

Abstract
Backgrounds & Aims:
Pre-cancerous metaplasia progression to dysplasia can increase the risk of gastric cancers. However, effective strategies to specifically target these pre-cancerous lesions are currently lacking. To address this, we aimed to identify key signaling pathways which are upregulated during metaplasia progression and critical for stem cell survival and function in dysplasia.
Methods:
To assess the response to chemotherapeutic drugs, we utilized metaplastic and dysplastic organoids derived from Mist1-Kras mice and twenty human pre-cancerous organoid lines established from gastric cancer patients. Phospho-antibody array analysis and scRNA-seq were performed to identify target cell populations and signaling pathways affected by Pyrvinium, a putative anti-cancer drug. Pyrvinium was administered to Mist1-Kras mice to evaluate drug effectiveness in vivo.
Results:
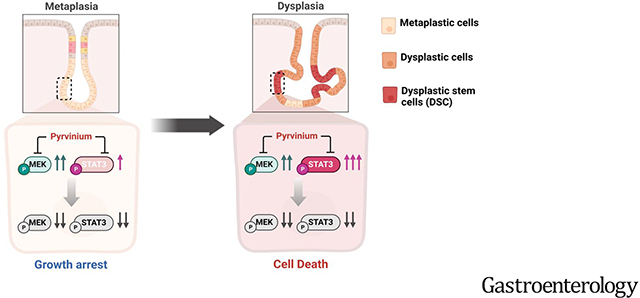
While pyrvinium treatment resulted in growth arrest in metaplastic organoids, it induced cell death in dysplastic organoids. Pyrvinium treatment significantly downregulated phosphorylation of ERK and STAT3 as well as STAT3-target genes. scRNA-seq data analyses revealed that Pyrvinium specifically targeted CD133+/CD166+ stem cell populations, as well as proliferating cells in dysplastic organoids. Pyrvinium inhibited metaplasia progression and facilitated the restoration of normal oxyntic glands in Mist1-Kras mice. Furthermore, Pyrvinium exhibited suppressive effects on the growth and survival of human organoids with dysplastic features, through simultaneous blocking the MEK/ERK and STAT3 signaling pathways.
Conclusions:
Through its dual blockade of MEK/ERK and STAT3 signaling pathways, Pyrvinium can effectively induce growth arrest in metaplasia and cell death in dysplasia. Therefore, our findings suggest that Pyrvinium is a promising chemotherapeutic agent for reprogramming the pre-cancerous milieu to prevent of gastric cancer development.
Keywords: Pyrvinium, Gastric cancer, Metaplasia, Dysplasia, Organoid, MEK/ERK, STAT3, Stem cells, Cancer Prevention
LAY SUMMARY:
Pyrvinium effectively induced cell death in pre-cancerous cells through a dual blockade of two different signaling pathways. Our results suggest that Pyrvinium may have potential as a therapeutic agent for treatment of gastric pre-cancerous lesions and prevention of gastric cancer development.
Graphical Abstract
Introduction
Gastric cancer is a significant global health concern, ranking as the fifth most common cancer and the third most common cause of cancer-related deaths.1 The risk of non-cardia gastric cancer is primarily associated with Helicobacter pylori (H. pylori) infection, which triggers a stepwise progression from atrophic gastritis, to intestinal metaplasia (IM), to dysplasia.2 Furthermore, evidence suggests that patients with IM or dysplasia may not benefit from H. pylori treatment on the risk of gastric cancer.3 The concept of a “point of no return” suggests that gastric cancers can occur after eradication of H. pylori if histologic changes are already advanced.4, 5 Patients with gastric cacner often present with varying degrees of pre-cancerous changes in gastric mucosa, such as IM and multifocal dysplastic glands.6 However, there are no therapeutic options available to treat asymptomatic patients with advanced pre-cancerous lesions. Therefore, there is an urgent need to identify therapeutic agents capable of effectively targeting these lesions.
We previously reported on the evolution of pyloric metaplasia (PM), IM, and dysplasia in the stomach of Mist1-CreERT2;LSL-Kras(G12D) mice (Mist1-Kras mice), which recapitulate the pre-neoplastic sequence in intestinal-type gastric carcinogenesis observed in human stomachs.7 We subsequently established metaplastic and dysplastic organoids from the Mist1-Kras mice exhibiting histological and molecular phenotypes similar to their human counterparts.8 We further identified a dysplastic stem cell (DSC) population, characterized by Trop2+/CD133+/CD166+ expression, and discovered that MEK inhibition can inhibit the growth of dysplastic organoids. More importantly, we found that Pyrvinium pamoate (Pyrvinium) induces apoptosis in dysplastic organoids.9
Pyrvinium has been used for over 70 years worldwide as an FDA-approved effective anthelminthic with a known safety profile.10 Recent pre-clinical studies have reported that Pyrvinium can impede the growth of various cancers, including colorectal, breast, lung, liver and pancreas cancers, and can inhibit mitochondrial function or crucial signaling pathways such as Wnt, PI3K/AKT, and Hedgehog.10 Notably, Pyrvinium has the capacity to selectively target and eliminate cancer stem cells in certain cancer types.11–13 Moreover, Pyrvinium has exhibited the capacity to enhance anti-tumor efficacy and overcome treatment resistance, when it is combined with chemotherapy drugs or radiation therapy.14, 15 In light of these promising anti-cancer effects, we evaluated unrecognized drug effects of Pyrvinium in pre-cancerous lesions and further explored its effects and underlying mechanisms in gastric metaplastic and dysplastic stages.
In this study, we aimed to identify signaling pathways, upregulated during metaplasia progression to dysplasia and altered by Pyrvinium treatment in metaplastic and dysplastic cells. We found that Pyrvinium treatment induced differential drug effects between metaplastic and dysplastic organoids due to differences in the basal STAT3 activity in these cell types. Also, Pyrvinium led to extensive cell death only in dysplastic organoids by specifically targeting dysplastic stem cells through a dual blockade of MEK/ERK and STAT3 signaling pathways. Our data not only demonstrate a robust potential for Pyrvinium suppression of metaplasia and dysplasia progression, but also provide promise in utilization of Pyrvinium for patients with pre-cancerous lesions to reduce cancer risk.
Materials and Methods
Organoid establishment, culture and drug treatment
Meta3 and Meta4 organoids were derived from Mist1-CreERT2Tg/+;LSL-K-ras(G12D)Tg/+ (Mist1-Kras) transgenic mice.7, 8 To establish human pre-cancerous organoids (hPCO), fresh tissues samples were obtained from patients with gastric cancer who underwent curative gastrectomy at Seoul National University Hospital (Institutional Review Board No. H-1806-166-954). All clinicopathological information, including Lauren classification, WHO classification, tumor-node-metastasis stages and microsatellite instability status is provided in Supplementary Table 1. Organoid lines were established as described in our previous study.9 In brief, approximately 5 cm mucosa strips were excised from gastric cancer towards the adjacent non-cancerous region in the direction to the corpus (Supplementary Figure 1). Each tissue piece was divided into two pieces for pathological examination and establishment of organoids. Dissociated glands were mixed with ice-cold Matrigel and plated in a 48-well plate, followed by adding Human IntestiCult medium (StemCell Technology). Meta3, Meta4, and hPCO were cultured in Cultrex® Reduced Growth Factor Basement Membrane Extract (R&D systems), Type R1 or Corning™ Matrigel™ Membrane Matrix (Thermo Fisher Scientific) with Mouse or Human IntestiCult medium (StemCell Technology) supplemented with 1% of penicillin/streptomycin (Gibco) in 48-well plates. Trametinib, Pyrvinium, SSTC3 and three STAT3 inhibitors (STAT3-IN-1, Sttatic, Cryptotanshinone, purchased from MedChemExpress) were dissolved in DMSO. Mouse and human organoids were cultured in Mouse or Human IntestiCult media containing either DMSO vehicle or certain concentrations of each drug for 3 days or 6 days, after they formed three-dimensional spherical structures. Phase-contrast images of organoids were obtained using the EVOS M7000 inverted microscope. Each experiment was conducted at least three times to ensure reproducibility.
Quantitative real-time PCR (qRT-PCR)
Total RNAs were extracted from Meta3 or Meta4 organoids using the Trizol (Invitrogen) reagent. Reverse-transcribed cDNA was synthesized using iScript gDNA clear cDNA synthesis kit (Bio-Rad). For measuring MDSC markers in Mist1-Kras mice treated with either DMSO or Pyrvinium, total RNAs were extracted from paraffin-embedded stomach sections using a RNeasy FFPE Kit (Qiagen). cDNA was prepared from 1 μg of total RNA with random hexamer primers (Promega) and the GoScript reverse transcription system (Promega). Quantitative RT-PCR was performed using iTaq Universal SYBR Green Supermix (Bio-Rad), a CFX96 Real-Time PCR Detection System (Bio-Rad) or StepOnePlus Real-Time PCR System (ThermoFisher, Applied Biosystems). TBP, β-actin or GAPDH was used to normalize gene expression levels. The primer sequences used for qRT-PCR are indicated in Supplementary Table 2. All reactions were performed in duplicate or triplicate and relative gene expression levels were obtained by the comparative 2−ΔΔCT method using CFX Maestro software (Bio-Rad) or StepOnePlus software version 2.3(Applied Biosystems).
Western blot
To extract proteins, organoids were dissolved in RIPA buffer (Sigma-Aldrich) containing protease and phosphatase inhibitors (Sigma-Aldrich). The protein concentration was measured using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). Ten μg of proteins from each sample were loaded to 4–20% Mini-PROTEAN® TGX™ Precast Protein Gels (Bio-Rad) and transferred onto PVDF (polyvinylidene difluoride) membranes (Millipore, Billerica, MA). Membranes were blocked with 5% Blotting-Grade Blocker (Bio-Rad) and applied with primary antibodies (Supplementary Table 3) at 4°C overnight. Immunoreactive bands were visualized using SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Scientific).
