

Abstract
Wilson disease (WD) is caused by mutations in the ATP7B gene that encodes a copper (Cu) transporting ATPase whose trafficking from the Golgi to endo-lysosomal compartments drives sequestration of excess Cu and its further excretion from hepatocytes into the bile. Loss of ATP7B function leads to toxic Cu overload in the liver and subsequently in the brain, causing fatal hepatic and neurological abnormalities. The limitations of existing WD therapies call for the development of new therapeutic approaches, which require an amenable animal model system for screening and validation of drugs and molecular targets.
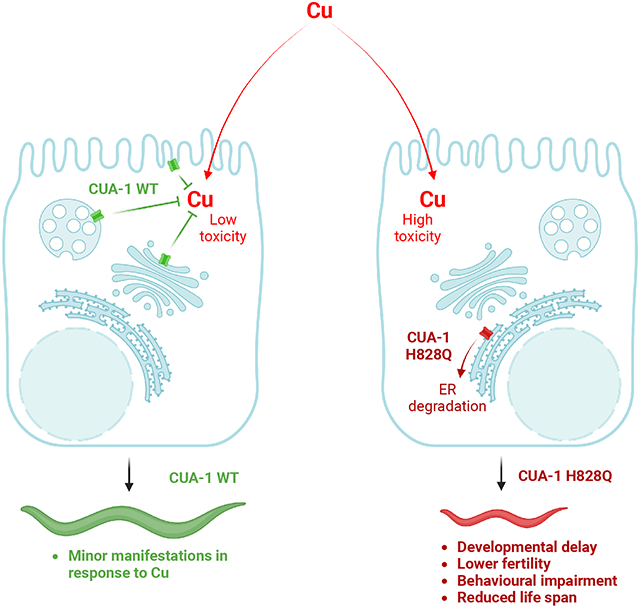
To achieve this objective, we generated a mutant C. elegans strain with a substitution of a conserved histidine (H828Q) in the ATP7B ortholog cua-1 corresponding to the most common ATP7B variant (H1069Q) that causes WD. cua-1 mutant animals exhibited very poor resistance to Cu compared to the wild-type strain. This manifested in a strong delay in larval development, a shorter lifespan, impaired motility, oxidative stress pathway activation, and mitochondrial damage. In addition, morphological analysis revealed several neuronal abnormalities in cua-1 mutant animals exposed to Cu.
Further investigation suggested that mutant CUA-1 is retained and degraded in the endoplasmic reticulum, similarly to human ATP7B-H1069Q. As a consequence, the mutant protein does not allow animals to counteract Cu toxicity. Notably, pharmacological correctors of ATP7B-H1069Q reduced Cu toxicity in cua-1 mutants indicating that similar pathogenic molecular pathways might be activated by the H/Q substitution and, therefore, targeted for rescue of ATP7B/CUA-1 function. Taken together, our findings suggest that the newly generated cua-1 mutant strain represents an excellent model for Cu toxicity studies in WD.
Keywords: ATP7B, CUA-1, copper homeostasis, Wilson disease, C. elegans, animal model, lysosomes, endoplasmic reticulum
Graphical Abstract
INTRODUCTION
Wilson disease (WD) is an autosomal recessive disorder of copper (Cu) metabolism manifesting predominantly with hepatic, neurological and psychiatric symptoms [1]. The ATP7B gene mutated in WD encodes a P1B-type Cu-transporting ATPase [2,3]. At normal Cu concentrations, ATP7B resides at the trans-Golgi network (TGN) where it transfers Cu into the Golgi lumen for incorporation into ceruloplasmin [4]. In response to elevated Cu, ATP7B traffics from the TGN to endo-lysosomal organelles where it drives sequestration of excess Cu and promotes its further biliary excretion via lysosomal exocytosis [5]. Loss of ATP7B function impairs biliary Cu excretion and Cu incorporation in ceruloplasmin. This leads to hepatic Cu overload, with subsequent damage of hepatic cells and accumulation of Cu in other organs including the brain, kidney, and eye. Without treatment, WD is fatal [1]. Approved treatments for WD include zinc salts and Cu chelators, which however have serious drawbacks in a large cohort of patients. This includes poor efficacy, intolerance, and severe side effects, among which worsening of neurological symptoms and blood vessel aneurisms represent the most dangerous [1]. As a result, there is a strong demand for novel therapeutic approaches.
In this context, several strategies have been explored for WD. One focused on genetic and pharmacological targeting of new molecular players/pathways to reduce Cu toxicity in ATP7B-deficient cells and animals. These included LRX receptors, autophagy, and epigenetic modifications [6-8]. Another approach relies on the functional rescue of mutant ATP7B variants with residual Cu-transporting activity. Usually, such ATP7B mutants (including the most frequent, H1069Q and R778L) fail to pass protein quality control checkpoints and are therefore, are subjected to ER retention and degradation. Over the last two decades, several components of the quality control (proteostatic) machinery have been identified and successfully targeted to normalize Cu homeostasis in cells expressing ATP7B mutants (Materia et al., 2012; D’Agostino et al., 2013; Concilli et al., 2016, 2020). Although new strategies have demonstrated an important therapeutic potential for WD, their translation into clinical use is significantly slowed down by the lack of an animal model that would allow rapid and massive evaluation of molecular targets and corresponding drugs reducing Cu toxicity.
Several mammalian WD models such as LEC rats, Atp7b−/− and toxic milk mice recapitulate the main features of the disease and have been used in preclinical studies [13]. However, evaluation of new drugs and/or their targets cannot be done on an extensive scale in such model systems due to the long time (usually several months) that is needed for analysis of treatment efficacy and due to stringent governmental regulations, that continue to limit number of animals for experiments and elevate costs. In this context C. elegans offers an excellent time and cost-effective option to study human diseases and, being an invertebrate, is favored to implement the principles of replacement, reduction, and refinement (3Rs) of experiments with mammals [14]. The C. elegans model has several distinct advantages, including a completely sequenced genome that shares extensive homology with that of mammals, ease of cultivation and storage, animal transparency, short life-cycle and lifespan, and affordable and rapid techniques for target gene modification aimed at null or transgenic animal creation [13,15,16]. Therefore, the impact of drug treatments or validation of corresponding molecular targets can be done in C. elegans in a very rapid and inexpensive way [15,16].
The C. elegans CUA-1 protein is an ortholog of the human Cu efflux transporters (ATP7A and B) and shares 42.6% identity with the human ATP7B protein [17]. In animals, two splice variants of cua-1 are expressed. CUA-1.1 apparently protects animals from Cu overload by sequestering Cu into lysosome-like organelles [17] and in this way resembles human ATP7B, which drives lysosomal sequestration of excess Cu in hepatic cells [5]. The other isoform, CUA-1.2, has been hypothesized to support Cu export from the intestine to peripheral tissues [17].
Considering the documented role of CUA-1 in sequestering excess Cu [17], we reasoned that generation of a C. elegans strain with a loss-of-function mutation in cua-1 might be helpful for studying Cu toxicity mechanisms in WD and their therapeutic targeting. Using gene editing, we targeted the cua-1 region encoding a conserved histidine residue in the nucleotide binding domain of the protein product and obtained the H828Q substitution, which mirrors the most frequent mutation of ATP7B in WD patients, H1069Q. As expected, the cua-1 mutant strain exhibited a significantly lower resistance to Cu compared to wild-type animals. Cu toxicity in the cua-1 mutant strain caused slower development, a reduced lifespan, and aberrant motility, and was attenuated by a Cu chelating agent. Furthermore, excess Cu led to dose-dependent changes in the behavioral phenotype of cua-1 mutant worms that could be detected after just 4h exposure and worsened with prolonged exposure to increased Cu concentrations. Notably, resistance of the cua-1 mutant strain to Cu was also improved by a reported corrector of WD-causing ATP7B-H1069Q mutant, indicating that this strain is also suitable for evaluation of mutant-correcting drug treatments in WD. Thus, our studies indicate that the newly generated cua-1 mutant strain represents a useful small animal model for investigation and therapeutic targeting of Cu toxicity mechanisms in Wilson disease and might be employed for cost-effective drug screens and validation.
RESULTS
Generation and characterization of a new cua-1 mutant strain
In addition to the sequence similarity of CUA-1 to human ATP7B, it also contains the main evolutionarily conserved domains that are required for Cu-transporting ATPase activity [17,18]. These include phosphorylation, nucleotide binding and auto-phosphatase domains as well as several motifs in the transmembrane domains that are needed for Cu transfer across membranes (Fig. 1A). The functional importance of these domains is underscored by the observation that numerous WD-causing mutations occur in these conserved regions [19]. One such mutation leads to a H/Q substitution at position 1069 in the nucleotide-binding domain of ATP7B and is found in about 50% of WD patients in Europe and North America [20]. To generate a similar substitution of the conserved histidine in CUA-1, CRISPR-Cas9 mediated gene editing was employed to obtain the cua-1(knu790[H828Q]) allele. Two specific sgRNAs were used to make Cas9-mediated breaks in exon 12 of cua-1 and a recoded DNA fragment with flanking homology arms was used as a repair template to insert a matching DNA sequence with a CAG (glutamine) instead of a CAT (histidine) codon (Fig. 1B, C). The presence of the mutation in the new C. elegans strain was confirmed by Sanger sequencing and by RT-PCR with specific primers that selectively amplify either native or mutant cua-1 (Fig. 1D). In the absence of excess Cu, quantitative behavioral phenotyping of the cua-1(knu790[H828Q]) mutant revealed no significant morphological or behavioral alterations compared to the wild-type (Fig. S1).
Figure 1. Generation of the H828Q substitution in CUA-1.

A. Three-dimensional representation of the human ATP7B and its C.elegans ortholog CUA-1. The proteins have eight transmembrane (TM) domains, which form a pore for transport of Cu across the membrane. The CPC motif in 6th transmembrane domain plays a key role in the metal translocation along the pore. ATP7B and CUA-1 contain also ATP-binding (blue), phosphatase (green), phosphorylation (red) domain and N-terminal tail with metal binding sites (orange balls). Arrows indicate conservative histidine in nucleotide binding domain, which is substituted in large cohort of WD patients and was targeted in cua-1(knu790[H828Q]) C.elegans strain. B. The panel shows a portion of exon 12 with coding triplets and site of H828Q substitution (red rectangle). The scheme below shows the alignament of donor DNA template (in violet), used for knockin generation, with recoded region (pink) and homology arms (green). The sights of sgRNA guided cut are shown in yellow. C. The scheme shows a portion of exon 12 with alignament of WT cua-1 with recoded region in the DNA template. The substituted nucleotides are highlighted in pink and result in silent mutations with exception of CAT (histidine) to CAG (glutamate) codon change. D. Agarose gel electrophoresis of RT-PCR products amplified from WT and cua-1(knu790[H828Q]) RNAs using either WT-specific or H828Q-specific cua-1 primers.
