

Abstract
At least seven cell death programs are activated during myocardial infarction (MI), but which are most important in causing heart damage is not understood. Two of these programs are mitochondrial-dependent necrosis and apoptosis. The canonical function of the pro-cell death BCL-2 family proteins BAX and BAK is to mediate permeabilization of the outer mitochondrial membrane during apoptosis allowing apoptogen release. BAX has also been shown to sensitize cells to mitochondrial-dependent necrosis, although the underlying mechanisms remain ill-defined. Genetic deletion of Bax or both Bax and Bak in mice reduces infarct size following reperfused myocardial infarction (MI/R), but the contribution of BAK itself to cardiomyocyte apoptosis and necrosis and infarction has not been investigated. In this study, we use Bak-deficient mice and isolated adult cardiomyocytes to delineate the role of BAK in the pathogenesis of infarct generation and post-infarct remodeling during MI/R and non-reperfused MI. Generalized homozygous deletion of Bak reduced infarct size ~50% in MI/R in vivo, which was attributable primarily to decreases in necrosis. Protection from necrosis was also observed in BAK-deficient isolated cardiomyocytes suggesting that the cardioprotection from BAK loss in vivo is at least partially cardiomyocyte-autonomous. Interestingly, heterozygous Bak deletion, in which the heart still retains ~28% of wild type BAK levels, reduced infarct size to a similar extent as complete BAK absence. In contrast to MI/R, homozygous Bak deletion did not attenuate acute infarct size or long-term scar size, post-infarct remodeling, cardiac dysfunction, or mortality in non-reperfused MI. We conclude that BAK contributes significantly to cardiomyocyte necrosis and infarct generation during MI/R, while its absence does not appear to impact the pathogenesis of non-reperfused MI. These observations suggest BAK may be a therapeutic target for MI/R and that even partial pharmacological antagonism may provide benefit.
Graphical Abstract
1. Introduction
Ischemic heart disease is the major cause of death worldwide with most deaths attributable to heart failure months to years following myocardial infarction (MI) [1]. Importantly, the major determinant of post-MI heart failure is the amount of damage sustained by the heart in the first 24 hours of infarction (infarct size) [2, 3]. Reperfusion has been firmly established to limit infarct size in humans [4], but its full benefit is undercut by reperfusion injury, reflecting oxidative stress, Ca2+ overload, and cardiomyocyte death [5]. Multiple cell death programs are activated during reperfused MI (MI/R), but which are most critical in causing myocardial damage is unclear [6, 7]. While early studies focused on apoptosis, it is now recognized that various regulated necrosis programs are important. These include mitochondrial permeability transition-dependent necrosis (discussed below) [6, 8–10]; necroptosis (a form of necrosis mediated by a receptor interacting protein kinase 3 (RIPK3) → mixed lineage kinase-like (MLKL) core pathway) [11–16]; ferroptosis (an iron-dependent form of necrosis involving hydroperoxidation of lipids in multiple cell membranes) [17–21]; pyroptosis (a gasdermin-dependent form of necrosis thought to occur mainly in inflammatory cells but which has also been reported in cardiomyocytes [22–24]); and autosis (an autophagy-dependent form of cell death involving the plasma membrane Na+/K+ ATPase) [25–27]. Mechanistic connections among these cell death programs and their functional importance remain poorly understood.
The key regulatory events in mitochondrial-dependent apoptosis and mitochondrial permeability transition-dependent necrosis take place at the outer and inner mitochondrial membranes, respectively. The defining event in apoptosis is mitochondrial outer membrane permeabilization (MOMP), allowing the release of cytochrome c and other apoptogens to the cytoplasm where they promote caspase activation [28, 29]. In contrast, the triggering event in mitochondrial permeability transition-dependent necrosis is Ca2+-induced opening of a channel in the inner mitochondrial membrane, the mitochondrial permeability transition pore (mPTP), whose identity remains unclear [6, 30–32]. The canonical regulators of MOMP are the BCL-2 family of proteins, which has 3 subclasses [28]: The first is comprised of pro-cell death BAX and BAK, highly homologous proteins that homo-and hetero-oligomerize in the outer mitochondrial membrane to promote MOMP. BAX translocates from cytosol to insert into outer mitochondrial membrane during apoptosis, while BAK resides constitutively at this membrane. The second subclass is the BH3-only proteins, which transduce death signals to BAX and BAK by directly interacting with and conformationally activating these proteins. The third subclass is made up of anti-cell death BCL-2 proteins, which both interact with and neutralize BAX and BAK and sequester BH3-only activators. In addition to its canonical role in mitochondrial-dependent apoptosis, BAX has also been demonstrated to sensitize mPTP opening and necrosis, although the precise mechanisms remain unclear [33, 34].
Germ line deletion of Bax has been shown to decrease infarct size during MI/R in isolated perfused hearts [35] and, to a lesser extent, following non-reperfused MI in vivo [36]. Additionally, mice with cardiomyocyte-specific deletion of Bax combined with germ line deletion of Bak manifest smaller infarcts following MI/R in vivo [33, 34]. However, the contribution of BAK itself to acute infarction has not been investigated in any of these models, nor has its possible involvement in post-infarct remodeling. In addition, it remains unknown in any cell type whether, in addition to its canonical role in apoptosis, BAK also regulates necrosis, similar to BAX. To address these questions, this study employed Bak−/− mice and isolated adult cardiomyocytes in models of MI/R and non-reperfused MI.
2. Methods
2.1. Animal use
All animal experiments were performed according to the animal experimental guidelines of the Animal Care and Use Committee of the Albert Einstein College of Medicine and conform with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Anesthesia: For MI/R and non-reperfused MI procedures, a combination of ketamine (100mg/kg IP) and xylazine (10mg/kg IP) was used. For echocardiography, inhaled isoflurane (1–2%) was used. Euthanasia was performed at the conclusion of each experiment using inhaled isoflurane (2%) followed by cervical dislocation. Our protocol also included early euthanasia for any animal in pain or distress, although no animals required this in the studies reported herein.
2.2. Mouse breeding and Bak genotyping
Germline Bak−/− mice were the gift of Drs. Tullia Lindsten and Craig B. Thompson (Memorial Sloan Kettering Cancer Center) [37]. These mice were propagated primarily as homozygotes but periodically bred back to heterozygosity to minimize genetic drift. Wild type mice obtained from heterozygote crosses were bred as homozygotes in parallel to provide controls. PCR of genomic DNA from tail was used for Bak genotyping as follows: primer 1: 5’-GGCTCTTCACCCCTTACATCAG-3’; primer 2: 5’-GTTTAGCGGGCCTGGCAACG-3’; and primer 3: 5’-GCAGCGCATCGCCTTCTATC-3’. PCR conditions are as described (https://www.jax.org/strain/004183). Primers 1 and 2 identify the wild type allele (540 nucleotides); primers 1 and 3 identify the mutant allele (400 nucleotides).
2.3. MI/R and non-reperfused MI models
Male and female, 12-14-week-old, wild type, Bak+/−, and Bak−/−, C57BL/6J mice were studied using previously described models of: (a) MI/R, consisting of 45 min myocardial ischemia followed by 4 h, 24 h, 7 d, 14 d, 21 d, and 28 d reperfusion [38]; and (b) non-reperfused MI, consisting of continuous myocardial ischemia for 24 h, 7 d, 14 d, 21 d, and 28 d [39]. In each case, Ischemia was induced by ligation of the left coronary artery (LCA). Rigor and reproducibility: The surgeon was blinded to genotype in all experiments involving assessment of infarct size. In the MI/R model, our predetermined criteria for inclusion of data from a given mouse in this analysis were: (a) evidence that reperfusion was successful by appearance of a red color in the blanched ischemic zone when the LCA suture was released; and (b) AAR/LV 45–65%. By these criteria, 97% of mice who underwent MI/R were included. In the infarct size experiments reported in Fig. 1C, D, mortality in the MI/R group was 5% (2/40 died) and was equal in wild type (1/20) and Bak−/− (1/20) groups. In the infarct size experiments reported in Fig. 2C, D, mortality in the MI/R group was 10% (3/30 died) and was equal in wild type (1/10), Bak+/− (1/10), and Bak−/− (1/10) groups. In the non-reperfused MI experiments reported in Fig. 5, the mortality was high in the MI group, as expected (see Fig. 4F), but equal in both genotypes. The number of mice studied is specified for each experiment.
Fig. 1. Effect of Bak deletion on infarct size in MI/R.
