

Abstract
Proteins exist as dynamic conformational ensembles. Here we suggest that the propensities of the conformations can be predictors of cell function. The conformational states that the molecules preferentially visit can be viewed as phenotypic determinants, and their mutations work by altering the relative propensities, thus the cell phenotype. Our examples include (i) inactive state variants harboring cancer driver mutations that present active state-like conformational features, as in K-Ras4BG12V compared to other K-Ras4BG12X mutations; (ii) mutants of the same protein presenting vastly different phenotypic and clinical profiles: cancer and neurodevelopmental disorders; and (iii), alterations in the occupancies of the conformational (sub)states influencing enzyme reactivity. Thus, protein conformational propensities can determine cell fate. They can also suggest the allosteric drugs efficiency.
Keywords: Conformational ensembles, cell fate, occupancy, cancer, RASopathies, neurodevelopmental disorders
Introduction
Recently, we called for a revision of the decades-old sequence-structure-function paradigm and replacing it by a modern sequence-conformational ensemble-function paradigm. We argued that such a revised outlook more accurately encapsulates the linkage between sequence and function, and especially, is required by the updated dynamic energy landscape [1].
Proteins are not rigid, neither in vitro nor in the cell. They fluctuate. They sample an ensemble of states, triggering conformational heterogeneity. The states are visited with different frequencies. High energy states are visited rarely, low energy states frequently. The smaller conformational fluctuations of stable folded proteins sample states around the average structure yielding more homogeneous ensembles. Unstable, or intrinsically disordered proteins that interconvert between a broad range of conformations with relatively low barriers between them, can present high conformational heterogeneity [2]. Clustering of the conformations [3], or describing the ensembles by their associated thermodynamic weights [4], can describe the proteins’ structural heterogeneity. The input can come from biophysical experiments, such as crystallography, single-molecule fluorescence (or Förster) resonance energy transfer (smFRET) experiments that measure distances at the 1-10 nM range, single-particle cryo-electron microscopy (cryo-EM), which can capture conformational transitions by observing single conformer in each condition, cryo-X-ray structural snapshots from multiple related structures [5], quantitative backbone structural dynamics by solution nuclear magnetic resonance (NMR) spectroscopy [6], computational models obtained from explicit solvent long time scales molecular dynamics (MD) simulations [7], and especially, MD/NMR combination [8,9]. As we and others have shown, this combination is powerful in clarifying the mechanism of autoinhibition [10] and in showing how oncogenic mutations can alter the dynamic nature of the K-Ras/calmodulin complex [8]. The frequencies of visits of close conformations can provide the propensities of the conformational states. Here we suggest that propensities of different states can express distinct protein and cell phenotypes (Figure 1 depicts the concept). High propensity of active state-like conformations of a mutational variant obtained from an inactive structure can point to a strong mutation with a consequent high chance of a transformed cell phenotype [7]. In a protein that can act in two vastly different phenotypes, cancer and neurodevelopmental disorders (NDDs), a stronger mutation with an active state profile may point to cancer. Our premise is that a strong mutation elicits strong signaling ([7] and references therein). However, on their own, single proteins, and single mutations do not determine cell phenotype. Homeostatic mechanisms, the types and locations of additional mutations, expression levels, cell type, timing, and chromatin structure, all play key roles [11,12].
Figure 1.
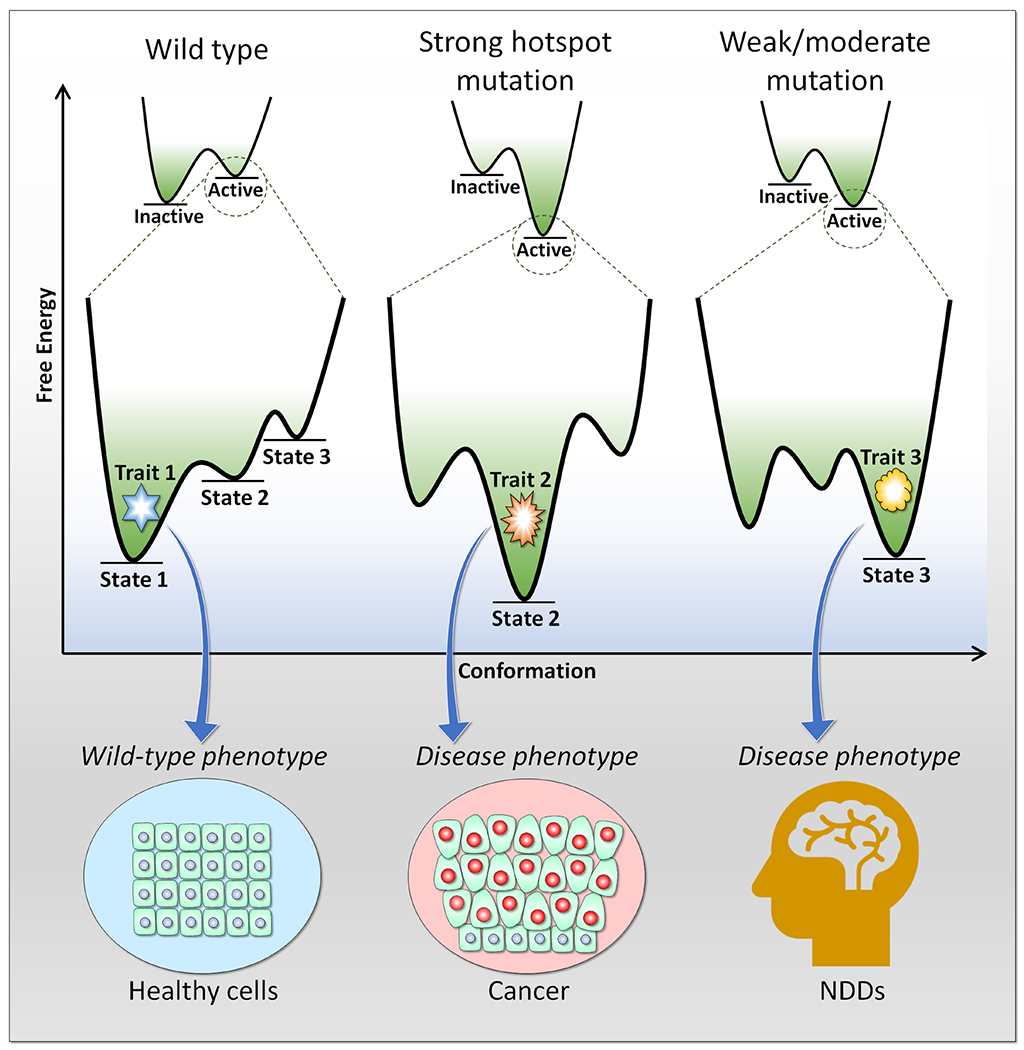
Free energy profiles of different conformational states expressing different phenotypic cell traits. For example, strong hotspot mutations alter the conformational states of the protein, generating a cancer disease phenotype, while weak/moderate mutations express a NDD disease phenotype.
