

SUMMARY
Somatic copy number gains are pervasive across cancer types, yet their roles in oncogenesis are insufficiently evaluated. This inadequacy is partly due to copy gains spanning large chromosomal regions, obscuring causal loci. Here, we employed organoid modeling to evaluate candidate oncogenic loci identified via integrative computational analysis of extreme copy gains overlapping with extreme expression dysregulation in The Cancer Genome Atlas. Subsets of “outlier” candidates were contextually screened as tissue-specific cDNA lentiviral libraries within cognate esophagus, oral cavity, colon, stomach, pancreas, and lung organoids bearing initial oncogenic mutations. Iterative analysis nominated the kinase DYRK2 at 12q15 as an amplified head and neck squamous carcinoma oncogene in p53−/− oral mucosal organoids. Similarly, FGF3, amplified at 11q13 in 41% of esophageal squamous carcinomas, promoted p53−/− esophageal organoid growth reversible by small molecule and soluble receptor antagonism of FGFRs. Our studies establish organoid-based contextual screening of candidate genomic drivers, enabling functional evaluation during early tumorigenesis.
In brief
Salahudeen et al. utilize organoid models to evaluate candidate outlier oncogenic loci within somatic copy number gain regions in TCGA datasets of esophagus, oral cavity, colon, stomach, pancreas, and lung cancer. Contextual screening within tissue-matched organoids validated oncogenic candidates, including DYRK2 and FGF3 in oral and esophageal cancer, respectively.
Graphical Abstract
INTRODUCTION
Somatic copy number aberrations (SCNAs) in the form of amplifications or deletions occur commonly in solid tumors.1,2 Amplifications have been successfully targeted by therapeutics such as trastuzumab,3 whereas deletions, such as MTAP,4 are currently the subject of therapeutic development for synthetic lethal interactions.5 However, a systematic understanding of the contribution of SCNAs to tumor biology and patient outcomes remains an aspirational goal.6 Furthermore, while recurrent amplified or deleted regions are increasingly delineated with large-scale genomic studies, many being prognostic, the oncogenic function of specific genes within amplicons is often unknown.
Several methods have been developed for the identification of recurrent SCNAs, including GISTIC2, which enables the delineation of focal alterations1,2 and RUBIC.7 However, copy number amplicons are often broad, spanning multiple megabases, impairing driver identification. To discover driver loci within SCNA, we bioinformatically identified candidate “outlier” genes demonstrating both high-level copy number alterations and concordant extreme expression dysregulation. The outlier approach is highly discriminatory, prioritizing candidate drivers within broad SCNA regions, highlighting known oncogenes and tumor suppressors and numerous novel candidates, as demonstrated in breast cancer,8 several of which have been functionally validated.9,10 Related methods have identified chromosomal rearrangements11 by comparing “normal” with dysregulated gene expression. However, to date, a systematic screen of candidate SCNA drivers across primary tissues has not been undertaken.
To functionally test hypotheses from genomic cancer analyses, immortalized cell lines or xenograft models are frequently used. Such systems have potential disadvantages of mono- or oligo-clonality (i.e., lack of tumor heterogeneity), secondary mutation burden in cell lines, limited tractability of patient-derived xenografts, and low throughput of genetically engineered mice. In contrast, in vitro organotypic culture of untransformed primary tissues as three-dimensional organoids offers a promising approach for driver oncogene validation.12 Indeed, organoids faithfully recapitulate multilineage differentiation and tissue architecture and yet retain experimental tractability for in vitro genetic and pharmacologic studies.13–23 We and others have initiated gastrointestinal malignancies by oncogene-engineering wild-type organoids from mouse13,14 and human tissues.23–26 Importantly, these cancer organoid models are generated by introduction of “first hit” oncogenic alleles into a normal wild-type genome, representing predominant drivers of the cognate cancer type. Subsequently, putative oncogenic loci can be overlaid and functionally assessed in a scalable, rapid, and reproducible manner.
Here, we exploited the tabula rasa background of a diverse range of first hit-engineered organoid models to rapidly interrogate the oncogenic potential of candidate amplified/overexpressed outlier loci across several solid tumor histologies. The transduction of lentiviral barcoded open reading frame (ORF) libraries, representing tumor subtype-specific SCNAs, into cognate tissue-specific organoid models then allowed systematic functional screening of oncogenicity, followed by iterative genetic and pharmacologic hit validation.
RESULTS
Pan-cancer bioinformatic identification of tumor outlier loci exhibiting matched extreme copy number alteration and expression
The analysis of copy number (CN) somatic events in cancer is impeded by broad extension of somatic CN events over the genome, thus increasing the likelihood of false positives. Many methods for CN driver identification analyze only DNA and thus include alterations that have no effect on expression. The approaches that use an integrative expression/CN methodology often include the expression information by identifying genes whose expression is correlated with CN values. This strategy has the limitation that regression between the CN and expression values is often polynomial and not evenly distributed between high and low CN values (i.e., coefficient between higher CN and higher expression is always higher than the lower CN and lower expression), thus potentially including false negatives. Building upon our prior studies,8 we sought to integrate gene expression information into our model to more accurately identify outliers. We propose that putative drivers are those with a modification in the CN landscape at a given position with a corresponding functional effect in gene expression.
To refine putative amplified SCNAs consequential to oncogenesis, we matched gene-based extreme CN events (amplifications and deletions) with extreme expression effects (Figure 1A, STAR Methods). To identify which samples fall in the overexpressed tail, we calculated the threshold for the 5% right and 5% left values in the theoretical distribution (represented by the vertical lines in Figure S1A). With these two thresholds, we identified expression outliers, which are the values in the real expression distribution that are greater than right threshold (overexpression outliers). We then matched overexpression outliers with extreme CN amplifications (defined as samples with a segmented CN value higher than six times the standard deviation) in sequencing data from The Cancer Genome Atlas (TCGA) datasets ESCA (esophageal squamous cell carcinoma), HNSC (head and neck squamous carcinoma), COAD (colon adenocarcinoma), PDAC (pancreatic ductal adenocarcinoma), LUAD (lung adenocarcinoma), and STAD (stomach adenocarcinoma) (Figures 1B–1G).
Figure 1. Overview of integrative analysis for expression and copy number amplification events as pan-cancer outliers.
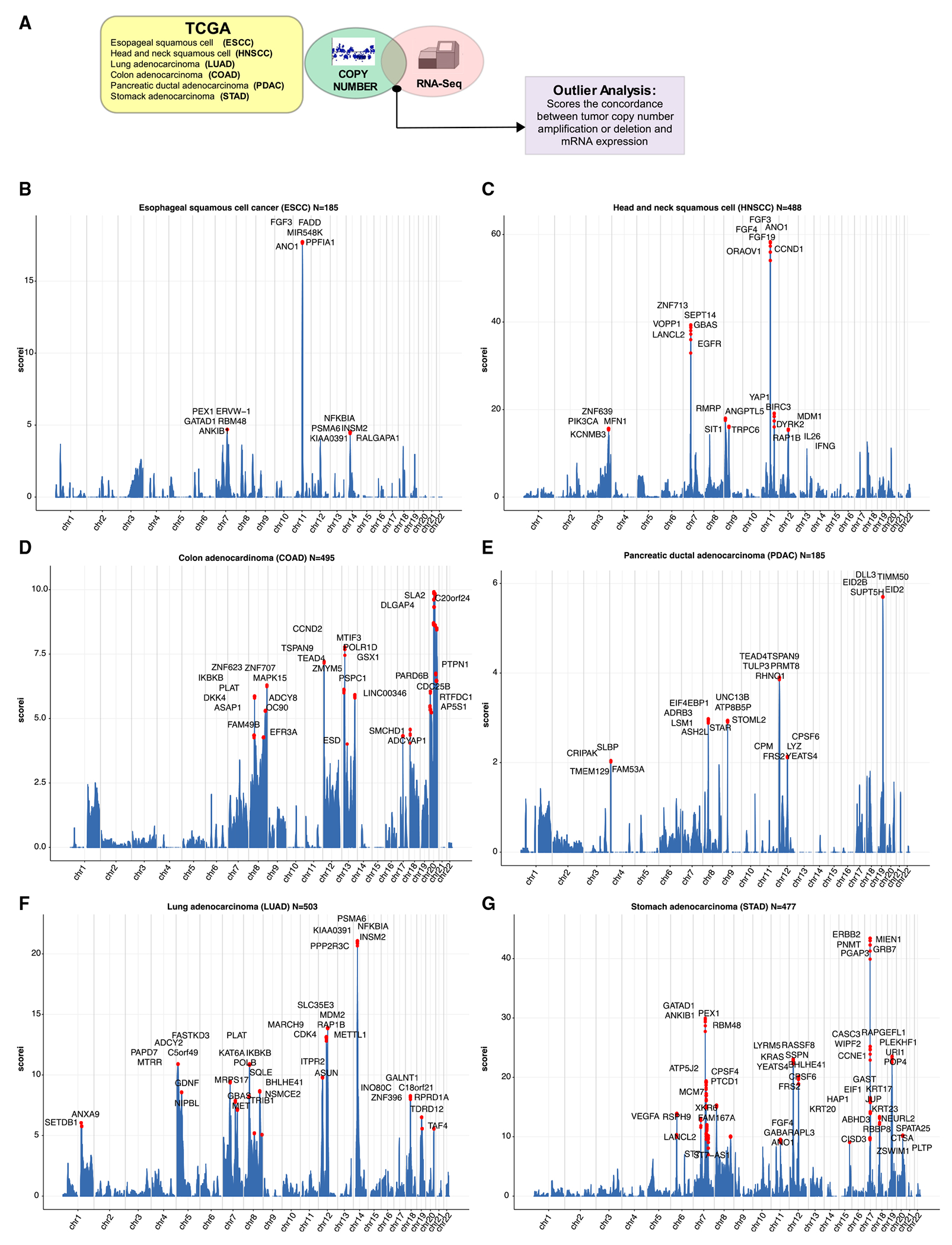
(A) Schematic of integrative analysis to nominate outlier gene candidates.
(B) Genomic landscape of selected TCGA esophageal squamous cell carcinoma (ESCC) outliers where y axis (iscore) represents the number of outliers for each specific gene.
(C) Genomic landscape of selected TCGA head and neck squamous cell carcinoma (HNSCC) outliers where y axis (iscore) represents the number of outliers for each specific gene.
(D) Genomic landscape of selected TCGA colon adenocarcinoma (COAD) outliers where y axis (iscore) represents the number of outliers for each specific gene.
(E) Genomic landscape of selected TCGA pancreatic ductal adenocarcinoma (PDAC) outliers where y axis (iscore) represents the number of outliers for each specific gene.
(F) Genomic landscape of selected TCGA NSCLC adenocarcinoma (LUAD) outliers where y axis (iscore) represents the number of outliers for each specific gene.
(G) Genomic landscape of selected TCGA stomach adenocarcinoma (STAD) outliers where y axis (iscore) represents the number of outliers for each specific gene.
Derivation of a pan-cancer panel of first hit-engineered organoid models
We then evaluated the aforementioned cancer type-specific candidate overexpression outliers in minimally transformed organoid models containing single or double oncogenic mutations, generated from the corresponding wild-type tissues of origin. This strategy was specifically designed to evaluate the contribution of outlier loci to earlier tumorigenesis, as opposed to testing their function in established tumors.
Primary epithelia isolated from human or mouse were cultured as previously described.13,26–28 We generated KrasG12D mouse pancreatic organoids by culturing LSL-KrasG12D29 pancreas as air-liquid interface (ALI) organoids and infected with adeno-virus-Cre-GFP to activate expression of latent KrasG12D13 (Figures S2A–S2B). Human wild-type gastric and colon organoids were grown under standard submerged methods.20,23,26 APC−/− colon organoids were generated by CRISPR-Cas923,24 and TP53R175H gastric organoids by stable lentivirus transduction (Figures S2C–S2F), both in the background of human wild-type organoids.
For this study, we also generated three organoid models corresponding to p53-null (p53−/−) oral squamous cell carcinoma, p53−/− esophageal squamous cell carcinoma (ESCC), and KrasG12D; p53−/− LUAD, described below. Mutational inactivation of the p53 tumor suppressor gene occurs at high frequency of primary HPV-negative head and neck squamous cell carcinomas (HNSCCs). We established normal oral mucosal (OM) organoids from mouse glossal epithelium cultured as ALI organoids (Figure 2A). Normal OM tissue consists of stratified squamous epithelium and connective tissue. In the basal layer, KI67, p63, and KRT5 are expressed.30,31 Mouse OM ALI organoids recapitulated squamous epithelial structures (Figure S3A). The basal cell marker KRT5 was expressed in the outer periphery, in which KI67+ cells were also detected, suggesting that cell proliferation occurred mainly at these sites (Figure S3A). These mouse OM organoids were maintained in ALI for more than 1 year.