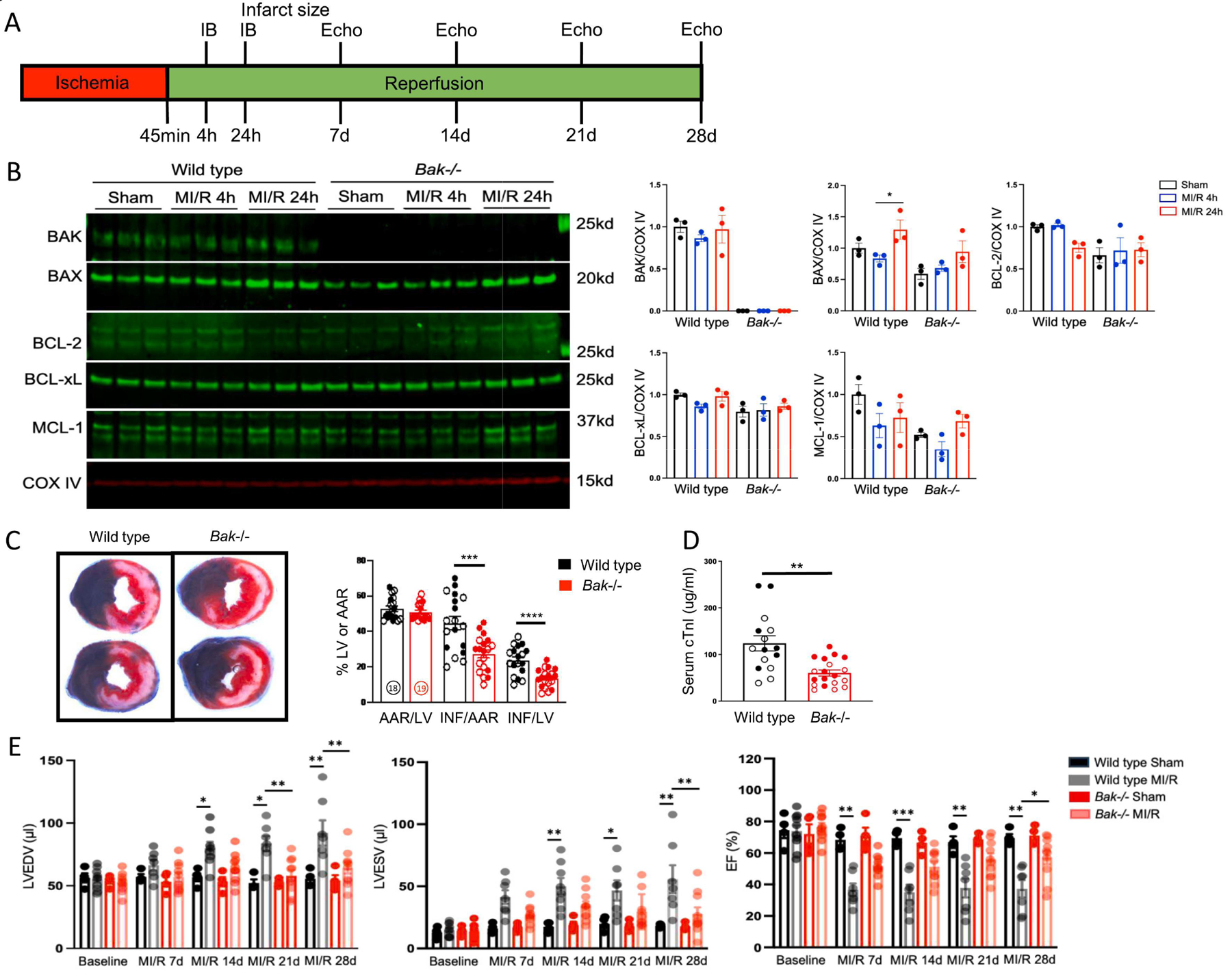
A. Schematic depicting MI/R protocol. B. Western blot analysis and quantification of the indicated proteins in cardiac mitochondria isolated from the left ventricular free wall of wild type and Bak−/− mice subjected to sham operation (harvested 24 h later) (black) or 45 min ischemia followed by 4 h (blue) or 24 h (red) reperfusion (N=3 male mice/group). C. Representative images and quantification of Evans blue dye and tetrazolium chloride-stained hearts from wild type (black; N=18 (9 male and 9 female)) and Bak−/− (red; N=19 (10 male and 9 female)) mice subjected to 45 min ischemia/24 h reperfusion. AAR/LV – area at risk/left ventricle. INF/LV – infarct size/ left ventricle. INF/AAR – infarct/area at risk. Males and females demarcated by filled and open circles, respectively. D. Serum concentrations of cardiac troponin I (cTnI) in wild type (black; N=15 (7 male and 8 female) and Bak−/− (red; N=18 (9 male and 9 female) mice subjected to 45 min ischemia/24 h reperfusion. E. Echocardiographically-determined left ventricular end-diastolic volume (LVEDV), left ventricular end-systolic volume (LVESV), and ejection fraction (EF) in wild type and Bak−/− mice at baseline and following sham operation or MI/R at the indicated time points (N=4 sham for each genotype, 7 wild type MI/R, 10 Bak−/− MI/R male mice). Wall thicknesses are shown in Fig. S2. Data – mean ± SEM. Statistic: Student’s t-test comparing 2 groups; ANOVA with Tukey’s post-test for ≥ 3 groups. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Fig. 2. Effect of partial depletion of BAK on infarct size.
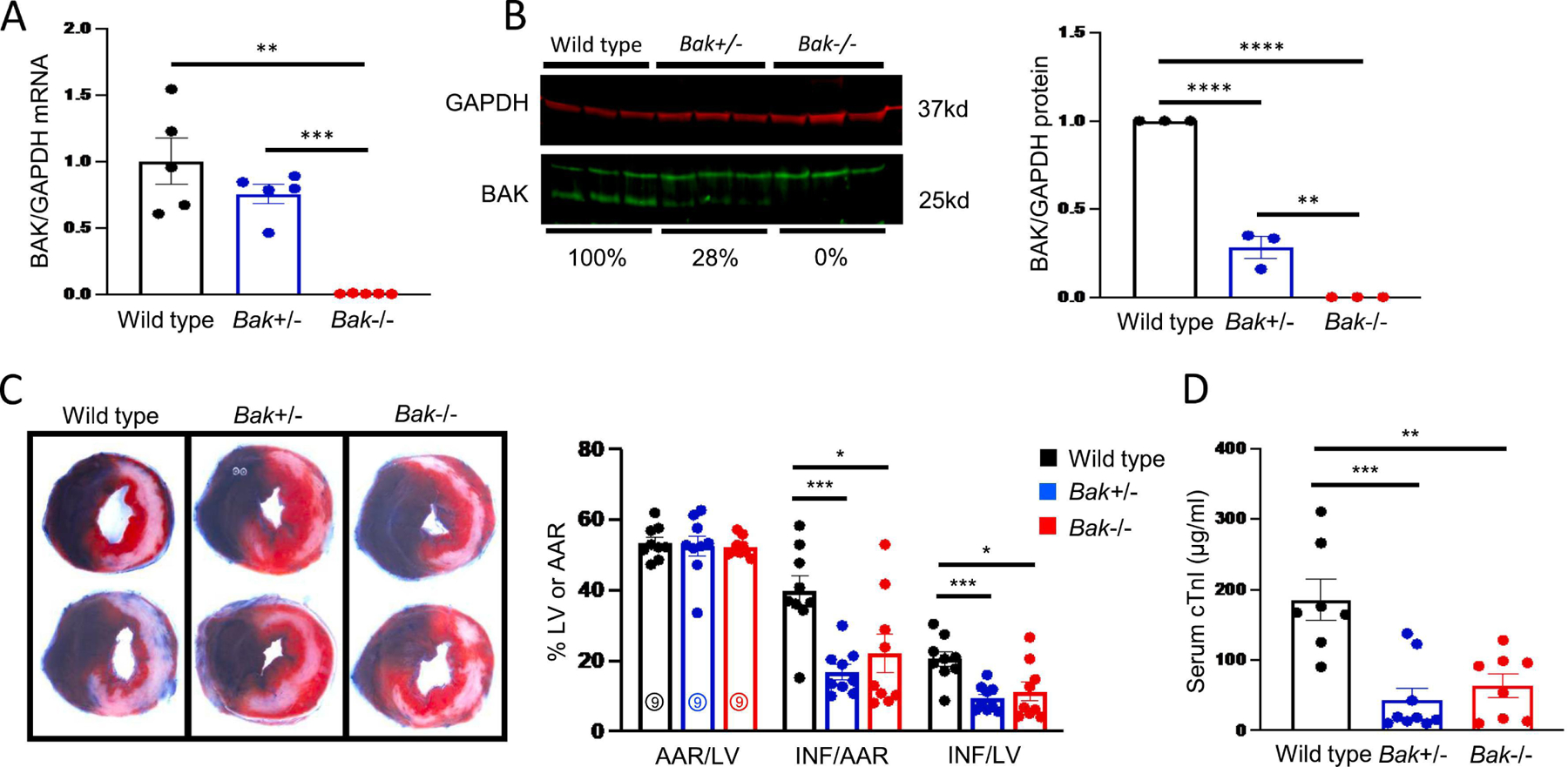
A. Quantification of BAK mRNA using qRT-PCR in hearts of wild type (black), Bak+/− (blue), and Bak−/− (red) mice (N = 5 males/group). B. Quantification of BAK protein levels by Western blot in hearts of wild type (black), Bak+/− (blue), and Bak−/− (red) mice (N = 3 males/group). C. Representative images and quantification of Evans blue dye and tetrazolium chloride-stained hearts from wild type (black; N = 9 males), Bak+/− (blue; N=9 males), and Bak−/− (red; N=9 males) mice subjected to 45 min ischemia/24 h reperfusion. D. Serum concentrations of cTnI in wild type (black; N = 7 males), Bak+/− (blue; N=9 males), and Bak−/− (red; N=8 males) mice following 45 min ischemia/24 h reperfusion. Data – mean ± SEM. Statistic: ANOVA with Tukey’s post-test. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Fig 5. BAK absence does not affect infarct size or post-infarct remodeling in non-reperfused MI.