The heterogeneity of conformational ensembles is important since distinct conformations may define functional specificities. Nuclear receptors that act as transcription factors through ligand-linked conformational changes provide one example [13]. Substitutions at a key position resulted in altered ligand specificities for multiple ligands through distinct favored conformations. The conformers’ populations are specifically allosterically shifted by the different mutations, altering their propensities. Atomistic MD simulations with enhanced sampling captured the resulting allosteric population shifts in the ensembles and correlated them with ligand-specific transcriptional activation. Mechanistically, pairwise cooperativities, where binding at the ‘receiving end’ is modulated solely by binding at the ‘initiating end’ is not an accurate representation [14]. Multiple steps are involved along the favored allosteric propagation pathways [15]. These binding events reflect multiple conformational selection steps including collectively modulated additional binding events involving concurrent regulation in the cell. Resolving the local energetic conflicts experienced during binding induces allosteric effects [16,17]. The dynamically interchanging conformations transform the propensities of the conformers in the ensemble. Allosteric population shifts in the ensembles promoted by covalent events are not limited to mutations, but also include posttranslational modifications, which can also alter function [18]. Finally, chaperones, the nanoscale molecular machines that recognize incompletely or incorrectly folded protein clients, arrest and assist in refolding them are a remarkable example of the link between changes in conformations and phenotypes [19,20].
We define propensities as the number of active molecules and suggest that determination of this number is significant for several reasons. Not only does it help in dissecting the relationships between the ensemble and protein (or RNA [21]) function. As we discuss below, it can help in determining, and predicting, the phenotype associated with the higher (lower) propensities. Within this framework, we recall the community effort of predicting protein function from its sequence [22] or structural features [23,24]. The relationship, and significance of heterogeneous conformational ensembles for function has increasingly been considered (e.g., [25–32]).
Here we underscore the significance and feasibility of predicting function from conformational propensities. In line with this, the effectiveness of allosteric drugs can be measured, and thus predicted, by the changes in propensities of the relevant conformations that they elicit.
Definitions and model overview
First, what is a “protein state” and what is a “protein conformation”? Both terms relate to protein ensembles with structures that are interconverting along time and conditions. Protein disorder is commonly referred to as the “disordered state”. Yet, when relating to protein shapes within an ensemble of “ordered” proteins, “state” and “conformation” are often used interchangeably. Here our definitions are based on the free energy landscape, and in line with the multiple interconverting molecules in the disordered state. In our definition, multiple shapes [(sub)states] located around the bottom of a minimum with minor differences in energy, which are separated by low kinetic barriers, constitute a conformational state (Figure 2), whereas conformational states are separated by higher barriers. An allosteric mutation can alter the relative stabilities of the states, and of the substates that they embody. Since a conformational state is described by bonded and nonbonded intramolecular interactions, different conformational states of the molecules would be impacted in different ways.
Figure 2.
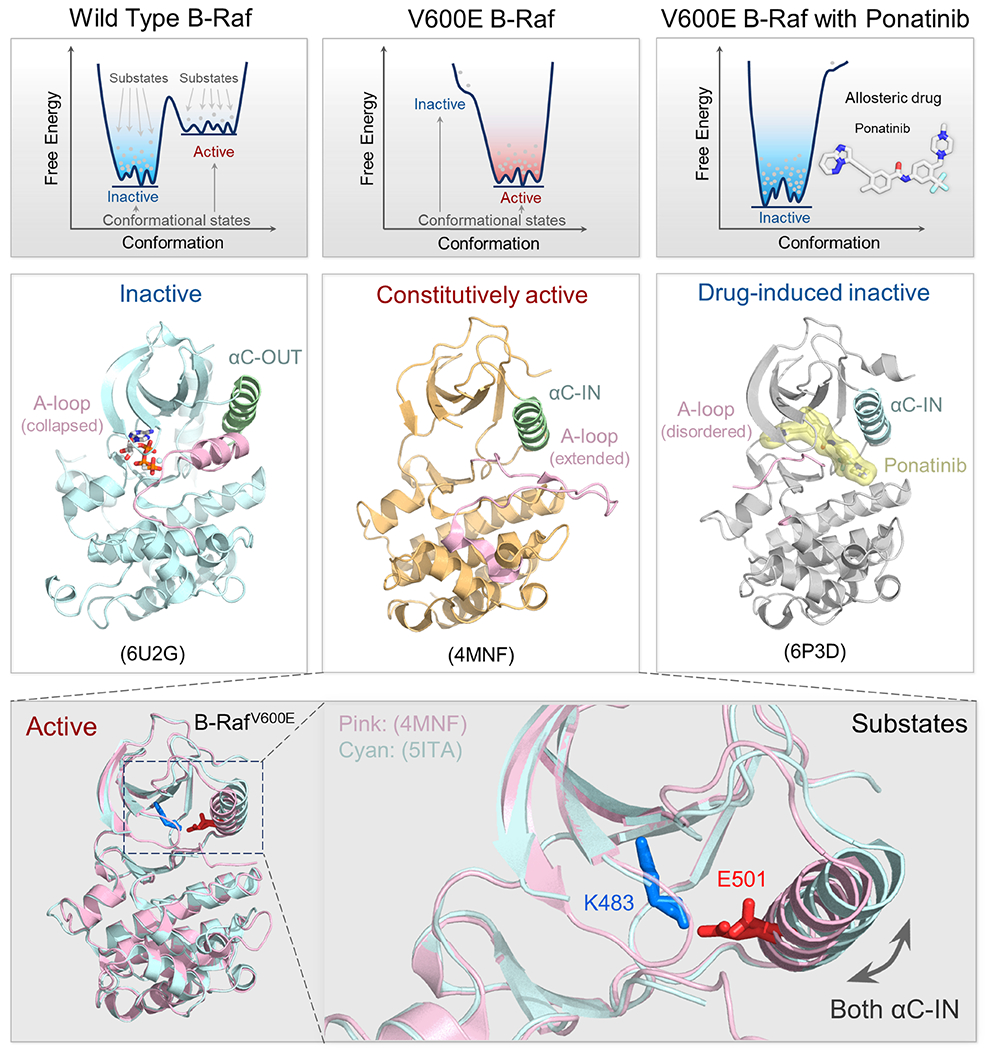
Illustration of the conformational states and substates of wild-type B-Raf, B-RafV600E, and allosterically inhibited B-RafV600E. Dynamic free energy landscapes and structures of B-Raf, B-RafV600E, and ponatinib bound B-RafV600E (top panels). The free energy shows the distribution and population of protein states. Each well represents a distinct conformational state (active or inactive), with the depth of the well indicating the stability of the state. Within each well, multiple dots indicate the population of substates that a protein can adopt. The barriers between these substates are lower than the barriers separating different conformational states. In the left column, wide type B-Raf primarily adopts an inactive state; in the middle column, B-RafV600E primarily shows an active state; in the right column, B-RafV600E bound to an allosteric drug ponatinib which disrupts its ability to phosphorylate, resulting in population shift towards the inactive state. Their predominant structures are shown below their respective free energy landscape plots (middle panels). Note that the V600E mutation shifts the relative stability of the protein from favoring the inactive state to the active state. We annotate the PDB code in each panel for reference. Within each conformational state, the protein can adopt a range of substates (bottom panels). For example, active B-RafV600E shows structural variations in the position of the αC helix. The important salt bridge between K483-E501 represents a key feature of the active protein kinase conformation.