Single cell RNA-sequencing (scRNA-seq)
To prepare cells from organoids for scRNA-seq, we followed the protocol outlined in the PIPseq Milli 3’ Single Cell Capture and Lysis User Guide (Fluent BioSciences). Simply, total 5000 cells in the Cell Suspension Buffer were added into Pre-templated Instant Partitions (PIPs) to capture mRNA for downstream processing. The resulting emulsions carrying captured mRNA were visually inspected by Fluent Biosciences before proceeding with downstream processing. Samples that passed inspection were processed according to the specifications of the PIPseq™ T2 Single Cell Sequencing Kit, including mRNA isolation, cDNA generation, cDNA amplification, sequencing-ready library preparation, library pooling, and sequencing. Sequencing of cDNA libraries was performed at the Vanderbilt Technologies for Advanced Genomics (VANTAGE) core facility using a standard operating procedure on an Illumina NovaSeq 6000 instrument, with a minimum sequencing depth of 25,000 reads per sample. The sequencing data was analyzed and processed using PIPseeker (Fluent BioSciences) and PartekFlow (Partek). Functional annotation of differentially expressed genes was carried out by PANTHER gene ontology (GO).
Mice and Drug treatment
Procedures involving mice in this study were carried out in accordance with protocols approved by the Institutional Animal Care and Use Committee at Vanderbilt University Medical Center. Experimental and control groups of Mist1-Kras mice were randomly assigned, and each experimental group contained 3 to 4 male and female mice, with littermates or age-matched mice being used for the study. Mice at 8 weeks of age were injected subcutaneously with 5 mg of tamoxifen (Sigma-Aldrich) in corn oil (Sigma-Aldrich) with 10% ethanol for three consecutive days. Three months after the tamoxifen injection, mice were treated with either DMSO or Pyrvinium (4mg/L) via drinking water for one or two weeks. Previously, Pyrvinium has usually been administered as a single dose of 5 mg/kg of body weight and can be repeated every 2 to 3 weeks for human patients with pinworm infection. In this experiment, mice were administered with the total amount of Pyrvinium, 3.52 mg/kg, over a 2-week period. This calculation is based on the average adult mouse weight of 23 grams and their daily water intake of 5.8 mL.
Quantification and Statistical Analysis
The diameters of mouse and human organoids were measured using ImageJ software (NIH) at 0 and 3 or 6 days after DMSO or drug treatment from four entire well images of each condition at 4x magnification. For counting live or dead cells, organoids were stained with Calcein AM for live cells and EthD-1 for dead cells 3 days after treatment and the number of EthD-1 positive organoids were manually counted from images taken from three wells of each condition at 4x magnification. All experiments, performed for statistical analysis, were repeated three times. The mean values from each condition were compared by Two-tailed Mann-Whitney test, One-way, Two-way ANOVA (with Tukey’s multiple comparison) or non-parametric Spearman correlation using Graphpad Prism Software 9.0.
Results
Pyrvinium selectively induces cell death of mouse dysplastic organoids
We utilized previously established metaplastic (Meta3) and dysplastic (Meta4) organoids8 to evaluate combined drug effects of Pyrvinium, a putative Casein Kinase 1α (CK1α) activator16, and Trametinib, a MEK inhibitor, in Meta3 and Meta4 organoids. Meta3 expressed SPEM cell markers, Aquaporin 5 (AQP5) and CD44v9, while Meta4 organoids were specifically positive for a dysplastic cell marker, TROP2, as well as AQP5 (Figure 1A). The Meta3 organoids displayed a significant reduction in size compared to control organoids treated with DMSO in response to Trametinib, Pyrvinium, and both (Figure 1B and D). In contrast, the Meta4 organoids showed varying responses to Trametinib and Pyrvinium. Trametinib treatment only suppressed dysplastic organoid growth and induced a significant decrease in organoid diameter in one Meta4 line, Meta4_2 (Figure 1C and E). Although other Meta 4 organoid lines showed no differences in size after the Trametinib treatment, the organoid cell heights were thinner compared with control organoids, indicating lower cellularity as shown in H&E images (Figure 1C). Pyrvinium treatment not only led to a reduction in organoid size but also extensive cell death in all three Meta4 lines, as shown by numerous pyknotic cells in phase-contrast images (Figure 1C) and confirmed by live/dead cell staining (Figure 1F). Interestingly, co-treatment with Trametinib and Pyrvinium did not provide any additive or synergistic effects on Meta4 organoids (Figure 1C and E). Taken together, these results suggest that Pyrvinium can induce cell death in dysplastic organoids and inhibit growth of metaplastic organoids, but MEK inhibition can only lead to growth arrest in both metaplastic and dysplastic organoids.
Figure 1. Pyrvinium induces cell death in mouse dysplastic organoids.
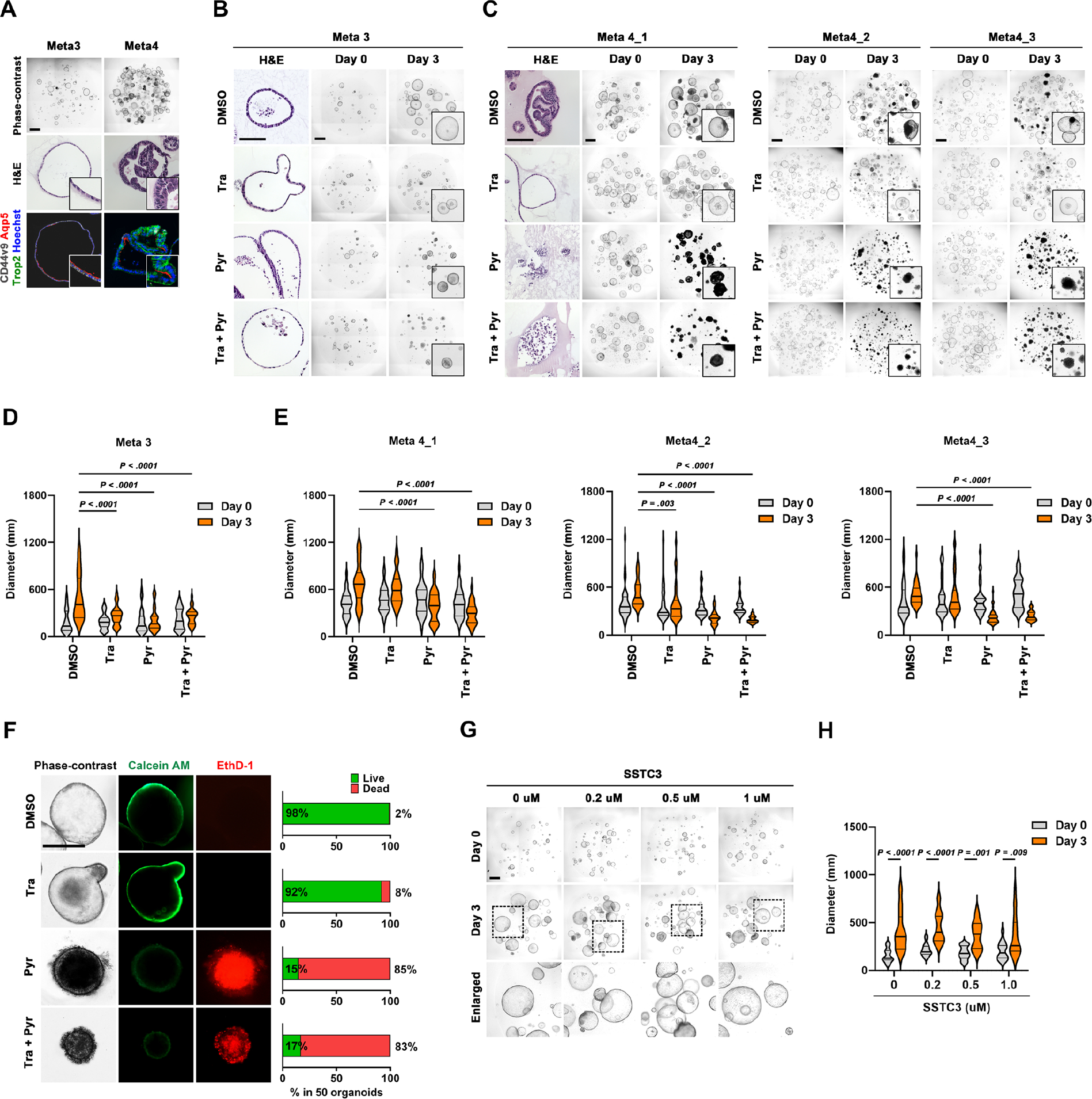
(A) Phase-contrast, H&E images, and co-immunostaining for CD44v9, AQP5, and TROP2 in Meta3 and Meta4 organoids. (B and C) H&E and phase-contrast images of Meta3 (B) and 3 different Meta4 organoids (C) treated with DMSO vehicle, 1μM of Trametinib (Tra), 100nM of Pyrvinium (Pyr) and their combinations for 3 days. (D and E) Quantitation of organoid diameters before and after treatment. (F) Phase-contrast images and live/dead (Calcein AM/EthD-1) cell staining with the percentages of live and dead cells in Meta4 organoid after treatment. (G and H) Phase-contrast images (G) and quantitation of diameters (H) of Meta4 organoids before and after treatment with 0, 0.2, 0.5, and 1 μM of SSTC3, a specific Casein Kinase 1α activator. Mean ± standard deviation. Two-way ANOVA with Tukey’s multiple comparisons. Scale bar = 500 μm.
In our previous study, we suggested that Pyrvinium might have controlled the dysplastic cell survival and maintenance through activation of CK1α and functioned as a downstream of inhibitor of the Wnt pathway.9 To further evaluate whether the CK1α activation is critical for the survival of dysplastic organoids, we treated Meta4 with SSTC3, a CK1α-specific activator17. Surprisingly, SSTC3 did not cause any growth suppression or cell death in Meta4 organoids (Figure 1G). Wnt-target genes were not downregulated after the Pyrvinium treatment and the expression of several genes, such as Axin2, Cdca4, Rnf43, and Znrf3, were actually increased 24 hours after treatment (Supplementary Figure 2). Therefore, our results indicate that Pyrvinium does not negatively impact the Wnt signaling pathway in dysplastic gastric cells.
Pyrvinium affects both ERK and STAT pathways in mouse metaplastic and dysplastic organoids.