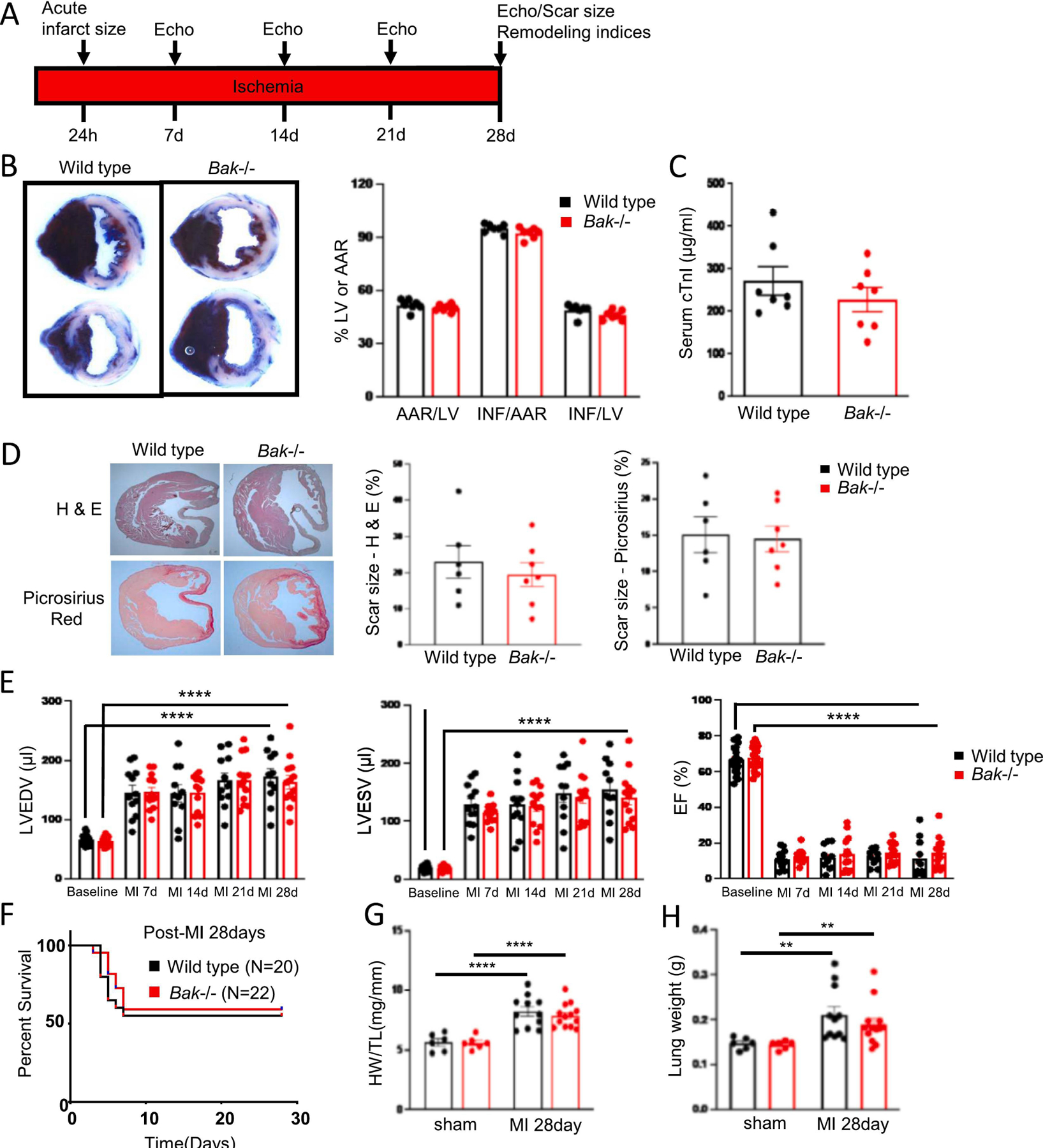
A. Schematic depicting non-reperfused MI protocol. B. Representative images and quantification of Evans blue dye and tetrazolium chloride-stained hearts from wild type (black) and Bak−/− (red) mice subjected to 24 h ischemia without reperfusion (N=7 males/group). C. Serum concentrations of cTnI in wild type (black) and Bak−/− (red) mice following 24 h ischemia without reperfusion (N=7 males/group). D. Cardiac sections stained with H & E and Picrosirius red 28 d post-non-reperfused MI and quantification of scar size in wild type (black; N=6 males) and Bak−/− (red; N=7 males). E. Echocardiographic assessment at baseline, 7 d, 14 d, 21 d, and 28 d post-non-reperfused MI in wild type and Bak−/− mice. (Baseline: wild type (black) N = 20 males, Bak−/− (red) N=22 males; 7 d: wild type N=12 males; Bak−/− 13 males; 14 d, 21 d, and 28 d: wild type N=11 males; Bak−/− N=13 males. Wall thicknesses are shown in Fig. S5. F. Kaplan-Meier survival curve of wild type and Bak−/− mice subjected to non-reperfused MI. At start: wild type (black) N=20 males; Bak−/− (red) N=22 males. G. LV hypertrophy 28 d following sham operation (wild type (black) N=6 males; Bak−/− (red) N=6 males) or non-reperfused MI (wild type (black) N=11 males; Bak−/− (red) N=13 males). H. Lung weights 28 d following sham operation or non-reperfused MI in wild type (black) and Bak−/− (red) mice (numbers of mice same as in G). Data – mean ± SEM. Statistic: Student’s t-test comparing 2 groups; ANOVA with Tukey’s post-test for ≥ 3 groups. ** P < 0.01, **** P < 0.0001.
Fig. 4. Assessment of BAK-mediated necrosis during MI/R.
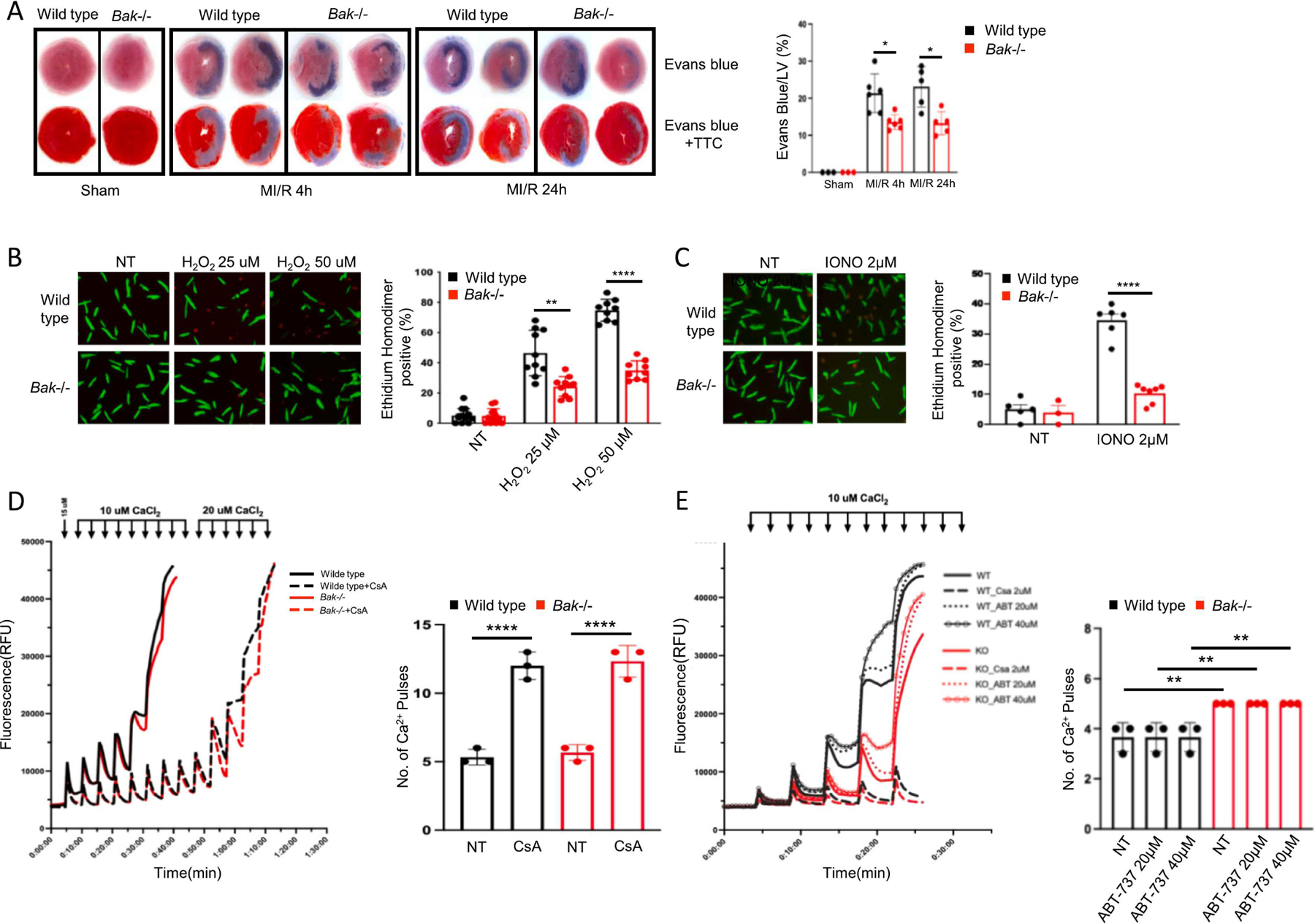
B. Representative images and quantification of necrosis assessed by staining with Evans blue dye administered pre-mortem at the time of reperfusion by itself (top) or combined with post-mortem TTC staining (bottom) in hearts of wild type (black) and Bak−/− (red) mice subjected to sham operation (harvested 24 h later) or 45 min ischemia and 4 h or 24 h reperfusion (N= 3 sham per genotype, 6 MI/R 4 h per genotype, 5 MI/R 24 h per genotype male mice). B. Isolated adult mouse cardiomyocytes from wild type (black) and Bak−/− (red) mice treated with the indicated concentrations of H2O2 and cell death scored 30 min later by ethidium homodimer staining of nucleus. C. Isolated adult mouse cardiomyocytes from wild type (black) and Bak−/− (red) mice treated with the indicated concentration of ionomycin and cell death scored 30 min later with ethidium homodimer. For B. and C, N = 3 independent experiments, each in duplicate with 2–3 fields scored per replicate. D, E. Two independent calcium retention assay experiments using cardiac mitochondria isolated from wild type or Bak−/− mice (N=3 mice of each genotype in each experiment). CsA and ABT-737 denote pretreatment of mitochondria with cyclosporine A (2 μ) or ABT-737 (at the indicated concentration) for 20 min. Data – mean ± SEM. Statistic: ANOVA with Tukey’s post-test. * P < 0.05, ** P < 0.01, **** P < 0.0001.
2.4. Quantification of infarct size by tetrazolium chloride/Evans blue dye staining
Area at risk (AAR) and infarct size (INF) were quantified using Evans blue dye and 2,3,5-triphenyltetrazolium chloride (TTC) staining, respectively, as previously described [38]. Briefly, mice were re-anesthetized, the LCA was reoccluded, and Evans blue dye (5% (w/v) in PBS; 500 μL) infused retrograde into the right carotid artery, thereby staining the non-ischemic myocardium blue. The heart was then rapidly excised and cut into 5 short-axis slices, which were incubated in TTC (1% (w/v)) for 10 min at 37 °C, following which staining was stopped by incubation in ice-cold saline. TTC is reduced by dehydrogenases from a white to red color in viable and metabolically active cells. Both sides of each slice were photographed, and each slice weighed. The percent of total left ventricular (LV) cross-sectional area on each side of the slice that was not blue (AAR) or was white (INF) was quantified using Image J. The values on the two sides were averaged and then a weighted average of all slices calculated based on the mass of each slice to provide AAR/LV, INF/LV, and INF/AAR. The number of mice is specified for each experiment.
2.5. Quantification of infarct size and cardiomyocyte necrosis using serum cardiac troponin I concentration
As a second approach, infarct size was quantified using the serum concentration of cardiac troponin I (cTnI), as is used clinically. The release of cTnI from cardiomyocytes reflects primarily necrosis rather than secretion [40]; thus, serum cTnI concentration provides measures of both infarct size and cardiomyocyte necrosis. Blood was collected from the thoracic cavity after heart excision, centrifuged at 3,000 x g, and serum collected and assayed for cTnI using the High Sensitivity Mouse Cardiac Troponin I ELISA kit (Life Diagnostics) according to the manufacturer’s recommendations. The number of mice studied is specified for each experiment.
