

Abstract
CD4+ T-cells play fundamental roles in orchestrating immune responses and tissue homeostasis. However, our inability to unbiasedly associate peptide HLA-II (Human Leukocyte Antigens class-II) complexes with their cognate TCRs (T-cell receptors) has hampered our understanding of CD4+ T-cell function and role in pathologies. Here, we introduce TScan-II, a highly sensitive genome-scale CD4+ antigen discovery platform. This platform seamlessly integrates the endogenous HLA-II antigen processing machinery in synthetic antigen presenting cells and TCR signaling in T-cells, enabling simultaneous screening of multiple HLA and TCRs. Leveraging genome-scale human, virome, and epitope mutagenesis libraries, TScan-II facilitates de novo antigen discovery and deep exploration of TCR specificity. We demonstrate TScan-II’s potential for basic and translational research by identifying a noncanonical antigen for a cancer-reactive CD4+ T-cell clone. Additionally, we identified two antigens for clonally expanded CD4+ T-cells in Sjögren’s disease, binding distinct HLAs and expressed in HLA-II positive ductal cells within affected salivary glands.
Graphical Abstract
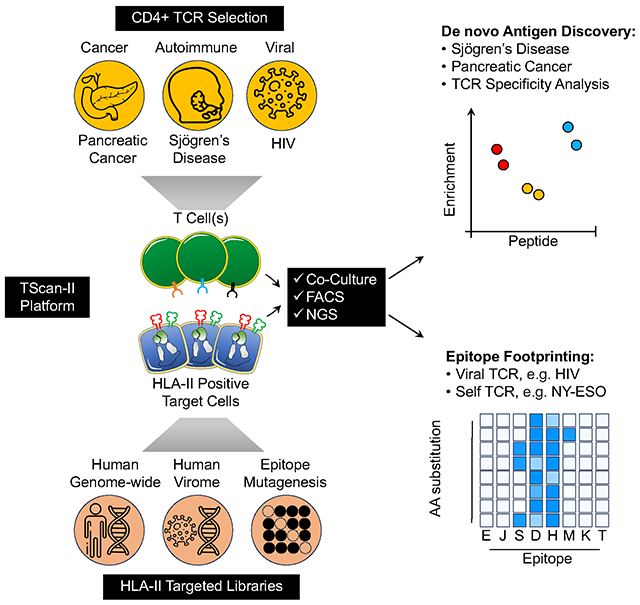
In Brief:
TScan-II is a genome-scale platform that integrates endogenous antigen processing and T-cell signaling, facilitating multiplex screens for the identification of physiologically relevant antigens recognized by CD4+ T cells. The unbiased nature of the platform allowed the identification of CD4+ T cell antigens in Sjögren’s disease and pancreatic cancer tumors.
Introduction
The adaptive immune system generates a long-lived memory of the pathogenic antigens it encounters through a set of highly variable cell surface receptors found on B and T lymphocytes1. However, the fundamental questions of which antigens are recognized remain incompletely understood. T-cells play essential roles in tissue homeostasis and protect against pathogens and cancer. T-cells recognize peptide antigens bound to HLA via their TCRs and are categorized into two subtypes, CD8+ and CD4+. CD8+ T-cells can be cytotoxic and directly destroy target cells following recognition of HLA-I bound antigens via release of granzyme and perforin. The peptides CD4+ cells recognize are generated by proteolysis of proteins in endosomes and lysosomes. This recognition stimulates cytokine secretion, activation, and differentiation into diverse T-cell subsets with tailored functions.
Recent advances in single-cell sequencing technologies have illuminated T-cells′ cellular and molecular diversity in healthy tissues, sites of infection, tumors, and inflamed autoimmune organs. However, what T-cells recognize in these contexts has remained elusive due to the lack of genome-scale high-throughput antigen discovery tools. Cell-based screening methods for CD8+ T-cells, such as T-Scan, have achieved proteome-wide success in identifying cognate antigens for TCRs from CD8+ cells, but methods for CD4+ T-cell antigen identification have not reached this level of sensitivity or throughput2. Initial attempts for CD4+ antigen discovery have mainly relied upon single-chain peptide HLA and, in instances where platforms can incorporate endogenous machinery, have been limited to a few thousand antigens, thus not genome-scale or unbiased3–8.
Developing an antigen discovery platform for CD4+ T-cells is complicated by the lower affinity of CD4+ TCRs for their cognate antigens, confinement of HLA-II expression to specialized cell types, and the complexity of HLA-II antigen processing machinery. To circumvent this, we sought to exploit the successes of the T-Scan platform in identifying CD8+ T-cell antigens for CD4+ T-cell antigens. TScan utilizes the granzyme-mediated cytolytic ability of CD8+ cells to activate a fluorescent reporter of granzyme B (GzB) protease activity. The target cells express a library of candidate antigens that are processed and presented endogenously on HLA-I molecules. Co-culture of the target cells with CD8+ T-cells of interest activates the GzB reporter in cells displaying cognate antigens, allowing enrichment of rare cognate target cells with high sensitivity and specificity2. Unfortunately, the T-Scan system lacks the HLA-II antigen processing and presentation machinery required for CD4+ T-cell screening and CD4+ cells do not typically produce GzB. Here, we report the development of TScan-II, a highly engineered complementary genome-scale platform for systematically discovering antigens productively recognized by TCRs from CD4+ T-cells. We applied this platform for the de novo discovery of CD4+ T-cell antigens in pancreatic cancer and Sjögren’s disease (SjD). The unbiased discovery of unsuspected specific antigens recognized by CD4+ T-cells helps unravel the mechanisms underlying cancer and autoimmunity.
Results
Development of Reporter Cell lines Capable of HLA-II Antigen Processing and Delivery
To build a high-throughput CD4+ antigen discovery platform capable of processing and presenting antigens at a genome-scale that incorporates endogenous T-cell signaling, we exploited a reporter of GzB activity. To do this, we had to overcome several technical challenges in both T-cells and target cells: 1) The need for HLA class II presentation machinery: Unlike HLA-I9, expression of HLA-II machinery is primarily limited to professional antigen-presenting cells (APC)10 which have low transduction rates and significantly lower brightness of the infrared GzB reporter, hampering utility for high-throughput screening. 2) The need to target endogenously expressed proteins of all types to HLA-II, which typically present peptides generated derived from endocytosed extracellular proteins undergoing proteolysis in lysosomal compartments10. This required development of a system for efficient HLA-II targeting and processing of endogenously expressed antigens. 3) The need for granzyme secretion for reporter activation: Unlike CD8+ cells, CD4+ cells, with some rare exceptions, do not typically possess GzB-mediated cytolytic activity; hence, we had to develop a platform for efficient CD4+ TCR-dependent secretion of GzB into target cells (Figure 1A).
Figure 1. Development of the TScan-II platform.
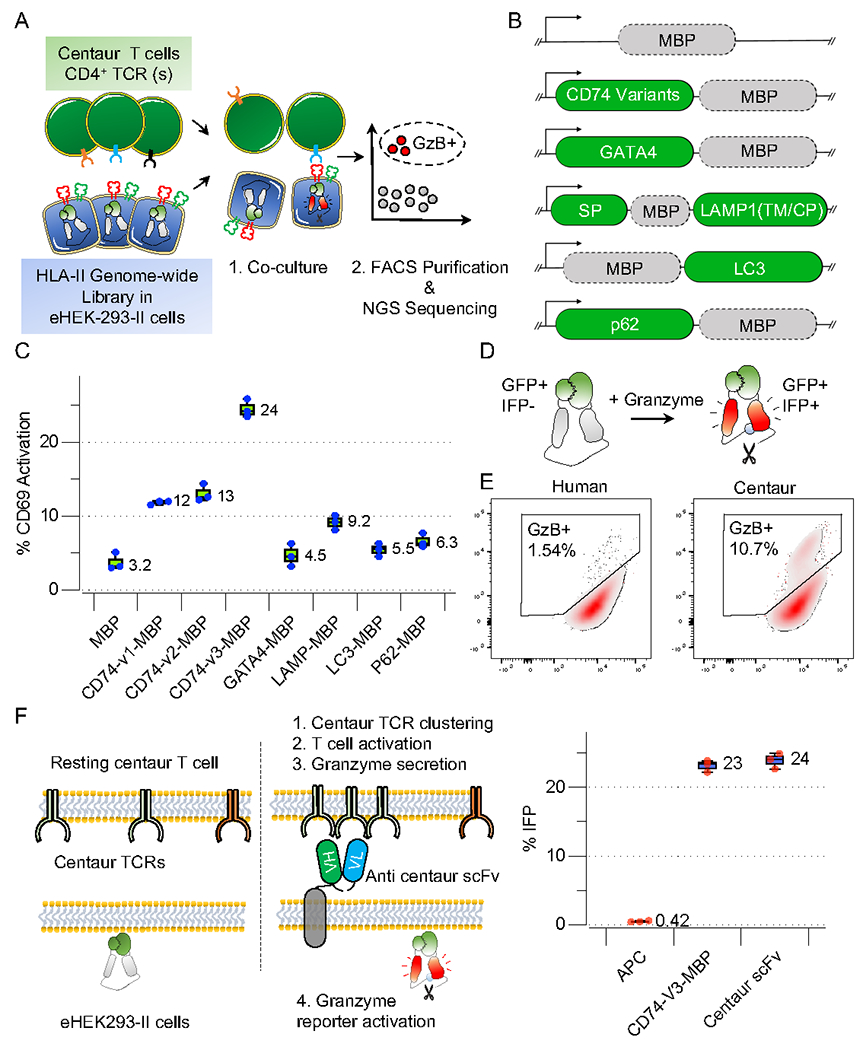
A) Conceptual layout of the CD4+ antigen discovery platform.
B) Schematic representation of fusion proteins developed to assess the presentation of full-length MBP fusions to CD74, GATA4, LAMP1 (SP: signal peptide, TM: transmembrane, CP: cytoplasmic), LC3, p62 on HLA-DRB1*1501.
C) Boxplots depicting activation levels (CD69) of Ob1.A12 TCR transduced SKW-3 cells following 6 h co-culture with eHEK-293-II cells with endogenously expressed full-length MBP fusions. The box is drawn around the inner quartile range, and the whiskers show minimum to maximum values for all plots shown here and below.
D) The GzB reporter is visually represented in a schematic, illustrating how GzB mediates the cleavage of the substrate sequence (VGPD→ŜGR) within IFPGZB-Hi. Cleavage leads to IFP fluorescence (red).
E) FACS histograms depicting GzB reporter activation in eHEK-293-II following co-culture with centaur T-cells. Target eHEK-293-II cells co-express the HLA-DRB1*1501 allele and full-length MBP fused to CD74-v3. Primary T-cells express the unmodified (left panel) or centaur Ob1.A12 TCR and CD4 co-receptor (right panel).
F) Frequency of eHEK-293-II cells that activate the GzB reporter in the presence of membrane-bound anti-centaur scFv or an endogenously processed full-length MBP fused to CD74-v3 following co-culture with Ob1.A12 centaur T-cells.
To develop a cell line capable of processing and presenting antigens on HLA-II cells like APCs, we engineered HEK293T cells. We mutated B2M via CRISPR/Cas9 B2M to avoid potential cross-reactivity with HLA-I presented antigens (Figure S1A)9. We engineered HEK293T cells to process and present antigens on HLA-II via viral expression of the HLA Class II transactivator (CIITA). CIITA plays an essential role in the transcription of classic HLA-II molecules such as HLA-DR, HLA-DP, and HLA-DQ and genes such as CD74, HLA-DM, and HLA-DO that are not only essential for regulating antigen processing but also for effective loading of epitopes on HLA-II11. HEK293T cells expressing high HLA-II levels (CIITA-HEK239T) were sorted and used as APCs (Figure S1A). HLA-II alleles were assembled as a single construct, where the HLA-β and HLAα chains were separated by 2A self-cleaving peptides (β-P2A-α) and transduced into CIITA-HEK239T-cells.
To optimize HLA-II antigen processing and presentation, we compared several strategies for lysosomal targeting, including fusions to LAMP1, CD74, LC3, p62, and GATA4 (Figure 1B). We used the low-affinity TCR (Ob.1A12), which recognizes a Myelin Basic Protein (MBP) epitope, for platform optimization12. Among these, a variant of CD74 was most efficient for MBP antigen trafficking and loading as measured by CD69 upregulation on cognate on Ob.1A12 TCR+ T-cells (Figure 1C). This segment of CD74 allows the proper folding and trafficking of HLA-II to the lysosome via dileucine (LI, IL/ML)-based endosomal targeting signals13. We further enhanced antigen presentation by removing its arginine-based ER retention motif (CD74-v3) (Figure 1C). We also evaluated the fusions using a high-affinity HIV TCR(F24) antigen pair14 and observed that CD74-v3 was the most efficient fusion for antigen presentation (Figure S1B). Furthermore, we showed that CD74-v3 fusion of long peptides antigens (90aa or 56aa) from different sources (CMV, BRAFV600E, and NY-ESO) on various HLA-II alleles could activate SKW3 T-cells harboring the cognate TCRs (Figure S1C).
In the original TScan-I GzB reporter, uses the GzB cleavage sequence (VGPD↓FGR) scaffold of infrared fluorescent protein (IFP) which disrupts IFP (IFPGZB) fluorescence2,15 that is restored upon GzB-mediated cleavage (Figure 1D). However, phenylalanine (F) exposure following GzB cleavage generates a potent N-degron that renders IFPGzB unstable, limiting its fluorescence16. Thus, phenylalanine was mutated to serine (VGPD↓SGR), generating a stable GzB reporter with increased fluorescence, IFPGzB-Hi (Figure S1E). We expressed a caspase-resistant version of the inhibitor of caspase-activated DNase (ICADCR) to prevent genomic DNA fragmentation following granzyme-induced cell death in targeted HEK293T cells. We termed the B2M null target cells expressing CIITA, HLA allele, HLA-II localized antigens, IFPGzB-Hi and ICADCR as “engineered HEK-293-II (eHEK-293-II)” cells (Figure S1K).
Development of T-cells capable of GzB Secretion Following Recognition of Cognate pHLA-II
We assessed the efficiency of GzB secretion into eHEK-293-II by introducing an intact CD4+ TCR into CD8+ cells. While the expression of CD4+ TCRs in CD8+ cells permitted target cell activation, the signal-to-noise ratio was insufficient for a genome-scale screening. This problem could be due to 1) the absence of CD4 in CD8+ T-cells; 2) mispairing of exogenously introduced CD4+ TCRs with endogenous TCRs, or 3) low affinities of CD4+ TCR for cognate epitopes on HLA-II relative to CD8+ TCRs for HLA-I. To circumvent these problems, we generated a hybrid TCR17–19 that reduces endogenous mispairing. We also introduced CD4 with the engineered TCRs into CD8+ cells for efficient recruitment of Lck kinase to the TCR-pHLA-II synapse20(Figure S1D). Co-culture of engineered CD8+ cells expressing the enhanced TCR and CD4 molecule with eHEK-293-II cells expressing the cognate epitope resulted in a ~10-fold increase of GzB reporter activation (Figure 1E). We termed these engineered cells “centaur” T-cells (Figure S1K).
Mutating the endogenous TCR beta chains showed no significant improvement (Figure S1F-G), indicating that CD3 competition is not limiting. We also examined the cell surface levels of several T-cell adhesion and co-stimulatory factors and found high levels of CD58 and CD155 on our eHEK-9T-II target cells but no CD80 expression. However, expressing CD80 yielded no improvement in the signal-to-noise ratio (Figure S1H). To assess whether the endogenous HEK-293T HLA-II alleles expressed following CIITA transduction impacted antigen detection, we mutated the endogenous HLA-II alleles (Figure S1I) but observed no discernable difference in the level of GzB reporter activity (Figure S1J)
To benchmark the relative efficiency of GzB delivery from centaur cells into targets, we developed a membrane-bound centaur-specific scFv expressing target cell that should maximally activate the target cells for comparison (Figure 1F). Strikingly, the percent of target cells activating the GzB reporter was comparable in cells expressing the MBP antigen to that of target cells expressing the membrane-bound anti-centaur scFv, suggesting that our antigen delivery and presentation strategy results in efficient centaur TCR activation (Figure 1F). The genetic engineering performed to generate eHEK293-II target cells and centaur T-cells is summarized schematically in Figure S1K.
Characterization and optimization of TScan-II using viral reactive TCRs
As an initial test assessing this antigen discovery platform, we focused on CD4+ responses in a rare group of human immunodeficiency virus (HIV) infected individuals known as “elite controllers” who maintain an undetectable HIV viral load in the absence of antiretroviral therapy21. To do this, we synthesized an HIV proteome-wide library, tiling across the entire proteomes of multiple different HIV strains, with each fragment barcoded twice via synonymous codons. We initially used a public CD4+ TCR (F24) that recognizes Gag293, the most immunoprevelant CD4+ epitope in the HIV capsid protein (Figure 2A)14. Our HIV library included two antigens that precisely matched the published epitope of this TCR. However, considering the library’s composition of multiple strains, additional variants of this epitope are also present within this library for a total of 29 related Gag fragments. These variants exhibit 1-7 amino acid variations relative to the reported epitope. This TScan-II screen consistently observed reproducible enrichment of the two published antigens across all seven replicas (Figure 2B, Figure S2A, and Table S1). Of interest, all 29 fragments that were significantly enriched relative to the input library of 2,493 fragments were variants of the published Gag peptide and the extent of their enrichment directly correlated to the number of amino acid differences relative to the published epitope (Figure S2C). Furthermore, there was a high level of concordance between both barcode fragments, affirming the reliability of the GAG epitope hits (Figure S2B). We confirmed the reactivity of F24 to these variants through co-culture experiments (Figure S2D). Thus, the TScan-II platform not only achieved 100% efficiency in identifying known TCR hits but also readily identified novel epitope variants from a complex library of antigens.
Figure 2. HIV genome-wide and Mutagenesis TScan-II screens.
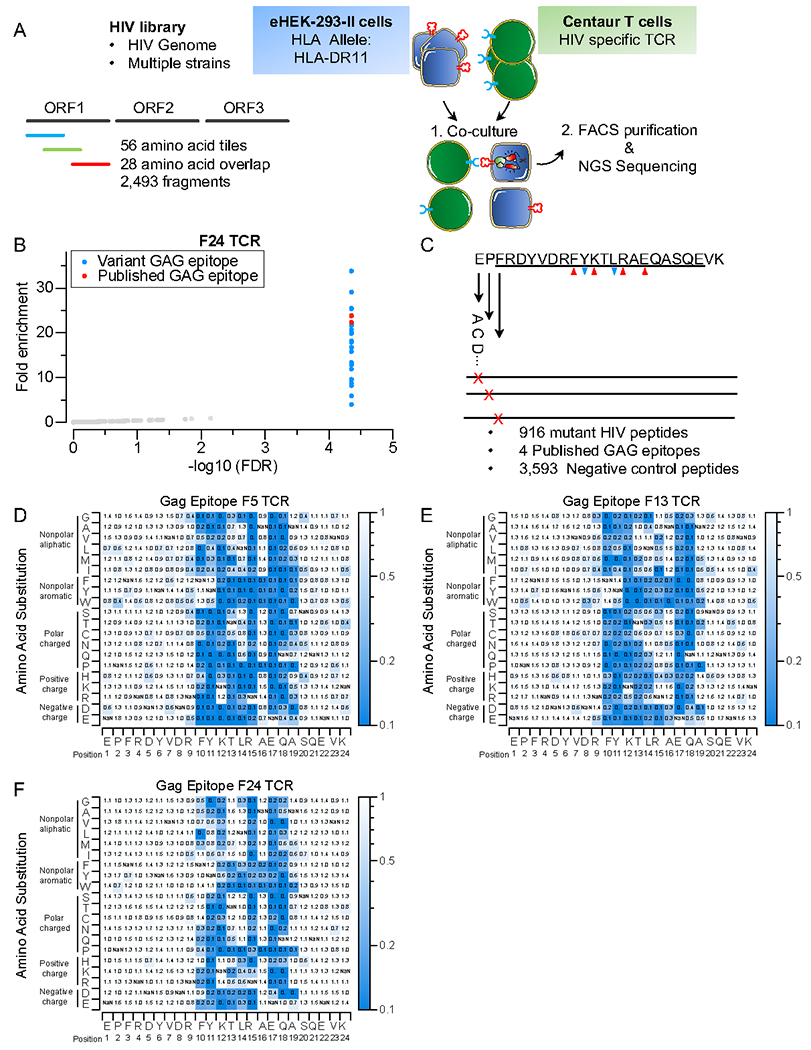
A) Schematic of TScan-II HIV genome-wide screen. The HIV library comprises 2,492 peptide fragments tiling across the genomes of multiple HIV strains in 56aa steps with 28aa overlap.
B) TScan-II screen using an HIV elite controller TCR against the HIV library. Each dot represents one peptide. The y-axis shows the geometric mean of the fold-change across seven replicates for screens with HLA-DRB1*1101 and the GAG-specific TCR (F24). Red dots represent peptides, with an epitope identical to the published Gag epitope for this TCR, blue dots represent variant Gag epitopes.
C) The GAGepitope saturation mutagenesis library. The library encodes 4 WT epitopes, 956 mutant Gag epitopes, and 3,593 negative controls. The downward blue arrows indicate anchor residues, while the upward red arrows indicate TCR-facing residues.
D, E, and F) Heatmap representation of comprehensive mutagenesis analysis of GAG epitope using 3 HIV elite controller TCRs, F5 (D), F13 (E), and F24(F). The heatmap value represents this mutant’s relative enrichment compared to the published (Gag) epitope.
