

Abstract
Thalassemias are among the most common hereditary diseases in the world because heterozygosity offers protection against malarial infection. Affected individuals have variable expression of alpha or beta chains that lead to their unbalanced utilization during hemoglobin formation, oxidative stress, and apoptosis of red cell precursors prior to maturation. Some individuals produce sufficient hemoglobin to survive but suffer the vascular stress imposed by chronic anemia and ineffective erythropoiesis. In other patients, mature red cell formation is insufficient and chronic transfusions are required—suppressing anemia and ineffective erythropoiesis but at the expense of iron overload. The cardiovascular consequences of thalassemia have changed dramatically over the previous five decades because of evolving treatment practices. This manuscript summarizes this evolution, focusing on complications and management pertinent to modern patient cohorts.
Keywords: arrhythmias, cardiovascular health, chelation, heart failure, iron overload, pulmonary hypertension, thalassemia
Graphical Abstract
Nontransfused thalassemia patients suffer vascular stress imposed by chronic anemia, ineffective erythropoiesis, and iron overload. Transfusion ameliorates the first two stressors while exacerbating the third. The cardiovascular consequences of thalassemia have changed dramatically over the previous five decades because of evolving treatment practices. This manuscript summarizes this evolution, focusing on complications and management pertinent to modern patient cohorts, with longer survival increasing arrhythmia burden and diastolic heart failure.
HEART DISEASE IN THE PRE-TRANSFUSION ERA
Prior to routine availability of safe, cross-matched blood, survival in thalassemia was governed by the patient’s native hemoglobin level. Ineffective erythropoiesis was overwhelming, leading to massive extramedullary hematopoiesis, splenomegaly, and bone marrow expansion(1). Compensatory hyperemia produced high output cardiac failure and pulmonary hypertension (2). For severe anemia, the increase in cardiac output was insufficient to maintain tissue oxygen delivery, leading to wide-spread tissue hypoxia (3). Somatic growth was blunted, and secondary sexual characteristics never developed (4). Pericarditis was common, although its pathophysiology was never well characterized (5). Death typically occurred in the first or second decade of life(2).
HEART DISEASE IN THE EARLY TRANSFUSION ERA
Routine transfusion therapy to suppress ineffective erythropoiesis was initiated in the 1950’s–1960’s, with the goal of improving quality of life. Transfusions were able to successfully suppress bony deformities and promote normal somatic growth in early childhood (4, 6). Unfortunately, without any effective iron removal strategies, endocrine, hepatic, and cardiac toxicity were inevitable, with death typically occurring in the second decade of life (7). Dilated cardiomyopathy was the most common cause of death, with tachyarrhythmias as an important co-morbidity. Acute and chronic pericarditis were common without evidence of constriction (5). A long asymptomatic period was typical, although electrocardiographic changes such as first-degree heart block and repolarization abnormalities were common. Higher order conduction abnormalities were also observed in advanced disease. Once either left or right heart failure symptoms presented, progression to death was rapid, even with optimal medical therapy.
INTRODUCTION OF IRON CHELATION
Sustained deferoxamine therapy first began to be applied in the early 1970’s, initially using intramuscular injection (8). After multiple studies demonstrated clinical efficacy (9, 10), deferoxamine therapy transitioned to standard of care therapy over the course of a decade. The cardioprotection of deferoxamine therapy was incontrovertible, however, success was incomplete. While life expectancy had improved, the median was only 20 years of age, with cardiac related deaths dominating (11). By the early 2000’s, the median age of death had increased to 30 years of age(12), and a cohort effect was noted with later cohorts having improved survival(13). Key predictors of survival mostly suggested chelation compliance as survival-limiting. Deferoxamine therapy was painful and the long, subcutaneous infusions were life-disruptive. Two widely cited studies implicated mean serum ferritin (14) and other markers of iron balance as key predictors for mortality (15), which was intuitive. This led to routine surveillance of liver iron by biopsy as a surrogate for cardiac risk. However, while a high liver iron continues to predict increased cardiac risk, many patients were still dying despite apparently adequate control of somatic iron stores. A subsequent single center study found that the number of deferoxamine infusions per week was the sole predictor of survival—independent of liver iron concentration (16). This study predated our understanding that the heart was uniquely vulnerable to labile plasma iron, which circulates whenever iron chelation is not present. Patients having two or more unprotected days per week were dying from heart disease regardless of their measured liver iron concentration.
MYOCARDITIS
The cardiac decompensation of patients with apparently excellent somatic iron control was puzzling leading to some speculation that these patients were dying from myocarditis (17). Unfortunately, myocarditis and iron cardiomyopathy can be phenotypically indistinguishable at times. Furthermore, biopsy-proven myocarditis was documented in a subset of thalassemia patients presenting with heart failure, and patients were enriched for certain HLA linkages, making immune-mediated cardiac destruction more plausible (18). However, only one center published this association, raising questions whether there might be regional genetic predisposition. Furthermore, myocarditis subsequently disappeared as a reported comorbidity as iron chelation and monitoring improved. Thus, it suggests that cardiac iron overload was at least a powerful risk factor, if not the primary causal mediator of these presumed cases of myocarditis, similar to its apparent role in pericarditis.
SUDEN CARDIAC DEATH
A similar argument can be made for the occurrence of sudden death in thalassemia patients. Registry studies implicated arrhythmias as the cause of death in up to 6% of patients(13). In one longitudinal cohort from Naples, 14/51 thalassemia major patients reportedly died from sudden cardiac death over a 21-year follow-up interval (19). The likely arrhythmia mechanism common to these deaths was Torsades De Pointes. which is a re-entrant ventricular tachycardia caused by dyssynchronous ventricular repolarization. Torsades De Pointes is inefficient in pumping blood and can rapidly degenerate into ventricular fibrillation. The patients who developed sudden cardiac death had significantly worse iron overload as well as greater QT dispersion. Although cardiac T2* data were not available in this cohort, it is likely that myocardial iron overload was the proximate cause of the repolarization abnormalities. Cardiac iron deposition has been demonstrated to particularly target iron channels modulating repolarization (20, 21), leading to QT prolongation(22) and dispersion(19), both risk factors for sudden cardiac death (19). Fortunately, with improved access to iron chelation and cardiac monitoring, the incidence of sudden cardiac death in thalassemia has markedly decreased.
ROLE OF MRI IN TAILORED THERAPY
The persistent mortality, despite access to iron chelation, reflected the intrinsic difficulties of maintaining compliance with deferoxamine therapy with the narrow window between cardiac symptoms and cardiac mortality. It was challenging to motivate patients or providers to intensify iron chelation therapies in asymptomatic patients with normal electrocardiograms and echocardiograms (ECG). Even when subclinical dysfunction was noted by radionuclide studies, it was challenging to maintain intensive chelation sufficiently long enough to guarantee survival(23). Introduction of the cardiac T2* technique in 2001(24), and its subsequent clinical validation from registry data(25) forever altered thalassemia management. Suddenly, evidence of significant cardiac iron deposition (T2* < 10) became sufficient evidence to escalate intensive iron chelation(26). By intervening early, cardiac decompensation could entirely be avoided, leading to a dramatic improvement in cardiac survival(27). Magnetic resonance imaging (MRI) also provided powerful insights into the underlying pathophysiology of cardiac iron loading, including the connection(28) and the disconnection (29) between liver and cardiac iron loading under different clinical situations, the importance of labile plasma iron control(30), and the painfully slow rate of cardiac iron clearance(29, 31, 32). For the first time, clinicians could modify their iron chelation strategies based on reliable, early markers of cardiac risk.