To identify what signaling pathways are controlled by Pyrvinium in Meta4 organoids, we conducted a phospho-array analysis using Meta3 and Meta4 organoids treated with either DMSO vehicle or Pyrvinium. Paired analysis between Meta3 and Meta4 organoids with or without Pyrvinium treatment displayed differential profiles of protein phosphorylation, which includes STAT, ERK signaling related transcription factor, protein kinase enzymes, NF-κB, and MAPK pathways (Figure 2A). In particular, the most prominent effect of Pyrvinium treatment was dephosphorylation of proteins related to the STAT pathway (Figure 2B). To identify potential proteins responsible for Pyrvinium-induced cell death in Meta4 organoids, we postulated that phosphoprotein candidates upregulated in Meta4 organoids would exhibit a significant reduction in phosphorylation upon Pyrvinium treatment. We selected top 10 protein candidates including p-MYCT358, p-PKCthetaT538, p-C-RafS43 p-STAT3Y705, p-Pyk2T580, p-Histone H3.1S10, p-CREBT100, p-Pyk2T881, p-SRFS77, and p-ERK1/2(p44/42)T202 (Figure 2C and D). As the organoids were derived from Mist1-Kras mice, it is not surprising that many proteins on the list are directly related to MAPK and PKC signaling pathways, with the exception of STAT3 (Figure 2E). Therefore, these results suggest that STAT3 signaling is upregulated in dysplastic cells and might be an important pathway for dysplastic cell survival.
Figure 2. Phospho-antibody array identifies downregulation of MAPK and STAT signaling pathways by Pyrvinium.
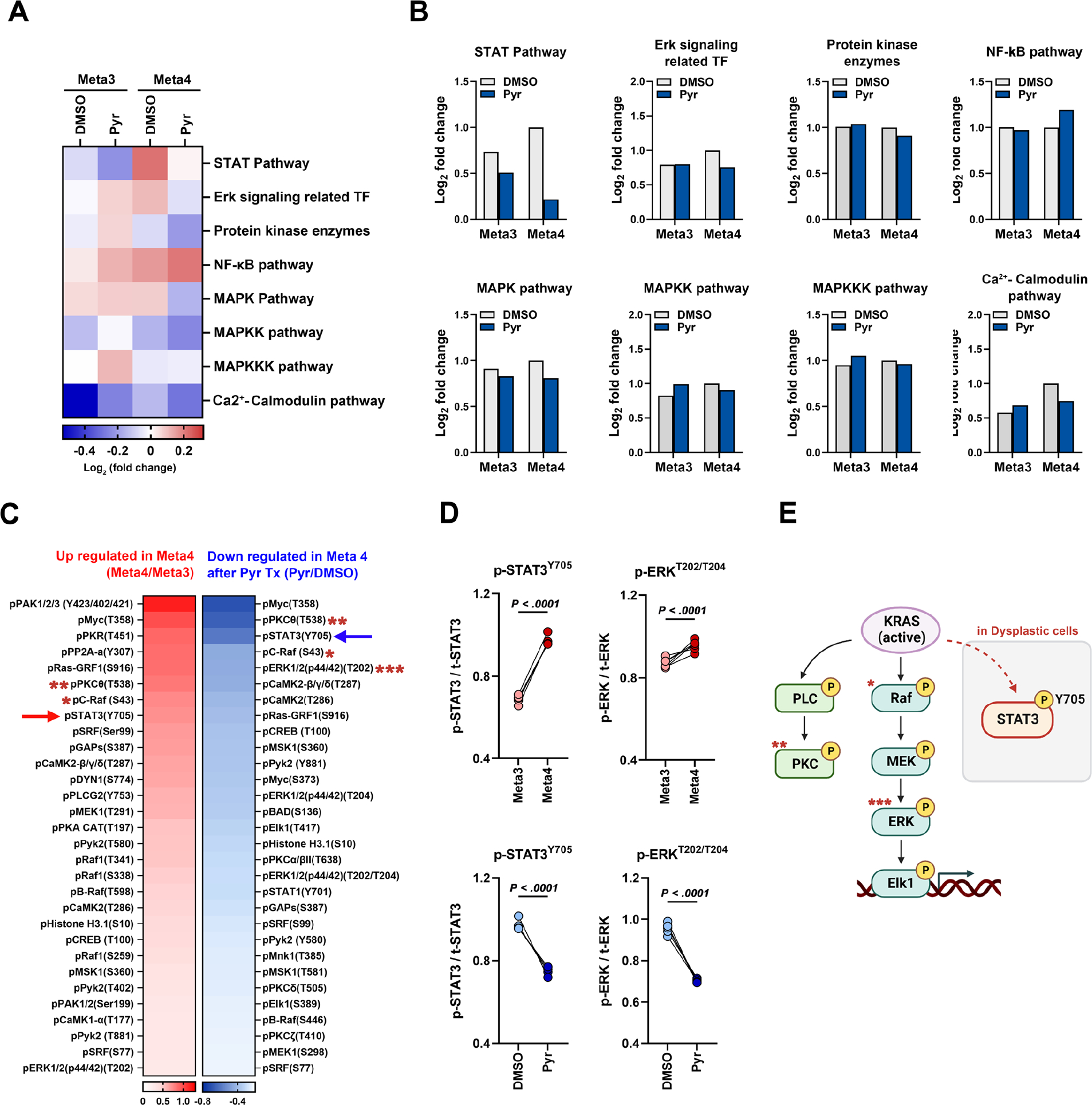
(A) Pathway enrichment analysis of differentially phosphorylated proteins in Meta3 and Meta4 organoids 1 day after Pyrvinium (Pyr) treatment using the Molecular Signatures Database (MSigDB) hallmark gene set collection. Color scale represents standardized log2(FDR) values. (B) Log2 fold change bar graphs of signaling pathways in Meta3 and Meta4 organoids after treatment. (C) Heat map showing upregulated or downregulated phospho-proteins in Meta4 organoids compared to Meta3. (D) Changes in phosphorylation of STAT3Y705 and ERKT202/T204 between Meta3 and Meta4 organoids (red, upper panel) or between DMSO and Pyr treatment in Meta4 organoids (blue, lower panel). Each square represents the signal ratio. (E) Schematic illustration of STAT3 and RAS/MAPK pathway affected by Pyr treatment in dysplastic organoids, created with BioRender.com. Mean ± standard deviation. Two-tailed Mann-Whitney test.
Pyrvinium induces cell death in dysplastic organoids through a dual-blockade of ERK and STAT3 signaling pathways
Dysregulated STAT3 signaling pathway has been strongly implicated in tumorigenesis through effects on cell growth, angiogenesis, immune system evasion, and prevention of apoptosis. 18 We therefore investigated how Pyrvinium regulates the STAT3 activity in dysplastic organoids. STAT3 activation is typically induced by phosphorylation on either a tyrosine residue (pSTAT3Y705) or a serine residue (p-STAT3S727). Our phospho-array data revealed a loss of STAT3Y705 phosphorylation after Pyrvinium treatment. Western blot analysis confirmed a significant reduction in the phosphorylation of STAT3Y705, but not STAT3S727, in Meta4 organoids (Figure 3A and B). Of note, a significant decrease in p-ERK levels was observed after Pyrvinium treatment, consistent with the results from the phospho-array analysis and Trametinib also led to a significant reduction in p-ERK levels, as expected (Figure 3A and B). These findings suggest that dual suppression of STAT3Y705 and ERK phosphorylation is necessary to induce cell death in dysplastic organoids. Meta3 organoids also displayed reduced phosphorylation of both STAT3Y705 and ERK upon Pyrvinium treatment (Figure 3C). However, the basal phosphorylation level of STAT3Y705 in Meta3 organoids was significantly lower than that of Meta4 organoids (Figure 3C–E) and the organoids did not exhibit any cell death, as shown previously 8.
Figure 3. Pyrvinium suppresses mouse dysplastic organoids through a double blockade of ERK and STAT3 signaling pathways.