Figure 2. Generation and characterization of murine p53−/− oral mucosa, esophageal, and KrasG12D p53−/− lung adenocarcinoma organoids.
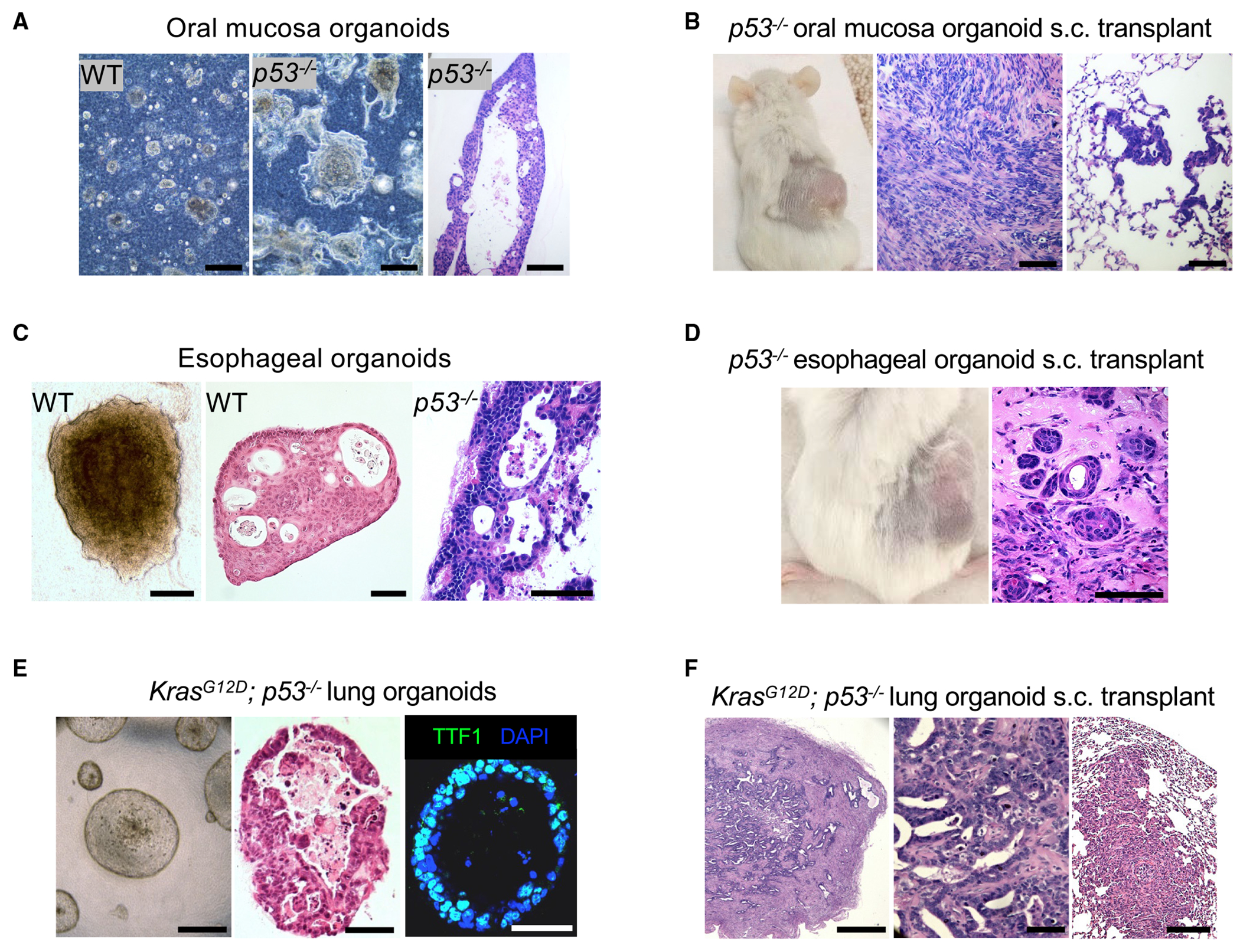
(A) Organoid bright-field imaging of wild-type (WT) (left panel) and p53−/− (middle panel) mouse oral mucosa organoids cultured in ALI for 7 days. H&E staining (right panel) of p53−/− mouse oral mucosa organoids cultured in ALI for 34 days. Scale bar: 100 μm.
(B) Tumor formation of p53−/− mouse oral mucosa organoids 7 months after implantation (right panel) and H&E of primary tumor (middle panel) and lung metastasis (right panel) (WT; n = 3, p53−/−; n = 3). Scale bar: 100 μm.
(C) Bright field (left panel) and H&E staining of wild-type (middle panel) versus p53−/− mouse esophageal organoids (right panel). Scale bar: 250 μm.
(D) p53−/− mouse esophageal organoid tumor formation (left panel) and H&E stain (right panel) 6 months post implantation. Scale bar: 250 μm.
(E) Phase contrast microscopy (left panel), H&E staining (middle panel), and TTF-1 immunofluorescence (right panel) of KrasG12D; p53−/− mouse lung organoids after 4 weeks of culture in basal F12 medium. Scale bar: 250 μm.
(F) H&E of primary tumor (left panel), high magnification (middle panel), and lung metastasis (right panel) 8 weeks post subcutaneous implantation of KrasG12D; p53−/− mouse lung organoids. Scale bars: 500, 100, and 500 μm, respectively.
Mouse OM organoids could also be grown in submerged formats where they exhibited similar organization as ALI OM organoids with an outer rim of KRT5+ KI67+ proliferative basal cells and could again be cultured for more than 1 year (Figure S3B). Human OM could be cultured for up to 6 weeks in submerged or collagen ALI formats and included KRT5+ ECAD+ basal cells and the notable presence of keratinization within the organoid lumens (Figures S3C and S3D). Upon lentiviral transduction with the oncogenic R175H allele of p53, these could be serially passaged up to 6 months without obvious signs of dysplasia (data not shown).
To initiate oncogenic transformation in vitro, mouse p53flox/flox mouse OM organoids were infected with adenovirus Cre-GFP to create a contextual p53−/− oral cancer model (Figure 2A). These p53−/− organoids exhibited a stratified cell arrangement with a thick KRT5+ cell layer and multilayered KI67+ cells upon long-term culture (Figure S4A). p53 deletion enhanced proliferation (Figure S4B), accompanied by Ki67 upregulation and Cdkn1a downregulation (Figure S4C). Furthermore, p53 deletion induced in vivo tumorigenicity and metastasis of mouse OM organoids (Figure 2B).
In addition to oral squamous cell carcinoma, we also generated ALI esophageal squamous organoids using the same p53flox/flox murine model (Figure 2C). Esophageal ALI organoids from p53flox/flox mice formed KRT5+ squamous epithelium with similar morphology to OM organoids (Figure 2C). Upon in vitro infection with adenovirus Cre-GFP, the resultant p53−/− esophageal organoids exhibited dysplasia (Figure 2C), maintenance of KRT5 (Figure S4D), and loss of p53 expression (Figure S4E). Further, we observed in vivo tumorigenicity when p53−− ESCC organoids were subcutaneously transplanted into immunodeficient mice with keratin pearl formation and other histologic features consistent with squamous cell carcinoma (Figure 2D).
Lastly, NSCLC LUAD organoids (Figure 2E) were generated from murine pulmonary parenchymal tissue using a protocol similar to our previous studies.13,28 Accordingly, ALI lung organoids were generated from unexcised LSL-KrasG12D; p53flox/flox mice.29,32 Upon infection with a negative control adenovirus expressing an immunoglobulin Fc fragment (Ad-Fc), these wild-type lung organoids proliferated in medium containing epidermal growth factor (EGF) and Noggin (EN) but not in basal F12 medium, similar to our prior studies in human lung organoids28 (Figure S4F). However, upon in vitro adenovirus Cre-GFP infection, the lung ALI organoids were converted to a KrasG12D; p53−/− genotype and exhibited proliferation in basal F12 medium (Figure 2E), expression of mutant KrasG12D and loss of p53 (Figures S4G and S2H), TTF-1 expression (Figure 2E), and in vivo tumorigenicity and metastases (Figure 2F).
Contextual tissue-specific functional screening of copy number amplification outliers
Having characterized these six tissue-specific, minimally transformed oncogenic models, we then overlaid integrative analysis of the expression outliers for each cognate cancer type (Figure 1) to computationally identify over 1,000 candidate amplification outliers (Table S1). From these loci, we selected 393 available full-length cDNAs from the CCSB-Broad lentivirus ORF collection33 having barcodes external to the ORFs, facilitating pooled screens. A custom cDNA ORF library was thus generated for each of the six cancer-specific outlier predictions, for infection of the corresponding minimally transformed tissue organoids. For instance, esophageal ESCC TCGA outliers were represented by an equivalent barcoded lentivirus cDNA ORF library, for infection into p53−/− esophageal organoids (Figures S1B and 3).
Lentiviruses were generated in arrayed format and pooled for equal titer amounts (STAR Methods). Loci encoding well-established oncogenic drivers were deliberately not included in our screen to avoid potentially dominant “jackpot” effects that could obscure contributions of novel outliers. Organoids were infected with the pooled cDNA ORF virus corresponding to the outliers for that histologic site and screened as independent biological replicates (STAR Methods), and aliquots from initial plating (t = 0) and after four passages (terminal time point) were collected. Next-generation sequencing (NGS) was performed to evaluate barcode enrichment at the terminal time point, typically at culture days 30–50, versus t = 0 (Figure 3; Table S2). Organoids were subjected to prolonged culture after pooled lentiviral infection and puromycin selection; barcodes were quantitated at each time point by NGS. Barcode consistency was observed across independent biological replicates despite expected bias for lentivirus with smaller cDNA ORFs favoring higher packaging and infectivity (Figures S5–S7; Table S2).
Figure 3. Screening pan-cancer candidate amplification outliers in organoids with corresponding tissue context.
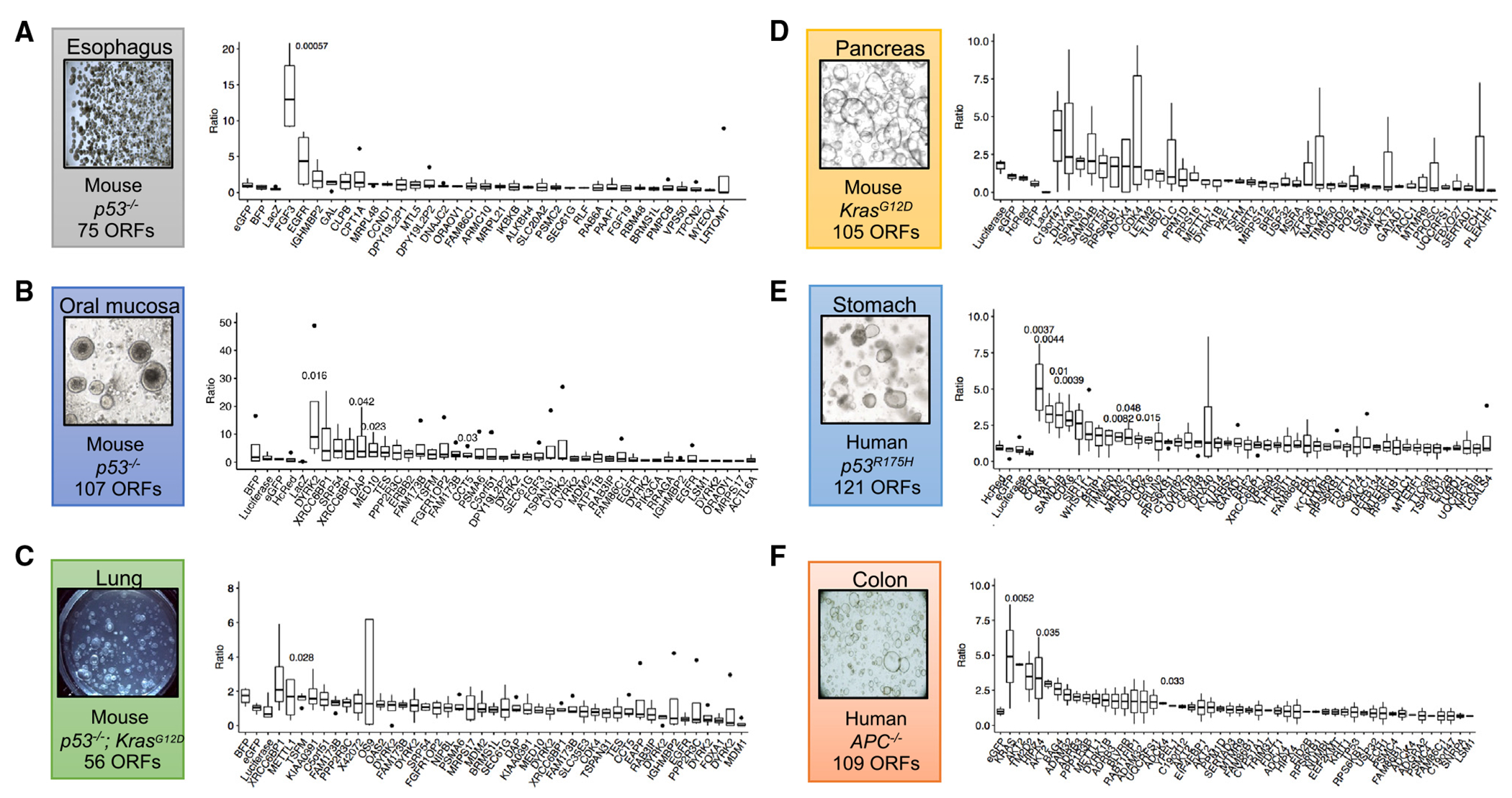
(A–F) Barcode ratios of terminal:initial time points from NGS demonstrating relative enrichments from pooled lentiviral ORF screens. Boxplots represent four technical replicates with the exception of (F), which was performed with two technical replicates. Terminal time points are as follows: A = day 53, B = day 52, C = day 32, D = day 50, E = day 55, and F = day 56.
