

Abstract
Background:
KRAS is among the most commonly mutated oncogenes in cancer, and previous studies have shown associations with survival in many cancer contexts. Evidence from both clinical observations and mouse experiments further suggests that these associations are allele- and tissue-specific. These findings motivate using clinical data to understand gene interactions and clinical covariates within different alleles and tissues.
Methods:
We analyze genomic and clinical data from the AACR Project GENIE Biopharma Collaborative for samples from lung, colorectal, and pancreatic cancers. For each of these cancer types, we report epidemiological associations for different KRAS alleles, apply principal component analysis to discover groups of genes co-mutated with KRAS, and identify distinct clusters of patient profiles with implications for survival.
Results:
KRAS mutations were associated with inferior survival in lung, colon, and pancreas, although the specific mutations implicated varied by disease. Tissue- and allele-specific associations with smoking, sex, age, and race were found. Tissue-specific genetic interactions with KRAS were identified by principal components analysis, which were clustered to produce five, four, and two patient profiles in lung, colon, and pancreas. Membership in these profiles was associated with survival in all three cancer types.
Conclusion:
KRAS mutations have tissue- and allele-specific associations with inferior survival, clinical covariates, and genetic interactions.
Impact:
Our results provide greater insight into the tissue- and allele-specific associations with KRAS mutations and identify clusters of patients that are associated with survival and clinical attributes from combinations of genetic interactions with KRAS mutations.
Introduction
KRAS encodes two, highly related 21 kD monomeric GTPases that function to control cellular behaviors in response to extracellular stimuli. The on/off state of the KRAS protein is determined by nucleotide binding, with the GTP-bound form existing in an active signaling conformation. Missense mutations in KRAS alter the homeostatic balance of GDP and GTP binding toward the active state, either by reducing GTP hydrolysis or by increasing the rate of GTP loading (1). KRAS mutations are most common in pancreatic ductal adenocarcinoma (PDAC), colorectal cancer (CRC), and lung adenocarcinoma (LUAD), the most common form of non-small cell lung cancer (NSCLC). In each of these diseases, codon 12 mutations predominate, accounting for nearly 90% of all KRAS mutations, although non-codon 12 mutations account for a significant proportion of KRAS alleles in some cancers (2). Experimental studies in mouse models have demonstrated that mutations at different K-RAS residues exhibit distinct properties at the molecular, cellular, and organismal levels (3,4).
Both the predictive and prognostic value of KRAS mutations has been studied extensively in many different cancer contexts. In NSCLC, previous studies have demonstrated that KRAS mutations are associated with worsened survival, particularly in adenocarcinomas (5–7). However, the clinical significance of these mutations is still sometimes considered controversial due to heterogeneity in study populations (6). In CRC, KRAS mutations have also been negatively associated with overall survival and relapse-free survival in metastatic cancers, with a more controversial role in non-metastatic cancers (8). Finally, in PDAC, the RAS signaling pathway has been implicated as a key oncogenic driver, with KRAS mutations appearing early in the development of this cancer (9).
In some epidemiological studies, the role of different KRAS alleles has been examined. For example, in CRC patients, codon 12 mutations are typically associated with worse overall survival, relative to patients with KRAS wild-type cancers, while codon 13 mutations are not, and moreover, CRC patients with codon 146 mutations appear to exhibit improved overall survival relative to patients with other mutations (10–14). These clinical observations are consistent with our preclinical studies of the relative effects of K-RasG12D, K-RasG13D, and K-RasA146T in mouse models (3,4).
We recently performed a comprehensive data analysis of different tumor types – including colorectal, pancreatic, and lung – to gain further insight into the mechanisms responsible for the varying clinical characteristics by mutant allele (2). This analysis revealed distinct, tissue-specific co-mutation networks for each KRAS allele, which suggests that gene interactions are both allele-specific and tissue-specific. With clinical data now available, these findings motivate the search for relationships between gene interactions and clinical covariates within different alleles and tissues. In this work, we analyze curated data for NSCLC, CRC, and PDAC tumor samples from the AACR Project GENIE Biopharma Collaborative, which provides extensive clinical covariates in addition to sequencing data. We report epidemiological associations within each cancer type for different KRAS alleles and apply a version of principal component analysis adapted for these data to discover groups of genes driving mutations with KRAS in each tissue, as well as clusters of patient profiles with implications for survival.
Materials and Methods
Study design and population
The AACR GENIE (Genomics Evidence Neoplasia Information Exchange) Project represents one of the largest public cancer genomic releases, consisting of over 154,000 sequenced samples from over 137,000 patients. These samples span 19 institutions and represent dozens of cancer types. Details on sample collection and targeted gene sequencing panels can be found in (15). Recently, the GENIE Project and 10 biopharmaceutical companies have formed the BioPharma Collaborative (BPC) in order to generate rich clinical data, including information about patients’ attributes, tumor characteristics, drug treatment exposures, and radiologic and clinical responses, to accompany the genomic information for 12 of these cancer cohorts. In this work, we use the curated BPC datasets for NSCLC, CRC, and PDAC, all of which were constructed using the PRISSMM (Pathology, Radiology, Imaging, Signs, Symptoms, Biomarker and MedOnc notes) data model. Further details on the data curation process can be found in (16).
The curated NSCLC dataset contains 2,014 tumor samples from 1,849 distinct patients (Supplementary Figure S1). For patients with multiple samples, we only included the earliest sequenced NSCLC sample. We further omitted any patients with missing values in at least one of the included clinical covariates, which are: institution at which the patient was located (0 missing values); sex (0 missing values); age at diagnosis (0 missing values); race, categorized as White, Asian, Black, or other (59 missing values); smoking history, binarized as ever or never (3 missing values); stage at diagnosis (126 missing values); and histology, categorized as adenocarcinoma, squamous cell cancer, or other (257 missing values). Finally, we omitted any patients whose sequencing report was obtained after death or censorship (64 samples). This resulted in a total of 1,436 patients included in our initial analysis. Further association analyses were then conducted within the subset of 521 patients with stage IV adenocarcinomas based on preliminary findings.
The curated CRC dataset contains 1,571 tumor samples from 1,500 distinct patients (Supplementary Figure S2). As with the NSCLC dataset, we only included the earliest sequenced CRC sample for each patient, and we omitted any patients with missing values in at least one of the following clinical covariates: institution at which the patient was located (0 missing values); sex (0 missing values); age at diagnosis (0 missing values); race, categorized as White, Asian, Black, or other (70 missing values); and stage at diagnosis (13 missing values). We also omitted any patients whose sequencing report was obtained after death or censorship (87 samples). This resulted in a total of 1,327 patients in our analysis.
The curated PDAC dataset contains 1,129 tumor samples from 1,109 distinct patients (Supplementary Figure S3). As before, we included the earliest sequenced PDAC sample for each patient and omitted those with missing values in at least one of the following covariates: institution at which the patient was located (0 missing values); sex (0 missing values); age at diagnosis (0 missing values); race, categorized as White, Asian, Black, or other (29 missing values); stage at diagnosis (8 missing values); and histology, categorized as adenocarcinoma and other (401 missing values). We also omitted 63 patients whose sequencing report was dated after their death or censorship. This resulted in a total of 650 patients in our analysis.
Statistical analysis
To evaluate the association between KRAS mutations and stage at diagnosis, as well as histology, in the NSCLC cohort, we used multivariate logistic regression models. The outcome variable in each case was whether or not a KRAS mutation of any kind was present. We report odds ratios (ORs) and the corresponding 95% confidence intervals (CIs). To evaluate the effect of KRAS mutations on survival, we used a Cox proportional hazards regression model. Because sequencing is not done exactly at the time of diagnosis, often occurring months or even years later, this dataset can be considered left-truncated (17). We accounted for this by using the counting process version of the Cox model, where the time from diagnosis to generation of the sequencing report was used as the entry time. The outcome variable was overall survival, in years, from time of diagnosis, and the exposure variable was whether or not a KRAS mutation was present. The proportional hazards assumption was assessed by testing for association between the scaled Schoenfeld residuals and time. Finally, we estimated the effect of KRAS mutations on survival within each subgroup, defined by stage at diagnosis or histology. We report hazards ratios (HRs) and the corresponding 95% CIs. All models included age at diagnosis, sex, and, when applicable, stage at diagnosis as covariates. Because many patients with early stage disease at diagnosis were only sequenced at time of recurrence, as a sensitivity analysis, we also re-fitted all models restricted to just the 834 patients for whom sequencing took place within 3 months of diagnosis.