The propensities of the conformational states relate to their relative energies. Apart from repressors, under physiological conditions, the number of molecules populating the active conformational state is lower than that in the inactive states. The enhanced sampling of a variant harboring a driver mutation temporally increases the number of molecules with active conformational state features; in enzymes, making it predisposed for catalysis, in small GTPases, making it predisposed for effector activation. The number of active molecules decides signaling strength, thus cell function. Effective allosteric drugs reduce the number active molecules. Cellular processes depend on interactions, which are mediated by intermolecular contacts. In turn, these depend on binding affinity and the number of available, binding-competent conformations, that is, the propensities of the relevant conformational states [33–35]. Mapped occupancies of active conformational states can help foretell cell fate by harnessing the free energy landscapes and conformational dynamics.
Prediction of functional cell states and cell fates
The functions of a large number of proteins remain unknown, lacking experimental and manual annotation [36]. Computational methods have been exploiting the data, developing, and applying functional prediction algorithms most commonly from sequences, using global and local (multiple) sequence alignments searching for, and interrogating homologs. ‘Function association matrices’ have also been used to annotate even remote homologs, and recently, sequence-based protein language models have been developed as well. Experimentally determined structural information was not used as much for protein function prediction largely due to its paucity as compared to known sequences. AlphaFold2 [37] and other recent machine language-based structural prediction methods, such as the fast Meta AI ESMFold [38], which are filling in the ‘dark matter’ of the protein universe, will likely alter this landscape. AlphaFold2 has been used in large-scale prediction of protein functions through heterogeneous feature fusion [36]. Protein 3D structural data are advantageous since structure is better conserved than sequence. Amino acid contact maps have been successfully exploited as well [24]. However, none of these directly capture conformational mechanisms like allostery, which are rooted in ensembles, and controlled by their dynamic distributions [39]. Allostery and signaling are properties of populations, whereas these methods predict structural snapshots ranked by a scoring function. To obtain conformational data they can be applied multiple times with different parameters; however, they will still not provide propensities which are influenced by kinetic barrier heights that separate the states.
The quintessential nature of predictions derived from propensities of conformational states differ from predictions of function from sequences in a number of ways. First, they provide a conformational profile of a protein, whose function is already known. Second, that conformational profile, which epitomizes conformations that have been preferentially visited, and their associated propensities, can be viewed as key phenotypic determinants of the cell. Conformational profiles provide a phenotypic resolution which neither sequences nor 3D structures are able to attain. Beyond the known function of the protein, through their propensities, they may describe protein action across time. Since they point to the number of molecules that exist in the active conformational state, this may allow us to consider questions such as, what is the extent of the protein activity. Further, if the protein can contribute to multiple diseases, as in the case of phosphoinositide 3-kinase (PI3K) lipid kinase and Src homology 2 (SH2) domain-containing tyrosine phosphatase-2 (SHP2) phosphatase acting in cancer and NDDs, the propensities may suggest which of these diseases is the one that is more likely to emerge from certain mutations or their combinations. The extent of protein activity and the related disease can provide a signature of the cell phenotype. Taken together, this posits the methodological challenge of how to obtain propensities, which could integrate with algorithms for prediction of function, for a more complete and relevant functional description.
Methods for observing and predicting propensities
Acquiring temporal, high resolution atomic-level ensemble models that accurately represent conformational heterogeneity is vital to understanding of how proteins and cells work [40]. Several experimental techniques, including time-resolved X-ray crystallography and cryo-EM [41–45], and spectroscopic methods [46,47], can trap conformations. However, their usefulness may fall short. The conformations need to be generated which is technically complex [47]. Modeling conformational variability at ambient temperatures directly from X-ray diffraction data has been challenging [40]. Recent advances in refinement of multiconformer ensemble models from multitemperature X-ray diffraction data have made the collection of high-quality heterogeneous diffraction data possible. Integrating automated sampling with manual refinement of diffraction datasets at different temperatures (313 to 363 K) resulted in multiconformer models, including their relative occupancies, and interconnections.