H828Q substitution in CUA-1 increases Cu toxicity in C. elegans
The lack of any severe behavioral phenotype in the cua-1(knu790[H828Q]) strain suggested that the mutant strain could adapt to environmental Cu levels. Such a phenomenon was observed for hepatic ATP7B-KO cells, which adapt to Cu present in the medium by down-regulating the high affinity Cu influx transporter CTR1 but rapidly die upon Cu challenge [8]. We reasoned that exposure to Cu might overcome any protective mechanisms in cua-1(knu790[H828Q]) so we tested whether an increase in Cu would result in pathological manifestations. Previous studies indicated that wild-type animals are resistant to Cu up to a concentration of 100 μM [17]. Therefore, increasing concentrations of CuCl2 up to 100 μM were added to the culture plates and the development of wild-type and cua-1(knu790[H828Q]) animals was evaluated after 3 days from the egg stage. Analysis of animal populations revealed a significant delay in cua-1(knu790[H828Q]) development upon Cu exposure (Fig. 2A, B). We found that Cu strongly reduced the number of mutant animals that reach adulthood in the population compared to wild-type animals (Fig. 2B). Keeping animals in Cu for a second generation had a more devastating impact on the mutant with practically no adults found at 100 μM CuCl2 (Fig. 2B). These findings indicate that H828Q substitution in CUA-1 results in reduced resistance to high Cu in becoming adults.
Figure 2. H828Q substitution in CUA-1 increases Cu toxicity in C. elegans.

A, B. Wild-type (WT) and mutant (H828Q) animals were directly grown in CuCl2-supplemented plates for 3 days (1st generation, F1 protocol; see Methods). Alternatively, 3 day interval of CuCl2 incubation was used for animals, whose parents were also grown in the presence of CuCl2 before egg laying (2nd generation, F2 protocol; see Methods). The plates were analyzed under the stereomicroscope (A) to quantify number of adults in the population (B). Both micrograms (A) and quantification (B) indicate that 100 μM Cu reduced number of mutant adults in the first generation, while even lower Cu concentrations significantly delayed development of mutants to adult stage in the second generation (** p<0.01, **** p<0.0001; two way ANOVA; n≥3 experiments). C. The graphs show survival curves of WT and mutant animals grown in the presence of different Cu concentrations. Both 50 and 100 μM Cu significantly shortened the lifespan of mutant animals (**** p<0.0001; Mantel-Cox test; n≥20 animals). D. Brood size was calculated for individual WT and mutant animals grown on plates containing 100 μM Cu. Quantification reveals reduction in the number of eggs deposited by mutants (**** p<0.0001; t-test; n=10 animals). E. The graph shows that daily rate of egg laying is reduced in mutant animals. F. Number of “bag worms” was calculated in the populations of WT and mutant animals grown on plates containing 100 μM Cu. The graph shows that mutant population contains more bag of worms (*** p<0.001, **** p<0.0001; two way ANOVA; n≥3 experiments). Scale bar: 1.2 mm (A)
In view of the lower tolerance of mutant animals to Cu, we further investigated the consequences of elevated Cu toxicity in the cua-1(knu790[H828Q]) strain. To this end we analyzed the lifespan and the fertility of mutant and wild-type animals exposed to Cu. We found that both 50 and 100 μM CuCl2 significantly reduced the lifespan of mutants compared to wild-type animals (Fig. 2C). Further, using 100 μM CuCl2 in brood size and egg-laying assays revealed that the fertility of mutant animals decreases upon exposure to Cu (Fig. 2D, E). Notably, some adult animals failed to release eggs upon exposure to Cu. As a result, hatching from the eggs occurred inside the parent leading to the death of the latter (the so called “bag-of-worms” phenotype, See. Movie 1). Quantification revealed a significantly higher number of bag-of-worms in the cua-1(knu790[H828Q]) strain compared to wild-type animals (Fig. 2F). In summary, the above results show that the H828Q substitution affects CUA-1 function and promotes Cu toxicity in C. elegans.
Next, we employed atomic absorption spectroscopy (AAS) to evaluate Cu levels in wild-type and mutant animals. Similar amounts of Cu were detected in both strains under control conditions and upon Cu overload (Fig. S2A), although cua-1(knu790[H828Q]) tended to accumulate Cu when exposed to 25 μM CuCl2. This suggests that elevated Cu toxicity in cua-1(knu790[H828Q]) animals is likely to be caused by a change in compartmentalization/trafficking rather than by an excess of the metal. We considered that some Cu excretion pathways could be activated to compensate for mutation in cua-1 and tried to target them with silver. The electrochemical similarity of Cu+ and Ag+ ions allows silver ions to be incorporated into the metabolic Cu pathways and replace Cu in some Cu-transporting proteins, thereby disrupting their function [21]. AAS revealed that silver treatment caused a substantial accumulation of Cu in cua-1(knu790[H828Q]) but not in wild-type animals (Fig. S2B, C), indicating a possible dysfunction of the Cu-transporting system in mutant animals. This also corresponded to poor survival and motility of cua-1(knu790[H828Q]) animals exposed to silver (Fig. S2D, E).
Cu causes oxidative stress and mitochondrial dysfunction in cua-1 mutant animals
Elevated oxidative stress is considered to be one of the mechanisms through which mounting Cu causes toxicity in WD due to the ability of this metal to generate reactive oxygen species (ROS). We used an oxidative stress marker (gst-4p::GFP) to test whether Cu induces an oxidative stress pathway response in the cua-1(knu790[H828Q]) strain. This sensor contains the promoter of the gst-4 gene, whose robust transactivation in response to different ROS-generating stimuli has been documented [22] and GFP as a reporter, which can be easily detected in living animals. The cua-1(knu790[H828Q]) strain was crossed with the pgst-4::GFP transgenic strain and subsequently investigated by microscopy under control and Cu-overload conditions. We found that untreated wild-type and cua-1(knu790[H828Q]) exhibited very weak and comparable expression of the gst-4p::GFP transgene. Exposure to Cu caused a moderate increase in the GFP signal in the wild-type strain, while GFP fluorescence was strongly elevated by Cu in cua-1(knu790[H828Q]) animals (Fig. 3A, B) indicating an induction of a response to oxidative stress. This induction was especially evident in intestinal cells (Fig. 3C arrows), where intensive Cu absorption is likely to occur. In line with these observations, a significant transactivation of the gst-4 gene in response to Cu was detected in the mutant strain by RNA-seq (see below), indicating that elevated gst-4 expression could be used as marker of Cu-induced oxidative stress in C. elegans.
Figure. 3. Copper induces oxidative stress and mitochondrial damage in cua-1(knu790[H828Q]) strain.

A-C. Wild type (WT) and mutant (H828Q) animals were crossed with C. elegans strain expressing oxidative s tress sensor gst-4p::GFP. Resulting strains were synchronized, grown for 48h and incubated with CuCl2 overnight. Then gst-4p::GFP signal was analyzed by fluorescent microscopy. False color images (A) and quantification (B) show elevated gst-4p::GFP fluorescence in mutants compared to WT animals (** p<0.01, t-test; n≥60 animals) indicating higher oxidative stress in mutant s train. Panel C show distribution of gst-4p::GFP across the animal body in cua-1(knu790[H828Q]) strain. Right image shows higher magnifications of the boxed area in the left image. Arrows indicate elevated gst-4p::GFP signal in the cells laying in the proximal portion of the gut. D, E. WT and H828Q animals were grown in control or CuCl2-supplemented plates for 3 days and then stained with mitochondrial transmembrane potential sensor TMRE, whose intensity was analyzed by fluorescent microscopy. False color images of TMRE signal (D) and quantification (C) show reduced TMRE fluorescence in Cu-treated mutants compared to WT animals (**** p<0.0001, *** p<0.001, two way ANOVA; n≥30 animals) indicating a higher degree of mitochondrial damage in mutants train upon exposure to Cu. Scale bars: 300 μM (A; C, left panel; D), 160 μM (C, right panel).
Higher sensitivity to Cu toxicity in the cua-1(knu790[H828Q]) strain also prompted us to evaluate the impact on mitochondria in mutant animals. Recent studies revealed mitochondria as the organelles that are most sensitive to Cu toxicity in WD cell and animal models [8,23]. To test whether mitochondria are affected by Cu in the cua-1(knu790[H828Q]) strain, we treated animals with a mitochondrial membrane potential probe (TMRE), which labels functional mitochondria and is excluded from damaged organelles. Microscopy analysis revealed a similar intensity of the TMRE signal in untreated WT and mutant animals, while exposure to Cu induced a significant reduction of the TMRE signal in cua-1(knu790[H828Q]) worms (Fig. 3D, E), indicating considerable impairment of mitochondrial function in the mutant strain.
Elevated Cu toxicity correlates with motility impairment and neuronal alterations in the cua-1 mutant strain
Wilson disease is associated in many patients with neurological symptoms, which include tremors, dystonia, and movement defects. Notably, the ATP7B H1069Q mutation frequently leads to neurological manifestations in patients [24]. Unfortunately, how Cu causes neurological aberrations and consequent mobility deterioration remains poorly understood because an animal WD model with significant behavioral impairment has not been available until recently (see discussion).
In this context we investigated the behavioral phenotype of the mutant. We used high-resolution multiworm tracking [25] to compare wild-type and cua-1(knu790[H828Q]) worms that we left untreated, exposed to excess Cu for 4 h, or reared on excess Cu for one/two generations (Fig. 4). From the resulting behavioral fingerprints (Fig. 4A), it is apparent that untreated wild-type and cua-1(knu790[H828Q]) animals have near-identicle behaviors (also see Fig. S1). However, mutant worms exposed to 100 μM Cu resulted in significant changes in behavior that were more pronounced at longer exposure times. Furthermore, there is a dose-dependent effect of increasing Cu concentrations on altering the behavior of worms reared on excess Cu (Fig. 4B). Movement away from the untreated strains in phenomic space, with increasing Cu concentrations, is more pronounced for cua-1(knu790[H828Q]) worms reared on excess Cu compared to wild-type animals. Together, these findings provide clear evidence of an increased sensitivity of cua-1(knu790[H828Q]) worms to the effects of excess Cu.