In all following analyses in the NSCLC cohort, we restricted our attention to patients with stage IV adenocarcinomas, based on the preliminary analysis above suggesting that KRAS mutations are only associated with survival in this subgroup. We assessed association between the presence of any KRAS mutation and each clinical covariate of interest via multivariate logistic regression models and report ORs and 95% CIs. We again assessed the effect of having any KRAS mutation on survival and the effect of KRAS mutations within each subgroup defined by each categorical covariate using Cox proportional hazards regression. Finally, we assessed the effects of having specific KRAS mutations on survival. We report HRs and 95% CIs. All models included age at diagnosis and sex as covariates.
We followed a similar strategy in the CRC and PDAC cohorts, but conducted all analyses in the full sample sets. To assess association between the presence of any KRAS mutations and each clinical covariate of interest, we used multivariate logistic regression models, and report ORs and 95% CIs. To assess the effect of having any KRAS mutations, as well as specific KRAS mutations, on survival, we used the counting process version of the Cox proportional hazards regression model, with the generation of the sequencing report defining entry time, and report HRs and 95% CIs. All models included age at diagnosis, stage at diagnosis, and sex as covariates. In the case of the PDAC cohort, we additionally included histology as a covariate. Finally, as a sensitivity analysis, we again re-fitted all models using just the 346 CRC samples and the 289 PDAC samples respectively whose sequencing reports were generated within three months of diagnosis.
Many of the statistical tests described above involve multiple comparisons. In such cases, we specifically report what we refer to as 95% Bonferroni CIs, where we actually compute (100 − 5/n)% CIs, with n denoting the number of comparisons. This is analogous to a Bonferroni correction of p-values, but in the setting of confidence intervals.
Bernoulli principal components analysis
We used a variant of GLM-PCA (18), which we refer to here as Bernoulli principal components analysis (B-PCA), to estimate latent principal components (PCs) that summarize signal and relationships in the mutation data. GLM-PCA is a generalization of principal components analysis to data with exponential family likelihoods and was initially developed for application to count data in single-cell RNA-sequencing. In B-PCA, we consider the special case of binary data, which is motivated by the representation of each gene of interest as a binary variable – either mutated or not – in our tumor sequencing samples.
Suppose we have an matrix , where is the number of tumor samples and is the number of genes, such that if gene is mutated in sample and 0 otherwise. We assume that each is distributed as a Bernoulli random variable with unobserved probability parameter . We further assume that signal in the mutation data can be described in terms of a smaller number of latent components, such that the matrix can be decomposed into the product of two lower-dimensional matrices. More specifically, to avoid constraints due to the range of , we model
where is a matrix of latent components and is an matrix representing the contributions of each component to each sample. We assume that , so that we are summarizing the signal in the mutation data in a smaller number of interpretable components.
We can estimate as the parameters maximizing the Bernoulli likelihood of our observed mutations . In particular, we use an alternating maximization procedure in which we first provide an initial guess and compute the estimate maximizing the likelihood conditional on . We then compute maximizing the likelihood conditional on , and repeat this procedure until the likelihood from one iteration to the next differs by less than a pre-specified tolerance . Maximization is done numerically using the library nloptr’s implementation of the subplex algorithm.
The stability of the resulting fit was assessed using a resampling procedure. In particular, in each of 20 iterations, we randomly resampled the patients with replacement, followed the same procedure to fit this model, and then computed the RV coefficient (19) between this new estimate of and the original estimate. The RV coefficient is a generalization of correlation to matrices, with values closer to 1 indicating better concordance. If the original fit is stable, we would expect the resulting set of RV coefficients to be close to 1, and if the original fit is unstable, we expect at least some values closer to 0.
We applied B-PCA to 27 genes in patients with stage IV adenocarcinoma in the NSCLC cohort, 48 genes in patients from the CRC cohort, and 35 genes in patients from the PDAC cohort. We included KRAS in both cases, and the remaining genes were selected as those that are (1) found to have at least 4% mutation frequency in the respective tissue in the GENIE data, and (2) were sequenced in at least 15% of samples. For PDAC, condition (1) was relaxed to 2% due to the lower number of genes with high mutational frequency. We excluded any patients from this analysis who were missing sequencing results in at least one of the remaining genes, which resulted in final totals of 449 NSCLC (specifically LUAD) patients, 1,128 CRC patients, and 604 PDAC patients.
To compute an initial guess , we estimated such that each is the proportion of reads for gene found to be altered in sample . We then ran standard PCA on , and used the resulting principal components as the starting guess. The number of components was chosen using a scree plot (20), which happened to yield for all three cohorts.
We also used the estimates from B-PCA to cluster patients within each cohort. This was done by computing Euclidean distances between each patient in the lower-dimensional B-PCA space, as described by the matrix , and running the -means algorithm with 50 random starts. We excluded the first principal component from this analysis because it summarizes the overall levels of mutations, which can be interpreted as related to mutational load. The number of clusters was chosen in each case as the value between 2 and 10 resulting in the lowest Bayesian Information Criterion (BIC) when a Cox proportional hazards regression model was fit with cluster identity as a predictor. This yielded 5, 4, and 2 clusters for the NSCLC, CRC, and PDAC cohorts respectively.
Software
All statistical analyses were performed in R version 4.1.3. Survival analyses were performed using the survival package version 3.4.0 with the coxph function to fit the Cox regression models and the cox.zph function to test the proportional hazards assumption. In the implementation of B-PCA, numerical maximization was done using nloptr version 2.0.3 with the NLOPT_LN_SBPLX algorithm.
Data availability
The NSCLC and CRC data are publicly available on Synapse at the Synapse ID syn27056172.
The PANC 1.1-consortium data are available upon request at this link: https://www.synapse.org/#!Synapse:syn26288991.
Results
KRAS and survival in the NSCLC cohort
The demographic characteristics of NSCLC with wild-type KRAS and those with any kind of KRAS mutation are summarized in Supplementary Table S1. We found that 31% of patients in this cohort carried a KRAS mutation. Among those with KRAS mutations, the majority (86%) of mutations occurred in codon 12, followed by 8% in codon 13, 3% in codon 61, and the remaining 3% in various other codons.
Table 1 summarizes the associations between having a KRAS mutation and each covariate of interest, as well as the effect of having a KRAS mutation on overall survival. In the entire sample set, we observed an association for inferior survival (HR = 1.25, 95% CI 1.06, 1.47) with the presence of a KRAS mutation, but there were several important interactions to consider. While there was no evidence of association between KRAS mutations and stage at diagnosis, suggesting they are equally likely to be found at any stage, KRAS mutations were only associated with survival among stage IV patients (Table 1). This was confirmed by testing for an interaction effect between stage at diagnosis and KRAS mutations in the counting process version of a Cox regression (interaction HR = 1.75, 95% CI = 1.01, 3.02), with sex and age at diagnosis also included in the model. There is potential selection bias when considering stage at diagnosis, since many patients with early stage disease at diagnosis were only sequenced at disease progression. However, the interaction effect between having a KRAS mutation and being stage IV at diagnosis persisted even when subsetting only to patients for whom sequencing took place within three months of diagnosis (interaction HR = 2.59, 95% CI = 1.00, 6.71). Furthermore, we found that KRAS mutations were more likely to be found in adenocarcinomas than in squamous cell cancers and also were only associated with survival in adenocarcinomas (Table 1).
Table 1.
Associations between KRAS mutation and covariates of interest, reported as ORs with 95% Bonferroni CIs (corrected over all tested covariates), and effects of having a KRAS mutation on survival in each subgroup, reported as HRs with 95% Bonferroni CIs (corrected within each category of covariates), in the NSCLC cohort. Only Stage IV adenocarcinomas are considered in quantities reported below the horizontal line.
| KRAS OR (Association with mutation occurrence and each covariate) | KRAS HR (Association with survival within each subgroup) | |
|---|---|---|
|
| ||
| Stage 1 | ||
| Stage I | Referent | 0.85 (0.44, 1.63) |
| Stage II | 1.05 (0.56, 1.91) | 1.43 (0.70, 2.90) |
| Stage III | 0.77 (0.46, 1.27) | 0.94 (0.58, 1.53) |
| Stage IV | 0.81 (0.54, 1.23) | 1.44 (1.10, 1.90) |
| Histology 2 | ||
| Squamous cell | Referent | 0.86 (0.30, 2.46) |
| Adenocarcinoma | 9.67 (3.89, 31.83) | 1.39 (1.11, 1.75) |
| Other | 7.51 (2.59, 27.05) | 0.87 (0.48, 1.57) |
|
| ||
| Institution 1 | ||
| MSK | Referent | 1.72 (1.13, 2.61) |
| DFCI | 1.38 (0.73, 2.60) | 1.63 (0.97, 2.73) |
| VICC | 1.43 (0.64, 3.12) | 1.62 (0.88, 2.98) |
| Sex 3 | ||
| Male | Referent | 1.71 (1.09, 2.67) |
| Female | 1.55 (0.86, 2.86) | 1.55 (1.11, 2.16) |
| Age (years) 4 | 1.04 (1.01, 1.07) | |
| Race 1 | ||
| White | Referent | 1.51 (1.11, 2.06) |
| Asian | 0.12 (0.01, 0.53) | 0.96 (0.14, 6.73) |
| Black | 1.09 (0.28, 3.64) | 25.91 (1.55, 433.56) |
| Other | 0.51 (0.02, 3.69) | 2.57 (0.04, 157.78) |
| Smoking History 1 | ||
| Never | Referent | 0.98 (0.30, 3.19) |
| Ever | 11.70 (4.52, 39.47) | 1.43 (1.07, 1.91) |
Age at diagnosis and sex included in the model.
