM106B with the lysosomal hydrolase cathepsin D in microtubule-associated protein 2 (MAP2)–positive neurons (fig. S5B, rows 1 and 2), glial fibrillary acidic protein–positive astrocytes (fig. S5B, row 3), and ionized calcium binding adaptor molecule 1–positive microglia (fig. S5B, row 4). These findings are consistent with the identification of full-length TMEM106B as an endolysosomal transmembrane protein with the core domain localized to the vesicle lumen (12–14). However, consistent with a recent report that observed no relationship between TMEM106B immunoreactivity by IHC and TMEM106B haplotype (21), immunolabeling with our TMEM106B core antibody was unable to differentiate between risk haplotype or disease status by IHC (fig. S6). We anticipate that this is due to the detection of full-length TMEM106B in addition to the sarkosyl-insoluble 29-kDa TMEM106B core. Hence, the development of additional antibodies or research tools may be needed to differentiate between these two species and define patterns of 29-kDa TMEM106B core deposition in the brain; current findings indicate that biochemical extraction is the only viable method to visualize and quantify the insoluble 29-kDa TMEM106B core at present.
TMEM106B core deposition associates with TDP-43 proteinopathy
To gain insight into the functional relevance of TMEM106B core accumulation, we next evaluated its relationship with TDP-43 pathology. FTLD-TDP can be classified into pathological subtypes on the basis of the morphology and distribution of TDP-43 inclusions (22, 23); we focused on the three most common subtypes (types A, B, and C) that collectively account for more than 95% of all FTLD-TDP cases (24) and examined their relationship with TMEM106B core accumulation. We observed higher TMEM106B core deposition in FTLD-TDP type A than type B or C, even after adjusting for both age at death and sex (Table 4). Because FTLD-TDP type A is the most common overall, is genetically associated with mutations in the progranulin (GRN) gene (25, 26), and is pathologically characterized by a greater diversity of TDP-43 inclusion types (including neuronal cytoplasmic inclusions, dystrophic neurites, neuronal intranuclear inclusions, glial inclusions, and perivascular inclusions) (27, 28), we speculated that higher TMEM106B core burden could be associated with more extensive TDP-43 pathology. To test this idea, we examined the relationship between the abundance of the sarkosyl-insoluble 29-kDa TMEM106B core fragment and the amount of urea-soluble pTDP-43, which revealed that TMEM106B accumulation was associated with higher pTDP-43 burden in both unadjusted analyses (β: 0.25, P = 0.01) and in a multivariable analysis adjusted for age at death and sex (β: 0.47, P < 0.0001) (Table 5). To further investigate the observed association between TMEM106B core deposition and TDP-43 burden, we performed double-label IF for TMEM106B and pTDP-43 in four FTLD-TDP cases but did not detect colocalization between the two proteins (fig. S7).
Table 4. Comparison of 29-kDa TMEM106B core accumulation and TDP-43 subtype in FTLD-TDP.
β, regression coefficient; CI, confidence interval; min, minimum; max, maximum. β values, 95% CIs, and P values result from unadjusted linear regression models or linear regression models adjusted for age at death and sex, where 29-kDa TMEM106B abundance (measured as intensity of the 29-kDa band on Western blot of the sarkosyl insoluble fraction) was considered on the base 10 logarithmic scale. β values are interpreted as the difference in the 29-kda TMEM106B abundance between FTLD-TDP type A cases and the indicated subtypes. P < 0.025 was considered statistically significant after correcting for multiple testing.
| Unadjusted analysis | Adjusted analyses | ||||
|---|---|---|---|---|---|
|
|
|||||
| TDP-43 type | Median 29-kDa TMEM106B levels (min, max) | β (95% CI) | P value | β (95% CI) | P value |
|
| |||||
| A | 47.64 (0, 470.6) | 0.00 (reference) | NA | 0.00 (reference) | NA |
|
| |||||
| B | 4.21 (0.13, 98.5) | −0.736 (−0.953 to −0.520) | <0.0001 | −0.545 (−0.763 to −0.327) | <0.0001 |
|
| |||||
| C | 13.25 (0, 148.9) | −0.456 (−0.703 to −0.209) | 0.0003 | −0.3946 (−0.631 to −0.159) | 0.001 |
Table 5. Associations of 29-kDa TMEM106B with pTDP-43 and tSTMN2 RNA in FTLD-TDP.
β, regression coefficient; CI, confidence interval; RIN, RNA integrity number in the frontal cortex; tSTMN2 RNA, truncated STMN2 RNA concentration in the frontal cortex; pTDP-43 protein, phosphorylated TDP-43 protein concentration in the frontal cortex. β values, 95% CIs, and P values result from unadjusted linear regression models or linear regression models adjusted for the indicated variables. Twenty-nine–kilodalton TMEM106B abundance, pTDP-43 burden, and tSTMN2 RNA were considered on the base 10 logarithmic scale. β values are interpreted as the associations in 29-kDa TMEM106B abundance and the indicated variables. P < 0.025 was considered statistically significant after correcting for multiple testing.
| Unadjusted analysis | Multivariable analysis | ||||
|---|---|---|---|---|---|
| β (95% CI) | P-value | β (95% CI) | P-value | Adjusted for | |
| pTDP-43 protein | 0.248 (0.051–0.446) | 0.0141 | 0.471 (0.288–0.653) | <0.0001 | Age at death and sex |
| tSTMN2 RNA | 0.638 (0.331–0.944) | <0.0001 | 0.758 (0.475–1.040) | <0.0001 | Age at death, sex and RIN |
Given that the pathological aggregation of TDP-43 is also associated with its nuclear clearance and loss of function resulting in defects in TDP-43–dependent splicing, we measured aberrant splicing of the TDP-43 target stathmin-2 (STMN2) to determine whether TDP-43 dysfunction was also associated with increased TMEM106B core deposition. Loss of TDP-43 function has been shown to lead to the aberrant incorporation of a premature stop codon in STMN2 (16, 29, 30), and we recently demonstrated that the abnormal production of truncated STMN2 (tSTMN2) transcripts provides a sensitive readout of TDP-43 dysfunction, accurately discriminating FTLD-TDP from patients with FTLD-tau in a well-characterized autopsy cohort (16). Consistent with the relationship between TMEM106B core deposition and pTDP-43 burden in FTLD-TDP, we also observed an association between the 29-kDa TMEM106B core and tSTMN2 transcript abundance in both unadjusted analyses (β: 0.64, P < 0.0001) and after adjusting for age at death, sex, and RNA integrity number (RIN) (β: 0.76, P < 0.0001) (Table 5). Collectively, these findings indicate that deposition of the 29-kDa sarkosyl-insoluble TMEM106B core fragment in FTLD-TDP is associated with rs3173615 risk genotype, as well as increased TDP-43 pathology and loss of function.