2.6. Quantification of myocardial necrosis with Evans blue dye administered pre-mortem
To complement the release of cTnI from cardiomyocytes to blood as a readout of necrosis (above), we also assessed myocardial necrosis by quantifying the staining of heart tissue by Evans blue dye (5 % (w/v), 500 μL) administered intraperitoneally immediately after reperfusion. Evans blue dye is bound by albumin, and tissue staining has been shown to correlate with the entry of dye into necrotic cells [41, 42]. The percentage of LV area stained by Evans blue dye administered pre-mortem was quantified using an approach analogous to that described above for post-mortem quantification using TTC staining. The number of mice studied is specified for each experiment.
2.7. TUNEL
Hearts were fixed in 3.7% formaldehyde in ionized zinc buffer (Z-fix; Anatech), embedded in paraffin, and 5 μM short-axis sections cut. Using sections from the apical third of the heart, apoptotic cells were identified with terminal deoxynucleotidyl transferase dUTP nick end labeling using the DeadEnd Fluorometric TUNEL System [43] (Promega) according to the manufacturer’s recommendations. Cardiomyocytes were identified by morphology and immunostaining for troponin T (Proteintech; 1:200). Nuclei were counterstained with DAPI. For the TUNEL signal to be scored as positive, it needed to co-localize with the DAPI signal. TUNEL-positive nuclei were quantified by manual counting as described below. Total nuclei were quantified using the default algorithms of the Intellesis Trainable Segmentation module of Zen 2.6 Pro software (Carl Zeiss Microscopy), an artificial intelligence-based system trained on different regions of the myocardium to recognize and count DAPI-stained round objects. The number of TUNEL-positive cardiomyocyte and non-cardiomyocyte nuclei/total nuclei was reported. The number of mice studied is specified for each experiment. For each heart, 10 randomly selected fields in the left ventricular free wall (corresponding to the AAR) were evaluated.
2.8. Cleaved caspase-3 immunostaining
Cleaved (active) caspase-3 immunostaining (Cell Signaling; 1:200 dilution) was performed on sections from same hearts used for TUNEL. Cardiomyocytes were identified by staining with wheat germ agglutinin (WGA: Thermofisher). Immunostaining of MEFs that were not treated or treated with staurosporine 2 μM for 2 h and fixed in paraformaldehyde (4% v/v) were used as controls.
2.9. Caspase-3/7 activity in cardiac lysates
Caspase-3/7 activity in homogenates of the left ventricular free (which includes infarct and peri-infarct ones) was assessed using the Caspase-Glo assay kit (Promega). This kit contains a substrate specific for caspases-3/7, carbobenzoxy-VAD, covalently linked with aminoluciferin. When cleaved by caspases-3/7, the aminoluciferin provides a substrate for luciferase resulting in luminescence. Heart homogenates were incubated with kit reagent for 30 min at room temperature according to the manufacturer’s instructions and luminescence measured in a plate-reading luminometer (Thermo Labsystems). The number of mice studied is specified for each experiment. Lysates of mouse embryonic fibroblast (MEFs) that were not treated or treated with staurosporine 2 μM for 4 h (50 μg) were used as controls.
2.10. RNA isolation and qRT-PCR
Following excision of the atria and great vessels, total RNA was extracted from mouse hearts using Trizol Reagent (ThermoFisher) according to manufacturer’s protocol. For baseline characterizations in mice not subjected to surgery, whole hearts homogenates were used. For mice subjected to sham opeartion or MI/R, left ventricular free wall homogenates were used. RNA was reverse transcribed to cDNA using SuperScript III First-Strand Synthesis System (ThermoFisher). The following primer pairs were used for amplification of cDNAs using qPCR: BAK forward 5’- GGGCCACTAATTCCCAGAAA- 3’, reverse 5’- TCCAAAGTAGCAGGAGTGTTG-3’; BAX forward 5’-GGAGATGAACTGGACAGCAATA-3’, reverse 5’- CAAAGTAGAAGAGGGCAACCA-3’; BCL-2 forward 5’-TTGGCCCTTCGGAGTTTAAT-3’, reverse 5’-GTCCTGTGATTCTCCCTTCTTC-3’; BCL-xL forward 5’- TGGACAATGGACTGGTTGAG-3’, reverse 5’- CCCTCTCTGCTTCAGTTTCTT-3’; MCL-1 forward 5’-CTGACTTCCCAGCTCACAAA-3’, reverse 5’- ACTCAGACCACATGCTTCTTC-3’; and GAPDH (internal control) forward 5’- CGACTTCAACAGCAACTC-3’, reverse 5’-GTAGCCGTATTCATTGTCAT-3’. qRT-PCR was performed in triplicate for each sample. Gene expression levels were calculated using the ΔΔCT method. The number of mice studied is specified for each experiment.
2.11. Protein isolation and immunoblotting
Following excision of the atria and great vessels, protein was extracted from heart using RIPA buffer (Thermo Fisher Scientific) supplemented with 10% protease inhibitor cocktail. For baseline characterizations in mice not subjected to surgery, whole hearts homogenates were used. For mice subjected to sham opeartion or MI/R, left ventricular free wall homogenates were used. Following homogenization, samples were centrifuged at 14,000xg for 10 min at 4°C, supernatant collected, and protein concentration measured following a standard BCA method. For isolation of mitochondrial protein, hearts were incubated in sucrose buffer (250 mM sucrose, 50 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 1mM EGTA) and homogenized and then centrifuged at 1000 x g to pellet unlysed cells and nuclei. The resulting supernatant was centrifuged at 10,000 x g, and the pellet containing mitochondria recovered. Protein (50ug) was then fractionated using SDS-PAGE (4–20%) followed by Western blotting. Western blotting was performed using antibodies to BAK (Cell Signaling Technology; dilution 1:500); BAX (Cell Signaling Technology; dilution 1:500); BCL-2 (Cell Signaling Technology; 1:500); BCL-xL (Cell Signaling Technology; dilution 1:1000); MCL-1 (Rockland; dilution 1:1000); cleaved caspase-3 (Cell Signaling; 1:500); COX IV (Thermofisher; dilution 1:5000); α-tubulin (ThermoFisher; dilution 1:1000); and GAPDH (Sigma; dilution 1:5000). Bands were identified using an infrared fluorescence secondary antibody and gels imaged on the Odyssey CLx Infrared Imaging System (LI-COR Biosciences) and quantified with Image J software. The number of mice studied is specified for each experiment.
2.12. Histological staining of hearts and determination of scar size
Hearts were fixed, paraffin-embedded, and sectioned as described above for TUNEL. Hematoxylin & eosin (H & E) staining and Picrosirius red staining were performed on 5 sections per heart spanning from the apex to the base. For each stain, the cross-sectional area of the infarct and the cross-sectional area of the entire LV (excluding cavity) were quantified in each section using Image J. Scar size, using each stain, was then calculated as: [(sum of the 5 infarct areas)/(sum of the 5 LV areas)] X 100 and expressed as a percentage of LV area. The number of mice studied is specified for each experiment.
2.13. Echocardiography
Echocardiography was performed under isoflurane (1–2%) anesthesia on a 37°C heated platform using a Vevo 2100 ultra-high frequency imaging system with a MS550D mouse transducer system (VisualSonics) as previously described [44]. Left ventricular internal dimensions and thicknesses of the interventricular septum and posterior wall were measured at end-diastole and end-systole in the short-axis and long-axis views for three continuous cardiac cycles. Data shown are averages of those in the long-axis view. The number of mice studied is specified for each experiment.
2.14. Induction and quantification of necrosis in isolated adult cardiomyocytes
Adult mouse cardiomyocytes were isolated from wild type and Bak−/− mice as described [45]. Cardiomyocytes were re-suspended in culture medium, seeded onto laminin (5 μg/mL)-coated tissue culture plates, and maintained in a humidified incubator with 5% CO2 at 37°C. Media was replaced with fresh, prewarmed media 1 h later. Four hours after that, cells were treated with H2O2 (25 or 50 μM) or ionomycin (2 μM) for 30 min. Cells were then incubated in calcein-AM (2 μM) and ethidium homodimer (2 μM) and visualized using an Axio Observer.Z1 microscope (Carl Zeiss) at ×100 magnification. Cell death was scored by the ability of ethidium homodimer, a cell-impermeable dye, to enter cells and stain nuclei. Total number of cells were quantified using the cell-permeable dye calcein-AM, which is cleaved by cellular esterases removing the acetomethoxy (AM) moiety and leaving the fluorescent calcein trapped in the cell. The percentage of ethidium homodimer-positive cells was reported. Three independent experiments were performed, each in duplicate. For each replicate, 2–3 randomly selected fields (~20–30 cells per field) were scored.
2.15. Calcium retention capacity assay
Cardiac mitochondria isolated from whole hearts of wild type and Bak−/− mice were placed in a buffer containing 125 mM KCl, 20 mM HEPES, 1 mM EGTA-Tris, 2 mM KH2PO4, 5 mM MgCl2, 15 mM NaCl, 1 mM K2EDTA, 0.1 mM malate, pH 7.1 for 5 min on ice to deplete Ca2+. Following centrifugation at 10,000 X g for 5 min at 4°C, the mitochondria pellet was resuspended in a buffer containing 120 mM KCl, 10 mM Tris-HCl, 5 mM MOPS, 5 mM KH2PO4, 0.01 mM EGTA-Tris, 10 mM glutamate, 5 mM malate, 1 μM Calcium Green-5N, pH 7.4) (ThermoFisher) and pipetted into black clear bottom 96-well plate. Mitochondria were then untreated or treated with 2 μM cyclosporine A (CSA; Sigma) or with 20 or 40 μM ABT-737 for 20 min protected from light. After the incubation period, fluorescence readouts (485 nm/535 nm, Ex/Em) were performed kinetically, at baseline and following addition of 10 mM CaCl2 boluses (2 μl of 1 M CaCl2 into 200 μL in well). This weas done simultaneously with a multichannel pipette and quantification performed using an Infinite M1000 multiwell plate reader (TECAN). This step was repeated until all groups showed a marked increase in Relative Fluorescence Units (RFU) not returning to baseline values, which is indicative of the loss of mitochondrial calcium retention capacity. In each of the two independent experiments reported herein, mitochondria from 3 mice were studied in each condition with ≥ 3 technical replicates for each measurement.
2.16. Statistics
Numbers of mice were chosen based on prior experience with MI/R and unreperfused MI models to be sufficient to identify statistically significant differences in infarct sizes and cell death parameters. Data are expressed as mean ± SEM. Two groups were compared using Student’s t-test. Three or more groups were compared using ANOVA with a Tukey post-test. Kaplan Meier mortality curves were compared using log-rank (Mantel -Cox) test. Differences were considered significant if P < 0.05.