Age at diagnosis, sex, and stage at diagnosis included in the model.
Age at diagnosis included in the model.
Sex included in the model.
Motivated by these findings, we restricted subsequent analyses to just stage IV adenocarcinoma patients. When considering survival within this group, the HR for having any KRAS mutation was 1.62 (95% CI 1.28, 2.04; test of proportional hazards p = 0.088). We found no associations with the patient’s institution, but KRAS mutations were more likely to be found in older patients and those with smoking history, and less likely to be found in patients of Asian descent (Table 1). Notably, these associations with age and Asian descent persist even when adjusting for smoking history as well (Supplementary Table S2). Although Asian patients have been reported to be more likely to have EGFR mutations, which are mutually exclusive with KRAS mutations, the association with Asian descent also still persists, though attenuated, when only considering those with wild-type EGFR (Supplementary Table S2).
We next examined the effects of specific KRAS mutations on survival within these patients with stage IV lung adenocarcinoma (Figure 1A, B). Of the four mutations occurring in at least 10 patients (G12C, G12V, G12D, and G13C), we found associations with inferior survival with G12C and G12V (test of proportional hazards p = 0.23). Note that neither of these associations are present when considering the whole sample, rather than just the stage IV adenocarcinomas (Supplementary Table S2). We also assessed whether the associations with smoking history and age at diagnosis were localized to specific KRAS mutations by testing for any association with mutation type among those with one of these four mutation types. We found that those with G12C mutations are substantially more likely to be smokers, relative to those with G12D mutations (Supplementary Table S2). However, there was no evidence that the association found earlier for age is localized to specific mutation types.
Figure 1.
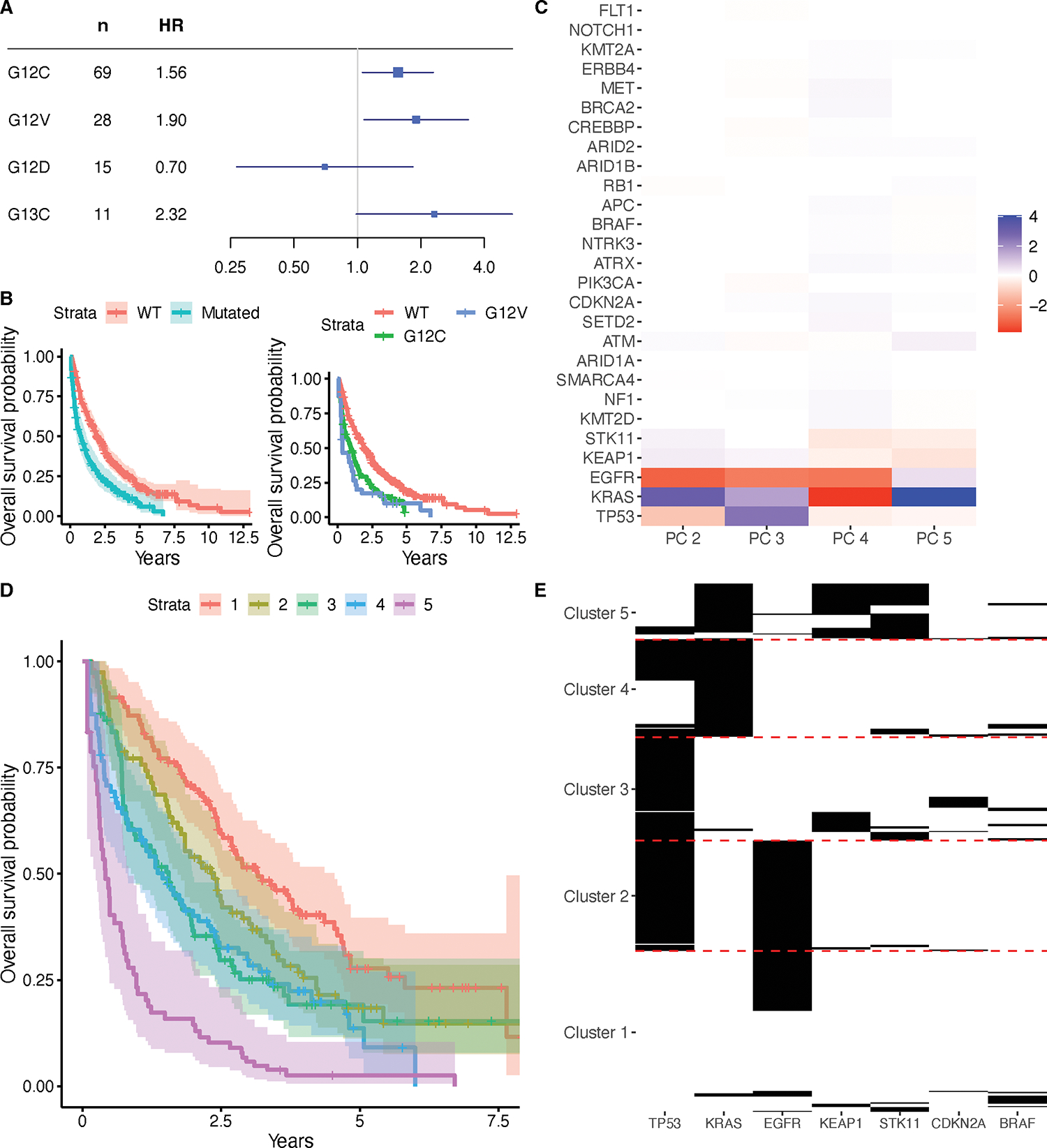
Associations of KRAS mutations with survival in the stage IV adenocarcinomas from the NSCLC cohort. (A) HRs with corresponding 95% Bonferroni CIs for each specific KRAS mutation type occurring in at least 10 patients, as assessed via Cox proportional hazards regression models adjusting for age and sex. (B) Kaplan-Meier curves with 95% CIs for patients with wild-type KRAS and patients with any kind of KRAS mutation, and Kaplan-Meier curves for patients with wild-type KRAS, patients with G12C mutations, and patients with G12V mutations. (C) Loadings matrix for the second through fifth principal component found from applying B-PCA to mutation data in the stage IV adenocarcinomas of the NSCLC cohort. (D) Kaplan-Meier curves comparing the patients in five clusters, derived from clustering the patient scores for the principal components. (E) Heatmap showing mutation patterns among the genes differentially mutated (FDR < 0.05) across the five clusters.
Co-mutations with KRAS and patient clusters in the NSCLC cohort
We previously demonstrated, using publicly available somatic genotyping data, that the genetic networks in KRAS mutant cancers are allele- and tissue-specific (2). At the time, however, matching clinical data were not available. To understand co-mutational relationships between KRAS and other genes of interest and how they relate to clinical outcomes, we applied B-PCA to the mutation data from the stage IV adenocarcinoma patients. We considered a subset of genes, including KRAS, which were selected to represent genes with relatively high mutation frequencies in these data (see Supplementary Table S3 for demographic characteristics of included and excluded patients). We ran B-PCA with five PCs (Figure 1C, Supplementary Table S4) and found this fit to be stable via a resampling strategy (RV coefficients ranged from 0.16 to 0.98, with quartiles of 0.50, 0.59, and 0.69). We then used the counting process version of Cox proportional hazards regression to examine the factor scores for the PCs, all in the same model and also including age at diagnosis and sex, for potential association with overall survival (Supplementary Table S2). This analysis revealed associations with inferior survival for both PC 2 (HR = 1.04, 95% Bonferroni CI = 1.00, 1.09, test for proportional hazards p = 0.011) and PC 3 (HR = 1.11; 95% Bonferroni CI = 1.05, 1.17; test for proportional hazards p = 0.23), which implies that patients with higher scores for these PCs had worse survival. Notably, both of these PCs has a positive loading for KRAS, so higher scores imply a higher probability of having a KRAS mutation. Analogously, we also found an association with improved survival for PC 4 (HR = 0.90; 95% Bonferroni CI = 0.84, 0.96; test for proportional hazards p = 0.64), which has a negative loading for KRAS. Hence, these associations are consistent with our previous finding that the presence of a KRAS mutation is associated with inferior survival. As a sensitivity analysis, we repeated this test with tumor mutational burden (TMB, computed as the total number of mutations among genes included in the B-PCA analysis for each sample) included as a covariate and found the same associations (Supplementary Table S2).