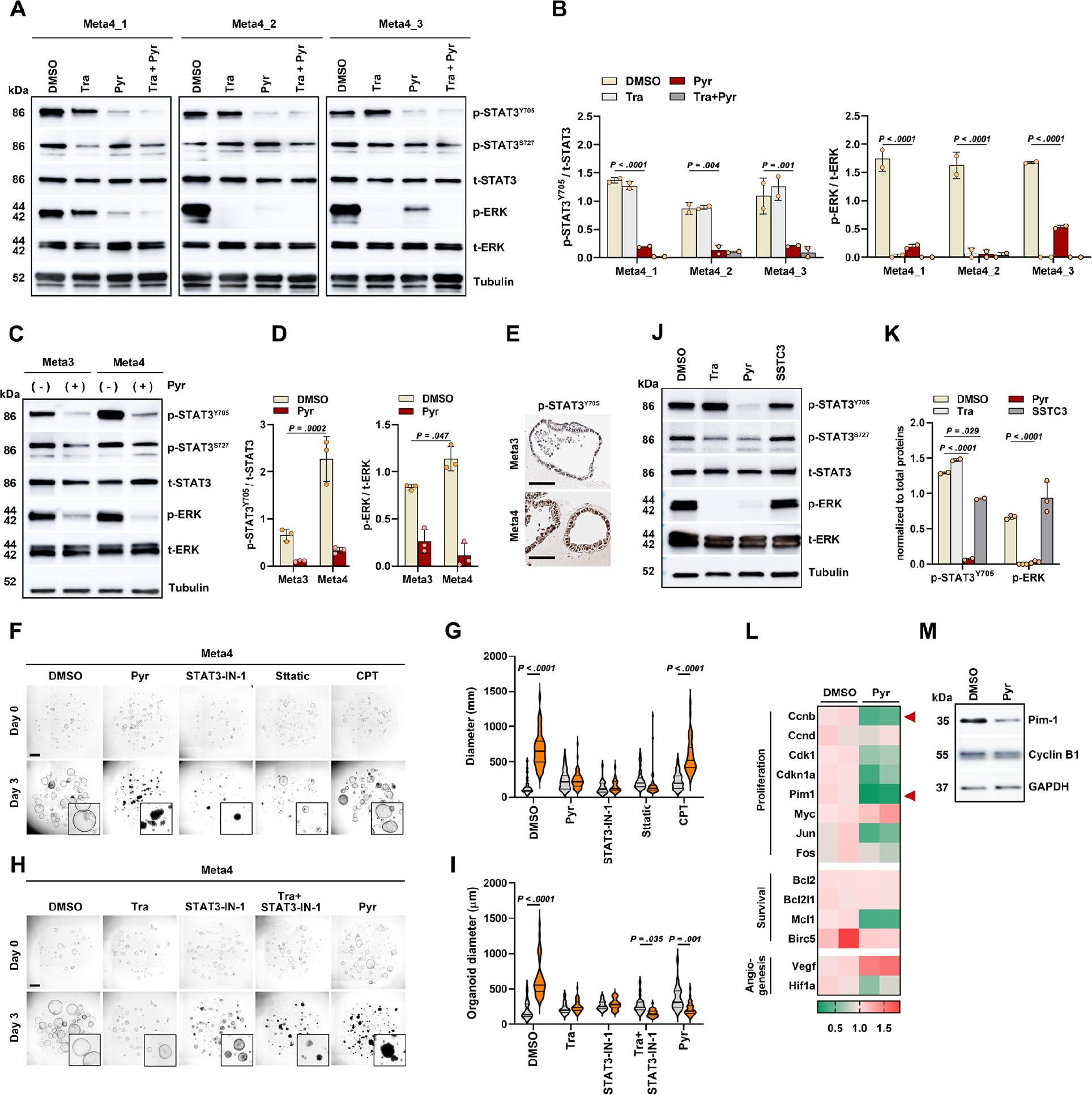
(A and B) Western blot analysis for phospho-STAT3 (p-STAT3) and phospho-ERK (p-ERK) in three Meta4 organoid lines after treatment of Trametinib (Tra), Pyrvinium (Pyr) and their combinations for 1 day (A) and ratios of p-STAT3/total STAT3 and p-ERK/total ERK (B). (C and D) Western blot for p-STAT3 and p-ERK in Meta3 and Meta4 organoids (C) and ratios of p-STAT3/total STAT3 and p-ERK/total ERK (D). (E) Immunostaining for p-STAT3 in Meta3 and Meta4 organoids. (F and G) Phase-contrast images of Meta4 organoids treated with DMSO, Pyr (100nM), STAT3-IN-1 (1μM), Stattic (2μM), and Cryptotanshinone (CPT) (5μM) for 3 days (F), and quantitation of organoid diameters before and after treatment (G). (H and I) Phase-contrast images of Meta4 organoids treated with DMSO, Trametinib (Tra, 1μM), STAT3-IN-1 (500nM), combination of Tra and STAT3-IN-1, and Pyr, for 3 days (H), and quantitation of organoid diameters before and after treatment (I). (J and K) Western blot for p-STAT3 and p-ERK after treatment of Tra, Pyr, and SSTC3 (J) and ratios of p-STAT3/t-STAT3 and p-ERK/t-ERK (K). (L) Heatmap of quantitative real time-PCR analysis for STAT3 target genes in Meta4 organoid treated with DMSO or Pyr for 1 day. Relative quantification of each gene expression level was normalized to β-actin gene expression. Color scale represents normalized mRNA expression values. n= 3 biological replicates. (M) Representative protein levels of Pim-1 and Cyclin-B1 after treatment. Mean ± standard deviation. One or two-way ANOVA with Tukey’s multiple comparisons. Scale bar = 500 μm
We additionally treated the Meta3 and Meta4 organoids with three different STAT3-specific inhibitors, including STAT3-IN-1, Sttatic, and Cryptotanshinone (CPT). All STAT3-specific inhibitors showed no influence on the survival and growth of Meta3 organoids (Supplementary Figure 3), indicating growth arrest induced by Pyrvinium is mediated only by suppression of MEK/ERK signaling in metaplastic cells. Two STAT3-specific inhibitors, STAT3-IN-1 and Sttatic, induced growth arrest in Meta4 organoids, but did not lead to cell death (Figure 3F and G). In contrast, co-treatment with Trametinib and STAT3-IN-1 resulted in significant cell death in Meta4 organoids, comparable to the effect of Pyrvinium (Figure 3h, i). We confirmed that SSTC3 treatment did not reduce p-STAT3 Y705 or p-ERK expression confirming that Pyrvinium action is not through CK1α activation (Figure 3J and K). We additionally examined changes in STAT3-related genes in response to Pyrvinium treatment. The expression of several STAT3-target genes, including Cyclin B1 and Pim-1, was downregulated after Pyrvinium treatment (Figure 3L and Supplementary Figure 4), which was also confirmed at the protein level (Figure 3M). Pim-1 is overexpressed in various human cancers and protects against cell apoptosis. 19, 20 Thus, the downregulation of Pim-1 might be associated with the induction of cell death in dysplastic cells. Consequently, our results indicate that a dual inhibition of both ERK and STAT3 signaling pathways is critical for dysplastic cell survival.
Pyrvinium targets Trop2+CD133+CD166+ dysplastic stem cells
To evaluate transcriptomic alterations and identify subpopulations of dysplastic organoids affected by Pyrvinium treatment, we performed single-cell RNA-sequencing using cells from Meta4 organoids treated with either DMSO or Pyrvinium for 1 day before organoids exhibited morphological disruption. Uniform Manifold Approximation and Projection (UMAP) demonstrated an almost complete separation between DMSO-treated and Pyrvinium-treated cells, indicating dramatic alterations in transcription elicited by Pyrvinium (Figure 4A). Five subpopulations were determined by unsupervised clustering combined with lineage specific markers including Stmn1 and Pcna for proliferative cells, TFF3 for differentiated intestinal-type cells, CD133 and CD166 for DSCs, Mal2 and Lgals3 for SPEM cells, and Ddit3 and Areg 21 for damaged cells (Figure 4B). Gene ontology analysis combined with PANTHER classification demonstrated distinct gene expression profiles among the groups (Figure 4C). Pyrvinium treatment led to increased catalytic and transporter activities, whereas decreased binding and transcription regulator activities. While many subpopulations such as proliferative cells, SPEM cells, and DSCs were found in DMSO-treated cells, over 73.9% of cells were damaged and mostly differentiated cells among Pyrvinium-treated cells (Figure 4D and E). Volcano plot and UMAP analysis indicated that several STAT3-target genes observed in DMSO-treated cells, such as Fos, Fosb, and Jun, were downregulated after Pyrvinium treatment (Figure 4F and G), while damage-related genes such as Dusp4, Ddit3 and Areg were upregulated in Pyrvinium-treated cells (Figure 4F and H). Furthermore, we found a substantial increase in both 7-AAD and Annexin V expression in the Pyrvinium-treated cells, confirming increased activities in necrosis and apoptosis in response to Pyrvinium (Figure 4I). It is important to note that the DSC subpopulation was not observed after Pyrvinium treatment. qRT-PCR analysis validated decreases in CD133 and CD166 gene expression (Figure 4J) and FACS analysis also demonstrated a significant decrease in CD133+CD166+ DSCs (Figure 4K). These findings indicate that Pyrvinium specifically targets DSCs as well as proliferating cells, leading to cell death in dysplastic organoids.
Figure 4. Pyrvinium targets stem cell population in mouse dysplastic organoids.
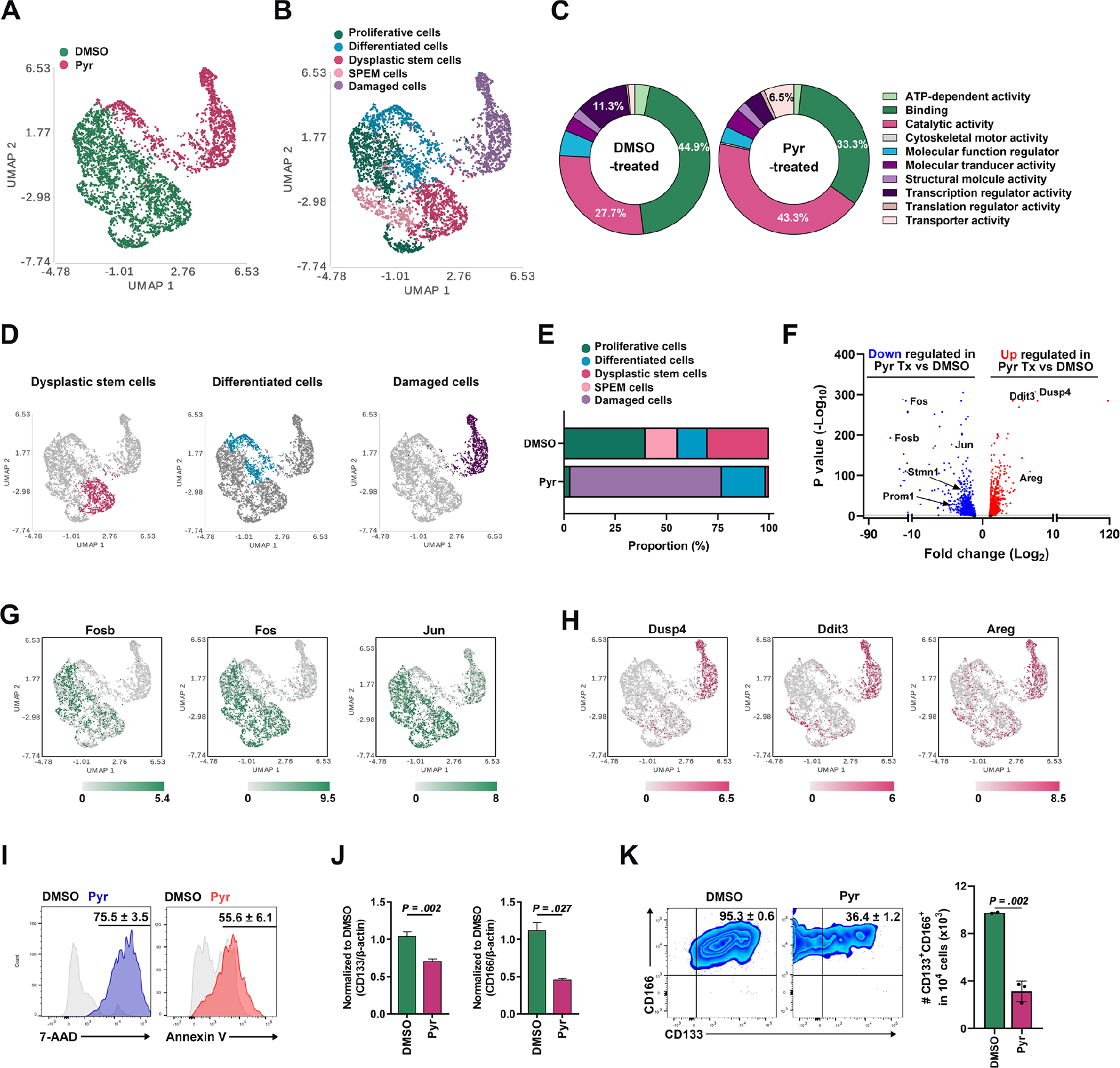
(A and B) A superimposed Uniform Manifold Approximation and Projection (UMAP) plot showing annotated clusters from DMSO-treated and Pyrvinium (Pyr)-treated Meta4 cells (A) and subpopulation-matched cluster overlay (B). (C) PANTHER gene ontology classification results based on top 300 differentially expressed genes in DMSO-treated and Pyr-treated Meta4 cells. (D and E) Clusters of dysplastic stem cells (DSCs), differentiated cells or damaged cells in a superimposed UMAP plot (D) and their proportions in DMSO-treated and Pyr-treated Meta4 samples (E). (F) Volcano plot of genes downregulated or upregulated in Pyr-treated vs DMSO-treated Meta4 cells. (G and H) STAT3-target genes upregulated (Fosb, Fos, and Jun) (G) or downregulated (Dusp4, Ddit3, and Areg) genes (H) in a superimposed UMAP plot. (I) FACS analysis of 7-AAD and annexin V expression in DMSO-treated and Pyr-treated Meta4 organoids. (J) Quantitative real time-PCR analysis showing relative expression of DSC markers, CD133 and CD166, in DMSO-treated vs Pyr-treated Meta4 organoids. (K) FACS analysis of the CD133 and CD166 positive cells in DMSO-treated and Pyr-treated Meta4 organoids. Mean ± standard deviation. Two-tailed Mann-Whitney test.