2.17. Data availability
The datasets generated and analyzed in this study are available from the corresponding author upon reasonable request.
3. Results
3.1. Baseline characterization of Bak−/− mice
Homozygous germline inactivation of Bax alone and both Bax and Bak in mice result in mild and severe developmental abnormalities respectively. In contrast, mice with homozygous germline deletion of Bak alone are developmentally normal and fertile [37, 46]. However, before employing Bak−/− mice in our studies, we confirmed that baseline body and organ weights (Table 1) and cardiac dimensions and function by echocardiography (Table 2) were normal. In addition, we confirmed that homozygous deletion of Bak resulted in the absence of detectable BAK mRNA and protein in the heart. In addition, baseline mRNA and protein levels of other pro-cell death (BAX) and anti-cell death (BCL-2, BCL-xL, MCL-1) BCL-2 family members were not changed except minimal decreases in BCL-2 protein in Bak−/− hearts (Fig S1).
Table 1.
Baseline body parameters
| Parameter | Wild type (N=20) | Bak−/− (N=22) | P value |
|---|---|---|---|
| Body weight (g) | 24.5±1.1 | 24.6±0.9 | 0.8165 |
| Tibial length (mm) | 19.3±0.7 | 19.7±0.5 | 0.5039 |
| Heart weight (mg) | 102±3.7 | 106±5.1 | 0.7137 |
| Lung weight (mg) | 132±4.7 | 137±6.3 | 0.5692 |
| Liver weight (mg) | 902±26.7 | 877±31.3 | 0.7774 |
| Spleen weight (mg) | 73±3.5 | 77±2.6 | 0.6932 |
| Kidney weight (mg) | 283±9.1 | 271±7.6 | 0.5637 |
| Brain weight (mg) | 433±17.3 | 426±22.9 | 0.8639 |
Data – mean ± SEM. Statistic: Student’s t-test.
Table 2.
Baseline echocardiographic parameters
| Parameter | Wild type (N=20) | Bak−/− (N=22) | P value |
|---|---|---|---|
| LVEDV (μl) | 65.7±1.8 | 63.2±1.6 | 0.2901 |
| LVESV (μl) | 20.3±1.0 | 19.7±0.9 | 0.6253 |
| EF (%) | 66.7±1.8 | 67.5±1.5 | 0.7284 |
| IVS(d) (mm) | 1.01±0.047 | 0.94±0.019 | 0.2119 |
| IVS(s) (mm) | 1.40±0.095 | 1.40±0.046 | 0.9872 |
| PW(d) (mm) | 0.86±0.072 | 0.86±0.043 | 0.9703 |
| PW(s) (mm) | 1.24±0.082 | 1.23±0.079 | 0.9763 |
LVEDV – left ventricular end diastolic volume; LVESD – left ventricular end systolic volume; EF – ejection fraction; IVS(d) – thickness interventricular septum in diastole; IVS(s) – thickness interventricular septum in systole; PW(d)– thickness posterior wall in diastole; PW(s)– thickness posterior wall in systole. Data – mean ± SEM. Statistics – Student’s t-test. Note: Baseline echocardiographic data in this table is the same data listed under baseline in Figs. 7E and S5.
3.2. BAK is critical for infarct generation during MI/R
To assess the role of BAK in infarct generation during MI/R, we subjected male and female wild type and Bak−/− mice to 45 min of myocardial ischemia induced by left coronary artery occlusion followed by 4 h, 24 h, 7 d, 14 d, 21 d, and 28 d of reperfusion (Fig 1A). We first investigated the effect of MI/R on levels of BAK and other BCL-2 proteins in isolated cardiac mitochondria because BAK resides primarily at mitochondria and the other BCL-2 proteins exert their cell death actions primarily at this organelle (Fig. 1B). Compared with sham operation, 45 min of ischemia followed by 4 h or 24 h reperfusion did not significantly affect levels of BAK, BAX, BCL-2, BCL-xL, or MCL-1, although there were modest increases in mitochondrial BAX levels in both genotypes comparing 24h versus 4 h reperfusion. We next assessed the effect of homozygous Bak deletion on infarct size at 24 h reperfusion, a time point when acute damage is complete (Fig. C, D). While the area at risk was similar between groups, infarct size was reduced 39% by tetrazolium staining (Fig 1C) and 51% by serum concentrationd of cTnI (Fig 1D) in Bak−/− mice as compared with wild type mice. No significant differences were noted between sexes. To determine the effects of reduction in infarct size resulting from BAK absence on cardiac function, we performed a longitudinal study in a separate cohort of mice subjected to sham operation or 45 min ischemia followed by 7d, 14 d, 21 d, and 28 d of reperfusion (Figs 1E and S2). Decreases in ejection fraction were evident in wild type mice as early as 7 d reperfusion and were accompanied by increases in left ventricular end-diastolic volume starting at 14 d reperfusion. Bak deletion resulted in marked improvement in both parameters. Taken together, these data demonstrate that BAK plays an important role in the pathogenesis of MI/R.
3.3. Effect of partial depletion of BAK protein on cardioprotection
To determine if there is a threshold or dose response effect of BAK levels in mediating cardiac damage during MI/R, we compared infarct sizes among wild type, Bak+/−, and Bak−/−. Levels of Bak mRNA and protein in the hearts of Bak+/− mice were 75% and 28%, respectively, of those of wild type mice; and, as previously noted, Bak mRNA and protein were undetectable in hearts of Bak−/− mice (Fig 2A, B). Compared to wild type mice, infarct size, as assessed by tetrazolium staining or serum cTnI concentrations, were reduced to the same extent in Bak+/− and Bak−/− mice (Fig 2C, D). These results demonstrate that 72% loss of BAK protein cardioprotects to the same extent as its complete absence.
3.4. Effect of BAK deletion on apoptosis
The canonical function of BAK and BAX is to homo- and hetero-oligomerize in the outer mitochondrial membrane to bring about MOMP, the defining event in mitochondrial-dependent apoptosis[28]. However, prior studies have demonstrated that BAX also functions to promote necrosis during MI/R as well as in other cell types [33, 34], although the underlying mechanisms remain unclear. Accordingly, we investigated the role of BAK in cardiomyocyte apoptosis and necrosis during MI/R using multiple orthogonal assays.
To assess apoptosis, we began with terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), which assesses the concentration of free 3’-hydroxyl groups in the sugar backbone of DNA [43] using tissues or cells. Increases in TUNEL can reflect internucleosomal DNA cleavage mediated by caspase-activated DNase during apoptosis [47]. Cardiomyocytes were identified by morphology and by co-staining for the cardiomyocyte marker troponin T, the specificity of which was demonstrated in control experiments substituting IgG for the primary antibody (Fig S3). The percentage of TUNEL-positive nuclei was minimal in hearts of wild type and Bak−/− mice subjected to sham operation (Figs 3 and S4). Marked increases in TUNEL-positive nuclei were observed in both cardiomyocytes and non-cardiomyocytes in both the infarct and peri-infarct zones in wild type mice subjected to 45 min ischemia followed by both 4h and 24 h reperfusion. The infarct and peri-infarct zones were differentiated in this analysis by the severe disruption of cardiomyocyte structure and dimming of the troponin T signal in the infarct. Compared to wild type mice, Bak−/− mice subjected MI/R exhibited substantial reductions in the percentage of TUNEL-positive nuclei in both cardiomyocytes and non-myocytes in both infarct and peri-infarct zones (5 mice at each time point).
Fig. 3. Assessment of BAK-mediated apoptosis during MI/R.