To interpret the mutational patterns encoded by these PCs and how they operate together, we clustered patient scores for the PCs into five clusters. This was the number of groups that minimized the BIC of the counting process version of the Cox proportional hazards regression model using cluster identity, age at diagnosis, and sex as predictors. We labeled these Clusters 1 through 5, in order of their median survival, and found, from the final version of this regression model (Supplementary Table S2), that membership in Clusters 3, 4, and 5 were associated with worse survival than Cluster 1 (Cluster 3 HR = 1.88, 95% Bonferroni CI = 1.20, 2.95; Cluster 4 HR = 1.80, 95% CI = 1.13, 2.86; Cluster 5 HR = 3.99, 95% Bonferroni CI = 2.41, 6.59; test for proportional hazards p = 0.089), as shown in Figure 1D. This was again repeated with TMB included as a covariate with similar results (Supplementary Table S2), with the exception that Cluster 4 is no longer significantly associated after multiple testing correction. We used univariate logistic regressions to assess whether each gene was differentially mutated in each cluster when compared to all other clusters (Supplementary Table S5). The mutational patterns among genes with significant differential mutations (FDR < 0.05) are visualized in Figure 1E, which can be used to characterize these groups of patients. In particular, Cluster 1 samples, which had the best survival, most commonly have either no mutations among these genes or an isolated EGFR mutation; Cluster 2 samples nearly all have both EGFR and TP53 mutations; Cluster 3 almost all has TP53 mutations and almost never KRAS mutations; Cluster 4 has all KRAS mutations and sometimes TP53 mutations; and Cluster 5, which had the worst survival, almost all has KRAS mutations with many accompanied by KEAP1 and/or STK11 mutations.
These patterns are suggestive of relationships between KRAS mutations and these genes, which were confirmed by Fisher tests. Namely, we found positive associations with STK11 (OR = 6.32, 95% Bonferroni CI = 2.95, 14.10) and KEAP1 (OR = 3.83, 95% Bonferroni CI = 1.79, 8.35). We also found negative associations between KRAS and EGFR (OR = 0.034, 95% Bonferroni CI = 0.0059, 0.11) as well as TP53 (OR = 0.56, 95% Bonferroni CI = 0.34, 0.92). Notably, KRAS and EGFR are nearly completely mutually exclusive, whereas KRAS and TP53 are negatively correlated but can still occur together. There was no evidence for the associations with EGFR or TP53 differing by the specific KRAS mutation type, from Fisher tests considering the four most common KRAS mutation types at the Bonferroni p = 0.05 level, but there were differences found with STK11 and KEAP1. Subsequent logistic regressions between STK11 or KEAP1 mutations and the specific KRAS mutation types revealed positive associations with G12C, G12V, and G13C mutations for STK11 and with G12C and G13C mutations for KEAP1 (Supplementary Table S4).
We also examined associations between cluster identities and our covariates of interest using univariate regression models (Supplementary Table S6). Patients in Clusters 3, 4, and 5 are much more likely to have a smoking history than patients in Cluster 1; this effect is particularly strong for Cluster 5, in which 46 out of 47 patients are smokers. This is consistent with our prior findings that STK11 and KEAP1 co-mutate with G12C, and that G12C mutations are associated with smoking. Patients in Clusters 4 and 5 were also, on average, 5.9 and 6.4 years older at diagnosis than patients in Cluster 1, respectively. Finally, patients in Cluster 2 are more likely to be Asian, while patients in Clusters 4 and 5 are more likely to be white, when compared to patients in Cluster 1. This is consistent with our previous finding that Asian patients are less likely to have KRAS mutations. There was no association between cluster identity and sex or institution. When additionally controlling for TMB as a sensitivity analysis (Supplementary Table S6), we find the same associations, expect patients in Cluster 5 are no longer significantly older and patients in Clusters 4 and 5 are no longer more likely to be white.
KRAS and survival in the CRC cohort
Supplementary Table S1 summarizes the demographic characteristics of samples with wild-type KRAS and those with KRAS mutations in the CRC cohort. In total, 44% of the cohort had a KRAS mutation of any kind. Of these patients, there was a greater spread of mutated codons than in the NSCLC cohort: 67% had a mutation in codon 12, 19% had a mutation in codon 13, 7% had a mutation in codon 146, 3% had a mutation in codon 61, and 4% had a mutation in another codon.
Associations between having a KRAS mutation and each covariate of interest, as well as the effect of having a KRAS mutation on overall survival, are summarized in Table 2. All models included age, sex, and stage at diagnosis as covariates. Among the covariates examined, there are no associations with KRAS mutations. Although there appears to only be an association with survival among patients who were stage IV at diagnosis, there was no evidence of an interaction effect (interaction HR = 1.08, 95% CI = 0.78, 1.50), using a Cox regression model that also included age at diagnosis and sex as covariates, which suggests the lack of association in the other stages could be attributed to lower sample sizes. We also repeated this analysis when restricting just to samples sequenced within 3 months of diagnosis, with the same result (interaction HR = 1.45, 95% CI = 0.40, 5.24).
Table 2.
Associations between having a KRAS mutation and each covariate of interest, reported as ORs with 95% Bonferroni CIs (corrected over all tested covariates), and effects of having a KRAS mutation on survival in each subgroup, reported as HRs with 95% Bonferroni CIs (corrected within each category of covariates), in the CRC cohort.
| KRAS OR (Association with mutation occurrence and each covariate) | KRAS HR (Association with survival within each subgroup) | |
|---|---|---|
|
| ||
| Institution | ||
| MSK | Referent | 1.22 (0.91, 1.64) |
| DFCI | 0.92 (0.65, 1.30) | 1.71 (1.24, 2.37) |
| VICC | 1.11 (0.69, 1.77) | 1.73 (1.12, 2.67) |
| Sex | ||
| Male | Referent | 1.50 (1.18, 1.92) |
| Female | 1.07 (0.78, 1.46) | 1.46 (1.12, 1.91) |
| Age (years) | 0.99 (0.98, 1.01) | |
| Race | ||
| White | Referent | 1.55 (1.25, 1.93) |
| Black | 1.41 (0.76, 2.52) | 1.00 (0.47, 2.11) |
| Asian | 0.92 (0.45, 1.86) | 1.47 (0.59, 3.69) |
| Other | 0.76 (0.22, 2.35) | 0.74 (0.059, 9.24) |
| Stage | ||
| Stage I | Referent | 1.80 (0.49, 6.62) |
| Stage II | 1.12 (0.53, 2.41) | 1.65 (0.81, 3.35) |
| Stage III | 1.09 (0.55, 2.23) | 1.37 (0.90, 2.07) |
| Stage IV | 1.27 (0.65, 2.54) | 1.49 (1.16, 1.92) |
In the entire sample, KRAS mutations are associated with inferior survival (HR = 1.48, 95% CI = 1.27, 1.74, test for proportional hazards p = 0.22). Because stage at diagnosis could be considered a possible intermediate factor in a causal pathway relating KRAS mutations to survival, we also repeated this analysis with stage at diagnosis excluded from the model, and found a similar association (HR = 1.45, 95% CI = 1.24, 1.70, test for proportional hazards p = 0.095). Next, we examined the effects of specific KRAS mutations on survival. Of the eight mutations occurring in at least ten patients (G12D, G12V, G13D, G12C, A146T, G12S, G12A, and Q61H), we found negative associations with survival for G12D and G12V (test of proportional hazards p = 0.35) (Figure 2A, B).
Figure 2.