Each library included ORFs that corresponded to genes whose expression were expected to have neutral effects to assess the technical quality of the screen and as a baseline to judge enrichment by a given candidate locus (Figures 3A–3F). Across screens, we observed ORFs expressing GFP, LACZ, BFP, and luciferase to have no significant enrichments between terminal endpoint and initial time points with the vast majority having fold change ratios < 2 (Figures 3A–3F). Furthermore, screen replicates exhibited both consistent distributions among all barcodes (Figures S5–S7) and tight distributions among technical screen replicates in the control neutral ORFs described above (Figure 3; Table S2).
Notably, outlier hits were functionally enriched in cell cycle processes (CDK4, CDK6 in gastric and pancreas) (Figures 3D and 3E), DNA repair (XRCC6BP1 in lung) (Figure 3C), and kinase signaling (KRAS, AKT in colon) (Figure 3F; Table S2). Interestingly, in p53R175H human gastric organoids, two of the top five outliers corresponded to CDK6. Among analyzing gastric cancer outliers with >2-fold change vs. t = 0, six of the top hits (DYRK1B, SAMD4B, MRPS12, SIRT2, EIF3K, and PAF1) all co-localized to chromosome 19q13.2. We focused on DYRK2 and FGF3 for validation in this study, given their role in oncogenic signal transduction pathways and their potentially druggable kinase activities.
DYRK2 and FGF3 are candidate amplified oncogenic drivers in oral and esophageal squamous cell carcinoma
Screens in the p53−/− OM organoid model identified the dual specificity tyrosine phosphorylation-regulated kinase 2 (DYRK2) as a highly ranked and significant hit (Figure 3B). DYRK2 amplification occurs in approximately 5% of HNSCC.34 DYRK2 is thought to be required for tumor growth via proteasome phosphorylation35 and induces p53-dependent apoptosis to DNA damage.36,37 Of the DYRK2 ORFs screened, we found greater enrichment of ORFs mapping to the isoform 2 transcript variant (Table S2). To iteratively validate the role of DYRK2, we infected p53−/− mouse OM organoids with DYRK2 isoform 2-expressing lentivirus (Figure 4A). DYRK2 overexpression enhanced proliferation of p53−/− OM organoids versus lentivirus eGFP-infected organoids in vitro (Figures 4B, S8A, and S8B) and promoted in vivo tumorigenicity upon subcutaneous transplantation (Figures 4C and 4D; Table S3).
Figure 4. DYRK2 and FGF3 overexpression induces proliferation and tumorigenicity of p53−/− mouse oral mucosa and esophageal organoids, respectively.
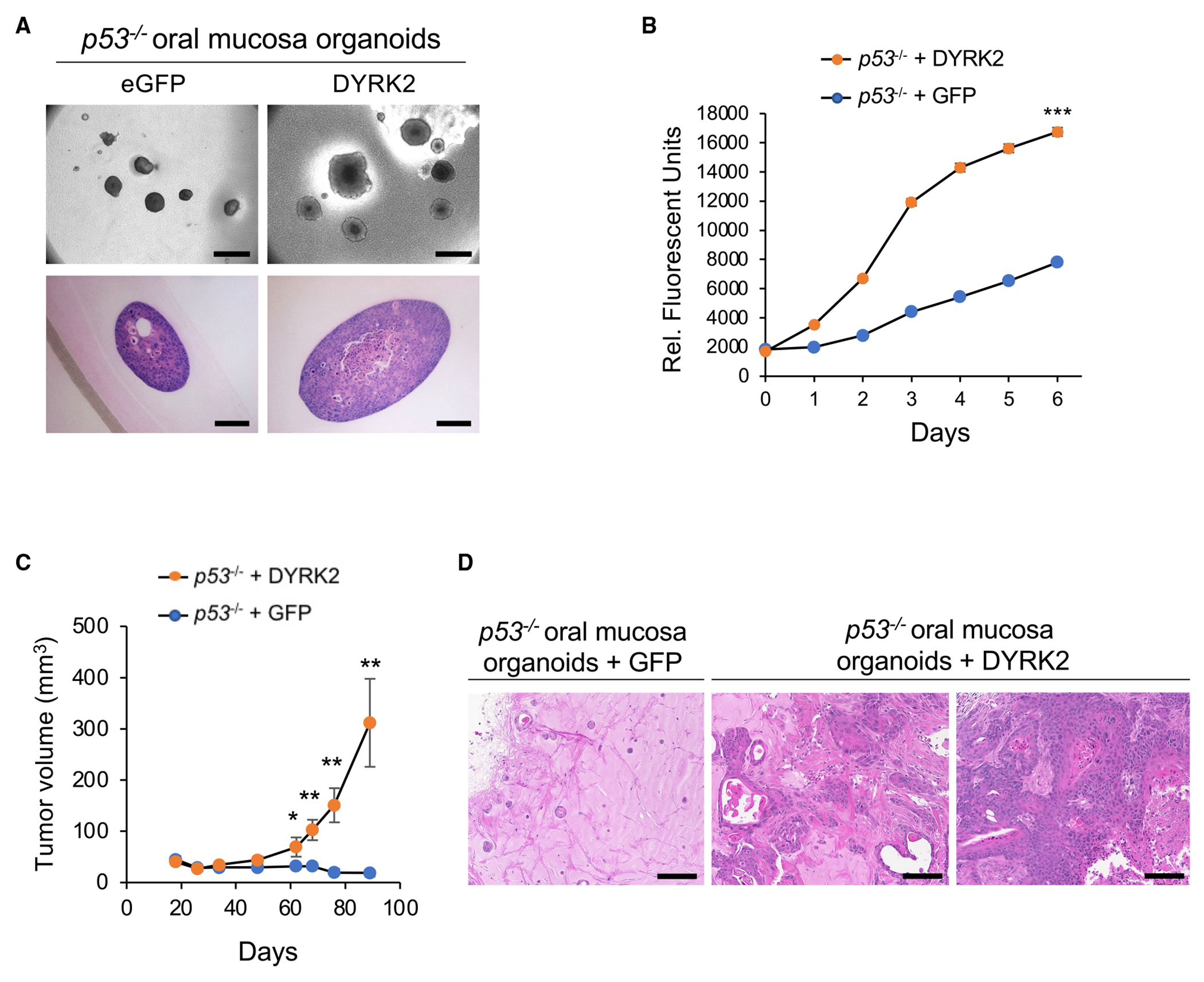
(A) Morphology of p53−/−; eGFP and p53−/−; DYRK2 organoids cultured in ALI for 14 days. Scale bar: bright field; 500 μm (top panel), H&E staining; 100 μm (bottom panel).
(B) In vitro proliferation of p53−/−; eGFP and p53−/−; DYRK2 organoids generated from oral mucosal tissue from a different donor mouse in ALI assessed by resazurin reduction. Data represent mean ± SEM. ***p < 0.001, two-tailed Student’s t test.
(C) Tumor volume of p53−/−; eGFP (n = 6) and p53−/−; DYRK2 (n = 7) organoids. Data represent mean ± SEM. *p < 0.05, **p < 0.01, two-tailed Student’s t test.
(D) H&E staining of p53−/−; eGFP and p53−/−; DYRK2 organoid tumors. Scale bar: 100 μm.
In p53−/− esophageal organoids, FGF3 exhibited the greatest overall effect across all replicates compared to a modest enrichment in CCND1 and EGFR, genes with known oncogenic effects upon upregulation (Figure 3A; Table S2). We then evaluated FGF3 by lentiviral expression in an independent esophageal p53−/− organoid line not used in the ORF screen (Figures 5A, 5B, and S8C), again confirming increased proliferation in serum-containing minimal F12 medium (Figure 5C). Given the strong proliferation phenotype in the absence of the mitogen EGF, we hypothesized that FGF3 overexpression could serve an EGF surrogate, and that FGF3 could act functionally as an autocrine growth factor in ESCC. Such a model was supported by ELISA detection of FGF3 secretion into organoid culture supernatants (Figure S8C).
Figure 5. Targeting oncogenic FGF3 amplification with soluble FGFR-ECD or FGFR-specific tyrosine kinase inhibitors in p53−/− mouse esophageal squamous organoids.
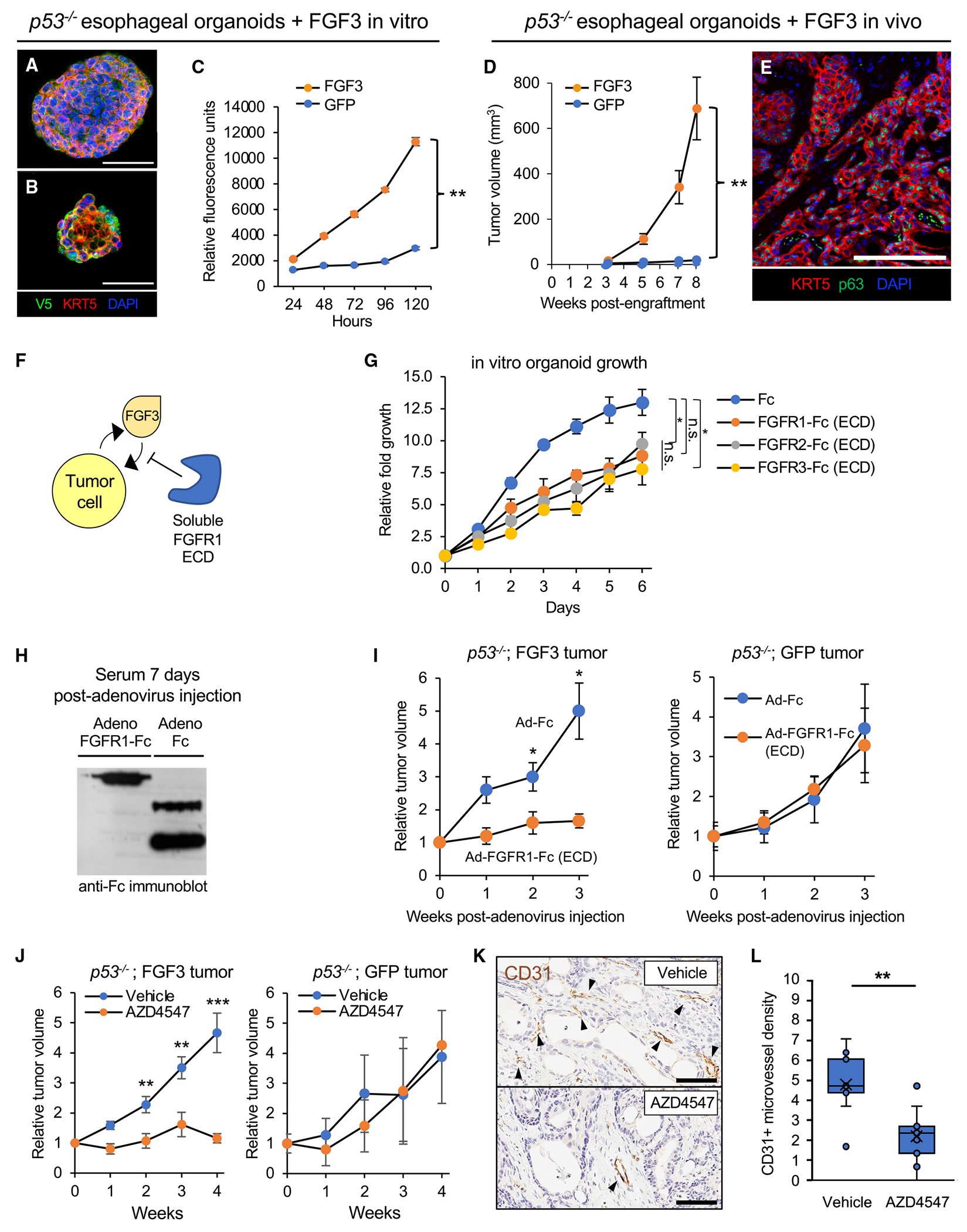
(A) Multicolor immunofluorescence of p53−/− esophageal organoids with lentiviral expression of FGF3 with a C-terminal V5 tag. Scale bar: 100 μm.