Figure 4. Behavioral phenotype of cua-1(knu790[H828Q]) vs wild-type worms.

A. Hierarchical clustering of behavioural fingerprints of wild-type (WT, grey) and cua-1(knu790[H828Q]) (H828Q, pink) worms, showing a stronger behavioral phenotype of H828Q worms compared to WT exposed to excess Cu. The heatmap shows the entire set of 8,289 behavioral features extracted by Tierpsy for: untreated worms, worms exposed to 100 μM CuCl2 for 4h (4h) and worms reared on 100 μM CuCl2 for one/two generations (F1/F2, respectively). The top dendrogram shows the relationship of the individual extracted features within the entire feature set, with the tracking period bar denoting when during image acquisition the feature was extracted: pre-stimulation (pink), during stimulation with blue light (blue), and post-stimulation (green). The left dendrogram shows the phenotypic similarity of the animals, with little difference observed between the untreated strains (bottom). However, H828Q worms reared for 1 or 2 generations on 100 μM Cu cluster separately from the other conditions, showing a distinct behavioral phenotype for mutant worms under these conditions. The color map (right) represents the normalized z-score of the features. B. Position of H828Q and WT worms in phenomic space with respect to the top principal components in the behavioral feature set. Arrows show the movement of both strains reared for one generation (squares) on plates supplemented with increasing concentrations of CuCl2 (25-100 μM) away from untreated worms (stars) in phenomic space as the concentration of Cu increases. Worms exposed to 100 μM CuCl2 for 4h (circles) and worms reared on 100 μM CuCl2 for two generations (triangles) are also included, and demonstrate that a 4h exposure to excess Cu does little to change the position of WT worms in phenomic space, but causes a significant movement of H828Q animals away from the untreated worms. Error bars represent standard error of the mean. C-E. The key behavioral features differing between H828Q and WT worms treated with 100 μM CuCl2. Individual points on the boxplots represent averaged values of 3 worms per well in the tracking plates. Pairwise statistically significant differences of behaviour were calculated between H828Q and WT worms cultured under the same conditions using block permutation t-tests (n = 10,000 permutations) and corrected for multiple comparisons using the Benjamini Hochberg correction (** p < 0.01, *** p < 0.001, **** p < 0.0001, ns p > 0.05). F. Attenuation of the photophobic escape response of H828Q worms reared for two generations on plates supplemented with 100 μM CuCl2. The photophobic escape response is characterized by the overall fraction of worms moving forward following stimulation with a 10s pulse of high intensity blue light (blue shading). Coloured lines represent averages of the detected fraction of worms moving forwards across all biological replicates and shaded areas represent the 95% confidence intervals. All behavioural experiments were conducted across 3 independent days and all data shown represents well averaged values (3 worms per well), with n ≥ 116 wells for each strain and condition.
As with untreated worms (Fig. S1), 4h exposure to 100 μM Cu caused no significant difference in the morphology of mutant vs wild-type worms. However, long exposure during development to excess Cu caused a significant reduction in the length of mutant worms compared to wild-type animals reared under the same conditions (Fig. 4C). A key phenotype of the mutant strain exposed to excess Cu was faster body bending characterized by an increased time derivative of midbody curvature. We observe a significant increase in the rate of change of midbody curvature of cua-1(knu790[H828Q]) animals exposed to 100 μM Cu after just 4h (Fig. 4D) and this phenotype became more significant for the mutant strain reared on 100 μM Cu for one or more generations. There was no change in the locomotion of crawling worms reared on excess Cu for one generation or less. However, there was a significant decrease in the overall speed (Fig. 4E) and an attenuation of the blue light escape response (fraction of forward moving worms following stimulation with high intensity blue light, Fig. 4F) for cua-1(knu790[H828Q]) worms compared to wild-type reared on 100 μM Cu for two generations.
We further tested motility impairment in cua-1(knu790[H828Q]) animals using a liquid motility assay, which requires higher energy expenditure. Wild-type and mutant animals were treated with Cu and their motility was evaluated by the thrashing assay, which estimates the frequency of lateral swimming (thrashing) movements [26]. The animals were placed in a liquid medium and their movement was video recorded under a stereomicroscope. Movies 2, 3 and Figures 5A, B demonstrate that cua-1(knu790[H828Q]) animals swam with a slower movement frequency compared to wild-type controls. Correspondingly, quantitative analysis of the movies using the wrMTrck ImageJ plugin revealed that Cu significantly affects mobility of mutant animals in a concentration dependent manner (Fig. 5C). Furthermore, analysis of the movement tracks demonstrated that Cu severely affects displacement of mutant animals (Fig. 5D).
Figure 5. Copper impairs motility and affects GABAergic neurons of cua-1(knu790[H828Q]) animals.

A-D. Wild-type (WT) and mutant (H828Q) animals were grown in CuCl2-supplemented plates for 3 days. Then the animals were transferred to water and their thrashing motility was analyzed (see Methods). Images in panel A show a last frames of representative time-lapse movies, which capture movement of WT or mutant animals (see Supplementary movies 2 and 3), while corresponding images in panel B show calculations of thrashes for each individual animal. Quantification (C) indicates that Cu caused a stronger reduction of thrashing rates in mutant strain compared to WT animals (**** p<0.0001; two way ANOVA; n≥25 animals). Panel D displays color-coded trajectories of individual WT and mutant animals (shown in panels A and B and movies 2 and 3) where each position of animal body is depicted in the color corresponding to the certain time point in the movie sequence (see color time scale bar). The images show that Cu significantly reduced displacement of mutant animals compared to WTs train. E-F. Wild type (WT) and mutant (H828Q) animals expressing marker of GABAergic neurons unc-47p::GFP were grown in plates containing 100μM CuCl2 for 3 days. Panel E1 shows low magnification view of neuron bodies (arrowheads) and commissures (arrows) in WT animal. Normal (E2) and branching (E3, arrow) commissures are shown at higher magnification. Panel F1 shows low magnification of neuronal bodies (arrowheads) and comissures (arrows) in mutant animal. Panels F2-F5 denonstrate commissure abnormalities including necklace-like blebbing (arrows), abnormal curvature (arrowheads) and branching (empty arrowheads). Graph (G) shows the number of branched commisures per animal in WT and mutant strains (**** p<0.0001, *** p<0.001; two way ANOVA; n≥60 animals). Scale bars : 2.4mm (A, B, D), 55μM (E1), 25 μM (E2,3), 50μM (F1, F5), 38μM (F2-4)
To understand whether motility impairment correlates with any neuronal abnormality, we investigated how Cu affects the morphology of GABAergic motor neurons in cua-1(knu790[H828Q]) animals. For this purpose, the cua-1(knu790[H828Q]) strain was crossed with a unc-47p::GFP transgenic strain, which expresses the GFP under a GABA-specific promoter. This allows GABAergic neurons to be highlighted by green fluorescence and observed in living animals. The ventral cord of C. elegans contains 19 GABAergic motor neurons, which extend commissures to the dorsal part across the body (Fig. 5E). We did not detect any significant neuronal alteration in untreated mutant animals compared to the wild-type strain, and exposure to 100 μM Cu did not result in a significant loss of neurons in either strain. However, we noted several neuronal commissure alterations, which were more frequent in cua-1(knu790[H828Q]) animals. This manifested in several phenotypes including aberrant branching, formation of necklace-like commissures and abnormal curvature (Fig. 5F). Some of these alterations were also detected in wild-type animals (Fig. 5E) but to a much lesser extent. Indeed, quantification revealed that the number of abnormally branching commissures increased in Cu-treated mutants but not in WT animals (Fig. 5G). Thus, Cu induces alterations in neuronal morphology and possibly, as a consequence, compromises motility of cua-1(knu790[H828Q]) animals.
RNA-seq reveals significant transcriptome modulation in the cua-1 mutant strain
We used RNA-seq analysis to compare transcriptomes of wild-type and cua-1(knu790[H828Q]) strains to investigate molecular pathways that might be activated by the H828Q mutation. Wild-type and mutant animals were synchronized, grown for 3 days and collected for mRNA isolation. RNA-seq analysis (GEO Reference number GSE234467) revealed a significant transactivation of 1,430 genes and downregulation of 1,367 transcripts in the cua-1(knu790[H828Q]) strain (Fig. 6A). First, we checked whether transcription of the main genes involved in Cu homeostasis was altered by H828Q substitution in CUA-1. We found that cua-1 transcripts were elevated in the H828Q strain (Fig. 5B) which is possibly a consequence of the degradation of mutant CUA-1 (see below) and the consequent risk of Cu accumulation, and thus represents a compensation mechanism. This was paralleled by transactivation of tyr-4 and pamn-1 genes (Fig. 6B) encoding orthologs of tyrosinase and PAM enzymes, whose activity requires ATP7A/B-mediated supply of Cu [19]. By contrast, the transcription of the Cu chaperone genes cuc-1 (ATOX1 ortholog) and cox-17 was downregulated (Fig. 6B). Next, we evaluated the expression of possible orthologs of the main human Cu influx transporter CTR1 (hCTR1). C. elegans expresses several genes that share 30-40% identity with hCTR1 [27]. The most characterized CTR1 candidate gene chca-1 (F58G6.9) did not show any significant modulation of mRNA levels in the cua-1(knu790[H828Q]) strain. However, we found the other two candidate CTR1 genes, F27C1.2 and K12C11.7 [27], were downregulated in the mutant (Fig. 6B), presumably to limit Cu influx and avoid Cu-mediated damage. Further gene ontology enrichment analysis of upregulated transcripts in mutant animals revealed several activated pathways, of which DNA repair and cation homeostasis might be of particular relevance (Fig. 6C). Induction of DNA repair was previously documented by us in ATP7B-KO hepatic cells (Polishchuk et al., 2019), while the group of upregulated cation homeostasis genes contained, apart from cua-1, several divalent cation transporters, which potentially could participate in metal detoxification. All the above changes in gene expression apparently help animals to adapt to a partial loss of cua-1 function and to survive in a medium with normal Cu levels (see discussion).