Identification of the insoluble TMEM106B interactome gives mechanistic insight into the role of TMEM106B deposition in disease pathogenesis
To define specific functional processes that might be influenced by aggregation of the TMEM106B filament core, we first optimized our antibody to immunoprecipitate TMEM106B from the sarkosyl-insoluble fraction from two FTLD-TDP cases with low or high 29-kDa TMEM106B accumulation and confirmed that our antibody efficiently immunoprecipitated both the 29-kDa and high–molecular weight species of TMEM106B (fig. S8A). Then, we isolated the sarkosyl-insoluble fraction from nine patients with FTLD-TDP with high 29-kDa TMEM106B core deposition, performed immunoprecipitation with either the TMEM106B antibody or an immunoglobulin G (IgG) antibody control, and subsequently identified core domain–interacting partners by MS. Using IgG as a negative control to exclude proteins that bind nonspecifically, we identified 1309 proteins that specifically interact with TMEM106B in the sarkosyl-insoluble fraction (adjusted P < 0.05) (Fig. 3A and data file S2). To investigate the functional impact of TMEM106B core deposition, we evaluated the association of insoluble TMEM106B interactors with cellular functions and disease by performing Gene Ontology (GO) (Fig. 3B and fig. S8B) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis (Fig. 3C) on the insoluble TMEM106B interactome (Fig. 3B and fig. S8B). The top GO Biological Process and KEGG terms when sorted by fold enrichment were “cytoplasmic translation” and “ribosome,” respectively. “Intracellular transport” and “endocytosis” were also highly enriched, as was the “pathways of neurodegeneration” KEGG term, suggesting that TMEM106B fibrils interact with proteins previously associated with pathogenic processes. To illustrate the relationships between proteins in pathways of interest, we created STRING diagrams of hits within the “endocytosis,” “ribosome,” and “pathways of neurodegeneration” KEGG pathways. Endocytic pathway hits included dynamin proteins (DNM1, DNM2, and DNM3) and epidermal growth factor receptor pathway substrate 15 (EPS15), both of which play important roles in clathrin-mediated endocytosis (31), as well as early endosome antigen 1 (EEA1) and Ras-associated binding (RAB) family members, which play important roles in vesicle trafficking and early endosome maturation (Fig. 3D). One of the most enriched hits in our experiment was kinesin family member 5B (K1F5B) [log2(fold change) = 4.97] (Fig. 3, A and D), a ubiquitously expressed subunit of the kinesin-1 complex critical for anterograde intracellular transport (32). Members of the retromer complex [vacuolar protein sorting-associated protein 26A (VPS26A), VPS35, and VPS29] were also present, however, suggesting that transport-associated proteins that interact with TMEM106B filaments are not limited to the newly formed endosome. Hits in the “ribosome” pathway consisted of both 40S and 60S ribosomal subunit–associated proteins. In the “pathways of neurodegeneration” network, we found that TDP-43 itself interacts with insoluble TMEM106B, as well as numerous autophagy-associated proteins whose mutations are associated with FTLD-TDP and amyotrophic lateral sclerosis (ALS), including TRAF family member associated NFKB activator (TANK)–binding kinase 1 (TBK1), optineurin, and valosin-containing protein (VCP). Of the hits unique to this network, huntingtin was the most highly enriched and the 11th most enriched protein in the assay overall [log2(fold change) = 3.79]. Last, we noted the presence of proteins that are regulated by TDP-43 at the RNA level—including cytoplasmic fragile X messenger ribonucleoprotein 1 interacting protein 2 (CYFIP2), which was the sixth most enriched hit in our assay. We therefore assessed the overlap between our insoluble TMEM106B interactome and two different datasets (33–35) of TDP-43 splicing targets and observed that 43 of the proteins that immunoprecipitated with TMEM106B filaments are known splicing targets of TDP-43 (fig. S8C). To validate our data, we first confirmed the presence of hits of interest in the P3 fraction of FTLD-TDP cases (fig. S9A) and subsequently confirmed that KIF5B coimmunoprecipitates with insoluble TMEM106B and is detectable in sarkosyl soluble and insoluble fractions (fig. S9, B and C). Overall, the identification of insoluble TMEM106B interactors uncovers biological pathways that may be altered in response to TMEM106B deposition, providing mechanistic insight into its role in FTLD-TDP pathogenesis.
Fig. 3. Insoluble TMEM106B interactome gives mechanistic insight into the role of TMEM106B deposition in disease pathogenesis for FTLD-TDP.
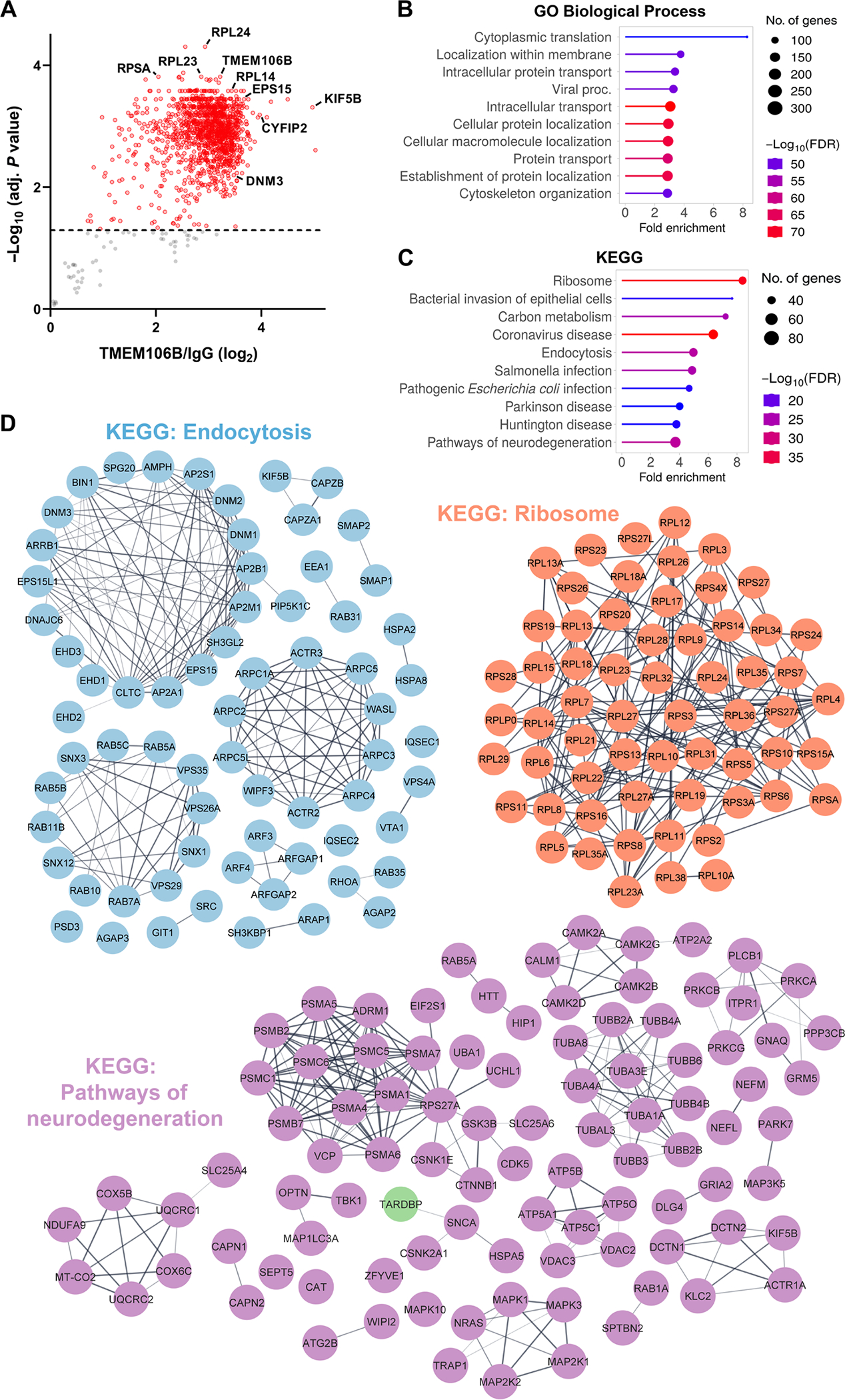
(A) Volcano plot of significantly enriched proteins that coimmunoprecipitated with TMEM106B from the sarkosyl-insoluble P3 fraction of nine FTLD-TDP CC/TT185 cases. Those with an adjusted P < 0.05 (cutoff marked by dashed line) are highlighted in red. (B and C) Top 10 GO Biological Process (B) and KEGG pathways (C) of significant hits in (A), sorted by fold enrichment. (D) STRING diagrams depicting proteins in the “endocytosis,”“ribosome,” and “pathways of neurodegeneration” KEGG pathways. Markov clustering (granularity/inflation parameter = 3) was used to visualize proteins with well-characterized functional relations in each pathway.