Mapping of CD4+ TCR epitope interface using saturated mutagenesis screens
To assess whether TScan-II can enable finely detailed mapping of CD4+ TCR epitopes, we generated a saturation mutagenesis library of the Gag epitope where each residue in the epitope is mutated to all other 19 amino acids (Figure 2C). Screens were performed using the three TCRs (F24, F3, and F5) reported in HIV elite controllers. As anticipated, only mutant GAG fragments enriched comparably to the WT GAG peptides and more strongly than all negative control peptides (Figure S2E, F, G, and Table S1). The mutants of GAG fragments can be broadly categorized into two groups based on their behavior in the screen. The first group consists of enriched mutant fragments, where the substitution of amino acids had either no or minimal effect on TCR or HLA binding. The second fragment group dropped out of the screen, indicating that these mutations affected residues critical for TCR recognition or HLA binding. To identify critical residues, we compared the enrichment of each mutant peptide to the WT peptides (Figure 2D, E, F). Notably, the crystal structure of the GAG epitope in complex with HLA-DR11 indicated that Tyrosine and Leucine (FRDYVDRFYKTLRAEQASQE) are anchor residues for HLA-DR1122, consistent with our studies in which Tyrosine could only be replaced by aromatic residues such as Phenylalanine or Tryptophan, while Leucine could be replaced primarily by residues with some hydrophobicity. This screen also revealed several non-anchor residues within the GAG epitope (FRDYVDRFYKTLRAEQASQE) as crucial for TCR activation. Based on structural data, these residues are involved in the binding of F24 and F13 TCRs. The correlation between the mutagenesis and the structural analysis further highlights the effectiveness of TScan-II in mapping the TCR and peptide-HLA interface. Interestingly, while each TCR had a unique epitope footprint, they each recognized all 29 naturally occurring epitope variants (Figure S2A). This flexibility in epitope recognition could contribute to the “elite controller” status of these individuals.
Virome-wide discovery of CD4+ TCR antigens
To examine if T-Scan-II can successfully identify cognate antigens from more complex libraries, we made a library tiled across the human virome (206 viral species and over 1,000 different strains and screened using the F24 CD4+ TCR (Figure 3A), which exhibited by far the most relaxed specificity (Figure 2F). Despite the 50-fold increase in library complexity, we observed enrichment of the antigens that precisely matched the published and variant epitopes described earlier (Figure 3B, Table S1). To assess whether the enriched fragments from the E2 protein of Human papillomavirus type 49 (HPV) were due to cross-reactivity or noise, we generated a second library of the top enriched fragments from the primary screen with additional overlapping tiles. All enriched fragments from the second screen were either from the HIV GAG protein or the HPV (Figure S3B, Table S1). The minimal epitope recognized within E2/HPV antigens (Figure 3D) contained residues that were either identical to the core HIV GAG epitope mapped in Figure 2F or were permitted substitutions. We validated the recognition of HPV epitope, further confirming that the original signals observed in the primary screen were valid hits and not noise (Figure 3D, Figure S3A). These findings demonstrate the capability of TScan-II to successfully identify specific CD4+ T-cell epitopes within a complex virome-wide library.
Figure 3. Virome-wide TScan-II screen.
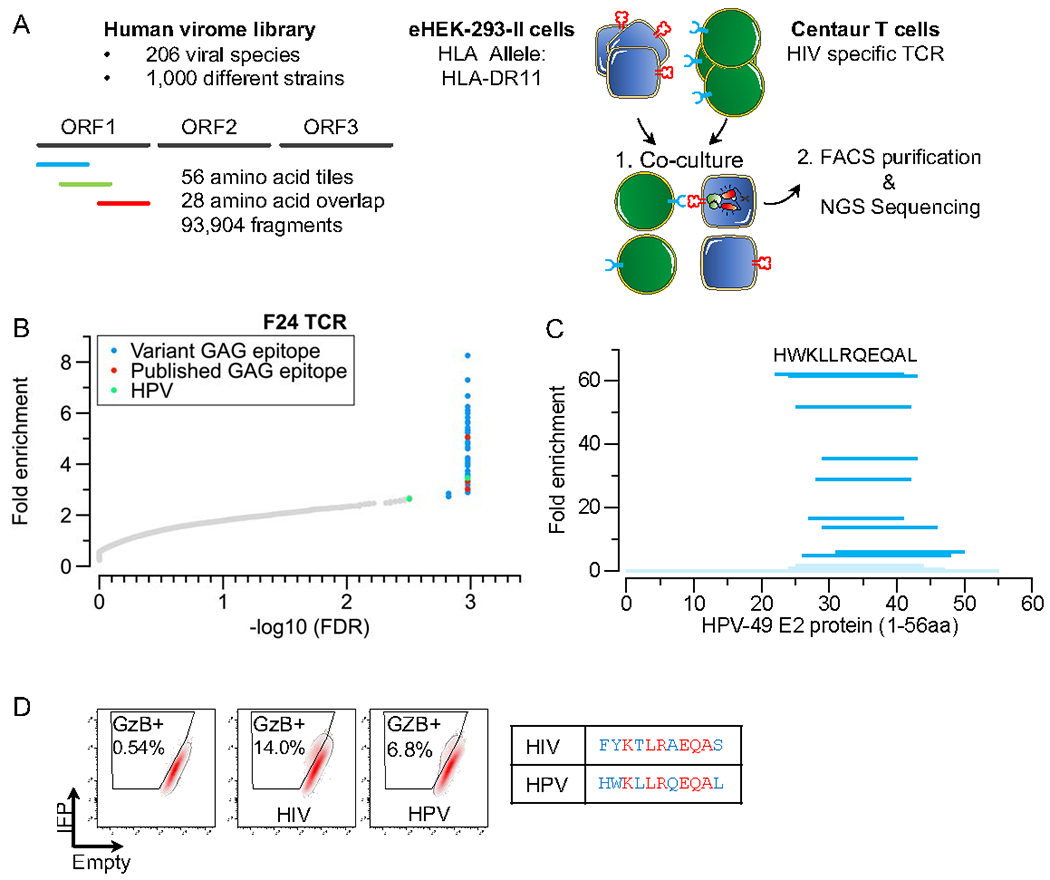
A) Schematic of the TScan-II human virome screen. The human virome library comprises 93,904 56aa fragments collectively tiled across the human virome (206 viral species, > 1,000 strains), including multiple HIV strains.
B) TScan-II screen of HIV elite controller F24 TCR with the virome library. Each dot represents one peptide, with the y-axis plotting the geometric mean of the fold-change across eight replicates against the HLA- DRB1*1101 allele. Red dots represent peptides with WT GAG epitope for this TCR; blue dots represent variant GAG epitopes, green dot represents a peptide from the HPV type 49.
C) TScan-II screen of a library of 12, 15, 18, and 20 aa fragments tiled at 1 AA intervals across the 56mer antigenic segments of HPV. The length of each dark blue indicates the scoring segment of HPV. Fragments in light blue did not score. The sequence depicted represents the minimal epitope.
D) FACS depicting reporter activation after a 6h co-culture with F24 T-cells. eHEK-293-II cells express the HLA-DRB1*1101 allele and a control peptide (left), HIV (middle), or HPV (right). Identical residues shared between the minimal HPV epitope and core GAG epitope are colored in red, while blue residues represent permitted amino acid substitutions based on Figure 2F.
Genome-scale discovery of self-reactive CD4+ TCR targets
To assess TCR targets at genome-scale, we examined a therapeutic TCR candidate for specificity. NY-ESO-1 (CTAG1B), a tumor-associated antigen, is expressed in various solid tumors but not in normal healthy tissues except for the thymus and testis. Given these unique characteristics, CTAG1B is considered an ideal target for adoptive cell therapy (ACT)23,24. Several CTAG1B TCRs are actively being assessed in clinical studies. While many therapeutic TCRs are thought to be specific initially, the complications observed in ACT patients associated with toxic off-targets reinforce the urgency of unbiased reactivity assessment of these TCRs25–27. Thus, we sought to assess the specificity of the HLA-DRB1*0401 (HLA-DRB4) restricted NY-ESO-1 TCR (3598-2)28.
We generated an HLA-II targeted library that tiled across the entire human proteome in 90-aa fragments with 45-aa overlap (Human Peptidome V2). To assess whether the platform can be multiplexed, we also introduced HLA-DRB1*0401 (HLA-DRB4), HLA-DRB1*03:01(HLA-DR3), and HLA-DQB1*02:01_HLA-DQA1*05(HLA-DQ2) into our target cells (Figure 4A). We performed a TScan-II screen and observed robust and reproducible enrichment of only six peptides in the library from 3 sets of ORFs (Figure 4B, Table S2), two of which were the only fragments containing the cognate CTAG1B epitope. Additionally, three overlapping fragments belonging to CTAG2 (NY-ESO-2) ORF were enriched. The CTAG2 and CTAG1B proteins are paralogous proteins that share a 14 AA region of substantial identity and similarity spanning the original CTAG1B epitope (Figure 4C). Another strongly enriching antigen across all replicas was a peptide from Titin (TTN), a protein highly abundant in muscle tissues. This epitope was similar to the NY-ESO-1 in a consecutive stretch of 5 amino acids and contained several conservative changes outside of that region (Figure 4C). In validation experiments, we observed reactivity as measured by granzyme secretion to each antigen but not to control antigens (Figure 4D). Interestingly, there was a direct correlation between the degree of TCR recognition and how tightly the peptide is predicted to bind HLA-DR429(Figure S4A). These observations indicate that the platform can readily identify known and novel CD4+ TCR self-antigens at genome-scale, even when target cells express multiple HLA-II alleles simultaneously. Thus, the TScan-II platform is amenable to HLA multiplexing, which is especially important in instances where the TCR restriction across the patient’s HLA alleles is unknown.
Figure 4. Human genome-wide screening using NY-ESO-specific CD4+ TCR.
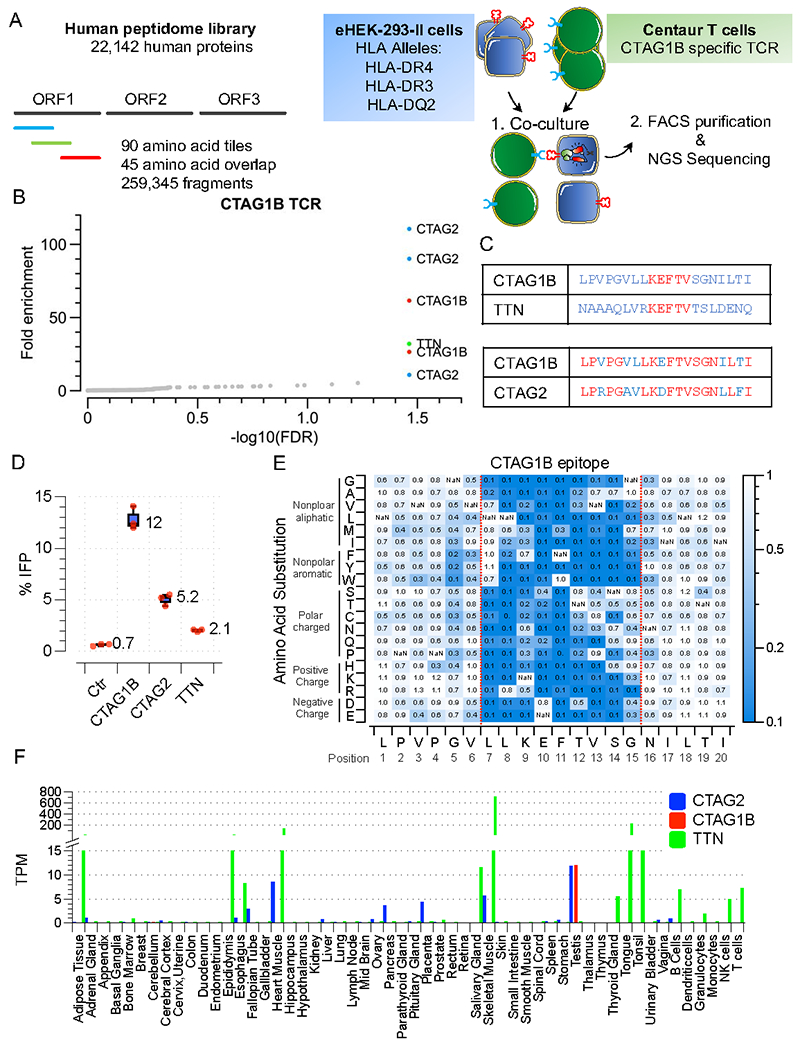
A) Schematic of TScan-II human genome-wide screen. The human genome-wide v2 library comprises 259,345 antigens that tile across the entire human proteome in 90 aa fragments with 45 aa overlap.
B) TScan-II Screen of CTAG1B CD4+ TCR (3598-2) against the human genome-wide v2 library. The eHEK-293-II cells express the HLA- DRB1*04, HLA-DRB1*03:01, and HLA-DQB1*02_HLA-DQA1*05 alleles.
C) Predicted 20 aa epitopes from the enriched 90aa peptides.
D) Validation of the identified antigens in target cells following 6 h co-culture with cells expressing CTAG1B TCR and eHEK-293-II cells expressing HLA- DRB1*04.
E) Heatmap representation of comprehensive mutagenesis analysis of the CTAG1B epitope in HLA- HLA- DRB1*04 eHEK-293-II cells using CTAG1B specific TCR (3598-2). The mutant epitopes are expressed in the context of a 56 aa fragment. Heatmaps are as in Figure 2.
F) Transcript expression levels for CTAG2, CTAG1B, and TTN from 61 human tissues based on data from three sources: HPA, GTEx, and FANTOM5. Bar graph with the y-axis plotting consensus normalized expression value (TPM, Transcripts Per Kilobase Million) and x-axis representing the multiple human tissues and blood cell types.
We mapped the TCR-CTAG1B interface using saturation mutagenesis (Figure S4B, Figure S4C, Table S2), generating a heatmap of critical residues within the epitope’s 10 amino acid core (L7-N16) (Figure 4E). By comparing critical residues with the predicted TTN epitope, we found that TTN contains an identical central core of residues (K9-V13) and differs from NY-ESO only by five amino acids in its outer core, each of which (L7V, L8R, S14T, G15S, N16L) are permissive to different degrees. Likewise, CTAG2 differs by only two residues with tolerated changes (L7V, E10D) explaining its cross-reactivity. An interesting takeaway is that mutagenesis to alanine in 3 key positions, V13, S14, and G15, had minimal effects, suggesting alanine scanning maps may be unreliable for predicting cross-reactivity. Unlike NY-ESO, CTAG2 and TTN have a broad expression profile across various healthy tissues (Figure 4F), which could be problematic in a clinical setting using this TCR for ACT27.
Characterization of pancreatic cancer reactive CD4+ TCRs
In a cancer setting, identifying the neo-antigens recognized by T-cells is approachable as they can be identified by DNA sequencing and tested, but self-antigens pose a much more difficult problem, limiting their widespread applicability for TCR-based immunotherapy. To explore TScan-II in this capacity, we extended our previous study of co-culturing HLA-II positive pancreatic cancer tumor organoids with autologous patient PBMCs30(Figure 5A). This approach resulted in the expansion of organoid-primed T-cell (opT) clones, of which approximately 90% of the TCRs originated from a single Th1 CD4+ T-cell clone (CXCR3+, CCR4−, CD69+, CD103+)30. Intriguingly, this CD4+ T-cell clone possessed a unique TCRβ chain and two distinct TCRα(α,α’) chains. Thus, we synthesized two TCRs, DHM6 and DHM7. Notably, only DHM6 TCR recognized the matched patient-derived but not a tumor organoid derived from a different patient (unmatched) (Figure 5SA).
Figure 5. Characterization of a pancreatic reactive TCRs.
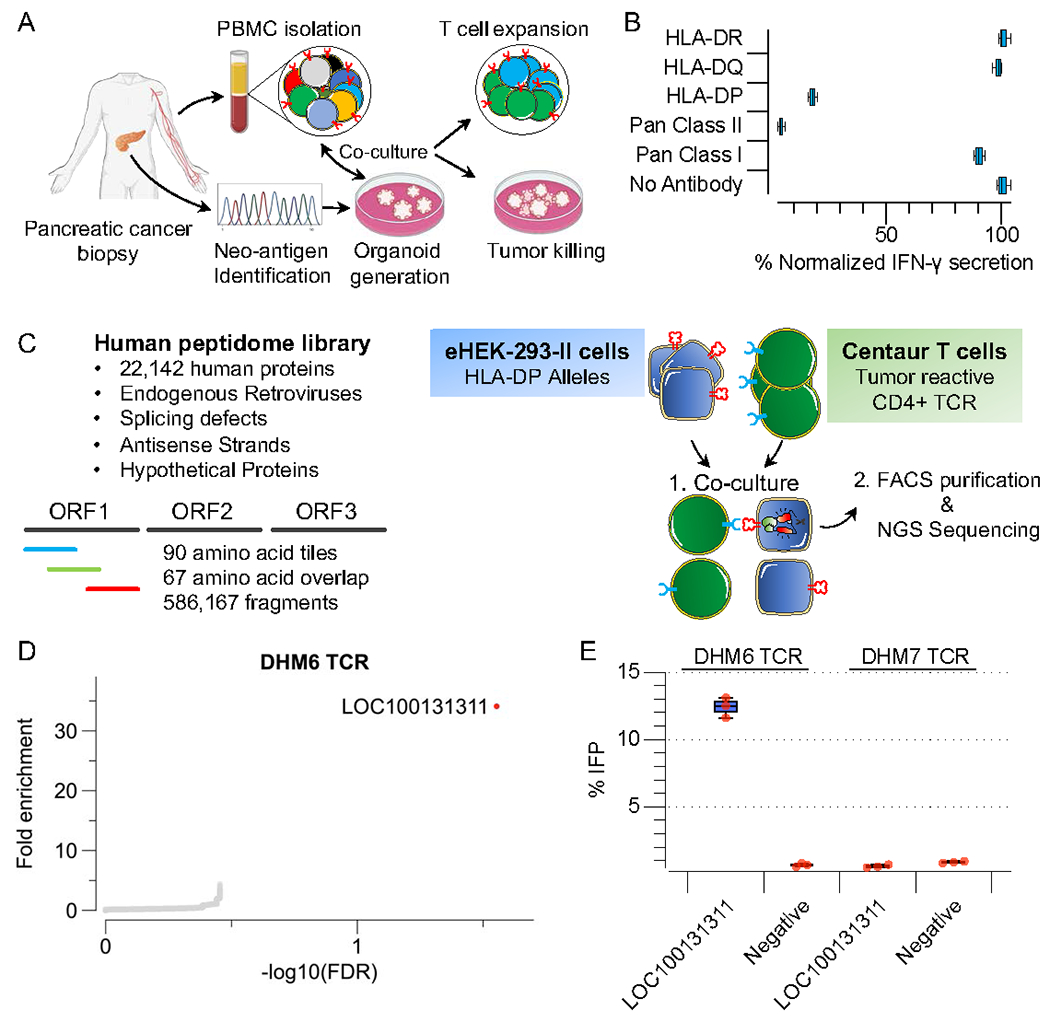
A) Schematic of the experimental workflow to derive and characterize pancreatic cancer tumor-reactive T-cell clones.
B) Characterizing tumor reactivity and HLA-II allele restriction of tumor-reactive TCRs using patient-derived organoids. Primary centaur T-cells were co-cultured with patient-derived organoids. IFN-γ secretion was used as a readout of T-cell activity. Pan-HLA Class I antibody (W6/32), Pan-HLA Class II antibody (Tu39), HLA-DR (L243), HLA-DP (B7/21), HLA-DQ (SPVL3) were used for blocking experiments.
C) Schematic of TScan-II human genome-wide screen. The human peptidome v3 genome-wide library comprises 586,167 antigens that tile across the entire human proteome, alternative translational products, endogenous retroviruses, splicing defects, antisense strands, and hypothetical proteins in 90 aa fragments with 67 aa overlap.
D) TScan-II screen of CD4+ TCR(DHM6) against the human peptidome v3 library and HLA-DP expressing library eHEK-293-II cells.
E) Validation of the identified antigens in target cells using the GzB reporter following 6h co-culture with DHM6 transduced T-cells and indicated 90mers antigens.
To identify the target of DHM6, we generated a patient-specific neo-antigen library by performing whole-exome sequencing of matched tumor and normal cell DNA from the pancreatic cancer patient and identified 54 nonsynonymous somatic mutations within tumor cells (Table S3, Figure S5B). We screened the TCR against the neo-antigen library and did not observe a reproducible enrichment of any of the peptide fragments, suggesting DHM6 likely does not recognize a neo-antigen (Figure S5C-D).
To examine if DHM6 TCR might recognize a self-antigen, we designed an expanded human peptidome library (Human Peptidome V3), which encompassed not only the annotated human proteome as used in the CTAG1B screen but also fragments derived from splicing defects such as intron retention and exon junction, alternative translation products, endogenous retroviruses, antisense strands, and hypothetical proteins covering 586.167 fragments (Figure 5C)31–37. Using this library, we performed a TScan-II screen using DHM6 T-cells (Figure 5C) and observed a robust enrichment of a peptide fragment from a hypothetical protein (LOC100131311) residing in the antisense strand of induced myeloid leukemia cell differentiation protein (MCL1) (Figure 5D, Table S3)38. Utilizing strand-specific gene expression analysis, we detected transcripts spanning the length of the antigenic fragment in 33% of assessed organoids, including the matching patient tumor (Figure S5F). Using a set of overlapping tiles, we mapped the epitope to a 15aa segment (Figure S5E). We deconvoluted the HLA-DP restriction of this antigen using target cells expressing each HLA-DP allele heterodimer (Figure S5G). Notably, these alleles can be found in up to 25% of individuals of African descent, particularly among black South Africans 39.
Tissue-infiltrated CD4+ T-cells in Sjögren’s disease recognize self-antigens.