Figure 6. RNA-seq reveals significant changes in the transcriptome of cua-1(knu790[H828Q]) C. elegans strain.

A. WT and mutant animals were grown in standard plates for 3 days and collected for RNA-seq. Volcano plot shows outcome of RNA-seq analysis with up (red) and down (green) regulated genes in mutant animals compared to WT controls. B. The graph shows changes in expression of individual Cu-related genes in cua-1(knu790[H828Q]) strain (**p<0.01, ***p<0.001; t-test; n=4 replicates). C. Gene ontology (GO) enrichment analysis was conducted for differently expressed genes in mutant transcriptome (see Methods). The graph shows GO categories significantly enriched in upregulated genes. D. WT and mutant animals were grown in plates containing 100μM Cu for 3 days and collected for RNA-seq. Volcano plot shows differences in transcriptional response of mutant strain to Cu compared to WT animals. E. Differently expressed genes in Cu-treated mutant animals were subjected to GO enrichment analysis. Significantly enriched GO categories in upregulated part of mutant transcriptome are shown in the graph. F. Differently expressed genes in Cu-treated mutant animals were subjected to KEGG pathway analysis (see Methods). The graph shows significantly enriched KEGG pathways in upregulated part of mutant transcriptome G, H. Heat maps shows expression levels of genes that belong to “defense response” GO category (G) and to “metabolic pathways” KEGG term (H) in WT and H828Q animals treated with Cu. Brackets indicate a group of gst genes.
Next, we compared the transcriptional response of cua-1 mutant and wild-type strains to elevated Cu (Fig. 6D). Surprisingly, this comparison did not reveal any significant change in expression of major Cu-transporting/binding genes. However, GO enrichment and KEGG pathways analysis revealed that Cu triggered a significant upregulation of genes involved in defense response and drug/xenobiotic metabolism (Fig. 6E, F). Notably, upregulated transcripts in these pathways corresponded to gst (glutathione S-transferase) genes (Fig. 6G, H, brackets), which participate in detoxification of metallo- and xenobiotics via their conjugation with reduced glutathione [29]. Activation of GST enzymes occurs during oxidative stress conditions that, as outlined above for the widely used oxidative stress reporter gst-4p::GFP, was induced in cua-1 mutants exposed to Cu. Correspondingly, RNA-seq revealed a significant induction of gst-4 by Cu in the cua-1(knu790[H828Q]) strain (Fig. 6G, H, brackets), indicating that mutant animals activate GST-mediated mechanisms to defend themselves from Cu overload and consequent oxidative stress. This was paralleled by transactivation of such metal stress response genes as cdr-2, F53A9.6, F53A9.7, F53A9.1 and haly-1 indicating elevated Cu toxicity in mutant animals (Suppl table 2; GO:0010035~response to inorganic substance).
The H828Q mutation causes CUA-1 retention and degradation in the ER
A growing body of evidence suggests that the H1069Q mutant of ATP7B undergoes significant retention and degradation in the ER [11,12,30]. This does not allow newly synthetized ATP7B-H1069Q to reach Cu excretion sites and leads to hepatic Cu accumulation in patients carrying this mutation. To understand whether substitution of the conserved histidine in CUA-1 recapitulates the behavior of ATP7B-H1069Q, a GFP-tagged version of WT and CUA-1H828Q was expressed in C. elegans intestinal cells and its intracellular distribution was analyzed using confocal microscopy. Previous studies suggest that CUA-1 resides at the plasma membrane under basal conditions and moves to lysosomal organelles in response to Cu [17]. This relocation of CUA-1 promotes sequestration of excess Cu into the lysosomal lumen [16]. We observed similar behavior of wild-type CUA-1. Figure 7A shows that under steady state conditions it is mainly at the cell membrane, independently of the expression level. Cu induced wild-type CUA-1 translocation to lysosomal structures (Fig. S3A, B). In contrast, a significant amount of CUA-1H828Q was detectable in the ER membrane network that comprised the nuclear envelope and expanded across the entire cell (Fig. 7B; Fig. S3C, D). To confirm the ER localization of the mutant protein, we crossed animals expressing GFP-CUA-1H828Q with a strain expressing the bona fide ER marker TRAM-1 fused with ChemOrange2. Confocal microscopy revealed an extensive overlap of mutant CUA-1 with the ER marker (Fig. 7C) indicating a significant retention of CUA-1H828Q in the ER. Notably, the degree of the ER retention correlated with higher expression levels of the mutant protein, while in the cells expressing lower amounts of CUA-1H828Q a leakage of the mutant protein to the plasma membrane was detected (Fig. 7D). This highly recapitulates the behavior of the ATP7B-H1069Q mutant, which fills the ER when overexpressed. Lower, and especially endogenous, expression levels of ATP7B-H1069Q allow the quality control machinery to rapidly eliminate the ER-retained pool of newly synthetized mutant protein, while residual amounts of ATP7B-H1069Q circumvent ER quality control checkpoints and traffic towards endo-lysosomal structures and the cell surface [12,30].
Figure 7. H828Q substitution causes ER retention and degradation of CUA-1.

A, B. GFP-tagged versions of WT (A) or H828Q (B) variants of CUA-1 were expressed in intestinal cells of C.elegans and investigated using confocal microscopy. The panels show GFP signal and its overlap with phase contrast images of the animal. Arrows in panel A indicate cells showing CUA-1(WT) at the plasma membrane. Arrows in panel B indicate cells exhibiting CUA-1(H828Q) both at the plasma membrane and in the ER membrane network. C. Confocal image shows that CUA-1(H828Q) overlaps with ER marker TRAM1-ChemOrange2 along ER membranes (arrows) and nuclear envelop (arrowheads). D, E. Localization of GFP-tagged CUA-1(H828Q) was analyzed in control animals (D) or animals incubated with 25μM MG132 (E). Arrows in panel D indicate cells with intense CUA-1(H828Q) signal along the plasma membrane and weak signal along ER network. Arrows in panel B indicate that MG132 treatment caused an increase in the intracellular ER signal of CUA-1(H828Q). F-I. The images show distribution of CUA-1(H828Q) in control (F, G) and MG132-treated (H, I) animals expressing ER marker TRAM1-ChemOrange2. Arrowheads in F1,2 and in H1,2 indicate the cells with high expression of CUA-1(H828Q), which shows substantial overlap with TRAM1. Arrows in in F1,2 and in H1,2 indicate cells with lower expression of CUA-1(H828Q), which is visible mainly at the plasma membrane in untreated animals (F1,2) and start to appear in the ER upon MG132 treatment (H1,2). Panels F3 and H3 show higher magnification of areas boxed in F2 and H3 respectively. Arrows in F3 and H3 indicate ER membranes with barely detectable CUA-1(H828Q) signal in untreated animals (F3) and elevated CUA-1(H828Q) fluorescence in animals exposed to MG132. Graphs in panels G and I represent intensity profiles of ER marker (TRAM1) and CUA-1(H828Q) signals calculated along the dash lines in panels F3 and H3 respectively. Peaks of CUA-1(H828Q) and ER marker coincide in MG132-treated cells (I) indicating accumulation of mutant CUA-1 in the ER. Scale bars : 30 μM (A; B; F1,2; H1,2), 11 μM (C, F3, H3), 20 μM (D, E).
This prompted us to investigate whether CUA-1H828Q is subjected to ER degradation. For this purpose, animals expressing the GFP-tagged version of the mutant protein were treated with the proteasome inhibitor MG132, which has been shown to suppress ER degradation of ATP7B-H1069Q [12,28,30]. We observed that in animals expressing low CUA-1H828Q levels, the mutant protein was hardly detectable in the ER and resided mainly at the cell membrane (Fig. 7C, F, G) while MG132-treated animals exhibited a detectable GFP-CUA-1H828Q signal in the ER regardless of the expression level (Fig. 7E, H, I). Thus, ER accumulation of CUA-1H828Q upon MG132 treatment suggests that a significant amount of the ER-retained mutant protein undergoes degradation.
Cu chelators and ATP7B-H1069Q mutant corrector reduce Cu toxicity in cua-1 mutant strain
To test whether cua-1(knu790[H828Q]) animals might be used as a model system to evaluate treatments that reduce Cu toxicity in WD, this mutant strain was exposed to small molecules whose therapeutic potential was documented in WD models and/or patients. Cu reducing agents, including Cu chelators, have been widely employed for WD treatment [1]. In addition, rescue of the ATP7B mutant function with correctors has recently emerged as a possible therapeutic strategy [11,12,28]. Therefore, we tested whether treatment with Cu chelators or ATP7B-H1069Q mutant correctors counteracts Cu toxicity in cua-1(knu790[H828Q]) animals. The Cu chelators BCS and TTM were selected for this purpose. BCS has been used already in C. elegans studies [17] while TTM is an approved drug for WD [1]. Each chelator was added to the culture plates at 50 μM or 100 μM in combination with 100 μM Cu. We found that BCS limited the impact of Cu on the development of mutant animals in a concentration-dependent manner, as did 50 μM TTM (Fig. 8A, B). This indicates that, like in WD, chelation agents significantly reduce Cu toxicity.
Figure 8. Copper chelators and p38 inhibitor reduce copper toxicity in cua-1(knu790[H828Q]) C.elegans strain.

A, B. Wild-type (WT) and mutant (H828Q) animals were grown for 3 days on plates, which were supplemented with 100 μM CuCl2 alone or combined with either 100 μM BCS or 50 μM TTM. The plates were analyzed under the stereomicroscope (A) to quantify number of adults in the population (B). Both micrograms (A) and quantification (B) indicate that both BCS (50μM and 100mM) and TTM (50μM) reduced impact of Cu on the development of mutant animals(** p<0.01, **** p<0.0001; two way ANOVA; n=3 experiments). C. Wild-type (WT) and mutant (H828Q) animals were grown for 62h on plates, which were supplemented with 100 μM CuCl2 only or in combination with inhibitor of p38 MAP kinases SB202190 at 5 or 10 μM concentration. The plates were analyzed to quantify number of adults in the population. Quantification shows that p38 inhibitor improves development of mutant animals grown in the presence of Cu (** p<0.01, *** p<0.001; one way ANOVA; n=3 experiments). Scale bar: 1.2 mm (A).