(B) Multicolor immunofluorescence of p53−/− esophageal organoids with lentiviral expression of GFP with a C-terminal V5 tag. Scale bar: 100 μm.
(C) In vitro proliferation of FGF3 versus GFP-expressing p53−/− esophageal organoids by resazurin reduction. Data represent mean ± SEM. **p < 0.01, two-tailed Student’s t test.
(D) In vivo tumor formation and growth upon subcutaneous transplantation of FGF3- versus GFP-expressing p53−/− esophageal organoids in immunodeficient NOG mice. Each group has n = 10 biological replicate mice. Data represent mean ± SEM. *p < 0.05, **p < 0.01, two-tailed Student’s t test.
(E) Multicolor immunofluorescence of p53−/−;FGF3 esophageal organoid subcutaneous tumor sections. Scale bar: 100 μm.
(F) Schematic of inhibition of FGF3-driven oncogenesis by soluble ligand-binding extracellular domains of FGF receptors.
(G) Proliferation of FGF3 p53−/− esophageal organoids upon incubation with Fc or FGFR 1, 2, or 3 ECD-Fc fusions at 10 μg/mL assessed by resazurin reduction. Data represent mean ± SEM. *p < 0.05, two-tailed Student’s t test.
(H) Circulating serum expression of FGFR1-Fc fusion protein after intravenous injection of immunodeficient NSG mice infected with the corresponding recombinant adenoviruses. Western blot, anti-Fc.
(I) Serial measurements of subcutaneous tumor growth subsequent to adenoviral infection of FGFR1 ECD-Fc versus Fc controls in FGF3-expressing (n = 6) (left panel) or GFP-expressing (n = 5) (right panel) p53−/− esophageal organoid subcutaneous tumors. Data represent mean ± SEM. *p < 0.05, two-tailed Student’s t test.
(J) Serial measurements of FGF3-expressing (left panel) or GFP-expressing (right panel) p53−/− esophageal organoid subcutaneous tumors subsequent to daily oral administration of vehicle or the FGFR inhibitor AZD4547. FGF3-expressing organoids have n = 9 biological replicate mice in each treatment group. GFP-expressing organoids have n = 8 in the vehicle treatment group and n = 7 in the AZD4547 treatment group. **p < 0.01, ***p < 0.001, two-tailed Student’s t test.
(K) Representative CD31 immunohistochemistry staining of subcutaneous tumors from FGF3-expressing p53−/− esophageal organoids in (E). Scale bar: 200 μm.
(L) Chalkley quantitation of CD31+ microvessel density in (F), where each group is three technical replicates for each mouse subcutaneous tumor. **p < 0.01, two-tailed Student’s t test.
We then investigated if FGF3 overexpression conferred a phenotype upon subcutaneous transplantation into immunodeficient mice. Paralleling in vitro observations, FGF3 overexpression in p53−/− esophageal organoids robustly induced tumorigenicity in vivo (Figures 5D, 5E, and S8D; Table S3). We then assessed if tumor growth could be attenuated by FGFR antagonism by scavenging secreted FGF3 via soluble ligand-binding FGFR ectodomain (ECD) (Figure 5F). Accordingly, recombinant ligand-binding extracellular domains of FGFR1–3 (FGFR1-Fc, FGFR2-Fc, FGFR3-Fc) inhibited growth of FGF3-overexpressing esophageal p53−/− organoids in vitro (Figure 5G). For in vivo assessment, we utilized an adenoviral vector (Ad-FGFR1-Fc, Data S1 file) for in vivo liver infection and hepatocyte secretion of the FGFR1-Fc ectodomain fusion protein into the circulation of mice (Figure 5H). Mice infected with FGFR1-Fc adenovirus but not a control adenovirus expressing an Fc immunoglobulin fragment alone (Ad-Fc) inhibited tumor growth in FGF3-overexpressing organoid tumors. Notably, control p53−/− esophageal organoids expressing GFP did not exhibit a response (Figures 5I and S9A).
Having established tumor suppression by circulating FGFR1-Fc, we then evaluated whether selective small molecule FGFR tyrosine kinase inhibitors, a drug class efficacious in treatment of FGFR-rearranged tumors,38 could also inhibit proliferation of ESCC overexpressing FGF3. Two first-generation pan-FGFR inhibitors, AZD4547 and BGJ-398, elicited dose-dependent inhibition of p53−/− ESCC organoid models overexpressing FGF3 with nanomolar EC50 values (Figure S9B). Interestingly, this response to FGFR inhibitors was masked when organoids were cultured in EGF (data not shown) possibly due to functional redundancy between these two growth factor classes. Accordingly, AZD4547 significantly reduced growth of FGF3-overexpressing p53−/− esophageal organoid tumors versus vehicle (Figures 5J and S9C; Table S3), Importantly, growth of eGFP tumors (i.e., without FGF3 overexpression) was not significantly different between AZD4547 and vehicle treatments (Figure 5J and S9C; Table S3). Given the importance of FGF signaling in angiogenesis, we tested if FGFR inhibition resulted in decreased tumor vasculature. FGF3-overexpressing organoid tumors exhibited decreased CD31+ microvessel density upon AZD4547 treatment compared to vehicle (Figures 5K and 5L; Table S3). Taken together, our findings support a role for FGF3 amplification and overexpression as a pro-tumorigenic alteration in the development of early esophageal squamous cancer.
DISCUSSION
Pan-cancer bioinformatics studies of TCGA and other genome-scale cancer surveys have proven informative but lack a laboratory counterpart of functional validation in an equivalent in vitro contextual experimental system.39 We present an approach that powerfully enables systematic interrogation of these multicancer datasets in cognate organoid screens matched to the tissue of origin. First, an integrative analysis of gene expression with SCNAs prioritized and nominated CN outliers with potential significance to oncogenesis. Second, these candidate outlier datasets were directly coupled to functional validation in primary organoid cultures from colon, stomach, pancreas, oral mucosa, lung, and esophagus, exploiting the availability of organoids from these tissues.
Here, contextual modeling where SCNA outlier candidates specific to a cancer histologic type were screened in matching tissue organoids possessing a pre-existing, predominant “first hit” for that malignancy: for instance, KRAS mutations in PDAC, APC in COAD, and p53 mutation in HNSCC, ESCC, LUAD, and STAD. This not only modeled the physiologic SCNA occurrence in the context of pervasive signature mutations, but it also leveraged the accelerated growth of such oncogene-engineered organoids versus their wild-type counterparts to amass sufficient starting material for barcoded screens. Further, the “bottom-up” nature of the current screening in a minimally transformed background conveyed potentially increased sensitivity to detect oncogenic loci, versus testing in patient-derived tumor organoids or cell lines where a multitude of pre-existing epi/genetic alterations could obscure effects of a given outlier candidate.
CN alterations are one of the most ubiquitous features of cancer genomes, yet a comprehensive understanding of their oncogenic contributions has yet to be achieved.6 Earlier studies have nominated SCNAs with high genomic resolution but have not considered concomitant gene expression alterations, thus masking functionally consequential regions. While chromosomal deletions involving canonical tumor suppressors such as TP53 or amplifications of growth factor receptor pathway components such as EGFR or ERBB2 are well understood, evaluation of other SCNAs has been modest. Functional genomics experiments in immortalized cancer cell lines have identified synthetic lethality events such as MTAP deletion and S-adenosyl methionine arginine methyltransferase4 or a recent preprint suggesting inhibition of PKMYT1 in CCNE1-amplified tumors.40 Large-scale functional genomics and small molecule screens facilitated by efforts such as the Cancer Dependency Map could yield further insights into SCNAs.39,41
Among SCNAs, we prioritized screening of outlier amplifications over deletions, reasoning that oncogenic hits would be amenable to pharmacologic inhibition. The lists of amplified outliers generated by our analysis serve as a basis for further comprehensive evaluation in a screening system of organoid models of early tumorigenesis. In our screening studies of a subset of the amplified outliers using cDNA ORFs, we were able to confirm functional enrichment of two pharmacologically tractable oncogenic amplifications in squamous cancers of the oral cavity and esophagus. In contrast to adenocarcinomas, squamous cancers exhibit relatively fewer actionable mutations.42–44 First, the tyrosine kinase DYRK2 strongly promoted in vitro proliferation and in vivo tumorigenicity of OM organoids, as a highly relevant model for HNSCC. DYRK2, belonging to the cyclin-dependent kinase, mitogen-activated protein kinase, glycogen synthase kinase, and CDC-like kinase (CMGC) superfamily, has been previously implicated in either tumor promotion or inhibition.37 DYRK2 could exhibit oncogenic activity in oral cancer due to an epi/genomic context that favors DYRK2 upregulation versus gain-of-function kinase alterations (e.g., KRAS, BRAF, EGFR, PIK3CA) seen in pancreatic, lung, colon, and other adenocarcinomas. Our studies suggest a potential dependency of DYRK2 amplifications in oral cancers that could be conceivably amenable to DYRK2 small molecule inhibition. Given conflicting models of DYRK2 action during tumor progression,37 additional studies will be required to definitely address DYRK2 function in oral cancer, such as with future development of potent and specific DYRK2 small molecule kinase inhibitors.
Secondly, we functionally confirmed FGF3, a known oncogene, as an amplified esophageal squamous cell cancer oncogene at 11q13 using p53−/− esophageal organoids. We used pharmacologic FGFR inhibition, encompassing both small molecule FGFR inhibitors and soluble FGFR1-3 ectodomain ligand sequestration as a functional tool to validate the specificity of the in vitro and in vivo esophageal organoid FGF3 proliferation phenotypes. However, FGFR small molecule inhibitors have received regulatory approval in biliary and bladder cancer.38 Thus, our preliminary findings of FGFR inhibitor in vivo and in vitro efficacy against FGF3-overexpressing esophageal organoids warrant further exploration and evaluation in FGF3-amplified patient-derived models of ESCC and other cancer types such as the CN-driven integrative breast cancer subgroup, IntClust2, which manifests prominent FGF3 amplification.45 Certainly, our data suggest autocrine FGF-FGFR signaling as a potential druggable oncogenic mechanism in esophageal squamous tumors harboring FGF3 amplification and overexpression.
Notably, EGFR and KRAS amplifications confer clinical sensitivity to inhibitors targeting corresponding pathways.46,47 In addition, since FGF3 is an embryonal FGF and is not significantly expressed in adult human tissues, FGF3 could be considered an oncofetal antigen susceptible to adoptive T cell therapies selective for cells presenting FGF3 peptides on class I major histocompatibility complex. Since >40% of ESCCs harbor 11q13 amplicon FGF3 amplifications,48 successful targeting of FGF3 in ESCC could represent a precision oncology strategy.49 Additional driver loci such as CCND1 are co-amplified with FGF3 at 11q13, although in our screen FGF3 displayed stronger effects than CCND1. Nevertheless, our data suggest that FGF3 could indeed cooperate with CCND1 to promote tumorigenesis.
Beyond the current investigations, our screens have identified numerous candidates for further study. TCGA STAD amplification outlier screening in p53R175H-overexpressing human gastric organoids revealed the highest scoring hit as CDK6, a commonly amplified gene widely implicated in carcinogenesis and a target for pharmacological inhibition. Senescence induction in gastric cancer cells by the CDK4/6 inhibitor palbociclib is reduced upon p53 knockdown, consistent with cooperation between p53 mutation and CDK6.50 Intriguingly, several highly ranked gastric cancer SCNA hits, namely DYRK1B, SAMD4B, MRPS12, SIRT2, EIF3K, and PAF1, co-localized to 19q13.2, suggesting a multifaceted role of this amplicon in the fitness of gastric cancer cells. As 19q13.2 amplification occurs in other cancer types including PDAC,51 lung,52 and breast,53,54 potential cooperation between these SCNA hits will be of interest.