DISCUSSION
In this study, we revealed that the minor allele at rs3173615 in the TMEM106B gene is associated with longer survival after symptom onset in patients with FTLD-TDP, suggesting that the TMEM106B protective haplotype associates with a less aggressive disease course. Moreover, we show that accumulation of the sarkosyl-insoluble 29-kDa TMEM106B core fragment is also associated with rs3173615 genotype, with homozygous carriers of the risk allele (CC/TT185) exhibiting robust TMEM106B core deposition in comparison with minimal accumulation observed in carriers of the protective genotype (GG/SS185). We also noted greater abundance of dimeric soluble TMEM106B in GG/SS185 compared with CC/TT185 carriers. Our results indicate that core accumulation in CC/TT185 individuals is not simply mediated by an increase in full-length TMEM106B expression. Although previous reports that quantified cryo-EM observations suggested that TMEM106B core aggregation correlated with aging (14), our findings provide compelling evidence that TMEM106B haplotype is the primary driver of TMEM106B core deposition. Our observation that the TMEM106B core also accumulates in aged, clinically normal control cases carrying the risk haplotype is consistent with the aforementioned age-related accumulation but does not exclude involvement of TMEM106B filament formation in the FTLD-TDP disease process. Rather, given that we found that aggregation of the TMEM106B filament core was associated with TDP-43–dependent mis-splicing and pathology in patients with FTLD-TDP, we anticipate that TMEM106B core deposition modulates susceptibility but does not directly induce TDP-43 proteinopathy. Moreover, because aggregation and dysfunction of TDP-43 is considered a key pathogenic driver in FTLD-TDP, our data suggest that decreased accumulation of the TMEM106B core may slow TDP-43 deposition and dysfunction, which could explain the association of the rs3173615 protective genotype with longer survival after symptom onset.
Given the complexity of the TMEM106B locus and that a number of SNPs are in LD with rs3173615 [reviewed in (9)], it is possible that other noncoding SNPs contribute to the phenotype associated with the TMEM106B risk haplotype. Thus, it will be important to investigate how exactly each TMEM106B SNP influences TMEM106B deposition. Nevertheless, we speculate that rs3173615 specifically modulates disease risk, given that the protein coding change associated with this variant (p.T185S) is located within the TMEM106B filament core sequence, and exogenous TMEM106BT185 exhibits increased lysosomal localization in comparison with exogenous TMEM106BS185 through a mechanism that may depend on changes in its glycosylation (18, 36). Moreover, although the amino acid change from threonine to serine seems relatively conserved, the threonine at position 185 may be essential to stabilize the β sheet structure of the core during fibril formation, because threonine has relatively high β sheet propensity (37–39). Consistent with this possibility, the Parkinson’s disease–associated A53T mutation in α-synuclein has been reported to enhance aggregation of α-synuclein relative to wild-type and other mutant forms of the protein (40–44). In addition, the 43-residue-long oligomer of amyloid-β (Aβ43) is capped by a threonine at its C-terminal end and was demonstrated to form protofibrils more rapidly and at lower concentrations than its 42- and 40-residue-long counterparts, Aβ42 and Aβ40. This observation was attributed in part to T43 mediating direct contact with protofibrillar aggregates (45). A study of amyloid fibrils formed by pancreatic-secreted protein, amylin, also revealed that substitution of serine for threonine enhanced filament aggregation and attributed to threonine’s methyl group restricting conformational freedom of the filament secondary structure, thus favoring tighter packing of adjacent β sheets (46). In light of these intriguing observations, future studies are needed to assess whether TMEM106BT185 similarly stabilizes filaments derived from the 29-kDa TMEM106B core fragment.
Histological evaluation of postmortem FTLD-TDP brain tissue from carriers of the homozygous protective versus risk genotypes could provide clarity as to whether altered subcellular localization in the endolysosomal pathway might account for reduced accumulation of the TMEM106BS185 core, although this analysis is complicated by the current inability of antibodies to differentiate between full-length and insoluble TMEM106B core filaments by IHC. A recently generated antibody targeting the TMEM106B core domain (amino acids 239 to 250) was reported to exhibit specificity for C-terminal fragments of TMEM106B under denaturing conditions (21, 47). However, given the observed discrepancy between TMEM106B core deposition measured by biochemical methods and TMEM106B abundance observed using histological methods (47)—a finding replicated herein—we anticipate that additional antibody validation will reveal that TMEM106B core antibodies also detect and immunolabel both full-length and soluble, nonaggregated core by IHC. As is, both our findings and those described by Vicente and colleagues (47) indicate that the biochemical detection of the TMEM106B core in the sarkosyl-insoluble fraction is currently the most effective and robust method to quantitatively assess the association of TMEM106B haplotype with TMEM106B fibril accumulation. Further, use of isogenically corrected induced pluripotent stem cell (iPSC)–derived neurons homozygous for the rs3173615 risk or protective variant may provide an innovative strategy to explore this idea in more depth and assess specifically whether p.T185S affects the lysosomal residence of TMEM106B.
We also observed a relationship between TMEM106B core deposition and TDP-43 dysfunction. Given emerging evidence on the role of the endolysosomal system in disease, it is tempting to speculate about the potential mechanistic link between these pathological factors. Consistent with this idea, we recently found that genetic or pharmacologic disruption of the endolysosomal pathway through either overexpression of dominant-negative Rab5Q79L or treatment with the PIKfyve inhibitor apilimod drove robust nuclear clearance and cytosolic aggregation of wild-type TDP-43 in human embryonic kidney 293T cells (48). A similar induction of TDP-43 pathology was observed when lysosome acidification was inhibited in iPSC-derived human motor neurons (49). These findings strongly imply that endolysosomal dysfunction could be a key driver of TDP-43 pathology, but whether TMEM106B core deposition occurs upstream or downstream of this dysfunction remains to be determined. It has been noted that the pH of the lysosomal environment reportedly favors TMEM106B fibril formation (12–14), but our interactome data suggest that the insoluble TMEM106B core interacts with proteins outside of the lysosome. Although these data appear contradictory, the intriguing observation that loss of progranulin is associated with reduced integrity of the endolysosomal membrane compartment (50) could offer a plausible scenario: We speculate that loss of progranulin downstream of pathogenic GRN mutations facilitates the release of TMEM106B core filaments from the endolysosomal compartment through a loss of membrane integrity, thereby driving key interactions identified here by MS. Moreover, because we demonstrate that the TMEM106B protective haplotype prevents core deposition, this working model could also explain the reduced penetrance of GRN mutations in carriers of the TMEM106B protective haplotype (51).
Our proteomic analyses revealed that some of the most enriched pathways in the insoluble TMEM106B interactome were those associated with ribosomal function, intracellular transport, and neurodegenerative disease, providing mechanistic insight into the consequences of TMEM106B core deposition. Many of these interactions may hint at a detrimental role for TMEM106B core aggregation in dysfunctional translation, protein localization, and protein quality control within the cell, creating exciting avenues for future study. TMEM106B is known to be important for the trafficking of endolysosomes (52, 53). The interaction of the insoluble core domain with Rab proteins and retromer components suggests that TMEM106B core aggregation might disrupt endosome maturation or sorting, processes that are also proposed to be disrupted in AD (54, 55) and could contribute to or exacerbate TDP-43 loss of function (56, 57). The identification of TBK1, optineurin, and VCP as interactors is also of interest, given their genetic link with the pathogenesis of FTD/ALS (58). The functions of all three proteins converge on protein degradation in the cell, because the coordinated activity of TBK1 and optineurin are critical for proper autophagy (59, 60), and VCP facilitates the proteasomal degradation of ubiquitinated substrates (61). TBK1 has also been shown to disrupt endosome maturation in human neurons (49), and we recently demonstrated that C9orf72 poly[glycine (GA)]–mediated sequestration of TBK1 results in endosomal dysfunction, pTDP-43 deposition, and neurodegeneration in mice (48). These phenotypes were exacerbated in animals expressing a disease-associated loss of function mutation in TBK1 (R228H) (48). Endolysosomes have also been shown to traffic ribosomes (62) and RNA granules (63) throughout the axon. Annexin A11 (ANXA11) has been shown to play a key role in such long-distance trafficking and was a significantly enriched hit within the insoluble TMEM106B interactome in our assay (P = 0.004). Specifically, ANXA11 was identified as the tether between RNA granules and lysosomes, and FTD/ALS-associated mutations in ANXA11 compromise the ability of ANXA11 to mediate docking of RNA granules to the lysosome and impair RNA “hitchhiking” in neurons (63). Mutations in the neuron-specific kinesin-1 subunit KIF5A have similarly been identified to cause ALS with FTD (64), further emphasizing the link between impaired intracellular trafficking and disease. KIF5B, another kinesin-1 subunit, was among the most abundant proteins associated with TMEM106B filaments in our study. Together, our interactome data hint at a potential complex involving TMEM106B, ribosomes/RNA granules, and trafficking lysosomes, indicating that aggregation of the TMEM106B core could sequester proteins associated with transport pathways in neurons, impairing RNA transport, local translation, and endolysosomal function in FTLD-TDP.