To correct cua-1(knu790[H828Q]) mutant phenotypes, we also used the p38 MAP kinase inhibitor SB202190. Our previous studies showed that this inhibitor reduces ER retention and degradation of ATP7B-H1069Q and, thereby, rescues its trafficking and ability to sequester/excrete excess Cu [28]. Moreover, SB202190 was effective in recovering other pathogenic ATP7B mutants including the frequent Eastern Asian variant R778L [28]. cua-1(knu790[H828Q]) animals were treated with the p38 inhibitor at 5 μM or 10 μM in combination with Cu. Subsequent analysis revealed that the p38 inhibitor significantly improved developmental progression of Cu-treated animals since the percentage of animals that reached the adult stage increased significantly in the population (Fig. 8C). These experiments suggest that pathogenic mechanisms caused by H/Q substitution are conserved in C. elegans and might be targeted by mutant correctors in drug screening.
DISCUSSION
C. elegans has emerged as a very efficient small animal model for drug discovery, including in the field of genetic diseases [15,31,32]. Low-cost maintenance, a fast life cycle, and simplicity of drug/dsRNA delivery make C. elegans an attractive tool for large scale drug tests/screenings [15,16,33]. This offers a very valuable alternative to vertebrate animals whose use in biomedical research is expected to be more and more limited due to stringent bioethical regulations [15]. In this study we evaluated a new cua-1(knu790[H828Q]) mutant strain in C. elegans as a model that might be helpful to study the impact of different treatments on Cu toxicity in Wilson disease. We found that although there is no robust behavioral phenotype for mutant worms in the absence of excess Cu, mutant animals exhibited very high sensitivity to Cu compared to the wild-type strain. This manifested in a delay in reaching adulthood, reduced lifespan and brood size, that can be reverted by Cu chelation, which is widely used for treatment of WD patients. Thus, like ATP7B loss, CUA-1 dysfunction (which is an ATP7B ortholog) leads to reduced Cu tolerance. This notion is in line with the previously reported ability of CUA-1 to detoxify Cu via its sequestration to lysosomal structures [17].
It is also important to note that CUA-1 operates as an ortholog of ATP7A, whose dysfunction causes Cu deficiency in Menkes disease. A mutation leading to a H/Q substitution in ATP7A has not been reported in Menkes patients, but an aberrant functional phenotype was described for an engineered ATP7A-H1086Q mutant [34]. Although cua-1(knu790[H828Q]) does not manifest any phenotype that would indicate Cu deficiency, the relevance of this strain for Menkes syndrome deserves to be explored by exposing the mutant worms to Cu-limiting conditions. This, however, requires an extensive separate study.
Our findings underscore the features of the cua-1(knu790[H828Q]) strain that resemble WD phenotypes which make it a very useful small animal model to study WD-relevant copper toxicity mechanisms. First, mutant worms show poor copper resistance like other cell and animal models of WD [7,35]. Second, the CUA-1H828Q mutant phenocopies the H1069Q mutant of ATP7B but not the H1086Q mutant of ATP7A in terms of trafficking and localization. Both CUA-1H828Q and ATP7B-H1069Q show extensive ER retention and degradation. By contrast, ATP7A-H1086Q is not subjected to any significant ER retention/degradation and is efficiently transported to the Golgi [34]. Finally, the disease-causing H/Q mutation was detected only in ATP7B (not in ATP7A). Thus, all treatments normalizing the localization and trafficking of the CUA-1H828Q mutant would have relevance only for WD patients carrying the H/Q mutation. It is hard to imagine any relevance of such correcting treatments for Menkes patients who have never been found to have a H/Q mutation in ATP7A.
Our findings indicate that elevated sensitivity of mutant strain to Cu might be caused by fast degradation of the CUA-1H828Q mutant protein, as previously documented for the most frequent human ATP7B mutation H1069Q [11,12,28,30]. Notably, a corrector of this human mutant ATP7B (a p38 inhibitor) reduced toxic impact of Cu treatment in the cua-1(knu790[H828Q]) strain. This suggests that at least some mechanisms of H/Q mutant retention in the ER and degradation are conserved in C. elegans. Therefore, the cua-1(knu790[H828Q]) mutant strain provides an opportunity for in vivo investigation/validation of mutant-correcting drugs that target machineries regulating expression and trafficking of mutant ATP7B proteins. In our view, this represents a very important feature of the cua-1(knu790[H828Q]) model because, despite a high frequency of the H1069Q ATP7B variant in the population, experimental systems endogenously expressing the H/Q mutation are rare. The only in vitro model available are hepatocyte-like cells derived from patients carrying the H1069Q mutation [30,34]. Recently, a Drosophila melanogaster model with an H778Q mutation in dATP7 (ortholog of human ATP7B) was developed [35]. However, the suitability of this fly strain for analysis of mutant-correcting drugs/treatments has yet to be tested. In this context, our cua-1(knu790[H828Q]) C. elegans model adds an important option for the evaluation of existing and novel experimental strategies for ATP7B-H1069Q rescue.
It is worth noting that another cua-1-deficient strain exists in C. elegans (VC672). This strain contains a deletion of the cua-1 region containing the last two transmembrane helices as well as an ATP-binding motif and a dileucine-based sorting signal. Unfortunately, this strain is not suitable for evaluation of correctors of mutant proteins because the extensive deletion in cua-1 suppresses the expression of the corresponding protein product [17,36]. Moreover, the use of this strain represents additional complexity because it has to be genetically balanced due to the embryonic lethality of cua-1 KO mutant animals [17,36]. By contrast, our cua-1(knu790[H828Q]) animals arrive to adult state unless treated with relatively high concentrations of Cu. This appears to be in line with the later disease onset and milder phenotype in patients with the H1069Q substitution in ATP7B compared to patients with null ATP7B mutations leading to lack of any ATP7B protein product [1].
It is known that ATP7B-H1069Q retains substantial Cu-transporting activity and can partially exit from the ER into the secretory pathway [12,30,38]. Thus, elevated expression of this mutant might be sufficient to compensate for its degradation and, thereby, to avoid toxic accumulation of Cu. Apparently this happens in our cua-1(knu790[H828Q]) strain because RNA-seq revealed higher cua-1 mRNA levels in mutant animals compared to the wild-type strain. In this case, elevated CUA-1H828Q expression levels might be sufficient to handle Cu in mutant animals. In addition, in the cua-1(knu790[H828Q]) strain, several compensatory pathways are activated to attenuate Cu toxicity and, therefore, to adapt to environmental Cu levels. This includes downregulation of the CTR1 ortholog genes F27C1.2 and K12C11.7 to limit influx of Cu from the extracellular space. Notably, a similar decrease in CTR1 mRNA levels was observed in ATP7B-deficient hepatic cells [8], which reduces CTR1 expression. This allows them to survive in culture medium containing low concentrations of Cu. Another adaptive mechanism shared by ATP7B-deficient cells and cua-1(knu790[H828Q]) animals includes activation of genes regulating DNA repair, which occurs in response to Cu-mediated DNA damage [8].
Although the above mechanisms allow mutant animals to survive under steady state conditions, they seem to be insufficient when the cua-1(knu790[H828Q]) strain is exposed to higher Cu concentrations. RNA-seq data suggest that neither upregulation of cua-1 nor downregulation of CTR1 orthologs occurred upon Cu overload to compensate for CUA-1H828Q dysfunction. Cu overload in the cua-1(knu790[H828Q]) strain induces a severe toxicity, which manifests in numerous phenotypes. First, elevated expression of the gst-4p::GFP sensor suggests that Cu-treated mutant animals are subjected to a strong oxidative stress. RNA-seq also indicated transactivation of several gst genes, which encode enzymes participating in detoxification of metals causing oxidative damage [29,39]. Oxidative stress sensor was particularly induced in intestinal cells, where CUA-1 sequesters Cu excess [16]. However, several lines of evidence suggest that Cu toxicity in cua-1(knu790[H828Q]) animals goes beyond the intestine. A high gst-4p::GFP signal revealed oxidative stress also in muscle cells, while poor labeling with TMRE indicated a loss of mitochondrial potential across the entire worm body indicating Cu-induced mitochondrial dysfunction also in extra-intestinal tissues in cua-1(knu790[H828Q]) animals. As a consequence, oxidative stress and mitochondrial dysfunction might contribute to the developmental delay and shorter lifespan of mutant animals. Finally, we observed significant damage to neurons in Cu-treated mutant animals that correlated with a significant impairment of motility.
Importantly, the presence of a clear behavioral/neurological phenotype in the cua-1(knu790[H828Q]) strain represents a very useful feature for investigating mechanisms causing neurological symptoms in WD patients. Neurological manifestations in WD include such movement disorders as tremors, parkinsonism, and dystonia, and occur in a very significant cohort (up to 68% in some centers) of patients. Interestingly, the ATP7B H1069Q mutation has been associated with neurological impairment since clinical studies have suggested that homozygosity of the H1069Q mutation leads to more frequent neurological presentations [24]. Considering the high frequency of the H1069Q mutation [20], having a simple animal model allowing study of neurological aberration caused by the H/Q substitution in ATP7B will be of great importance. Most WD animal models are limited to hepatic manifestations while behavioral/neurological phenotypes are rare and include late onset mild manifestations in toxic milk mice and movement impairment in Atp7b knockout zebrafish [13,40]. Both models, however, do not express the H/Q mutant of ATP7B and would not be helpful for evaluating therapeutic strategies based on mutant correction. Here, we characterized a robust, dose-dependent behavioral phenotype for cua-1(knu790[H828Q]) worms in the presence of excess Cu. Given that we can detect significant changes in mutant behavior after just 4h exposure to elevated Cu levels in a 96-well plate assay, these phenotypes are tractable for high-throughput screens to identify compounds which rescue changes in mutant behavior as a result of Cu toxicity. Finally, the cua-1(knu790[H828Q]) strain adds a unique opportunity to study and target the neurodegenerative mechanisms associated with the most frequent human mutation in Wilson disease, thus taking advantage of the multiple opportunities offered by C. elegans as an animal model for neurodegenerative disorders. This includes but is not limited to the ease of following neural degeneration in a living and transparent organisms with a small number of neurons (302) and the complete knowledge of neuronal connectome at the electron microscopic level.
In summary, our findings indicate that the cua-1(knu790[H828Q]) strain we generated in C. elegans represents a useful small animal model for investigation and therapeutic targeting of Cu toxicity mechanisms driving the pathogenesis of Wilson disease.