In almost all autoimmune diseases, there is a pressing need to understand the antigenic landscape of tissue-infiltrating T-cells40, a key prerequisite for understanding the underlying etiology and for developing immune-modulatory therapies. One such autoimmune disease is Sjögren’s disease (SjD), a chronic systemic archetypal and heterogeneous autoimmune disease with special targeting of the exocrine glands41. Its etiology is unknown, but it appears to engage the adaptive immune system, as evidenced by focal lymphocytic infiltration of the salivary (SG) and lacrimal glands. The lymphocytic foci in SG of SjD patients are predominantly CD4+ T-cells42–44 with an antigen-experienced phenotype45–48. Additionally, ductal cells of the SG in SjD patients express high levels of HLA-II and are surrounded by CD4+ T-cells and B cells46,48–52. In response to IFNγ exposure, the SG ductal cells express T-cell co-stimulatory molecules such as CD80, CD86, CD40, and ICOSL46,53–56. Moreover, historically, an association between the expanded C*07:01–B*08:01–DRB1*03:01–DQB1*02:01 haplotype and the development of SjD was suggested in patients of European ancestry57–59–60,61. Collectively these observations raise the question of whether any of the clonally-expanded infiltrating T-cells are indeed self-reactive.
We employed TScan-II to assess whether SG-expanded CD4+ T-cells can recognize a self-antigen present in our human genome-wide library (Figure 6A) by profiling a large cohort of published SG-infiltrating CD4+ lymphocytes from 15 SjD patients that shared the HLA-DRB1*03:01, HLA-DQB1*02:01 alleles (Table S4)47. Twenty TCR pairs across five SjD patients were ultimately chosen for subsequent genome-scale TScan-II screening according to the strategy detailed in the methods (Table S4). Interestingly, while in normal T-cells, up to one-third express two distinct TCRα chains, approximately 50% of the SjD T-cells did 60,62–64. In most cases, following the transduction of TCRs which were sourced from T cells that expressed two TCRα chains (α or α’), we could only detect a high level of expression for one of the two TCR pairs (TCRβ-α or TCRβ-α’) on the cell surface of primary T cells (Figure S6A).
Figure 6. Human genome-wide TScan-II screens of clonally expanded CD4+ T-cells in SjD.
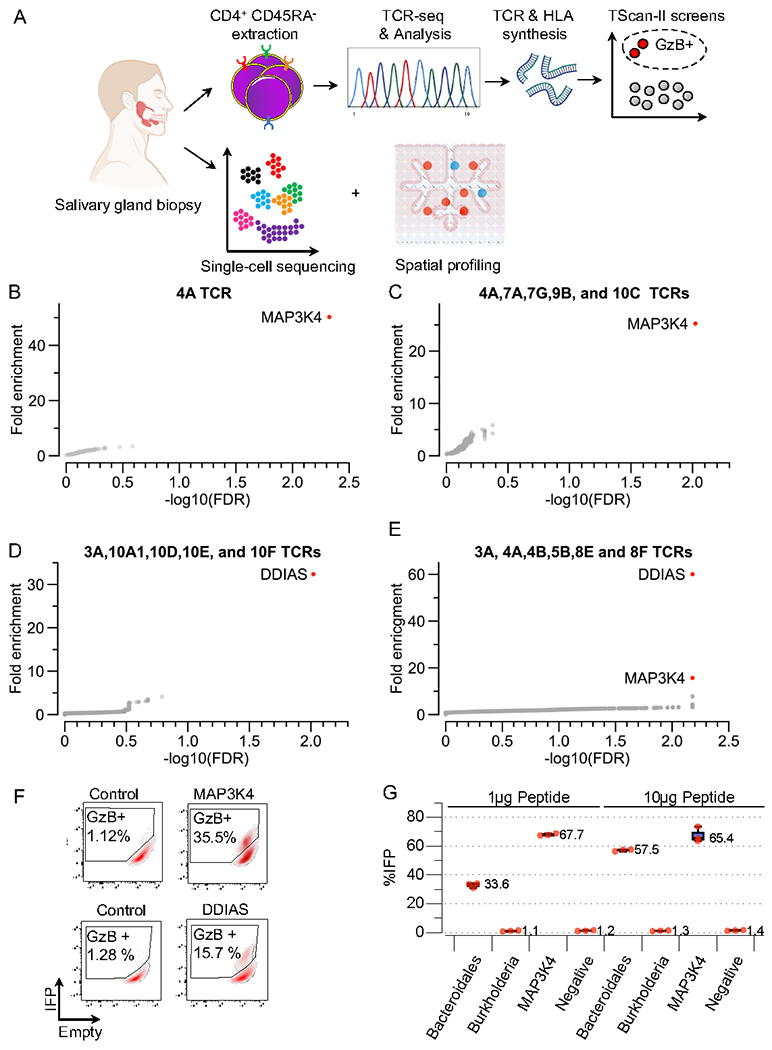
A) Schematic representation of the experimental workflow to characterize clonally expanded CD4+ T-cell clones in SjD patients.
B-E) TScan-II screens were performed against the human genome-wide v2 library with eHEK-293-II cells expressing HLA-DRB1*04, HLA-DRB1*03:01, and HLA-DQB1*02_HLA-DQA1*05. B) Pooled TScan-II screen of 5 TCRs 4A,7A,7G,9B, and 10C C) TScan-II screen of 4A TCR. D) Pooled TScan-II screen using 5 TCRs 3A,10A1,10D,10E, and 10F. E) Pooled TScan-II using 6 TCRs 3A,4A,9C,8A,8B and 5B.
F) FACS plots depicting activation of GzB reporter following a 6 h co-culture experiment with 3A and 4A centaur T-cells towards the indicated cognate antigens.
G) Relative activation of the GzB reporter in cells pulsed with the indicated peptide fragment cocultured with 4A T-cells.
We performed the first genome-wide screen using a mixture of 5 centaur T-cell lines pooled, each expressing a unique TCR (4A,7A,7G,9B, and 10C). We observed enrichment of two adjacent peptide fragments from Mitogen-Activated Protein Kinase Kinase Kinase 4 (MAP3K4/MTK1) (Figure 6B, Table S5, Table S6). Deconvolution identified TCR4A (Subject 4 SSA+/SSB−) and HLA-DR3 restriction (Figure 6F, S6B). We performed a second genome-wide TScan-II screen using only 4A T-cells (Figure 6C, Table S5, Table S6) and identified the same two MAP3K4 fragments. Henceforth, the remaining genome-wide screens were performed using pools of 5 TCRs. We identified a second antigen reactivity from the additional three genome-wide screens, this time to DNA Damage Induced Apoptosis suppressor (DDIAS), using a collection of 5 TCRs (3A,10A1,10D,10E, and 10F) (Figure 6D, Table S5, Table S6). Deconvolution revealed TCR3A (Subject 3 SSA−/SSB−) and HLA-DQ2 restriction (Figure 6F, Figure S6C). To further assess the multiplexing capability, we performed a screen with 3A and 4A within a pool of six total T-cell clones and detected both MAP3K4 and DDIAS, thereby demonstrating the system’s ability to identify specific TCR-antigen interactions even in the presence of multiple active T-cell clones (Figure 6E, Table S5, S6). Further experiments confirmed the original two antigens, and none of the other 18 TCRs identified additional antigens even when all six patient-specific HLA-II alleles were included in the screens (Figure S6D, Figure S6E, Table S5, and Table S6).
Identification of a cross-reactive Bacteroidales peptide
Little is known about how tolerance is broken in autoimmune diseases. One hypothesis proposes that cross-reactive T-cells, capable of recognizing microbial and self-peptides, play a role in the development of autoimmune disorders65–67. Thus, we took advantage of the knowledge of the minimal core residues sufficient to activate both TCRs (Figures S6F-S6I) to bioinformatically impute viral and bacterial epitopes that may activate the 3A and 4A TCRs. We identified a peptide fragment from the TonB-dependent receptor protein of Bacteroidales that can readily activate TCR 4A (Figure 6G). Intriguingly, an increase in the abundance of Bacteroidales has been reported in the oral microbial flora of SjD patients compared to healthy controls, although the clinical significance of this finding is not known68. Further investigation is needed to assess the significance of this finding to SjD pathogenesis.
Autoimmune antigens in SjD are expressed in HLA-II expressing salivary gland cells
To assess the expression of antigens in the SG of SjD patients, we used single-cell RNA sequencing data of the minor SG biopsies collected from SjD patients fulfilling the classification criteria for SjD (Figure 7A). As expected, we observed a clear increase in the abundance of B-cell and T-cell infiltration within SGs of SjD patients compared to non-SjD controls (Figure 7B). In the epithelial compartment, MAP3K4 was expressed in the acinar, ductal, and myoepithelial cells within the SG (Figure 7C). DDIAS expression was mostly limited to the ductal cells of the SG (Figure 7C). Both MAP3K4 and DDIAS are expressed at higher levels in the ducal cells of SjD patients (Figure S7A), consistent with studies showing that the expression of DDIAS is upregulated in the SG of SjD patients69,70. To assess the spatial expression relative to the ductal cells and lymphocytic infiltrates, we used the Visium platform (10X Genomics). Visium provides unbiased, coarse-grained, spatially resolved (~55 um) transcriptomic information but cannot resolve individual cells. However, in direct support of our scRNAseq results, we observed partial co-location of expression of MAP3K4 and DDIAS with markers of ductal cells (i.e., S100A2 and CFTR)71. Furthermore, we detected transcripts for both DDIAS and MAP3K4 in areas of lymphocytic inflammation (arrows Figure S7B).
Figure 7. Single-cell expression profiling of CD4+ antigens and HLA-II machinery within the salivary gland.
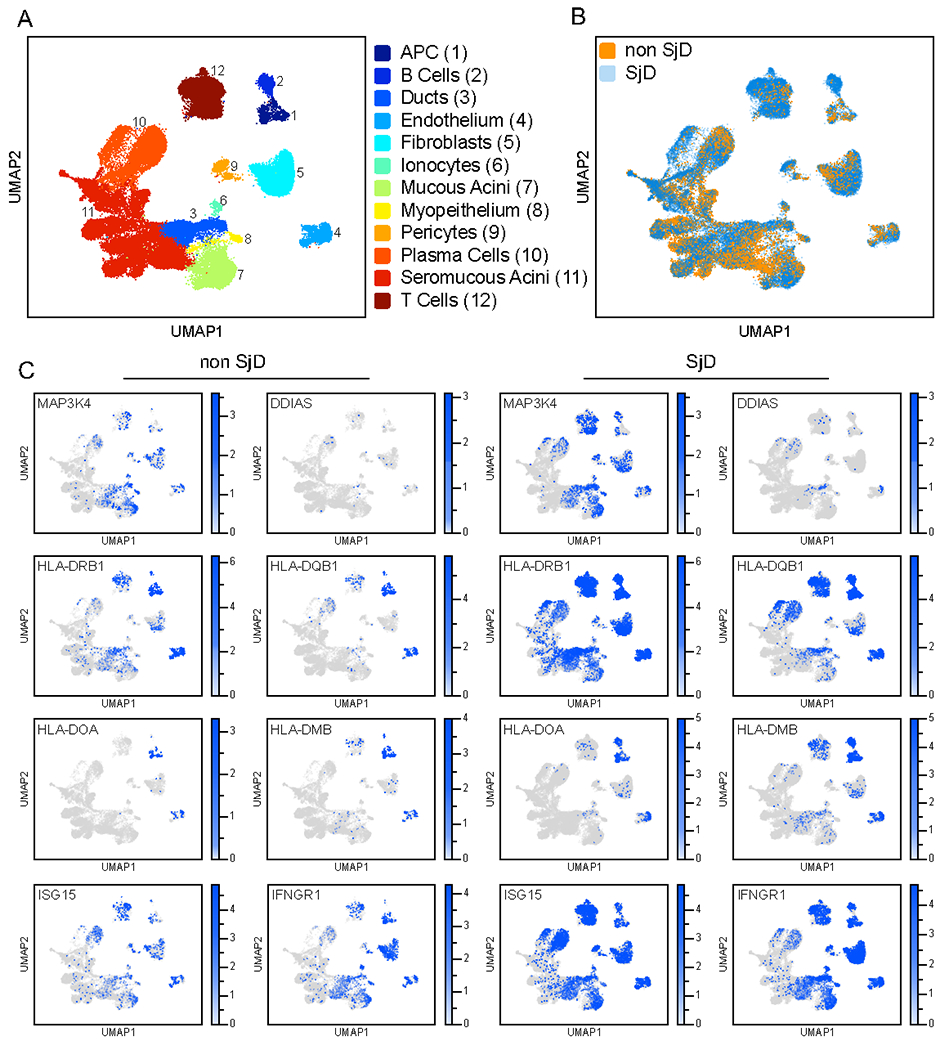
A) UMAP visualization of scRNA-seq data from dissociated SGs of healthy and SjD. Cells from minor SG from n = 6 subjects who did not fulfill ACR 2016 classification criteria and were otherwise healthy and n = 7 SjD subjects who fulfilled American College of Rheumatology Classification Criteria were integrated into a single object containing 12 clusters. Selective clusters are highlighted by their functional annotation.
B) UMPA plot depicting the abundance of SG cells in non-SjD (Healthy) and SjD subjects overlaid on the UMAP plot depicted in A.
C) Expression of various HLA-II marker genes and MAP3K4 and DDIAS among non-SjD and SjD patients overlaid on the UMAP plot depicted in A.
Given the expansion of CD4+ cells in the SjD SG, a central question is where the autoimmune targets encounter HLA-II expression. Is it in the associated APCs in the inflammatory infiltrates, or could it also be on the salivary gland tissues themselves? In healthy SGs, we found HLA-II expression on B-cells, macrophages, and dendritic cells, as expected. However, HLA-II was also expressed within the SG’s mucous acini and ductal cells (Figure 7C) and is markedly further increased in the ductal cells of SjD patients (Figure S7A). A main characteristic of focal lymphocytic infiltration in SjD patients is the periductal arrangement of CD4+ cells52,72. Importantly, ductal cells express both MAP3K4 and DDIAS. Hence, multiple cell types within the salivary gland are likely competent to present HLA-II antigens during disease progression. We also detected INFGR1 and ISG15 gene expression within the ductal cells; therefore, the observed upregulation of the HLA-II within the ductal cells could likely result from chronic salivary gland inflammation and interferon (Figure 7A, Figure S7A). Collectively, we observed that both MAP3K4 and DDIAS are expressed within specific populations of cells in the SG that are most likely capable of processing and presenting HLA-II antigens, which could contribute to SjD.
Discussion
The immune system’s primary function is to maintain tissue homeostasis by recognizing and eliminating pathogens and damaged cells. Uncovering which antigens are recognized has remained largely elusive, partially due to the absence of high-throughput tools to probe T-cell specificity. Here, we describe TScan-II, a genome-scale platform for the de novo identification of CD4+ TCR antigens. TScan-II employs endogenous HLA-II processing machinery to process antigens from full-length ORFs or long peptide fragments, thus greatly expanding the complexity of antigenic libraries. The coupling of these complex libraries with a reporter of T-cell activity having a high signal-to-noise ratio resulted in the enrichment of scarce HLA-II antigens. We applied T-Scan-II to decipher the antigen specificity of disease-relevant CD4+ TCRs in infectious disease, cancer, and autoimmunity. In our proof of principle screens, TScan-II was capable of identifying cognate antigens from viral and self-reactive CD4+ TCRs and provided specificity insights for both. Interestingly, in both HIV and NY-ESO screens, the identified cross-reactive epitopes had minimal homology to the original epitope, underscoring the difficulty of predicting TCR specificities in the absence of unbiased genome-scale approaches. The ease with which the platform can be multiplexed at both T-cell and HLA levels allows this tool to be highly customizable to various basic and translational applications, enabling genome-scale antigen discovery for CD4+ T-cells.
The unbiased nature of TScan-II makes it an ideal tool for the dissection of the antigenic landscape associated with CD4+ cells within the TME. In this regard, we employed patient-derived organoids to assess the reactivity of CD4+ T-cell clones toward HLA-II-positive pancreatic cancer tumors. Taking an unbiased approach with a library covering unconventional transcripts and translation products in addition to the entire proteome allowed us to discover this tumor-reactive TCR identified an epitope originating from an uncharacterized gene expressed within matching patient tumor organoids. The ability of TScan-II to find different classes of antigens within the non-canonical immunopeptidome showcases its utility in cancer research.
In the autoimmune context, we applied TScan-II to SjD and examined the antigen specificity of clonally expanded CD4+ T-cells within the SG patients. By screening 20 expanded clones from 5 SjD patients, we identified self-antigens that can readily and potently activate two of the most expanded clones from two individuals. A key question concerns how HLA- II self-antigens are presented to CD4+ cells in SjD. There are two possibilities: 1) Antigens can be phagocytosed and presented by APC cells within the SG, or 2) They can be presented directly to CD4+ T-cells by HLA-II positive SG cells. Ductal epithelial cells within the SG have been circumstantially implicated in presenting self-antigens to CD4+ cells. Using single-cell data, we detected HLA-II expression on ductal cells within the SG of healthy individuals, which were considerably upregulated within the inflamed salivary gland of SjD. Importantly, both DDIAS and MAP3K4 were expressed by the HLA-II positive ductal cells, consistent with the possibility that both antigens can be processed and presented by ductal epithelial cells directly to CD4+ T-cells. Spatial transcriptomics showed that both antigens were expressed in areas within the SG with high lymphocyte density. Importantly, peptides spanning the length of DDIAS and MAP3K4 proteins have been previously identified in HLA-II ligandome datasets from bone marrow, kidney, and lung 73,74. Collectively, not only can DDIAS and MAP3K4 be processed and presented on HLA-II alleles, but they are also expressed in cells expressing HLA-II and reside in the neighborhood of periductal immune infiltrates within the SG. How cells directly present peptides from their cytoplasmic proteins on HLA-II molecules is unknown but could be an internal form of presentation employing bulk autophagy, or peptides transported into the ER may be able to load onto HLA-II molecule prior to transport to the lysosomes10. These specific TCRs may aid in further testing of the HLA-II antigen presentation on SG in the future.
Several autoimmune diseases, such as RA and type 1 diabetes, a subset of patients display a strong association with certain HLA-II alleles75–78. Historically, studies in SjD have identified associations with the HLA-II locus, which were later corroborated by GWAS studies among SjD patients57–61,79,80. In Europeans, the C*07:01–B*08:01-DRB1*03:01–DQB1*02:01 haplotype exhibits the strongest genetic association with SjD 60,61,79. In this study, we identified self-antigens restricted to both of these two HLA-II alleles. Is this an unusual case where two different HLA-II alleles that happen to be linked each can independently contribute to SjD pathogenesis, or could another mechanism also be involved? During the course of our studies, several publications suggested that the causative genetic element in the C*07:01–B*08:01–DRB1*03:01–DQB1*02:01 haplotype may actually be due to the C4 gene81–83. This extended haplotype is in strong linkage disequilibrium (LD) with a C4A null allele in populations of European origin 81,84,85. The same haplotype has also been linked to SLE57–60,86. Recombinants that separate the HLA from the C4 locus have shown that C4 is the likely cause of SLE81,87,88. In addition, heterozygous C2 deficiency—when present in combination with a low C4A copy number—substantially increased the risk of SLE and primary SjD by 10-fold compared to individuals with 2 C4A copies and normal C283. The complement system plays a critical role in the clearance of cellular debris, such as apoptotic cell fragments and bacterial pathogens. The impaired clearance of cellular debris, pathogens, and immune complexes due to the absence or lower levels of complement could, in turn, result in the accumulation of antigen fragments (clearance hypothesis) 89–91. Lupus-prone mice expressing the human C4A protein have an improved ability to clear apoptotic cells, exhibit lower levels of self-reactive B-cells, and decreased levels of autoantibodies targeting SSA, relative to mice expressing human C4B, providing supporting evidence for the clearance hypothesis 92. This hypothesis would be consistent with our finding on the cross-reactive epitope in Bacteroidales. Furthermore, GWAS studies have shown that a subset of SjD and SLE patients of European descent exhibit HLA-II associations independent of C4 allele status93,94. In particular, variations in the XL9 region in different haplotypes contribute to an increase in HLA-II gene expression95–97. Assuming the accuracy of these findings, the association between disease susceptibility and either lower C4 protein levels in one haplotype or the increase in HLA-II expression in others would result in a pro-immunogenic increased amount of self-antigen presentation rather than a common antigen specificity among affected SjD individuals95. As a result, multiple self-antigens expressed in the exocrine glands, each with varying HLA-II restrictions, are likely to contribute to the CD4+ T-cell mediated pathogenesis observed in SjD. The divergent HLA-II restriction of the two self-antigens identified here is consistent with the above hypothesis. The simultaneous involvement of complement and CD4+ T-cells presents a complex scenario where the chronic interplay of innate and adaptive immune systems synergize to initiate and advance SjD, culminating in the heterogeneous clinical presentations witnessed among patients. Future studies probing the antigenic landscape of expanded T-cell clones across a large cohort of patients with SjD and multiple autoimmune diseases will further shed light on commonalities and differences among antigen governing pathogenesis.
Collectively, through the various applications described here, we provide evidence that the TScan-II platform can facilitate the identification of TCR-epitope relationships in infectious diseases, cancer, and autoimmunity and will serve as an avenue for the dissection of many more such relationships in the future.
Limitations of the study
TScan-II has several limitations. First, it relies on the genetic encoding of candidate antigens, which does not include certain antigens, such as CD1-bound lipids. Secondly, in its current form, it is unlikely to present peptides with post-translational modifications (PTMs). One potential approach to address this limitation is the use of full-length proteins or engineering modifiers, such as protein kinases and citrullination enzymes, to reside in the endoplasmic reticulum. Finally, this unbiased screen revealed that many TCRs do not recognize a self-antigen, an observation that may not actually be a limitation of the platform per se but possibly a feature of the biology of T-cell infiltration in SjD. Among the possible explanations are that the self-antigen target is absent from our library or that the TCRs are not self-reactive but instead recognize epitopes derived from bacterial, fungal, viral, or food sources, given the exposure of the salivary gland to the environment.
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Stephen Elledge (selledge@genetics.med.harvard.edu).
Materials availability
Information and requests for resources and reagents may be directed to the lead contact.