The unexpected identification of TDP-43 and its splicing targets within the insoluble TMEM106B interactome (Fig. 3, fig. S8C) suggests that insoluble TMEM106B core deposits may sequester TDP-43 targets, providing a mechanism by which TMEM106B filaments could exacerbate the consequences of TDP-43 loss of function. Among the TDP-43 targets, CYFIP2 was the most enriched and among the top hits overall. Originally identified as a binding partner of the fragile X messenger ribonucleoprotein (65), CYFIP2 has since been reported to associate with transporting ribonucleoproteins in developing neurons, potentially helping to repress translation as RNA granules are trafficked along the axon (66). The potential for TMEM106B core–mediated sequestration of CYFIP2 may thus link TMEM106B fibril formation, translation, and TDP-43 function. DNM1 was another TDP-43 target identified and was also expectedly a member of the “endocytosis” KEGG pathway, given its role in endosome membrane fission (67). Its presence in both categories is further evidence that TMEM106B core aggregation and TDP-43 dysfunction may act to disrupt similar pathways. Future studies are needed to explore the functional relationship between TMEM106B and TDP-43 targets.
Although the findings presented provide insight into the involvement of TMEM106B core deposition in modulating disease risk and TDP-43 proteinopathy, there are limitations associated with the current study that should be acknowledged. Curiously, we did not observe a difference in survival between the CC/TT185 and CG/TS185 genotypes, despite seeing differences in core accumulation between these groups. This finding may suggest that TMEM106B core deposition acts in a dominant manner, with the mere presence of TMEM106B filaments sufficient to increase disease risk and additional accumulation over this threshold exerting relatively marginal effects on disease course. However, future studies are needed to thoroughly test this idea. In addition, it will be important to validate current findings in an additional FTLD-TDP autopsy cohort.
Another limitation is our inability to identify the exact subcellular localization of TMEM106B core filaments, although the identification of interacting partners of insoluble TMEM106B described here will facilitate characterization of TMEM106B filament distribution in future studies. iEM could also be used to provide insight into this important question, but this is difficult in postmortem human brain tissue, given that cell and organelle membranes are not well preserved. An additional technical consideration regarding the standard autopsy protocol (left side of the brain is placed in fixative and the right side of the brain is frozen) may be particularly important to keep in mind for investigations associated with TMEM106B, because a large population–based study using magnetic resonance imaging in cognitively normal individuals found that TMEM106B risk haplotype was associated with asymmetric temporal lobe atrophy affecting predominantly the left hemisphere (68). This finding raises the intriguing possibility that TMEM106B core deposition may also be asymmetric, particularly if core deposition proves to be a key driver in modulating susceptibility to degeneration. In addition, the nature of TMEM106B immunoprecipitation from the sarkosyl-insoluble fraction disrupts tissue architecture, precluding the identification of spatially preserved protein interactions. Although we believe that the observed enrichment of protein groups linked to specific biological pathways (such as translation or transport) reflects identification of legitimate core domain interactors, the use of proximity labeling methods to complement our MS results would be a powerful way to validate which TMEM106B core interactions occur in the native environment of diseased cells.
Given that the rs3173615 major allele was similarly associated with increased risk in AD with limbic-predominant age-related TDP-43 encephalopathy neuropathologic change (LATE-NC) (10, 69–72), future studies will need to explore whether an analogous effect of genotype on TMEM106B core deposition would be observed in patients with AD either in the presence or in the absence of LATE-NC. A recent study observed increased TMEM106B immunoreactivity in the frontal cortices and hippocampi of individuals with LATE-NC (73), although the biochemical characterization of 29-kDa TMEM106B core deposition and relationship with rs3173615 genotype in LATE-NC has yet to be assessed. Our work has linked TMEM106B core deposition to TDP-43 pathology and disease progression but does not provide definitive evidence that TMEM106B filaments influence TDP-43 pathology, although this is certainly a major goal of future experimental studies. Note that TMEM106B core filaments were detected in a wide range of other neurodegenerative diseases, including AD, corticobasal degeneration, dementia with Lewy bodies, progressive supranuclear palsy, and more (12–14). Given the observed connection between the TMEM106B core fragment and TDP-43 pathology in our data and the fact that neurodegeneration-associated proteins were detected in the insoluble TMEM106B interactome, it would be interesting to determine whether core deposition could also influence the accumulation of other aggregation-prone proteins. It would also be informative to determine whether differences in the TMEM106B interactome exist across these diseases and in the healthy brain, which could clarify whether TMEM106B fibril formation has conserved biological consequences or whether there are unique effects of core domain aggregation in different pathologic contexts. Overall, although there are still unanswered questions in our understanding of the role of TMEM106B in neurodegenerative disease risk and progression, the findings presented herein provide insight into TMEM106B core deposition and suggest that contrary to initial supposition, accumulation of TMEM106B core filaments is not benign. Instead, the finding that the sarkosyl-insoluble 29-kDa TMEM106B core fragment associates with TMEM106B risk haplotype in FTLD-TDP, as well as with TDP-43 pathology and loss of function, indicates that core deposition may be central to the mechanism by which TMEM106B haplotype modulates disease risk. These findings also have several implications for future clinical studies, providing evidence that TMEM106B haplotype is an essential factor to consider and control for in clinical cohorts of patients with FTD.
MATERIALS AND METHODS
Study design
The goals of this study were to (i) investigate the relationship between TMEM106B rs3173615 genotype and survival after disease onset and TMEM106B aggregation in a postmortem FTLD-TDP cohort and (ii) identify protein interactors of insoluble TMEM106B in the FTLD-TDP brain. We included all FTLD-TDP cases in the Mayo Clinic Brain Bank for which there was frozen tissue available for both genotyping and biochemical extraction of the sarkosyl-insoluble fraction. Initial antibody validation was performed on FTLD-TDP cases confirmed to be postive for TMEM106B filaments by cryo-EM, with subsequent analysis of 29-kDa TMEM106B accumulation by immunoblotting in the complete FTLD-TDP postmortem cohort that was performed by a technician blinded to rs3173615 genotype. To validate immunoblotting results by MS, a subset of FTLD-TDP type A cases were selected (including all eight individuals homozygous for the protective allele along with heterozygous and homozygous carriers of the risk allele that were matched for sex, age, and mutation status). To identify the TMEM106B filament interactome, we selected nine FTLD-TDP cases with the highest 29-kDa TMEM106B accumulation and performed the immunoprecipitation with either a TMEM106B antibody or IgG control for each case. All MS analysis was performed in a blinded manner. Written informed consent was obtained from all individuals (or their legal next of kin if they were unable to give written consent) before study entry, and all protocols were approved by the Mayo Clinic Institutional Review Board.
Statistics
All statistical analyses were performed in GraphPad Prism 9 (with the exception of data presented in Fig. 2, G and H). The association between survival after onset and TMEM106B rs3173615 status in FTD cases was performed using single-variable (unadjusted) analysis and multivariable linear regression models (adjusted for age at death and sex). Associations with 29-kDa TMEM106B accumulation in the FTLD-TDP postmortem cohort was also conducted using single-variable (unadjusted) analysis and multivariable linear regression models (adjusted for age at death and sex). Twenty-nine–kilodalton TMEM106B protein, pTDP-43 protein, and tSTMN2 RNA amounts were analyzed on the base 10 logarithmic scale because of their skewed distributions. β values and 95% confidence intervals (CIs) were interpreted as the difference in the means between all FTLD-TDP cases combined or the individual TMEM106B rs3173615 genotypes (using the homozygous risk TMEM106B rs3173615 genotype as the reference group). Significant P values are based on the number of comparisons performed and are provided in the legend for each table. In Fig. 2 (B, E, and F), statistical analysis was performed using a one-way analysis of variance (ANOVA) with Dunn’s multiple comparisons. In Fig. 2C, statistical analysis was performed using a Student’s t test. In Fig. 2 (G and H), statistical analysis was performed in R using a simple linear regression (least-squares method) to fit the line and Pearson coefficient to assess the correlation.
Supplementary Material
Acknowledgments:
We would like to thank the patients and their families for participation in this work. Human postmortem brain samples and associated data were provided by the Mayo Clinic Brain Bank (including but not limited to Mayo ADRC, ALLFTD, and P01NS084974). We thank E. Fox and E. Dammer (Emory School of Medicine) for discussions.
