

Abstract
A novel budding yeast species was isolated from a soil sample collected in the United States of America. Phylogenetic analyses of multiple loci and phylogenomic analyses conclusively placed the species within the genus Pichia. Strain yHMH446 falls within a clade that includes Pichia norvegensis, Pichia pseudocactophila, Candida inconspicua, and Pichia cactophila. Whole genome sequence data were analyzed for the presence of genes known to be important for carbon and nitrogen metabolism, and the phenotypic data from the novel species were compared to all Pichia species with publicly available genomes. Across the genus, including the novel species candidate, we found that the inability to use many carbon and nitrogen sources correlated with the absence of metabolic genes. Based on these results, Pichia galeolata sp. nov. is proposed to accommodate yHMH446T (= NRRL Y-64187= CBS 16864). This study shows how integrated taxogenomic analyses can add mechanistic insight to species descriptions.
Graphical Abstract
The discovery, formal description, and taxogenomic analysis of a novel species of Pichia revealed correlations between genome content and traditional physiological assays.
Introduction
The fungal subphylum Saccharomycotina consists of over 1000 known species and at least 84 recognized genera, including the well-known genera Candida and Saccharomyces. The genus Pichia was described in 1904 by Hansen (Hansen 1904; Kurtzman 2011). Pichia species, as well as Candida species affiliated with this genus, are widely distributed and have been isolated from multiple environments, including soil, fermented products, and cacti (Kurtzman, Fell and Boekhout 2011; Opulente et al. 2018). The species Pichia kudriavzevii (syn. Issatchenkia orientalis syn. Candida krusei) is an opportunistic pathogen and one of the leading causes of yeast infections in humans (Pelletier et al. 2005; Douglass et al. 2018; Opulente et al. 2019), as well as being considered as a potential industrial producer of organic acids (Tran et al. 2019). Despite this broad ecological diversity, many Pichia species grow on a limited number of carbon sources. On average, they can only use six of the commonly tested carbon sources, whereas Saccharomycotina species can use an average of 15 carbon sources (Kurtzman, Fell and Boekhout 2011; Opulente et al. 2018). For example, glucosides (e.g. maltose, sucrose, and lactose) and hexoses other than glucose (e.g. galactose) are generally not utilized by Pichia (Kurtzman 2011).
During a survey of yeast biodiversity throughout the United States of America, we identified a candidate novel species of Pichia through yeast enrichment and isolation protocols. The candidate novel species was isolated from a soil sample from Pavilion Key in the Everglades, Florida. The isolate was first identified to be a candidate for a novel species through sequencing the ITS and LSU regions of their rDNA locus. With the cost of whole genome sequencing decreasing and the availability of publicly available genomes increasing, whole genome sequence data can be integrated into taxonomy to further solidify species placement and yield additional mechanistic insights, such as the genetic bases of phenotypic traits or a genotype-phenotype map. Recently, a handful of papers have begun using genomic methods to identify and describe novel species (Libkind et al. 2020; Čadež et al. 2021). The development of more formal taxogenomic pipelines would further facilitate these approaches. Here, we used whole genome sequencing and taxogenomic analyses of this novel species to provide further support for its placement, identify novel genome characteristics, and correlate traits with genome content.
Materials and methods
Species isolation and identification
The strain representing the candidate novel species examined in this study is listed in Table S1; all additional species are included in Table S2. The isolation and identification of the strain in this study was done according to the enrichment protocols in Sylvester et al. (2015) with modifications adopted by Spurley et al. (2022). The strain yHMH446 was part of a broader survey project, which sampled different substrates (e.g., soil, fruits, and bark) across the United States to isolate and identify different yeast species (Sylvester et al. 2015; Haase et al. 2017; Opulente et al. 2019; Spurley et al. 2022). The strain yHMH446 was both collected and isolated by Max A. B. Haase. The soil sample was from Pavilion Key in the Everglades, FL (GPS: 25.707884, −81.351863). This sample was also incubated at 30०C in liquid SC (Yeast Nitrogen Base w/o Amino acids, Ammonium sulfate, or Glucose 6.7g/L; Ammonium Sulfate 5g/L; Drop-out mix 1.7g/L) medium in 0.8% glucose. Upon visualization of growth, a second round of enrichment was done in liquid SC medium until growth was visible. The culture was then plated to yeast-peptone-dextrose agar (YPDA – Yeast Extract 10g/L, Peptone 20g/L, Glucose 20g/L, Agar 20g/L), and unique colony morphologies were picked for species identification using Sanger sequencing of the internal transcribed spacer region (ITS) and the LSU region of the rDNA locus (Taylor et al. 2000). After our initial species identification using the ITS region and LSU region, we also included the gene encoding transcriptional elongation factor 1-α (TEF1) in downstream analyses to determine phylogenetic relatedness and distinctness.