MATERIALS AND METHODS
C. elegans culture and strains
Nematodes were grown and handled following standard procedures, under uncrowded conditions, at 20°C, on NGM (Nematode Growth Medium) agar plates seeded with Escherichia coli strain OP50 [41]. CuCl2 was used as the source for copper supplementation in NGM dishes where indicated. Wild-type (WT) animals used in this work were C. elegans variety Bristol, strain N2. A list of all other strains used in the study is provided in Supplementary Table 3.
Generation of a C. elegans strain with a H828Q substitution in CUA-1
The mutant Cua-1(knu781[H828Q]) strain carrying a substitution of the conservative histidine (H828Q) in CUA-1 was generated by Nemametrix company (Eugene, OR, USA) using CRISPR-CAS9 technology. Two sgRNAs (sgRNA1: AAGCTTCAGTGGCTCCGGAG and sgRNA2: TCCGATTGGAAATGCGGTAG) were selected based on predictive algorithms and relative C. elegans genomic position. 44 bp were recoded between the cut sites of the two sgRNAs. Most were silent recoding except for the amino acid change from H to Q at amino acid 828. The recoded region was flanked with 35 bp homology arms. The 114 bp Donor Homology Sequence (ODN) and both cas9 sgRNAs were injected into N2 worms for the insertion of the donor homology and resultant point mutation in exon 12 of Y76A2A.2a. To isolate animals harboring the H828Q mutation, PCR amplification was performed using a single reverse primer (5’ - CATGGAATCGGGAAGTGTTCGG - 3’) and two forward primers annealing with the wild type (5’ - AGCTTTGTCGGAGCATCCGATT - 3’) or the mutant (5’ – ACAGCCAATCGGAAACG - 3’) sequence. Homozygosity was confirmed using Sanger sequencing. Two independent lines were generated and out-crossed to the N2 strain to eliminate a potential off-target mutation and since they showed an equivalent phenotype, we choose one of them that in this work we design as strain NA2098 cua-1(knu781[H828Q]).
Treatment with copper, silver and drugs
To evaluate Cu toxicity in the WT and cua-1(knu781[H828Q]) strains, the animals were grown in NGM plates supplemented with CuCl2 at different concentrations ranging from 25 to 100 μM. To assess the short-term impact of Cu on animal behavior, first generation (F1) and second generation (F2) treatment protocols were used. In the short-term protocol, synchronized young adults were moved to Cu-containing plates for 4 h and then analyzed using several behavioral assays (see below). In the F1 protocol, synchronized animals were grown on control or copper containing NGM plates for 3 days and the percentage of adults in each condition was analyzed. In the F2 protocol, F1 animals were grown with or without copper for 4 days and synchronized hermaphrodites were picked onto fresh plates (control or copper-containing) and allowed to lay eggs for 2 h before being removed. F2 animals that hatched from these eggs were grown for 3 days on control or copper-containing dishes before subsequent analyses described below. Silver toxicity in WT and cua-1(knu781[H828Q]) strains was evaluated by growing synchronized animals in plates containing Ag nanoparticles (produced chemically by reduction of silver nitrate) at concentrations of 0.5 μg/ml or 1 μg/ml. Animal development, life span, and thrashing motility were evaluated to assess the impact of silver treatment. The ability of Cu chelators (BCS and TTM) or p38 MAPK inhibitor (SB202190) to reduce Cu toxicity in cua-1(knu781[H828Q]) strains was assessed by adding these drugs in combination with Cu to NGM plates containing synchronized animals. BCS and TM were used at a concentration of 50 or 100 μM c, while SB202190 was used at a concentration of 5 or 10 μM. The impact of drugs on the development of Cu-treated mutant animals was assesses by quantification of the number of adults in the population.
Larval development
WT and cua-1(knu781[H828Q]) animals were treated according to the above protocols. After treatment, the plates were placed under a Leica M205 FA stereomicroscope and the percentage of adults in the plates was quantified. A delay or failure of transition to the adult stage was employed as a reliable readout of copper (or silver) toxicity, while the effectiveness of Cu chelators or the p38 inhibitor to counteract Cu toxicity was evaluated, on the basis of their ability to overcome this developmental delay.
Life span
WT and mutant cua-1(knu781[H828Q]) animals were synchronized, allowing 5-6 adults to lay eggs for 2 h. At stage L4, worms were passed onto new plates (control or containing CuCl2 at different concentrations) and moved to a new plate every 2 days during their reproductive period (to prevent contamination by progeny). Dead and surviving animals were scored every 2 days, until all animals had died. The animals were scored as dead when they did not respond to head or tail touch. Similar analysis was performed for animals treated with Ag nanoparticles.
Brood size and egg laying
Synchronized L4 hermaphrodites from WT and mutant cua-1(knu781[H828Q]) strains were picked up and transferred singly onto a plate with or without copper. Each animal was allowed to lay eggs and then transferred to a new NGM plate every 12 h until the egg laying period was complete. The total number of eggs were used to calculate the brood size of the strain. For the egg laying experiments, the number of eggs laid on each plate was calculated every 24 h until no more eggs were laid.
Worm bagging
Synchronized WT and cua-1(knu781[H828Q]) animals grown with or without copper for 14 days were analyzed every day under a stereomicroscope. Animals with a “Bag of worms” (BOW) phenotype were identified as containing larvae in their interior. This developmental defect occurs due to abnormal retention and subsequent hatching of eggs inside the parental body. BOW nematodes were removed from the plates and quantified as a percentage from the total number of animals in each plate.
Thrashing assay
WT and cua-1(knu781[H828Q]) animals were transferred to M9 buffer for analysis of their thrashing motility. Thrashing is defined as the lateral swimming movement in which the worm body bends from one C-shape to the opposite Ɔ-shape and vice versa (Buckingham and Sattelle 2009). Synchronized young adults were assayed. After 5 min in the M9 buffer, time-lapse images of animal movement were captured for 30 sec at M205 FA stereomicroscope (Leica) equipped with IC80hd camera (Leica) controlled by Las X imaging software (Leica) [42]. The frequency of thrashing moves was determined in time-lapse image sequences using wrMTrck plugin of ImageJ or in house Worm-Turn software (https://github.com/alexey-romanov/worm-turn). Thrashing analysis was also performed for animals treated with Ag nanoparticles.
Quantitative behavioral phenotyping
Quantitative phenotyping experiments were performed by recording young adult worms crawling across an agar surface using our multi-camera worm tracker and high-content behavioral phenotypes were then extracted with Tierpsy, using methods previously described in detail [25,43,44]. Briefly, age-synchronized populations of worms were achieved by the bleach synchronization of unsynchronized gravid adults (dx.doi.org/10.17504/protocols.io.2bzgap6). L1 worms were then refed onto NGM-only or NGM plates supplemented with 25-100 μM CuCl2 and incubated at 20°C until they reached young adulthood (as described above). Tracking plates consisted of square 96-well plates (Whatman UNIPLATE, WHAT-77011651) filled with 200 μl of no peptone NGM-only (dx.doi.org/10.17504/protocols.io.bvian4ae), or no peptone NGM supplemented with CuCl2 (25-100 μM). When animals reached adulthood, worms were washed in M9 and, using a COPAS 500 Flow Pilot (dx.doi.org/10.17504/protocols.io.bvkbn4sn), dispensed into the wells of tracking plates (3 worms per well) containing the same concentration of CuCl2 used to rear the worms. To track the behavior of worms exposed to excess Cu for 4 h, worms reared on NGM-only plates were dispensed into the wells of tracking plates supplemented with 100 μM CuCl2. Plates were then incubated at 20°C for 3.5 h, before being transferred to the tracker for a further 30 mins to habituate prior to tracking. Videos were acquired in a temperature-controlled room (20°C) at 25 frames per second and a resolution of 12.4 μm px−1. Following a methodology previously described in detail [25], three videos were taken sequentially: a 5-min pre-stimulus video; a 6-min blue light recording with three 10-s blue light pulses starting at 60, 160, and 260 s (to test photophobic escape response), and a 5-min post-stimulus recording. Videos were tracked and segmented with Tierpsy Tracker [45], using previously detailed protocols and parameters [44]. Following published methods [43], a set of 3,076 predefined behavioral features [46] for each video were obtained on a per-track basis and then averaged across tracks to produce a single feature vector for each well. Statistically significant differences in the mutant feature sets obtained under each experimental condition were compared to wild-type animals reared under the same conditions using block permutation t-tests. Python (version 3.9.2) was used to perform the analysis, using n = 10,000 permutations that were shuffled within, but not between, the independent days of image acquisition. The p-values were then corrected for multiple comparisons using the Benjamini-Hochberg procedure to control the false discovery rate at 5%. All codes used are available at: https://github.com/Tom-OBrien/H828Q_phenotyping , and a detailed step-by-step protocol for experimental procedures is also available (dx.doi.org/10.17504/protocols.io.n2bvj84bbgk5/v1).
Atomic adsorption spectroscopy
C. elegans populations from WT and cua-1(knu781[H828Q]) strains were grown from eggs in liquid culture (with or without copper or silver) for 5 days. On the 6th day, liquid culture was centrifuged for 3 min at 3000 x g to precipitate worms. Then, samples were kept on ice for 20 min to collect nematodes of stage L1/L2. Samples were washed 4-5 times with M9 buffer (3 min at 3000 x g + 20 min on ice) to remove bacteria. After that C. elegans were collected in Eppendorf tubes, vacuum dried, and dissolved in HNO3. Then, the samples were cleared by centrifugation and the concentrations of metals were measured using atomic absorption spectrometry (AAS) with electrothermal atomization and the Zeeman correction of nonselective absorption using a ZEEnit 650p spectrometer (Analytic Jena) with automatic sampling duplication.
TMRE labelling
To assess the impact of Cu on mitochondria, WT and cua-1(knu781[H828Q]) animals were synchronized from eggs and grown for 16 h on control and Cu-containing plates. Cu-treated and control animals were then incubated with 0.1 μM of the mitochondrial membrane potential dye TMRE (Thermo Fisher Scientific) for 16 h and then images were captured on a Leica DM5500 microscope. The intensity of TMRE signal was quantified using ImageJ software.