Funding:
This work was supported by Mayo Clinic Foundation (to L.P. and D.W.D.), the Liston Family Foundation (to L.P.), Mayo Clinic Center for Regenerative Medicine (to Z.K.W.), Mayo Clinic Neuroscience-Focused Research Team (to Z.K.W.), National Institutes of Health/National Institute on Aging [ADRC 5P30AG0062677 (to L.P., R.C.P., and D.W.D.), ALLFTD U19AG063911 (to B.F.B., L.P., D.W.D., R.C.P., D.S.K., and N.R.G.-R.), R01AG063780 (to C.N.C.), R01AG071513 (to C.N.C.), R01AG065219 (to C.N.C.), RF1NS120992 (to M.P. and K.A.J.), R01AG34791 (to K.A.J.), R01AG062171 (to L.P.), and RF1AG062077 (to L.P.)], National Institutes of Health/National Institute of Neurological Disorders and Stroke [U54NS123743 (to L.P. and M.P.), R35NS097273 (to L.P.), P01NS084974 (to L.P., Y.-J.Z., and D.W.D.), U01AG061357 (to N.T.S.), U01NS110438 (to L.P.), U01NS110438 (to L.P.), R01NS117461 (to Y.-J.Z.), and R21NS127331 (to Y.-J.Z.)], Target ALS Foundation (to L.P., Y.-J.Z., and M.P.), Alzheimer’s Association Zenith Award (to L.P.), Alzheimer’s Association Strategic Fund (to. L.P.), and Cure Alzheimer’s Fund (to L.P. and C.N.C.). In addition, this work was also supported by gifts from the Sol Goldman Charitable Trust (to Z.K.W.), the Donald G. and Jodi P. Heeringa Family (to Z.K.W.), the Haworth Family Professorship in Neurodegenerative Diseases fund (to Z.K.W.), and the Albertson Parkinson’s Research Foundation (to Z.K.W.).
Footnotes
Competing interests: R.C.P. has advisory roles for Roche Inc., Genentec Inc., Eli Lilly and Company, Eisai Inc., and Nestle Inc. B.F.B. performs consulting activities for the Tau Consortium (funded by the Rainwater Charitable Foundation) and also receives institutional research grant support for clinical trials from the following: Alector, Biogen, Transposon, Cognition Therapeutics, EIP Pharma, and GE Healthcare. N.T.S. and D.M.D. are both cofounders and consultants for Emtherapro Inc. Z.K.W. serves as principal investigator (PI) or co-PI on Biohaven Pharmaceuticals Inc. (BHV4157–206) and Vigil Neuroscience Inc. [VGL101–01.002, VGL101–01.201, positron emission tomography (PET) tracer development protocol, Csf1r biomarker and repository project, and ultrahigh field magnetic resonance imaging in the diagnosis and management of colony stimulating factor 1 receptor–related adult-onset leukoencephalopathy with axonal spheroids and pigmented glia] projects/grants. He serves as co-PI of the Mayo Clinic APDA Center for Advanced Research and as an external advisory board member for the Vigil Neuroscience Inc. and as a consultant on neurodegenerative medical research for Eli Lilly and Company. All other authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. The TMEM106b core antibody can be obtained by contacting L.P. and establishing a material transfer agreement with Mayo Clinic (https://redcap.vanderbilt.edu/surveys/?s=NMRA8TLFM7). Code used for data analysis was deposited in Zenodo (DOI: 10.5281/zenodo.10233187).
REFERENCES AND NOTES
- 1.Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF, Frontotemporal lobar degeneration : A consensus on clinical diagnostic criteria. Neurology 51, 1546–1554 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T, TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Neumann M, Kwong LK, Truax AC, Vanmassenhove B, Kretzschmar HA, Van Deerlin VM, Clark CM, Grossman M, Miller BL, Trojanowski JQ, Lee VM-Y, TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J. Neuropathol. Exp. Neurol. 66, 177–183 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Gallagher MD, Suh E, Grossman M, Elman L, McCluskey L, Van Swieten JC, Al-Sarraj S, Neumann M, Gelpi E, Ghetti B, Rohrer JD, Halliday G, Van Broeckhoven C, Seilhean D, Shaw PJ, Frosch MP, Alafuzoff I, Antonell A, Bogdanovic N, Brooks W, Cairns NJ, Cooper-Knock J, Cotman C, Cras P, Cruts M, De Deyn PP, DeCarli C, Dobson-Stone C, Engelborghs S, Fox N, Galasko D, Gearing M, Gijselinck I, Grafman J, Hartikainen P, Hatanpaa KJ, Highley JR, Hodges J, Hulette C, Ince PG, Jin LW, Kirby J, Kofler J, Kril J, Kwok JB, Levey A, Lieberman A, Llado A, Martin JJ, Masliah E, McDermott CJ, McKee A, McLean C, Mead S, Miller CA, Miller J, Munoz DG, Murrell J, Paulson H, Piguet O, Rossor M, Sanchez-Valle R, Sano M, Schneider J, Silbert LC, Spina S, van der Zee J, Van Langenhove T, Warren J, Wharton SB, White CL 3rd, Woltjer RL, Trojanowski JQ, Lee VM, Van Deerlin V, Chen-Plotkin AS, TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol. 127, 407–418 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Blitterswijk M, Mullen B, Nicholson AM, Bieniek KF, Heckman MG, Baker MC, DeJesus-Hernandez M, Finch NA, Brown PH, Murray ME, Hsiung GY, Stewart H, Karydas AM, Finger E, Kertesz A, Bigio EH, Weintraub S, Mesulam M, Hatanpaa KJ, White CL 3rd, Strong MJ, Beach TG, Wszolek ZK, Lippa C, Caselli R, Petrucelli L, Josephs KA, Parisi JE, Knopman DS, Petersen RC, Mackenzie IR, Seeley WW, Grinberg LT, Miller BL, Boylan KB, Graff-Radford NR, Boeve BF, Dickson DW, Rademakers R, TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol. 127, 397–406 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cruchaga C, Graff C, Chiang HH, Wang J, Hinrichs AL, Spiegel N, Bertelsen S, Mayo K, Norton JB, Morris JC, Goate A, Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch. Neurol. 68, 581–586 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, Dejesus-Hernandez M, Crook R, Hunter T, Ghidoni R, Benussi L, Crook J, Finger E, Hantanpaa KJ, Karydas AM, Sengdy P, Gonzalez J, Seeley WW, Johnson N, Beach TG, Mesulam M, Forloni G, Kertesz A, Knopman DS, Uitti R, White CL 3rd, Caselli R, Lippa C, Bigio EH, Wszolek ZK, Binetti G, Mackenzie IR, Miller BL, Boeve BF, Younkin SG, Dickson DW, Petersen RC, Graff-Radford NR, Geschwind DH, Rademakers R, TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology 76, 467–474 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, Dickson DW, Rademakers R, Boeve BF, Grossman M, Arnold SE, Mann DM, Pickering-Brown SM, Seelaar H, Heutink P, van Swieten JC, Murrell JR, Ghetti B, Spina S, Grafman J, Hodges J, Spillantini MG, Gilman S, Lieberman AP, Kaye JA, Woltjer RL, Bigio EH, Mesulam M, Al-Sarraj S, Troakes C, Rosenberg RN, White CL 3rd, Ferrer I, Llado A, Neumann M, Kretzschmar HA, Hulette CM, Welsh-Bohmer KA, Miller BL, Alzualde A, Lopez de Munain A, McKee AC, Gearing M, Levey AI, Lah JJ, Hardy J, Rohrer JD, Lashley T, Mackenzie IR, Feldman HH, Hamilton RL, Dekosky ST, van der Zee J, Kumar-Singh S, Van Broeckhoven C, Mayeux R, Vonsattel JP, Troncoso JC, Kril JJ, Kwok JB, Halliday GM, Bird TD, Ince PG, Shaw PJ, Cairns NJ, Morris JC, McLean CA, DeCarli C, Ellis WG, Freeman SH, Frosch MP, Growdon JH, Perl DP, Sano M, Bennett DA, Schneider JA, Beach TG, Reiman EM, Woodruff BK, Cummings J, Vinters HV, Miller CA, Chui HC, Alafuzoff I, Hartikainen P, Seilhean D, Galasko D, Masliah E, Cotman CW, Tunon MT, Martinez MC, Munoz DG, Carroll SL, Marson D, Riederer PF, Bogdanovic N, Schellenberg GD, Hakonarson H, Trojanowski JQ, Lee VM-Y, Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat. Genet. 42, 234–239 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicholson AM, Rademakers R, What we know about TMEM106B in neurodegeneration. Acta Neuropathol. 