Overall, we have demonstrated the robust application of primary organoid culture to validate candidate cancer driver datasets in pooled barcoded screening formats, using amplified outlier loci as proof of principle. Future efforts could adapt this multi-tissue organoid framework to CRISPR variants such as dCas9 fusions to transcriptional activators or histone demethylases.55–57 The use of exclusively human organoid models and progressively miniaturized formats22 could be further combined with systematic functional evaluation of additional tissue organoid types, genomic regions, and classes of genetic alterations. The continued exploration of organoid-based functional genomics screens, as described here, has significant application to oncology as well as more broadly to human disease in general.
Limitations of the study
Our study has numerous limitations. First and foremost, any functional insights regarding amplification outliers are limited to early tumorigenesis and do not demonstrate necessity in established cancers. Further gain-of-function investigations of candidate loci in independent oncogene-engineered organoid models, and conversely loss of function in organoids from established cancers, will be required to robustly verify reproducibility and relevance to human malignancy. Bioinformatic outlier identification could be improved by considering tumor cell purity and stromal composition from TCGA data to avoid false negative calls. Candidate lentiviral cDNA ORF expression is subject to packaging limits where large inserts may compromise titer resulting in technical bias, desired full-length cDNAs either may not be available or may not represent the correct isoform, and uniform library representation may be cumbersome and costly. For example, negative control luciferase and BFP ORFs achieved 2-fold enrichment in pancreatic and lung screens respectively. This negative control enrichment, while not observed for other negative control ORFs, may reflect the stochastic nature of independent biological replicates as well as the limitations of cDNA ORFs unable to provide for robust statistical analysis as can be achieved in ultradeep sgRNA and shRNA screens with hundreds of control elements able to set a robust baseline. Our study was also limited to a subset of candidate outliers and thus a more comprehensive, saturating analysis with multiple follow-up validation studies would be more ideal. These limitations further underscore the need for functional screening technologies utilizing uniform pooled libraries with comprehensive coverage, which may be achieved with variant CRISPR-Cas9 technologies. Finally, the use of murine models, while experimentally robust, introduces interspecies complexity that could be overcome by equivalent human organoids.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ameen A. Salahudeen (ameen@uic.edu).
Materials availability
Organoids lines generated in this study are available under Material Transfer Agreement.
Data and code availability
Outlier screening data has been deposited at the CTD^2 data portal and are publicly available as of the date of the publication. The DOI is listed in the key resource table.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resource table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| DYRK2 antibody | Cell Signaling | 8143; RRID:AB_10889932 |
| GAPDH (D16H11) Rabbit mAb | Cell Signaling | 5174S; RRID:AB_10622025 |
| Peroxidase AffiniPure Goat Anti-Rabbit IgG (H + L) | Jackson ImmunoResearch | 711-035-152; RRID: AB_10015282 |
| Mouse anti-E-cadherin | BD Biosciences | BD610182; RRID:AB_397581 |
| Alexa Fluor 647 labeled-anti-CK5 | Abcam | ab193895; RRID:AB_2728796 |
| Rat-anti-Ki67 | eBiosciences | 14-5698-82; RRID:AB_10854564 |
| Cy3-conjugated AffiniPure goat anti-mouse IgG (H + L) | Jackson ImmunoResearch | 115-165-062; RRID:AB_2338685 |
| Cy3 Cy3-conjugated AffiniPure goat anti-rat IgG (H + L) | Jackson ImmunoResearch | 112-165-167; RRID:AB_2338251 |
| Peroxidase AffiniPure Goat Anti-Mouse IgG, Fcγ subclass 2a specific | Jackson ImmunoResearch | 115-035-206; RRID:AB_2338514 |
| Bacterial and virus strains | ||
| p53R175H lentivirus | Ken Scott laboratory | KS5 |
| Ad-Cre-GFP | University of Iowa Vector Core | 1174 |
| Adenovirus Fc | Stanford University | N/A |
| DYRK2 lentivirus | Genetic Perturbation Platform - Broad Institute | TRCN489007 |
| CCSB-Broad Lentiviral Expression Library | The Broad ORF collection | N/A |
| Biological samples | ||
| p53R175H gastric organoids | Stanford University | N/A |
| KrasG12D pancreatic organoids | Stanford University | N/A |
| Human APC−/− colon organoids | Stanford University | N/A |
| p53−/− oral squamous cell carcinoma organoids | Stanford University | N/A |
| p53−/− esophageal squamous cell carcinoma organoids | Stanford University | N/A |
| KrasG12D; p53−/− lung adenocarcinoma | Stanford University | N/A |
| Oral mucosa (OM) mouse organoids | Stanford University | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| cOmplete™, Mini Protease Inhibitor Cocktail | Sigma Aldrich | 4693124001 |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | ThermoFisher Scientific | 34580 |
| Fluoro-Gel II with DAPI | Electron Microscopy Sciences | 17985–50 |
| HEPES | Thermo Fisher Scientific | BP299100 |
| Nicotinamide | Sigma | N0636-500G |
| GlutaMAX™ Supplement | ThermoFisher Scientific | 31980030 |
| N-acetyl-l-cysteine | Sigma-Aldrich | A7250-100G |
| B27 supplement | Invitrogen | 17504–001 |
| A83-01 | Tocris | 2939 |
| Recombinant human EGF | Peprotech | AF-100-15 |
| Recombinant human Noggin | Peprotech | 120-10-C |
| 2-mercaptoethanol | ThermoFisher Scientific | 21985023 |
| DTT | Sigma | 10197777001 |
| Penicillin-Streptomycin-Glutamine (100X) | Life Technologies | 10378016 |
| Recombinant Human FGFR1a (IIIc) Fc | Peprotech | 160–02 |
| Recombinant Human FGFR2a (IIIc) Fc | Peprotech | 160–03 |
| Recombinant Human FGFR3 (IIIc) Fc | Peprotech | 160–05 |
| Critical commercial assays | ||
| RNeasy Plus Mini Kit | Qiiagen | 74104 |
| iScript Reverse Transcription Supermix | BioRad | 1708841 |
| FGF3 ELISA | Aviva Systems Biology | OKEH02512 |
| Hs FGF3 Taqman Assay | ThermoFisher | 4448892 |
| Hs beta-actin Taqman Assay | ThermoFisher | 401846 |
| Deposited data | ||
| Outlier Barcode Sequencing Data | NCBI BioProject | BioProject: PRJNA1032711 |
| TCGA Expression Data | Firehose, Broad Institute | http://gdac.broadinstitute.org/ |
| Broad Genomic Perturbation Platform | Broad Institute | https://portals.broadinstitute.org/gpp/public/ |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | CRL-11268 |
| Experimental models: Organisms/strains | ||
| Il2rgtm1Sug/JicTac (NOG) Immunodeficient mice | Taconic | NOG-F |
| C57BL/6NTac mice | Taconic | B6-F |
| p53flox/flox mice | Jackson Labs | 008462 |
| LSL-KrasG12D mice | Jackson Labs | 008179 |
| Oligonucleotides | ||
| Tp53 Forward Primer: tgaggttcgtgtttgtgcct | Stanford PAN Facility | N/A |
| Tp53 Reverse Primer: gcagttcagggcaaaggact | Stanford PAN Facility | N/A |
| Ki67 Forward Primer: agctctgcagtctccacaac | Stanford PAN Facility | N/A |
| Ki67 Reverse Primer: ctctgcctcgtgactgtgtt | Stanford PAN Facility | N/A |
| Cdkn1a Forward Primer: ttgtcgctgtcttgcactct | Stanford PAN Facility | N/A |
| Cdkn1a Reverse Primer: tttcggccctgagatgttcc | Stanford PAN Facility | N/A |
| DYRK2 Forward Primer: cagtgctcacgacacaacca | Stanford PAN Facility | N/A |
| DYRK2 Reverse Primer: ccgtctatgaatgctgtccag | Stanford PAN Facility | N/A |
| Gapdh Forward Primer: tgaacgggaagctcactgg | Stanford PAN Facility | N/A |
| Gapdh Reverse Primer: tccaccaccctgttgctgta | Stanford PAN Facility | N/A |
| APC gRNA: gtttgagctgtttgaggagg | Stanford PAN Facility | N/A |
| Recombinant DNA | ||
| px330 CRISPR Plasmid | AddGene | 42230 |
| Software and algorithms | ||
| Excel | Microsoft | https://www.microsoft.com/en-us/microsoft-365/excel |
| Powerpoint | Microsoft | https://www.microsoft.com/en-us/microsoft-365/powerpoint |
| Prism 8.2.0 | GraphPad | https://www.graphpad.com |
| RStudio | RStudio | https://rstudio.com |
| Custom Code | Zenodo | https://github.com/NCICCGPO/CTD2-Network/tree/main/Stanford/amplification_outliers; https://doi.org/10.5281/zenodo.8383516 |
| R 4.0.1 | R | https://www.r-project.org/ |
| Other | ||
| LUCENTBLUE X-ray FILM, 8 × 10cm FOR CHEMILUMINESCENCE | E&K Scientific | EK-5129 |
| iScript™ Reverse Transcription Supermix | Bio-Rad | 1708841 |
| Power SYBR™ Green PCR Master Mix | Thermofisher Scientific | 4367659 |
| Cultrex Rat Collagen I, Lower Viscosity (3 mg/mL) | Trevigen | 3443-100-01 |
| AlamarBlue Cell Viability Reagent | Life Technologies | DAL1100 |
| Citrate Buffer, pH 6.0, Antigen Retrieval Solution | Millipore Sigma | C9999 |
| Normal donkey serum | Jackson ImmunoResearch | 017-000-121 |
| Trevigen Cultrex Reduced Growth Factor BME Type 2 | R&D Systems | 3533-005-02 |
| RIPA Buffer | Thermofisher Scientific | 89900 |
| 4%–12% Bis-Tris Gels | Thermofisher Scientific | |
| TrypLE™ Express (1X), no Phenol Red | Life Technologies | 12604021 |
| DNase I | Worthington | LS006328 |
| Collagenase Type 4 5x50mg | Worthington | LS004212 |
| DNeasy Blood and Tissue Kit | Qiagen | 69506 |
| Polybrene Infection/Transfection Reagent | Sigma | TR-1003-G |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Human specimens
Normal tissues were obtained through the Stanford Tissue Bank from patients undergoing surgical procedures at Stanford Health Care (SHC). All experiments utilizing human material were approved by the SHC Institutional Review Board and performed under protocol #28908. Written informed consent for research was obtained from donors prior to tissue procurement. Analysis of influence of gender identity upon experiments was not performed.
Mouse models
Female C57BL/6 mice or NOG-E mice (no sex differences were observed) were used for organoid model generation and subcutaneous tumor implantation (mice were obtained from Taconic Biosciences) in accordance with NIH and Stanford Administrative Panel on Laboratory animal Care (APLAC). Mice were group housed and littermates were randomly selected for experimentation at 4–8 weeks of age. Animals were maintained on a 12-h light/dark cycle, in a temperature- and humidity-controlled room with food and water.
Organoid derivation and culture
Mouse esophagus, tongue/oral mucosa and pancreas were dissected from female 6–8-week-old p53flox/flox mice and mouse peripheral lung from 6–8-week-old female LSL-KrasG12D; p53flox/flox mice29,32 and minced into small pieces. Minced tissues were embedded in collagen gel in air-liquid interface (ALI).13,14 2 × 108 pfu adenovirus Ad-Fc or Ad-Cre-GFP (University of Iowa Vector Core) per 500 μL medium were added on top of the inner dish collagen I (Wako) to activate KrasG12D expression and delete p53. Established organoids were maintained in ALI. Culture medium for mouse oral mucosa, esophagus and lung organoids was advanced DMEM/F12 supplemented with 1 mM HEPES, 10 mM nicotinamide, Glutamax, 1 mM N-acetylcysteine, B27, 0.5 μM A83-01, PSQ, 50 ng/mL recombinant human EGF, 100 ng/mL recombinant human NOGGIN. Pancreas organoids were cultured in WENR media (see below). Human gastric and colon organoids were generated from deidentified surgical specimens from Stanford Hospital under an approved IRB protocol following established methods and grown in a submerged format in WENR media20,23 in BME-2 extracellular matrix (R&D Systems).26,28 The R175H mutant of p53 was transduced by lentivirus into wild type human gastric organoids. APC null colon organoids were generated as previously described.20,23,26 For DYRK2 validation experiments, lentivirus DYRK2 (TRCN489007, Broad ORF collection) or eGFP control (BRDN559466, Broad ORF collection) was infected into p53−/− mouse oral mucosa organoids in log phase using spinfection and subjected to puromycin selection.26,28 For FGF3 validation experiments, an independent p53−/− esophageal organoid line was infected by lentiviral eGFP control or FGF3 (TRCN477770, Broad ORF collection), as above. All cultures were maintained at 37°C at 5% CO2 and 95% relative humidity. All lines used in this study tested negative for mycoplasma.