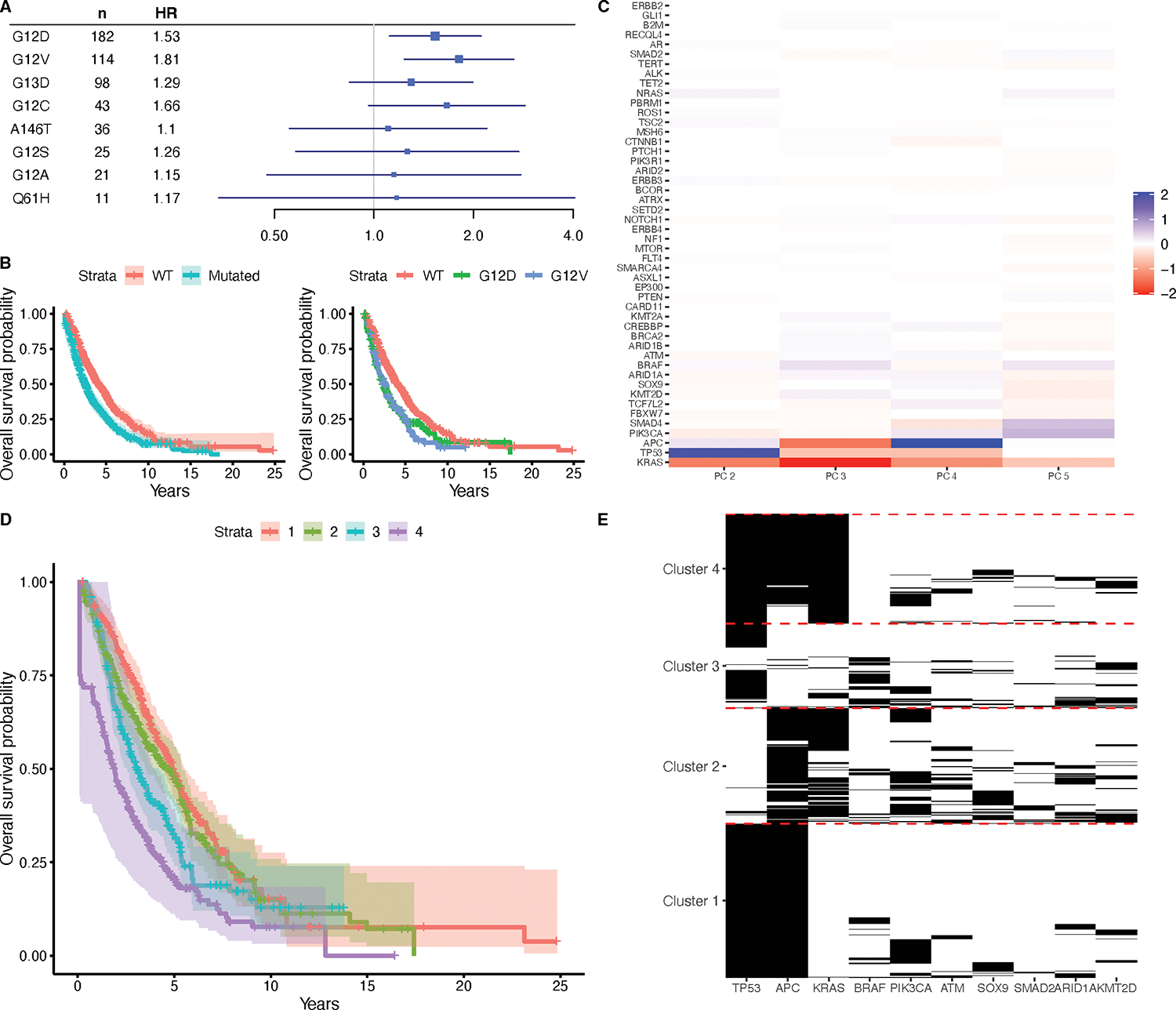
Associations of KRAS mutations with survival in the CRC cohort. (A) HRs with corresponding 95% Bonferroni CIs for each specific KRAS mutation type occurring in at least 10 patients, as assessed via Cox proportional hazards regression models adjusting for age, sex, and stage at diagnosis. (B) Kaplan-Meier curves with 95% confidence intervals for patients with wild-type KRAS and patients with any kind of KRAS mutation, and Kaplan-Meier curves for patients with wild-type KRAS, and patients with G12D or G12V mutations. (C) Loadings matrix for the second through fifth principal component found from applying B-PCA to mutation data in the CRC cohort. (D) Kaplan-Meier curves comparing the patients in four clusters, derived from clustering the patient scores for the principal components. (E) Heatmap showing mutation patterns among the top 10 genes differentially mutated (FDR < 0.05) across the four clusters.
Co-mutations with KRAS and patient clusters in the CRC cohort
We also applied B-PCA to the mutation data from the CRC cohort. Supplementary Table S3 summarizes the demographic characteristics of included and excluded patients from this analysis; notably, all the VICC patients were excluded due to insufficient sequencing of the selected genes in the panels used at this institution. We chose to use five PCs (Figure 2C, Supplementary Table S4), and our resampling strategy confirmed a stable fit (RV coefficients ranged from 0.67 to 0.99, with quartiles of 0.91, 0.93, and 0.97). We then examined the PCs for associations between the corresponding scores and overall survival using the counting process version of a Cox regression model (Supplementary Table S2). This was done as in the NSCLC analysis, but additionally including stage at diagnosis in the model. This revealed a protective effect for PC 4 (HR = 0.93, 95% Bonferroni CI = 0.89, 0.97, test for proportional hazards p = 0.02). Since PC 4 has a negative loading for KRAS, this is again consistent with our general finding that KRAS mutations are associated with inferior survival. This effect remained even after including TMB in the model (Supplementary Table S2).
We again interpreted the mutational patterns in these PCs by clustering patient scores into four clusters, labeled in order of their median survival (Figure 2D). We found that membership in Clusters 2, 3, and 4 are associated with worse survival than Cluster 1 (Cluster 2 HR = 1.39, 95% Bonferroni CI = 1.02, 1.88; Cluster 3 HR = 1.64, 95% Bonferroni CI = 1.20, 2.25; Cluster 4 HR = 1.81, 95% Bonferroni CI = 1.35, 2.42; test for proportional hazards p = 0.086), which was consistent even with TMB included in the model (Supplementary Table S2). This was also assessed as in NSCLC, with the addition of stage at diagnosis in the model. We then again used univariate logistic regressions to assess whether each gene was differentially mutated in each cluster when compared to all other clusters (Supplementary Table S5), and the top 10 genes with significant differential mutations (FDR < 0.05) are visualized in Figure 2E. Most patients in Cluster 1, which had the best survival, have APC and TP53 mutations, but very few have KRAS mutations. By contrast, the patients in Cluster 2 frequently have both APC and KRAS mutations, but rarely with TP53. The patients in Cluster 3 often have just TP53 mutations, and finally, the patients in Cluster 4, which have the worst survival, most often have all three of TP53, APC, and KRAS mutations. These results suggest a complex interplay between KRAS, TP53, and APC mutations, with different combinations associated with different implications for survival.
Finally, we again examined associations between cluster identities and our covariates of interest using univariate regression models, which revealed differences with race and institution that persisted even when including TMB in the model (Supplementary Table S6). In particular, patients in Cluster 4 were more likely to be Black than those in Cluster 1. Although our analysis described earlier did not reveal differences in survival across race when considering KRAS mutations alone, this suggests that there may some kind of association when considering mutational patterns more cohesively. In addition, patients in Cluster 3 were more likely to belong to MSK than DFCI as compared to patients in Cluster 1, but don’t have any other apparent demographic difference. Because institution effects have otherwise been minimal in the analyses presented, this may be due to some subtle difference in patient population.
KRAS and survival in the PDAC cohort
Supplementary Table S1 summarizes the demographic characteristics of samples with wild-type KRAS and those with KRAS mutations in the PDAC cohort, of which 89% had a KRAS mutation of any kind. This is the largest proportion of the three cancer types considered. Of these patients, the majority (91%) specifically had a codon 12 mutation. The next most common was a codon 61 mutation (8%), and the rest of the patients had codon 13 or other mutations.
In Table 3, the associations between having a KRAS mutation and each covariate of interest, as well as the effect of having a KRAS mutation on overall survival, are summarized. All models were adjusted for age, sex, stage at diagnosis, and histology. In the entire sample, KRAS mutations are associated with inferior survival (HR = 1.88, 95% CI = 1.38, 2.55, test for proportional hazards p = 0.48). This association is very similar when removing stage at diagnosis from the model (HR = 1.84, 95% CI = 1.36, 2.49, test for proportional hazards p = 0.21). Among the covariates examined, KRAS mutations are associated with older age at diagnosis (Table 3), with more mutations among older patients. While there were some apparent differences in survival when considering subgroups by institution or race, these have very wide confidence intervals and could be explained by the smaller sample sizes in this cohort, which is less than half the size of the other two tissue types.
Table 3.