High resolution atomistic, long time scales conformational sampling by MD simulations, or Monte Carlo, can usefully predict and capture temporal propensities, as well as the kinetic barriers for conformational switching [41,48–50]. However, high barriers require lengthy time scales for exhaustive sampling of the conformational space, unless a priori constrained by experimental measurements [51,52]. Emerging integrated machine learning–MD methods may help in large scale systems, as well as enhanced sampling techniques [31,53–56]. MD simulations can describe molecular mechanisms by exploiting empirical potentials, which can be improved by machine learning approaches. As we discuss below, in our hands, conformational behavior of protein variants observed in explicit solvent MD simulations could distinguish between disease outcomes even though the differences may be moderate, pointing to the challenges in sampling and the requirements for longer time scales, or emerging superior sampling approaches, as the above methods aim to accomplish.
The K-Ras4B mutational variants, cancer and NDDs, and enzyme examples
Sampling of conformational space in vivo is influenced by the cellular environment, which is challenging to capture. However, conformational studies in solution may still unravel puzzling in vivo consequences. We surmised that the differences in the propensities of conformational states of K-Ras4B variants could be a key factor in the differences in the rates of GTP hydrolysis, nucleotide exchange rates, and selectivity for plasma membrane phospholipids, thus in oncogenic aggressiveness [57]. KRAS is the most highly mutated RAS gene in human cancer, causing various cancer phenotypes in different organs (Figure 3a). Among the oncogenic mutations at the active site, G12 is the most highly populated, followed by G13 and Q61. To test our conjecture, we considered two strong K-Ras4B mutations, K-Ras4BG12V and K-Ras4BG12D (Figure 3b). The first is the strongest K-Ras4B mutation. It is the most aggressive and chemotherapy resistant. The second is the most frequent and the key mutation in pancreatic adenocarcinoma. Both single residue substitutions decelerate intrinsic and catalyzed GTP hydrolysis, retaining the protein in a constitutively active state, promoting strong cell proliferation. The crystal structures of both mutants are similar, providing no clue to their differential oncogenic behavior. To discern why the difference in the aggressiveness of the mutations, we collected NMR data and performed explicit solvent MD simulations of the active and inactive conformations, aiming to explore its conformational sampling, thus occupancies [7]. We observed that the two mutational variants exhibit distinct conformational dynamics in their GDP-bound states, which in the wild-type protein is the inactive state. As expected from constitutive activating mutations, even in this state, they often visit active-like conformations, resembling that of the active GTP-bound state. However, their conformational profiles differed. K-Ras4BG12V visited active-like conformations much more frequently than the K-Ras4BG12D did. Inspection of the conformational details explained the reason: The fluctuating interactions of the aliphatic sidechain of the Val12 with the Switch II region of K-Ras4BG12V-GDP differ from those of Asp12 in GDP-bound state, which is not observed in the crystal structure, where the contacts in the crystalline state constrain the dynamics by stabilizing the Switch I region of the protein. Thus, the differences in the G12 mutants’ conformations can be explained by the contacts with the Switch II region which stabilize the active-like state, resolving the differential oncogenic aggressiveness conundrum.
Figure 3.
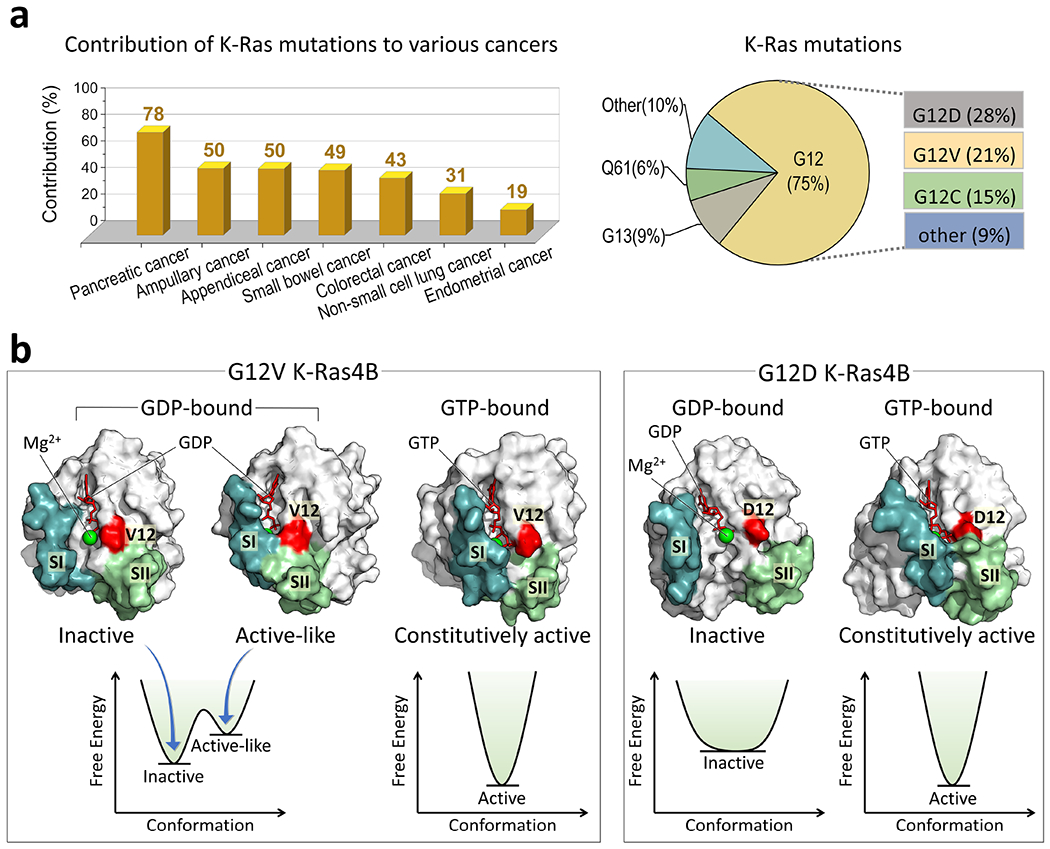
(a) K-Ras mutations can drive cancers. Analysis of the AACR cancer cohort (GENIE Cohort v13.1-public) reveals that K-Ras mutations have high associations with pancreatic, ampullary, appendiceal, small bowel, colorectal, non-small cell lung, and endometrial cancers. The most prevalent K-Ras mutation sites are at codon 12, 13, and 61, with G12 mutations being the most frequent, accounting for ~75% of all K-Ras mutations. Among these mutations, G12D is the most common (~28%), followed by G12V (~23%) and G12C (~15%). (b) K-Ras4B encompasses two critical regions, Switch I (SI) and Switch II (SII). In the inactive GDP-bound state, the two regions are separated (referred as the open SI-SII conformation), which prevents the K-Ras interactions with its effectors. In the active GTP-bound state, these two regions come into closer proximity (referred as the closed SI-SII conformation), favorable for the effector binding. Oncogenic mutations in K-Ras4B can shift the equilibrium towards the active state. The G12V and G12D mutants have a high population in the GTP-bound state with the closed SI-SII conformation, and a low population in the GDP-bound state with the open SI-SII conformation. The G12V mutation induces a more potent activation of K-Ras compared to the G12D mutation. This difference in activation strength may arise from the distinct dynamic ensembles of the two mutants. In the GDP-bound state, K-Ras carrying the most aggressive G12V mutation visits frequently the active-like conformation, featuring instances of SI and SII separation. This further amplifies the likelihood of downstream effectors binding, thereby intensifying the signal transduction.