ORAL IRON CHELATION
MRI would not have had such a powerful impact on survival in thalassemia if it were not temporally paired with greater access to oral iron chelation. Deferiprone was developed in the laboratory of Dr. Victor Hoffbrand and gained increasing traction in the UK and Italy during the 1990’s. In 2001, Anderson et al. reported a striking difference in cardiac iron deposition in patients treated with deferiprone in comparison with case-matched patients treated with deferoxamine(33). Registry data from Italy also suggested a powerful survival benefit of deferiprone therapy(34). The cardiac efficacy of deferiprone, in monotherapy(35) and in combination(36), was subsequently verified in randomized controlled trials. The safety and efficacy of combined deferiprone and deferoxamine therapy was also verified in preclinical cardiac iron loading(26) as well as overt heart failure(32, 37), leading to incorporation of combined chelation therapy in the American Heart Association Consensus Guidelines for Iron Cardiomyopathy(38).
However, deferiprone was not a magic bullet. Deferiprone at 75 mg/g places fewer than 50% of patients into negative iron balance(39). Furthermore, agranulocytosis occurs in some patients leading to drug discontinuation. Fortunately, the oral iron chelator deferasirox entered clinical trials in 2002 (40) and was approved for clinical use in November, 2005. Deferasirox has completely replaced deferoxamine in younger patients and continues to be the most frequently prescribed iron chelator in the world. Approval of the tablet formulation in 2015 improved its tolerability and compliance. Although deferasirox does not remove iron as quickly from the heart as deferiprone because of its high plasma binding(35, 41), its longer plasma half-life offers excellent cardiac prophylaxis. Renal dysfunction represents the most common dose-limiting complication including acute renal failure, Fanconi’s syndrome, glomerular damage, and proteinuria (https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/206910s000lbl.pdf).
The existence of three US Food and Drug Administration-approved chelators has created multiple options(37, 42, 43) and dosing strategies for combination therapy(44, 45). Clinicians can now tailor-make their chelation strategies to the patient’s organ iron loading, organ toxicity, and ability to tolerate certain therapies. The era of personalized iron chelation has arrived and is drastically modifying the risk of pericarditis, myocarditis, iron cardiomyopathy, and sudden death in thalassemia patients. Although compliance with iron chelation therapy remains an ongoing challenge, longitudinal studies demonstrate a profound era-effect, with markedly improved cardiac and all-cause survival in thalassemia patients born with access to modern chelation and monitoring practices (46).
POST IRON CARDIOMYOPATHY ERA
Nobody escapes this world alive. While premature cardiac death has plummeted with improvements in iron chelation and MRI surveillance, death from other complications of thalassemia have increased (47). Even when not directly fatal, non-cardiac complications may manifest cardiac consequences. The most common and serious complication is type 2 diabetes mellitus, which typically presents in middle age and increases in prevalence every decade thereafter (48). Diabetes has many important cardiovascular effects, including vascular oxidative stress, adverse vascular remodeling, cardiac hypertrophy, and microvasculature destruction. The sum total of these contributions is accelerated vascular aging.
Other endocrinopathies common in thalassemia can also impact the heart. Hypothyroidism (49), hypoparathyroidism (50), low growth hormone (51), and low testosterone (52) can reinforce ventricular dysfunction of any mechanisms. Vitamin (B1, D, folate), mineral (selenium, zinc), and metabolic co-factor (carnitine) deficiencies are common in thalassemia and can cause or worsen ventricular dysfunction and should be investigated in any patient with unexplained ventricular dysfunction (53–55). Hepatitis C is common in many older thalassemia patients transfused prior to complete validation of the blood supply. Chronic hepatitis C can produce a smoldering low-level myocarditis (56) associated with myocardial scarring on MRI (57, 58) and rhythm disturbances, as well as predisposition to diabetes (59). Thus, complete cardiac protection includes many organ systems outside the purview of the cardiologist, requiring a multidisciplinary approach.
PULMONARY HYPERTENSION
Pulmonary hypertension refers to increased pressure in the vasculature supplying the lungs. Criteria for diagnosis of pulmonary hypertension were recently revised downward, with a systolic pulmonary artery pressure > 20 torr on cardiac catheterization representing early disease(60). Screening is usually performed by ECG, based upon the velocity of the tricuspid regurgitation (TR) (leakage) jet. In practice, a TR velocity below 2.5 m/s is normal, a velocity greater than 3.0 m/s is abnormal, and 2.5–3.0 m/s is borderline and requires placement into a clinical context(61). Pulmonary hypertension is typically grouped according the WHO classification(62). Type 1 reflects a primary increase in pulmonary arterial tone. Type 2 results from increases in pulmonary artery pressure secondary to increases in left atrial pressure from left ventricular systolic or diastolic dysfunction. Type 3 pulmonary hypertension occurs from chronic pulmonary disease, Type 4 pulmonary hypertension occurs from chronic thromboembolic disease, and Type 5 is used to describe pulmonary hypertension from systemic diseases such as thalassemia with mixed pathophysiologies. Pulmonary hypertension may be thought of as the interaction between abnormal mechanical and chemical stressors (Figure 1) (63).
FIGURE 1.
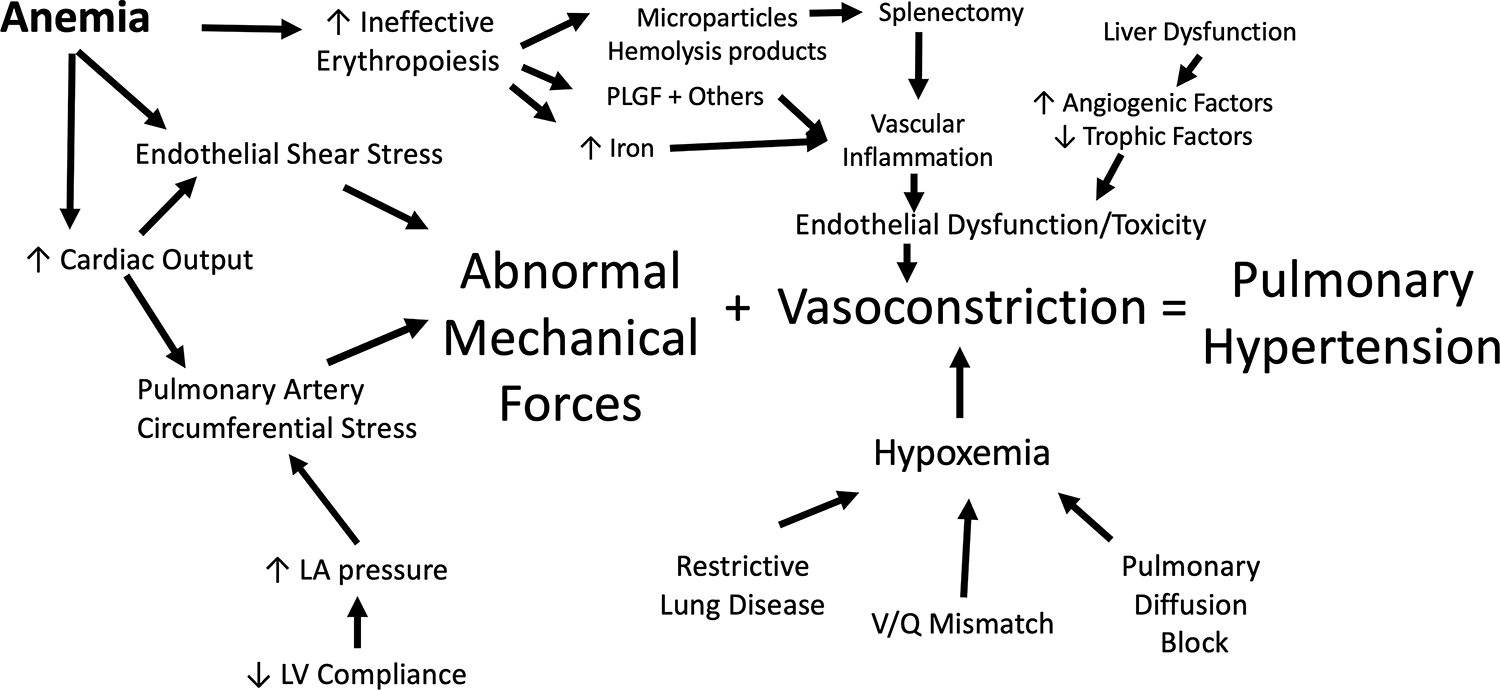
Schematic representing the multiple physical and chemical contributors to pulmonary hypertension in thalassemia as well as their proximate mediators. Abbreviations: LA, left atrial; LV, left ventricular; PLGF, placentally derived growth factor.