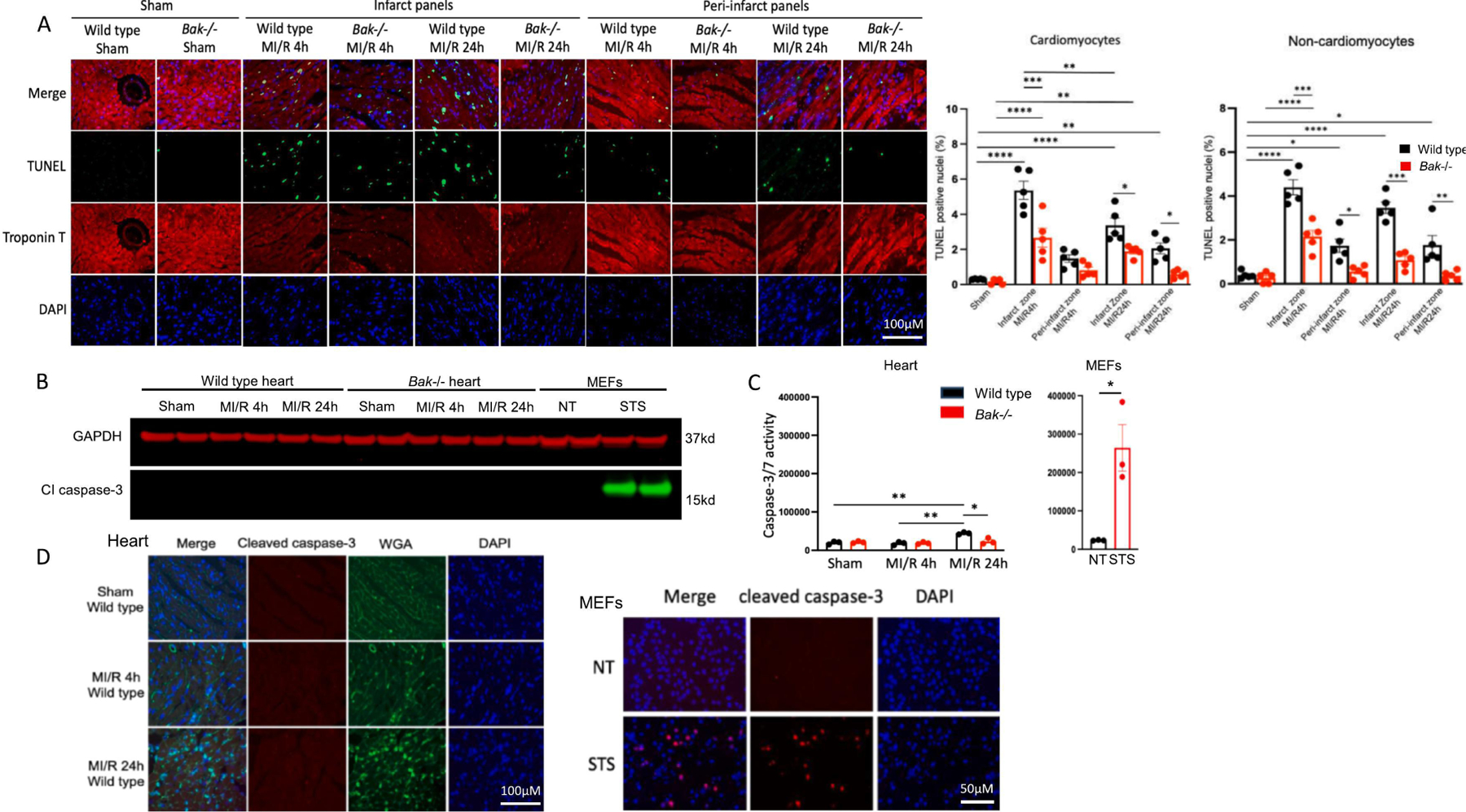
A. Micrographs and quantification of TUNEL in the left ventricular free wall of wild type (black) and Bak−/− (red) mice subjected to sham operation (harvested 24 h later) or 45 min ischemia and 4 h or 24 h reperfusion. Image shown is representative of hearts of N=5 males/group. B. Immunoblot analysis and quantification of cleaved caspase-3 in the left ventricular free wall homogenates of wild type and Bak−/− mice subjected to sham operation (harvest 24 h later) or 45 min ischemia and 4 h or 24 h reperfusion (N=2 males/group) and in MEFs untreated or treated with staurosporine (2 μM) for 4 h. C. Caspase-3/7 enzymatic activity in left ventricular free wall homogenates of wild type (black) and Bak−/− (red) mice (N=3 males/group) and in MEFs untreated or treated with staurosporine (2 μM) for 4 h. D. Cleaved caspase-3 immunostaining in left ventricular free wall of wild type and Bak−/− mice (image shown is representative of hearts of N=5 males/group) and in MEFs untreated or treated with staurosporine (2 μM) for 4 h. Data – mean ± SEM. Statistic: Student’s t-test comparing 2 groups; ANOVA with Tukey’s post-test for ≥ 3 groups. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
These above data could be interpreted as showing that cardiomyocyte and non-myocyte apoptosis during MI/R is BAK-dependent. However, TUNEL is not completely specific for apoptosis [48–51]. Moreover, evolution of concepts in the cell death field and new data pertaining to the heart have led to questions as to whether apoptosis contributes significantly to cardiac cell death and infarct generation during MI/R. Regarding the former, apoptosis was initially thought to occur through both caspase-dependent and independent mechanisms. However, following the recognition of regulated necrosis programs, the current definition of apoptosis requires dependence on effector caspases (caspases-3, −6, and −7) [52]. Additionally, simultaneous deletion of caspases-3 and −7 in mice (Nkx2.5-Cre mediated caspase-3 deletion combined with germ line caspase-7 deletion) has been demonstrated to have no effect on or the percentage of TUNEL-positive cardiomyocytes or infarct size as determined by TTC/Evans blue staining during MI/R [53]. Accordingly, we decided to investigate further before concluding that the decreases in TUNEL during MI/R resulting from Bak deletion prove reduction in BAK-dependent cardiac cell apoptosis.
Since caspase-6 has a limited repertoire of substrates [54], almost all instances of apoptosis involve activation of caspases-3 and/or −7, which have overlapping substrate specificities and cleave multiple cellular proteins [52, 55]. Accordingly, we began by immunoblotting homogenates of the left ventricular free wall, which encompasses the infarct zone, with an antibody that recognizes neoepitopes revealed by caspase-3 cleavage. Cleaved caspase-3 could not be detected in wild type or Bak−/− mice subjected to sham operation or 45 min ischemia followed by 4 h or 24 h reperfusion (Fig. 3B). As a control, we employed MEFs not treated or treated with staurosporine, a small molecule used widely to induce apoptosis. Lysates from MEFs treated with staurosporine exhibited abundant cleaved caspase-3.
We next assessed caspase-3/7 enzymatic activity on the same homogenates. Similar to the immunoblotting results, the enzymatic activity of caspases-3/7 was quite low in wild type and Bak−/− left ventricular free wall homogenates from mice subjected sham operation or to 45 min ischemia followed by 4 h reperfusion (Fig. 3C). While there was a small increase in enzymatic activity at the 24 h reperfusion time point, this was small compared with that in staurosporine-treated MEFs.
Because the immunoblotting and enzymatic activity assays above employ cardiac homogenates, we considered the possibility that these assays might not be sensitive enough to detect signal coming from the more limited population of cells dying within the infarct. Accordingly, we next immunostained heart sections containing the infarct zone for cleaved caspase-3 co-staining with wheat germ agglutinin (WGA) to identify cardiomyocytes. While the WGA staining outlined intact cardiomyocyte sarcolemma in sham operated hearts, plasma membrane structure became progressively more disrupted during ischemia followed by reperfusion of increasing duration (Fig. 3D). Importantly, no cleaved caspase-3 immunostaining was detected in the infarct zone at either reperfusion time point. In contrast, cleaved caspase-3 immunostaining was easily detectable in staurosporine-treated MEFs.
Taken together, these data do not demonstrate caspase-3/7 activation during MI/R in either wild type or Bak−/− mice. While triple knockouts of caspase-3/6/7 will be needed to definitively settle the issue of whether apoptosis plays a role in MI/R, our current data set suggests that the TUNEL signal in cardiomyocytes and non-myocytes during MI/R may not reflect apoptosis. Accordingly, the decreases in TUNEL resulting from Bak deletion cannot be interpreted to indicate that BAK is mediating apoptosis in this model.
3.5. Effect of BAK deletion on necrosis
Serum concentrations of cTnI reflect the release of this contractile protein from cardiomyocytes into the blood. Although some literature suggests that troponin can be secreted, most troponin release during MI/R is thought to reflect loss of cardiomyocyte plasma membrane integrity [40], the defining feature of necrosis, and is the reason that serum cTnI concentrations are used clinically to diagnose MI. The fact that we observed that Bak deletion reduces serum cTnI concentrations during MI (Figs.1D and 2D) suggest that BAK is impacting necrosis. To test this more directly, we injected Evans blue dye intraperitoneally at the time of reperfusion. This dye binds serum proteins, and its uptake by cardiac cells provides a proxy for loss of plasma membrane integrity [41, 42] indicative of necrosis. While no Evans blue staining was observed in the hearts of sham operated mice, we observed Evans blue staining of cardiac sections, which overlapped precisely with absence of tetrazolium staining, in hearts of mice subjected to 45 min ischemia and 4 h or 24 h reperfusion, (Fig. 4A). Deletion of Bak decreased Evans blue staining of these areas by 37% at both time points. These data demonstrate that a significant portion of the necrosis during MI/R is BAK-dependent.
To further investigate whether BAK impacts cardiomyocyte-intrinsic necrosis signaling, we next studied primary adult cardiomyocytes isolated from wild type and Bak−/− mice. These cells were treated with varying concentrations of hydrogen peroxide or the Ca2+ ionophore ionomycin. Cell viability was assessed using loss of plasma membrane integrity at the very early time point of 30 min following treatments, a readout indicative of necrosis . BAK absence reduced both hydrogen peroxide-induced and ionomycin-induced cardiomyocyte necrosis by ~53% (Fig. 4B, C). These data demonstrate that BAK mediates cardiomyocyte necrosis and suggest that at least a portion of the cardioprotection provided by Bak deletion during MI/R in vivo involves cardiomyocyte-intrinsic signaling.
Multiple necrosis programs have been reported to be activated in cardiomyocytes during MI/R including mitochondrial permeability transition-dependent necrosis [6, 8–10], necroptosis [11–16], ferroptosis [17–21], pyroptosis [22–24], and autosis [25–27]. Although mitochondria play potential roles in several, they are most central to mitochondrial permeability transition-dependent necrosis. Because BAK resides primarily at mitochondria, we next investigated the effect of Bak deletion on mitochondrial permeability transition with the calcium retention capacity (CRC) assay performed on isolated cardiac mitochondria from wild type and Bak−/− mice. In the first experiment involving mitochondria from 3 mice per genotype, we observed no difference in the Ca2+ load needed to trigger mitochondrial Ca2+ release. In addition, the same degree of de-sensitization was observed following pretreatment with cyclosporine A, a small molecule that inhibits mitochondrial permeability transition (Fig. 4D). In the second experiment, which also involved 3 mice per genotype, we observed that one extra 10 mM Ca2+ pulse was needed to precipitate permeability transition in the Bak−/−, compared with wild type, mitochondria (Fig. 4E). Although this was statistically significant, we point out that this is a minimal difference in this assay. In an attempt to unmask a more marked difference between wild type and Bak−/− mitochondria, we also included in the second experiment groups pre-treated with ABT-737, a small molecule inhibitor of the anti-cell death proteins BCL-2 and BCL-xL. However, ABT-737 did not sensitize Ca2+-induced permeability transition in mitochondria of either genotype. Taken together, these data indicate that Bak deletion has little to no effect on mitochondrial permeability transition. The interpretation of this finding relative to the ability of BAK absence to limit necrotic cell death is discussed below.
3.6. BAK absence does not reduce infarct size or post-infarct remodeling in non-reperfused MI
We next investigateed whether BAK is critical for infarction generation or post-infarct remodeling in non-reperfused MI. To model the latter, we subjected wild type and Bak−/− mice to permanent LCA occlusion followed by sacrifice at various time points (Fig. 5A). Despite comparable AARs, 24 h of ischemia without reperfusion resulted in much larger infarcts, as expected, than did 45 min ischemia followed by 24 h reperfusion (compare Figs. 5B with 1C). Deletion of Bak did not attenuate infarct size at the 24 h time point as assessed by tetrazolium staining or serum cTnI concentrations (Fig. 5B, C). We also assessed scar size after 28 d of ischemia using hematoxylin and eosin and picrosirius red staining (Fig. 5D). At this time point, scar size reflects both the acute ischemic damage sustained in the first 24 h and the effects of subsequent cardiac remodeling. Scar sizes were similar in wild type and Bak−/− mice. Since cell death has been shown to contribute to cardiac remodeling [56, 57], we also investigated the effect of Bak deletion on cardiac dimensions and function 7 d, 14 d, 21 d and 28 d of ischemia, mouse mortality, and post-mortem heart and lung weights. No differences were observed between wild type and Bak−/− mice (Fig. 5E–I). We conclude that BAK absence does not affect infarct size or post-infarct remodeling and dysfunction in non-reperfused MI.