Pyrvinium treatment inhibits metaplasia progression to dysplasia in the mouse stomach.
Next, we investigated whether the STAT3 signaling pathway is upregulated during metaplasia progression to dysplasia in vivo. At 3 months after tamoxifen injection in Mist1-Kras mice, stomachs predominantly exhibited metaplastic glands, with an appearance of dysplastic glands becoming increasingly prevalent by four months.7 We therefore treated the mice at 3 months after tamoxifen injection and evaluated whether Pyrvinium could ameliorate metaplasia progression to dysplasia (Figure 5A). One or two weeks after either DMSO or Pyrvinium treatment, gastric mucosa in each group of mice was examined. Acid-secreting parietal cells gradually repopulated the corpus mucosa in mice treated with Pyrvinium (Figure 5B and C). Conversely, the number of hyperplastic foveolar glands with UEA1 and Ki-67 positive proliferating cells significantly decreased (Figure 5D and E), along with a significant decrease in the number of dysplastic glands (Figure 5F and G). Furthermore, Pyrvinium treatment resulted in downregulation of p-STAT3 and Pim-1 (Figure 5H), as observed in dysplastic organoids (Figure 3L and M). We additionally conducted a comprehensive examination of major organs in wild-type mice treated with either DMSO or Pyrvinium and confirmed that Pyrvinium did not induce any noticeable histopathological abnormalities (Supplementary Figure 5). We further explored alterations in the immune microenvironment after Pyrvinium treatment. Mice treated with DMSO, exhibited various inflammatory cell infiltration, such as lymphocytes and macrophages, in the lamina propria. Pyrvinium treatment induced a dramatic decrease in the CD4+ T cell population (Figure 5I), but it did not show a significant difference in CD8+ T or CD19+ B cells (Figure 5I and J). M2-macrophages have been shown to promote progression of SPEM and IM7, 22, and both NK cells and CD163-positive M2-macrophages significantly decreased in Pyrvinium-treated mice (Figure 5K–M). While the total number of CD3+ T cells decreased in Pyrvinium-treated mice, the relative ratio of CD3+CD25+ regulatory T cell number significantly increased (Supplementary Figure 6) and the expression of myeloid-derived suppressor cell (MDSC)-related markers tended to increase, although the changes were not significant (Supplementary Figure 7). These results suggest that Pyrvinium effectively inhibits metaplasia progression to dysplasia and promotes the regeneration of normal gastric mucosa in vivo by contributing an anti-inflammatory microenvironment.
Figure 5. Pyrvinium treatment inhibits metaplasia progression to dysplasia in vivo.
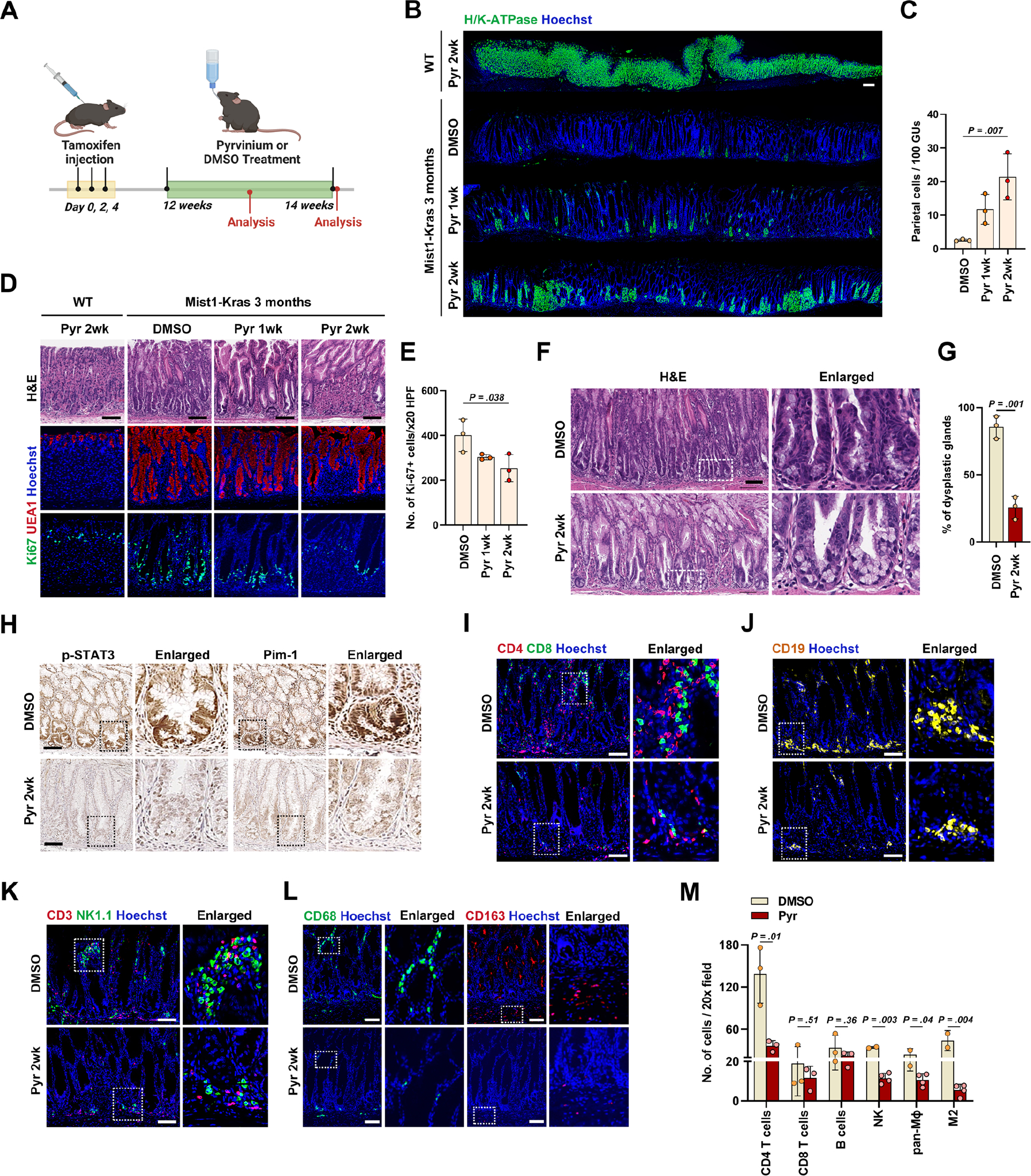
(A) Experimental scheme of Pyrvinium (Pyr) treatment in the Mist1-Kras mice at 3 months after Tamoxifen injection created with BioRender.com. (B) Panoramic view of immunostaining for H/K-ATPase in gastric corpus from wild type mice treated with Pyr for 2 weeks and Mist1-Kras mice treated with DMSO, Pyr for 1 week or 2 weeks. (C) Quantitation of parietal cell-containing gastric units (GUs) per 100 GUs. (D) H&E (top) and immunostaining for UEA1 (middle) and Ki-67 (bottom) in the wild type mice treated with Pyr for 2 weeks and Mist1-Kras mice treated with DMSO or Pyr for 1 or 2 weeks. (E) Quantitation of Ki-67-positive cells per 20X field. (F) Representative H&E images of gastric corpus from Mist1-Kras mice treated with DMSO or Pyr for 2 weeks. (G) Percentages of dysplastic glands in remaining hyperplastic glands. (H) Immunostaining for phospho-STAT3 (p-STAT3) and Pim-1 in gastric corpus from Mist1-Kras mice treated with DMSO or Pyr for 2 weeks. (I and J) Co-immunostaining for CD4 and CD8 (I) and immunostaining for CD19 (J) in the gastric corpus from Mist1-Kras mice treated with DMSO or Pyr for 2 weeks. (K and L) Co-immunostaining for CD3 and NK1.1 (K) and CD68 or CD163 (L) in the gastric corpus from Mist1-Kras mice treated with DMSO or Pyr for 2 weeks. (M) Quantitation of various immune cells per 20X field in Mist1-Kras mice treated with DMSO or Pyr. Mean ± standard deviation. Unpaired t-test or one-way ANOVA with Tukey’s multiple comparisons. Scale bar = 100 μm.
Pyrvinium inhibits growth and survival of human pre-cancerous cells with dysplastic features.