METHOD DETAILS
Computational amplified/upregulated outlier analysis
TCGA expression data (rsem genes normalized rnaseqv2) and copy number data (hg19 nocnv SNP6 array data) were downloaded for lung adenocarcinoma (LUAD), esophageal carcinoma (ESCA, filtered to retain only the squamous samples), head and neck squamous carcinoma (HNSC), pancreatic adenocarcinoma (PAAD), colon adenocarcinoma (COAD) and stomach adenocarcinoma (STAD) from Firehose (http://gdac.broadinstitute.org/). We considered a segment amplified if the segment value was higher than the median value of all segments of the sample plus six times the standard deviation of the central quantiles of the values of the sample (0.25–0.75). Amplified segments were matched with the position of the gene (hg19) to assign gene-level values. For gene expression data, values were normalized using a log2 (adding a unit to the original count) transformation. In order to identify expression outliers, we first estimated from the tumor expression data a theoretical normal distribution for each gene using the function fitdistr from the MASS package. From this theoretical distribution we extract the 5% and 95% quantiles using the function qnorm. Those quantiles defined the lower and upper threshold, expression values below the 5% threshold were consider down regulation outliers and values above 95% threshold were considered up regulation outliers. Finally, we called a given gene an outlier if it was simultaneously amplified and an upregulated expression outlier following the approach from Curtis et al.8 Outlier distributions were plotted with the ggbio package, and transitions across genes were smoothed using the smooth.spline function.
Tissue contextual pooled screening of putative outlier genes
Organoids were either removed from matrix with either TrypLE or collagenase type IV as previously described.26,27 Upon matrix removal, organoids were further digested into single cell suspensions with TrypLE for 20 min at 37°C. Cells were centrifuged at 600 x g for 3 min, then washed and incubated with 100 Kunitz Units DNase I in 1 mL of Advanced DMEM/F12 for 15 min at room temperature. Cells were then counted and 1000 cells per ORF construct were infected via spinfection as independent biological replicate screening experiments (up to n = 4; see Table S2). Each biological replicate was resuspended in complete culture media +8 μg/mL polybrene, plus pooled lentivirus library with equal weighting inclusive of negative controls (MOI = 0.8) to a total volume of 250 μL per replicate in a 48 well plate. Plates were centrifuged for 1 h at 32 °C at 600 x g, then allowed to recover for 4–6 h at 37°C in a cell incubator. Spinoculated cells were then plated in ECM/Matrigel at 100,000 cells per 50 μL droplet in complete media. Each biological replicate was cultured separately including subsequent passaging and gDNA harvesting. Each replicate was allowed to grow for 96 h and then transferred either to ALI or maintained in ECM per above culture conditions. After 96 h, cultures were then subjected to puromycin selection at IC90 concentrations established by prior dose response studies. After 96 h of puromycin selection, the cultures were digested into single cell suspensions and half the biomass was snap frozen as a representation of the initial screen timepoint. Cultures were then passaged serially upon confluence as previously described26,27 and screens were terminated after the fourth passage.
Barcode amplification and Deep Sequencing
Snap frozen cell pellets, corresponding to independent biological replicates, were extracted with DNeasy (Qiagen) following the manufacturer’s protocol. 10 μg of genomic DNA was subjected to a one step PCR strategy per the Broad Genomic Perturbation Platform protocol (https://portals.broadinstitute.org/gpp/public/resources/protocols). Briefly, a pool of Illumina P5 primers were pooled and combined with a barcode library specific library primer. PCR products were gel extracted and subjected to Bioanalyzer analysis to assess for sample purity and quantified by Qubit. Libraries representing individual biological replicate samples were then pooled and Deep Sequencing was performed on an Illumina MiSeq instrument configured 300 | 8 | 0 | 300 bp with Nextera XT format. Fastq files were processed using the R package ShortRead58 and ORF barcodes were counted as a DNAStringSet from the BioString R package,59 and reverse barcodes were counted using the reverseComplement function. Barcodes (both forward and reverse) were counted using the vcountPattern function with an error limit of one single mismatch in forward and reverse strand reads combined.
ORF enrichment analysis
The proportion of barcodes for each gene/sample were calculated by dividing the barcode counts by the total read counts of the NGS library. We then calculated the ratio of enrichment for each gene by dividing the proportion of barcodes at the terminal timepoint by the proportion of barcodes at the initial timepoint. In order to avoid division by zero, genes were filtered for any sample proportions less than 0.0003 in the initial timepoints. Barcodes were plotted in rank order by genes with the median value of the ratio, separating the controls from the putative drivers. The significance of the increase in counts for each ORF was evaluated using a one-side t test comparing the final timepoint vs. 0 timepoint.
Immunoblotting
Lysate preparation and immunoblot analyses were performed using standard methods. Briefly, cells were harvested in RIPA buffer containing protease inhibitor cocktail (cOmplete Mini, Roche) and centrifuged at 5000 × g for 10 min to remove debris. Protein concentration was assessed using BCA Kit (Bio-Rad). Samples were supplemented with SDS sample buffer containing 5% 2-mercaptomethanol and 100 mM DTT. NuPAGE 4%–12% Bis-Tris Gels (ThermoFisher Scientific) were used for SDS-PAGE, then transferred to PVDF membranes (EMD Millipore). Membranes were blocked with 5% non-fat dry milk in TBS/0.05% Triton X-(TBST/milk). Membranes were incubated with primary antibodies: anti-DYRK2 antibody (#8143, Cell Signaling) and anti-GAPDH (#5174, Cell Signaling) diluted in TBST/milk overnight at 4°C. HRP-conjugated anti-Rabbit IgG (H + L) antibody (111-035-003, Jackson ImmunoResearch) were incubated for 1h at room temperature. Bound antibodies were visualized using SuperSignal West Pico Chemiluminescent Substrates (ThermoFisher Scientific) and exposure of AccuRay Blue X-ray Films (E&K Scientific).
qRT-PCR
Total RNA was isolated from cells using RNeasy (QIAGEN) and cDNA was synthesized using iScript Reverse Transcription Supermix (Bio-Rad). qRT-PCR was performed to measure specific gene expression with Power SYBR Green assay (Applied Biosystems) or TaqMan assays (ThermoFisher) as indicated in the key resource table. Relative RNA expression was calculated using standard curve method and normalized by Gapdh or Actb.
Proliferation assays and in vitro small molecule studies
Organoids were dissociated into single cells. 5000 cells were plated with 5–10 μL of Matrigel in flat-bottom 96 well plates or 10 μL of collagen in round-bottom 96 well plates. 10% AlamarBlue was added to wells and incubated for 4 h at 37 °C at indicated days. Fluorescence (Ex/Em = 530/590 nm) was measured in a Biotek plate reader according to the manufacturer’s protocols. Small molecule treatments were carried out as dose response studies in triplicate 72 h after seeding.
Immunofluorescence staining
Paraffin-embedded sections were incubated in citrate antigen retrieval solution and blocked with 10% normal donkey serum. Sections were incubated with the following primary antibodies: mouse anti-E-cadherin (BD610182, BD Biosciences), Alexa Fluor 647-labeled anti-KRT5 (ab193895, Abcam), rat-anti-Ki67 (14-5698-82, eBioscience) overnight at 4°C. Sections were washed and subsequently incubated with the following secondary antibodies: Cy3-conjugated AffiniPure goat anti-mouse IgG (H + L) (115-165-062, Jackson ImmunoResearch), Cy3-conjugated AffiniPure goat anti-rat IgG (H + L) (112-165-167, Jackson ImmunoResearch). Sections were washed and mounted with Fluoro-Gel II with DAPI (Cat. 17985-50, Electron microscopy sciences). Sections were imaged by a Zeiss Axio-Imager Z1 with ApoTome attachment or a Leica SP6 inverted confocal microscope as previously described.28
In vivo organoid transplantation
All animal experimental procedures were approved by an IACUC protocol. Subcutaneous transplantations were performed in NOG-E mice (Taconic) as previously described.13 Briefly, organoid cells were dissociated into single cell suspensions and mixed in 100 μL of Matrigel and 105-106 cells were subcutaneously injected into the flank. Tumor formation was assessed by palpation and tumors were measured by digital calipers for length, width, and height and ellipsoid volumes were calculated as previously described.13 Animals were euthanized at predefined endpoints or morbidity criteria and tumor and tissue samples were freshly fixed in formalin and embedded in paraffin.
In vivo adenovirus injection
106 GFP-expressing p53−/− mouse esophageal organoid cells or 105 FGF3-expressing p53−/− mouse esophageal organoid cells were subcutaneously implanted as above in 20 mice. Mice were monitored for serial tumor measurement and mice with measurable tumors were randomized to achieve a mean of 100 mm3 tumor volume. 5 × 108 pfu of adenovirus expressing FGFR1-ECD-Fc, encoding the soluble extracellular ligand-binding domain of human FGFR1 fused to C-terminal mouse IgG2α Fc (Ad FGFR1-ECD-Fc), or control adenovirus expressing mouse IgG2α Fc (Ad Fc) was injected i.v. retroorbitally and serum was analyzed by Western blot for expression of FGFR1-ECD-Fc or Fc using anti-IgG2α Fc (Jackson ImmunoResearch). All adenoviral inserts were cloned into the E1 region of E1−E3− Ad strain 5 by homologous recombination and amplified in in 293 cells followed by CsCl2 gradient purification of virus, as previously described.60–63
FGF3 ELISA
Organoid conditioned media from log phase growth GFP or FGF3 expressing p53−/− esophageal organoid cells were assessed according to the manufacturer’s protocol (Aviva Biosciences catalog # OKEH02512).
AZD4547 in vivo treatment
Mice were subcutaneously transplanted with either FGF3- or GFP-expressing p53−/− esophageal organoid cells and tumor volumes were serially measured and mice were randomized as above. Mice were treated with either an oral suspension of AZD4547 or vehicle daily as previously described64 where AZD4547 was compounded in 1% vol:vol Tween-80 at 2.5 mg/mL and animals were dosed daily at 12.5 mg/kg via oral gavage. Tumors were serially measured as above.
Quantitation of CD31+ microvessel density
Immunohistochemistry for CD31 (MAB1398Z, EMD Millipore) was performed on 5 μm FFPE tissue sections using proteinase K antigen retrieval as described.65 Microvessel density was scored using the Chalkley method as previously described.66,67 Briefly, FFPE sections of mouse tumors from FGF3 expressing p53−/− esophageal organoid subcutaneous transplants were subjected to CD31 IHC staining. The mean of three CD31+ hotspots at 20× magnification were scored on the overlap of 25 random points on an ocular grid.
QUANTIFICATION AND STATISTICAL ANALYSIS
When relevant, experiment statistical testing is reported in method details and in figure legends. Statistical testing was performed in R or excel and the type of test and p values are reported in figure legends at thresholds such as 0.05, 0.01. Figure legends indicate data values, error bars and for in vivo experiments, number of animals in experimental groups.
Supplementary Material
Highlights.
Integrative analysis of extreme somatic copy number gains and extreme expression dysregulation
Screening of “outlier” oncogenes in diverse organoid models of early tumorigenesis
Validation of DYRK2 and FGF3 as oncogenic candidates in oral and esophageal squamous cancers
Demonstration of FGFR inhibitor efficacy in engineered esophageal squamous cancers with FGF3 copy gain
ACKNOWLEDGMENTS
We thank members of the Kuo laboratory and the CTD^2 consortium for helpful discussions. We thank Scott Younger for supervising the design and construction of lentiviral library pools. This work was supported by the NCI Cancer Target Discovery and Development (CTD^2) Network (U01CA217851, C.J.K and C.C.; U01CA176058, W.C.H.). Support was also provided by NIH K08DE027730 and D.R. discretionary funds to A.A.S., AEI RYC2019-026576-I, “LaCaixa” Foundation LCF/PR/PR17/51120011 to J.A.S., and NIH U54CA224081, NIH U01CA199241, Emerson Collective, Ludwig Cancer Research, and Stand Up To Cancer to C.J.K. This manuscript is dedicated to the memories of Dr. Daniela Gerhard and Dr. Kenneth Scott.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113355.