4. Discussion
This study provides the first evidence that BAK contributes critically to infarct generation during MI/R. Generalized deletion of Bak reduced infarct following 45 min ischemia/24 h reperfusion size by 39% and 51%, as assessed by TTC/Evans blue staining and serum cTnI concentrations, respectively. By comparison, a prior study showed that generalized Bax deletion reduced infarct size by 49% in isolated perfused hearts subjected to 30 min global ischemia/2 h reperfusion [35]. While BAK and BAX exhibit redundancy with respect MOMP in simple cell systems [58], the fact that loss of either BAK or BAX reduces infarct size demonstrate the opposite of redundancy in MI/R. These data suggest that both proteins are needed for a shared function and/or each protein possesses additional distinct – yet to be determined – functions that contribute importantly to MI/R. In a previous study, we observed that combined cardiomyocyte-specific deletion of Bax with generalized deletion of Bak reduced infarct size ~44% [34]. The similar magnitudes of reductions in infarct size with combined Bax/Bak deletion (~44%), individual deletion of Bax (49%), and individual deletion of Bak (~39–51%) favors the hypothesis that both proteins are needed for a shared function rather than each possessing distinct functions important for infarction. However, given the inherent experiment to experiment variability in the MI/R model, direct comparisons of single and double knockouts will be needed to dissect these possibilities precisely.
It was unexpected that Bak+/− mice, whose cardiac levels of BAK are 28% of wild type, would be cardioprotected to the same extent as Bak−/− mice whose hearts lack BAK. This finding suggests a threshold effect in which higher levels of BAK are needed for its full cardiac-damaging effects during MI/R. This finding also has potential therapeutic implications as it suggests that less than complete pharmacological inhibition of BAK may suffice to reduce infarct size.
In addition to its effects on overall infarct size, a second important result in this study is that BAK absence decreases necrotic cell death during MI/R. This was illustrated in vivo by reductions in cTnI released from cardiomyocytes into blood and, conversely, by decreased uptake into cardiac cells of Evans blue dye infused at the time of reperfusion – both assays reflecting plasma membrane dysfunction, the defining feature of necrosis. Additionally, we observed reductions in necrosis in isolated Bak−/− adult cardiomyocytes also using loss of plasma membrane integrity at a very early time point (30 min) as the readout. This is the first demonstration that, similar to BAX [33, 34], BAK mediates necrosis. These isolated cardiomyocyte studies demonstrate cell autonomous effects of BAK loss on cardiomyocyte necrosis suggesting that the cardioprotection resulting from BAK loss during MI/R in vivo is at least partially cardiomyocyte autonomous.
The molecular mechanisms by which BAK and other BCL-2 proteins mediate necrosis are poorly understood and remain an area of active study. Two mechanisms have been advanced for BAX-mediated necrosis including BAX functioning as an outer mitochondrial membrane component of the mitochondrial permeability transition pore [33] and BAX sensitizing necrosis through promoting mitochondrial fusion [34], the latter a known non-cell death function of BAX [59]. Unlike BAX, which resides in an inactive conformation in cytosol and achieves full activation upon translocating to mitochondria, BAK is constitutively localized primarily at mitochondria. Accordingly, we hypothesized that BAK-mediated necrosis may involve the specific cell death program termed mitochondrial permeability transition-dependent necrosis. However, in contrast to the effects of Bax deletion on cardiomyocyte necrosis and infarction during MI/R, our CRC results show that loss of BAK has minimal to no effect on mitochondrial permeability transition. These results are in agreement with a previous study [33]. This observation does not mean that BAK does not contribute to mitochondrial permeability transition because combined loss of BAK and BAX markedly inhibits mitochondrial permeability [33]. Further work will be needed to understand why BAK and BAX are redundant with respect to this mitochondrial permeability transition but not redundant with respect to cell death.
While regulated cell death accounts for a substantial portion of cardiomyocyte loss during MI/R, a role for regulated death programs in non-reperfused MI, while reported [36, 60, 61], remains controversial. Nevertheless, since not all instances of MI in humans are eligible for, or able to be treated with, reperfusion therapy, we also studied homozygous Bak deletion in a non-reperfused context. In contrast to MI/R, we observed that absence of BAK did not reduce infarct size in non-reperfused MI, measured acutely at 24 hours using tetrazolium/Evan blue staining or at 28 d by scar size. Moreover, although regulated cell death has been implicated in the transition to heart failure [56, 57, 61], we observed no effect of BAK absence on post-MI cardiac remodeling or dysfunction.
In summary, we have demonstrated that BAK contributes significantly to cardiomyocyte necrosis and infarct generation during MI/R but is not required for infarct generation or postinfarct remodeling in non-reperfused MI.
Supplementary Material
Fig. S1. Baseline mRNA and protein levels of BCL-2 family proteins in hearts of wild type and Bak−/− mice
A. Quantification of baseline levels of the indicated transcripts by qRT-PCR in hearts of wild type (black) and Bak−/− (red) mice (N = 5 male mice/group). B. Western blot and quantification of baseline levels of the indicated proteins in hearts of wild type (black) and Bak−/− (red) mice. (N=4 male mice/group in top immunoblot and 3 mice/group in bottom immunoblot). Statistic: Student’s t-test. ** P < 0.01, **** P < 0.0001.
Fig. S2. Left ventricular wall thicknesses in wild type and Bak−/− mice in MI/R study in Fig 1E.
Echocardiographically-determined interventricular septal thicknesses in diastole (IVSd) and systole (IVSs) and left ventricular posterior wall thicknesses in diastole (LVPWd) and systole (LVPWs) in wild type and Bak−/− mice at baseline and following sham operation or MI/R at the indicated time points (N=4 sham-operated for each genotype, 7 wild type MI/R, 10 Bak−/− MI/R male mice). Data – mean ± SEM. Statistic: ANOVA with Tukey’s post-test for ≥ 3 groups. * P < 0.05. Remainder of echocardiographic data are shown in Fig. 1E.
Fig. S3. Control immunostaining studies for troponin T and TUNEL
Micrograph of heart section showing negative staining for troponin T when IgG (3.5 μg/ml) substituted for troponin T primary antibody (top). Micrograph of heart section treated with DNase I (10 units/ml) showing positive TUNEL signal (bottom).
Fig. S4. Larger images of TUNEL shown in Fig 3A.
Micrographs of TUNEL-stained nuclei in infarct zone of wild type mice subjected to 45 min ischemia followed by 4 h or 24 h reperfusion to show cardiomyocyte (white arrows) and non-cardiomyocyte (yellow arrows) TUNEL. Note that TUNEL signal (green) overlaps with DAPI (blue) to give an aqua color on the merged image. This image is representative of hearts of N=5 males/group.
Fig. S5. Left ventricular wall thicknesses in wild type and Bak−/− mice in non-reperfused MI study in Fig 5E.
Echocardiographically-determined interventricular septal thicknesses in diastole (IVSd) and systole (IVSs) and left ventricular posterior wall thicknesses in diastole (LVPWd) and systole (LVPWs) in wild type and Bak−/− mice at baseline and following sham operation and non-reperfused MI at the indicated time points. Baseline: wild type (black) N=20 males; Bak−/− (red) N=22 males; 7 d: wild type N= 12 males; Bak−/− 13 males; 14 d, 21 d, and 28 d: wild type N=11 males; Bak−/− N=13 males. Data – mean ± SEM. Statistic: ANOVA with Tukey’s post-test for ≥ 3 groups. **** P < 0.0001. Remainder of echocardiographic data are shown in Fig. 5E.
Highlights.
BAK contributes critically to infarct generation during reperfused MI.
BAK mediates necrosis in reperfused MI.
Partial reduction in BAK levels provides substantial cardioprotection in reperfused MI suggesting that even less than complete BAK antagonism may be therapeutically beneficial.
BAK absence does not impact acute cardiac damage or post-infarct remodeling in non-reperfused myocardial infarction.
Acknowledgements
We thank Joshua L. Axelrod and Felix G. Liang for advice in the experiments.
Funding
This work was supported by NIH grants R01HL164772, R01HL157319, and R01HL159062 to RNK. RNK was partially supported by The Dr. Gerald and Myra Dorros Chair in Cardiovascular Disease. We thank the Wilf Family for their continuing support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
The authors declare no conflicts of interest relating to this work.