It is well-known that non-tumor-bearing mucosa surrounding gastric cancer frequently harbors metaplastic or dysplastic glands with varying degrees of genetic or epigenetic abnormalities, a phenomenon known as “field cancerization”.23 To investigate the inhibitory effects of Pyrvinium on the growth and survival of these human pre-cancerous lesions, we established twenty human pre-cancerous organoid (hPCO) lines from gastric mucosa adjacent to gastric cancer obtained from surgical specimens. The hPCO lines were characterized based on morphology and expression profiles of AQP5 and CD44v9 as metaplastic cell markers and TROP2 as a dysplastic cell marker (Figure 6E). Following treatment with Trametinib, Pyrvinium, and their combination for 6 days, we measured the diameters of organoids and examined organoid death, categorized by scores, ranging from 1 to 4 (where 0 represents no death, and 1, 2, 3, and 4 correspond to 0–10%, 10–50%, 50–90%, and >90% deceased organoids, respectively). Based on these results, we classified the hPCO lines into three categories: sensitive (score 4), moderate (score 2 or 3), and resistant (score 0 or 1) groups (Figure 6E). Five Pyrvinium-sensitive hPCO lines displayed significant organoid death, while nine Pyrvinium-resistant hPCO lines remained mostly alive with no or a few dead cells and six Pyrvinium-moderate lines exhibited mild to moderate extent of cell death (Figure 6 and Supplementary Figure 8). Three representative images from Pyrvinium-sensitive hPCO lines (hPCO-2, hPCO-3, and hPCO-34) are shown in Figure 6A, and we confirmed the organoid death by live/dead cell staining using the hPCO-34 line (Supplementary Figure 9). Pyrvinium induced organoid death in Pyrvinium-sensitive lines, but co-treatment with Trametinib did not show any additional effects, as observed in the mouse dysplastic organoids (Figure 1C). Interestingly, Pyrvinium-sensitive lines did not respond to Trametinib and continuously grew for 6 days, which is in contrast to the growth inhibition observed in mouse organoids after Trametinib treatment (Figure 6A and B). In addition, Pyrvinium-resistant hPCO lines (hPCO-12, hPCO-19, and hPCO31) did not die, but still exhibited growth arrest after the treatment with Pyrvinium and/or Trametinib (Figure 6C and D). We additionally evaluated whether the drug responses in different human organoids are associated with their histological and molecular characteristics (Figure 6E). Remarkably, we observed that Pyrvinium-sensitive hPCO lines exhibited positive correlations with dysplastic morphology and high levels of TROP2 expression, whereas they demonstrated negative correlations with AQP5 and CD44v9 expression (Figure 6F–H). Collectively, our findings suggest that the Pyrvinium effects are significantly associated with dysplastic features of hPCO lines.
Figure 6. Pyrvinium targets human pre-cancerous organoid (hPCO) with dysplastic features.
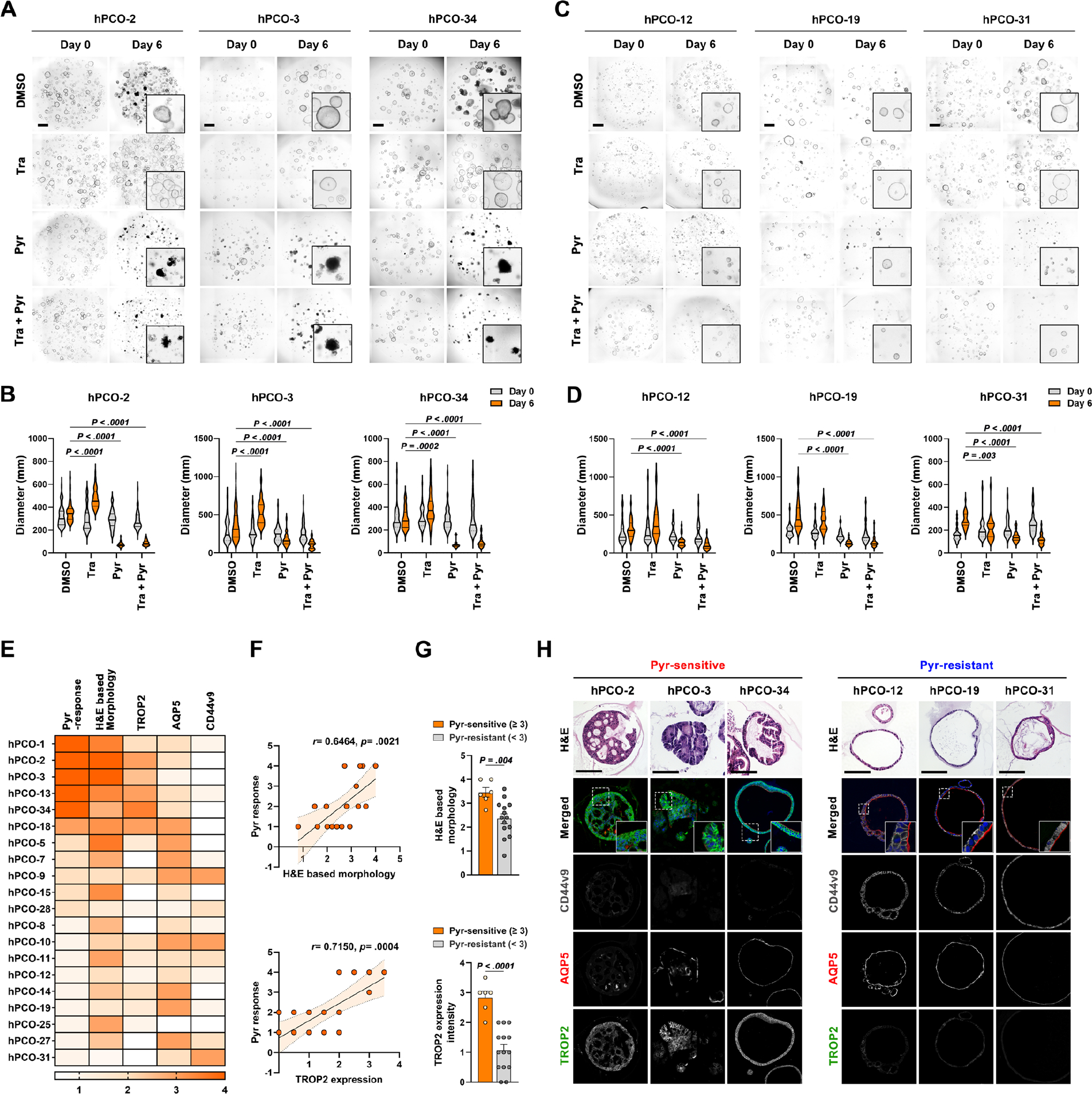
(A and B) Representative phase-contrast images of three Pyrvinium (Pyr)-sensitive hPCO lines treated with DMSO, 1uM of Trametinib (Tra), 100nM of Pyrvinium and their combinations for 6 days (A) and quantitation of organoid diameters before and after treatment (B). (C and D) Phase-contrast images of three Pyr-resistant hPCO lines treated with DMSO, 1uM of Trametinib (Tra), 100nM of Pyrvinium and their combinations for 6 days (C) and quantitation of organoid diameters before and after treatment (D). (E) Heatmap displaying Pyr response, morphology, and expression of TROP2, AQP5, and CD44v9 in twenty hPCO lines. (F) Correlation graphs of Pyr response with morphology and TROP2 expression in hPCO lines. (G) Bar graphs showing morphology or TROP2 expression scores between Pyr-sensitive and – resistant hPCO lines. (H) Representative H&E images and co-immunostaining for CD44v9, AQP5, and TROP2 in three Pyr-sensitive and -resistant hPCO lines. Mean ± standard deviation. Spearman’s correlation coefficient, unpaired t-test or Two-way ANOVA with Tukey’s multiple comparisons. Scale bar = 500 μm.
Pyrvinium blocks both ERK and STAT3 signaling pathways in human dysplastic cells.
We further examined the relationship between the Pyrvinium responsiveness and the STAT3 and ERK signaling pathway activation in hPCO lines. We examined changes in phosphorylation levels of STAT3Y705 and ERK in three Pyrvinium-sensitive and -resistant hPCO lines after Pyrvinium treatment. Pyrvinium treatment decreased both p-STAT3Y705 and p-ERK levels in all three Pyrvinium-sensitive hPCO lines (Figure 7A and B), whereas no significant differences were observed in the Pyrvinium-resistant group (Figure 7C and D). Similar to our findings in mouse organoids, upregulation in STAT3 Y705 phosphorylation was also observed in the Pyrvinium-sensitive hPCO lines compared to the Pyrvinium-resistant group (Figure 7E–7G), as well as in the gastric tissues used for the hPCO establishment (Figure 7H). We additionally treated hPCO lines with three STAT3-specific inhibitors and assessed their effects on the growth and survival in hPCO lines. Although two STAT3 inhibitors, Sttatic or CPT, but not STAT3-IN-1, arrested organoid growth in Pyrvinium-sensitive hPCO-2 line, the organoids survived until 6 days after the treatment (Figure 7I and J). In contrast, Pyrvinium-resistant hPCO-31 did not respond to all STAT3 inhibitors (Figure 7K and L). Thus, these results suggest that Pyrvinium can effectively suppress the growth and survival of hPCOs by simultaneously inhibiting both STAT3 and ERK signaling pathways.
Figure 7. Pyrvinium induces the cell death in dysplastic human pre-cancerous (hPCO) organoids by blocking ERK and STAT3 signaling pathways.