DECLARATION OF INTERESTS
A.A.S. has served as a consultant for Boehringer Ingelheim and Pharmacosmos and is a former employee of Tempus Labs. C.J.K. is a scientific advisory board member for Surrozen, Inc., Mozart Therapeutics, and NextVivo. C.C. has served as a scientific advisory board member/consultant for Genentech, Grail, DeepCell, Nanostring, and Viosera. W.C.H. is a consultant for Thermo Fisher, Solasta Ventures, MPM Capital, KSQ Therapeutics, Tyra Biosciences, Frontier Medicine, Jubilant Therapeutics, RAPPTA Therapeutics, Hexagon Bio, Serinus Biosciences, Function Oncology, and Calyx.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
REFERENCES
- 1.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905. 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhsng CZ, Wala J, Mermel CH, et al. (2013). Pan-cancer patterns of somatic copy number alteration. Nat. Genet 45, 1134–1140, [doi]. 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oh DY, and Bang YJ (2020). HER2-targeted therapies - a role beyond breast cancer. Nat. Rev. Clin. Oncol 17, 33–48. 10.1038/s41571-019-0268-3. [DOI] [PubMed] [Google Scholar]
- 4.Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE, Vazquez F, Weir BA, Fitzgerald ME, Tanaka M, et al. (2016). MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 351, 1214–1218. 10.1126/science.aad5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang A, Garraway LA, Ashworth A, and Weber B (2020). Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov 19, 23–38. 10.1038/s41573-019-0046-z. [DOI] [PubMed] [Google Scholar]
- 6.Kristensen VN, Lingærde OC, Russnes HG, Vollan HKM, Frigessi A, and Børresen-Dale AL (2014). Principles and methods of integrative genomic analyses in cancer. Nat. Rev. Cancer 14, 299–313. 10.1038/nrc3721. [DOI] [PubMed] [Google Scholar]
- 7.van Dyk E, Hoogstraat M, Ten Hoeve J, Reinders MJT, and Wessels LFA (2016). RUBIC identifies driver genes by detecting recurrent DNA copy number breaks. Nat. Commun 7, 12159. 10.1038/ncomms12159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. (2012). The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352. 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanchez-Garcia F, Villagrasa P, Matsui J, Kotliar D, Castro V, Akavia UD, Chen BJ, Saucedo-Cuevas L, Rodriguez Barrueco R, Llobet-Navas D, et al. (2014). Integration of genomic data enables selective discovery of breast cancer drivers. Cell 159, 1461–1475. 10.1016/j.cell.2014.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, Natrajan R, Marchio C, Iorns E, Mackay A, et al. (2010). FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 70, 2085–2094. 10.1158/0008-5472.CAN-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, et al. (2005). Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 310, 644–648. 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 12.Lo YH, Karlsson K, and Kuo CJ (2020). Applications of Organoids for Cancer Biology and Precision Medicine. Nat. Can. (Ott.) 1, 761–773. 10.1038/s43018-020-0102-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li X, Nadauld L, Ootani A, Corney DC, Pai RK, Gevaert O, Cantrell MA, Rack PG, Neal JT, Chan CWM, et al. (2014). Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat. Med 20, 769–777. 10.1038/nm.3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nadauld LD, Garcia S, Natsoulis G, Bell JM, Miotke L, Hopmans ES, Xu H, Pai RK, Palm C, Regan JF, et al. (2014). Metastatic tumor evolution and organoid modeling implicate TGFBR2 as a cancer driver in diffuse gastric cancer. Genome Biol. 15, 428. 10.1186/s13059-014-0428-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salahudeen AA, and Kuo CJ (2015). Toward recreating colon cancer in human organoids. Nat. Med 21, 215–216. 10.1038/nm.3818. [DOI] [PubMed] [Google Scholar]
- 16.Broutier L, Mastrogiovanni G, Verstegen MM, Francies HE, Gavarró LM, Bradshaw CR, Allen GE, Arnes-Benito R, Sidorova O, Gaspersz MP, et al. (2017). Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med 23, 1424–1435. 10.1038/nm.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Francies HE, Barthorpe A, McLaren-Douglas A, Barendt WJ, and Garnett MJ (2019). Drug Sensitivity Assays of Human Cancer Organoid Cultures. Methods Mol. Biol 1576, 339–351. 10.1007/7651_2016_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang L, Holtzinger A, Jagan I, BeGora M, Lohse I, Ngai N, Nostro C, Wang R, Muthuswamy LB, Crawford HC, et al. (2015). Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med 21, 1364–1371. 10.1038/nm.3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Francies HE, Secrier M, Perner J, Miremadi A, Galeano-Dalmau N, Barendt WJ, Letchford L, Leyden GM, Goffin EK, et al. (2018). Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeutics. Nat. Commun 9, 2983. 10.1038/s41467-018-05190-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sato T, Stange DE, Ferrante M, Vries RGJ, Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J, Siersema PD, and Clevers H (2011). Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772. 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 21.van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J, Taylor-Weiner A, Kester L, et al. (2015). Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945. 10.1016/j.cell.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Du Y, Li X, Niu Q, Mo X, Qui M, Ma T, Kuo CJ, and Fu H (2020). Development of a miniaturized 3D organoid culture platform for ultra-high-throughput screening. J. Mol. Cell Biol 12, 630–643. 10.1093/jmcb/mjaa036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y, Watanabe T, Kanai T, and Sato T (2015). Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med 21, 256–262. 10.1038/nm.3802. [DOI] [PubMed] [Google Scholar]
- 24.Drost J, van Jaarsveld RH, Ponsioen B, Zimberlin C, van Boxtel R, Buijs A, Sachs N, Overmeer RM, Offerhaus GJ, Begthel H, et al. (2015). Sequential cancer mutations in cultured human intestinal stem cells. Nature 521, 43–47. 10.1038/nature14415. [DOI] [PubMed] [Google Scholar]
- 25.Artegiani B, van Voorthuijsen L, Lindeboom RGH, Seinstra D, Heo I, Tapia P, López-Iglesias C, Postrach D, Dayton T, Oka R, et al. (2019). Probing the Tumor Suppressor Function of BAP1 in CRISPR-Engineered Human Liver Organoids. Cell Stem Cell 24, 927–943.e6. 10.1016/j.stem.2019.04.017. [DOI] [PubMed] [Google Scholar]
- 26.Lo YH, Kolahi KS, Du Y, Chang CY, Krokhotin A, Nair A, Sobba WD, Karlsson K, Jones SJ, Longacre TA, et al. (2021). A CRISPR/Cas9-engineered ARID1A-deficient human gastric cancer organoid model reveals essential and non-essential modes of oncogenic transformation. Cancer Discov. 11, 1562–1581. 10.1158/2159-8290.CD-20-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, Liu IH, Chiou SH, Salahudeen AA, Smith AR, et al. (2018). Organoid Modeling of the Tumor Immune Microenvironment. Cell 175, 1972–1988.e16, S0092-8674(18)31513-7 [pii]. 10.1016/j.cell.2018.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salahudeen AA, Choi SS, Rustagi A, Zhu J, van Unen V, de la O SM, Flynn RA, Margalef-Català M, Santos AJM, Ju J, et al. (2020). Progenitor identification and SARS-CoV-2 infection in human distal lung organoids. Nature 588, 670–675. 10.1038/s41586-020-3014-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. (2003). Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450. S153561080300309X [pii]. [DOI] [PubMed] [Google Scholar]
- 30.Ebrahimi M, and Botelho M (2017). Adult Stem Cells of Orofacial Origin: Current Knowledge and Limitation and Future Trend in Regenerative Medicine. Tissue Eng. Regen. Med 14, 719–733. 10.1007/s13770-017-0078-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones KB, Furukawa S, Marangoni P, Ma H, Pinkard H, D’Urso R, Zilionis R, Klein AM, and Klein OD (2019). Quantitative Clonal Analysis and Single-Cell Transcriptomics Reveal Division Kinetics, Hierarchy, and Fate of Oral Epithelial Progenitor Cells. Cell Stem Cell 24, 183–192.e8. 10.1016/j.stem.2018.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, and Jacks T (2005). The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 65, 10280–10288. [DOI] [PubMed] [Google Scholar]
- 33.Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, et al. (2011). A public genome-scale lentiviral expression library of human ORFs. Nat. Methods 8, 659–661. 10.1038/nmeth.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cancer Genome Atlas Network (2015). Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582. 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo X, Wang X, Wang Z, Banerjee S, Yang J, Huang L, and Dixon JE (2016). Site-specific proteasome phosphorylation controls cell proliferation and tumorigenesis. Nat. Cell Biol 18, 202–212. 10.1038/ncb3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taira N, Nihira K, Yamaguchi T, Miki Y, and Yoshida K (2007). DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol. Cell 25, 725–738. 10.1016/j.molcel.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 37.Tandon V, de la Vega L, and Banerjee S (2021). Emerging roles of DYRK2 in cancer. J. Biol. Chem 296, 100233. 10.1074/jbc.REV120.015217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weaver A, and Bossaer JB (2021). Fibroblast growth factor receptor (FGFR) inhibitors: A review of a novel therapeutic class. J. Oncol. Pharm. Pract 27, 702–710. 10.1177/1078155220983425. [DOI] [PubMed] [Google Scholar]
- 39.Hahn WC, Bader JS, Braun TP, Califano A, Clemons PA, Druker BJ, Ewald AJ, Fu H, Jagu S, Kemp CJ, et al. (2021). An expanded universe of cancer targets. Cell 184, 1142–1155. 10.1016/j.cell.2021.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gallo D, Young JTF, Fourtounis J, Martino G, Álvarez-Quilón A, Bernier C, Duffy NM, Papp R, Roulston A, Stocco R, et al. (2022). CCNE1 amplification is synthetic lethal with PKMYT1 kinase inhibition. Nature 604, 749–756. 10.1038/s41586-022-04638-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boehm JS, Garnett MJ, Adams DJ, Francies HE, Golub TR, Hahn WC, Iorio F, McFarland JM, Parts L, and Vazquez F (2021). Cancer research needs a better map. Nature 589, 514–516. 10.1038/d41586-021-00182-0. [DOI] [PubMed] [Google Scholar]
- 42.Gandara DR, Riess JW, and Lara PN Jr. (2018). In Search of an Oncogene Driver for Squamous Lung Cancer. JAMA Oncol. 4, 1197–1198. 10.1001/jamaoncol.2018.0774. [DOI] [PubMed] [Google Scholar]
- 43.Mountzios G, Rampias T, and Psyrri A (2014). The mutational spectrum of squamous-cell carcinoma of the head and neck: targetable genetic events and clinical impact. Ann. Oncol 25, 1889–1900. 10.1093/annonc/mdu143. [DOI] [PubMed] [Google Scholar]
- 44.Salem ME, Puccini A, Xiu J, Raghavan D, Lenz HJ, Korn WM, Shields AF, Philip PA, Marshall JL, and Goldberg RM (2018). Comparative Molecular Analyses of Esophageal Squamous Cell Carcinoma, Esophageal Adenocarcinoma, and Gastric Adenocarcinoma. Oncol. 23, 1319–1327. 10.1634/theoncologist.2018-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rueda OM, Sammut SJ, Seoane JA, Chin SF, Caswell-Jin JL, Callari M, Batra R, Pereira B, Bruna A, Ali HR, et al. (2019). Dynamics of breast-cancer relapse reveal late-recurring ER-positive genomic subgroups. Nature 567, 399–404. 10.1038/s41586-019-1007-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Catenacci DVT, Moya S, Lomnicki S, Chase LM, Peterson BF, Reizine N, Alpert L, Setia N, Xiao SY, Hart J, et al. (2021). Personalized Antibodies for Gastroesophageal Adenocarcinoma (PANGEA): A Phase II Study Evaluating an Individualized Treatment Strategy for Metastatic Disease. Cancer Discov. 11, 308–325. 10.1158/2159-8290.CD-20-1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wong GS, Zhou J, Liu JB, Wu Z, Xu X, Li T, Xu D, Schumacher SE, Puschhof J, McFarland J, et al. (2018). Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat. Med 24, 968–977. 10.1038/s41591-018-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cancer Genome Atlas Research Network; Analysis Working Group: Asan University; BC Cancer Agency; Brigham and Women’s Hospital; Broad Institute, Brown University; Case Western Reserve University; Dana-Farber Cancer Institute, Duke University (2017). Integrated genomic characterization of oesophageal carcinoma. Nature 541, 169–175. 10.1038/nature20805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abnet CC, Arnold M, and Wei WQ (2018). Epidemiology of Esophageal Squamous Cell Carcinoma. Gastroenterology 154, 360–373. 10.1053/j.gastro.2017.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Valenzuela CA, Vargas L, Martinez V, Bravo S, and Brown NE (2017). Palbociclib-induced autophagy and senescence in gastric cancer cells. Exp. Cell Res 360, 390–396. 10.1016/j.yexcr.2017.09.031. [DOI] [PubMed] [Google Scholar]