The most important mechanical force is the high cardiac output resulting from chronic anemia. This increases the volumetric flow rate and the distending pressure to the lung vasculature while decreasing the blood viscosity and resulting shear stress on the vascular endothelia(64). Back pressure to the pulmonary veins increases and is worsened by any concomitant ventricular systolic or diastolic dysfunction (Type 2 pulmonary hypertension). The chemical stressors on the pulmonary vasculature primarily result from accelerated red cell precursor production and destruction (ineffective erythropoiesis)(65), as well as activation of the coagulation cascade (66). Developing erythroblasts produce placentally derived growth factor (PLGF) and other powerful signaling molecules(67), leading to increased endothelin 1 signaling(68). Primary overexpression of PLGF in mice results in pulmonary hypertension(69). Premature apoptosis of red cells leads to increased circulating hemoglobin, heme, and heme-containing microparticles (70, 71) that are powerful oxidants and scavengers of the vascular vasodilator nitric oxide. Oxidative stress leads to uncoupled nitric oxide synthase activity, promoting intimal hyperplasia, increased vascular tone, and unfavorable remodeling. Hemolysis also releases arginase which catabolizes the key substrate for nitric oxide production, further favoring uncoupled nitric oxide synthase activity and fibrosis(72). The spleen and liver are the primary protection against circulating vasoactive substances through phagocytosis and detoxification. Not surprisingly, splenectomy is a powerful risk factor for pulmonary hypertension in thalassemia (73). While the use of splenectomy is decreasing, particularly in thalassemia major, some continue to advocate its use to lower transfusion requirements. End stage liver disease, whether from iron overload, hepatitis, or some combination, is also a powerful predictor of pulmonary hypertension.
Since erythropoietic drive and splenectomy are such powerful risk factors, it is not surprising that patients with thalassemia intermedia are at much higher risk of pulmonary hypertension than thalassemia major (74, 75). In contrast, we found no patient with documented pulmonary hypertension in a cohort of 60 well-transfused, adult, thalassemia major patients, although seven had borderline TR velocities and disrupted nitric oxide metabolism (76). The critical role of ineffective erythropoiesis in pulmonary hypertension is particularly important(73) when considering the risks and benefits of drugs that impact red cell maturation. The activin-trap inhibitor, luspatercept, facilitates stability of endogenously produced red blood cells in patients with thalassemia major, leading to significant reductions of transfusion volume in some patients(77). However, subsequent lowering of transfusion volume has been associated with extramedullary erythropoiesis in some(78), suggesting released suppression of ineffective erythropoiesis.
Borderline TR velocities reflect a disease spectrum that can often be managed by modifications in the hematological management, as well as correction of secondary contributors such as nighttime hypoxia or chronic thromboembolism. However, catheter-documented pulmonary hypertension is a remarkably lethal disease(79, 80). Although some patients respond well to monotherapy with PGE5 inhibitors(81, 82), thalassemia patients with documented pulmonary hypertension require urgent referral to a pulmonary hypertension specialist for tailored, multidrug therapies and careful monitoring(61).
ATRIAL ARRHYTHMIAS
The left and right atrium are stressed in thalassemia patients for multiple reasons. Volume overload from anemia and pressure overload from diastolic dysfunction trigger progressive atrial dilation (83–86). Iron overload and myocarditis (which can be clinically silent) create scars that bar normal electrical conduction and can poison atrial systolic and diastolic function (87, 88). This sets up a substrate for re-entrant atrial arrhythmias, including intra-atrial reentrant tachycardia, atrial fibrillation, and chaotic atrial rhythm, which are increasingly common in adults with thalassemia (89). These rhythms may present with tachycardia, palpitations, or exercise intolerance. Loss of atrio-ventricular synchrony reduces cardiac output, particularly in the presence of diastolic dysfunction. The rhythms are often paroxysmal in nature at first but will tend to persist over time. The greatest danger of these rhythms is embolic stroke. The loss of coordinated atrial activity allows thrombus to form within the atrium, often in the atrial appendages. Left atrial thrombi have the potential to embolize to any systemic organ, with the brain having the greatest consequences.
Atrial arrhythmias can also be clinically silent, especially when nonsustained. Thus, annual screening with ambulatory ECG is essential for adults over the age of 35 or 40 years. These devices have become much more convenient to wear and interpret than traditional “Holter” monitors. Self-monitoring is also now possible with many sophisticated wearable or easily portable devices. Nonsustained arrythmias will typically be treated with anticoagulants to prevent stroke risk. More sustained rhythms require the care of an electrophysiologist, either with antiarrhythmic medication or radiofrequency ablation to interrupt the re-entrant rhythm circuit within the atrium.
While arrhythmias often occur in iron cardiomyopathy(38), as well as some patients with asymptomatic decreases in cardiac T2* (25), the bulk of adult-onset arrhythmias occur without detectable iron in the ventricle (normal cardiac T2*). However, the atria are too thin for accurate detection of iron overload, so it is possible that atrial iron deposition could be reinforcing abnormal rhythm generation. Thus, strict adherence to iron chelation is always advisable when atrial arrythmias are present even if a causal role cannot be established.
HEART FAILURE WITH PRESERVED EJECTION FRACTION
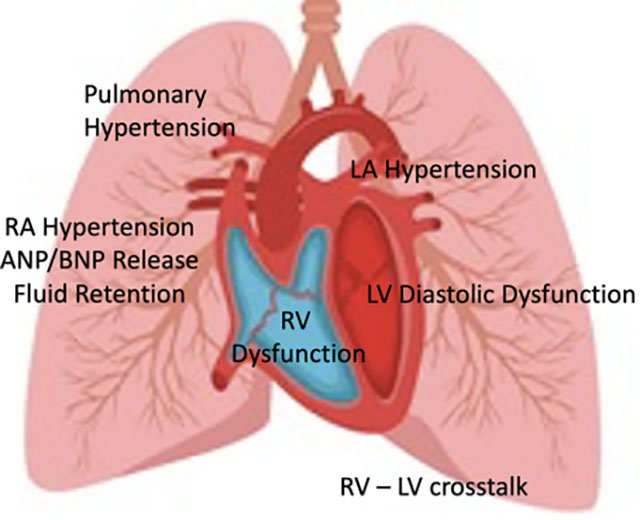
Heart failure is defined as the inability of the heart to pump sufficient blood to maintain metabolic demands. Classically, this has occurred when the heart muscle has been damaged and cannot squeeze vigorously; this is known as heart failure with reduced ejection fraction (HFrEF). Iron cardiomyopathy, myocarditis, and nutritional deficiencies typically present as HFrEF. Heart failure with preserved ejection fraction (HFpEF) occurs when the heart becomes too stiff to fill easily, leading to back pressure in the lungs (pulmonary edema), pulmonary hypertension, and right heart failure (Figure 2). While HFrEF is still more common than HFpEF in thalassemia, the prevalence of HFpEF has been steadily rising as the life expectancy of thalassemia improves and HFrEF is prevented (90). While there are a myriad of contributing factors to HFpEF, the final common pathway is that heart becomes preload limited. Essentially, that means that heart filling pressures become too high when cardiac demand is increased, limiting heart output and oxygen delivery. This can occur because the heart is too small or too stiff, leading to a small stroke volume despite normal systolic function. Sometimes the heart filling pressures will be acceptable under resting conditions but will increase dramatically under exercise conditions because normal compensatory improvements in diastolic ventricular function fail to occur. The rise in left sided filling pressures present as breathlessness with exertion, and the rise in right sided filling pressures manifest as abdominal discomfort and peripheral edema. On ECG, there is typically atrial enlargement, abnormal diastolic function, and mild elevations in pulmonary artery pressure; systolic ventricular function is typically normal.