132, 639–651 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu L, De Jager PL, Yang J, Trojanowski JQ, Bennett DA, Schneider JA, The TMEM106B locus and TDP-43 pathology in older persons without FTLD. Neurology 84, 927–934 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mao F, Robinson JL, Unger T, Posavi M, Amado DA, Elman L, Grossman M, Wolk DA, Lee EB, Van Deerlin VM, Porta S, Lee VMY, Trojanowski JQ, Chen-Plotkin AS, TMEM106B modifies TDP-43 pathology in human ALS brain and cell-based models of TDP-43 proteinopathy. Acta Neuropathol. 142, 629–642 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang A, Xiang X, Wang J, Lee C, Arakhamia T, Simjanoska M, Wang C, Carlomagno Y, Zhang G, Dhingra S, Thierry M, Perneel J, Heeman B, Forgrave LM, DeTure M, DeMarco ML, Cook CN, Rademakers R, Dickson DW, Petrucelli L, Stowell MHB, Mackenzie IRA, Fitzpatrick AWP, Homotypic fibrillization of TMEM106B across diverse neurodegenerative diseases. Cell 185, 1346–1355.e15 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang YX, Cao Q, Sawaya MR, Abskharon R, Ge P, DeTure M, Dickson DW, Fu JY, Ogorzalek Loo RR, Loo JA, Eisenberg DS, Amyloid fibrils in FTLD-TDP are composed of TMEM106B and not TDP-43. Nature 605, 304–309 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schweighauser M, Arseni D, Bacioglu M, Huang M, Lovestam S, Shi Y, Yang Y, Zhang W, Kotecha A, Garringer HJ, Vidal R, Hallinan GI, Newell KL, Tarutani A, Murayama S, Miyazaki M, Saito Y, Yoshida M, Hasegawa K, Lashley T, Revesz T, Kovacs GG, van Swieten J, Takao M, Hasegawa M, Ghetti B, Spillantini MG, Ryskeldi-Falcon B, Murzin AG, Goedert M, Scheres SHW, Age-dependent formation of TMEM106B amyloid filaments in human brains. Nature 605, 310–314 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan Y, Zhao Q, Xia W, Tao Y, Yu W, Chen M, Liu Y, Zhao J, Shen Y, Sun Y, Si C, Zhang S, Zhang Y, Li W, Liu C, Wang J, Li D, Generic amyloid fibrillation of TMEM106B in patient with Parkinson’s disease dementia and normal elders. Cell Res. 32, 585–588 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prudencio M, Humphrey J, Pickles S, Brown A-L, Hill SE, Kachergus JM, Shi J, Heckman MG, Spiegel MR, Cook C, Song Y, Yue M, Daughrity LM, Carlomagno Y, Jansen-West K, de Castro CF, De Ture M, Koga S, Wang Y-C, Sivakumar P, Bodo C, Candalija A, Talbot K, Selvaraj BT, Burr K, Chandran S, Newcombe J, Lashley T, Hubbard I, Catalano D, Kim D, Propp N, Fennessey S; NYGC ALS Consortium, Fagegaltier D, Phatnani H, Secrier M, Fisher EMC, Oskarsson B, van Blitterswijk M, Rademakers R, Graff-Radford NR, Boeve BF, Knopman DS, Petersen RC, Josephs KA, Thompson EA, Raj T, Ward M, Dickson DW, Gendron TF, Fratta P, Petrucelli L, Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J. Clin. Investig. 130, 6080–6092 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prudencio M, Gonzales PK, Cook CN, Gendron TF, Daughrity LM, Song Y, Ebbert MTW, van Blitterswijk M, Zhang YJ, Jansen-West K, Baker MC, DeTure M, Rademakers R, Boylan KB, Dickson DW, Petrucelli L, Link CD, Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum. Mol. Genet. 26, 3421–3431 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lang CM, Fellerer K, Schwenk BM, Kuhn PH, Kremmer E, Edbauer D, Capell A, Haass C, Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J. Biol. Chem. 287, 19355–19365 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen-Plotkin AS, Unger TL, Gallagher MD, Bill E, Kwong LK, Volpicelli-Daley L, Busch JI, Akle S, Grossman M, Van Deerlin V, Trojanowski JQ, Lee VM, TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J. Neurosci. 32, 11213–11227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson ECB, Carter EK, Dammer EB, Duong DM, Gerasimov ES, Liu Y, Liu J, Betarbet R, Ping L, Yin L, Serrano GE, Beach TG, Peng J, De Jager PL, Haroutunian V, Zhang B, Gaiteri C, Bennett DA, Gearing M, Wingo TS, Wingo AP, Lah JJ, Levey AI, Seyfried NT, Large-scale deep multi-layer analysis of Alzheimer’s disease brain reveals strong proteomic disease-related changes not observed at the RNA level. Nat. Neurosci. 25, 213–225 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perneel J, Neumann M, Heeman B, Cheung S, Van den Broeck M, Wynants S, Baker M, Vicente CT, Faura J, Rademakers R, Mackenzie IRA, Accumulation of TMEM106B C-terminal fragments in neurodegenerative disease and aging. Acta Neuropathol. 145, 285–302 (2023). [DOI] [PubMed] [Google Scholar]
- 22.Sampathu DM, Neumann M, Kwong LK, Chou TT, Micsenyi M, Truax A, Bruce J, Grossman M, Trojanowski JQ, Lee VM, Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am. J. Pathol. 169, 1343–1352 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davidson Y, Kelley T, Mackenzie IR, Pickering-Brown S, Du Plessis D, Neary D, Snowden JS, Mann DM, Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol. 113, 521–533 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, Mann DM, Dickson DW, Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. 122, 137–153 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Josephs KA, Ahmed Z, Katsuse O, Parisi JF, Boeve BF, Knopman DS, Petersen RC, Davies P, Duara R, Graff-Radford NR, Uitti RJ, Rademakers R, Adamson J, Baker M, Hutton ML, Dickson DW, Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J. Neuropathol. Exp. Neurol. 66, 142–151 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Mackenzie IR, Baker M, Pickering-Brown S, Hsiung GY, Lindholm C, Dwosh E, Gass J, Cannon A, Rademakers R, Hutton M, Feldman HH, The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain 129, 3081–3090 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Josephs KA, Stroh A, Dugger B, Dickson DW, Evaluation of subcortical pathology and clinical correlations in FTLD-U subtypes. Acta Neuropathol. 118, 349–358 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mackenzie IR, Baborie A, Pickering-Brown S, Du Plessis D, Jaros E, Perry RH, Neary D, Snowden JS, Mann DM, Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: Classification and relation to clinical phenotype. Acta Neuropathol. 112, 539–549 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klim JR, Williams LA, Limone F, San Juan IG, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, Moccia R, Cassel SH, Chen K, Wainger BJ, Woolf CJ, Eggan K, ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 22, 167–179 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Melamed Z, Lopez-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, Freyermuth F, McMahon MA, Beccari MS, Artates JW, Ohkubo T, Rodriguez M, Lin N, Wu D, Bennett CF, Rigo F, Da Cruz S, Ravits J, Lagier-Tourenne C, Cleveland DW, Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 22, 180–190 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doherty GJ, McMahon HT, Mechanisms of endocytosis. Annu. Rev. Biochem. 78, 857–902 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Serra-Marques A, Martin M, Katrukha EA, Grigoriev I, Peeters CA, Liu Q, Hooikaas PJ, Yao Y, Solianova V, Smal I, Pedersen LB, Meijering E, Kapitein LC, Akhmanova A, Concerted action of kinesins KIF5B and KIF13B promotes efficient secretory vesicle transport to microtubule plus ends. eLife 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu EY, Russ J, Cali CP, Phan JM, Amlie-Wolf A, Lee EB, Loss of nuclear TDP-43 is associated with decondensation of LINE retrotransposons. Cell Rep. 27, 1409–1421.