Data and code availability
Single-cell RNA-seq data have been deposited in the NIH Database of Genotypes and Phenotypes (dbGaP). The accession information is listed in the Key Resources Table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD3 antibody (OKT3) | Thermo Fisher Scientific | Cat: 14-0037-82 |
| anti-human TCR α/β Antibody | Biolegend | Cat: 306711 |
| Alexa Fluor 647 anti-human CD8 Antibody | Biolegend | Cat: 311410 |
| Brilliant Violet 421 ™ anti-human CD80 Antibody | Biolegend | Cat: 305222 |
| Alexa Fluor® 647 anti-human CD19 Antibody | Biolegend | Cat: 363039 |
| Biotin anti-mouse TCR β chain Antibody | Biolegend | Cat: 109204 |
| APC anti-human CD8 Antibody | Biolegend | Cat: 344722 |
| Alexa Fluor® 647 anti-human HLA-DR, DP, DQ Antibody | Biolegend | Cat: 361704 |
| FITC anti-human HLA-DQ Antibody | Biolegend | Cat: 361503 |
| Alexa Fluor® 647 anti-human CD69 Antibody | Biolegend | Cat: 310918 |
| APC anti-human CD58 (LFA-3) Antibody | Biolegend | Cat: 330917 |
| APC anti-human CD155 (PVR) Antibody | Biolegend | Cat: 337617 |
| Alexa Fluor® 647 anti-human CD54 Antibody | Biolegend | Cat: 353113 |
| FITC anti-mouse TCR β chain Antibody | Biolegend | Cat: 109206 |
| Alexa Fluor® 647 anti-human CD3 Antibody | Biolegend | Cat: 317312 |
| Bacterial and Virus Strains | ||
| One Shot® Stbl3™ Chemically Competent E. coli | Thermo Fisher Scientific | Cat: C7373-03 |
| ElectroMAX™ DH10B™ Cells | Thermo Fisher Scientific | Cat: 18290015 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Interleukin-2, human (hIL-2) 25ug | Sigma (Millipore) | Cat: HIL2-RO |
| Gateway™ LR Clonase™ II Enzyme mix | Thermo Fisher Scientific | Cat:11791100 |
| Gateway™ BP Clonase™ II Enzyme mix | Thermo Fisher Scientific | Cat:11789100 |
| Q5® High-Fidelity 2X Master Mix | New England Biolabs | Cat:M0492L |
| Lenti-X Concentrator | Takara Bio | 631232 |
| Puromycin Dihydrochloride | Thermo Fisher Scientific | Cat:A1113803 |
| Polybrene | Thermo Fisher Scientific | Cat:TR-1003-G |
| Superscript™ IV First-Strand Synthesis System with ezDNase™ Enzyme | Thermo Fisher Scientific | Cat:18091150 |
| Ambion™ RNase H, from E. coli, 10 U/μL | Thermo Fisher Scientific | Cat:AM2293 |
| Benzonase Nuclease HC, Purity > 99% | Thermo Fisher Scientific | Cat:71206-3 |
| Nourseothricin Sulfate (Streptothricin Sulfate) | Gold Biotechnology | Cat:N-500-1 |
| NEBuilder® HiFi DNA Assembly Master Mix | New England Biolabs | Cat:E2621L |
| Dynabeads® Human T-Activator CD3/CD28 | Thermo Fisher Scientific | Cat:11132D |
| jetPRIME | Polyplus | Cat:114-07 |
| Critical Commercial Assays | ||
| GeneJET Genomic DNA Purification Kit | Thermo Fisher Scientific | Cat:K0721 |
| RNeasy Plus Mini Kit | Qiagen | Cat:74134 |
| Anti-Biotin Microbeads Miltenyi | Miltenyi Biotec | Cat:130-090-485 |
| RosetteSep™ Human CD8+ T Cell Enrichment Cocktail | Stem Cell Technologies | Cat:15023 |
| Deposited Data | ||
| Singel-Cell-RNA-Seq | Yin, 2021 Huang and Perez, 2021 This Study | dbGAP: Phs002446.v1 .p |
| Experimental Models: Cell Lines | ||
| Lenti-X™ 293T Cell Line | Takara Bio | 632180 |
| eHEK-293-II | This Study | N/A |
| HLA-II expressing eHEK-293-II | This Study | N/A |
| Jurkat, Clone E6-1 | ATCC | Cat:TIB-152 |
| SKW-3 Cell line | Gift from Julia Etchin at Dana Farber Cancer Institue | N/A |
| Oligonucleotides | ||
| See Table S7 | This Study | N/A |
| Recombinant DNA | ||
| pHAGE-CMV:DEST-PGK: ICADCR. | This Study | N/A |
| pHAGE-CMV:IFPGzB-Hi-PGK: ICADCR | This Study | N/A |
| pHAGE-TREX-CIITA-PGK-Neomycin | This Study | N/A |
| pHAGE-EF1L-anti-CD3 scFv-PGK-Puromycin | This Study | N/A |
| pHAGE-EF1L-anti-H57-597-scFv-PGK- Puromycin | This Study | N/A |
| pHAGE-TREX-DEST-PGK- Blasticidin | This Study | N/A |
| pHAGE-TREX-DEST-PGK-Hygromycin | This Study | N/A |
| pHAGE-TREX-DEST-PGK-Nourseothricin | This Study | N/A |
| pHAGE-EF1a-DEST-PGK-floxed-mCD19 | This Study | N/A |
| pHAGE-EF1:DEST-PGK: CD4 | This Study | N/A |
| pHAGE-CMV-CD74_V3-DEST-Puromycin | This Study | N/A |
| Human Peptidome in pHAGE-CMV-CD74_V3-DEST | This Study | N/A |
| Human Virome in pHAGE-CMV-CD74 V3-DEST | This Study | N/A |
| Tiling Library in pHAGE-CMV-CD74 V3-DEST | This Study | N/A |
| HIV Library in pHAGE-CMV-CD74 V3-DEST | This Study | N/A |
| Mutagenesis Library in pHAGE-CMV-CD74_V3-DEST | This Study | N/A |
| Software and Algorithms | ||
| Bowtie | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| Cutadapt | Martin, 2011 | http://cutadapt.readthedocs.io/en/stable/ |
| Cell Ranger v3.0.1 | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Scanpy | Wolf, 2018 | https://github.com/scverse/scanpy |
| Seurat | Sajita, 2015 | https://github.com/satijalab/seurat |
| Python v3.7 | Python Software Foundation | https://www.python.org |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Human Participants
Pancreatic cancer subject recruitment:
Peripheral blood and tumor tissue samples were obtained from individuals diagnosed with metastatic pancreatic cancer. Specifically, the TCRs examined in this study and the corresponding tumor organoids were derived from patient 38 (p38) (52 year old male, self-reported Caucasian) diagnosed with stage IV pancreatic ductal adenocarcinoma. The study protocol was approved by the Dana-Farber Cancer Institute Institutional Review Board (IRB), Boston, Massachusetts, and was conducted in accordance with the principles of the Declaration of Helsinki. The samples were collected between 2018-2020.
SjD subject recruitment for single-cell RNAseq:
All subjects were seen at the NIH Clinical Center Dental Clinic and provided informed consent to NIH Central IRB approved protocol, 15-D-0051 (NCT02327884, PI: Warner) prior to any study procedures. Subjects were evaluated comprehensively following standardized clinical protocols by a multidisciplinary clinical research team, including oral medicine/clinical oral pathology, rheumatology, and ophthalmology, as previously reported 98. Subjects were classified according to the 2016 SjD classification criteria99,100. 6 subjects who did not fulfill SjD 2016 classification criteria(aged 40 to 56, comprising 4 females and 2 male all of White ethnicity) and were otherwise healthy, and 7 SjD (aged 43 to 75, comprising 6 females and 1 male, with the ethnic distribution of 4 Black, 2 Hispanic, and 1 White) subjects who fulfilled American College of Rheumatology Classification Criteria99,100 were ultimately chosen for scRNA-seq.
SjD subject recruitment for 10X Visium:
Labial minor salivary gland biopsies were performed on two individuals (Patient 1: 47 years old, male, white, non-Hispanic, Patient 2 Adult, 56 years old, female, female, white, non-Hispanic.) attending the Oklahoma Sjögren’s Research Clinic who met the 2016 SjD classification for SjD99,100. This study was approved by the Oklahoma Medical Research Foundation Institutional Review Board, IRB 06-12, IRB 07-12, and IRB 11-18. The socioeconomic status of the participants was not determined for this study.
The socioeconomic status of the participants was not determined for this study.
Cell lines
HEK293T cell lines (Takara Biosciences) were cultured in DMEM GlutaMAX™ (Life Technologies) media.
Primary T cells
Primary T-cells were cultured in either RPMI (Life Technologies) or ImmunoCult-XF (Stemcell Technologies) plus 50U/ml IL-2 (Sigma). DMEM GlutaMAX™ and RPMI media were supplemented with 10% (v/v) FBS (Hyclone) and 1% (v/v) Penicillin/Streptomycin (Life Technologies).
METHOD DETAILS
T-cell extraction and expansion
Apheresis blood samples (Brigham and Women’s Hospital) were used for T-cell extraction using the RosetteSep CD8 purification kit (StemCell). The purity of CD8+ extraction was assessed by CD8 (Biolegend) and CD4(Biolegend) antibody staining and FACS. Apheresis blood samples were also used for primary blood mononuclear cells (PBMCs) extraction. PBMCs were purified using a Ficoll (Cytiva Life Sciences) gradient. PBMC cells were subsequently irradiated by 75 Gy ionizing radiation.
For expansion, 1E6 centuar T-cells were grown in the presence of irradiated PBMCs(20E6) plus 50U/ml IL-2 (Sigma), and 0.1μg/ml anti-CD3 antibody (Life technologies).
T-cell transduction
1E6 primary CD8+ cells were plated per well in a 24-well plate and stimulated with a 1:1 ratio of Dynabeads Human T-Activator CD3/CD28 (Life Technologies). 24 h following activation, T-cells were transduced with 150ul of concentrated lentivirus expressing centaur TCRs.
48 h following Dynabeads activation cells, beads were removed. 72 h following transduction, transduced cells were sorted using a biotinylated mTCR antibody (Clone: H57-597) and MACS® Columns (Miltenyi Biotec). The purification efficiency of transduced T-cells was assessed using the mTCR (Clone: H57-597) and CD4 (Clone: OCT4) antibodies.
Lentiviral production
Lentivirus was produced through co-transfection of lentiviral vectors and packing plasmids (Tat, Rev, Gag-Pol, and VSV-G) into HEK293T-cells (Takara Biosciences). All transfection into HEK293T was done using jetPRIME® (Polyplus). Lentiviral supernatants were collected 48 h post-transfection; for HEK293T transduction, lentivirus was added to cells in the presence of 8 ug/ml Polybrene. Selection of transduced target cells was performed using Hygromycin (200 μg/ml, Puromycin (2 μg/ml), Blasticidin (10 μg/ml), or Nourseothricin (400 μg/ml). Primary T-cells that were transduced with lentivirus in the absence of Polyberne.
Knockout of endogenous B2M gene
To knock out the endogenous B2M in HEK293T-cells, CRISPR/Cas9 ribonucleoprotein (RNP) complexes were used. In brief, three synthetic gRNAs (TrueGuideTM) were mixed individually with Cas9 protein (TrueCutTM Cas9 Protein v2) and delivered via electroporation (NeonTM Transfection) into HEK293T-cells. MHC-negative cells were sorted using the Pan-MHC (Biolegend, Clone W6/32) and B2M (Biolegend, Clone 2M2) antibodies. To achieve a pure B2M population, cells were FACS sorted three times.
B2M-sgRNA-1: AGTCACATGGTTCACACGGC
B2M-sgRNA-2: TCACGTCATCCAGCAGAGAA
B2M-sgRNA-3: TGAAGATGCCGCATTTGGAT
Mutation of endogenous HLA-II genes
To mutate out the endogenous HEK293T HLA-II alleles (DPB1*04:01:01, DQA1*01:02:01, DQB1*06:02:01, DRB1*15:01:01) that are expressed following CIITA transduction CRISPR/Cas9 ribonucleoprotein (RNP) complexes were used. In brief, gRNAs targeting either HLA-DRB, HLA-DP, or HLA-DQ (IDT) were mixed individually with Cas9 protein (TrueCutTM Cas9 Protein v2) and delivered sequentially via electroporation (NeonTM Transfection) into CIITA transduced HEK293T-cells. HLA-II negative cells were sorted sequentially using HLA-DR (L243), HLA-DP (B7/21), or HLA-DQ (SPVL3) antibodies.
HLA-DRB-sgRNA-1: TTTGCCATGTAGGAACCTCC AGG
HLA-DQB-sgRNA-1: CAGACACAACTACGGGGTTG TGG
HLA-DQA-sgRNA-1: ATTTGGAGGTTTTGACCCGC AGG
HLA-DPB-sgRNA-1: CACGTGACGGATTTCTACCC AGG
HLA-DPA-sgRNA-1: CGTCACGTGGCTGTGCAACG GGG
Knockout of endogenous TCR/β gene
To knock out the endogenous TCRb in primary CD8+ cells, CRISPR/Cas9 ribonucleoprotein (RNP) complexes were used. In brief, primary CD8+ were initially activated using magnetic CD3/CD28 beads (Invitrogen Dynabeads). Two synthetic gRNAs (TrueGuideTM) were mixed individually with Cas9 protein (TrueCutTM Cas9 Protein v2) and delivered via electroporation (NeonTM Transfection) into primary CD8+ cells. Cells null for TCR β (Clone: IP26) were FACS sorted and subsequently transduced with lentivirus to constitutively express centaur TCRs. Cells that stained positive with mTCR (Clone: H57-597) and CD4 (Clone: OCT4) and negative TCR (Clone: IP26) were sorted and used for downstream studies.
TRBC-sgRNA-1: GCAGTATCTGGAGTCATTGA
TRBC-sgRNA-2: GGAGAATGACGAGTGGACCC
Generation of CIITA and IFPGzB-Hi ICADCR constructs
The ICADCR cassette was cloned downstream of the PGK promoter to create pHAGE-CMV:DEST-PGK: ICAPCR. Using site-directed mutagenesis, the GzB cleavage sequence (VGPD↓FGR) of IFPGzB was mutated to VGPD↓ŜSGR to generate the IFPGzB-Hi reporter. The IFPGzB-Hi reporter was cloned by Gateway cloning (Thermo Fisher) into pHAGE-CMV:DEST-PGK: ICADCR to generate pHAGE-CMV:IFPGzB-Hi-PGK: ICADCR. CIITA (Uniprot: P33076) cDNA was cloned into pHAGE-TREX-Dest-PGK-Neomycin expression construct using Gateway cloning.
Generation of membrane bound anti-CD3 scFv
The heavy and light chains of a monoclonal antibody clone: OKT3, were assembled as a single construct and separated by a flexible linker composed of Glycine and Serine (VH-linker-VL). For membrane targeting, we added an N-terminal CD8α signal peptide (P01732.1 : 1-21aa), followed by a myc-tag (EQKLISEEDL) to assess membrane surface expression. To tether the scFv to the cellular membrane, we added a C-terminal PDGFR transmembrane peptide (P09619.1512-561aa). The final construct was synthesized as a gBlocks (IDT) and cloned into the pDONR221 vector (Invitrogen) using Gateway cloning. The membrane-bound anti-CD3 scFv_was ultimately cloned into pHAGE-EF1L-DEST-PGK-Puromycin. The construct was introduced in eHEK-293-II cells expressing either the IFPGzB(low)) or IFPGzB(Hi) genes via lentiviral transduction.
Generation of membrane-bound anti-centaur scFv
The heavy and light chains of a monoclonal antibody clone: H57-597, were assembled as a single construct and separated by a flexible linker composed of Glycine and Serine (VL-linker-VH). For membrane targeting, we added an N-terminal CD8α signal peptide (P01732.1: 1-21aa), followed by a myc-tag (EQKLISEEDL) to assess membrane surface expression. To tether the scFv to the cellular membrane, we added a C-terminal PDGFR transmembrane peptide (P09619.1512-561aa). The final construct was synthesized as a gBlocks (IDT) and cloned into the pDONR221 vector (Invitrogen) using Gateway cloning. The membrane-bound scFv was ultimately cloned into pHAGE-EF1L-DEST-PGK-Puromycin. The construct was introduced in eHEK-293-II cells via lentiviral transduction.
HLA allele design and cloning
All protein sequences for HLA-II alleles were retrieved from IPD-IMGT/HLA (EMBL-EBI). Both HLA-II α and β chains were assembled as a single construct, where the HLA-β and HLAα chains were separated by a 2A self-cleaving peptide (β-P2A-α). Sequences were reverse translated, synthesized as gBlocks (IDT), and cloned into the pDONR221 vector (Invitrogen) using Gateway cloning. All HLA alleles were inserted in one of 4 below lentiviral destination vectors via Gateway cloning.
pHAGE-TREX-DEST-PGK- Blasticidin
pHAGE-TREX-DEST-PGK-Hygromycin
pHAGE-TREX-DEST-PGK-Nourseothricin
pHAGE-EF1a-DEST-PGK-floxed-mCD19
Combinations of alleles cloned into expression vectors encoding different selection markers were introduced via lentiviral transduction into eHEK-293-II cell lines. Transduced cells were selected based on the presence of a drug selection marker or cell surface markers.
CD4+ TCR design and cloning
The human CD4 gene (P01730) was cloned downstream of the PGK promoter to create the pHAGE-EF1:DEST-PGK: CD4 lentiviral vector. All T-cell receptors (TCRs) were engineered as hybrid TCRs, in which the entire human α and β V(D)J regions were preserved and grafted on the mutant mouse α and β constant regions, respectively. The mouse transmembrane and cytoplasmic parts were mutated to concurrently 1) remove a degron within the TCR alpha to increase TCR abundance, 2) enhance TCR association with CD3 for more efficient T-cell signaling, and 3) improve specific pairing of the exogenous TCRs, so mispairing with endogenous TCRs is reduced. Hybrid α and β TCR sequences were then combined into a single fragment separated by a 2A self-cleaving peptides sequence (TCRβ-P2A-TCRα). The hybrid TCRs were codon optimized and subsequently cloned into a pDONR221 entry clone and then transferred into the pHAGE-EF1:DEST-PGK: CD4 vector using Gateway cloning.
Validation experiments
To validate hits from our genome-wide screens with purified peptides, eHEK-293-II target cells were plated at ~2E5 in a 48-well plate in DMEM GlutaMAX™. 16 h later, cells were pulsed with the 1uM of purified peptide for 30 min in a 37C incubator. Centaur T-cells were added to target cells in RPMI plus IL-2. The co-cultured cells were incubated for 8-12h and analyzed for GzB reporter activation by FACS.
Purified peptides used
MAP3K4 Peptide 1: LRKDLEIAAEFRLSA
MAP3K4 Peptide 2: EIAAEFRLSAPVRDL
MAP3K4 Peptide 3: EIAAEFRLSA
MAP3K4 Peptide 4: LRKDLEIAAEFRLSAPVRDL
DDIAS Peptide1: SLNKFLAVLESEIAV
DDIAS Peptide2: ESLNKFLAVLESEIAV
DDIAS Peptide3: FSESLNKFLAVLESEIAV
DDIAS Peptide4: LNKFLAVLESEIAVTQADVS
For endogenously expressed antigens, peptide fragments of interest were reverse translated and synthesized as DNA fragments (gBlocks Integrated DNA Technologies), and Gateway cloned into the pHAGE-CMV-CD74_V3-DEST-Puromycin lentiviral vector. DNA constructed was packaged into lentivirus and introduced into eHEK-293-II target cells. Transduced target cells were co-cultured with centaur T-cells in RPMI plus IL-2. The co-cultured cells were incubated for 8 h and analyzed for GzB reporter activation by FACS.
Library design, cloning, and transduction
Human virome and peptidome libraries: The human peptidome V2 library of 259,345 fragments tiling across the entire human proteome in 90-aa fragments with 45-aa overlap. The human peptidome V3 library (~350,000) was designed to complement our human peptidome V2 library. This library comprises 586,167 fragments that tile across the entire human proteome in 90-aa fragments with 67-aa overlaps. The human virome library comprised 93,904 tiles of 56-aa fragments collectively tiled across the human virome (206 viral species and over 1,000 different strains). Human peptidome libraries (V2 and V3) and virome libraries were cloned into the pHAGE-CMV-CD74_V3-DEST-Puromycin lentiviral vector using Gateway cloning. A 1000x representation was maintained during each cloning step to provide uniform library coverage. For screens with the human peptidome V2 library, 200E6 eHEK-293-II cells were transduced with the lentivirus at an MOI of 5 (5000x library coverage). Each screen was performed using seven to eight replicates (200E6 eHEK-293-II cells, 5000x library representation per replicate) and was co-cultured with centaur T-cells for a minimum of 8 h. The human peptidome V3 library, 300E6 eHEK-293-II cells, were transduced with the lentivirus at an MOI of 5 (2500x library coverage) and selected with puromycin (1ug/ml). Each screen was performed using seven to eight replicates (300E6 eHEK-293-II cells, 2500x library representation per replicate). The human virome library was transduced into 100E6 eHEK-293-II cells at MOI of 3 (3000x library coverage). Each screen was performed using seven replicates (100E6 eHEK-293-II cells, 3000x library representation per replicate). Following the co-culture of centaur cells with target cells, IFPGzB-Hi positive cells (Alexa-700 channel) were sorted on a SONY MA900 FACS machine.
To generate an HIV library, we included ORFs from 10 different HIV strains as annotated in the Uniprot database. ORFs were tiled as 56 aa fragments with 28 aa overlaps between adjacent tiles. Every fragment in the library was barcoded twice using synonymous aa codons. Twist Biosciences synthesized the 2,493-fragment library.
To generate epitopes mutagenesis libraries, epitopes were encoded in the context of 56 aa fragments. For NY-ESO and HIV mutagenesis library design, each amino acid in the annotated epitope (20 aa) was mutated to the other 19 amino acids. For MAP3K4 and DDIAS, each amino acid in the 50 aa fragment was mutated to all other 19 amino acids. Each peptide was reverse translated with synonymous aa codons. Each library also included four unmutated fragments that served as positive controls. To include negative controls during TScan-II screens, multiple mutagenesis libraries were synthesized as a single oligo pool.