FIGURE 2.
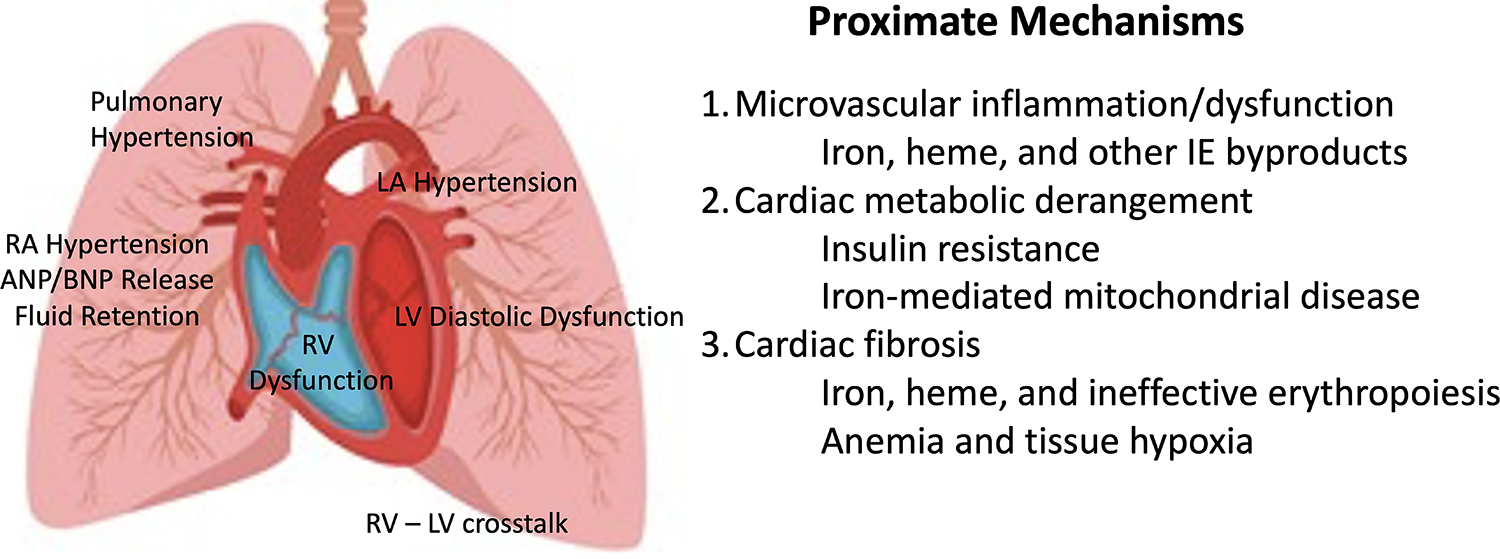
(Left) Pictorial representation of HFpEF physiology. Left ventricular diastolic dysfunction causes high left ventricular (LV) filling pressures, left atrial (LA) hypertension, pulmonary hypertension, poor right ventricular (RV) filling through RV–LV cross-talk, poor RV systolic function because of increased afterload, leading to increased right atrial (RA) pressures and naturetic protein release (ANP and BNP). (Right) Suspected mechanisms for the left ventricular changes in the thalassemia HFpEF phenotype. Many circulating oxidative and inflammatory mediators damage the microvasculature, derange myocardial energetics, and promote interstitial fibrosis. Abbreviations: ANP, Atrial natriuretic peptide; BMP, B-type natriuretic peptide; HFpEF, heart failure with preserved ejection fraction.
The molecular basis for HFpEF is poorly understood. Cardiac microvascular destruction and dysfunction are common, increasing the diffusing distance for oxygen to exercising myocytes (91). Angina can occur, but myocyte distribution is patchy and diffuse rather than residing in a typical myocardial arterial territory. The cause of the arterial destruction is myriad and includes hypoxia from chronic anemia; oxidative stress of vascular endothelia from circulating free iron, heme, and microparticles; circulating inflammatory mediators; and imbalances of the coagulation system. As previously discussed, poor glucose control is a powerful vascular stressor in any individual and a powerful risk factor for HFpEF in the general population. Insulin resistance triggers metabolic changes in the heart’s utilization of fat and glucose as well as impairing the heart’s vascular reactivity.
HFpEF remains remarkably difficult to treat (92). Aldosterone, angiotension, angiotensin-neprilysin, sodium-glucose cotransporter 2, and angiotensin converting enzyme inhibition have all been tried with mixed results. Diuretics may provide some symptomatic relief but are not disease modifying. Current guidelines support continued hypertension control and angiotensin 2 blockers, with sodium-glucose cotransporter 2 and angiotensin-neprilysin inhibitors as 2A and 2B indications (93). Antifibrotic agents are under clinical trials (94). But prevention remains the mainstay for HFpEF, as discussed in the following section.
HEART HEALTH IN THE POST IRON CARDIOMYOPATHY ERA
The victory against iron cardiomyopathy now allows thalassemia patients to have near normal life expectancy. But both ineffective erythropoiesis and iron overload cause vascular oxidative stress, effectively accelerating the aging process. While there is no magic bullet to arrest vascular oxidative stress, there are well-known lifestyle modifications that improve vascular health. The most important of these is regular exercise. While there are no controlled trials of exercise programs in thalassemia, the anecdotal evidence is powerful and the data from non-thalassemia patients is irrefutable. The mechanisms of exercise benefits on vascular health are complex and multifactorial, including improved insulin sensitivity, downregulation of sympathetic tone and renin–angiotensin–aldosterone activity, improved blood pressure, and better sleep quality. While the American Heart Association endorses minimum standards of 150 minutes per week of moderate to strenuous exercise (https://health.gov/our-work/nutrition-physical-activity/physical-activity-guidelines), only two in five Americans achieve this goal because of demands of work, lack of access to exercise facilities, and the seemingly unattainable nature of the goal. There are two keys to success: 1) recognition that every little bit helps—something is always better than nothing, and 2) the best exercise is the one that can be sustained, day in and day out. Humans are built to walk, and it can be done almost anywhere. But finding something enjoyable (or at least tolerable) and accessible is the key to sustainability.
Regular exercise must be complemented by a good diet and glucose control. Portion control is the simplest mechanism for improvement, which can be challenging in a country like the United States that supersizes everything. Eating breakfast at home and bringing one’s own lunch to work can be simple mechanisms to control both the quantity and quality of two thirds of daily meals. Limiting simple carbohydrates, one of the mainstays of processed foods, is important for stabilizing insulin sensitivity and beta cell function. Fruits and vegetables provide important antioxidants as well as diet diversity. Given the consumptive nature of chronic anemia, it is challenging to maintain adequate vitamin and mineral sufficiency from diet alone. Standard multivitamins are recommended, and supplements of vitamin C, zinc, selenium, and B-vitamins should be considered. For those patients with type 2 diabetes, or pre-diabetes, the care of an endocrinologist and affiliated dietary counselor is essential.