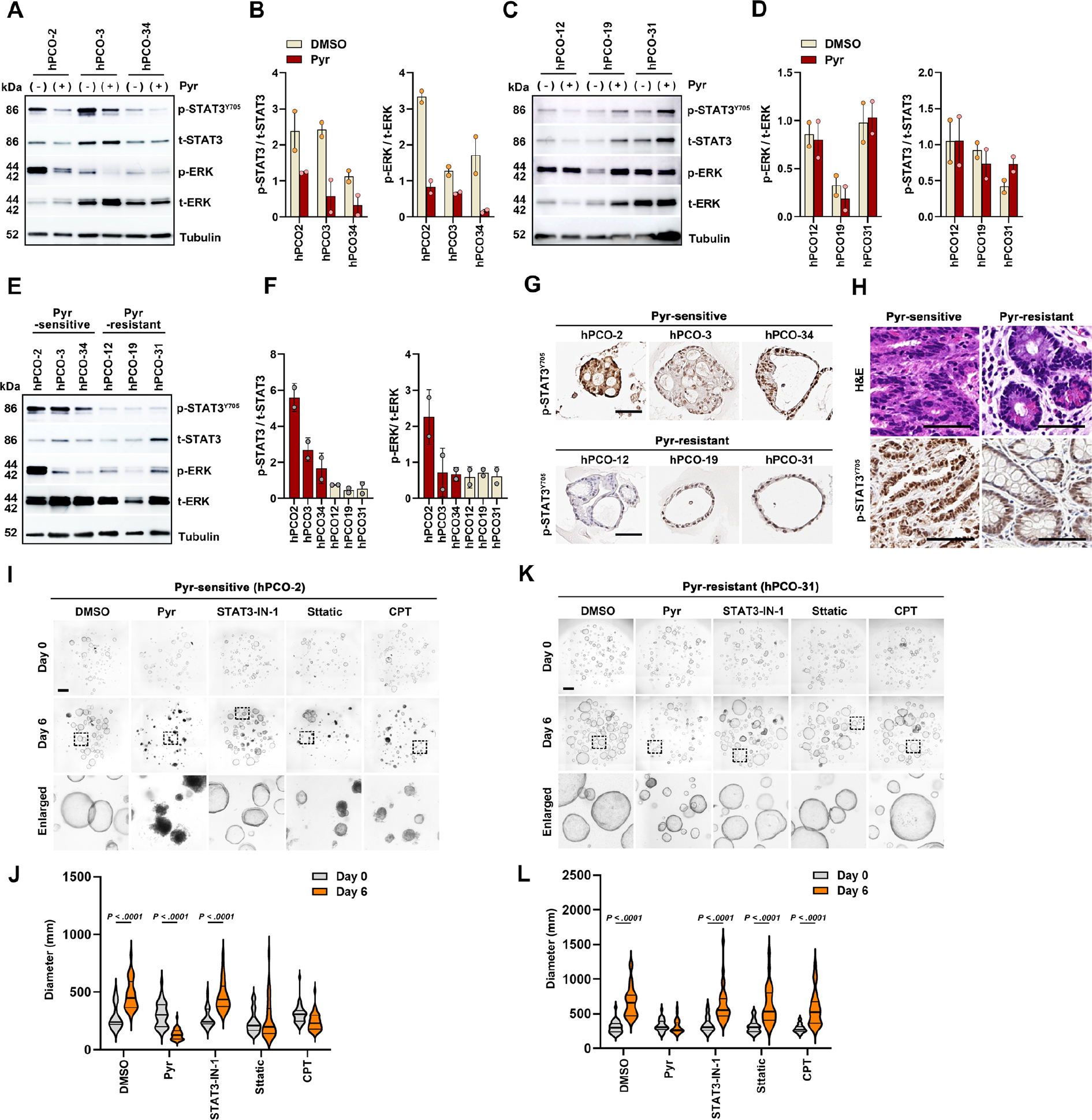
(A and B) Western blot analysis for p-STAT3 and p-ERK in three Pyr-sensitive hPCO lines after Pyr treatment (A), and ratios of p-STAT3/total STAT3 (t-STAT3) and p-ERK/total ERK (t-ERK) (B). (C and D) Western blot for p-STAT3 and p-ERK in three Pyr-resistant hPCO lines after Pyr treatment (C) and ratios of p-STAT3/t-STAT3 and p-ERK/t-ERK (D). (E and F) Western blot for basal p-STAT3 and p-ERK levels in Pyr-sensitive and -resistant hPCO lines (E), and ratios of p-STAT3/t-STAT3 and p-ERK/t-ERK (F). (G) Representative immunostaining images of p-STAT3 in Pyr-sensitive or – resistant hPCO lines. (H) H&E and p-STAT3 immunostaining images of gastric tissues, used for hPCO line establishment. (I and J) Phase-contrast images of Pyr-sensitive hPCO-2 treated with DMSO, Pyr, STAT3-IN-1, Sttatic, and Cryptotanshinone (CPT) for 6 days (I) and quantitation of organoid diameters before and after treatment (J). (K and L) Phase-contrast images of Pyr-resistant hPCO-31 treated with each drug for 6 days (K) and quantitation of organoid diameters (L). Scale bar = 500 μm. Mean ± standard deviation. Two-way ANOVA (with Tukey test for pairwise comparisons.
Discussion
Long-lasting tissue injury can lead to multifocal occurrences of pre-cancerous lesions, which accumulate genetic alterations over time and eventually develop multiple cancer foci in the stomach. Both severe metaplastic and dysplastic lesions are believed to be irreversible.24 Therefore, it is imperative to develop preventive therapeutic approaches that directly target these pre-cancerous lesions. However, few investigations have been performed in this area, due to the inherent difficulties in studying gastric pre-cancerous cell expansion and progression in humans. In this study, we took advantage of mouse and human pre-cancerous organoids and defined alterations in signaling pathways between metaplastic and dysplastic stages. We found that the STAT3 signaling pathway is dysregulated in the dysplastic stage and this caused different drug responses to Pyrvinium. It is important to note that Pyrvinium has been suggested as a Wnt pathway inhibitor by activating CK1α, which can regulate colon cancer cell survival.16 Also, several other signaling pathways, such as Hippo, hedgehog, and PI3K, can be regulated by Pyrvinium in various cancers.25–27 We have previously reported that Pyrvinium negatively controls the Wnt signaling pathway in dysplasia. In the present study, we report that Pyrvinium can negatively control the phosphorylation of ERK and STAT3 Y705. Also, a dual blockade of both STAT3 and MEK/ERK signaling pathways, not through a direct CK1α activation, leads to extensive cell death in dysplastic organoids (Figure 1G and Supplementary Figure 10).
Numerous data have demonstrated therapeutic effects of MEK inhibitors in various cancers.28 Our previous and present studies demonstrate suppressive effects of the MEK inhibitor, Trametinib, on the growth in mouse dysplastic cells. However, most human pre-cancerous organoid (hPCO) lines did not respond to Trametinib and hPCO lines with dysplastic features grew normally. hPCO lines were established from heterogeneous gastric cancer patient tissues and showed relatively lower phosphorylation levels of ERK. We found that both Pim-1 mRNA and protein levels are significantly downregulated in dysplastic organoids after Pyrvinium treatment. The PIM kinase family is overexpressed in various human cancers, and plays a crucial role in tumor progression and protection from cell apoptosis.29 Also, STAT3 can upregulate PIM1 gene transcription20 and the PIM family is required to maintain ERK signaling in prostate cancer19. We previously identified dysplastic stem cells, Trop2+CD133+CD166+ cells, first present in the dysplastic stage in both mouse and human. Single cell-RNAseq data analysis revealed that Pyrvinium specifically targets the dysplastic stem cells. Furthermore, Pyrvinium regressed in vivo metaplastic progression to dysplasia 2 weeks after Pyrvinium treatment in the Mist1-Kras mouse stomachs. Therefore, the drug response to Pyrvinium in pre-cancerous lesions does not only depend on the activation of both ERK and STAT3 pathways, but also the presence of dysplastic stem cells.
Strong activation of the STAT3 signaling pathway upon MEK inhibition has been reported in KRAS mutant cancer cells and a dual inhibition of MEK and STAT3 has been demonstrated to be more effective to treat oncogene-addicted cells.18, 30–32 Similarly, we observed high phosphorylation levels of STAT3 in both mouse and human dysplastic cells. It has been suggested that Pyrvinium’s dramatic anticancer activity is mediated by its suppressive action on the STAT3 pathway in KRAS-mutant lung cancer and leukemia.33, 34 Our study demonstrated that STAT3 inhibition in dysplastic organoids induced various responses and was much less effective in inducing cell death, compared with Pyrvinium treatment. All these findings suggest that a dual blockade of MEK and STAT3 is required in the treatment of dysplasia in the stomach. In contrast, Pyrvinium treatment did not induce apoptosis in metaplastic cells and only led to the growth arrest in both mouse and human metaplastic organoid lines, suggesting a lower likelihood of potential adverse effects on tissue regeneration. Therefore, our study provides evidence for Pyrvinium as a potential therapeutic candidate that targets dysplastic stem cells and can prevent gastric pre-cancer progression to gastric cancer. Since gastric metaplasia is considered as a pre-cancerous lesion associated with dysplasia development, it would be crucial to reduce the risk of overgrowth of metaplasia without compromising its protective effect.
Since STAT3 and MAPK pathways are also involved in immune cell proliferation and activation35, 36, it is important to note the potential impact of Pyrvinium on immune cells. Also, previous reports have highlighted the crucial role of various immune cells, such as type 2 innate lymphoid cells, M2 macrophages and MDSC, during gastric metaplasia development and progression.37–41 Although we observed a global decrease in immune cell populations after Pyrvinium treatment, our current study is limited in providing direct evidence of Pyrvinium effects on immune cells and subsequent alterations of inflammatory cytokines/chemokines. Therefore, further studies focusing on the direct effects of Pyrvinium on immune cells are necessary to gain a more comprehensive understanding of suppressive roles of Pyrvinium in gastric pre-cancerous lesions.
Development of direct pharmacological interventions for targeting pre-cancerous lesions represents a promising strategy for cancer prevention. However, identifying specific signaling pathways that are crucial for high-risk pre-cancerous lesions remains one of the most challenging issues in this field. Our study concludes that MEK/ERK and STAT3 signaling pathways are differentially activated in gastric pre-cancerous lesions. Also, Pyrvinium exerts a dual blockade of MEK/ERK and STAT3, inducing significant cell death in STAT3 dysregulated dysplastic cells. Taken together, the repurposing of Pyrvinium may present a meaningful therapeutic opportunity to reprogram the gastric mucosa in patients who harbor gastric pre-cancerous lesions and prevent gastric cancer development.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT:
Gastric metaplasia and dysplasia are associated with an increased risk of developing gastric cancer, a deadly disease with limited treatment options. However, understanding of signaling pathways involved in the progression of these pre-cancerous lesions is currently limited and there is a lack of effective therapeutic agents that specifically target these lesions.
NEW FINDINGS:
Simultaneous suppression of MEK/ERK and STAT3 signaling pathways by Pyrvinium induces growth arrest in metaplastic cells and cell death in dysplastic cells. Also, Pyrvinium specifically targets stem cells present in dysplasia.
LIMITATIONS:
While our study identified MEK/ERK and STAT3 signaling as crucial pathways for pre-cancerous lesion growth and survival, it is important to note that these pathways likely interact with other signaling networks associated with gastric cancer development. Therefore, further investigations are necessary to fully comprehend the molecular intricacies of these lesions and to develop more targeted and effective therapeutic strategies.