To generate tilling libraries for the identification of minimal epitopes, antigenic fragments of 10, 15, 18, and 20 aa in length with 1-aa overlap that spanned the entire length of antigenic segments of HIV, HPV, MAP3K4, and DDIAS were generated. The smaller fragments were incorporated in the context of 200 bp DNA fragments; immediately following the short epitope sequence, we introduced a stop codon to generate epitopes of the desired length. The libraries were synthesized by Twist Biosciences.
The HIV, mutagenesis, and tiling libraries were PCR amplified using primers with BP recombination site overhangs and cloned into pDONR221, then cloned into the pHAGE-CMV-CD74 V3-DEST-Puromycin lentiviral vector using Gateway cloning. A 1000x representation was maintained during the BP and LR cloning steps to provide uniform library coverage. For screens with these libraries, eHEK-293-II cells were transduced with the lentivirus at an MOI of 0.2. Each screen was performed using seven to six replicates (1000x library representation per replicate) and was co-cultured with centaur T-cells (E: T ~0.5) for a minimum of 6h.
TScan-II NGS sequencing
Sorted IFPGzB-Hi positive eHEK293-II cells were mixed with 5E5 carrier Jurkat (Clone E6-1, ATCC Cat# TIB-152) before genomic DNA (gDNA) purification. gDNA was purified using the GeneJet gDNA purification kit (Thermo Fisher).
For human virome and peptidome libraries, gDNA of 100x representation of the target cells was used as input for each screen. For smaller libraries (10,000 oligos or less), gDNA from 1000X representation of target cells was used as input.
Multiplexed libraries were generated for Illumina sequencing using a 3-step PCR protocol. All PCRs were performed with 1-4μg of template gDNA using the Q5® High-Fidelity 2X Master Mix (New England Biolabs). PCR1 was performed using TScan-II PCR-1 primers that flanked the antigen cassette (TScan-II_PCR1_Forward CTGTACCAACAACAAGGTCGGC; TScan-II_PCR1_Reverse CTGTTTGACCGGTTTCGCTACC). PCR1 samples were all pooled, and 1μ1 was used as input for PCR2. PCR2 forward primers were designed to create a stagger sequence between the priming site and the Illumina sequence. The 8 TScan-II_PCR2_forward primers were pooled in equal ratios. PCR2 reaction was performed using the pooled forward primers (TScan-II_PCR2_Forward: tccctacacgacgctcttccgatct[]GTTTGTACAAAAAAGCAGG, [] represents either t, ct, gct, agct, cagct, acagct or tacagct ; TScan-II_PCR2_Reverse : gtgactggagttcagacgtgtgctcttccgatctGCCTTATTCCAAGCGGCTTC). PCR was performed to PCR3 to add on the sequencing adaptors and sample-specific indices. One μl of PCR2 was used as input for PCR3 (TScan-II_PCR3_Forward : aatgatacggcgaccaccgagatctacactcttTCCCTACACGACGCTCTTCCG; TScan-II_PCR3_Reverse: CAAGCAGAAGACGGCATACGAGAT[]GTGACTGGAGTTCAGACGTGT “[]” represents a 7bp unique sample index). After PCR3, samples were pooled based on their desired read count, and gel was extracted using the NucleoSpin® Gel and PCR Clean-up kit (Takara Biosciences). Samples were sequenced on the Illumina NextSeq machine.
TScan-II data analysis
The design of libraries was performed in Python using customized codes. NGS Illumina data were analyzed using Bowtie, Samtools, and Cutadapt tools on a Linux server. In brief, PCR adaptors used during the cloning of the libraries were trimmed using Cutadapt. The trimmed reads were subsequently aligned using Bowtie against our customized libraries. DataGraph, Python, and Excel were used for data analysis. The relative enrichment of each fragment in each replica was calculated by dividing the factional abundance of the reads in the sorted samples relative to the fractional abundance in the input samples. The geometrical mean across all 6-8 replicas was calculated for plotting.
Characterization of organoid-reactive T-cells
The generation and culture of the organoids was done according to previously established protocols 30,101. The culture media was replaced every 4 days, and the organoids used in all experiments were between passages 7 and 12.PBMCs were isolated from the peripheral blood of patient 38 (pt38) using Ficoll-Paque density gradient centrifugation. The isolated PBMCs were cultured for 10 days in human T-cell medium (HTM) consisting of serum-free medium (CellGernix, 20801-0100), 10% human AB serum (Innovative Research, IPLA-SerAB-13458), human IL-2 (1000 IU/mL, Prospec, cyt-209), human IL-15 (10 ng/mL, Prospec, cyt-230), human IL-21 (10 ng/mL, Prospec, cyt-408), 1% penicillin-streptomycin (Gibco, 15140-122), 1% amphotericin B solution (Sigma, A2942), and ciprofloxacin (Fisher Scientific, 50255729).After 10 days of culture, 100,000 PBMCs were co-cultured with autologous tumor organoids in a 96-well flat bottom plate (Falcon, 353072) using HTM. Each well contained 100,000 cells of autologous tumor organoids. The co-cultures were maintained for 7 days. On day 7, the cells were transferred to a new 24-well plate (Falcon, 353047), and the PBMCs were stimulated again with autologous organoids at a ratio of 1:1 for an additional 7 days. This step resulted in the generation of organoid-primed T-cells (opT). The phenotype of opT-cells was determined using flow cytometry and CytoF analysis(pt38, JITC, 2021).
The recognition of tumor cells by opT-cells was confirmed using the M30 enzyme-linked immunosorbent assay (ELISA). The assay was performed to evaluate the reactivity of opT-cells against tumor cells. To test if the engineered-T-cells recognize tumor organoids, 0.1 million organoids per well were co-cultured with 0.1 million engineered-T-cells in a HTM in a 96-well flat bottom plate. After 24h and 48h co-culture, the supernatants were collected and tested for IFN-γ secretion by ELISA (Mabtech, 3420-1H-20). For the HLA class I and HLA class II blocking experiment, purified antibodies without azide (final 10ug/ml) were used. The antibodies used include Pan-HLA Class I antibody (clone W6/32, Biolegend, 311428), Pan-HLA Class II antibody (clone Tu39, BD Pharmingen, 555556), HLA-DR (L243), HLA-DP (B7/21), HLA-DQ (SPVL3).
The expanded CD4+ T-cell clone possessed a unique TCRβ chain and two distinct TCRα(α,α’) chains. To assess which TCR pairs (TCRβ-TCRα or TCRβ-TCRα’) can recognize the tumor organoids, we synthesized the two TCRs (DHM6 & DHM7) using the single TCRβ chain matched with either TCRα or TCRα’ chains and introduced each into primary T-cells. TCR-transduced T-cells were co-cultured with the tumor-derived organoids in the presence of HLA-I and class II antibodies. DHM7 was inactive, but the DHM6 TCR exhibited a substantial and distinguishable recognition of the corresponding patient-derived organoids (matched), as evidenced by the secretion of IFN-γ. Importantly, DHM6 did not exhibit reactivity to a pancreatic cancer organoid derived from a different patient’s (unmatched) tumor sample. Using a pan HLA-II blocking antibody, we confirmed that the DHM6 recognition of the tumor organoid is HLA-II dependent Furthermore, using a set of HLA-II allele-specific (HLA-DR, DP, or DQ) blocking antibodies, we identified HLA-DP as the alleles mediating DHM6 recognition of the tumor organoid.
To sequence the TCRα and β chains, genomic DNA extracted from opT-cells was used for TCR sequencing using the Adaptive Biotechnologies platform. Deep sequencing was performed for both TCRα and β chains. The obtained sequencing data were analyzed using immunoSEQ Analyzer V.3.0. The top-ranking CDR3β sequence was found to have a frequency of 90.4%. The corresponding matched CDR3α chain was identified based on the sequencing data ratio.
Pancreatic cancer neo-antigen screen
Each oligo was designed to encode 41 amino acid fragments, encompassing the mutation flanked by 20 amino acids on either side. The WT peptide fragments and fragments encoding mutations from non-matching patients serve as negative controls in our patient-specific neo-antigen screen. All peptide fragments within the library were barcoded four times using synonymous amino acid codons. The TScan-II screen of DHM6 TCR was performed in eHEK-293II target cells expressing the patient-specific HLA-DP alleles, and the neo-antigen library.
Characterization of SjD T-cells
We profiled a large cohort of published SG-infiltrating CD4+ lymphocytes from 10 SjD patients that shared the HLA-DRB1*03:01 and HLA-DQB1*02:01 alleles47. To allow for a more in-depth analysis, we also complemented this dataset with TCR sequences SG-infiltrating memory CD4+ lymphocytes from 5 additional SjD patients. Twenty TCR pairs across five SjD patients were ultimately chosen for subsequent genome-scale TScan-II screening. Nineteen of these TCRs stratified for antigen discovery were i) directly sourced from SjD patient salivary gland samples, ii) exhibited a memory-like phenotype (CD45RA−), and iii) were clonally expanded within the tissue. These findings are prototypic characteristics of T cell antigen recognition. A direct correlation between the degree of clonal expansion of these specific TCRs and the severity of phenotypes observed among SjD patients further supports their significance 47. In addition to the clonally expanded T cells, one additional CD4+ TCR was included based on the presence of a shared CDR3β motif identified across patients through GLIPH analysis102. This shared motif was found specifically among the CDR3β of CD4+ T cells present in the salivary gland infiltrates of 3 SjD patients but was absent in non-SjD patients. We synthesized each of the 20 TCRs separately as a TCRβ-α pair. In the instances where a single CD4+ cell expressed two TCRα chains (α or α’) and a single TCRβ, we synthesized two separate TCR constructs, each with the single TCRβ chain and two different TCRα chains (TCRβ-α or TCRβ-α’).
SjD TScan-II screens
To identify potential self-antigens that SjD-associated TCRs may target, we initially generated a set of twenty centaur T-cell lines by transducing each line with a unique TCR construct. Only transductions that yielded high levels of centaur TCR expression were subsequently purified using MACS® Columns for further TScan-II screens. This stringent selection process ensured that all the T-cell lines used in the screens had an optimal expression of the transduced TCR, thus maximizing the sensitivity and accuracy of antigen discovery using the TScan-II platform. We then expressed HLA-DR3 and HLA-DQ2 alleles shared across all 5 patients into eHEK-293 target cells transduced with our HLA-II targeted human peptidome V2 library. For pooled TScan-II screens, a mixture of 5-6 centaur T-cell lines was pooled in equal ratios a, each expressing a unique TCR from different patients. Target cells expressing the human peptidome were then co-cultured with the mixture of centaur T-cells at an effector-to-target ratio (E: T) of ~1.
Salivary gland single-cell data tissue acquisition
Minor salivary glands (MSG) (N=13; n=7 SjD and n=6 NV) tissues were removed following standard methods and immediately placed in ice-cold RPMI and dissociated using the Miltenyi Multi-tissue Dissociation Kit A. Single-cell suspensions were serially filtered through 70- and 30-μm filters and rinsed with 1× Hanks’ buffered salt solution. Cells were centrifuged at 300 g for 10 min at 4 °C and washed once with 1× Hanks’ buffered salt solution. Cell counting and viability were determined using a Trypan blue exclusion assay. Additional glands from these patients were submitted for histopathological assessment, including focus scoring.
Salivary gland scRNA-seq
Single-cell suspensions targeting approximately 5,000-10,000 cells were loaded onto a 10x Genomics Chromium Next GEM Chip B in the NIDCR/NIDCD Genomics and Computational Biology Core Facility. After cell capture, single-cell library preparation was performed following the instructions for the 10x Chromium Next GEM Single Cell 3’ kit v3 (10x Genomics). The libraries were pooled and sequenced on four lanes of a NextSeq500 sequencer (Illumina), adopting the read configuration indicated by the manufacturer.
Salivary gland scRNA-seq data processing
Read processing was performed using the 10x Genomics workflow. Cell Ranger v3.0.1 Single-Cell Software Suite was used for demultiplexing, barcode assignment, and UMI quantification (http://software.10xgenomics.com/single-cell/overview/welcome). Sequencing reads were aligned to the hg38 reference genome (Genome Reference Consortium Human Build 38) using a pre-built annotation package from the 10x Genomics website (https://www.10xgenomics.com/). Samples were demultiplexed using the ‘cell ranger mkfastq’ function, and gene count matrices were generated using the ‘cellranger count’ function. The single-cell data were analyzed scanpy. Filtering was performed using the standard quality control steps.
Salivary gland scRNA-seq data analysis
Single-cell data sets were integrated from published data 71,103 and unpublished data generated by the Warner Lab (dbGAP: phs002446.v1.p1). Cells from minor SG from n = 6 subjects who did not fulfill ACR 2016 classification criteria and were otherwise healthy and n = 7 SjD subjects who fulfilled American College of Rheumatology Classification Criteria99,100 were integrated into a single object containing 12 clusters.
10X Visium tissue harvest
Harvested tissue was washed with saline (Henry Schein, #8900026), blotted on dry gauze, and transferred into a cryomold half-filled with OCT compound (VWR, #25608-930), then finished with additional OCT. Cryomolds were initially frozen by placement on a tray filled with 2-methyl butane (VWR, #100504-954) on dry ice. After the initial freeze, the cryomolds were moved and stored at −80°C for long-term storage.
10X Visium slides preparation, and sequencing
Cryosectioning was performed using a CryoStar™ NX70 Cryostat (ThermoFisher #14-071-401) with an Accu-Edge Low profile microtome blade (Sakura #4689) and following 10X Genomics protocol. Optimal permeabilization of 27 minutes was established. Single sections (10μm) from four subjects were placed on gene expression slides (10X Genomics, #1000187). After cryosectioning, the cut side of the cryoblock was placed facedown into the cryoblock pre-filled with a small amount of fresh, partially frozen OCT. Using partially frozen OCT preserved the cut surface and minimized warming of the cut tissue. After tissue placement on each 10X Genomics slide, slides were placed in a slide mailer and stored inside resealable bags at −80°C for up to 4 days. Fixation and H&E staining were performed according to 10X Genomics CG000160 Rev A protocol. Gene expression (GEX) libraries were prepared using the Visium Spatial Gene Expression kit (10X Genomics, #1000187) and Dual Index Kit TT Set A (10X Genomics #1000215) according to the 10X Genomics protocol. Qubit dsDNA HS Assay kit (ThermoFisher #Q32854) and Qubit 2.0 Fluorometer (ThermoFisher #Q32866) were used for sequencing library quantifications. All amplified cDNAs, and sequencing libraries were quality-controlled using Agilent 2200 or 4200 TapeStation (Agilent #G2964AA or #G2991AA). HighSensitivity D5000 ScreenTapes/Reagents (Agilent #5067-5592/5067-5593) were used to quantify amplified cDNAs. HighSensitivity D1000 ScreenTapes/Reagents (Agilent #5067-5584/5067-5585) were used to quantify GEX libraries. Based on the tissue coverage area (range: 3 - 40 %; average: 14.5%), the sequencing depth per sample was established, and libraries pools were made targeting 50,000 read pairs per spot. Sequencing was performed on Illumina NovaSeq S4 PE150.
10X Visium data analysis
Visium raw data from minor salivary gland samples were converted into fastq files using spaceranger mkfastq function of 10x Genomics software (v1.3.1). Reads were subsequently mapped to the human reference genome (GRCH38) using the STAR method, and the spatial feature counts were generated using the spaceranger count function. Spaceranger count data were loaded as a Seurat object using the Load10X_Spatial function in Seurat (v4.0.6)104. The SpatialFeaturePlot function of Seurat was used to visualize the expression of marker genes.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details for all experiments can be found in the figure legends. For TScan-II screens, the calculation of p-values was done by permutation testing of the geometric mean across enrichment values. Specifically, low input count peptides were first excluded due to high variance in estimates of enrichment. To the remaining peptides, pseudocounts of 1 were added to each library (sort and input libraries) before normalizing: first to library size, then to input normalized counts. These enrichment values were then permuted (n=1,000,000 replicates) within each sort condition to generate a background distribution for the geometric mean of each peptide. False discovery rate correction was done by the Benjamini-Hochberg method. For screens with two barcodes corresponding to the same peptide, where both barcodes had >50 counts in the input library, combined p-values were computed as above after merging enrichment values between barcodes.
Supplementary Material
Figure S1. Optimization of Antigen processing and TCR signaling for the TScan-II platform, related to Figure 1
A) FACS histograms depicting the sequential engineering of eHEK-293-II cells. CRISPR/Cas9 ribonucleoprotein (RNP) was used to mutate the B2M gene. Cells that did not express B2M (Clone 2M2) or HLA-A, B, or C (Clone W6/32) on their cell surface were sorted and subsequently transduced with lentivirus to express the MHC-II transactivator (CIITA constitutively). Cells that stably expressed CIITA and had high levels of HLA-DR/DP/DQ (Clone: Tü39) were sorted and used for downstream studies.
B) Boxplots depicting activation levels of F24 TCR transduced SKW-3 cells following co-culture with HLA-DR11 expressing eHEK-293-II cells in the presence of endogenously expressed and processed HIV antigens. SKW-3 were identified using CD3, and their activation was measured via the early activation marker CD69 following co-culture with target cells. The line in the box plots indicates the median. The box is drawn around the inner quartile range, and the whiskers show minimum to maximum values.
C) FACS plots depicting activation levels of TCR transduced SKW-3 cells following co-culture with eHEK-293-II cells in the absence (lower panel) or presence (upper panel) of endogenously expressed (Fused to CD74-v3) and processed antigens. TCRs tested include an HLA- DRB1*0701 restricted Cytomegalovirus specific (left), HLA-DQB1*03:02, DQA1*03:01 restricted BRAFV600E (middle), or HLA- DRB1*04 restricted NY-ESO (CTAG1B) antigen.
D) FACS histograms depicting the generation of centaur primary T-cells. Primary CD8+ T-cells are transduced with lentivirus to express the modified CD4+TCRs and the glycoprotein CD4. Cells stained positive with mTCR (Clone: H57-597) and CD4 (Clone: OCT4) were sorted and used for downstream studies.
E) The frequency of cells that activate the GzB reporter in eHEK-293-II cells expressing either IFPGzB (VGPD→FGR) or IFPGzB-Hi (VGPD→SGR) upon co-culture with primary CD8+ T-cells stimulated with a membrane-bound anti-CD3 antibody (OK3T).
F) Fluorescence-activated cell sorting plots depicting mutation of endogenous TCR beta chain in primary T-cells. CRISPR/Cas9 ribonucleoprotein (RNP) was used to mutate the TCR beta gene. Cells null for TCR beta (Clone: IP26) were sorted and subsequently transduced with lentivirus to constitutively express centaur TCR. Cells that stained positive with mTCR (Clone: H57-597) and CD4 (Clone: OCT4) and negative hTCR (Clone: IP26) were sorted and used for downstream studies.
G) The frequency of target eHEK-293-II cells that activate the GzB reporter was evaluated after co-culture with primary T-cells transduced with centaur (Ob1.A12). The centaur T-cells used in this study were either wild-type (WT) or TCR null (sgTCR-B). The target eHEK-293-II cells utilized co-expressed the HLA-DRB1*15:01 allele and either full-length Myelin Basic Protein (MBP) fused to CD74-v3 or a control protein fused to CD74-v3.
H) The frequency of target eHEK-293-II that activate the GzB reporter in the presence or absence of CD80 following co-culture with centaur (Ob1.A12) transduced primary. Target eHEK-293-II cells either co-expressed the HLA-DRB1*15:01 allele along with CD80 and full-length Myelin Basic Protein or a control protein fused to CD74-v3.
I) FACS histograms depicting the mutation of HLA-II alleles. CRISPR/Cas9 ribonucleoprotein (RNP) was used to mutate the HLA-DRB, HLA-DPB, and HLA-DQB genes. Cells null for HLA-DR/DP/DQ (Clone: Tü39) were sorted and used for downstream analysis depicted in J.
J) The frequency of target cells that activate the GzB reporter following co-culture with centaur (F24) transduced primary. Target eHEK-293-II cells assessed were either null or expressed the endogenous HLA-II alleles. All target cells expressed the HLA-DRB1*1101 allele, along with either the cognate GAG antigen or a control protein fused to CD74-v3.
K) The schematic representation of genetic engineering performed to generate eHEK293-II target cells and centaur T-cells.
Figure S2. TScan-II screens using the HIV CD4+ TCRs, related to Figure 2
A) Alignment of the enriched fragment from the screen TScan-II HIV genome-wide screen depicted in Figure 2B. The published GAG epitope sequence is highlighted above the alignment.
B) Reproducibility of internal replicates of the genome-wide screen depicted in Figure 2B. Each dot represents one peptide fragment, with the x and y axes plotting the geometric mean of the fold-change of each DNA barcode across seven replicates. Red dots represent peptides from an HIV strain with an epitope identical to the published epitope for this TCR. The blue dots represent peptide fragments from variant GAG epitopes.
C) Fold enrichment of Gag variants from the TScan-II screen depicted in Figure 2B. The x-axis represents the number of variations, including amino acid substitutions or deletions, relative to the published Gag epitope found within the enriched fragments.
D) Relative activation of the GzB reporter in cells pulsed with HIV peptides co-cultured with F24 T-cells. The line in the box plots indicates the median. Variant A: FRDYVDRFFRVLRAEQASQE, Variant B: FRDYVDRFYKTLRAEQASQD, Variant C: FRDYVDRFFKTLRAEQATQD, Variant D: FRDYVDRFFKTLRAEQAT, Variant E: FRDYVDRFYKTLRAEQATQE. The box is drawn around the inner quartile range, and the whiskers show minimum to maximum values.