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seddighi S, Qi YA, Brown AL, Wilkins OG, Bereda C, Belair C, Zhang Y, Prudencio M, Keuss MJ, Khandeshi A, Pickles S, Hill SE, Hawrot J, Ramos DM, Yuan H, Roberts J, Sacramento EK, Shah SI, Nalls MA, Colon-Mercado J, Reyes JF, Ryan VH, Nelson MP, Cook C, Li Z, Screven L, Kwan JY, Shantaraman A, Ping L, Koike Y, Oskarsson BR, Staff N, Duong DM, Ahmed A, Secrier M, Ule J, Jacobson S, Rohrer J, Malaspina A, Glass JD, Ori A, Seyfried NT, Maragkakis M, Petrucelli L, Fratta P, Ward MS, Mis-spliced transcripts generate de novo proteins in TDP-43-related ALS/FTD. bioRxiv 2023.01.23.525149 [Preprint]. 23 January 2023. 10.1101/2023.01.23.525149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma XR, Prudencio M, Koike Y, Vatsavayai SC, Kim G, Harbinski F, Briner A, Rodriguez CM, Guo C, Akiyama T, Schmidt HB, Cummings BB, Wyatt DW, Kurylo K, Miller G, Mekhoubad S, Sallee N, Mekonnen G, Ganser L, Rubien JD, Jansen-West K, Cook CN, Pickles S, Oskarsson B, Graff-Radford NR, Boeve BF, Knopman DS, Petersen RC, Dickson DW, Shorter J, Myong S, Green EM, Seeley WW, Petrucelli L, Gitler AD, TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature 603, 124–130 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nicholson AM, Finch NA, Wojtas A, Baker MC, Perkerson RB 3rd, Castanedes-Casey M, Rousseau L, Benussi L, Binetti G, Ghidoni R, Hsiung GY, Mackenzie IR, Finger E, Boeve BF, Ertekin-Taner N, Graff-Radford NR, Dickson DW, Rademakers R, TMEM106B p.T185S regulates TMEM106B protein levels: Implications for frontotemporal dementia. J. Neurochem. 126, 781–791 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chou PY, Fasman GD, Conformational parameters for amino acids in helical, β-sheet, and random coil regions calculated from proteins. Biochemistry 13, 211–222 (1974). [DOI] [PubMed] [Google Scholar]
- 38.Munoz V, Serrano L, Intrinsic secondary structure propensities of the amino acids, using statistical phi-psi matrices: Comparison with experimental scales. Proteins 20, 301–311 (1994). [DOI] [PubMed] [Google Scholar]
- 39.Stanger HE, Syud FA, Espinosa JF, Giriat I, Muir T, Gellman SH, Length-dependent stability and strand length limits in antiparallel β-sheet secondary structure. Proc. Natl. Acad. Sci. U.S.A. 98, 12015–12020 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT Jr., Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. U.S.A. 97, 571–576 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flagmeier P, Meisl G, Vendruscolo M, Knowles TP, Dobson CM, Buell AK, Galvagnion C, Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of α-synuclein aggregation. Proc. Natl. Acad. Sci. U.S.A. 113, 10328–10333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greenbaum EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, Axelsen PH, Giasson BI, The E46K mutation in α-synuclein increases amyloid fibril formation. J. Biol. Chem. 280, 7800–7807 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Heise H, Celej MS, Becker S, Riedel D, Pelah A, Kumar A, Jovin TM, Baldus M, Solid-state NMR reveals structural differences between fibrils of wild-type and disease-related A53T mutant α-synuclein. J. Mol. Biol. 380, 444–450 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Ostrerova-Golts N, Petrucelli L, Hardy J, Lee JM, Farer M, Wolozin B, The A53T α-synuclein mutation increases iron-dependent aggregation and toxicity. J. Neurosci. 20, 6048–6054 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Conicella AE, Fawzi NL, The C-terminal threonine of Aβ43 nucleates toxic aggregation via structural and dynamical changes in monomers and protofibrils. Biochemistry 53, 3095–3105 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang ST, Lin Y, Spencer RK, Thomas MR, Nguyen AI, Amdursky N, Pashuck ET, Skaalure SC, Song CY, Parmar PA, Morgan RM, Ercius P, Aloni S, Zuckermann RN, Stevens MM, Sequence-dependent self-assembly and structural diversity of islet amyloid polypeptide-derived β-sheet fibrils. ACS Nano 11, 8579–8589 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vicente CT, Perneel J, Wynants S, Heeman B, Van den Broeck M, Baker M, Cheung S, Faura J, Mackenzie IRA, Rademakers R, C-terminal TMEM106B fragments in human brain correlate with disease-associated TMEM106B haplotypes. Brain 146, 4055–4064 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shao W, Todd TW, Wu Y, Jones CY, Tong J, Jansen-West K, Daughrity LM, Park J, Koike Y, Kurti A, Yue M, Castanedes-Casey M, Del Rosso G, Dunmore JA, Zanetti Alepuz D, Oskarsson B, Dickson DW, Cook CN, Prudencio M, Gendron TF, Fryer JD, Zhang YJ, Petrucelli L, Two FTD-ALS genes converge on the endosomal pathway to induce TDP-43 pathology and degeneration. Science 378, 94–99 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hao J, Wells MF, Niu G, San Juan IG, Limone F, Fukuda A, Leyton-Jaimes MF, Joseph B, Qian M, Mordes DA, Budnik B, Dou Z, Eggan K, Loss of TBK1 activity leads to TDP-43 proteinopathy through lysosomal dysfunction in human motor neurons. bioRxiv 2021.10.11.464011 [Preprint]. 12 October 2021. 10.1101/2021.10.11.464011. [DOI] [Google Scholar]
- 50.Logan T, Simon MJ, Rana A, Cherf GM, Srivastava A, Davis SS, Low RLY, Chiu CL, Fang M, Huang F, Bhalla A, Llapashtica C, Prorok R, Pizzo ME, Calvert MEK, Sun EW, Hsiao-Nakamoto J, Rajendra Y, Lexa KW, Srivastava DB, van Lengerich B, Wang J, Robles-Colmenares Y, Kim DJ, Duque J, Lenser M, Earr TK, Nguyen H, Chau R, Tsogtbaatar B, Ravi R, Skuja LL, Solanoy H, Rosen HJ, Boeve BF, Boxer AL, Heuer HW, Dennis MS, Kariolis MS, Monroe KM, Przybyla L, Sanchez PE, Meisner R, Diaz D, Henne KR, Watts RJ, Henry AG, Gunasekaran K, Astarita G, Suh JH, Lewcock JW, DeVos SL, G., Rescue of a lysosomal storage disorder caused by Grn loss of function with a brain penetrant progranulin biologic. Cell 184, 4651–4668.e25 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perneel J, Manoochehri M, Huey ED, Rademakers R, Goldman J, Case report: TMEM106B haplotype alters penetrance of GRN mutation in frontotemporal dementia family. Front. Neurol. 14, 1160248 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luningschror P, Werner G, Stroobants S, Kakuta S, Dombert B, Sinske D, Wanner R, Lullmann-Rauch R, Wefers B, Wurst W, D’Hooge R, Uchiyama Y, Sendtner M, Haass C, Saftig P, Knoll B, Capell A, Damme M, The FTLD risk factor TMEM106B regulates the transport of lysosomes at the axon initial segment of Motoneurons. Cell Rep. 30, 3506–3519.e6 (2020). [DOI] [PubMed] [Google Scholar]