Thalassemia patients have three other unique vascular stressors that require careful long-term management: anemia, ineffective erythropoiesis, and iron overload. Hemoglobin levels and ineffective erythropoiesis are inversely related to one another. When transfusional intensity is insufficient, ineffective erythropoiesis promotes cardiac dilation and high output(95), vascular thrombosis(75), elastin degeneration(96), and pulmonary hypertension(75). However, current standards for transfusional intensity may change in the coming years because of agents that modify red cell maturation(77). It is absolutely essential that thalassemia patients be followed, at least annually, by hematologists who are aware of these subtleties and will monitor for pulmonary hypertension, extramedullary hematopoiesis, and other potential complications of inadequate suppression of ineffective erythropoiesis.
But the single-most important cardiovascular protective measure that thalassemia patients must maintain is meticulous attention to their iron chelation. Since most patients have fully saturated transferrin, every hour that iron chelation is not present results in circulating, oxidatively active iron species that damage vascular endothelia(97–99). Even if exposure to these labile iron species is insufficient to load the heart on MRI, vascular aging is accelerated through oxidative stress. Fortunately, the most common iron chelator, deferasirox, has a long half-life, providing round-the-clock coverage for most patients, improving endothelial function(100). However, bioavailability is lower in some patients(101) for whom divided dosing is well tolerated and provides more sustained cardiovascular coverage(102). Despite their shorter half-lives, deferiprone and deferoxamine also improve nitric oxide synthase function and endothelial reactivity(36, 103, 104), thus the optimal chelation strategies for endothelial protection are not fully characterized.
CONCLUSION
The spectrum of cardiovascular disease in thalassemia have morphed dramatically in the past five decades, from a universally fatal disease in childhood, to premature cardiovascular death, to a nearly normal lifespan. Yesterday’s successes have brought on new challenges. Many older thalassemia patients have multiorgan damage from partially treated iron overload that interacts unfavorably with the normal vascular aging process. Even in young and optimally-treated patients, the underlying thalassemia physiology and iron overload are vascular stressors that must be carefully managed by experienced hematologists. Fortunately, much of the knowledge learned from managing vascular aging in non-thalassemia patients comes to bear. Thus, the modern thalassemia patient has the tools and knowledge to maintain their health, albeit with sustained effort and vigilance.
ACKNOWLEDGMENTS
This work was supported by grants from the National Heart Lung and Blood Institute (1 RO1 HL075592-01A1), the National Institute of Diabetes, Digestive, and Kidney Diseases (1 R01DK097115-01A1), the Center for Disease Control (1 U01 DD000309-1), and the National Center for Research Resources (UL1 TR001855-02). Portions of this work were also supported by the Saban Research Institute and by Research Support in Kind from Philips Medical Systems.
Footnotes
COMPETING INTERESTS
Dr. John Wood is a consultant for Agios, Celgene/BMS, Hillhurst, and Pharmacosmos. He has also served as a scientific advisor to Bluebirdbio, Chiesi, Regeneron, and Vifor. He has research support from Philips Medical Systems, Additional Ventures, and the National Institutes of Health.
REFERENCES
- 1.Nittis S Erythroblastic anemias of childhood with special reference to the mechanism of the production of the bone changes. Bulletin NY Med Coll. 1950;13:78–90. [PubMed] [Google Scholar]
- 2.Engle MA. Cardiac involvement in Cooley’s anemia. Ann N Y Acad Sci. 1964;119:694–702. [DOI] [PubMed] [Google Scholar]
- 3.Engle MA, Erlandson M, Smith CH. Late Cardiac Complications of Chronic, Severe, Refractory Anemia with Hemochromatosis. Circulation. 1964;30:698–705. [DOI] [PubMed] [Google Scholar]
- 4.Wolman IJ. Transfusion Therapy in Cooley’s Anemia: Growth and Health as Related to Long-Range Hemoglobin Levels. A Progress Report. Ann N Y Acad Sci. 1964;119:736–47. [DOI] [PubMed] [Google Scholar]
- 5.Master J, Engle MA, Stern G, Smith CH. Cardiac complications of chronic, severe, refractory anemia with hemochromatosis. I. Acute pericarditis of unkown etiology. J Pediatr. 1961;58:455–63. [DOI] [PubMed] [Google Scholar]
- 6.Piomelli S, Karpatkin MH, Arzanian M, Zamani M, Becker MH, Geneiser N, et al. Hypertransfusion regimen in patients with Cooley’s anemia. Ann N Y Acad Sci. 1974;232(0):186–92. [DOI] [PubMed] [Google Scholar]
- 7.Engle MA, Erlandson M, Smith CH. Late cardiac complications of chronic, sever, refractory anemia. Circulation. 1964;30:698–705. [DOI] [PubMed] [Google Scholar]
- 8.Seshadri R, Colebatch JH, Gordon P, Ekert H. Long-term administration of desferrioxamine in thalassaemia major. Arch Dis Child. 1974;49(8):621–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marcus RE, Davies SC, Bantock HM, Underwood SR, Walton S, Huehns ER. Desferrioxamine to improve cardiac function in iron-overloaded patients with thalassemia major. Lancet. 1984;1(8373):392–3. [DOI] [PubMed] [Google Scholar]
- 10.Ehlers KH, Giardina PJ, Lesser ML, Engle MA, Hilgartner MW. Prolonged survival in patients with beta-thalassemia major treated with deferoxamine. J Pediatr. 1991;118(4 ( Pt 1)):540–5. [DOI] [PubMed] [Google Scholar]
- 11.Zurlo MG, De Stefano P, Borgna-Pignatti C, Di Palma A, Piga A, Melevendi C, et al. Survival and causes of death in thalassaemia major. Lancet. 1989;2(8653):27–30. [DOI] [PubMed] [Google Scholar]
- 12.Modell B, Khan M, Darlison M. Survival in beta-thalassaemia major in the UK: data from the UK Thalassaemia Register. Lancet. 2000;355(9220):2051–2. [DOI] [PubMed] [Google Scholar]
- 13.Borgna-Pignatti C, Rugolotto S, De Stefano P, Zhao H, Cappellini MD, Del Vecchio GC, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica. 2004;89(10):1187–93. [PubMed] [Google Scholar]
- 14.Olivieri NF, Brittenham GM. Iron-chelating therapy and the treatment of thalassemia. Blood. 1997;89(3):739–61. [PubMed] [Google Scholar]
- 15.Brittenham GM, Griffith PM, Nienhuis AW, McLaren CE, Young NS, Tucker EE, et al. Efficacy of deferoxamine in preventing complications of iron overload in patients with thalassemia major. New England Journal of Medicine. 1994;331(9):567–73. [DOI] [PubMed] [Google Scholar]
- 16.Gabutti V, Piga A. Results of long-term iron-chelating therapy. Acta Haematol. 1996;95(1):26–36. [DOI] [PubMed] [Google Scholar]
- 17.Kremastinos DT, Tiniakos G, Theodorakis GN, Katritsis DG, Toutouzas PK. Myocarditis in beta-thalassemia major. A cause of heart failure. Circulation. 1995;91(1):66–71. [DOI] [PubMed] [Google Scholar]