Physiological characterization
Microscopic examination and determination of physiological characteristics were done by standard methods (Kurtzman, Fell and Boekhout 2011). Growth tests were conducted in liquid media over 5 weeks at 25°C. Initial growth was performed in Yeast Nitrogen Base (YNB – Yeast Nitrogen Base w/o AA, AS, or glucose 6.7g/L; Ammonium Sulfate 5g/L) with 0.1% glucose. This culture was then inoculated into test media, and growth was assessed over 4 weeks using the protocols in Kurtzman et al. (2011). Two sequential rounds of growth in plastic tubes on a benchtop were done for both carbon and nitrogen source testing to ensure the yeasts were not using stores of carbon or nitrogen sources from the initial culturing step. Even using 0.1% glucose YNB media, we have found some yeasts (not included in this study) to display residual growth carbon sources that are not seen after the second round of growth, suggesting some yeasts use residual stores to grow.
Formation of true hyphae and pseudohyphae was investigated using the Dalmau plate method on YPD agar plates (Kurtzman, Fell and Boekhout 2011). Ascospore formation was investigated by growing strain yHMH446 on GPYA, 5% malt extract agar (MEA), and YPDA at 10°C and 22°C for 2 months; plates were examined every week under a microscope. The ascospore image (Figure 3D) was edited in photoshop to remove a blue tint that was an artifact of the camera. This was accomplished by setting the color saturation to zero. We also increased the contrast of the image to 100% and the brightness to six in photoshop. The original image is available also available (Figure S1).
Figure 3:
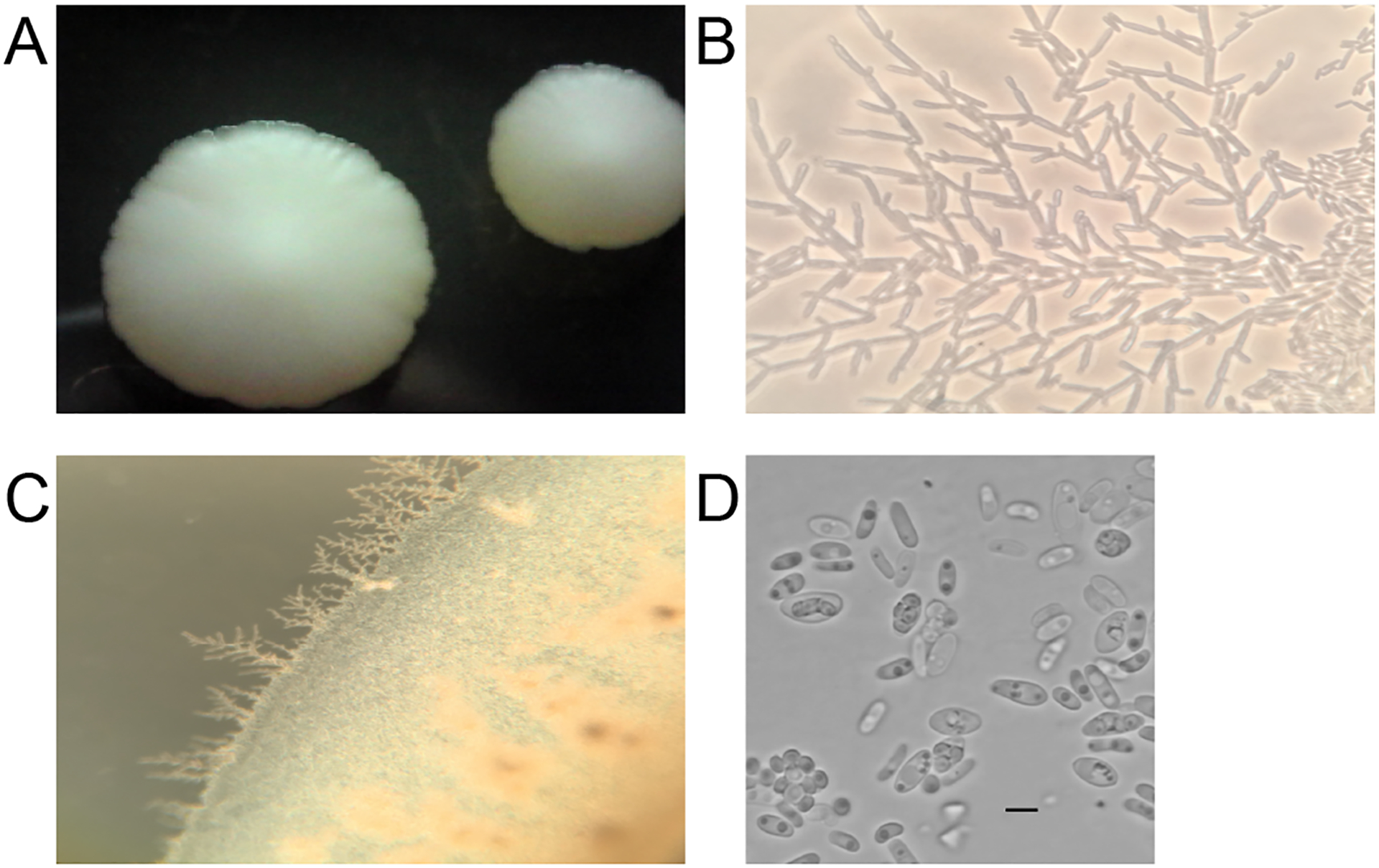
A) Colony morphology on YPD, B-C) pseudohyphal formation at 22°C on Dalmau plates, and D) hat-shaped ascospores of Pichia galeolata sp. nov. (yHMH446) grown on GPYA at 22°C. Bars = 5μm. Color saturation of this panel was decreased to zero to remove an artifact that colored the whole image blue.