Associations between having a KRAS mutation and each covariate of interest, reported as ORs with 95% Bonferroni CIs (corrected over all tested covariates), and effects of having a KRAS mutation on survival in each subgroup, reported as HRs with 95% Bonferroni CIs (corrected within each category of covariates), in the PDAC cohort.
| KRAS OR (Association with mutation occurrence and each covariate) | KRAS HR (Association with survival within each subgroup) | |
|---|---|---|
|
| ||
| Institution | ||
| MSK | Referent | 1.74 (1.13, 2.68) |
| DFCI | 0.97 (0.39, 2.73) | 2.90 (1.00, 8.42) |
| VICC | 1.41 (0.40, 7.83) | 2.79 (0.49, 15.82) |
| Sex | ||
| Male | Referent | 2.05 (1.27, 3.31) |
| Female | 1.26 (0.61, 2.66) | 1.74 (1.04, 2.91) |
| Age (years) | 1.03 (1.00, 1.07) | |
| Race | ||
| White | Referent | 1.89 (1.25, 2.85) |
| Asian | 0.57 (0.15, 3.22) | 1.03 (0.16, 6.73) |
| Black | 1.83 (0.33, 37.60) | 1.40 (0.09, 22.00) |
| Other | 0.75 (0.05, 130.39) | NA |
| Stage | ||
| Stage I | Referent | 2.86 (0.30, 27.70) |
| Stage II | 2.43 (0.48, 9.43) | 1.67 (0.92, 3.01) |
| Stage III | 2.50 (0.40, 14.30) | 3.30 (0.74, 14.77) |
| Stage IV | 3.43 (0.66, 13.92) | 1.67 (0.91, 3.05) |
| Histology | ||
| Adenocarcinoma | Referent | 1.79 (1.25, 2.56) |
| Other | 0.75 (0.18, 5.37) | 7.26 (1.18, 44.63) |
We also examined the effects of specific KRAS mutations on survival in the PDAC cohort (Figure 3A, B). There were five mutations occurring in at least ten patients (G12D, G12V, G12R, Q61H, Q61R), of which G12D, G12V, and G12R all had negative associations with survival (test for proportional hazards p = 0.73). We also assessed for associations between these mutations and age at diagnosis, and did not find any evidence of localization to specific mutation types among those with one of these five mutations (Supplementary Table S2).
Figure 3.
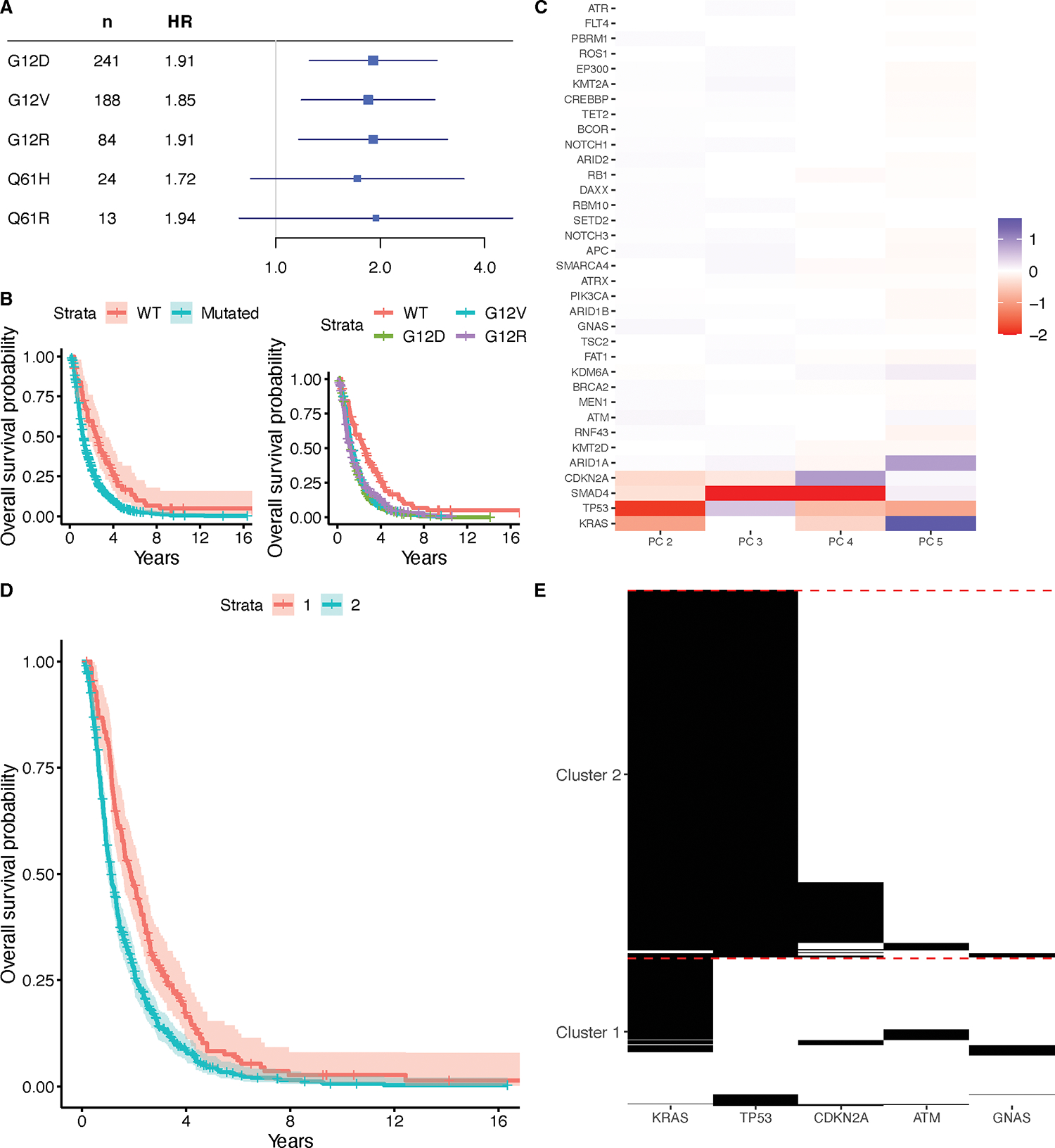
Associations of KRAS mutations with survival in the PDAC cohort. (A) HRs with corresponding 95% Bonferroni CIs for each specific KRAS mutation type occurring in at least 10 patients, as assessed via Cox proportional hazards regression models adjusting for age, sex, histology, and stage at diagnosis. (B) Kaplan-Meier curves with 95% confidence intervals for patients with wild-type KRAS and patients with any kind of KRAS mutation, and Kaplan-Meier curves for patients with wild-type KRAS, and patients with G12V, Q61H, G12D, or G12R mutations. (C) Loadings matrix for the second through fifth principal component found from applying B-PCA to mutation data in the PDAC cohort. Negative loadings are capped at −2 for visualization purposes. (D) Kaplan-Meier curves comparing the patients in two clusters, derived from clustering the patient scores for the principal components. (E) Heatmap showing mutation patterns among the genes with top loadings in each of the two clusters.
Co-mutations with KRAS and patient clusters in the PDAC cohort
We applied B-PCA to the mutation data from the PDAC cohort. Characteristics of the included and excluded patients are summarized in Supplementary Table S3. Although most of the excluded patients were treated at DFCI, due to the selection of genes, there were still a substantial number of DFCI patients remaining in this analysis, and the demographic characteristics were otherwise similar. We chose to use five PCs (Supplementary Table S4), and our resampling strategy confirmed a stable fit (RV coefficients range from 0.82 to 1.00, with all three quartiles at 1.00). The PCs were examined for associations between the corresponding scores and overall survival using the counting process version of a Cox regression model, including age at diagnosis, sex, stage at diagnosis, and histology in the model (Supplementary Table S2), which revealed a protective effect for PC 2 (HR = 0.92, 95% Bonferroni CI = 0.89, 0.99, test for proportional hazards p = 0.0012) and an association with inferior survival with PC 5 (HR = 1.09, 95% Bonferroni CI = 1.01, 1.17, test for proportional hazards p = 0.013) (Figure 3C). PC 2 has a negative loading for KRAS and PC 5 has a positive loading, which are again consistent with our general finding that KRAS mutations are associated with inferior survival. It should be noted that there are possible violations of the proportional hazards assumption here, and so these results should be interpreted with caution. These results remain the same when including TMB as a covariate (Supplementary Table S2).
We clustered patients by their mutational patterns into two groups (Figure 3D) and found that membership in Cluster 2 is associated with worse survival than in Cluster 1 (HR = 1.48, 95% CI = 1.20, 1.81, test for proportional hazards p = 0.049). This was assessed as in CRC, but also including histology in the model; the association persists when including TMB as a covariate (HR = 1.33, 95% CI = 1.07, 1.66, test for proportional hazards p = 0.045). Univariate logistic regression models were again used to identify genes with differential mutations between these two clusters (Supplementary Table S5), with significantly differential genes (FDR < 0.05) visualized in Figure 3E. Patients in Cluster 1 can predominantly be characterized as having KRAS mutations without TP53 mutations, while patients in Cluster 2 predominantly had both KRAS and TP53 mutations, suggesting worse survival when both of these genes are mutated. Finally, we tested for associations between cluster identity and our covariates of interest using univariate regression models, with and without TMB (Supplementary Table S6), which showed no relationships.