- 18.Kremastinos DT, Flevari P, Spyropoulou M, Vrettou H, Tsiapras D, Stavropoulos-Giokas CG. Association of heart failure in homozygous beta-thalassemia with the major histocompatibility complex. Circulation. 1999;100(20):2074–8. [DOI] [PubMed] [Google Scholar]
- 19.Russo V, Rago A, Pannone B, Papa AA, Di Meo F, Mayer MC, et al. Dispersion of repolarization and beta-thalassemia major: the prognostic role of QT and JT dispersion for identifying the high-risk patients for sudden death. Eur J Haematol. 2011;86(4):324–31. [DOI] [PubMed] [Google Scholar]
- 20.Kim E, Giri SN, Pessah IN. Iron(II) is a modulator of ryanodine-sensitive calcium channels of cardiac muscle sarcoplasmic reticulum. Toxicol Appl Pharmacol. 1995;130(1):57–66. [DOI] [PubMed] [Google Scholar]
- 21.Kuryshev YA, Brittenham GM, Fujioka H, Kannan P, Shieh CC, Cohen SA, et al. Decreased sodium and increased transient outward potassium currents in iron-loaded cardiac myocytes. Implications for the arrhythmogenesis of human siderotic heart disease. Circulation. 1999;100(6):675–83. [DOI] [PubMed] [Google Scholar]
- 22.Detterich J, Noetzli L, Dorey F, Bar-Cohen Y, Harmatz P, Coates T, et al. Electrocardiographic consequences of cardiac iron overload in thalassemia major. Am J Hematol. 2012;87(2):139–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis BA, Porter JB. Long-term outcome of continuous 24-hour deferoxamine infusion via indwelling intravenous catheters in high-risk beta-thalassemia. Blood. 2000;95(4):1229–36. [PubMed] [Google Scholar]
- 24.Anderson LJ, Holden S, Davis B, Prescott E, Charrier CC, Bunce NH, et al. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. European Heart Journal. 2001;22(23):2171–9. [DOI] [PubMed] [Google Scholar]
- 25.Kirk P, Roughton M, Porter JB, Walker J, Tanner M, Patel J, et al. Cardiac T2* magnetic resonance for prediction of cardiac complications in thalassemia major. JCMR. 2009;11(6):O2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Origa R, Bina P, Agus A, Crobu G, Defraia E, Dessi C, et al. Combined therapy with deferiprone and desferrioxamine in thalassemia major. Haematologica. 2005;90(10):1309–14. [PubMed] [Google Scholar]
- 27.Modell B, Khan M, Darlison M, Westwood MA, Ingram D, Pennell DJ. Improved survival of thalassaemia major in the UK and relation to T2* cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2008;10(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wood JC, Kang BP, Thompson A, Giardina P, Harmatz P, Glynos T, et al. The effect of deferasirox on cardiac iron in thalassemia major: impact of total body iron stores. Blood. 2010;116(4):537–43. [DOI] [PubMed] [Google Scholar]
- 29.Noetzli LJ, Carson SM, Nord AS, Coates TD, Wood JC. Longitudinal analysis of heart and liver iron in thalassemia major. Blood. 2008;112(7):2973–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wood JC, Glynos T, Thompson A, Giardina P, Harmatz P, Kang BP, et al. Relationship between labile plasma iron, liver iron concentration and cardiac response in a deferasirox monotherapy trial. Haematologica. 2011;96(7):1055–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson LJ, Westwood MA, Holden S, Davis B, Prescott E, Wonke B, et al. Myocardial iron clearance during reversal of siderotic cardiomyopathy with intravenous desferrioxamine: a prospective study using T2* cardiovascular magnetic resonance. British Journal of Haematology. 2004;127(3):348–55. [DOI] [PubMed] [Google Scholar]
- 32.Porter JB, Wood J, Olivieri N, Vichinsky EP, Taher A, Neufeld E, et al. Treatment of heart failure in adults with thalassemia major: response in patients randomised to deferoxamine with or without deferiprone. J Cardiovasc Magn Reson. 2013;15(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anderson LJ, Wonke B, Prescott E, Holden S, Walker JM, Pennell DJ. Comparison of effects of oral deferiprone and subcutaneous desferrioxamine on myocardial iron concentrations and ventricular function in beta-thalassaemia. Lancet. 2002;360(9332):516–20. [DOI] [PubMed] [Google Scholar]
- 34.Borgna-Pignatti C, Cappellini MD, De Stefano P, Del Vecchio GC, Forni GL, Gamberini MR, et al. Cardiac morbidity and mortality in deferoxamine- or deferiprone-treated patients with thalassemia major. Blood. 2006;107(9):3733–7. [DOI] [PubMed] [Google Scholar]
- 35.Pennell DJ, Berdoukas V, Karagiorga M, Ladis V, Piga A, Aessopos A, et al. Randomized controlled trial of deferiprone or deferoxamine in beta-thalassemia major patients with asymptomatic myocardial siderosis. Blood. 2006;107(9):3738–44. [DOI] [PubMed] [Google Scholar]
- 36.Tanner MA, Galanello R, Dessi C, Smith GC, Westwood MA, Agus A, et al. A randomized, placebo-controlled, double-blind trial of the effect of combined therapy with deferoxamine and deferiprone on myocardial iron in thalassemia major using cardiovascular magnetic resonance. Circulation. 2007;115(14):1876–84. [DOI] [PubMed] [Google Scholar]
- 37.Tanner MA, Galanello R, Dessi C, Smith GC, Westwood MA, Agus A, et al. Combined chelation therapy in thalassemia major for the treatment of severe myocardial siderosis with left ventricular dysfunction. J Cardiovasc Magn Reson. 2008;10(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pennell DJ, Udelson JE, Arai AE, Bozkurt B, Cohen AR, Galanello R, et al. Cardiovascular function and treatment in beta-thalassemia major: a consensus statement from the American Heart Association. Circulation. 2013;128(3):281–308. [DOI] [PubMed] [Google Scholar]
- 39.Taher A, Sheikh-Taha M, Koussa S, Inati A, Neeman R, Mourad F. Comparison between deferoxamine and deferiprone (L1) in iron-loaded thalassemia patients. Eur J Haematol. 2001;67(1):30–4. [DOI] [PubMed] [Google Scholar]
- 40.Nisbet-Brown E, Olivieri NF, Giardina PJ, Grady RW, Neufeld EJ, Sechaud R, et al. Effectiveness and safety of ICL670 in iron-loaded patients with thalassaemia: a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet. 2003;361(9369):1597–602. [DOI] [PubMed] [Google Scholar]
- 41.Pennell DJ, Porter JB, Cappellini MD, El-Beshlawy A, Chan LL, Aydinok Y, et al. Efficacy of deferasirox in reducing and preventing cardiac iron overload in beta-thalassemia. Blood. 2010;115(12):2364–71. [DOI] [PubMed] [Google Scholar]
- 42.Lal A, Porter J, Sweeters N, Ng V, Evans P, Neumayr L, et al. Combined chelation therapy with deferasirox and deferoxamine in thalassemia. Blood Cells Mol Dis. 2013;50(2):99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Totadri S, Bansal D, Bhatia P, Attri SV, Trehan A, Marwaha RK. The deferiprone and deferasirox combination is efficacious in iron overloaded patients with beta-thalassemia major: A prospective, single center, open-label study. Pediatr Blood Cancer. 2015;62(9):1592–6. [DOI] [PubMed] [Google Scholar]
- 44.Link G, Konijn AM, Breuer W, Cabantchik ZI, Hershko C. Exploring the “iron shuttle” hypothesis in chelation therapy: effects of combined deferoxamine and deferiprone treatment in hypertransfused rats with labeled iron stores and in iron-loaded rat heart cells in culture. J Lab Clin Med. 2001;138(2):130–8. [DOI] [PubMed] [Google Scholar]