In the second example, same protein single mutation variants can encode the vastly different phenotypic and clinical profiles of cancer and NDDs, leading us to query how [12,58]? To resolve this mystery, we selected key cancer mutations (E76K and D61Y/V) in SHP2 phosphatase (Figure 4) which regulates MAPK, and compared them with Noonan syndrome (NS, a RASopathy, a group of NDDs [59–62]) mutations of the same residues (E76D and D61G). We observed that the cancer drivers could induce a shift in the SHP2 ensemble toward the active state. As to the RASopathy mutations, they presented only limited conformational transitions, thus are less likely to promote proliferation. Thus, again, as the K-Ras4B variants above, conformational behavior and propensities of occupying active states-like conformations can predict not only mutation strength and clinical relevance, but are also capable in distinguishing, thus forecasting, disease outcome.
Figure 4.
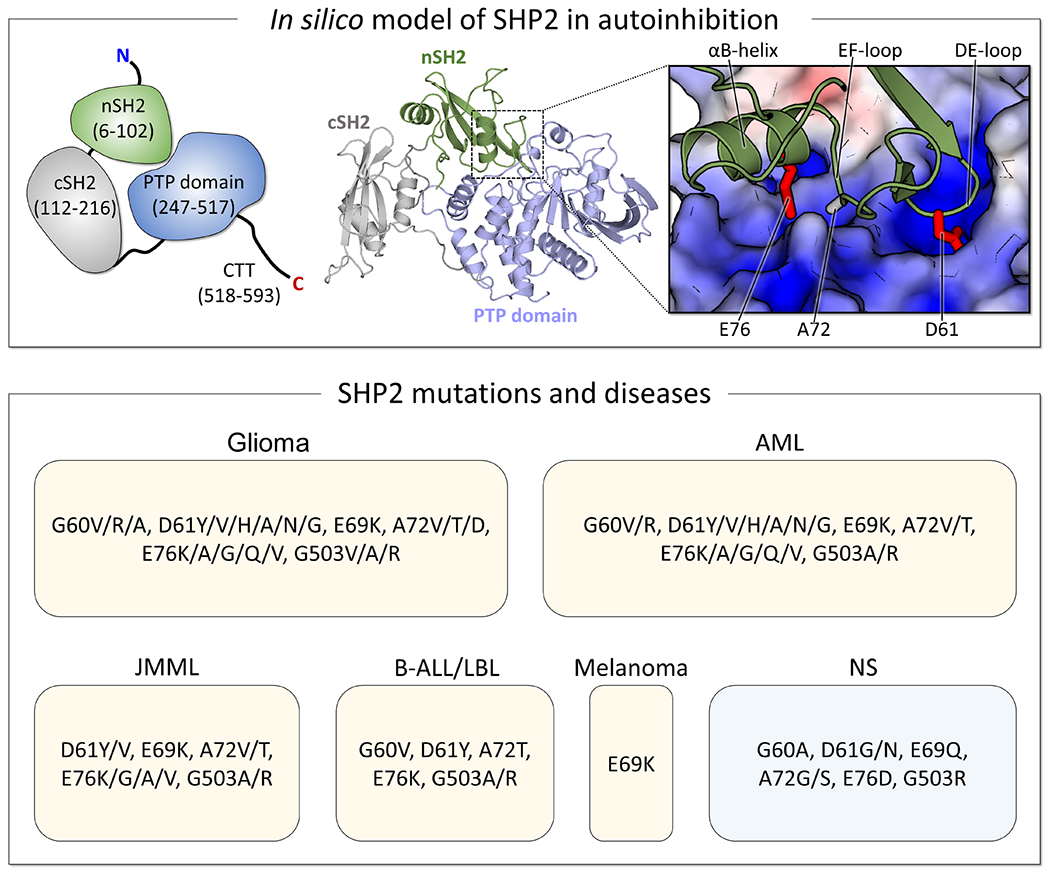
SHP2, a tyrosine phosphatase, consists of the nSH2, cSH2, and PTP domains. In its autoinhibited state, the nSH2 domain tightly binds to the catalytic cleft of the PTP domain, with D61 and E76/A72 orienting towards the active and allosteric sites, respectively. Analysis of the TCGA and GENIE databases identifies hotspot mutation sites as G60, D61, E69, A72, and E76 in the nSH2 domain, and G503 in the PTP domain. Mutations at these sites can drive various cancers, including juvenile myelomonocytic leukemia (JMML), acute myeloid leukemia (AML), B lymphoblastic leukemia/lymphoma (B-ALL/LBL), glioma, and melanoma, as well as neurodevelopmental disorders (NDD) like Noonan syndrome (NS). Interestingly, some mutations are shared between cancer and NS, but they are significantly rare in cancer cases. Some mutations at the hotspot mutation sites for several representative cancers and NS are listed.
Our third example [5] involves substituting a tyrosine in the ketosteroid isomerase (KSI) enzyme with phenylalanine, changing the bound and the reactive ensemble, but with the ninefold rate decrease arising from a weakened hydrogen bond, thus conformational dynamics, which impacts protein function and enzyme catalysis. Finally, conformational variants can influence aggregation [63,64].