Genome sequencing
Genomic DNA was isolated from strain yHMH446 using a phenol:chloroform extraction method (Shen et al. 2018). The strain was sequenced using the Illumina HiSeq platform (2×250) as described previously (Shen et al. 2018). Short read data are available at SRR16974481 for strain yHMH446.
Taxogenomic analysis
Genome assembly
Paired-end Illumina DNA sequencing reads were used to generate whole genome assemblies using the meta-assembler iWGS v1.1 (Zhou et al. 2016). The “best” assembly was chosen for each species based on genome size and N50 value (Table S3). The genome assembly for strain yHMH446 is available at JANIWQ000000000. Genome quality was assessed by quantifying their completeness based on the expected gene content of the Benchmarking Universal Single-Copy Orthologs (BUSCO) (v5.2.2) (Simão et al. 2015; Waterhouse et al. 2017). We used a set of 1711 genes inferred by BUSCO to be universal in Saccharomycotina. GenBank numbers for the ITS, LSU, and TEF1 sequences for yHMH446 are OL583853, OL583873, and OM328112.
Phylogenetic analyses
Both whole genome and multilocus phylogenetic analyses were performed to assess species relatedness (Table S2). Phylogenetic analyses of the complete LSU and ITS regions of the rDNA locus and the TEF1 gene were conducted using MEGA (version 10) (Kumar et al. 2018). The rDNA locus and the TEF1 genes for strain yHMH446 were obtained and assembled from raw paired-end reads using the HybPiper pipeline (v1.2) (Johnson et al. 2016). All species with publicly available sequences were obtained from YeastIP (Weiss et al. 2013), GenBank, and from the Westerdijk Fungal Biodiversity Institute. These sequences were aligned using the default parameters for CLUSTALW in MEGA. We constructed a maximum likelihood phylogeny using the default setting in MEGA, which uses the Tamura-Nei model of evolution with uniform rates among sites, with 1000 bootstrap replicates.
Amino acid sequences for BUSCO genes that were present across all species were aligned using MAFFT. In total, 935 protein-coding sequences were used for this analysis. The alignments were concatenated and used to assess phylogenomic placement. Species placement was determined using RAxML (v) with Saccharomyces cerevisiae as an outgroup. We ran RAxML using the model PROTGAMMAWAG (n = 100) and calculated the maximum likelihood tree with 1000 bootstraps.
Gene Trait Analyses
Gene presence was detected using TBLASTX searches of query sequences (Tables S4, S5) from characterized pathways in model organisms (e.g., Saccharomyces cerevisiae) for both the novel species described here and for publicly available genomes of species in the genus Pichia (Shen et al. 2018). We used an e-value cutoff of 10−10 to infer gene presence. We also condensed the MAL12 and IMA1–4 genes into a single group (IMA/MAL) since these genes are closely related paralogs (Voordeckers et al. 2012; Opulente et al. 2018).
Results and Discussion
Phylogenetic analyses
Multilocus analysis suggested that strain yHMH446 was sister to the clade containing Pichia cactophila (CBS 6926), Candida inconspicua (CBS 180), Pichia norvegensis (CBS 6564), and Pichia pseudocactophila (CBS 6929) (Figure 1A & Figure S2, Table S7). We found 1344 complete BUSCO genes in yHMH446 (Figure 1B, Table S8). Whole genome sequence data were limited, so we cannot determine the closest relative to yHMH446; however, our results indicated that the species with the most similar published genomes are P. norvegenesis and C. inconspicua (Figure 1C, Table S9) (Shen et al. 2018). Comparisons of the LSU sequences between yHMH446 and its closest relatives showed that known species differed considerably from yHMH446 in nucleotide sequences and indels, well beyond the 1% divergence often applied as a threshold for describing a novel species (Table S6). Combined, these results suggest that strain yHMH446 is a representative of a candidate for a novel Pichia species based on the phylogenetic species concept. Although only a single strain is currently available, its growth at unusually high temperatures (see below) and the publication of a genome sequence justifies its description. We propose Pichia galeolata sp. nov. to accommodate this strain.