- 45.Vlachodimitropoulou Koumoutsea E, Garbowski M, Porter J. Synergistic intracellular iron chelation combinations: mechanisms and conditions for optimizing iron mobilization. Br J Haematol. 2015;170(6):874–83. [DOI] [PubMed] [Google Scholar]
- 46.Forni GL, Gianesin B, Musallam KM, Longo F, Rosso R, Lisi R, et al. Overall and complication-free survival in a large cohort of patients with beta-thalassemia major followed over 50 years. Am J Hematol. 2023;98(3):381–7. [DOI] [PubMed] [Google Scholar]
- 47.Voskaridou E, Kattamis A, Fragodimitri C, Kourakli A, Chalkia P, Diamantidis M, et al. National registry of hemoglobinopathies in Greece: updated demographics, current trends in affected births, and causes of mortality. Ann Hematol. 2019;98(1):55–66. [DOI] [PubMed] [Google Scholar]
- 48.Gamberini MR, Fortini M, De Sanctis V, Gilli G, Testa MR. Diabetes mellitus and impaired glucose tolerance in thalassaemia major: incidence, prevalence, risk factors and survival in patients followed in the Ferrara Center. Pediatr Endocrinol Rev. 2004;2 Suppl 2:285–91. [PubMed] [Google Scholar]
- 49.De Sanctis V, De Sanctis E, Ricchieri P, Gubellini E, Gilli G, Gamberini MR. Mild subclinical hypothyroidism in thalassaemia major: prevalence, multigated radionuclide test, clinical and laboratory long-term follow-up study. Pediatr Endocrinol Rev. 2008;6 Suppl 1:174–80. [PubMed] [Google Scholar]
- 50.Tsironi M, Korovesis K, Farmakis D, Deftereos S, Aessopos A. Hypocalcemic heart failure in thalassemic patients. Int J Hematol. 2006;83(4):314–7. [DOI] [PubMed] [Google Scholar]
- 51.Erfurth EM, Holmer H, Nilsson PG, Kornhall B. Is growth hormone deficiency contributing to heart failure in patients with beta-thalassemia major? Eur J Endocrinol. 2004;151(2):161–6. [DOI] [PubMed] [Google Scholar]
- 52.Tsironi M, Deftereos S, Andriopoulos P, Farmakis D, Meletis J, Aessopos A. Reversal of heart failure in thalassemia major by combined chelation therapy: a case report. Eur J Haematol. 2005;74(1):84–5. [DOI] [PubMed] [Google Scholar]
- 53.Claster S, Wood JC, Noetzli L, Carson SM, Hofstra TC, Khanna R, et al. Nutritional deficiencies in iron overloaded patients with hemoglobinopathies. Am J Hematol. 2009;84(6):344–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wood JC, Claster S, Carson S, Menteer JD, Hofstra T, Khanna R, et al. Vitamin D deficiency, cardiac iron and cardiac function in thalassaemia major. Br J Haematol. 2008;141(6):891–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Witte KK, Clark AL, Cleland JG. Chronic heart failure and micronutrients. J Am Coll Cardiol. 2001;37(7):1765–74. [DOI] [PubMed] [Google Scholar]
- 56.Matsumori A, Shimada T, Chapman NM, Tracy SM, Mason JW. Myocarditis and heart failure associated with hepatitis C virus infection. J Card Fail. 2006;12(4):293–8. [DOI] [PubMed] [Google Scholar]
- 57.Pepe A, Positano V, Capra M, Maggio A, Pinto CL, Spasiano A, et al. Myocardial scarring by delayed enhancement cardiovascular magnetic resonance in thalassaemia major. Heart. 2009;95(20):1688–93. [DOI] [PubMed] [Google Scholar]
- 58.Pepe A, Meloni A, Borsellino Z, Cuccia L, Borgna-Pignatti C, Maggio A, et al. Myocardial fibrosis by late gadolinium enhancement cardiac magnetic resonance and hepatitis C virus infection in thalassemia major patients. J Cardiovasc Med (Hagerstown). 2015;16(10):689–95. [DOI] [PubMed] [Google Scholar]
- 59.Sanctis VD, Soliman A, Yassin M. Iron Overload and Glucose Metabolism in Subjects with beta-thalassaemia Major : An Overview. Curr Diabetes Rev. 2013;9(4):332–41. [DOI] [PubMed] [Google Scholar]
- 60.Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wood JC. Pulmonary hypertension in thalassemia: a call to action. Blood. 2022;139(13):1937–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ryan JJ, Thenappan T, Luo N, Ha T, Patel AR, Rich S, et al. The WHO classification of pulmonary hypertension: A case-based imaging compendium. Pulm Circ. 2012;2(1):107–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wood JC. Cardiac complications in thalassemia major. Hemoglobin. 2009;33 Suppl 1:S81–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vaclavu L, Baldew ZAV, Gevers S, Mutsaerts H, Fijnvandraat K, Cnossen MH, et al. Intracranial 4D flow magnetic resonance imaging reveals altered haemodynamics in sickle cell disease. Br J Haematol. 2018;180(3):432–42. [DOI] [PubMed] [Google Scholar]
- 65.Morris CR, Kim HY, Trachtenberg F, Wood J, Quinn CT, Sweeters N, et al. Risk factors and mortality associated with an elevated tricuspid regurgitant jet velocity measured by Doppler-echocardiography in thalassemia: a Thalassemia Clinical Research Network report. Blood. 2011;118(14):3794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Singer ST, Kuypers FA, Styles L, Vichinsky EP, Foote D, Rosenfeld H. Pulmonary hypertension in thalassemia: association with platelet activation and hypercoagulable state. Am J Hematol. 2006;81(9):670–5. [DOI] [PubMed] [Google Scholar]
- 67.Perelman N, Selvaraj SK, Batra S, Luck LR, Erdreich-Epstein A, Coates TD, et al. Placenta growth factor activates monocytes and correlates with sickle cell disease severity. Blood. 2003;102(4):1506–14. [DOI] [PubMed] [Google Scholar]
- 68.Patel N, Gonsalves CS, Malik P, Kalra VK. Placenta growth factor augments endothelin-1 and endothelin-B receptor expression via hypoxia-inducible factor-1 alpha. Blood. 2008;112(3):856–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sundaram N, Tailor A, Mendelsohn L, Wansapura J, Wang X, Higashimoto T, et al. High levels of placenta growth factor in sickle cell disease promote pulmonary hypertension. Blood. 2010;116(1):109–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Westerman M, Porter JB. Red blood cell-derived microparticles: An overview. Blood Cells Mol Dis. 2016;59:134–9. [DOI] [PubMed] [Google Scholar]
- 71.Kheansaard W, Phongpao K, Paiboonsukwong K, Pattanapanyasat K, Chaichompoo P, Svasti S. Microparticles from beta-thalassaemia/HbE patients induce endothelial cell dysfunction. Sci Rep. 2018;8(1):13033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morris CR, Vichinsky EP. Pulmonary hypertension in thalassemia. Ann N Y Acad Sci. 2010;1202:205–13. [DOI] [PubMed] [Google Scholar]
- 73.Karimi M, Musallam KM, Cappellini MD, Daar S, El-Beshlawy A, Belhoul K, et al. Risk factors for pulmonary hypertension in patients with beta thalassemia intermedia. Eur J Intern Med. 2011;22(6):607–10. [DOI] [PubMed] [Google Scholar]
- 74.Aessopos A, Farmakis D. Pulmonary hypertension in beta-thalassemia. Ann N Y Acad Sci. 2005;1054:342–9. [DOI] [PubMed] [Google Scholar]