Discussion
KRAS mutations are common in the most lethal cancers and have been associated with specific clinical outcomes in some studies, but not in others. For example, patients with advanced CRC who are treated with standard-of-care chemotherapy experience inferior responses if their cancer expresses mutant KRAS (21). Recent work has also described clinical characteristics and genetic interactions with KRAS in multiple cancer types, including the three examined here, which supports many of our findings on allele- and tissue-specific prevalence and co-mutational relationships (22). However, by analyzing the GENIE data, which links genomic information with highly curated clinical attributes, we were able to identify tissue-specific latent factors summarizing complex interactions across genes; form clusters of patient profiles that associate with survival; and glean additional novel insights related to clinical covariates, KRAS mutations, and patient outcomes.
We found a number of clinical outcomes supported by prior studies. In particular, we identified a negative association between KRAS mutations and survival in all three cancer types examined. We further recapitulated tissue- and allele-specific relationships (2) by showing specific associations with G12C and G12V mutations in LUAD (23), G12D and G12V mutations in CRC (21,24–26), and G12D, G12V, and G12R mutations in PDAC (9). One previous study had found codon 13 mutations to result in worse outcomes than codon 12 mutations in NSCLC (27). This finding was not reproduced here, but could be explained by the much smaller sample size of patients with codon 13 mutations in that study, as well as their focus on a specific treatment context.
Association analyses with the presence of KRAS mutations also yielded some interesting results, such as relationships with older age in both NSCLC and PDAC, and additional relationships with smoking history and race in NSCLC. Some, but not all, of these relationships were localized to specific mutation types. Previous studies support these associations in NSCLC (5), namely the increased incidence of KRAS mutations in older patients, smokers, and patients of non-Asian descent, as well as the localization of the association with smoking history to the G12C mutation. There are also some associations that have been previously reported in the literature but not recapitulated here. For example, prior work has found an association between Black race and the presence of KRAS mutations in CRC (8), but we did not find such an association here. This could potentially be attributed to the small sample size of Black patients in this predominantly white study population, leading to lower power in finding such associations. We did, however, find an association with Black race and membership to one of our learned clusters in CRC, which could suggest a specific relationship with combinations of KRAS mutations with those of other genes.
Through a version of principal components analysis adapted to these data, we additionally found tissue-specific genetic interactions and clusters of patient profiles associated with survival. The NSCLC, CRC, and PDAC cohorts were respectively described by five, four, and two clusters each, which summarize complex interactions across KRAS and other genes. In NSCLC, KRAS mutations were almost exclusively seen in the two clusters with worst survival, which were then differentiated primarily by whether TP53 was mutated, or whether STK11 and/or KEAP1 were mutated. Notably, the latter had the worst survival of all the clusters. Though not identical, there are important similarities between these clusters and those reported by (28), which is evidence of a reproducible signal. Moreover, these clustering results suggest both co-mutational and mutually exclusive relationships between KRAS and other genes, confirmed by Fisher tests, which recapitulate previously reported relationships (2,5). This again supports that these clusters constitute a biologically meaningful partitioning of patients. The cluster structures found in CRC and PDAC also centered around interactions with KRAS. The CRC clusters were strongly differentiated by different combinations of three genes, namely KRAS, TP53, and APC. In PDAC, where the majority of patients had KRAS mutations, the two clusters were instead primarily differentiated by whether or not TP53 was also mutated. All together, these clusters shed insight into the tissue-specific combinations of genes critical in distinguishing groups of patients with different survival levels.
The value of these clusters is further underscored by the clinical insights obtained from examining the associated covariates. In NSCLC, cluster membership was associated with smoking history, age, and race, all in ways consistent with our association analyses with the presence of KRAS mutations alone. However, in CRC, cluster membership revealed a new association with race that was not present when only considering KRAS mutations. This suggests clinical relationships that are specific to particular combinations of mutations, and otherwise may not have been uncovered from a simpler analysis. Hence, our approach was able to translate tissue-specific genetic interactions into meaningful patient clusters that connect different sets of mutations to different survival risks and clinical profiles.
It should be noted that a few associations were no longer present when including TMB in the model as a sensitivity analysis. Such results should be interpreted with care. It is possible that those particular findings were confounded by the total number of mutations present in each sample; for example, this could occur if a cluster simply contained all of the patients with the greatest number of mutations, regardless of what those mutations were. However, because we use panel data here and hence the total number of mutations is typically very small, patients in clusters that are enriched or depleted for certain mutations will also likely have differing TMBs from the other clusters. Cluster 4 in the NSCLC analysis was the only one that was no longer associated with survival after conditioning on TMB, but we also observed significant associations between membership in this cluster and the enrichment of specific mutations. As a result, TMB may simply be sharing a common effect with this cluster, resulting in a loss of signal, rather than acting as a true confounder in this analysis. Nevertheless, such findings should be considered carefully.
There are also some important limitations to this analysis that must be acknowledged. First, tumor purity information was not available for the samples in these cohorts, so we were not able to control for this factor in our analysis. This could contribute noise to the data that could in turn increase the variance of our estimates and obscure potential associations. Second, it is important to note that the study population had some imbalances, notably in terms of race. The large majority of patients were white, which means that there may not always have been high power to detect interactions or associations with respect to race.
Third, the negative associations found between KRAS mutations and survival do not automatically imply a causal relationship. In particular, as alluded to above, treatment exposures play a key role here. KRAS mutations are mutually exclusive with or negatively correlated with several genes, such as EGFR, that have targeted treatments available. As a result, the association with inferior survival may be at least partially attributed to the lack of targeted treatments for those without mutations in these genes. Further work is needed to disentangle these effects.
Fourth, the sampling scheme in this cohort poses some statistical challenges. Patient samples were often sequenced months or even years after diagnosis, which introduces left truncation because some patients might have died before they would have hypothetically entered the cohort. We accounted for this by using the counting process version of the Cox proportional hazards regression model, in which the time from diagnosis to sequencing was used as the entry point into the cohort. However, a further complication is that the time of sequencing is not always random; in particular, patients with early stage disease at diagnosis may not have samples sequenced until and if they worsen. This introduces a form of selection bias in which, for instance, patients with early stage disease at diagnosis might never enter the cohort if they never recur. This challenge has been described previously (29), currently without a clear consensus on how to address it. We mitigated the effects of this bias by conducting sensitivity analyses in which we only considered samples sequenced within three months of diagnosis. While this helps prevent some biased conclusions, this approach is also limiting due to the loss of power in the reduced sample. Hence, further methodological development is needed.
Finally, it should be noted that the B-PCA model used here represents a trade-off in model complexity and interpretability. This model has only two parameters – one describing the combinations of genes most relevant to each latent factor, and one describing the contributions of each latent factor to each tumor sample – which enables an explicit and highly interpretable description of the learned latent space. However, it is important to acknowledge that this is a linear model, and hence can only detect linear interactions across genes. In this work, we use B-PCA as an exploratory tool to extract the most relevant such interactions driving variation in the mutation data, to which end we did not find the linearity assumption to be limiting. In particular, our results both recapitulated known genetic interactions and formed clusters of patients highly associated with survival, even though no survival data was inputted to the model. If, however, the goal is to assess and detect any non-linear interactions across genes, future work could instead employ other techniques tailored to this aim. It should also be noted that our approach does not consider temporal events, and thus does not model mutations as sequentially acquired. Extensions of our work could leverage ideas from phylogenetic inference to describe genetic interactions within a temporal process.
Supplementary Material
Acknowledgements
The authors would like to acknowledge the American Association for Cancer Research and its financial and material support in the development of the AACR Project GENIE registry, as well as members of the consortium for their commitment to data sharing. Electronic health records were curated, and patient outcomes were defined using the PRISSMM™ (Pathology, Radiology, Imaging, Signs, Symptoms, BioMarker, Medical Oncology Notes) phenomic data system. PRISSMM™ is a set of phenomic data standards and tools for characterization and communication of structured information about cancer status and treatment outcomes for patients with solid tumors. This work was supported by grants from the NIH (R01CA178017 and R01CA232372 to K.M. Haigis and R35GM131802 and R01HG005220 to R. A. Irizarry), from the NSF (DGE1745303 to I. N. Grabski), and an award from the Cancer Research UK Grand Challenge and the Mark Foundation to the SPECIFICANCER team (of which K.M. Haigis is a member). This work was also supported, in part, by a grant from John and Georgia DallePezze to Memorial Sloan Kettering Cancer Center as well as the Ning Zhao & Ge Li Family Initiative for Lung Cancer Research and New Therapies (to G.J. Riely). Interpretations are the responsibility of study authors.