Oxidative stress assay
To analyze oxidative stress, we employed the CL2166 dvIs19 [(pAF15)gst-4p::GFP::NLS] strain, which expresses GFP under the gst-4 promoter. This strain was crossed with cua-1(knu781[H828Q]), thereby allowing visualization of gst-4p::GFP, an oxidative stress sensor, in mutant animals. Synchronized WT and mutant nematodes expressing gst-4p::GFP were grown for 2 days, exposed to 100 μM CuCl2 overnight, and investigated under a Leica DM5500 microscope. The intensity of the gst-4p::GFP signal was quantified using ImageJ software.
Analysis of GABAergic neurons
To assess the impact of Cu on GABAergic neurons, the EG1285 oxls12 [unc-47p::GFP; lin-15(+)] C. elegans strain was crossed with cua-1(knu781[H828Q]). The oxls12 strain expresses GFP under the promoter of unc-47 gene, which is specifically expressed in GABergic neurons and allows these neurons to be easily tracked in WT and mutant animals due to the constant presence of the GFP signal. Analysis of the morphology of GABAergic neurons was done in control and Cu-treated nematodes using a Leica DM5500 microscope and the number of branches per neuron was quantified.
RNA-seq and bioinformatics analysis
Synchronized WT or mutant animals were grown on 100-mm NGM plates for three days with or without copper. Then, the animals were washed three times with M9 buffer, resuspended in Trizol reagent, and flash frozen with liquid nitrogen. Frozen worms were ground in a dry ice-cooled mortar and pestle and total RNA was extracted using a chloroform/ethanol protocol. Four replicates were prepared for each experimental condition.
Total RNA from each replicate was quantified using the Qubit 4.0 fluorimetric Assay (Thermo Fisher Scientific). Libraries were prepared from 125 ng of total RNA using the NEGEDIA Digital mRNA-seq research grade sequencing service (Next Generation Diagnostic srl; [47]) which included library preparation, quality assessment and sequencing on a NovaSeq 6000 sequencing system using a single-end, 100 cycle strategy (Illumina Inc.). The raw data were analyzed by Next Generation Diagnostic srl proprietary NEGEDIA Digital mRNA-seq pipeline (v2.0) which involves a cleaning step by quality filtering and trimming, alignment to the reference genome and counting by gene. The raw expression data were normalized, analyzed, and visualized by Rosalind HyperScale architecture (OnRamp BioInformatics, Inc). The C. elegans ce11 reference genome was used for analyzing RNA-seq data.
The threshold for statistical significance chosen for differential expression was False Discovery Rate (FDR) < 0,05. Gene Ontology enrichment analysis (GOEA) and Functional Annotation Clustering analyses were performed on differentially expressed genes (DEGs) using DAVID Bioinformatic Resources (Huang et al., 2009), restricting the output to Biological Process terms (BP_FAT). The threshold for statistical significance of GOEA was FDR < 0.1 and Enrichment Score ≥ 1.5. The ‘Kyoto Encyclopedia of Genes and Genomes’ (KEGG Pathway) analyses was also performed. The threshold for statistical significance for the KEGG Pathway analyses was FDR < 0.1). The RNA-seq dataset was deposited in GEO repository with GSE234467 reference number.
Analysis of CUA-1 localization
The [vha-6p::CUA-1.1::GFP::unc-54 3’ UTR] plasmid encoding a GFP-tagged version of CUA-1 was subjected to point mutagenesis to generate H828Q substitution in CUA-1.1::GFP. The construct [vha-6p::CUA-1.1[H828Q]::GFP::unc-54 3’ UTR] was generated by QuikChange Multi site-directed mutagenesis kit (Agilent Technologies) using oligonucleotides designed to insert a matching DNA sequence with CAG (glutamine) instead of CAT (histidine) codon. The oligonucleotides used were Forward 5’ - CTGAAGCTTTGTCGGAGCAGCCGATTGGAAATGC - 3’ and Reverse 5’ GCATTTCCAATCGGCTGCTCCGACAAAGCTTCAG - 3’. Both WT and mutant constructs of CUA-1.1::GFP were microinjected at a concentration of 50 ng/μL in wild type animals. The co-injection marker [unc-122p::mCherry] driving mCherry expression in the coelomocytes was used to easily identify and select injected animals. The distribution of WT and H828Q variants of CUA-1.1::GFP was investigated in animals under basal conditions or after overnight exposure to 100 μM CuCl2. Transport of CUA-1.1::GFP variants to lysosomal compartments was assessed after incubation of animals in 2 μM Lysotracker Red (Invitrogen) for 16 h. To evaluate the degree of CUA-1 mutant retention and degradation in the ER, animals expressing CUA-1.1(H828Q)::GFP were crossed with VK2664 strain, expressing the ER marker tram-1 fused with CemOrange2 [nhx-2p::CemOrange2::tram-1 + myo-2p::GFP]. Then, the animals were treated with MG132 (25 μM for 16 h), which inhibits ER-associated degradation, and the impact of treatment on abundance of the CUA-1.1(H828Q)::GFP in the ER was analyzed. For imaging of CUA-1.1::GFP variants, the animals were immobilized in 0.01% tetramisole hydrochloride (Sigma-Aldrich) on 4% agar pads and observed under a LSM700 confocal microscope (Zeiss) using a 63X oil immersion objective and appropriate filter sets for GFP, CemOrange2, and Lysotracker Red.
Statistical analysis
Statistical significance was determined using t-test for pairwise comparison or using one-way or two-way ANOVA followed by Tukey’s post hoc test in GraphPad Prism, Version 9 (GraphPad Software). The same software was used to generate survival curves, whose statistical comparison was conducted using Log-Rank (Mantel Cox) test. All data are presented as mean ± S.D., and a p values less than 0.05 were considered to be statistically significant.
Supplementary Material
Synopsis.
Wilson disease (WD)-causing mutations in the copper transporter ATP7B frequently disrupt its trafficking causing copper toxicosis in the liver and brain. The lack of small animal models represents an obstacle for high throughput screening for targets and drugs counteracting copper toxicity in WD. Here we report that substitution of a conserved histidine impairs trafficking of CUA-1, the C. elegans ortholog of ATP7B, and promotes copper toxicity in the mutant strain. This offers a new time and cost-effective animal model to study and target copper toxicity in WD.
ACKNOWLEDGEMENTS
We would like to thank CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440) for strains; WormBase; and Prof. K. Shen (Stanford University, USA) for plasmids. This study received support from following projects/agencies/organizations: #NEXTGENERATIONEU (NGEU) funded by the Ministry of University and Research (MUR), National Recovery and Resilience Plan (NRRP), project MNESYS (PE0000006) – A Multiscale integrated approach to the study of the nervous system in health and disease (DN. 1553 11.10.2022); Telethon Italy (grant #TGM22CBDM05 to RSP); European Joint Project – Rare Diseases (EJP-RD WilsonMed grant to RSP); National Institutes of Health (grant ID: R01 DK129599 to BEK); Italian National Wilson Disease Organization (grant to RSP); National Research Council of Italy (CNR)(grants BioMemory-Network of scientific collections for bio-monitoring, biodiversity conservation, agri-food and environmental sustainability, and well- being, https://biomemory.cnr.it, DiSBA; NUTR-AGE-FOE2019-DSB.AD004.271; and NUTR-AGE-FOE2021-DM MUR n. 844 del 16/07/2021 to EDS); Russian Science Foundation (grants # 22-24-00762 to LVP and # 20-74-10087 to EYI); CNR/RFBR (Russian Foundation for Basic Research) Collaboration Program, Italy and Russia (grant 18-515-7811 to EDS and LVP; grant number 20-515-7813 to AI and EYI); European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Grant agreement No. 714853 to AEXB); Medical Research Council (grant MC-A658-5TY30 to AEXB). We would like to acknowledge Advanced Microscopy and Imaging Core (TIGEM) and Bioinformatics Core (Tigem) for help with data acquisition and analysis, Julian Ceron Madrigal (Hospital Duran i Reynals, Barcelona, Spain) and Cathal Wilson (Tigem) for critical reading of the manuscript, Ciro Talotti for technical help.