- 53.Schwenk BM, Lang CM, Hogl S, Tahirovic S, Orozco D, Rentzsch K, Lichtenthaler SF, Hoogenraad CC, Capell A, Haass C, Edbauer D, The FTLD risk factor TMEM106B and MAP6 control dendritic trafficking of lysosomes. EMBO J. 33, 450–467 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qureshi YH, Berman DE, Marsh SE, Klein RL, Patel VM, Simoes S, Kannan S, Petsko GA, Stevens B, Small SA, The neuronal retromer can regulate both neuronal and microglial phenotypes of Alzheimer’s disease. Cell Rep. 38, 110262 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simoes S, Neufeld JL, Triana-Baltzer G, Moughadam S, Chen EI, Kothiya M, Qureshi YH, Patel V, Honig LS, Kolb H, Small SA, Tau and other proteins found in Alzheimer’s disease spinal fluid are linked to retromer-mediated endosomal traffic in mice and humans. Sci. Transl. Med. 12, eaba6334 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schwenk BM, Hartmann H, Serdaroglu A, Schludi MH, Hornburg D, Meissner F, Orozco D, Colombo A, Tahirovic S, Michaelsen M, Schreiber F, Haupt S, Peitz M, Brustle O, Kupper C, Klopstock T, Otto M, Ludolph AC, Arzberger T, Kuhn PH, Edbauer D, TDP-43 loss of function inhibits endosomal trafficking and alters trophic signaling in neurons. EMBO J. 35, 2350–2370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tang FL, Zhao L, Zhao Y, Sun D, Zhu XJ, Mei L, Xiong WC, Coupling of terminal differentiation deficit with neurodegenerative pathology in Vps35-deficient pyramidal neurons. Cell Death Differ. 27, 2099–2116 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scarian E, Fiamingo G, Diamanti L, Palmieri I, Gagliardi S, Pansarasa O, The role of VCP mutations in the Spectrum of amyotrophic lateral sclerosis-frontotemporal dementia. Front. Neurol 13, 841394 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oakes JA, Davies MC, Collins MO, TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain 10, 5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Slowicka K, Vereecke L, van Loo G, Cellular functions of optineurin in health and disease. Trends Immunol. 37, 621–633 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Meyer H, Bug M, Bremer S, Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 14, 117–123 (2012). [DOI] [PubMed] [Google Scholar]
- 62.Cioni JM, Lin JQ, Holtermann AV, Koppers M, Jakobs MAH, Azizi A, Turner-Bridger B, Shigeoka T, Franze K, Harris WA, Holt CE, Late endosomes act as mRNA translation platforms and sustain mitochondria in axons. Cell 176, 56–72.e15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liao YC, Fernandopulle MS, Wang G, Choi H, Hao L, Drerup CM, Patel R, Qamar S, Nixon-Abell J, Shen Y, Meadows W, Vendruscolo M, Knowles TPJ, Nelson M, Czekalska MA, Musteikyte G, Gachechiladze MA, Stephens CA, Pasolli HA, Forrest LR, St George-Hyslop P, Lippincott-Schwartz J, Ward ME, RNA granules hitchhike on lysosomes for long-distance transport, using annexin A11 as a molecular tether. Cell 179, 147–164.e20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saez-Atienzar S, Dalgard CL, Ding J, Chio A, Alba C, Hupalo DN, Wilkerson MD, Bowser R, Pioro EP, Bedlack R, Traynor BJ, Identification of a pathogenic intronic KIF5A mutation in an ALS-FTD kindred. Neurology 95, 1015–1018 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schenck A, Bardoni B, Moro A, Bagni C, Mandel JL, A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP-related proteins FXR1P and FXR2P. Proc. Natl. Acad. Sci. U.S.A. 98, 8844–8849 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cioni JM, Wong HH, Bressan D, Kodama L, Harris WA, Holt CE, Axon-axon interactions regulate topographic optic tract sorting via CYFIP2-dependent WAVE complex function. Neuron 97, 1078–1093.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Imoto Y, Raychaudhuri S, Ma Y, Fenske P, Sandoval E, Itoh K, Blumrich EM, Matsubayashi HT, Mamer L, Zarebidaki F, Sohl-Kielczynski B, Trimbuch T, Nayak S, Iwasa JH, Liu J, Wu B, Ha T, Inoue T, Jorgensen EM, Cousin MA, Rosenmund C, Watanabe S, Dynamin is primed at endocytic sites for ultrafast endocytosis. Neuron 110, 2815–2835.e13 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Adams HH, Verhaaren BF, Vrooman HA, Uitterlinden AG, Hofman A, van Duijn CM, van der Lugt A, Niessen WJ, Vernooij MW, Ikram MA, TMEM106B influences volume of left-sided temporal lobe and interhemispheric structures in the general population. Biol. Psychiatry 76, 503–508 (2014). [DOI] [PubMed] [Google Scholar]
- 69.Dugan AJ, Nelson PT, Katsumata Y, Shade LMP, Boehme KL, Teylan MA, Cykowski MD, Mukherjee S, Kauwe JSK, Hohman TJ, Schneider JA; Alzheimer’s Disease Genetics Consortium, Fardo DW, Analysis of genes (TMEM106B, GRN, ABCC9, KCNMB2, and APOE) implicated in risk for LATE-NC and hippocampal sclerosis provides pathogenetic insights: A retrospective genetic association study. Acta Neuropathol. Commun. 9, 152 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Murray ME, Cannon A, Graff-Radford NR, Liesinger AM, Rutherford NJ, Ross OA, Duara R, Carrasquillo MM, Rademakers R, Dickson DW, Differential clinicopathologic and genetic features of late-onset amnestic dementias. Acta Neuropathol. 128, 411–421 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nelson PT, Wang WX, Partch AB, Monsell SE, Valladares O, Ellingson SR, Wilfred BR, Naj AC, Wang LS, Kukull WA, Fardo DW, Reassessment of risk genotypes (GRN, TMEM106B, and ABCC9 variants) associated with hippocampal sclerosis of aging pathology. J. Neuropathol. Exp. Neurol. 74, 75–84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rutherford NJ, Carrasquillo MM, Li M, Bisceglio G, Menke J, Josephs KA, Parisi JE, Petersen RC, Graff-Radford NR, Younkin SG, Dickson DW, Rademakers R, TMEM106B risk variant is implicated in the pathologic presentation of Alzheimer disease. Neurology 79, 717–718 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Neumann M, Perneel J, Cheung S, Van den Broeck M, Nygaard H, Hsiung GR, Wynants S, Heeman B, Rademakers R, Mackenzie IRA, Limbic-predominant age-related TDP-43 proteinopathy (LATE-NC) is associated with abundant TMEM106B pathology. Acta Neuropathol. 146, 163–166 (2023). [DOI] [PubMed] [Google Scholar]
- 74.Reilly L, Peng L, Lara E, Ramos D, Fernandopulle M, Pantazis CB, Stadler J, Santiana M, Dadu A, Iben J, Faghri F, Nalls MA, Coon SL, Narayan P, Singleton AB, Cookson MR, Ward ME, Qi YA, A fully automated FAIMS-DIA proteomic pipeline for high-throughput characterization of iPSC-derived neurons. bioRxiv 2021.11.24.469921 [Preprint]. 25 November 2021. 10.1101/2021.11.24.469921. [DOI] [Google Scholar]
- 75.Modeste E, Ping L, Watson CM, Duong DM, Dammer EB, Johnson ECB, Roberts BR, Lah JJ, Levey AI, Seyfried NT, Quantitative proteomics of cerebrospinal fluid from African Americans and Caucasians reveals shared and divergent changes in Alzheimer’s disease. bioRxiv 2022.12.07.519393 [Preprint]. 8 December 2022. 10.1101/2022.12.07.519393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.He T, Liu Y, Zhou Y, Li L, Wang H, Chen S, Gao J, Jiang W, Yu Y, Ge W, Chang H-Y, Fan Z, Nesvizhskii AI, Guo T, Sun Y, Comparative evaluation of proteome discoverer and fragpipe for the TMT-based proteome quantification. J. Proteome Res. 21, 3007–3015 (2022). [DOI] [PubMed] [Google Scholar]
- 77.Käll L, Canterbury JD, Weston J, Noble WS, MacCoss MJ, Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 (2007). [DOI] [PubMed] [Google Scholar]
- 78.da Veiga Leprevost F, Haynes SE, Avtonomov DM, Chang H-Y, Shanmugam AK, Mellacheruvu D, Kong AT, Nesvizhskii AI, Philosopher: A versatile toolkit for shotgun proteomics data analysis. Nat. Methods 17, 869–870 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nesvizhskii AI, Keller A, Kolker E, Aebersold R, A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 (2003). [DOI] [PubMed] [Google Scholar]
- 80.Dammer EB, Seyfried NT, Johnson ECB, Batch correction and harmonization of—Omics datasets with a tunable median polish of ratio. Front. Syst. Biol. 3, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. The TMEM106b core antibody can be obtained by contacting L.P. and establishing a material transfer agreement with Mayo Clinic (https://redcap.vanderbilt.edu/surveys/?s=NMRA8TLFM7). Code used for data analysis was deposited in Zenodo (DOI: 10.5281/zenodo.10233187).