E, F, and G) The TScan-II screens corresponding to the heatmap representation depicted in Figures 2D, E, and F. eHEK-293-II cells were transduced and selected to express the HLA- DRB1*1101 allele and primary T-cells to express the HIV specific TCRs from an elite controller patient. The mutagenesis library comprises 4,509 fragments, each being 56 aa in length. Each peptide is also barcoded twice using different amino acid codons. Each dot represents one peptide, with the y-axis plotting the geometric mean of the fold-change of each peptide across six replicates. Fold enrichment is defined as the ratio of the peptide abundance in the sorted population relative to the input library. Red and blue dots represent the first and second replicate (barcodes) of the HIV mutagenesis fragment. The darker and lighter colored dots are HIV mutant peptides that respectively enriched or dropped out.
Figure S3. HIV and HPV epitope mapping using the TScan-II platform, related to Figure 3
A) Validation of the identified antigens in target cells using the GzB reporter following co-culture with HIV (F24) TCR transduced primary T-cells. The box plots depict the frequency of target cells that activated the GzB reporter a 6 h co-culture experiment. eHEK-293-II cells were transduced and selected to express the HLA- DRB1*1101 allele. Antigens assessed include a control peptide (left), HIV GAG protein 289-336 aa (middle), or HPV regulatory E2 protein 29-84 aa (right). All antigens were delivered into target cells via lentiviral transduction. The line in the box plots indicates the median. The box is drawn around the inner quartile range, and the whiskers show minimum to maximum values.
B) TScan-II screen of the HIV elite controller F24 TCR using a second library of the top 7,000 enriched fragments from the primary screen (Figure 3B). eHEK-293-II cells were transduced and selected to express the HLA- DRB1*1101 allele, and primary T-cells to express the HIV-specific TCR (F24). Blue dots represent peptides from the HIV GAG protein. Green dots represent peptide fragments from the HPV regulatory E2 protein.
Figure S4. TScan-II mutagenesis screen using NY-ESO CD4+ TCR. Related to Figure 4
A) Predicated affinity of each of identified epitopes for the HLA-DRB1*04 allele is plotted (y-axis) vs. the level of T-cells activation as measured by GzB delivery into target cells (x-axis). Epitope binding affinity was measured using sliding tiles of 15 amino acids in length on the NetMHCII server.
B) Design of the CTAG1B epitope comprehensive mutagenesis library. Every position in the CTAG1B epitope (LPVPGVLLKEFTVSGNILTI) is mutated to each of the 19 alternative amino acids. The epitopes are expressed in the context of a 56 aa fragment c-terminal CD74-v3 tag. The two amino acids, N-terminal and C-terminal to the epitope, are also mutated in the 56 aa versions. The library encodes for 4 WT epitopes and 916 mutant CTAG1B epitopes, and 3,593 negative controls.
C) TScan-II screen of CTAG1B CD4+ TCR (3598-2) against a mutagenesis library. The mutagenesis library comprises 4,509 fragments, each being 56 aa in length. Each peptide is also barcoded twice using different amino acid codons. Each dot represents one peptide, with the y-axis plotting the geometric mean of the fold-change of each peptide across eight replicates. Fold enrichment is defined as the ratio of the peptide abundance in the sorted population relative to the input library. eHEK-293-II cells were transduced and selected to express the HLA-DRB1*04 allele, and primary T-cells to express the CTAG1B specific centaur TCR. Red and blue dots represent the CTAG1B mutagenesis fragment’s first and second barcodes (Replica). The darker and lighter colored dots are CTAG1B mutant peptides enriched or dropped out, respectively.
Figure S5. Characterization of a pancreatic reactive TCRs related to Figure 5
A) Characterizing tumor reactivity of tumor-expanded TCRs using patient-derived organoids.TCR-transduced PBMCs co-cultured with either autologous (matched) or non-autologous (unmatched) organoids. IFN-γ secretion was used as a readout of T-cell activity.
B) Schematic of TScan-II human neo-antigen library. The patient neo-antigen library comprises 1,404 fragments that tile across patient mutations with flanking sequences as 41 amino acid fragments. The WT peptide fragments and fragments encoding mutations from non-matching patients are negative controls in our library. All peptide fragments within the library were barcoded four times using synonymous amino acid codons.
C and D) TScan-II screen of pancreatic reactive DHM6 and DHM7 TCR against the neoantigen library. Each dot represents one peptide, with the y axis plotting the geometric mean of the fold-change of each peptide across eight replicates. Fold enrichment is defined as the ratio of the peptide abundance in the sorted population relative to the input library. eHEK-293-II cells were transduced and selected to express the patient HLA-DP alleles (DPB1*17 and DPB1*105). Blue dots represent four barcoded fragments, each encoding the patient mutant fragments. Gray dots represent either WT or mutations from non-matching patients and collectively act as negative controls.
E) Assessment of DHM6 reactivity towards overlapping fragments of LOC100131311. The box plots depict the frequency of target cells that activated the GzB reporter following a 6 h co-culture experiment. eHEK-293-II cells expressing a control protein were used as negative controls.
F) Total RNA was extracted from pancreatic cancer organoids derived from several patients (P3, P6, P10, P19, P34, P38 (matching patient). The extracted RNA was utilized for strand-specific cDNA synthesis specific to LOC100131311. The resulting cDNA was then employed to evaluate the expression of LOC100131311 using PCR.
G) Deconvolution of HLA restriction of DHM6 TCR. The box plots depict the results of GzB reporter activation following a 6 h co-culture experiment. Primary T-cells were transduced with DHM6 TCR and were co-cultured independently with eHEK-293-II target cells expressing the antigenic fragment and DPB1*105 or DPB1*17 alleles.
Figure S6. Validation and mapping of DDIAS and MAP3K4 antigens, related to Figure 6
A) FACS histograms depicting the initial transduction rate of primary T-cells using lentivirus encoding two TCRs with identical CDR3β chains matched with two different CDR3α(α or α’) chains from the same clonally expanded CD4+ T-cells stained with mTCR (Clone: H57-597).
B) Deconvolution of HLA restriction and TCR reactivity towards MAP3K4 antigen. The box plots depict the results of GzB reporter activation following a 6 h co-culture experiment. Primary T-cells were transduced with 4A,7A,7G,9B, and 10C TCRs and were co-cultured independently with eHEK-293-II target cells expressing MAP3K4 antigenic fragment and HLA-DQB1*02_HLA-DQA1*05 or HLA-DRB1*03:01.
C) Deconvolution of HLA restriction and TCR reactivity towards DDIAS antigen. The box plots depict the results of GzB reporter activation following a 6hr co-culture experiment. Primary T-cells were transduced with 3A,10A1,10D,10E, and 10F TCRs and were co-cultured independently with eHEK-293-II target cells expressing DDIAS antigenic fragment and HLA-DQB1*02_HLA-DQA1*05 or HLA-DRB1*03:01.
D) TScan-II screen of the 4A TCR using a second library of the top 2,200 enriched fragments from the primary screen shown in Figure 5C. Each dot represents one peptide, with the y-axis plotting the geometric mean of the fold-change of each peptide across six replicates. eHEK-293-II cells express 6 of patient 4 HLA-II alleles: HLA-DRB1*03:01, HLA-DRB1*13:0, HLA-DQB1*02:01_HLA-DQA1*05:01, HLA-DQB1*06:03_HLA-DQA1*01:03, HLA-DPB1*04:01_HLA-DPA1*01:03.
E) TScan-II screen of the 3A TCR using a second library of the top ~5500 enriched fragments from the primary screen shown in Figure 5D. Each dot represents one peptide, with the y-axis plotting the geometric mean of the fold-change of each peptide across 6 replicates. eHEK-293-II cells express HLA-DQB1*02_HLA-DQA1*05 and HLA-DRB1*03:01.
F and G) TScan-II screen of a library of 15 and 18 aa fragments with 1 aa overlap spanned the entire length of 90 aa antigenic segments of MAP3K4 and DDIAS. The length of each line along the x-axis indicates the corresponding region of the MAP3K4 covered by each peptide according to a pairwise protein alignment; the y-axis corresponds to the fold enrichment of that fragment. Fragments in light blue did not score while darker blue indicates the scoring peptides. The amino acid sequence depicted represents the minimal epitope characterized.
H) Relative activation of the GzB reporter in cells pulsed with 1 ug of MAP3K4 peptides co-cultured with 4A T-cells. Peptide 1: LRKDLEIAAEFRLSA, Peptide 2: EIAAEFRLSAPVRDL, Peptide 3: EIAAEFRLSA, Peptide 4: LRKDLEIAAEFRLSAPVRDL. The line in the box plots indicates the median. The box is drawn around the inner quartile range, and the whiskers show minimum to maximum values.
I) Relative activation of the GzB reporter in cells pulsed with DDIAS peptides co-cultured with 3A T-cells. The line in the box plots indicates the median. Peptide 1: SLNKFLAVLESEIAV, Peptide 2: ESLNKFLAVLESEIAV, Peptide 3: FSESLNKFLAVLESEIAV, Peptide 4: LNKFLAVLESEIAVTQADVS. The box is drawn around the inner quartile range, and the whiskers show minimum to maximum values.
Figure S7. Assessment of antigens and HLA-II transcript abundance within the salivary gland, related to Figure 7.
A) Plot showing the expression of select genes across ductal cells (as defined in Figure 7A) in both healthy individuals and SJD patients. Each dot represents the expression of each gene in an individual cell, p values were determined by a two-tailed t-test and are shown with asterisks, *<0.05, **<0.01, ***<0.001.
B) The spatial expression pattern DDIAS relative ductal and T-cells in the SG of three SjD subjects was assessed using the 10X Visium transcriptomics. Spotplot representing either ductal cell markers (mean normalized expression of S100A2 and CFTR) or T-cell markers (mean normalized expression of CD3D, CD3E, CD3G, and CD45).
Table S1. All HIV TScan II screening data related to Figures 2, 2S, 3 and 3S.
(A) HIV genome-wide library TScan-II screen data.
(B) HIV epitope mutagenesis TScan-II screen data.
(C) HIV virome wide TScan-II screen data.
(D) HIV virome secondary TScan-II screen data.
Table S2. All CTAG1B TScan II screening data related to Figures 4.
(A) CTAG1B human genome-wide TScan-II screen data.
(B) CTAG1B epitope mutagenesis TScan-II screen data.
Table S3. Pancreatic cancer neo-antigen and TScan-II screen tables related to Figure 5.
(A) Nonsynonymous somatic mutations within pancreatic cancer tumor cells.
(B) Pancreatic cancer human genome-wide TScan-II screen data.
Table S4. SjD TCR selection tables related to Figure 6.
(A) TCRβ sequences described in PBMID 27358913.
(B) TCRβ sequences from newly characterized SjD subjects in this study.
(C) CD4+ TCRs screened using TScan-II.
(D) TCR specificity groups with shared TCR sequences from more than three individuals identified by GLIPH2 analysis.
Table S5. SjD TScan-II screen data related to Figures 6 and 6S.
(A) Pooled human genome-wide TScan-II screen of 5 TCRs 4A,7A,7G, 9B, and 10C
(B) Human genome-wide TScan-II screen of 4A TCR.
(C) Human genome-wide TScan-II screen using 5 TCRs 3A,10A1,10D,10E, and 10F.
(D) Pooled human genome-wide TScan-II screen using 6 TCRs 3A,4A,9C,8A,8B and 5B.
(E) Secondary T-II screen data of 4A TCR.
(F) Secondary T-II screen data of 3A TCR.
Table S6. Enrichment of MAP3K4 and DDIAS peptides across various genome-wide screens related to Figure 6.
Table S7. Oligonucleotide Sequences, related to STAR Methods.
Highlights:
TScan-II finds the cognate and cross-reactive antigens of self and viral CD4+ TCRs
Saturated mutagenesis screens facilitate mapping of the CD4+ TCR epitope interface
CD4+ cells in Sjögren’s disease recognize self-antigens on HLA-II positive ductal cells
TScan-II finds antigens within the non-canonical immunopeptidome for cancer TCRs
Acknowledgments
This research was supported by The Sjögren’s Foundation (M.H.D), Howard Hughes Medical Institute Investigator (S.J.E.), Department of Defense BC171184 (S.J.E.), U24AI118633 (S.J.E), 5R01 AR073855 (C.J.L) and 5R01 AR074310 (A.D.F), NIDCR Division of Intramural Research Award DE000704 (B.M.W), the NIDCR Sjogren’s Clinical Investigations Team, the NIDCR/NIDCD Genomics and Computational Biology Core, NIH training grant T32 HG002295 (NK). S.K.M was supported by Kim and Judy Davis. The authors would also like to thank David Owen for comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
M.H.D., T.K., and S. J.E. are patent holders on the TScan-I and TScan-II technology. S. J.E. is a founder of TSCAN Therapeutics, ImmuneID, MAZE Therapeutics, and Mirimus. S.J.E. serves on the scientific advisory board of TSCAN Therapeutics, and MAZE Therapeutics. In accordance with Partners HealthCare’s conflict of interest policies, the Partners Office for Interactions with Industry has reviewed S.J.E.’s financial interest in TSCAN and determined that it creates no significant risk to the welfare of participants in this study or to the integrity of the research.
References
- 1.Flajnik MF, and Kasahara M (2010). Origin and evolution of the adaptive immune system: genetic events and selective pressures. Nat Rev Genet 11, 47–59. 10.1038/nrg2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kula T, Dezfulian MH, Wang CI, Abdelfattah NS, Hartman ZC, Wucherpfennig KW, Lyerly HK, and Elledge SJ (2019). T-Scan: A Genome-wide Method for the Systematic Discovery of T Cell Epitopes. Cell 178, 1016–1028.e13. 10.1016/j.cell.2019.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Birnbaum ME, Mendoza JL, Sethi DK, Dong S, Glanville J, Dobbins J, Ozkan E, Davis MM, Wucherpfennig KW, and Garcia KC (2014). Deconstructing the peptide-MHC specificity of T cell recognition. Cell 157, 1073–1087. 10.1016/j.cell.2014.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joglekar AV, and Li G (2020). T cell antigen discovery. Nature Methods. 10.1038/s41592-020-0867-z. [DOI] [PubMed] [Google Scholar]
- 5.Sharma G, Rive CM, and Holt RA (2019). Rapid selection and identification of functional CD8+ T cell epitopes from large peptide-coding libraries. Nat Commun 10, 4553. 10.1038/s41467-019-12444-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li G, Bethune MT, Wong S, Joglekar AV, Leonard MT, Wang JK, Kim JT, Cheng D, Peng S, Zaretsky JM, et al. (2019). T cell antigen discovery via trogocytosis. Nat Methods 16, 183–190. 10.1038/s41592-018-0305-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kisielow J, Obermair F-J, and Kopf M (2019). Deciphering CD4+ T cell specificity using novel MHC-TCR chimeric receptors. Nat Immunol 20, 652–662. 10.1038/s41590-019-0335-z. [DOI] [PubMed] [Google Scholar]
- 8.Lee MN, and Meyerson M (2021). Antigen identification for HLA class I- and HLA class II-restricted T cell receptors using cytokine-capturing antigen-presenting cells. Sci Immunol 6, eabf4001. 10.1126/sciimmunol.abf4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blum JS, Wearsch PA, and Cresswell P (2013). Pathways of antigen processing. Annu Rev Immunol 31, 443–473. 10.1146/annurev-immunol-032712-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roche PA, and Furuta K (2015). The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol 15, 203–216. 10.1038/nri3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reith W, LeibundGut-Landmann S, and Waldburger J-M (2005). Regulation of MHC class II gene expression by the class II transactivator. Nat Rev Immunol 5, 793–806. 10.1038/nril708. [DOI] [PubMed] [Google Scholar]
- 12.Wucherpfennig KW, and Hafler DA (1995). A review of T-cell receptors in multiple sclerosis: clonal expansion and persistence of human T-cells specific for an immunodominant myelin basic protein peptide. Ann NY Acad Sci 756, 241–258. 10.1111/j.1749-6632.1995.tb44522.x. [DOI] [PubMed] [Google Scholar]
- 13.Schröder B. (2016). The multifaceted roles of the invariant chain CD74 — More than just a chaperone. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1863, 1269–1281. 10.1016/j.bbamcr.2016.03.026. [DOI] [PubMed] [Google Scholar]
- 14.Benati D, Galperin M, Lambotte O, Gras S, Lim A, Mukhopadhyay M, Nouël A, Campbell K-A, Lemercier B, Claireaux M, et al. (2016). Public T cell receptors confer high-avidity CD4 responses to HIV controllers. J Clin Invest 126, 2093–2108. 10.1172/JCI83792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.To T-L, Piggott BJ, Makhijani K, Yu D, Jan YN, and Shu X (2015). Rationally designed fluorogenic protease reporter visualizes spatiotemporal dynamics of apoptosis in vivo. Proc Natl Acad Sci USA 112, 3338–3343. 10.1073/pnas.l502857112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Timms RT, Zhang Z, Rhee DY, Harper JW, Koren I, and Elledge SJ (2019). A glycine-specific N-degron pathway mediates the quality control of protein N-myristoylation. Science 365, eaaw4912. 10.1126/science.aaw4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soetandyo N, Wang Q, Ye Y, and Li L (2010). Role of intramembrane charged residues in the quality control of unassembled T-cell receptor α-chains at the endoplasmic reticulum. Journal of Cell Science 123, 1031–1038. 10.1242/jcs.059758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, and Morgan RA (2006). Enhanced Antitumor Activity of Murine-Human Hybrid T-Cell Receptor (TCR) in Human Lymphocytes Is Associated with Improved Pairing and TCR/CD3 Stability. Cancer Res 66, 8878–8886. 10.1158/0008-5472.CAN-06-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, and Morgan RA (2007). Enhanced Antitumor Activity of T Cells Engineered to Express T-Cell Receptors with a Second Disulfide Bond. Cancer Res 67, 3898–3903. 10.1158/0008-5472.CAN-06-3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Courtney AH, Lo W-L, and Weiss A (2018). TCR Signaling: Mechanisms of Initiation and Propagation. Trends in Biochemical Sciences 43, 108–123. 10.1016/j.tibs.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lambotte O, Boufassa F, Madec Y, Nguyen A, Goujard C, Meyer L, Rouzioux C, Venet A, Delfraissy J-F, and SEROCO-HEMOCO Study Group (2005). HIV Controllers: A Homogeneous Group of HIV-1--Infected Patients with Spontaneous Control of Viral Replication. Clinical Infectious Diseases 41, 1053–1056. 10.1086/433188. [DOI] [PubMed] [Google Scholar]
- 22.Galperin M, Farenc C, Mukhopadhyay M, Jayasinghe D, Decroos A, Benati D, Tan LL, Ciacchi L, Reid HH, Rossjohn J, et al. (2018). CD4+ T cell–mediated HLA class II cross-restriction in HIV controllers. Science Immunology 3, eaat0687. 10.1126/sciimmunol.aat0687. [DOI] [PubMed] [Google Scholar]
- 23.Thomas R, Al-Khadairi G, Roelands J, Hendrickx W, Dermime S, Bedognetti D, and Decock J (2018). NY-ESO-1 Based Immunotherapy of Cancer: Current Perspectives. Front Immunol 9, 947–947. 10.3389/fimmu.2018.00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park TS, Groh EM, Patel K, Kerkar SP, Lee C-CR, and Rosenberg SA (2016). Expression of MAGE-A and NY-ESO-1 in Primary and Metastatic Cancers. J Immunother 39, 1–7. 10.1097/CJI.0000000000000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, Grand F, Brewer JE, Gupta M, Plesa G, et al. (2013). Identification of a Titin-Derived HLA-A1-Presented Peptide as a Cross-Reactive Target for Engineered MAGE A3-Directed T Cells. Science Translational Medicine 5, 197ra103–197ra103. 10.1126/scitranslmed.3006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, et al. (2013). Cancer Regression and Neurological Toxicity Following Anti-MAGE-A3 TCR Gene Therapy. Journal of Immunotherapy 36, 133. 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, et al. (2013). Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122, 863–871. 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poncette L, Chen X, Lorenz FK, and Blankenstein T (2019). Effective NY-ESO-1-specific MHC II-restricted T cell receptors from antigen-negative hosts enhance tumor regression. J Clin Invest 129, 324–335. 10.1172/JCI120391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reynisson B, Alvarez B, Paul S, Peters B, and Nielsen M (2020). NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Research 48, W449–W454. 10.1093/nar/gkaa379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meng Q, Xie S, Gray GK, Dezfulian MH, Li W, Huang L, Akshinthala D, Ferrer E, Conahan C, Perea Del Pino S, et al. (2021). Empirical identification and validation of tumor-targeting T cell receptors from circulation using autologous pancreatic tumor organoids. J Immunother Cancer 9, e003213. 10.1136/jitc-2021-003213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chong C, Müller M, Pak H, Harnett D, Huber F, Grun D, Leleu M, Auger A, Arnaud M, Stevenson BJ, et al. (2020). Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat Commun 11, 1293. 10.1038/s41467-020-14968-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Q, Laverdure J-P, Lanoix J, Durette C, Côté C, Bonneil É, Laumont CM, Gendron P, Vincent K, Courcelles M, et al. (2020). Proteogenomics Uncovers a Vast Repertoire of Shared Tumor-Specific Antigens in Ovarian Cancer. Cancer Immunology Research 8, 544–555. 10.1158/2326-6066.CIR-19-0541. [DOI] [PubMed] [Google Scholar]