Footnotes
Author’s Disclosures: Dr. Riely has been an uncompensated consultant to Daiichi, Lilly, Pfizer, Merck, Verastem, Novartis, Flatiron Health, and Mirati. He has institutional research support from Mirati, Lilly, Takeda, Merck, Roche, Pfizer, and Novartis. Dr. Haigis has research support from Novartis.
References
- 1.Haigis KM. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017;3(10):686–97 doi 10.1016/j.trecan.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cook JH, Melloni GEM, Gulhan DC, Park PJ, Haigis KM. The origins and genetic interactions of KRAS mutations are allele- and tissue-specific. Nat Commun 2021;12(1):1808 doi 10.1038/s41467-021-22125-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Poulin EJ, Bera AK, Lu J, Lin YJ, Strasser SD, Paulo JA, et al. Tissue-Specific Oncogenic Activity of KRAS(A146T). Cancer Discov 2019;9(6):738–55 doi 10.1158/2159-8290.CD-18-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson CW, Lin YJ, Reid D, Parker J, Pavlopoulos S, Dischinger P, et al. Isoform-Specific Destabilization of the Active Site Reveals a Molecular Mechanism of Intrinsic Activation of KRas G13D. Cell reports 2019;28(6):1538–50 e7 doi 10.1016/j.celrep.2019.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El Osta B, Behera M, Kim S, Berry LD, Sica G, Pillai RN, et al. Characteristics and Outcomes of Patients With Metastatic KRAS-Mutant Lung Adenocarcinomas: The Lung Cancer Mutation Consortium Experience. J Thorac Oncol 2019;14(5):876–89 doi 10.1016/j.jtho.2019.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roman M, Baraibar I, Lopez I, Nadal E, Rolfo C, Vicent S, et al. KRAS oncogene in non-small cell lung cancer: clinical perspectives on the treatment of an old target. Molecular cancer 2018;17(1):33 doi 10.1186/s12943-018-0789-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao J, Han Y, Li J, Chai R, Bai C. Prognostic value of KRAS/TP53/PIK3CA in non-small cell lung cancer. Oncol Lett 2019;17(3):3233–40 doi 10.3892/ol.2019.10012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scott A, Goffredo P, Ginader T, Hrabe J, Gribovskaja-Rupp I, Kapadia MR, et al. The Impact of KRAS Mutation on the Presentation and Prognosis of Non-Metastatic Colon Cancer: an Analysis from the National Cancer Database. J Gastrointest Surg 2020;24(6):1402–10 doi 10.1007/s11605-020-04543-4. [DOI] [PubMed] [Google Scholar]
- 9.Luo J KRAS mutation in pancreatic cancer. Semin Oncol 2021;48(1):10–8 doi 10.1053/j.seminoncol.2021.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Margonis GA, Kim Y, Spolverato G, Ejaz A, Gupta R, Cosgrove D, et al. Association Between Specific Mutations in KRAS Codon 12 and Colorectal Liver Metastasis. JAMA Surg 2015;150(8):722–9 doi 10.1001/jamasurg.2015.0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imamura Y, Morikawa T, Liao X, Lochhead P, Kuchiba A, Yamauchi M, et al. Specific mutations in KRAS codons 12 and 13, and patient prognosis in 1075 BRAF wild-type colorectal cancers. Clin Cancer Res 2012;18(17):4753–63 doi 10.1158/1078-0432.CCR-11-3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blons H, Emile JF, Le Malicot K, Julie C, Zaanan A, Tabernero J, et al. Prognostic value of KRAS mutations in stage III colon cancer: post hoc analysis of the PETACC8 phase III trial dataset. Ann Oncol 2014;25(12):2378–85 doi 10.1093/annonc/mdu464. [DOI] [PubMed] [Google Scholar]
- 13.Janakiraman M, Vakiani E, Zeng Z, Pratilas CA, Taylor BS, Chitale D, et al. Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer research 2010;70(14):5901–11 doi 10.1158/0008-5472.CAN-10-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taieb J, Le Malicot K, Shi Q, Penault-Llorca F, Bouche O, Tabernero J, et al. Prognostic Value of BRAF and KRAS Mutations in MSI and MSS Stage III Colon Cancer. Journal of the National Cancer Institute 2017;109(5) doi 10.1093/jnci/djw272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Consortium APG. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov 2017;7(8):818–31 doi 10.1158/2159-8290.CD-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lavery JA, Lepisto EM, Brown S, Rizvi H, McCarthy C, LeNoue-Newton M, et al. A Scalable Quality Assurance Process for Curating Oncology Electronic Health Records: The Project GENIE Biopharma Collaborative Approach. JCO Clin Cancer Inform 2022;6:e2100105 doi 10.1200/CCI.21.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown S, Lavery JA, Shen R, Martin AS, Kehl KL, Sweeney SM, et al. Implications of Selection Bias Due to Delayed Study Entry in Clinical Genomic Studies. JAMA Oncol. 2022. Feb 1;8(2):287–291. doi: 10.1001/jamaoncol.2021.5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Townes FW, Hicks SC, Aryee MJ, Irizarry RA. Feature selection and dimension reduction for single-cell RNA-Seq based on a multinomial model. Genome Biol 2019;20(1):295 doi 10.1186/s13059-019-1861-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdi H RV coefficient and congruence coefficient. Encyclopedia of measurement and statistics 2007;849:853. [Google Scholar]
- 20.Cattell RB. The scree test for the number of factors. Multivariate behavioral research. 1966;1(2):245–76 doi 10.1207/s15327906mbr0102_10. [DOI] [PubMed] [Google Scholar]
- 21.Zocche DM, Ramirez C, Fontao FM, Costa LD, Redal MA. Global impact of KRAS mutation patterns in FOLFOX treated metastatic colorectal cancer. Frontiers in genetics 2015;6:116 doi 10.3389/fgene.2015.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JK, Sivakumar S, Schrock AB, Madison R, Fabrizio D, Gjoerup O, et al. Comprehensive pan-cancer genomic landscape of KRAS altered cancers and real-world outcomes in solid tumors. NPJ Precision Oncology 2022;6(1):1–14 doi 10.1038/s41698-022-00334-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. Journal of the National Cancer Institute 2012;104(3):228–39 doi 10.1093/jnci/djr523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allegra CJ, Rumble RB, Hamilton SR, Mangu PB, Roach N, Hantel A, et al. Extended RAS Gene Mutation Testing in Metastatic Colorectal Carcinoma to Predict Response to Anti-Epidermal Growth Factor Receptor Monoclonal Antibody Therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015;34(2):179–185. J Clin Oncol 2015. doi 10.1200/JCO.2015.63.9674. [DOI] [PubMed] [Google Scholar]
- 25.Mao C, Huang YF, Yang ZY, Zheng DY, Chen JZ, Tang JL. KRAS p.G13D mutation and codon 12 mutations are not created equal in predicting clinical outcomes of cetuximab in metastatic colorectal cancer: a systematic review and meta-analysis. Cancer 2013;119(4):714–21 doi 10.1002/cncr.27804. [DOI] [PubMed] [Google Scholar]
- 26.De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-Bianchi A, Tu D, Siena S, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 2010;304(16):1812–20 doi 10.1001/jama.2010.1535. [DOI] [PubMed] [Google Scholar]
- 27.Metro G, Chiari R, Duranti S, Siggillino A, Fischer MJ, Giannarelli D, et al. Impact of specific mutant KRAS on clinical outcome of EGFR-TKI-treated advanced non-small cell lung cancer patients with an EGFR wild type genotype. Lung Cancer 2012;78(1):81–6 doi 10.1016/j.lungcan.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 28.Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. (2015). Co- occurring Genomic Alterations Define Major Subsets of KRAS-Mutant Lung Adenocarcinoma with Distinct Biology, Immune Profiles, and Therapeutic Vulnerabilities. Cancer discovery, 5(8), 860–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kehl KL, Schrag D, Hassett MJ, Uno H. Assessment of Temporal Selection Bias in Genomic Testing in a Cohort of Patients With Cancer. JAMA Netw Open. 2020. Jun 1;3(6):e206976. doi: 10.1001/jamanetworkopen.2020.6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The NSCLC and CRC data are publicly available on Synapse at the Synapse ID syn27056172.
The PANC 1.1-consortium data are available upon request at this link: https://www.synapse.org/#!Synapse:syn26288991.