Figure 1:
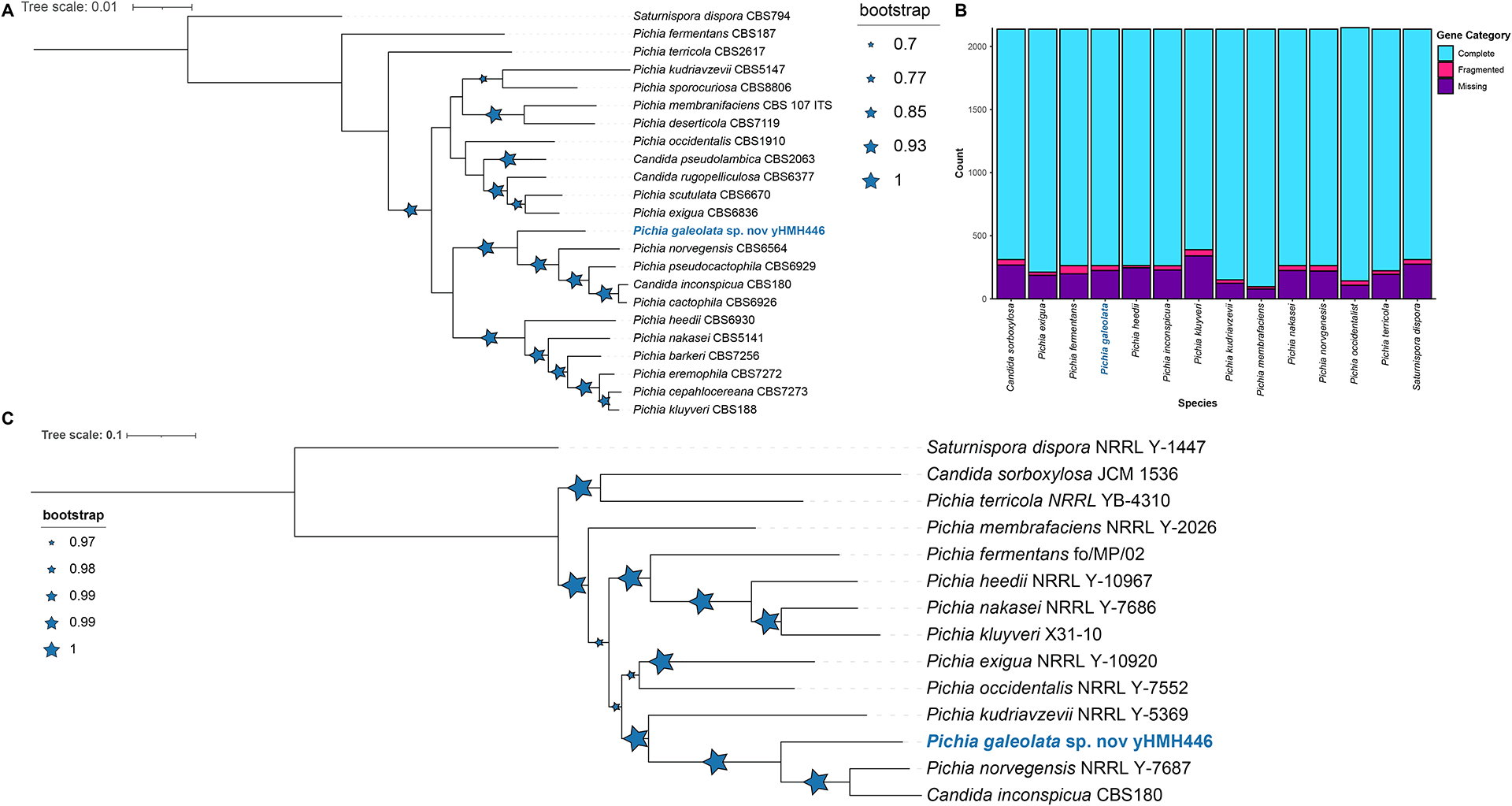
Both the concatenated ITS, D1/D2, and TEF1 phylogenetic placement and whole genome sequence analyses support the designation of strain yHMH446 as novel species in the genus Pichia. A) Maximum likelihood tree for 22 Pichia species based on concatenated D1/D2, ITS, and TEF1 sequences. Bootstrap values (n = 1000, reported as recovery frequencies) are indicated by blue stars; larger stars represent higher bootstrap values. B) BUSCO analyses for Pichia species whose genomes have been sequenced, including strain yHMH446. The bar graphs indicate that our novel genomes are of similar quality to publicly available genomes. C) Phylogenetic placement of strain yHMH446 obtained from a concatenated alignment of 935 conserved, single-copy orthologous genes. Saturnispora dispora was included as the outgroup. Bootstrap values (n = 1000, reported as recovery frequencies) are indicated by blue stars; larger stars represent higher bootstrap values.
Genomic and phenotypic analyses
We tested the candidate novel species for growth at temperatures up to 45°C. In general, most species in the genus Pichia do not grow at temperatures higher than 42°C. However, at least one strain of P. kudriavzevii (CBS 2911) is known to grow at 45°C. The results of our temperature tests indicate that yHMH446 can grow in temperatures up to 45°C. While the higher temperature growth is unusual, strains of P. cactophila, P. norvegensis, P. pseudocactophila, and C. inconspicua can all grow at 42°C (https://wi.knaw.nl/).