- 51.Kuuselo R, Simon R, Karhu R, Tennstedt P, Marx AH, Izbicki JR, Yekebas E, Sauter G, and Kallioniemi A (2010). 19q13 amplification is associated with high grade and stage in pancreatic cancer. Genes Chromosomes Cancer 49, 569–575. 10.1002/gcc.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim TM, Yim SH, Lee JS, Kwon MS, Ryu JW, Kang HM, Fiegler H, Carter NP, and Chung YJ (2005). Genome-wide screening of genomic alterations and their clinicopathologic implications in non-small cell lung cancers. Clin. Cancer Res 11, 8235–8242. 10.1158/1078-0432.CCR-05-1157. [DOI] [PubMed] [Google Scholar]
- 53.Basu A, and Lambring CB (2021). Akt Isoforms: A Family Affair in Breast Cancer. Cancers 13, 3445. 10.3390/cancers13143445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bellacosa A, de Feo D, Godwin AK, Bell DW, Cheng JQ, Altomare DA, Wan M, Dubeau L, Scambia G, Masciullo V, et al. (1995). Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int. J. Cancer 64, 280–285. 10.1002/ijc.2910640412. [DOI] [PubMed] [Google Scholar]
- 55.Nunez JK, Chen J, Pommier GC, Cogan JZ, Replogle JM, Adriaens C, Ramadoss GN, Shi Q, Hung KL, Samelson AJ, et al. (2021). Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell. 10.1016/j.cell.2021.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M, and Weissman JS (2016). Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife 5, e19760. 10.7554/eLife.19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. (2014). Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661. 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morgan M, Anders S, Lawrence M, Aboyoun P, Pagès H, and Gentleman R (2009). ShortRead: a bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics 25, 2607–2608. 10.1093/bioinformatics/btp450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pagès H, A. P, Gentleman R, and DebRoy S (2021). Biostrings: Efficient Manipulation of Biological Strings, R package version 2.60.2 10.18129/B9.bioc.Biostrings. [DOI] [Google Scholar]
- 60.Chang J, Mancuso MR, Maier C, Liang X, Yuki K, Yang L, Kwong JW, Wang J, Rao V, Vallon M, et al. (2017). Gpr124 is essential for blood-brain barrier integrity in central nervous system disease. Nat. Med 23, 450–460. 10.1038/nm.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kuhnert F, Mancuso MR, Shamloo A, Wang HT, Choksi V, Florek M, Su H, Fruttiger M, Young WL, Heilshorn SC, and Kuo CJ (2010). Essential regulation of CNS angiogenesis by the orphan G protein-coupled receptor GPR124. Science 330, 985–989. 10.1126/science.1196554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuo CJ, Farnebo F, Yu EY, Christofferson R, Swearingen RA, Carter R, von Recum HA, Yuan J, Kamihara J, Flynn E, et al. (2001). Comparative evaluation of the antitumor activity of antiangiogenic proteins delivered by gene transfer. Proc. Natl. Acad. Sci. USA 98, 4605–4610. 10.1073/pnas.081615298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yan KS, Janda CY, Chang J, Zheng GXY, Larkin KA, Luca VC, Chia LA, Mah AT, Han A, Terry JM, et al. (2017). Non-equivalence of Wnt and R-spondin ligands during Lgr5(+) intestinal stem-cell self-renewal. Nature 545, 238–242. 10.1038/nature22313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gavine PR, Mooney L, Kilgour E, Thomas AP, Al-Kadhimi K, Beck S, Rooney C, Coleman T, Baker D, Mellor MJ, et al. (2012). AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. 72, 2045–2056. 10.1158/0008-5472.CAN-11-3034. [DOI] [PubMed] [Google Scholar]
- 65.Fairweather M, Heit YI, Buie J, Rosenberg LM, Briggs A, Orgill DP, and Bertagnolli MM (2015). Celecoxib inhibits early cutaneous wound healing. J. Surg. Res 194, 717–724. 10.1016/j.jss.2014.12.026. [DOI] [PubMed] [Google Scholar]
- 66.Fox SB, Leek RD, Weekes MP, Whitehouse RM, Gatter KC, and Harris AL (1995). Quantitation and prognostic value of breast cancer angiogenesis: comparison of microvessel density, Chalkley count, and computer image analysis. J. Pathol 177, 275–283. 10.1002/path.1711770310. [DOI] [PubMed] [Google Scholar]
- 67.Vermeulen PB, Gasparini G, Fox SB, Colpaert C, Marson LP, Gion M, Beliën JAM, de Waal RMW, Van Marck E, Magnani E, et al. (2002). Second international consensus on the methodology and criteria of evaluation of angiogenesis quantification in solid human tumours. Eur. J. Cancer 38, 1564–1579. 10.1016/s0959-8049(02)00094-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Outlier screening data has been deposited at the CTD^2 data portal and are publicly available as of the date of the publication. The DOI is listed in the key resource table.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resource table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| DYRK2 antibody | Cell Signaling | 8143; RRID:AB_10889932 |
| GAPDH (D16H11) Rabbit mAb | Cell Signaling | 5174S; RRID:AB_10622025 |
| Peroxidase AffiniPure Goat Anti-Rabbit IgG (H + L) | Jackson ImmunoResearch | 711-035-152; RRID: AB_10015282 |
| Mouse anti-E-cadherin | BD Biosciences | BD610182; RRID:AB_397581 |
| Alexa Fluor 647 labeled-anti-CK5 | Abcam | ab193895; RRID:AB_2728796 |
| Rat-anti-Ki67 | eBiosciences | 14-5698-82; RRID:AB_10854564 |
| Cy3-conjugated AffiniPure goat anti-mouse IgG (H + L) | Jackson ImmunoResearch | 115-165-062; RRID:AB_2338685 |
| Cy3 Cy3-conjugated AffiniPure goat anti-rat IgG (H + L) | Jackson ImmunoResearch | 112-165-167; RRID:AB_2338251 |
| Peroxidase AffiniPure Goat Anti-Mouse IgG, Fcγ subclass 2a specific | Jackson ImmunoResearch | 115-035-206; RRID:AB_2338514 |
| Bacterial and virus strains | ||
| p53R175H lentivirus | Ken Scott laboratory | KS5 |
| Ad-Cre-GFP | University of Iowa Vector Core | 1174 |
| Adenovirus Fc | Stanford University | N/A |
| DYRK2 lentivirus | Genetic Perturbation Platform - Broad Institute | TRCN489007 |
| CCSB-Broad Lentiviral Expression Library | The Broad ORF collection | N/A |
| Biological samples | ||
| p53R175H gastric organoids | Stanford University | N/A |
| KrasG12D pancreatic organoids | Stanford University | N/A |
| Human APC−/− colon organoids | Stanford University | N/A |
| p53−/− oral squamous cell carcinoma organoids | Stanford University | N/A |
| p53−/− esophageal squamous cell carcinoma organoids | Stanford University | N/A |
| KrasG12D; p53−/− lung adenocarcinoma | Stanford University | N/A |
| Oral mucosa (OM) mouse organoids | Stanford University | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| cOmplete™, Mini Protease Inhibitor Cocktail | Sigma Aldrich | 4693124001 |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | ThermoFisher Scientific | 34580 |
| Fluoro-Gel II with DAPI | Electron Microscopy Sciences | 17985–50 |
| HEPES | Thermo Fisher Scientific | BP299100 |
| Nicotinamide | Sigma | N0636-500G |
| GlutaMAX™ Supplement | ThermoFisher Scientific | 31980030 |
| N-acetyl-l-cysteine | Sigma-Aldrich | A7250-100G |
| B27 supplement | Invitrogen | 17504–001 |
| A83-01 | Tocris | 2939 |
| Recombinant human EGF | Peprotech | AF-100-15 |
| Recombinant human Noggin | Peprotech | 120-10-C |
| 2-mercaptoethanol | ThermoFisher Scientific | 21985023 |
| DTT | Sigma | 10197777001 |
| Penicillin-Streptomycin-Glutamine (100X) | Life Technologies | 10378016 |
| Recombinant Human FGFR1a (IIIc) Fc | Peprotech | 160–02 |
| Recombinant Human FGFR2a (IIIc) Fc | Peprotech | 160–03 |
| Recombinant Human FGFR3 (IIIc) Fc | Peprotech | 160–05 |
| Critical commercial assays | ||
| RNeasy Plus Mini Kit | Qiiagen | 74104 |
| iScript Reverse Transcription Supermix | BioRad | 1708841 |
| FGF3 ELISA | Aviva Systems Biology | OKEH02512 |
| Hs FGF3 Taqman Assay | ThermoFisher | 4448892 |
| Hs beta-actin Taqman Assay | ThermoFisher | 401846 |
| Deposited data | ||
| Outlier Barcode Sequencing Data | NCBI BioProject | BioProject: PRJNA1032711 |
| TCGA Expression Data | Firehose, Broad Institute | http://gdac.broadinstitute.org/ |
| Broad Genomic Perturbation Platform | Broad Institute | https://portals.broadinstitute.org/gpp/public/ |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | CRL-11268 |
| Experimental models: Organisms/strains | ||
| Il2rgtm1Sug/JicTac (NOG) Immunodeficient mice | Taconic | NOG-F |
| C57BL/6NTac mice | Taconic | B6-F |
| p53flox/flox mice | Jackson Labs | 008462 |
| LSL-KrasG12D mice | Jackson Labs | 008179 |
| Oligonucleotides | ||
| Tp53 Forward Primer: tgaggttcgtgtttgtgcct | Stanford PAN Facility | N/A |
| Tp53 Reverse Primer: gcagttcagggcaaaggact | Stanford PAN Facility | N/A |
| Ki67 Forward Primer: agctctgcagtctccacaac | Stanford PAN Facility | N/A |
| Ki67 Reverse Primer: ctctgcctcgtgactgtgtt | Stanford PAN Facility | N/A |
| Cdkn1a Forward Primer: ttgtcgctgtcttgcactct | Stanford PAN Facility | N/A |
| Cdkn1a Reverse Primer: tttcggccctgagatgttcc | Stanford PAN Facility | N/A |
| DYRK2 Forward Primer: cagtgctcacgacacaacca | Stanford PAN Facility | N/A |
| DYRK2 Reverse Primer: ccgtctatgaatgctgtccag | Stanford PAN Facility | N/A |
| Gapdh Forward Primer: tgaacgggaagctcactgg | Stanford PAN Facility | N/A |
| Gapdh Reverse Primer: tccaccaccctgttgctgta | Stanford PAN Facility | N/A |
| APC gRNA: gtttgagctgtttgaggagg | Stanford PAN Facility | N/A |
| Recombinant DNA | ||
| px330 CRISPR Plasmid | AddGene | 42230 |
| Software and algorithms | ||
| Excel | Microsoft | https://www.microsoft.com/en-us/microsoft-365/excel |
| Powerpoint | Microsoft | https://www.microsoft.com/en-us/microsoft-365/powerpoint |
| Prism 8.2.0 | GraphPad | https://www.graphpad.com |
| RStudio | RStudio | https://rstudio.com |
| Custom Code | Zenodo | https://github.com/NCICCGPO/CTD2-Network/tree/main/Stanford/amplification_outliers; https://doi.org/10.5281/zenodo.8383516 |
| R 4.0.1 | R | https://www.r-project.org/ |
| Other | ||
| LUCENTBLUE X-ray FILM, 8 × 10cm FOR CHEMILUMINESCENCE | E&K Scientific | EK-5129 |
| iScript™ Reverse Transcription Supermix | Bio-Rad | 1708841 |
| Power SYBR™ Green PCR Master Mix | Thermofisher Scientific | 4367659 |
| Cultrex Rat Collagen I, Lower Viscosity (3 mg/mL) | Trevigen | 3443-100-01 |
| AlamarBlue Cell Viability Reagent | Life Technologies | DAL1100 |
| Citrate Buffer, pH 6.0, Antigen Retrieval Solution | Millipore Sigma | C9999 |
| Normal donkey serum | Jackson ImmunoResearch | 017-000-121 |
| Trevigen Cultrex Reduced Growth Factor BME Type 2 | R&D Systems | 3533-005-02 |
| RIPA Buffer | Thermofisher Scientific | 89900 |
| 4%–12% Bis-Tris Gels | Thermofisher Scientific | |
| TrypLE™ Express (1X), no Phenol Red | Life Technologies | 12604021 |
| DNase I | Worthington | LS006328 |
| Collagenase Type 4 5x50mg | Worthington | LS004212 |
| DNeasy Blood and Tissue Kit | Qiagen | 69506 |
| Polybrene Infection/Transfection Reagent | Sigma | TR-1003-G |