REFERENCES
- 1.Członkowska A, Litwin T, Dusek P, Ferenci P, Lutsenko S, Medici V, Rybakowski JK, Weiss KH, Schilsky ML. Nature Reviews Disease Primers article: Wilson disease. Nat Rev Dis Primers 2018;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet [Internet] 1993;5:327–337. Available from: https://www.ncbi.nlm.nih.gov/pubmed/8298639 [DOI] [PubMed] [Google Scholar]
- 3.Tanzi RE, Petrukhin K, Chernov I, Pellequer JL, Wasco W, Ross B, Romano DM, Parano E, Pavone L, Brzustowicz LM, et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet [Internet] 1993;5:344–350. Available from: https://www.ncbi.nlm.nih.gov/pubmed/8298641 [DOI] [PubMed] [Google Scholar]
- 4.Terada K, Nakako T, Yang XL, Iida M, Aiba N, Minamiya Y, Nakai M, Sakaki T, Miura N, Sugiyama T. Restoration of holoceruloplasmin synthesis in LEC rat after infusion of recombinant adenovirus bearing WND cDNA. J Biol Chem [Internet] 1998;273:1815–1820. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9430732 [DOI] [PubMed] [Google Scholar]
- 5.Medici V, Kieffer DA, Shibata NM, Chima H, Kim K, Canovas A, Medrano JF, Islas-Trejo AD, Kharbanda KK, Olson K, Su RJ, Islam MS, Syed R, Keen CL, Miller AY, Rutledge JC, Halsted CH, LaSalle JM. Wilson Disease: Epigenetic effects of choline supplementation on phenotype and clinical course in a mouse model. Epigenetics 2016;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamilton JP, Koganti L, Muchenditsi A, Pendyala VS, Huso D, Hankin J, Murphy RC, Huster D, Merle U, Mangels C, Yang N, Potter JJ, Mezey E, Lutsenko S. Activation of liver X receptor/retinoid X receptor pathway ameliorates liver disease in Atp7B(−/−) (Wilson disease) mice. Hepatology [Internet] 2016;63:1828–1841. Available from: https://www.ncbi.nlm.nih.gov/pubmed/26679751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Polishchuk EV, Merolla A, Lichtmannegger J, Romano A, Indrieri A, Ilyechova EY, Concilli M, De Cegli R, Crispino R, Mariniello M, Petruzzelli R, Ranucci G, Iorio R, Pietrocola F, Einer C, Borchard S, Zibert A, Schmidt HH, Di Schiavi E, Puchkova LV, Franco B, Kroemer G, Zischka H, Polishchuk RS. Activation of Autophagy, Observed in Liver Tissues From Patients With Wilson Disease and From ATP7B-Deficient Animals, Protects Hepatocytes From Copper-Induced Apoptosis. Gastroenterology 2019;156. [DOI] [PubMed] [Google Scholar]
- 8.Materia S, Cater MA, Klomp LW, Mercer JF, La Fontaine S. Clusterin and COMMD1 independently regulate degradation of the mammalian copper ATPases ATP7A and ATP7B. J Biol Chem [Internet] 2012;287:2485–2499. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22130675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D’Agostino M, Lemma V, Chesi G, Stornaiuolo M, Cannata Serio M, D’Ambrosio C, Scaloni A, Polishchuk R, Bonatti S. The cytosolic chaperone alpha-crystallin B rescues folding and compartmentalization of misfolded multispan transmembrane proteins. J Cell Sci [Internet] 2013;126:4160–4172. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23843626 [DOI] [PubMed] [Google Scholar]
- 10.Concilli M, Iacobacci S, Chesi G, Carissimo A, Polishchuk R. A systems biology approach reveals new endoplasmic reticulum-associated targets for the correction of the ATP7B mutant causing Wilson disease. Metallomics 2016;8. [DOI] [PubMed] [Google Scholar]
- 11.Concilli M, Petruzzelli R, Parisi S, Catalano F, Sirci F, Napolitano F, Renda M, Galietta LJV, Bernardo D Di, Polishchuk RS. Pharmacoproteomics pinpoints HSP70 interaction for correction of the most frequent Wilson disease-causing mutant of ATP7B. Proc Natl Acad Sci U S A 2020;117:32453–32463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reed E, Lutsenko S, Bandmann O. Animal models of Wilson disease. J Neurochem [Internet] 2018;146:356–373. Available from: https://www.ncbi.nlm.nih.gov/pubmed/29473169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Schiavi E, Andrenacci D. Invertebrate Models of Kallmann Syndrome: Molecular Pathogenesis and New Disease Genes. Curr Genomics 2013;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giunti S, Andersen N, Rayes D, De Rosa MJ. Drug discovery: Insights from the invertebrate Caenorhabditis elegans. Pharmacol Res Perspect. 2021;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Rocco M, Galosi S, Lanza E, Tosato F, Caprini D, Folli V, Friedman J, Bocchinfuso G, Martire A, Di Schiavi E, Leuzzi V, Martinelli S. Caenorhabditis elegans provides an efficient drug screening platform for GNAO1-related disorders and highlights the potential role of caffeine in controlling dyskinesia. Hum Mol Genet 2022;31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chun H, Sharma AK, Lee J, Chan J, Jia S, Kim BE. The intestinal copper exporter CUA-1 is required for systemic copper homeostasis in Caenorhabditis elegans. Journal of Biological Chemistry 2017;292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Polishchuk EV, Concilli M, Iacobacci S, Chesi G, Pastore N, Piccolo P, Paladino S, Baldantoni D, vanIJzendoorn SCD, Chan J, Chang CJ, Amoresano A, Pane F, Pucci P, Tarallo A, Parenti G, Brunetti-Pierri N, Settembre C, Ballabio A, Polishchuk RS. Wilson Disease Protein ATP7B Utilizes Lysosomal Exocytosis to Maintain Copper Homeostasis. Dev Cell 2014;29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta A, Lutsenko S. Evolution of copper transporting ATPases in eukaryotic organisms. Curr Genomics [Internet] 2012;13:124–133. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23024604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev [Internet] 2007;87:1011–1046. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=17615395 [DOI] [PubMed] [Google Scholar]
- 20.Gomes A, Dedoussis GV. Geographic distribution of ATP7B mutations in Wilson disease. Ann Hum Biol [Internet] 2016;43:1–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26207595 [DOI] [PubMed] [Google Scholar]
- 21.Puchkova LV, Broggini M, Polishchuk EV, Ilyechova EY, Polishchuk RS. Silver Ions as a Tool for Understanding Different Aspects of Copper Metabolism. Nutrients 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Link CD, Johnson CJ. Reporter transgenes for study of oxidant stress in Caenorhabditis elegans. In: Methods in Enzymology. 2002. [DOI] [PubMed] [Google Scholar]
- 23.Lichtmannegger J, Leitzinger C, Wimmer R, Schmitt S, Schulz S, Kabiri Y, Eberhagen C, Rieder T, Janik D, Neff F, Straub BK, Schirmacher P, DiSpirito AA, Bandow N, Baral BS, Flatley A, Kremmer E, Denk G, Reiter FP, Hohenester S, Eckardt-Schupp F, Dencher NA, Adamski J, Sauer V, Niemietz C, Schmidt HH, Merle U, Gotthardt DN, Kroemer G, Weiss KH, Zischka H. Methanobactin reverses acute liver failure in a rat model of Wilson disease. J Clin Invest [Internet] 2016;126:2721–2735. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27322060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gromadzka G, Schmidt HHJ, Genschel J, Bochow B, Rodo M, Tarnacka B, Litwin T, Chabik G, Członkowska A. p.H1069Q mutation in ATP7B and biochemical parameters of copper metabolism and clinical manifestation of Wilson’s disease. Movement Disorders 2006;21. [DOI] [PubMed] [Google Scholar]
- 25.Barlow IL, Feriani L, Minga E, McDermott-Rouse A, O’Brien TJ, Liu Z, Hofbauer M, Stowers JR, Andersen EC, Ding SS, Brown AEX. Megapixel camera arrays enable high-resolution animal tracking in multiwell plates. Commun Biol 2022;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsalik EL, Hobert O. Functional mapping of neurons that control locomotory behavior in Caenorhabditis elegans. J Neurobiol 2003;56. [DOI] [PubMed] [Google Scholar]
- 27.Yuan S, Sharma AK, Richart A, Lee J, Kim BE. CHCA-1 is a copper-regulated CTR1 homolog required for normal development, copper accumulation, and copper-sensing behavior in Caenorhabditis elegans. Journal of Biological Chemistry 2018;293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chesi G, Hegde RN, Iacobacci S, Concilli M, Parashuraman S, Festa BP, Polishchuk EV, Di Tullio G, Carissimo A, Montefusco S, Canetti D, Monti M, Amoresano A, Pucci P, van de Sluis B, Lutsenko S, Luini A, Polishchuk RS. Identification of p38 MAPK and JNK as new targets for correction of Wilson disease-causing ATP7B mutants. Hepatology 2016;63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhattacharjee A, Chakraborty K, Shukla A. Cellular copper homeostasis: current concepts on its interplay with glutathione homeostasis and its implication in physiology and human diseases. Metallomics [Internet] 2017;9:1376–1388. Available from: https://www.ncbi.nlm.nih.gov/pubmed/28675215 [DOI] [PubMed] [Google Scholar]
- 30.Parisi S, Polishchuk EV, Allocca S, Ciano M, Musto A, Gallo M, Perone L, Ranucci G, Iorio R, Polishchuk RS, Bonatti S. Characterization of the most frequent ATP7B mutation causing Wilson disease in hepatocytes from patient induced pluripotent stem cells. Sci Rep 2018;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hopkins CE, Brock T, Caulfield TR, Bainbridge M. Phenotypic screening models for rapid diagnosis of genetic variants and discovery of personalized therapeutics. Mol Aspects Med 2023;91:101153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan S, Korolnek T, Kim BE. Oral Elesclomol Treatment Alleviates Copper Deficiency in Animal Models. Front Cell Dev Biol 2022;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iyer S, Sam FS, DiPrimio N, Preston G, Verheijen J, Murthy K, Parton Z, Tsang H, Lao J, Morava E, Perlstein EO. Repurposing the aldose reductase inhibitor and diabetic neuropathy drug epalrestat for the congenital disorder of glycosylation PMM2-CDG. DMM Disease Models and Mechanisms 2019;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petris MJ, Voskoboinik I, Cater M, Smith K, Kim BE, Llanos RM, Strausak D, Camakaris J, Mercer JFB. Copper-regulated trafficking of the Menkes disease copper ATPase is associated with formation of a phosphorylated catalytic intermediate. Journal of Biological Chemistry 2002;277. [DOI] [PubMed] [Google Scholar]
- 35.Mi X, Li Z, Yan J, Li Y, Zheng J, Zhaung Z, Yang W, Gong L, Shi J. Activation of HIF-1 signaling ameliorates liver steatosis in zebrafish atp7b deficiency (Wilson’s disease) models. Biochim Biophys Acta Mol Basis Dis 2020;1866. [DOI] [PubMed] [Google Scholar]
- 36.Overeem AW, Klappe K, Parisi S, Kloters-Planchy P, Matakovic L, du Teil Espina M, Drouin CA, Weiss KH, van IjSCD. Pluripotent stem cell-derived bile canaliculi-forming hepatocytes to study genetic liver diseases involving hepatocyte polarity. J Hepatol [Internet] 2019;Available from: https://www.ncbi.nlm.nih.gov/pubmed/30965071 [DOI] [PubMed] [Google Scholar]
- 37.Mercer SW, Wang J, Burke R. In Vivo Modeling of the Pathogenic Effect of Copper Transporter Mutations That Cause Menkes and Wilson Diseases, Motor Neuropathy, and Susceptibility to Alzheimer’s Disease. J Biol Chem [Internet] 2017;292:4113–4122. Available from: https://www.ncbi.nlm.nih.gov/pubmed/28119449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Consortium TC elegans DM. Large-Scale Screening for Targeted Knockouts in the Caenorhabditis elegans Genome The C. elegans Deletion Mutant Consortium*. G3 (Bethesda) 2012;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van den Berghe PV, Stapelbroek JM, Krieger E, de Bie P, van de Graaf SF, de Groot RE, van Beurden E, Spijker E, Houwen RH, Berger R, Klomp LW. Reduced expression of ATP7B affected by Wilson disease-causing mutations is rescued by pharmacological folding chaperones 4-phenylbutyrate and curcumin. Hepatology [Internet] 2009;50:1783–1795. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19937698 [DOI] [PubMed] [Google Scholar]
- 40.Pearson SA, Cowan JA. Glutathione-coordinated metal complexes as substrates for cellular transporters. Metallomics. 2021;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.