Conclusions
Structures captured in crystals cannot point to the cell phenotype. Despite their vast merit, crystal structures capture static structural snapshots that are populated under the conditions of the experiments. As to single cell transcriptomics, if followed and measured at different cell states and time frames, it can point to the cell’s phenotype [65], including our example when differentiating between cancer cells and NDDS, where we observed different levels at different cell states [66].
Here we suggested that propensities of conformational states obtained by detailed MD simulations can do this too. Each method has its pluses and drawbacks. Single cell transcriptomics require time, laboratory set-up, tools and are costly. Simulations are handicapped by hurdles too. Still, they are more affordable and with recent developments in machine language/AI assistance, they get faster, increasingly accomplishing their aims [67].
Here our linchpin theory is that propensities of protein conformational states may powerfully predict cell phenotypes [1], and we believe that constructions of such software tools are feasible, albeit challenging. Data of mutation outcomes are available, and MD simulations are becoming increasingly routine. We further note that since effective allosteric drugs can also bias ensembles, their action may be captured as well. We offer that eventually, prediction of cell function from conformational ensembles can extend prediction of protein function and is worth considering.
Acknowledgements
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261201500003I. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This Research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Nussinov R, Liu Y, Zhang W, Jang H: Protein conformational ensembles in function: roles and mechanisms. RSC Chem Biol 2023:doi: 10.1039/D1033CB00114H. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This paper proposed revising the sequence-structure-function paradigm to sequence-conformational ensemble-function. The decades-old mindset implicitly considered that for each sequence there exists a single, well-organized protein structure. Yet, as this paper indicated, to sustain cell life, function requires (i) that there be more than a single structure, (ii) that there be switching between the structures, and (iii) that the structures be incompletely organized. Proteins are always interconverting between conformational states with varying energies. The more stable the conformation the more populated it is.
- 2.Kutlu Y, Ben-Tal N, Haliloglu T: Global Dynamics Renders Protein Sites with High Functional Response. J Phys Chem B 2021, 125:4734–4745. [DOI] [PubMed] [Google Scholar]
- 3.Sayilgan JF, Haliloglu T, Gonen M: Protein dynamics analysis identifies candidate cancer driver genes and mutations in TCGA data. Proteins 2021, 89:721–730. [DOI] [PubMed] [Google Scholar]
- 4.Thomasen FE, Lindorff-Larsen K: Conformational ensembles of intrinsically disordered proteins and flexible multidomain proteins. Biochem Soc Trans 2022, 50:541–554. [DOI] [PubMed] [Google Scholar]
- 5.Yabukarski F, Doukov T, Pinney MM, Biel JT, Fraser JS, Herschlag D: Ensemble-function relationships to dissect mechanisms of enzyme catalysis. Sci Adv 2022, 8:eabn7738. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Using the enzyme ketosteroid isomerase (KSI) as an example, this paper demonstrates the need for conformational ensembles to understand function. It further postulates that Ensemble-function studies will have an integral role in understanding enzymes and in meeting the future goals of a predictive understanding of enzyme catalysis and engineering new enzymes
- 6.Hansen AL, Xiang X, Yuan C, Bruschweiler-Li L, Bruschweiler R: Excited-state observation of active K-Ras reveals differential structural dynamics of wild-type versus oncogenic G12D and G12C mutants. Nat Struct Mol Biol 2023, 10.1038/s41594-023-01070-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grudzien P, Jang H, Leschinsky N, Nussinov R, Gaponenko V: Conformational Dynamics Allows Sampling of an “Active-like” State by Oncogenic K-Ras-GDP. J Mol Biol 2022, 434:167695. [DOI] [PubMed] [Google Scholar]; •• K-Ras4B has two two strong mutations, K-Ras4BG12V and K-Ras4BG12D. The first is the strongest K-Ras4B mutation. It is the most aggressive and chemotherapy resistant. The second is the most frequent and the key mutation in pancreatic adenocarcinoma. The authors ask why this difference. Exploiting molecular dynamics simulations and NMR they showed that the mutational variants differ in the conformational dynamics in their GDP-bound states, which in the wild-type protein is the inactive state. K-Ras4BG12D visited active-like conformations much more frequently than the K-Ras4BG12V did, and they explained why, resolving the differential oncogenic aggressiveness conundrum.
- 8.Abdelkarim H, Leschinsky N, Jang H, Baneijee A, Nussinov R, Gaponenko V: The dynamic nature of the K-Ras/calmodulin complex can be altered by oncogenic mutations. Curr Opin Struct Biol 2021, 71:164–170. [DOI] [PubMed] [Google Scholar]
- 9.Jang H, Banerjee A, Chavan T, Gaponenko V, Nussinov R: Flexible-body motions of calmodulin and the farnesylated hypervariable region yield a high-affinity interaction enabling K-Ras4B membrane extraction. J Biol Chem 2017, 292:12544–12559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chavan TS, Jang H, Khavrutskii L, Abraham SJ, Banerjee A, Freed BC, Johannessen L, Tarasov SG, Gaponenko V, Nussinov R, et al.: High-Affinity Interaction of the K-Ras4B Hypervariable Region with the Ras Active Site. Biophys J 2015, 109:2602–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nussinov R, Tsai CJ, Jang H: A New View of Activating Mutations in Cancer. Cancer Res 2022, 82:4114–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nussinov R, Tsai CJ, Jang H: How can same-gene mutations promote both cancer and developmental disorders? Sci Adv 2022, 8:eabm2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khan SH, Braet SM, Koehler SJ, Elacqua E, Anand GS, Okafor CD: Ligand-induced shifts in conformational ensembles that describe transcriptional activation. Elife 2022, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This paper provides a platform that allows structural characterization of physiologically-relevant conformational ensembles, which may also enable to design and predict transcriptional responses in novel ligands. The authors investigate ligand-induced conformational changes using a reconstructed, ancestral nuclear receptor. By making substitutions at a key position, they engineer receptor variants with altered ligand specificities.
- 14.Biddle JW, Martinez-Corral R, Wong F, Gunawardena J: Allosteric conformational ensembles have unlimited capacity for integrating information. Elife 2021, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nussinov R, Tsai CJ, Jang H: Allostery, and how to define and measure signal transduction. Biophys Chem 2022, 283:106766. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This paper innovates by asking and suggesting “What is productive signaling? How to define it, how to measure it, and most of all, what are the parameters that determine it? Further, what determines the strength of signaling from an upstream to a downstream node in a specific cell?” It notes that these questions have either not been considered or not entirely resolved despite their significance.