Using taxogenomic approaches to describe novel species provides additional resources to explore trait-gene associations to better understand the complexities and differences in carbon and nitrogen metabolism and to begin to build a genotype-phenotype map (Libkind et al. 2020). We looked at trait and gene presence and absence across publicly available genomes and the genome of our novel species candidate (Tables S4, S5). Our analyses contained multiple sets of genes that can be used in future taxogenomic analyses, including the pleiotropic PGM1 gene, which can be used as a positive control. PGM1 has many functions beyond its role in galactose metabolism, and it is therefore more likely to be present and conserved among species (Kuang et al. 2018). Thus, it can be used as a control to ensure analyses are working properly. We have also included a set of genes associated with the utilization of nitrate and nitrite (YNR1, YNI1, YNA1, YNA2, and YNT ) (Shen et al. 2018), as well as multiple genes whose associations with carbon utilization traits have been established in S. cerevisiae and other model systems. Across the majority of the Pichia species examined, the genes responsible for the utilization of galactose (GAL1, GAL7, and GAL10), maltose (MALx1, MALx2, and MALx3), and sucrose and raffinose (SUC2) were absent (Figure 2). In most cases, these missing genes were consistent with the carbon sources that cannot be utilized by these species. For example, no species grew on maltose, and most MAL genes were missing across all Pichia species. In contrast, 40% of species grew on xylose, and all species have homologs of genes known to be associated with xylose utilization (XYL1, XYL2, and XYL3). The inability of yeasts to grow on xylose, despite the presence of utilization genes, has been previously suggested to be due to the regulation of genes, redox imbalance, or enzyme specificity (Riley et al. 2016; Nalabothu et al. 2022).
Figure 2:
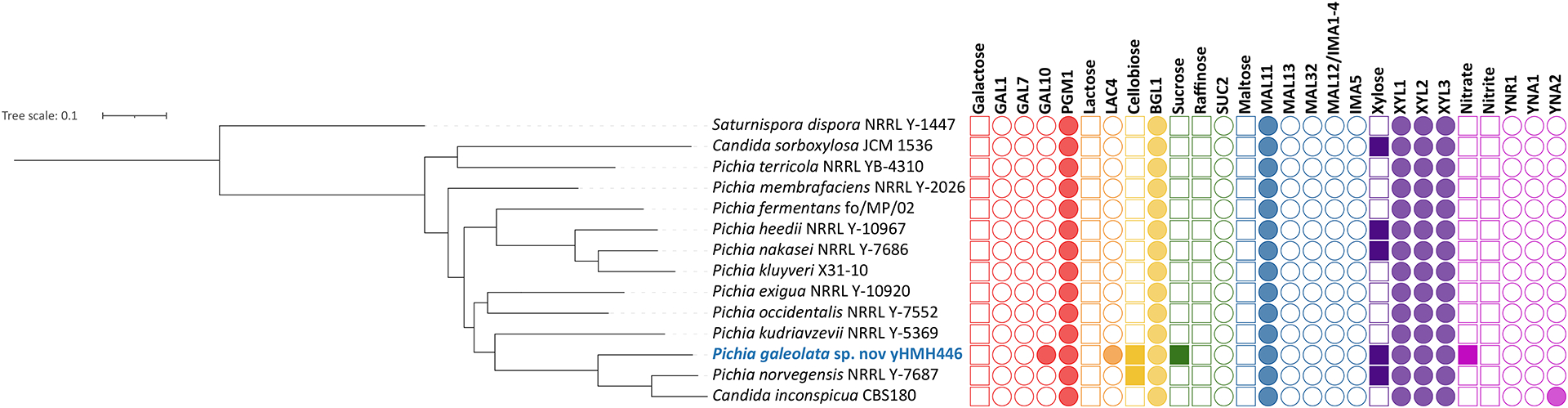
Metabolic traits and their underlying genes for the carbon sources galactose, cellobiose, sucrose, raffinose, maltose, L-sorbose, and xylose. Filled in boxes indicate either the ability to utilize a carbon source or gene presence. Gene presence was determined by BLAST analyses.
In some cases, we found the possible presence of a gene in a species that cannot grow on the carbon or nitrogen sources associated with the gene (e.g. MAL11 and YNR1). Discordance between genotype and phenotype could potentially occur for multiple reasons, including too lenient of a threshold used for gene presence, protein-coding or non-coding regulatory differences affecting gene function, or mutations that inactivate the gene. In the case of the MAL11, the gene presence met the e-value cutoff for strain yHMH446, but both the percent identities and lengths of the sequence alignments were lower in the species than they were for true positives (Table S5), suggesting this hit likely does not play roles in maltose metabolism. The YNR1 gene had a similar pattern; both sequence alignment lengths and percent identities were lower in the species than they were for true positives (Table S5). These false-positive results suggest that, while BLAST with e-value cutoffs can be used to detect genes, other measures more tailored to specific gene families should also be developed. Despite this handful of false positives, assessing gene-trait associations in taxogenomic studies is a useful practice that provides insight into the genomic bases of the physiological traits traditionally scored by taxonomists. In addition to providing further support and validation for previously explored carbon and nitrogen gene-trait associations, it simultaneously flags other metabolic genes and traits where alternative mechanisms and correlations need to be investigated by future research to address gaps and discrepancies in the budding yeast genotype-phenotype map.