- 33.Ouspenskaia T, Law T, Clauser KR, Klaeger S, Sarkizova S, Aguet F, Li B, Christian E, Knisbacher BA, Le PM, et al. (2022). Unannotated proteins expand the MHC-I-restricted immunopeptidome in cancer. Nat Biotechnol 40, 209–217. 10.1038/s41587-021-01021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiang R, Ma L, Yang M, Zheng Z, Chen X, Jia F, Xie F, Zhou Y, Li F, Wu K, et al. (2021). Increased expression of peptides from non-coding genes in cancer proteomics datasets suggests potential tumor neoantigens. Commun Biol 4, 1–12. 10.1038/s42003-021-02007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Starck SR, and Shastri N (2011). Non-conventional sources of peptides presented by MHC class I. Cell. Mol. Life Sci 68, 1471–1479. 10.1007/s00018-011-0655-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang S, Mao C, and Liu S (2019). Peptides encoded by noncoding genes: challenges and perspectives. Sig Transduct Target Ther 4, 1–12. 10.1038/s41392-019-0092-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lv D, Chang Z, Cai Y, Li J, Wang L, Jiang Q, Xu K, Ding N, Li X, Xu J, et al. (2022). TransLnc: a comprehensive resource for translatable lncRNAs extends immunopeptidome. Nucleic Acids Research 50, D413–D420. 10.1093/nar/gkab847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adinarayana KPS, Sravani TS, and Hareesh C (2011). A database of six eukaryotic hypothetical genes and proteins. Bioinformation 6, 128–130. 10.6026/97320630006128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gonzalez-Galarza FF, McCabe A, Santos E.J.M. dos, Jones J, Takeshita L, Ortega-Rivera ND, Cid-Pavon GMD, Ramsbottom K, Ghattaoraya G, Alfirevic A, et al. (2020). Allele frequency net database (AFND) 2020 update: gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Research 48, D783–D788. 10.1093/nar/gkzl029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamradt T, and Mitchison NA (2001). Tolerance and Autoimmunity. N Engl J Med 344, 655–664. 10.1056/NEJM200103013440907. [DOI] [PubMed] [Google Scholar]
- 41.Bloch KJ, Buchanan WW, Wohl MJ, and Bunim JJ (1965). SJOEGREN’S SYNDROME. A CLINICAL, PATHOLOGICAL, AND SEROLOGICAL STUDY OF SIXTY-TWO CASES. Medicine (Baltimore) 44, 187–231. [PubMed] [Google Scholar]
- 42.Adamson TC 3rd, Fox RI, Frisman DM, and Howell FV (1983). Immunohistologic analysis of lymphoid infiltrates in primary Sjogren’s syndrome using monoclonal antibodies. The Journal of Immunology 130, 203–208. 10.4049/jimmunol.130.1.203. [DOI] [PubMed] [Google Scholar]
- 43.Christodoulou MI, Kapsogeorgou EK, and Moutsopoulos HM (2010). Characteristics of the minor salivary gland infiltrates in Sjögren’s syndrome. J Autoimmun 34, 400–407. 10.1016/j.jaut.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 44.Kapsogeorgou EK, Christodoulou MI, Panagiotakos DB, Paikos S, Tassidou A, Tzioufas AG, and Moutsopoulos HM (2013). Minor salivary gland inflammatory lesions in Sjogren syndrome: do they evolve? J Rheumatol 40, 1566–1571. 10.3899/jrheum.130256. [DOI] [PubMed] [Google Scholar]
- 45.Skopouli FN, Fox PC, Galanopoulou V, Atkinson JC, Jaffe ES, and Moutsopoulos HM (1991). T cell subpopulations in the labial minor salivary gland histopathologic lesion of Sjögren’s syndrome. J Rheumatol 18, 210–214. [PubMed] [Google Scholar]
- 46.Mingueneau M, Boudaoud S, Haskett S, Reynolds TL, Nocturne G, Norton E, Zhang X, Constant M, Park D, Wang W, et al. (2016). Cytometry by time-of-flight immunophenotyping identifies a blood Sjögren’s signature correlating with disease activity and glandular inflammation. J Allergy Clin Immunol 137, 1809–1821.e12. 10.1016/jjaci.2016.01.024. [DOI] [PubMed] [Google Scholar]
- 47.Joachims ML, Leehan KM, Lawrence C, Pelikan RC, Moore JS, Pan Z, Rasmussen A, Radfar L, Lewis DM, Grundahl KM, et al. (2016). Single-cell analysis of glandular T cell receptors in Sjögren’s syndrome. JCI Insight 1. 10.1172/jci.insight.85609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moutsopoulos HM, Hooks JJ, Chan CC, Dalavanga YA, Skopouli FN, and Detrick B (1986). HLA-DR expression by labial minor salivary gland tissues in Sjögren’s syndrome. Ann Rheum Dis 45, 677. 10.1136/ard.45.8.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fox RI, Bumol T, Fantozzi R, Bone R, and Schreiber R (1986). Expression of histocompatibility antigen HLA-DR by salivary gland epithelial cells in Sjögren’s syndrome. Arthritis Rheum 29, 1105–1111. 10.1002/art.1780290908. [DOI] [PubMed] [Google Scholar]
- 50.Thrane PS, Halstensen TS, Haanaes HR, and Brandtzaeg P (1993). Increased epithelial expression of HLA-DQ and HLA-DP molecules in salivary glands from patients with Sjögren’s syndrome compared with obstructive sialadenitis. Clin Exp Immunol 92, 256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsunawaki S, Nakamura S, Ohyama Y, Sasaki M, Ikebe-Hiroki A, Hiraki A, Kadena T, Kawamura E, Kumamaru W, Shinohara M, et al. (2002). Possible function of salivary gland epithelial cells as nonprofessional antigen-presenting cells in the development of Sjögren’s syndrome. J Rheumatol 29, 1884–1896. [PubMed] [Google Scholar]
- 52.Molina C, Alliende C, Aguilera S, Kwon Y-J, Leyton L, Martínez B, Leyton C, Pérez P, and González M-J (2006). Basal lamina disorganisation of the acini and ducts of labial salivary glands from patients with Sjögren’s syndrome: association with mononuclear cell infiltration. Annals of the Rheumatic Diseases 65, 178–183. 10.1136/ard.2004.033837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Manoussakis MN, Dimitriou ID, Kapsogeorgou EK, Xanthou G, Paikos S, Polihronis M, and Moutsopoulos HM (1999). Expression of B7 costimulatory molecules by salivary gland epithelial cells in patients with Sjögren’s syndrome. Arthritis Rheum 42, 229–239. . [DOI] [PubMed] [Google Scholar]
- 54.Kapsogeorgou EK, Moutsopoulos HM, and Manoussakis MN (2001). Functional expression of a costimulatory B7.2 (CD86) protein on human salivary gland epithelial cells that interacts with the CD28 receptor, but has reduced binding to CTLA4. J Immunol 166, 3107–3113. 10.4049/jimmunol.166.5.3107. [DOI] [PubMed] [Google Scholar]
- 55.Ping L, Ogawa N, and Sugai S (2005). Novel role of CD40 in Fas-dependent apoptosis of cultured salivary epithelial cells from patients with Sjögren’s syndrome. Arthritis Rheum 52, 573–581. 10.1002/art.20789. [DOI] [PubMed] [Google Scholar]
- 56.Gong Y-Z, Nititham J, Taylor K, Miceli-Richard C, Sordet C, Wachsmann D, Bahram S, Georgel P, Criswell LA, Sibilia J, et al. (2014). Differentiation of follicular helper T cells by salivary gland epithelial cells in primary Sjögren’s syndrome. J Autoimmun 51, 57–66. 10.1016/j.jaut.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 57.Fye KH, Terasaki PI, Michalski JP, Daniels TE, Opelz G, and Talal N (1978). Relationship of Hla-Dw3 and Hla-B8 to Sjögren’S Syndrome. Arthritis & Rheumatism 21, 337–342. 10.1002/art.1780210308. [DOI] [PubMed] [Google Scholar]
- 58.Chused TM, Kassan SS, Opelz G, Moutsopoulos HM, and Terasaki PI (1977). Sjögren’s Syndrome Associated with HLA-Dw3. New England Journal of Medicine 296, 895–897. 10.1056/NEJM197704212961602. [DOI] [PubMed] [Google Scholar]
- 59.Harley JB, Reichlin M, Arnett FC, Alexander EL, Bias WB, and Provost TT (1986). Gene interaction at HLA-DQ enhances autoantibody production in primary Sjögren’s syndrome. Science 232, 1145–1147. 10.1126/science.3458307. [DOI] [PubMed] [Google Scholar]
- 60.Lessard CJ, Li H, Adrianto I, Ice JA, Rasmussen A, Grundahl KM, Kelly JA, Dozmorov MG, Miceli-Richard C, Bowman S, et al. (2013). Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren’s syndrome. Nat Genet 45, 1284–1292. 10.1038/ng.2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khatri B, Tessneer KL, Rasmussen A, Aghakhanian F, Reksten TR, Adler A, Alevizos I, Anaya J-M, Aqrawi LA, and Baecklund E (2022). Genome-wide association study identifies Sjögren’s risk loci with functional implications in immune and glandular cells. Nature Communications 13, 4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Elliott JI, and Altmann DM (1995). Dual T cell receptor alpha chain T cells in autoimmunity. Journal of Experimental Medicine 182, 953–959. 10.1084/jem.182.4.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hinz T, Weidmann E, and Kabelitz D (2001). Dual TCR-expressing T lymphocytes in health and disease. Int Arch Allergy Immunol 125, 16–20. 10.1159/000053792. [DOI] [PubMed] [Google Scholar]
- 64.Schuldt NJ, and Binstadt BA (2019). Dual TCR T Cells: Identity Crisis or Multitaskers? The Journal of Immunology 202, 637–644. 10.4049/jimmunol.1800904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Davies JM (1997). Molecular mimicry: Can epitope mimicry induce autoimmune disease? Immunology & Cell Biology 75, 113–126. 10.1038/icb.1997.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cusick MF, Libbey JE, and Fujinami RS (2012). Molecular mimicry as a mechanism of autoimmune disease. Clin Rev Allergy Immunol 42, 102–111. 10.1007/sl2016-011-8294-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Leech S. (1998). Molecular mimicry in autoimmune disease. Archives of Disease in Childhood 79, 448–451. 10.1136/adc.79.5.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou S, Cai Y, Wang M, Yang W-D, and Duan N (2018). Oral microbial flora of patients with Sicca syndrome. Molecular Medicine Reports 18, 4895–4903. 10.3892/mmr.2018.9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Verstappen GM, Kroese FGM, and Bootsma H (2021). T cells in primary Sjögren’s syndrome: targets for early intervention. Rheumatology 60, 3088–3098. 10.1093/rheumatology/kez004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Horvath S, Nazmul-Hossain ANM, Pollard RPE, Kroese FGM, Vissink A, Kallenberg CGM, Spijkervet FKL, Bootsma H, Michie SA, Gorr SU, et al. (2012). Systems analysis of primary Sjögren’s syndrome pathogenesis in salivary glands identifies shared pathways in human and a mouse model. Arthritis Res Ther 14, R238. 10.1186/ar4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang N, Pérez P, Kato T, Mikami Y, Okuda K, Gilmore RC, Conde CD, Gasmi B, Stein S, Beach M, et al. (2021). SARS-CoV-2 infection of the oral cavity and saliva. Nat Med 27, 892–903. 10.1038/s41591-021-01296-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Verstappen GM, Pringle S, Bootsma H, and Kroese FGM (2021). Epithelial–immune cell interplay in primary Sjögren syndrome salivary gland pathogenesis. Nat Rev Rheumatol 17, 333–348. 10.1038/s41584-021-00605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cai Y, Lv D, Li D, Yin J, Ma Y, Luo Y, Fu L, Ding N, Li Y, Pan Z, et al. (2023). IEAtlas: an atlas of HLA-presented immune epitopes derived from non-coding regions. Nucleic Acids Research 51, D409–D417. 10.1093/nar/gkac776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marcu A, Bichmann L, Kuchenbecker L, Kowalewski DJ, Freudenmann LK, Backert L, Mühlenbruch L, Szolek A, Lübke M, Wagner P, et al. (2021). HLA Ligand Atlas: a benign reference of HLA-presented peptides to improve T-cell-based cancer immunotherapy. J Immunother Cancer 9, e002071. 10.1136/jitc-2020-002071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee KH, Wucherpfennig KW, and Wiley DC (2001). Structure of a human insulin peptide-HLA-DQ8 complex and susceptibility to type 1 diabetes. Nat Immunol 2, 501–507. 10.1038/88694. [DOI] [PubMed] [Google Scholar]
- 76.Noble JA, Valdes AM, Cook M, Klitz W, Thomson G, and Erlich HA (1996). The role of HLA class II genes in insulin-dependent diabetes mellitus: molecular analysis of 180 Caucasian, multiplex families. Am J Hum Genet 59, 1134–1148. [PMC free article] [PubMed] [Google Scholar]
- 77.Hu X, Deutsch AJ, Lenz TL, Onengut-Gumuscu S, Han B, Chen W-M, Howson JMM, Todd JA, de Bakker PIW, Rich SS, et al. (2015). Additive and interaction effects at three amino acid positions in HLA-DQ and HLA-DR molecules drive type 1 diabetes risk. Nat Genet 47, 898–905. 10.1038/ng.3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee H-S, Jia X, Alfredsson L, Padyukov L, Klareskog L, Worthington J, et al. (2012). Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet 44, 291–296. 10.1038/ng.l076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Taylor KE, Wong Q, Levine DM, McHugh C, Laurie C, Doheny K, Lam MY, Baer AN, Challacombe S, Lanfranchi H, et al. (2017). Genome-Wide Association Analysis Reveals Genetic Heterogeneity of Sjögren’s Syndrome According to Ancestry. Arthritis Rheumatol 69, 1294–1305. 10.1002/art.40040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li Y, Zhang K, Chen H, Sun F, Xu J, Wu Z, Li P, Zhang L, Du Y, Luan H, et al. (2013). A genome-wide association study in Han Chinese identifies a susceptibility locus for primary Sjögren’s syndrome at 7q11.23. Nat Genet 45, 1361–1365. 10.1038/ng.2779. [DOI] [PubMed] [Google Scholar]
- 81.Kamitaki N, Sekar A, Handsaker RE, de Rivera H, Tooley K, Morris DL, Taylor KE, Whelan CW, Tombleson P, Loohuis LMO, et al. (2020). Complement genes contribute sex-biased vulnerability in diverse disorders. Nature 582, 577–581. 10.1038/s41586-020-2277-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lundtoft C, Pucholt P, Martin M, Bianchi M, Lundström E, Eloranta M-L, Sandling JK, Sjöwall C, Jönsen A, Gunnarsson I, et al. (2022). Complement C4 Copy Number Variation is Linked to SSA/Ro and SSB/La Autoantibodies in Systemic Inflammatory Autoimmune Diseases. Arthritis & Rheumatology 74, 1440–1450. 10.1002/art.42122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lundtoft C, Sjöwall C, Rantapää-Dahlqvist S, Bengtsson AA, Jönsen A, Pucholt P, Wu YL, Lundström E, Eloranta M-L, Gunnarsson T, et al. (2022). Strong Association of Combined Genetic Deficiencies in the Classical Complement Pathway With Risk of Systemic Lupus Erythematosus and Primary Sjögren’s Syndrome. Arthritis & Rheumatology 74, 1842–1850. 10.1002/art.42270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Briggs DC, Welsh K, Pereira RS, and Black CM (1986). A strong association between null alleles at the C4A locus in the major histocompatibility complex and systemic sclerosis. Arthritis & Rheumatism 29, 1274–1277. 10.1002/art.1780291014. [DOI] [PubMed] [Google Scholar]
- 85.Kumar A, Kumar P, and Schur PH (1991). DR3 and nonDR3 associated complement component C4A deficiency in systemic lupus erythematosus. Clinical Immunology and Immunopathology 60, 55–64. 10.1016/0090-1229(91)90111-M. [DOI] [PubMed] [Google Scholar]
- 86.Langefeld CD, Ainsworth HC, Graham DSC, Kelly JA, Comeau ME, Marion MC, Howard TD, Ramos PS, Croker JA, Morris DL, et al. (2017). Transancestral mapping and genetic load in systemic lupus erythematosus. Nat Commun 8, 16021. 10.1038/ncomms16021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Steinsson K, Jónsdóttir S, Arason GJ, Kristjánsdóttir H, Fossdal R, Skaftadóttir I, and Árnason A (1998). A study of the association of HLA DR, DQ, and complement C4 alleles with systemic lupus erythematosus in Iceland. Annals of the Rheumatic Diseases 57, 503–505. 10.1136/ard.57.8.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kristjánsdóttir H, Bjarnadóttir K, Hjálmarsdóttir IB, Gröndal G, Arnason A, and Steinsson K (2000). A study of C4AQ0 and MHC haplotypes in Icelandic multicase families with systemic lupus erythematosus. J Rheumatol 27, 2590–2596. [PubMed] [Google Scholar]
- 89.Carroll MC (2004). A protective role for innate immunity in systemic lupus erythematosus. Nat Rev Immunol 4, 825–831. 10.1038/nril456. [DOI] [PubMed] [Google Scholar]
- 90.White S, and Rosen A (2003). Apoptosis in systemic lupus erythematosus. Curr Opin Rheumatol 15, 557–562. 10.1097/00002281-200309000-00006. [DOI] [PubMed] [Google Scholar]
- 91.Atkinson JP (1989). Complement deficiency: predisposing factor to autoimmune syndromes. Clin Exp Rheumatol 7 Suppl 3, S95–101. [PubMed] [Google Scholar]
- 92.Simoni L, Presumey J, van der Poel CE, Castrillon C, Chang SE, Utz PJ, and Carroll MC (2020). Complement C4A Regulates Autoreactive B Cells in Murine Lupus. Cell Rep 33, 108330. 10.1016/j.celrep.2020.108330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Majumder P, Gomez JA, and Boss JM (2006). The Human Major Histocompatibility Complex Class II HLA-DRB1 and HLA-DQA1 Genes Are Separated by a CTCF-binding Enhancer-blocking Element *. Journal of Biological Chemistry 281, 18435–18443. 10.1074/jbc.M601298200. [DOI] [PubMed] [Google Scholar]
- 94.Raj P, Rai E, Song R, Khan S, Wakeland BE, Viswanathan K, Arana C, Liang C, Zhang B, Dozmorov I, et al. Regulatory polymorphisms modulate the expression of HLA class II molecules and promote autoimmunity. eLife 5, e12089. 10.7554/eLife.12089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vyse TJ, and Tsao BP (2022). Complement C4, the Major Histocompatibility Complex, and Autoimmunity. Arthritis & Rheumatology 74, 1318–1320. 10.1002/art.42119. [DOI] [PubMed] [Google Scholar]
- 96.Kawasaki A, Kusumawati PA, Kawamura Y, Kondo Y, Kusaoi M, Amano H, Kusanagi Y, Itoh K, Fujimoto T, Tamura N, et al. (2023). Genetic dissection of HLA-DRB1*15:01 and XL9 region variants in Japanese patients with systemic lupus erythematosus: primary role for HLA-DRB1*15:01. RMD Open 9, e003214. 10.1136/rmdopen-2023-003214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Harley ITW, and Sawalha AH (2022). Systemic lupus erythematosus as a genetic disease. Clin Immunol 236, 108953. 10.1016/j.clim.2022.108953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Warner BM, Baer AN, Lipson EJ, Allen C, Hinrichs C, Rajan A, Pelayo E, Beach M, Gulley JL, Madan RA, et al. (2019). Sicca Syndrome Associated with Immune Checkpoint Inhibitor Therapy. Oncologist 24, 1259–1269. 10.1634/theoncologist.2018-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shiboski CH, Shiboski SC, Seror R, Criswell LA, Labetoulle M, Lietman TM, Rasmussen A, Scofield H, Vitali C, Bowman SJ, et al. (2017). 2016 American College of Rheumatology/European League Against Rheumatism Classification Criteria for Primary Sjögren’s Syndrome: A Consensus and Data-Driven Methodology Involving Three International Patient Cohorts. Arthritis Rheumatol 69, 35–45. 10.1002/art.39859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shiboski CH, Shiboski SC, Seror R, Criswell LA, Labetoulle M, Lietman TM, Rasmussen A, Scofield H, Vitali C, Bowman SJ, et al. (2017). 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjögren’s syndrome: A consensus and data-driven methodology involving three international patient cohorts. Ann Rheum Dis 76, 9–16. 10.1136/annrheumdis-2016-210571. [DOI] [PubMed] [Google Scholar]
- 101.Huang L, Holtzinger A, Jagan I, BeGora M, Lohse I, Ngai N, Nostro C, Wang R, Muthuswamy LB, Crawford HC, et al. (2015). Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat Med 21, 1364–1371. 10.1038/nm.3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Huang H, Wang C, Rubelt F, Scriba TJ, and Davis MM (2020). Analyzing the Mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genome-wide antigen screening. Nat Biotechnol 38, 1194–1202. 10.1038/s41587-020-0505-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yin H, Pranzatelli TJF, French BN, Zhang N, Warner BM, Chiorini JA, and NIDCD/NIDCR Genomics and Computational Biology Core (2021). Sclerosing Sialadenitis Is Associated With Salivary Gland Hypofunction and a Unique Gene Expression Profile in Sjögren’s Syndrome. Front Immunol 12, 699722. 10.3389/fimmu.2021.699722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Optimization of Antigen processing and TCR signaling for the TScan-II platform, related to Figure 1
A) FACS histograms depicting the sequential engineering of eHEK-293-II cells. CRISPR/Cas9 ribonucleoprotein (RNP) was used to mutate the B2M gene. Cells that did not express B2M (Clone 2M2) or HLA-A, B, or C (Clone W6/32) on their cell surface were sorted and subsequently transduced with lentivirus to express the MHC-II transactivator (CIITA constitutively). Cells that stably expressed CIITA and had high levels of HLA-DR/DP/DQ (Clone: Tü39) were sorted and used for downstream studies.
B) Boxplots depicting activation levels of F24 TCR transduced SKW-3 cells following co-culture with HLA-DR11 expressing eHEK-293-II cells in the presence of endogenously expressed and processed HIV antigens. SKW-3 were identified using CD3, and their activation was measured via the early activation marker CD69 following co-culture with target cells. The line in the box plots indicates the median. The box is drawn around the inner quartile range, and the whiskers show minimum to maximum values.
C) FACS plots depicting activation levels of TCR transduced SKW-3 cells following co-culture with eHEK-293-II cells in the absence (lower panel) or presence (upper panel) of endogenously expressed (Fused to CD74-v3) and processed antigens. TCRs tested include an HLA- DRB1*0701 restricted Cytomegalovirus specific (left), HLA-DQB1*03:02, DQA1*03:01 restricted BRAFV600E (middle), or HLA- DRB1*04 restricted NY-ESO (CTAG1B) antigen.