CLINICAL RESEARCH RELEVANCE:
Our preclinical data strongly support Pyrvinium as a promising candidate drug for preventing the gastric cancer development in patients with pre-cancerous lesions. These findings suggest that Pyrvinium may offer a valuable therapeutic option for reducing the gastric cancer burden, a major global health concern. Further clinical trials are warranted to investigate the efficacy and safety of Pyrvinium in human subjects.
BASIC RESEARCH RELEVANCE:
Our investigation identified a distinctive pattern of upregulation in MEK/ERK and STAT3 signaling pathways between metaplasia and dysplasia and a significant upregulation in STAT3 phosphorylation in both murine and human dysplastic cells. These findings suggest that the presence of distinct molecular signatures, associated with the transition from metaplasia to dysplasia, underscores the importance of identifying novel therapeutic targets for gastric cancer precursors.
Grant support:
This work was supported by National Institutes of Health (NIH) grants R37 CA244970 and R01 CA272687 and VICC GI SPORE (P50 CA236733), the AGA Research Foundation’s AGA-R. Robert & Sally Funderburg Research Award in Gastric Cancer (AGA2022-32-01), and the Gastric Cancer Foundation (to E.C.) and grants from a Department of Veterans Affairs Merit Review Award IBX000930, DOD CA190172, and NIH R01 DK101332 (to J.R.G). Core Services performed through Vanderbilt University Medical Center’s Digestive Disease Research Center (P30 DK058404) and Vanderbilt Ingram Cancer Center (P30 CA068485) with imaging in the Vanderbilt Digital Histology Shared supported by a VA Shared Instrumentation grant (1IS1BX003097). This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT, 2021R1C1C1011172 and 2020R1I1A1A01069168) (to B.J. and H.K.)
Abbreviations:
- AQP
aquaporin 5
- CK1α
casein kinase 1 alpha
- DMSO
dimethyl sulfoxide
- DSC
dysplastic stem cell
- EthD-1
ethidium homodimer-1
- FDA
food and drug administration
- FFPE
formaldehyde-fixed paraffin-embedded
- H&E
hematoxylin and eosin
- hPCO
human pre-cancerous organoid
- IM
intestinal metaplasia
- qRT-PCR
quantitative real-time PCR
- scRNA-seq
single cell RNA-sequencing
- SPEM
spasmolytic polypeptide-expressing metaplasia
- STAT3
Signal transducer and activator of transcription 3
- UMAP
Uniform Manifold Approximation and Projection
Footnotes
Conflict of interest disclosure statement: The authors have no conflicts of interest.
Transcript Profiling: Data and code are available by request. The scRNA-seq datasets reported in this paper have been deposited in the NCBI Gene Expression Omnibus (GEO) database under accession codes GSE240890.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Transparency Statement:
The data, analytical methods, and study materials will be made available to other researchers.
References
- 1.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 2.Smyth EC, Nilsson M, Grabsch HI, et al. Gastric cancer. The Lancet 2020;396:635–648. [DOI] [PubMed] [Google Scholar]
- 3.Chen H-N, Wang Z, Li X, et al. Helicobacter pylori eradication cannot reduce the risk of gastric cancer in patients with intestinal metaplasia and dysplasia: evidence from a meta-analysis. Gastric cancer 2016;19:166–175. [DOI] [PubMed] [Google Scholar]
- 4.Correa P Helicobacter pylori and gastric carcinogenesis. The American journal of surgical pathology 1995;19:S37–43. [PubMed] [Google Scholar]
- 5.Suerbaum S, Michetti P. Helicobacter pylori infection. New England Journal of Medicine 2002;347:1175–1186. [DOI] [PubMed] [Google Scholar]
- 6.Cho SJ, Choi I, Kook MC, et al. Staging of intestinal-and diffuse-type gastric cancers with the OLGA and OLGIM staging systems. Alimentary pharmacology & therapeutics 2013;38:1292–1302. [DOI] [PubMed] [Google Scholar]
- 7.Choi E, Hendley AM, Bailey JM, et al. Expression of activated Ras in gastric chief cells of mice leads to the full spectrum of metaplastic lineage transitions. Gastroenterology 2016;150:918–930. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Min J, Vega PN, Engevik AC, et al. Heterogeneity and dynamics of active Kras-induced dysplastic lineages from mouse corpus stomach. Nature communications 2019;10:5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Min J, Zhang C, Bliton RJ, et al. Dysplastic Stem Cell Plasticity Functions as a Driving Force for Neoplastic Transformation of Precancerous Gastric Mucosa. Gastroenterology 2022;163:875–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schultz CW, Nevler A. Pyrvinium pamoate: Past, present, and future as an anti-cancer drug. Biomedicines 2022;10:3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venugopal C, Hallett R, Vora P, et al. Pyrvinium targets CD133 in human glioblastoma brain tumor–initiating cells. Clinical Cancer Research 2015;21:5324–5337. [DOI] [PubMed] [Google Scholar]
- 12.Xiang W, Cheong JK, Ang SH, et al. Pyrvinium selectively targets blast phase-chronic myeloid leukemia through inhibition of mitochondrial respiration. Oncotarget 2015;6:33769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Datta S, Sears T, Cortopassi G, et al. Repurposing FDA approved drugs inhibiting mitochondrial function for targeting glioma-stem like cells. Biomedicine & Pharmacotherapy 2021;133:111058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mologni L, Brussolo S, Ceccon M, et al. Synergistic effects of combined Wnt/KRAS inhibition in colorectal cancer cells. PloS one 2012;7:e51449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dhir T, Schultz CW, Jain A, et al. Abemaciclib is effective against pancreatic cancer cells and synergizes with HuR and YAP1 inhibition. Molecular Cancer Research 2019;17:2029–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thorne CA, Hanson AJ, Schneider J, et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nature chemical biology 2010;6:829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li B, Orton D, Neitzel LR, et al. Differential abundance of CK1α provides selectivity for pharmacological CK1α activators to target WNT-dependent tumors. Science signaling 2017;10:eaak9916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee H-J, Zhuang G, Cao Y, et al. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer cell 2014;26:207–221. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Anderson PD, Luo W, et al. Pim1 kinase is required to maintain tumorigenicity in MYC-expressing prostate cancer cells. Oncogene 2012;31:1794–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahata S, Sahoo PK, Pal R, et al. PIM1/STAT3 axis: A potential co-targeted therapeutic approach in triple-negative breast cancer. Medical Oncology 2022;39:74. [DOI] [PubMed] [Google Scholar]
- 21.Zaiss DM, Gause WC, Osborne LC, et al. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 2015;42:216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petersen CP, Weis VG, Nam KT, et al. Macrophages promote progression of spasmolytic polypeptide-expressing metaplasia after acute loss of parietal cells. Gastroenterology 2014;146:1727–1738. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Curtius K, Wright NA, Graham TA. An evolutionary perspective on field cancerization. Nature Reviews Cancer 2018;18:19–32. [DOI] [PubMed] [Google Scholar]
- 24.Yoon H Atrophic gastritis and intestinal metaplasia. Helicobacter pylori 2016:515–520. [Google Scholar]
- 25.Carrella D, Manni I, Tumaini B, et al. Computational drugs repositioning identifies inhibitors of oncogenic PI3K/AKT/P70S6K-dependent pathways among FDA-approved compounds. Oncotarget 2016;7:58743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li B, Fei DL, Flaveny CA, et al. Pyrvinium attenuates Hedgehog signaling downstream of smoothened. Cancer research 2014;74:4811–4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Basu D, Reyes-Mugica M, Rebbaa A. Histone acetylation-mediated regulation of the Hippo pathway. PloS one 2013;8:e62478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Y, Adjei AA. The clinical development of MEK inhibitors. Nature reviews Clinical oncology 2014;11:385–400. [DOI] [PubMed] [Google Scholar]
- 29.Tursynbay Y, Zhang J, Li Z, et al. Pim-1 kinase as cancer drug target: An update. Biomedical reports 2016;4:140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoon YK, Kim HP, Han SW, et al. KRAS mutant lung cancer cells are differentially responsive to MEK inhibitor due to AKT or STAT3 activation: implication for combinatorial approach. Molecular Carcinogenesis: Published in cooperation with the University of Texas MD Anderson Cancer Center 2010;49:353–362. [DOI] [PubMed] [Google Scholar]
- 31.Zhao C, Xiao H, Wu X, et al. Rational combination of MEK inhibitor and the STAT3 pathway modulator for the therapy in K-Ras mutated pancreatic and colon cancer cells. Oncotarget 2015;6:14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ryan MB, Corcoran RB. Therapeutic strategies to target RAS-mutant cancers. Nature reviews Clinical oncology 2018;15:709–720. [DOI] [PubMed] [Google Scholar]
- 33.Zheng Z-Y, Chu M-Y, Lin W, et al. Blocking STAT3 signaling augments MEK/ERK inhibitor efficacy in esophageal squamous cell carcinoma. Cell Death & Disease 2022;13:496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Decroocq J, Birsen R, Montersino C, et al. RAS activation induces synthetic lethality of MEK inhibition with mitochondrial oxidative metabolism in acute myeloid leukemia. Leukemia 2022;36:1237–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nature reviews cancer 2009;9:798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arthur JSC, Ley SC. Mitogen-activated protein kinases in innate immunity. Nature Reviews Immunology 2013;13:679–692. [DOI] [PubMed] [Google Scholar]
- 37.Meyer AR, Engevik AC, Madorsky T, et al. Group 2 innate lymphoid cells coordinate damage response in the stomach. Gastroenterology 2020;159:2077–2091. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Salvo C, Pastorelli L, Petersen CP, et al. Interleukin 33 triggers early eosinophil-dependent events leading to metaplasia in a chronic model of gastritis-prone mice. Gastroenterology 2021;160:302–316. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding L, Li Q, Chakrabarti J, et al. MiR130b from Schlafen4+ MDSCs stimulates epithelial proliferation and correlates with preneoplastic changes prior to gastric cancer. Gut 2020;69:1750–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ding L, Chakrabarti J, Sheriff S, et al. Toll-like Receptor 9 pathway mediates Schlafen+-MDSC polarization during Helicobacter-induced gastric metaplasias. Gastroenterology 2022;163:411–425. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ding L, Sheriff S, Sontz RA, et al. Schlafen4+-MDSC in Helicobacter-induced gastric metaplasia reveals role for GTPases. Frontiers in Immunology 2023;14:1139391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data, analytical methods, and study materials will be made available to other researchers.