Description of Pichia galeolata sp. nov.
D. A. Opulente, Q. K. Langdon, M. Jarzyna, Ke. V. Buh, M. A. B. Haase, M. Groenew., Hittinger
Growth on glucose-peptone-yeast extract agar:
After 7 days of growth at 22°C on YPD, cells are spherical to elongated. The cells occur singly and range from 2.0 – 15.0 × 2.0 – 5.7μm in size (Figure 3D).
Growth in 2% glucose-yeast extract-peptone agar:
After 7 days, colonies are cream, dull and ridged, butryous, and flat with undulate margin (Figure 3A). After 7 days at 22°C, pseudohyphae are formed under the cover glass on a Dalmau plate with YPD agar (Figure 3B–C).
Ascospore formation:
Ascospore formation was observed after 7 days on GPYA at 22°C. Asci are unconjugated and persistent, and they form two to four hat shaped ascospores within an ascus. (Figure 3D).
Fermentation:
Glucose is fermented, but galactose, maltose, raffinose, sucrose, lactose, and xylose are not (Table 1).
Table 1:
Physiological characteristics of strain yHMH446.
| yHMH446 | P. norvegensis | ||
|---|---|---|---|
| Carbon Assimilation | Glucose | + | + |
| Mannose | − | ND | |
| Fructose | − | ND | |
| Inulin | w | − | |
| Sucrose | w | − | |
| Raffinose | − | − | |
| Melibiose | − | − | |
| Galactose | − | − | |
| Lactose | − | − | |
| Trehalose | − | − | |
| Maltose | − | − | |
| Melezitose | − | − | |
| Methyl-alpha-D-glucoside | − | − | |
| Cellobiose | w | + | |
| Salicin | − | − | |
| L-Sorbose | w | − | |
| Rhamnose | − | − | |
| Xylose | w | − | |
| L-Arabinose | − | − | |
| D-Arabinose | − | − | |
| Ribose | − | − | |
| Methanol | − | − | |
| Ethanol | − | + | |
| Glycerol | w | + | |
| Adonitol | − | ND | |
| Erythritol | − | − | |
| Xylitol | − | − | |
| Galactitol | − | − | |
| Mannitol | − | − | |
| Sorbitol | − | ND | |
| Myo-inositol | − | − | |
| DL-Lactate | + | w | |
| Succinate | + | + | |
| Citrate | − | w | |
| Gluconate | − | − | |
| Glucosamine | + | + | |
| N-Acetyl-D-glucosamine | − | − | |
| Hexadecane | − | − | |
| Nitrogen | Creatinine | − | ND |
| Nitrate | − | − | |
| Nitrite | + | ND | |
| Lysine | + | + | |
| Allantoin | + | ND | |
| Fermentation | Glucose | + | s |
| Galactose | − | − | |
| Sucrose | − | − | |
| Maltose | − | − | |
| Lactose | − | − | |
| Raffinose | − | − | |
| Xylose | − | ND | |
| Temerapture | 22°C | + | ND |
| 30°C | + | ND | |
| 37°C | + | ND | |
| 42°C | + | ND | |
| 45°C | + | ND | |
| 50°C | − | ND |
Assimilation of carbon compounds:
Growth occurs with glucose, inulin (weak), sucrose (weak), cellobiose (weak), L-sorbose (weak), xylose (weak), glycerol (weak), DL-lactate, succinate, and glucosamine. . Growth is absent with mannose, fructose, raffinose, melibiose, galactose, lactose, trehalose, maltose, melezitose, methyl-alpha-D-glucoside, salicin, rhamnose, L-arabinose, D-arabinose, ribose, methanol, ethanol, adonitol, erythritol, xylitol, galactitol, mannitol, sorbitol, myo-inositol, citrate, gluconate, N-acetyl-D-glucosamine, and hexadecane (Table 1).
Assimilation of nitrogen compounds:
Growth occurs with nitrite, allantoin, and lysine. Growth is absent with creatinine and nitrate (Table 1).
Temperature growth:
Growth is observed at 22°C, 30°C, 37°C, 42°C, and 45°C. Growth is absent at 50°C (Table 1).
Holotype:
yHMH446 was isolated from soil collected from Pavilion Keys in Everglades, FL, USA (GPS: 25.707884, −81.351863) and is preserved in a metabolically inactive state in the yH strain collection at the University of Wisconsin-Madison. Ex-type strains are CBS 16864 and NRRL Y-64187.