- 16.Guzovsky AB, Schafer NP, Wolynes PG, Ferreiro DU: Localization of Energetic Frustration in Proteins. Methods Mol Biol 2022, 2376:387–398. [DOI] [PubMed] [Google Scholar]
- 17.Kumar A, Jernigan RL: Ligand Binding Introduces Significant Allosteric Shifts in the Locations of Protein Fluctuations. Front Mol Biosci 2021, 8:733148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nussinov R, Tsai CJ, Xin F, Radivojac P: Allosteric post-translational modification codes. Trends Biochem Sci 2012, 37:447–455. [DOI] [PubMed] [Google Scholar]
- 19.Horovitz A, Reingewertz TH, Cuellar J, Valpuesta JM: Chaperonin Mechanisms: Multiple and (Mis)Understood? Annu Rev Biophys 2022, 51:115–133. [DOI] [PubMed] [Google Scholar]
- 20.Horovitz A, Fleisher RC, Mondal T: Double-mutant cycles: new directions and applications. Curr Opin Struct Biol 2019, 58:10–17. [DOI] [PubMed] [Google Scholar]
- 21.Ding J, Lee YT, Bhandari Y, Schwieters CD, Fan L, Yu P, Tarosov SG, Stagno JR, Ma B, Nussinov R, et al. : Visualizing RNA conformational and architectural heterogeneity in solution. Nat Commun 2023, 14:714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kilinc M, Jia K, Jernigan RL: Improved global protein homolog detection with major gains in function identification. Proc Natl Acad Sci U S A 2023, 120:e2211823120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gligorijevic V, Renfrew PD, Kosciolek T, Leman JK, Berenberg D, Vatanen T, Chandler C, Taylor BC, Fisk IM, Vlamakis H, et al. : Structure-based protein function prediction using graph convolutional networks. Nat Commun 2021, 12:3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kagaya Y, Flannery ST, Jain A, Kihara D: ContactPFP: Protein function prediction using predicted contact information. Front Bioinform 2022, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koren G, Meir S, Holschuh L, Mertens HDT, Ehm T, Yahalom N, Golombek A, Schwartz T, Svergun DI, Saleh OA, et al. : Intramolecular structural heterogeneity altered by long-range contacts in an intrinsically disordered protein. Proc Natl Acad Sci U S A 2023, 120:e2220180120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abdollahi H, Prestegard JH, Valafar H: Computational modeling multiple conformational states of proteins with residual dipolar coupling data. Curr Opin Struct Biol 2023, 82:102655. [DOI] [PubMed] [Google Scholar]
- 27.Motiwala Z, Sandholu AS, Sengupta D, Kulkarni K: Wavelet coherence phase analysis decodes the universal switching mechanism of Ras GTPase superfamily. iScience 2023, 26:107031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goretzki B, Wiedemann C, McCray BA, Schafer SL, Jansen J, Tebbe F, Mitrovic SA, Noth J, Cabezudo AC, Donohue JK, et al. : Crosstalk between regulatory elements in disordered TRPV4 N-terminus modulates lipid-dependent channel activity. Nat Commun 2023, 14:4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sora V, Tiberti M, Beltrame L, Dogan D, Robbani SM, Rubin J, Papaleo E: PyInteraph2 and PyInKnife2 to Analyze Networks in Protein Structural Ensembles. J Chem Inf Model 2023, 10.1021/acs.jcim.3c00574. [DOI] [PubMed] [Google Scholar]
- 30.Conev A, Rigo MM, Devaurs D, Fonseca AF, Kalavadwala H, de Freitas MV, clementi C, Zanatta G, Antunes DA, Kavraki LE: EnGens: a computational framework for generation and analysis of representative protein conformational ensembles. Brief Bioinform 2023, 10.1093/bib/bbad242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oues N, Dantu SC, Patel RJ, Pandini A: MDSubSampler: a posteriori sampling of important protein conformations from biomolecular simulations. Bioinformatics 2023, 39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madhurima K, Nandi B, Munshi S, Naganathan AN, Sekhar A: Functional regulation of an intrinsically disordered protein via a conformationally excited state. Sci Adv 2023, 9:eadh4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fischer M, Coleman RG, Fraser JS, Shoichet BK: Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery. Nat Chem 2014, 6:575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parvin JD, McCormick RJ, Sharp PA, Fisher DE: Pre-bending of a promoter sequence enhances affinity for the TATA-binding factor. Nature 1995, 373:724–727. [DOI] [PubMed] [Google Scholar]
- 35.Ken ML, Roy R, Geng A, Ganser LR, Manghrani A, Cullen BR, Schulze-Gahmen U, Herschlag D, Al-Hashimi HM: RNA conformational propensities determine cellular activity. Nature 2023, 617:835–841. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This paper focuses on RNA conformational propensities. It establishes the role of ensemble-based conformational propensities in cellular activity, with an example of a cellular process driven by a rare and short-lived RNA conformational state of HIV-1 TAR RNA.