- 75.Musallam KM, Cappellini MD, Wood JC, Motta I, Graziadei G, Tamim H, et al. Elevated liver iron concentration is a marker of increased morbidity in patients with beta thalassemia intermedia. Haematologica. 2011;96(11):1605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meloni A, Detterich J, Pepe A, Harmatz P, Coates TD, Wood JC. Pulmonary hypertension in well-transfused thalassemia major patients. Blood Cells Mol Dis. 2015;54(2):189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cappellini MD, Viprakasit V, Taher AT, Georgiev P, Kuo KHM, Coates T, et al. A Phase 3 Trial of Luspatercept in Patients with Transfusion-Dependent beta-Thalassemia. N Engl J Med. 2020;382(13):1219–31. [DOI] [PubMed] [Google Scholar]
- 78.Alashkar F, Klump H, Lange CP, Proske P, Schussler M, Yamamoto R, et al. Luspatercept, a two-edged sword in beta-thalassemia-associated paravertebral extramedullary hematopoietic masses (EHMs). Eur J Haematol. 2022;109(6):664–71. [DOI] [PubMed] [Google Scholar]
- 79.Derchi G, Balocco M, Bina P, Caruso V, D’Ascola DG, Littera R, et al. Efficacy and safety of sildenafil for the treatment of severe pulmonary hypertension in patients with hemoglobinopathies: results from a long-term follow up. Haematologica. 2014;99(2):e17–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pinto VM, Musallam KM, Derchi G, Graziadei G, Giuditta M, Origa R, et al. Mortality in beta-thalassemia patients with confirmed pulmonary arterial hypertension on right heart catheterization. Blood. 2022;139(13):2080–3. [DOI] [PubMed] [Google Scholar]
- 81.Derchi G, Forni GL, Formisano F, Cappellini MD, Galanello R, D’Ascola G, et al. Efficacy and safety of sildenafil in the treatment of severe pulmonary hypertension in patients with hemoglobinopathies. Haematologica. 2005;90(4):452–8. [PubMed] [Google Scholar]
- 82.Morris CR, Kim HY, Wood JC, Porter JB, Klings ES, Trachtenberg FL, et al. Sildenafil therapy in thalassemia patients with doppler-defined risk for pulmonary hypertension. Haematologica. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Karamanou AG, Hamodraka ES, Vrakas SC, Paraskevaidis I, Lekakis I, Kremastinos DT. Assessment of left ventricular and atrial diastolic function using two-dimensional (2D) strain imaging in patients with beta-thalassemia major. Eur J Haematol. 2014;92(1):59–65. [DOI] [PubMed] [Google Scholar]
- 84.Kostopoulou AG, Tsiapras DP, Chaidaroglou AS, De Giannis DE, Farmakis D, Kremastinos DT. The pathophysiological relationship and clinical significance of left atrial function and left ventricular diastolic dysfunction in beta-thalassemia major. Am J Hematol. 2014;89(1):13–8. [DOI] [PubMed] [Google Scholar]
- 85.Derchi G, Bellone P, Forni GL, Lupi G, Jappelli S, Randazzo M, et al. Cardiac involvement in thalassaemia major: altered atrial natriuretic peptide levels in asymptomatic patients. Eur Heart J. 1992;13(10):1368–72. [DOI] [PubMed] [Google Scholar]
- 86.Spirito P, Lupi G, Melevendi C, Vecchio C. Restrictive diastolic abnormalities identified by Doppler echocardiography in patients with thalassemia major. Circulation. 1990;82(1):88–94. [DOI] [PubMed] [Google Scholar]
- 87.Li W, Coates T, Wood JC. Atrial dysfunction as a marker of iron cardiotoxicity in thalassemia major. Haematologica. 2008;93(2):311–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Monte I, Capodanno D, Nicolosi E, Licciardi S, Talini E, Di Bello V. Atrial and ventricular function in thalassemic patients with supra-ventricular arrhythmias. Heart Int. 2009;4(1):e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Malagu M, Marchini F, Fiorio A, Sirugo P, Clo S, Mari E, et al. Atrial Fibrillation in beta-Thalassemia: Overview of Mechanism, Significance and Clinical Management. Biology (Basel). 2022;11(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mancuso L, Vitrano A, Mancuso A, Sacco M, Ledda A, Maggio A. Left Ventricular Diastolic Dysfunction in beta-Thalassemia Major with Heart Failure. Hemoglobin. 2018;42(1):68–71. [DOI] [PubMed] [Google Scholar]
- 91.Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62(4):263–71. [DOI] [PubMed] [Google Scholar]
- 92.Omote K, Verbrugge FH, Borlaug BA. Heart Failure with Preserved Ejection Fraction: Mechanisms and Treatment Strategies. Annu Rev Med. 2022;73:321–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM, et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2022;79(17):1757–80. [DOI] [PubMed] [Google Scholar]
- 94.Lewis GA, Pearce K, Williams SG, Schelbert EB, Macnab A, Miller CA. The utility of cardiovascular imaging in heart failure with preserved ejection fraction: diagnosis, biological classification and risk stratification. Heart Fail Rev. 2021;26(3):661–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Aessopos A, Farmakis D, Karagiorga M, Voskaridou E, Loutradi A, Hatziliami A, et al. Cardiac involvement in thalassemia intermedia: a multicenter study. Blood. 2001;97(11):3411–6. [DOI] [PubMed] [Google Scholar]
- 96.Aessopos A, Farmakis D, Loukopoulos D. Elastic tissue abnormalities resembling pseudoxanthoma elasticum in beta thalassemia and the sickling syndromes. Blood. 2002;99(1):30–5. [DOI] [PubMed] [Google Scholar]
- 97.Kartikasari AE, Georgiou NA, Visseren FL, van Kats-Renaud H, van Asbeck BS, Marx JJ. Intracellular labile iron modulates adhesion of human monocytes to human endothelial cells. Arterioscler Thromb Vasc Biol. 2004;24(12):2257–62. [DOI] [PubMed] [Google Scholar]
- 98.Stoyanova E, Trudel M, Felfly H, Lemsaddek W, Garcia D, Cloutier G. Vascular endothelial dysfunction in beta-thalassemia occurs despite increased eNOS expression and preserved vascular smooth muscle cell reactivity to NO. PLoS One. 2012;7(6):e38089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cheung YF, Chan GC, Ha SY. Arterial stiffness and endothelial function in patients with beta-thalassemia major. Circulation. 2002;106(20):2561–6. [DOI] [PubMed] [Google Scholar]
- 100.Cheung YF, Chan GC, Ha SY. Effect of deferasirox (ICL670) on arterial function in patients with beta-thalassaemia major. Br J Haematol. 2008;141(5):728–33. [DOI] [PubMed] [Google Scholar]
- 101.Chirnomas D, Smith AL, Braunstein J, Finkelstein Y, Pereira L, Bergmann AK, et al. Deferasirox pharmacokinetics in patients with adequate versus inadequate response. Blood. 2009;114(19):4009–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pongtanakul B, Viprakasit V. Twice daily deferasirox significantly improves clinical efficacy in transfusion dependent thalassaemias who were inadequate responders to standard once daily dose. Blood Cells Mol Dis. 2013;51(2):96–7. [DOI] [PubMed] [Google Scholar]
- 103.Sriwantana T, Vivithanaporn P, Paiboonsukwong K, Rattanawonsakul K, Srihirun S, Sibmooh N. Deferiprone increases endothelial nitric oxide synthase phosphorylation and nitric oxide production. Can J Physiol Pharmacol. 2018;96(9):879–85. [DOI] [PubMed] [Google Scholar]
- 104.Duffy SJ, Biegelsen ES, Holbrook M, Russell JD, Gokce N, Keaney JF, Jr., et al. Iron chelation improves endothelial function in patients with coronary artery disease. Circulation. 2001;103(23):2799–804. [DOI] [PubMed] [Google Scholar]