Etymology:
The epithet gal.e.ol.a’ta, L. adj. nom, galeolata, helmet-shaped, but modified with “ol” to reflect the small size of the ascospores, which look like small helmets (Figure 3D).
Databases:
The genome of this species described below was deposited at DDBJ/ENA/GenBank under the accession JANIWQ000000000. The version described in this paper is version one. The Mycobank no. is 838264.
Conclusions
Pichia galeolata sp. nov. has been formally described and placed within the genus Pichia. Following the trend of most species of the genus Pichia, P. galeolata grows on a limited range of carbon sources. Whole genome sequence analyses suggest that their limited metabolic breadths are the result of the absence of genes known to be important for these metabolic traits.
Whole genome sequence data is rapidly becoming available for budding yeasts, and employing similar taxogenomic approaches to produce robust phylogenies, explore gene-trait associations, and build genotype-phenotype maps will soon be broadly conceivable and fruitful, especially when done in conjunction with formal species descriptions. Phylogenetic analyses using the LSU, ITS, and additional protein-coding gene regions will continue to be a valuable tool in species descriptions, as will traditional physiological assays. Nonetheless, we conclude that deploying taxogenomic approaches to describe new yeast species will advance our understanding of budding yeasts metabolic, phylogenetic, and genetic diversity.
Supplementary Material
Table S1: Isolation information for strain yHMH446.
Table S2: Phylogenetic analyses were performed on both whole genome and multi-locus data. This table identifies the data used to analyze each species.
Table S3: Whole genome assembly statistics and BUSCO analyses for strain yHMH446.
Table S4: Gene presence/absence results from TBLASTX, including a description of the genes analyzed and their sources.
Table S5: Results from TBLASTX for all genes in Table S4.
Table S6: Comparison of LSU sequences between strain yHMH446 and closely related species.
Table S7: BUSCO analyses for Pichia species, including publicly available genomes and strain yHMH446.
Table S8: Newick tree of Pichia species for multi-locus analysis.
Table S9: Newick tree of Pichia species for whole genome analysis.
Figure S2: Single-locus analyses, which encompass more of the genus Pichia, also support the designation of strain yHMH446 as novel species in the genus Pichia. A) Maximum likelihood tree for 28 Pichia species based on ITS sequences. Bootstrap values (n = 1000, reported as recovery frequencies) are indicated by blue stars; larger stars represent higher bootstrap values. B) Maximum likelihood tree for 28 Pichia species based on D1/D2 sequences. Bootstrap values (n = 1000) are indicated by blue stars; larger stars represent higher bootstrap values.
Figure S1: Original image of ascospore formation used in Figure 3D.
Take Away.
The novel species Pichia galeolata is discovered and formally described.
The genome sequence of P. galeolata is reported.
Taxogenomic analysis shows how genome content correlates with traditional physiological assays.
Acknowledgments:
We the University of Wisconsin Biotechnology Center DNA Sequencing Facility for providing DNA sequencing facilities and services; Jim McKeown for Latin advice; Maudy Smith for providing the photos showing the vegetative yeast cells, asci, and ascospores; Jacek Kominek for uploading the whole genome assemblies to GenBank; and the late Cletus P. Kurtzman for yeast taxonomy advice. This material is based upon work supported by the National Science Foundation under Grant Nos. DEB-1253634, DEB-1442148, DEB-2110403, and DGE-1256259; the USDA National Institute of Food and Agriculture Hatch Project No. 1020204, and in part by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science No. DE-SC0018409). QKL was also partly supported by the Predoctoral Training Program in Genetics, funded by the National Institutes of Health (5T32GM007133). CTH is a Pew Scholar in the Biomedical Sciences and H. I. Romnes Faculty Fellow, supported by the Pew Charitable Trusts and Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation (WARF), respectively.
Data Availability
All gene and genome sequence data have been deposited in GenBank under the accession numbers described above. All other data are contained within the manuscript and its supplements.