- 36.Zheng R, Huang Z, Deng L: Large-scale predicting protein functions through heterogeneous feature fusion. Brief Bioinform 2023, 10.1093/bib/bbad243. [DOI] [PubMed] [Google Scholar]
- 37.Ma W, Zhang S, Li Z, Jiang M, Wang S, Lu W, Bi X, Jiang H, Zhang H, Wei Z: Enhancing Protein Function Prediction Performance by Utilizing AlphaFold-Predicted Protein Structures. J Chem Inf Model 2022, 62:4008–4017. [DOI] [PubMed] [Google Scholar]
- 38.Lin Z, Akin H, Rao R, Hie B, Zhu Z, Lu W, Smetanin N, Verkuil R, Kabeli O, Shmueli Y, et al. : Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379:1123–1130. [DOI] [PubMed] [Google Scholar]
- 39.Nussinov R, Zhang M, Liu Y, Jang H: AlphaFold, Artificial Intelligence (AI), and Allostery. J Phys Chem B 2022, 126:6372–6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Du S, Wankowicz SA, Yabukarski F, Doukov T, Herschlag D, Fraser JS: Refinement of Multiconformer Ensemble Models from Multi-temperature X-ray Diffraction Data. bioRxiv 2023, 10.1101/2023.05.05.539620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sala D, Engelberger F, McHaourab HS, Meiler J: Modeling conformational states of proteins with AlphaFold. Curr Opin Struct Biol 2023, 81:102645. [DOI] [PubMed] [Google Scholar]
- 42.Schmidt M: Time-Resolved Macromolecular Crystallography at Pulsed X-ray Sources. Int J Mol Sci 2019, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Branden G, Neutze R: Advances and challenges in time-resolved macromolecular crystallography. Science 2021, 373. [DOI] [PubMed] [Google Scholar]
- 44.Maeots ME, Enchev RI: Structural dynamics: review of time-resolved cryo-EM. Acta Crystallogr D Struct Biol 2022, 78:927–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amann SJ, Keihsler D, Bodrug T, Brown NG, Haselbach D: Frozen in time: analyzing molecular dynamics with time-resolved cryo-EM. Structure 2023, 31:4–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lento C, Wilson DJ: Subsecond Time-Resolved Mass Spectrometry in Dynamic Structural Biology. Chem Rev 2022, 122:7624–7646. [DOI] [PubMed] [Google Scholar]
- 47.Selenko P: Quo Vadis Biomolecular NMR Spectroscopy? Int J Mol Sci 2019, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leman JK, Weitzner BD, Lewis SM, Adolf-Bryfogle J, Alam N, Alford RF, Aprahamian M, Baker D, Barlow KA, Barth P, et al. : Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat Methods 2020, 17:665–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sala D, Giachetti A, Rosato A: Insights into the Dynamics of the Human Zinc Transporter ZnT8 by MD Simulations. J Chem Inf Model 2021, 61:901–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sala D, Giachetti A, Rosato A: An atomistic view of the YiiP structural changes upon zinc(II) binding. Biochim Biophys Acta Gen Subj 2019, 1863:1560–1567. [DOI] [PubMed] [Google Scholar]
- 51.Matsunaga Y, Sugita Y: Use of single-molecule time-series data for refining conformational dynamics in molecular simulations. Curr Opin Struct Biol 2020, 61:153–159. [DOI] [PubMed] [Google Scholar]
- 52.Cerofolini L, Fragai M, Ravera E, Diebolder CA, Renault L, Calderone V: Integrative Approaches in Structural Biology: A More Complete Picture from the Combination of Individual Techniques. Biomolecules 2019, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Allison JR: Computational methods for exploring protein conformations. Biochem Soc Trans 2020, 48:1707–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]; • An excellent description of a compendium of methods for exploring protein conformations.
- 54.Wang Y, Lamim Ribeiro JM, Tiwary P: Machine learning approaches for analyzing and enhancing molecular dynamics simulations. Curr Opin Struct Biol 2020, 61:139–145. [DOI] [PubMed] [Google Scholar]
- 55.Buslaev P, Aho N, Jansen A, Bauer P, Hess B, Groenhof G: Best Practices in Constant pH MD Simulations: Accuracy and Sampling. J Chem Theory Comput 2022, 18:6134–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ghorbani M, Prasad S, Klauda JB, Brooks BR: Variational embedding of protein folding simulations using Gaussian mixture variational autoencoders. J Chem Phys 2021, 155:194108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meng M, Zhong K, Jiang T, Liu Z, Kwan HY, Su T: The current understanding on the impact of KRAS on colorectal cancer. Biomed Pharmacother 2021, 140:111717. [DOI] [PubMed] [Google Scholar]
- 58.Nussinov R, Yavuz BR, Arici MK, Demirel HC, Zhang M, Liu Y, Tsai CJ, Jang H, Tuncbag N: Neurodevelopmental disorders, like cancer, are connected to impaired chromatin remodelers, PI3K/mTOR, and PAK1-regulated MAPK. Biophys Rev 2023, 15:163–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dunnett-Kane V, Burkitt-Wright E, Blackhall FH, Malliri A, Evans DG, Lindsay CR: Germline and sporadic cancers driven by the RAS pathway: parallels and contrasts. Ann Oncol 2020, 31:873–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hebron KE, Hernandez ER, Yohe ME: The RASopathies: from pathogenetics to therapeutics. Dis Model Mech 2022, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ney G, Gross A, Livinski A, Kratz CP, Stewart DR: Cancer incidence and surveillance strategies in individuals with RASopathies. Am J Med Genet C Semin Med Genet 2022, 190:530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kang M, Lee YS: The impact of RASopathy-associated mutations on CNS development in mice and humans. Mol Brain 2019, 12:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li X, Ye M, Wang Y, Qiu M, Fu T, Zhang J, Zhou B, Lu S: How Parkinson’s disease-related mutations disrupt the dimerization of WD40 domain in LRRK2: a comparative molecular dynamics simulation study. Phys Chem Chem Phys 2020, 22:20421–20433. [DOI] [PubMed] [Google Scholar]
- 64.Li F, Chen Y, Tang Y, Liu X, Wei G: Dissecting the Effect of ALS Mutation G335D on the Early Aggregation of the TDP-43 Amyloidogenic Core Peptide: Helix-to-beta-Sheet Transition and Conformational Shift. J Chem Inf Model 2023, 63:3579–3590. [DOI] [PubMed] [Google Scholar]
- 65.Zheng Y, Carrillo-Perez F, Pizurica M, Heiland DH, Gevaert O: Spatial cellular architecture predicts prognosis in glioblastoma. Nat Commun 2023, 14:4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yavuz BR, Arici MK, Demirel HC, Tsai CJ, Jang H, Nussinov R, Tuncbag N: Neurodevelopmental disorders and cancer networks share pathways; but differ in mechanisms, signaling strength, and outcome. bioRxiv 2023, 10.1101/2023.04.16.536718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Agajanian S, Alshahrani M, Bai F, Tao P, Verkhivker GM: Exploring and Learning the Universe of Protein Allostery Using Artificial Intelligence Augmented Biophysical and Computational Approaches. J Chem Inf Model 2023, 63:1413–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Here the authors overview the learning of the universe of protein allostery using artificial intelligence augmented biophysical and computational approaches. They further provide an outlook of future directions suggesting that AI-augmented biophysical and computer simulation approaches are beginning to transform studies of protein allostery helping in drug discovery.