D) FACS histograms depicting the generation of centaur primary T-cells. Primary CD8+ T-cells are transduced with lentivirus to express the modified CD4+TCRs and the glycoprotein CD4. Cells stained positive with mTCR (Clone: H57-597) and CD4 (Clone: OCT4) were sorted and used for downstream studies.
E) The frequency of cells that activate the GzB reporter in eHEK-293-II cells expressing either IFPGzB (VGPD→FGR) or IFPGzB-Hi (VGPD→SGR) upon co-culture with primary CD8+ T-cells stimulated with a membrane-bound anti-CD3 antibody (OK3T).
F) Fluorescence-activated cell sorting plots depicting mutation of endogenous TCR beta chain in primary T-cells. CRISPR/Cas9 ribonucleoprotein (RNP) was used to mutate the TCR beta gene. Cells null for TCR beta (Clone: IP26) were sorted and subsequently transduced with lentivirus to constitutively express centaur TCR. Cells that stained positive with mTCR (Clone: H57-597) and CD4 (Clone: OCT4) and negative hTCR (Clone: IP26) were sorted and used for downstream studies.
G) The frequency of target eHEK-293-II cells that activate the GzB reporter was evaluated after co-culture with primary T-cells transduced with centaur (Ob1.A12). The centaur T-cells used in this study were either wild-type (WT) or TCR null (sgTCR-B). The target eHEK-293-II cells utilized co-expressed the HLA-DRB1*15:01 allele and either full-length Myelin Basic Protein (MBP) fused to CD74-v3 or a control protein fused to CD74-v3.
H) The frequency of target eHEK-293-II that activate the GzB reporter in the presence or absence of CD80 following co-culture with centaur (Ob1.A12) transduced primary. Target eHEK-293-II cells either co-expressed the HLA-DRB1*15:01 allele along with CD80 and full-length Myelin Basic Protein or a control protein fused to CD74-v3.
I) FACS histograms depicting the mutation of HLA-II alleles. CRISPR/Cas9 ribonucleoprotein (RNP) was used to mutate the HLA-DRB, HLA-DPB, and HLA-DQB genes. Cells null for HLA-DR/DP/DQ (Clone: Tü39) were sorted and used for downstream analysis depicted in J.
J) The frequency of target cells that activate the GzB reporter following co-culture with centaur (F24) transduced primary. Target eHEK-293-II cells assessed were either null or expressed the endogenous HLA-II alleles. All target cells expressed the HLA-DRB1*1101 allele, along with either the cognate GAG antigen or a control protein fused to CD74-v3.
K) The schematic representation of genetic engineering performed to generate eHEK293-II target cells and centaur T-cells.
Figure S2. TScan-II screens using the HIV CD4+ TCRs, related to Figure 2
A) Alignment of the enriched fragment from the screen TScan-II HIV genome-wide screen depicted in Figure 2B. The published GAG epitope sequence is highlighted above the alignment.
B) Reproducibility of internal replicates of the genome-wide screen depicted in Figure 2B. Each dot represents one peptide fragment, with the x and y axes plotting the geometric mean of the fold-change of each DNA barcode across seven replicates. Red dots represent peptides from an HIV strain with an epitope identical to the published epitope for this TCR. The blue dots represent peptide fragments from variant GAG epitopes.
C) Fold enrichment of Gag variants from the TScan-II screen depicted in Figure 2B. The x-axis represents the number of variations, including amino acid substitutions or deletions, relative to the published Gag epitope found within the enriched fragments.
D) Relative activation of the GzB reporter in cells pulsed with HIV peptides co-cultured with F24 T-cells. The line in the box plots indicates the median. Variant A: FRDYVDRFFRVLRAEQASQE, Variant B: FRDYVDRFYKTLRAEQASQD, Variant C: FRDYVDRFFKTLRAEQATQD, Variant D: FRDYVDRFFKTLRAEQAT, Variant E: FRDYVDRFYKTLRAEQATQE. The box is drawn around the inner quartile range, and the whiskers show minimum to maximum values.
E, F, and G) The TScan-II screens corresponding to the heatmap representation depicted in Figures 2D, E, and F. eHEK-293-II cells were transduced and selected to express the HLA- DRB1*1101 allele and primary T-cells to express the HIV specific TCRs from an elite controller patient. The mutagenesis library comprises 4,509 fragments, each being 56 aa in length. Each peptide is also barcoded twice using different amino acid codons. Each dot represents one peptide, with the y-axis plotting the geometric mean of the fold-change of each peptide across six replicates. Fold enrichment is defined as the ratio of the peptide abundance in the sorted population relative to the input library. Red and blue dots represent the first and second replicate (barcodes) of the HIV mutagenesis fragment. The darker and lighter colored dots are HIV mutant peptides that respectively enriched or dropped out.
Figure S3. HIV and HPV epitope mapping using the TScan-II platform, related to Figure 3
A) Validation of the identified antigens in target cells using the GzB reporter following co-culture with HIV (F24) TCR transduced primary T-cells. The box plots depict the frequency of target cells that activated the GzB reporter a 6 h co-culture experiment. eHEK-293-II cells were transduced and selected to express the HLA- DRB1*1101 allele. Antigens assessed include a control peptide (left), HIV GAG protein 289-336 aa (middle), or HPV regulatory E2 protein 29-84 aa (right). All antigens were delivered into target cells via lentiviral transduction. The line in the box plots indicates the median. The box is drawn around the inner quartile range, and the whiskers show minimum to maximum values.
B) TScan-II screen of the HIV elite controller F24 TCR using a second library of the top 7,000 enriched fragments from the primary screen (Figure 3B). eHEK-293-II cells were transduced and selected to express the HLA- DRB1*1101 allele, and primary T-cells to express the HIV-specific TCR (F24). Blue dots represent peptides from the HIV GAG protein. Green dots represent peptide fragments from the HPV regulatory E2 protein.
Figure S4. TScan-II mutagenesis screen using NY-ESO CD4+ TCR. Related to Figure 4
A) Predicated affinity of each of identified epitopes for the HLA-DRB1*04 allele is plotted (y-axis) vs. the level of T-cells activation as measured by GzB delivery into target cells (x-axis). Epitope binding affinity was measured using sliding tiles of 15 amino acids in length on the NetMHCII server.
B) Design of the CTAG1B epitope comprehensive mutagenesis library. Every position in the CTAG1B epitope (LPVPGVLLKEFTVSGNILTI) is mutated to each of the 19 alternative amino acids. The epitopes are expressed in the context of a 56 aa fragment c-terminal CD74-v3 tag. The two amino acids, N-terminal and C-terminal to the epitope, are also mutated in the 56 aa versions. The library encodes for 4 WT epitopes and 916 mutant CTAG1B epitopes, and 3,593 negative controls.
C) TScan-II screen of CTAG1B CD4+ TCR (3598-2) against a mutagenesis library. The mutagenesis library comprises 4,509 fragments, each being 56 aa in length. Each peptide is also barcoded twice using different amino acid codons. Each dot represents one peptide, with the y-axis plotting the geometric mean of the fold-change of each peptide across eight replicates. Fold enrichment is defined as the ratio of the peptide abundance in the sorted population relative to the input library. eHEK-293-II cells were transduced and selected to express the HLA-DRB1*04 allele, and primary T-cells to express the CTAG1B specific centaur TCR. Red and blue dots represent the CTAG1B mutagenesis fragment’s first and second barcodes (Replica). The darker and lighter colored dots are CTAG1B mutant peptides enriched or dropped out, respectively.
Figure S5. Characterization of a pancreatic reactive TCRs related to Figure 5
A) Characterizing tumor reactivity of tumor-expanded TCRs using patient-derived organoids.TCR-transduced PBMCs co-cultured with either autologous (matched) or non-autologous (unmatched) organoids. IFN-γ secretion was used as a readout of T-cell activity.
B) Schematic of TScan-II human neo-antigen library. The patient neo-antigen library comprises 1,404 fragments that tile across patient mutations with flanking sequences as 41 amino acid fragments. The WT peptide fragments and fragments encoding mutations from non-matching patients are negative controls in our library. All peptide fragments within the library were barcoded four times using synonymous amino acid codons.
C and D) TScan-II screen of pancreatic reactive DHM6 and DHM7 TCR against the neoantigen library. Each dot represents one peptide, with the y axis plotting the geometric mean of the fold-change of each peptide across eight replicates. Fold enrichment is defined as the ratio of the peptide abundance in the sorted population relative to the input library. eHEK-293-II cells were transduced and selected to express the patient HLA-DP alleles (DPB1*17 and DPB1*105). Blue dots represent four barcoded fragments, each encoding the patient mutant fragments. Gray dots represent either WT or mutations from non-matching patients and collectively act as negative controls.
E) Assessment of DHM6 reactivity towards overlapping fragments of LOC100131311. The box plots depict the frequency of target cells that activated the GzB reporter following a 6 h co-culture experiment. eHEK-293-II cells expressing a control protein were used as negative controls.
F) Total RNA was extracted from pancreatic cancer organoids derived from several patients (P3, P6, P10, P19, P34, P38 (matching patient). The extracted RNA was utilized for strand-specific cDNA synthesis specific to LOC100131311. The resulting cDNA was then employed to evaluate the expression of LOC100131311 using PCR.
G) Deconvolution of HLA restriction of DHM6 TCR. The box plots depict the results of GzB reporter activation following a 6 h co-culture experiment. Primary T-cells were transduced with DHM6 TCR and were co-cultured independently with eHEK-293-II target cells expressing the antigenic fragment and DPB1*105 or DPB1*17 alleles.
Figure S6. Validation and mapping of DDIAS and MAP3K4 antigens, related to Figure 6
A) FACS histograms depicting the initial transduction rate of primary T-cells using lentivirus encoding two TCRs with identical CDR3β chains matched with two different CDR3α(α or α’) chains from the same clonally expanded CD4+ T-cells stained with mTCR (Clone: H57-597).
B) Deconvolution of HLA restriction and TCR reactivity towards MAP3K4 antigen. The box plots depict the results of GzB reporter activation following a 6 h co-culture experiment. Primary T-cells were transduced with 4A,7A,7G,9B, and 10C TCRs and were co-cultured independently with eHEK-293-II target cells expressing MAP3K4 antigenic fragment and HLA-DQB1*02_HLA-DQA1*05 or HLA-DRB1*03:01.
C) Deconvolution of HLA restriction and TCR reactivity towards DDIAS antigen. The box plots depict the results of GzB reporter activation following a 6hr co-culture experiment. Primary T-cells were transduced with 3A,10A1,10D,10E, and 10F TCRs and were co-cultured independently with eHEK-293-II target cells expressing DDIAS antigenic fragment and HLA-DQB1*02_HLA-DQA1*05 or HLA-DRB1*03:01.
D) TScan-II screen of the 4A TCR using a second library of the top 2,200 enriched fragments from the primary screen shown in Figure 5C. Each dot represents one peptide, with the y-axis plotting the geometric mean of the fold-change of each peptide across six replicates. eHEK-293-II cells express 6 of patient 4 HLA-II alleles: HLA-DRB1*03:01, HLA-DRB1*13:0, HLA-DQB1*02:01_HLA-DQA1*05:01, HLA-DQB1*06:03_HLA-DQA1*01:03, HLA-DPB1*04:01_HLA-DPA1*01:03.
E) TScan-II screen of the 3A TCR using a second library of the top ~5500 enriched fragments from the primary screen shown in Figure 5D. Each dot represents one peptide, with the y-axis plotting the geometric mean of the fold-change of each peptide across 6 replicates. eHEK-293-II cells express HLA-DQB1*02_HLA-DQA1*05 and HLA-DRB1*03:01.
F and G) TScan-II screen of a library of 15 and 18 aa fragments with 1 aa overlap spanned the entire length of 90 aa antigenic segments of MAP3K4 and DDIAS. The length of each line along the x-axis indicates the corresponding region of the MAP3K4 covered by each peptide according to a pairwise protein alignment; the y-axis corresponds to the fold enrichment of that fragment. Fragments in light blue did not score while darker blue indicates the scoring peptides. The amino acid sequence depicted represents the minimal epitope characterized.
H) Relative activation of the GzB reporter in cells pulsed with 1 ug of MAP3K4 peptides co-cultured with 4A T-cells. Peptide 1: LRKDLEIAAEFRLSA, Peptide 2: EIAAEFRLSAPVRDL, Peptide 3: EIAAEFRLSA, Peptide 4: LRKDLEIAAEFRLSAPVRDL. The line in the box plots indicates the median. The box is drawn around the inner quartile range, and the whiskers show minimum to maximum values.
I) Relative activation of the GzB reporter in cells pulsed with DDIAS peptides co-cultured with 3A T-cells. The line in the box plots indicates the median. Peptide 1: SLNKFLAVLESEIAV, Peptide 2: ESLNKFLAVLESEIAV, Peptide 3: FSESLNKFLAVLESEIAV, Peptide 4: LNKFLAVLESEIAVTQADVS. The box is drawn around the inner quartile range, and the whiskers show minimum to maximum values.
Figure S7. Assessment of antigens and HLA-II transcript abundance within the salivary gland, related to Figure 7.
A) Plot showing the expression of select genes across ductal cells (as defined in Figure 7A) in both healthy individuals and SJD patients. Each dot represents the expression of each gene in an individual cell, p values were determined by a two-tailed t-test and are shown with asterisks, *<0.05, **<0.01, ***<0.001.
B) The spatial expression pattern DDIAS relative ductal and T-cells in the SG of three SjD subjects was assessed using the 10X Visium transcriptomics. Spotplot representing either ductal cell markers (mean normalized expression of S100A2 and CFTR) or T-cell markers (mean normalized expression of CD3D, CD3E, CD3G, and CD45).
Table S1. All HIV TScan II screening data related to Figures 2, 2S, 3 and 3S.
(A) HIV genome-wide library TScan-II screen data.
(B) HIV epitope mutagenesis TScan-II screen data.
(C) HIV virome wide TScan-II screen data.
(D) HIV virome secondary TScan-II screen data.
Table S2. All CTAG1B TScan II screening data related to Figures 4.
(A) CTAG1B human genome-wide TScan-II screen data.
(B) CTAG1B epitope mutagenesis TScan-II screen data.
Table S3. Pancreatic cancer neo-antigen and TScan-II screen tables related to Figure 5.
(A) Nonsynonymous somatic mutations within pancreatic cancer tumor cells.
(B) Pancreatic cancer human genome-wide TScan-II screen data.
Table S4. SjD TCR selection tables related to Figure 6.
(A) TCRβ sequences described in PBMID 27358913.
(B) TCRβ sequences from newly characterized SjD subjects in this study.
(C) CD4+ TCRs screened using TScan-II.
(D) TCR specificity groups with shared TCR sequences from more than three individuals identified by GLIPH2 analysis.
Table S5. SjD TScan-II screen data related to Figures 6 and 6S.
(A) Pooled human genome-wide TScan-II screen of 5 TCRs 4A,7A,7G, 9B, and 10C
(B) Human genome-wide TScan-II screen of 4A TCR.
(C) Human genome-wide TScan-II screen using 5 TCRs 3A,10A1,10D,10E, and 10F.
(D) Pooled human genome-wide TScan-II screen using 6 TCRs 3A,4A,9C,8A,8B and 5B.
(E) Secondary T-II screen data of 4A TCR.
(F) Secondary T-II screen data of 3A TCR.
Table S6. Enrichment of MAP3K4 and DDIAS peptides across various genome-wide screens related to Figure 6.
Table S7. Oligonucleotide Sequences, related to STAR Methods.
Data Availability Statement
Single-cell RNA-seq data have been deposited in the NIH Database of Genotypes and Phenotypes (dbGaP). The accession information is listed in the Key Resources Table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD3 antibody (OKT3) | Thermo Fisher Scientific | Cat: 14-0037-82 |
| anti-human TCR α/β Antibody | Biolegend | Cat: 306711 |
| Alexa Fluor 647 anti-human CD8 Antibody | Biolegend | Cat: 311410 |
| Brilliant Violet 421 ™ anti-human CD80 Antibody | Biolegend | Cat: 305222 |
| Alexa Fluor® 647 anti-human CD19 Antibody | Biolegend | Cat: 363039 |
| Biotin anti-mouse TCR β chain Antibody | Biolegend | Cat: 109204 |
| APC anti-human CD8 Antibody | Biolegend | Cat: 344722 |
| Alexa Fluor® 647 anti-human HLA-DR, DP, DQ Antibody | Biolegend | Cat: 361704 |
| FITC anti-human HLA-DQ Antibody | Biolegend | Cat: 361503 |
| Alexa Fluor® 647 anti-human CD69 Antibody | Biolegend | Cat: 310918 |
| APC anti-human CD58 (LFA-3) Antibody | Biolegend | Cat: 330917 |
| APC anti-human CD155 (PVR) Antibody | Biolegend | Cat: 337617 |
| Alexa Fluor® 647 anti-human CD54 Antibody | Biolegend | Cat: 353113 |
| FITC anti-mouse TCR β chain Antibody | Biolegend | Cat: 109206 |
| Alexa Fluor® 647 anti-human CD3 Antibody | Biolegend | Cat: 317312 |
| Bacterial and Virus Strains | ||
| One Shot® Stbl3™ Chemically Competent E. coli | Thermo Fisher Scientific | Cat: C7373-03 |
| ElectroMAX™ DH10B™ Cells | Thermo Fisher Scientific | Cat: 18290015 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Interleukin-2, human (hIL-2) 25ug | Sigma (Millipore) | Cat: HIL2-RO |
| Gateway™ LR Clonase™ II Enzyme mix | Thermo Fisher Scientific | Cat:11791100 |
| Gateway™ BP Clonase™ II Enzyme mix | Thermo Fisher Scientific | Cat:11789100 |
| Q5® High-Fidelity 2X Master Mix | New England Biolabs | Cat:M0492L |
| Lenti-X Concentrator | Takara Bio | 631232 |
| Puromycin Dihydrochloride | Thermo Fisher Scientific | Cat:A1113803 |
| Polybrene | Thermo Fisher Scientific | Cat:TR-1003-G |
| Superscript™ IV First-Strand Synthesis System with ezDNase™ Enzyme | Thermo Fisher Scientific | Cat:18091150 |
| Ambion™ RNase H, from E. coli, 10 U/μL | Thermo Fisher Scientific | Cat:AM2293 |
| Benzonase Nuclease HC, Purity > 99% | Thermo Fisher Scientific | Cat:71206-3 |
| Nourseothricin Sulfate (Streptothricin Sulfate) | Gold Biotechnology | Cat:N-500-1 |
| NEBuilder® HiFi DNA Assembly Master Mix | New England Biolabs | Cat:E2621L |
| Dynabeads® Human T-Activator CD3/CD28 | Thermo Fisher Scientific | Cat:11132D |
| jetPRIME | Polyplus | Cat:114-07 |
| Critical Commercial Assays | ||
| GeneJET Genomic DNA Purification Kit | Thermo Fisher Scientific | Cat:K0721 |
| RNeasy Plus Mini Kit | Qiagen | Cat:74134 |
| Anti-Biotin Microbeads Miltenyi | Miltenyi Biotec | Cat:130-090-485 |
| RosetteSep™ Human CD8+ T Cell Enrichment Cocktail | Stem Cell Technologies | Cat:15023 |
| Deposited Data | ||
| Singel-Cell-RNA-Seq | Yin, 2021 Huang and Perez, 2021 This Study | dbGAP: Phs002446.v1 .p |
| Experimental Models: Cell Lines | ||
| Lenti-X™ 293T Cell Line | Takara Bio | 632180 |
| eHEK-293-II | This Study | N/A |
| HLA-II expressing eHEK-293-II | This Study | N/A |
| Jurkat, Clone E6-1 | ATCC | Cat:TIB-152 |
| SKW-3 Cell line | Gift from Julia Etchin at Dana Farber Cancer Institue | N/A |
| Oligonucleotides | ||
| See Table S7 | This Study | N/A |
| Recombinant DNA | ||
| pHAGE-CMV:DEST-PGK: ICADCR. | This Study | N/A |
| pHAGE-CMV:IFPGzB-Hi-PGK: ICADCR | This Study | N/A |
| pHAGE-TREX-CIITA-PGK-Neomycin | This Study | N/A |
| pHAGE-EF1L-anti-CD3 scFv-PGK-Puromycin | This Study | N/A |
| pHAGE-EF1L-anti-H57-597-scFv-PGK- Puromycin | This Study | N/A |
| pHAGE-TREX-DEST-PGK- Blasticidin | This Study | N/A |
| pHAGE-TREX-DEST-PGK-Hygromycin | This Study | N/A |
| pHAGE-TREX-DEST-PGK-Nourseothricin | This Study | N/A |
| pHAGE-EF1a-DEST-PGK-floxed-mCD19 | This Study | N/A |
| pHAGE-EF1:DEST-PGK: CD4 | This Study | N/A |
| pHAGE-CMV-CD74_V3-DEST-Puromycin | This Study | N/A |
| Human Peptidome in pHAGE-CMV-CD74_V3-DEST | This Study | N/A |
| Human Virome in pHAGE-CMV-CD74 V3-DEST | This Study | N/A |
| Tiling Library in pHAGE-CMV-CD74 V3-DEST | This Study | N/A |
| HIV Library in pHAGE-CMV-CD74 V3-DEST | This Study | N/A |
| Mutagenesis Library in pHAGE-CMV-CD74_V3-DEST | This Study | N/A |
| Software and Algorithms | ||
| Bowtie | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| Cutadapt | Martin, 2011 | http://cutadapt.readthedocs.io/en/stable/ |
| Cell Ranger v3.0.1 | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Scanpy | Wolf, 2018 | https://github.com/scverse/scanpy |
| Seurat | Sajita, 2015 | https://github.com/satijalab/seurat |
| Python v3.7 | Python Software Foundation | https://www.python.org |