References
- Čadež N, Bellora N, Ulloa R et al. Hanseniaspora smithiae sp. nov., a Novel Apiculate Yeast Species From Patagonian Forests That Lacks the Typical Genomic Domestication Signatures for Fermentative Environments. Front Microbiol 2021;12:679894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglass AP, Offei B, Braun-Galleani S et al. Population genomics shows no distinction between pathogenic Candida krusei and environmental Pichia kudriavzevii: One species, four names. PLOS Pathog 2018;14:e1007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase MAB, Kominek J, Langdon QK et al. Genome sequence and physiological analysis of Yamadazyma laniorum f.a. sp. nov. and a reevaluation of the apocryphal xylose fermentation of its sister species, Candida tenuis. FEMS Yeast Res 2017;17:fox019–fox019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen EC. Grundlinien zur systematik der Saccharomyceten. Zentralbl Bakt Parasit Infekt Zweite Abteilung 1904;12:529–38. [Google Scholar]
- Johnson MG, Gardner EM, Liu Y et al. HybPiper: Extracting coding sequence and introns for phylogenetics from high‐throughput sequencing reads using target enrichment. Appl Plant Sci 2016;4:1600016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang MC, Kominek J, Alexander WG et al. Repeated Cis-Regulatory Tuning of a Metabolic Bottleneck Gene during Evolution. Mol Biol Evol 2018;35:1968–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Li M et al. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 2018;35:1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtzman C, Fell JW, Boekhout T. The Yeasts: A Taxonomic Study. Elsevier, 2011. [Google Scholar]
- Kurtzman CP. Pichia EC Hansen (1904). The Yeasts. Elsevier, 2011, 685–707. [Google Scholar]
- Libkind D, Čadež N, Opulente DA et al. Towards yeast taxogenomics: lessons from novel species descriptions based on complete genome sequences. FEMS Yeast Res 2020;20:foaa042. [DOI] [PubMed] [Google Scholar]
- Nalabothu RL, Fisher KJ, LaBella AL et al. Codon optimization improves the prediction of xylose metabolism from gene content in budding yeasts. Mol Biol Evol 2023;40:msad111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opulente DA, Langdon QK, Buh KV et al. Pathogenic budding yeasts isolated outside of clinical settings. FEMS Yeast Res 2019;19:foz032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opulente DA, Rollinson EJ, Bernick-Roehr C et al. Factors driving metabolic diversity in the budding yeast subphylum. BMC Biol 2018;16:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier R, Alarie I, Lagacé R et al. Emergence of disseminated candidiasis caused by Candida krusei during treatment with caspofungin: case report and review of literature. Med Mycol 2005;43:559–64. [DOI] [PubMed] [Google Scholar]
- Riley R, Haridas S, Wolfe KH et al. Comparative genomics of biotechnologically important yeasts. Proc Natl Acad Sci 2016;113:9882–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X-X, Opulente DA, Kominek J et al. Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum. Cell 2018;175:1533–1545.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015;31:3210–2. [DOI] [PubMed] [Google Scholar]
- Spurley WJ, Fisher KJ, Langdon QK et al. Substrate, temperature, and geographical patterns among nearly 2000 natural yeast isolates. Yeast 2022;39:55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvester K, Wang Q-M, James B et al. Temperature and host preferences drive the diversification of Saccharomyces and other yeasts: A survey and the discovery of eight new yeast species. FEMS Yeast Res 2015;15:fov002. [DOI] [PubMed] [Google Scholar]
- Taylor JW, Jacobson DJ, Kroken S et al. Phylogenetic species recognition and species concepts in fungi. Fungal Genet Biol 2000;31:21–32. [DOI] [PubMed] [Google Scholar]
- Tran VG, Cao M, Fatma Z et al. Development of a CRISPR/Cas9-Based Tool for Gene Deletion in Issatchenkia orientalis. mSphere 2019;4:e00345–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voordeckers K, Brown CA, Vanneste K et al. Reconstruction of ancestral metabolic enzymes reveals molecular mechanisms underlying evolutionary innovation through gene duplication. PLOS Biol 2012;10:e1001446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse RM, Seppey M, Simão FA et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol 2017;35:543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S, Samson F, Navarro D et al. YeastIP: a database for identification and phylogeny of Saccharomycotina yeasts. FEMS Yeast Res 2013;13:117–25. [DOI] [PubMed] [Google Scholar]
- Zhou X, Peris D, Kominek J et al. In silico Whole Genome Sequencer and Analyzer (iWGS): a computational pipeline to guide the design and analysis of de novo genome sequencing studies. G3 Genes, Genomes, Genet 2016;6:3655–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Isolation information for strain yHMH446.
Table S2: Phylogenetic analyses were performed on both whole genome and multi-locus data. This table identifies the data used to analyze each species.
Table S3: Whole genome assembly statistics and BUSCO analyses for strain yHMH446.
Table S4: Gene presence/absence results from TBLASTX, including a description of the genes analyzed and their sources.
Table S5: Results from TBLASTX for all genes in Table S4.
Table S6: Comparison of LSU sequences between strain yHMH446 and closely related species.
Table S7: BUSCO analyses for Pichia species, including publicly available genomes and strain yHMH446.
Table S8: Newick tree of Pichia species for multi-locus analysis.
Table S9: Newick tree of Pichia species for whole genome analysis.
Figure S2: Single-locus analyses, which encompass more of the genus Pichia, also support the designation of strain yHMH446 as novel species in the genus Pichia. A) Maximum likelihood tree for 28 Pichia species based on ITS sequences. Bootstrap values (n = 1000, reported as recovery frequencies) are indicated by blue stars; larger stars represent higher bootstrap values. B) Maximum likelihood tree for 28 Pichia species based on D1/D2 sequences. Bootstrap values (n = 1000) are indicated by blue stars; larger stars represent higher bootstrap values.
Figure S1: Original image of ascospore formation used in Figure 3D.
Data Availability Statement
All gene and genome sequence data have been deposited in GenBank under the accession numbers described above. All other data are contained within the manuscript and its supplements.