References
- [1].Tsao CW, Aday AW, Almarzooq ZI, Anderson CAM, Arora P, Avery CL, Baker-Smith CM, Beaton AZ, Boehme AK, Buxton AE, Commodore-Mensah Y, Elkind MSV, Evenson KR, Eze-Nliam C, Fugar S, Generoso G, Heard DG, Hiremath S, Ho JE, Kalani R, Kazi DS, Ko D, Levine DA, Liu J, Ma J, Magnani JW, Michos ED, Mussolino ME, Navaneethan SD, Parikh NI, Poudel R, Rezk-Hanna M, Roth GA, Shah NS, St-Onge MP, Thacker EL, Virani SS, Voeks JH, Wang NY, Wong ND, Wong SS, Yaffe K, Martin SS, American E Heart Association Council on, C. Prevention Statistics, S. Stroke Statistics, Heart Disease and Stroke Statistics-2023 Update: A Report From the American Heart Association, Circulation (2023). [DOI] [PMC free article] [PubMed]
- [2].Jenca D, Melenovsky V, Stehlik J, Stanek V, Kettner J, Kautzner J, Adamkova V, Wohlfahrt P, Heart failure after myocardial infarction: incidence and predictors, ESC Heart Fail 8(1) (2021) 222–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gerber Y, Weston SA, Enriquez-Sarano M, Berardi C, Chamberlain AM, Manemann SM, Jiang R, Dunlay SM, Roger VL, Mortality Associated With Heart Failure After Myocardial Infarction: A Contemporary Community Perspective, Circ Heart Fail 9(1) (2016) e002460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hausenloy DJ, Yellon DM, Targeting Myocardial Reperfusion Injury--The Search Continues, N Engl J Med 373(11) (2015) 1073–5. [DOI] [PubMed] [Google Scholar]
- [5].Murphy E, Steenbergen C, Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury, Physiol Rev 88(2) (2008) 581–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN, Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease, Physiol Rev 99(4) (2019) 1765–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jia XF, Liang FG, Kitsis RN, Multiple Cell Death Programs Contribute to Myocardial Infarction, Circ Res 129(3) (2021) 397–399. [DOI] [PubMed] [Google Scholar]
- [8].Robichaux DJ, Harata M, Murphy E, Karch J, Mitochondrial permeability transition pore-dependent necrosis, J Mol Cell Cardiol 174 (2023) 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y, Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death, Nature 434(7033) (2005) 652–8. [DOI] [PubMed] [Google Scholar]
- [10].Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD, Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death, Nature 434(7033) (2005) 658–62. [DOI] [PubMed] [Google Scholar]
- [11].Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, Carano RA, Cao TC, van Bruggen N, Bernstein L, Lee WP, Wu X, DeVoss J, Zhang J, Jeet S, Peng I, McKenzie BS, Roose-Girma M, Caplazi P, Diehl L, Webster JD, Vucic D, RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury, Cell Death Differ 23(9) (2016) 1565–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tummers B, Green DR, Mechanisms of TNF-independent RIPK3-mediated cell death, Biochem J 479(19) (2022) 2049–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Weinlich R, Oberst A, Beere HM, Green DR, Necroptosis in development, inflammation and disease, Nat Rev Mol Cell Biol 18(2) (2017) 127–136. [DOI] [PubMed] [Google Scholar]
- [14].Guo X, Chen Y, Liu Q, Necroptosis in heart disease: Molecular mechanisms and therapeutic implications, J Mol Cell Cardiol 169 (2022) 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang T, Zhang Y, Cui MY, Jin L, Wang YM, Lv FX, Liu YL, Zheng W, Shang HB, Zhang J, Zhang M, Wu HK, Guo JJ, Zhang XQ, Hu XL, Cao CM, Xiao RP, CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis, Nat Med 22(2) (2016) 175–182. [DOI] [PubMed] [Google Scholar]
- [16].Parks RJ, Menazza S, Holmstrom KM, Amanakis G, Fergusson M, Ma H, Aponte AM, Bernardi P, Finkel T, Murphy E, Cyclophilin D-mediated regulation of the permeability transition pore is altered in mice lacking the mitochondrial calcium uniporter, Cardiovasc Res 115(2) (2019) 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Stockwell BR, Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications, Cell 185(14) (2022) 2401–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jiang X, Stockwell BR, Conrad M, Ferroptosis: mechanisms, biology and role in disease, Nat Rev Mol Cell Biol 22(4) (2021) 266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Li W, Feng G, Gauthier JM, Lokshina I, Higashikubo R, Evans S, Liu X, Hassan A, Tanaka S, Cicka M, Hsiao HM, Ruiz-Perez D, Bredemeyer A, Gross RW, Mann DL, Tyurina YY, Gelman AE, Kagan VE, Linkermann A, Lavine KJ, Kreisel D, Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation, J Clin Invest 129(6) (2019) 2293–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fang X, Wang H, Han D, Xie E, Yang X, Wei J, Gu S, Gao F, Zhu N, Yin X, Cheng Q, Zhang P, Dai W, Chen J, Yang F, Yang HT, Linkermann A, Gu W, Min J, Wang F, Ferroptosis as a target for protection against cardiomyopathy, Proc Natl Acad Sci U S A 116(7) (2019) 2672–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Baba Y, Higa JK, Shimada BK, Horiuchi KM, Suhara T, Kobayashi M, Woo JD, Aoyagi H, Marh KS, Kitaoka H, Matsui T, Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes, Am J Physiol Heart Circ Physiol 314(3) (2018) H659–H668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F, Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death, Nature 526(7575) (2015) 660–5. [DOI] [PubMed] [Google Scholar]
- [23].Shi H, Gao Y, Dong Z, Yang J, Gao R, Li X, Zhang S, Ma L, Sun X, Wang Z, Zhang F, Hu K, Sun A, Ge J, GSDMD-Mediated Cardiomyocyte Pyroptosis Promotes Myocardial I/R Injury, Circ Res 129(3) (2021) 383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gao Y, Shi H, Dong Z, Zhang F, Sun A, Ge J, Current knowledge of pyroptosis in heart diseases, J Mol Cell Cardiol 171 (2022) 81–89. [DOI] [PubMed] [Google Scholar]
- [25].Nah J, Zhai P, Huang CY, Fernandez AF, Mareedu S, Levine B, Sadoshima J, Upregulation of Rubicon promotes autosis during myocardial ischemia/reperfusion injury, J Clin Invest 130(6) (2020) 2978–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liu Y, Shoji-Kawata S, Sumpter RM Jr., Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, Shaw SY, Clarke PG, Puyal J, Levine B, Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia, Proc Natl Acad Sci U S A 110(51) (2013) 20364–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ikeda S, Zablocki D, Sadoshima J, The role of autophagy in death of cardiomyocytes, J Mol Cell Cardiol 165 (2022) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR, The BCL-2 family reunion, Mol Cell 37(3) (2010) 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kale J, Osterlund EJ, Andrews DW, BCL-2 family proteins: changing partners in the dance towards death, Cell Death Differ 25(1) (2018) 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I, Lippe G, Bernardi P, Dimers of mitochondrial ATP synthase form the permeability transition pore, Proc Natl Acad Sci U S A 110(15) (2013) 5887–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Carroll J, He J, Ding S, Fearnley IM, Walker JE, Persistence of the permeability transition pore in human mitochondria devoid of an assembled ATP synthase, Proc Natl Acad Sci U S A 116(26) (2019) 12816–12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Karch J, Bround MJ, Khalil H, Sargent MA, Latchman N, Terada N, Peixoto PM, Molkentin JD, Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD, Sci Adv 5(8) (2019) eaaw4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM, Martinez-Caballero S, Osinska H, Cheng EH, Robbins J, Kinnally KW, Molkentin JD, Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice, Elife 2 (2013) e00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Whelan RS, Konstantinidis K, Wei AC, Chen Y, Reyna DE, Jha S, Yang Y, Calvert JW, Lindsten T, Thompson CB, Crow MT, Gavathiotis E, Dorn GW, 2nd, B. O'Rourke, R.N. Kitsis, Bax regulates primary necrosis through mitochondrial dynamics, Proc Natl Acad Sci U S A 109(17) (2012) 6566–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hochhauser E, Kivity S, Offen D, Maulik N, Otani H, Barhum Y, Pannet H, Shneyvays V, Shainberg A, Goldshtaub V, Tobar A, Vidne BA, Bax ablation protects against myocardial ischemia-reperfusion injury in transgenic mice, Am J Physiol Heart Circ Physiol 284(6) (2003) H2351–9. [DOI] [PubMed] [Google Scholar]
- [36].Hochhauser E, Cheporko Y, Yasovich N, Pinchas L, Offen D, Barhum Y, Pannet H, Tobar A, Vidne BA, Birk E, Bax deficiency reduces infarct size and improves long-term function after myocardial infarction, Cell Biochem Biophys 47(1) (2007) 11–20. [DOI] [PubMed] [Google Scholar]
- [37].Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, MacGregor GR, Thompson CB, The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues, Mol Cell 6(6) (2000) 1389–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, Kimura H, Chow CW, Lefer DJ, Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function, Proc Natl Acad Sci U S A 104(39) (2007) 15560–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Calvert JW, Elston M, Nicholson CK, Gundewar S, Jha S, Elrod JW, Ramachandran A, Lefer DJ, Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice, Circulation 122(1) (2010) 11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Amgalan D, Pekson R, Kitsis RN, Troponin release following brief myocardial ischemia: apoptosis versus necrosis, JACC Basic Transl Sci 2(2) (2017) 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Miller DL, Li P, Dou C, Armstrong WF, Gordon D, Evans blue staining of cardiomyocytes induced by myocardial contrast echocardiography in rats: evidence for necrosis instead of apoptosis, Ultrasound Med Biol 33(12) (2007) 1988–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yao L, Xue X, Yu P, Ni Y, Chen F, Evans Blue Dye: A Revisit of Its Applications in Biomedicine, Contrast Media Mol Imaging 2018 (2018) 7628037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Gavrieli Y, Sherman Y, Ben-Sasson SA, Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation, J Cell Biol 119(3) (1992) 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dann MM, Clark SQ, Trzaskalski NA, Earl CC, Schepers LE, Pulente SM, Lennord EN, Annamalai K, Gruber JM, Cox AD, Lorenzen-Schmidt I, Seymour R, Kim KH, Goergen CJ, Mulvihill EE, Quantification of murine myocardial infarct size using 2-D and 4-D high-frequency ultrasound, Am J Physiol Heart Circ Physiol 322(3) (2022) H359–H372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ackers-Johnson M, Li PY, Holmes AP, O'Brien SM, Pavlovic D, Foo RS, A Simplified, Langendorff-Free Method for Concomitant Isolation of Viable Cardiac Myocytes and Nonmyocytes From the Adult Mouse Heart, Circ Res 119(8) (2016) 909–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Knudson CM, Tung KS, Tourtellotte WG, Brown GA, Korsmeyer SJ, Bax-deficient mice with lymphoid hyperplasia and male germ cell death, Science 270(5233) (1995) 96–9. [DOI] [PubMed] [Google Scholar]
- [47].Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X, Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade, Cell 91(4) (1997) 479–89. [DOI] [PubMed] [Google Scholar]
- [48].Al-Lamki RS, Skepper JN, Loke YW, King A, Burton GJ, Apoptosis in the early human placental bed and its discrimination from necrosis using the in-situ DNA ligation technique, Hum Reprod 13(12) (1998) 3511–9. [DOI] [PubMed] [Google Scholar]
- [49].Fickert P, Trauner M, Fuchsbichler A, Zollner G, Wagner M, Marschall HU, Zatloukal K, Denk H, Oncosis represents the main type of cell death in mouse models of cholestasis, J Hepatol 42(3) (2005) 378–85. [DOI] [PubMed] [Google Scholar]
- [50].Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R, In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note, Hepatology 21(5) (1995) 1465–8. [DOI] [PubMed] [Google Scholar]
- [51].Saraste A, Morphologic criteria and detection of apoptosis, Herz 24(3) (1999) 189–95. [DOI] [PubMed] [Google Scholar]
- [52].Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, Baehrecke EH, Barlev NA, Bazan NG, Bernassola F, Bertrand MJM, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Boya P, Brenner C, Campanella M, Candi E, Carmona-Gutierrez D, Cecconi F, Chan FK, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Cohen GM, Conrad M, Cubillos-Ruiz JR, Czabotar PE, D'Angiolella V, Dawson TM, Dawson VL, De Laurenzi V, De Maria R, Debatin KM, DeBerardinis RJ, Deshmukh M, Di Daniele N, Di Virgilio F, Dixit VM, Dixon SJ, Duckett CS, Dynlacht BD, El-Deiry WS, Elrod JW, Fimia GM, Fulda S, Garcia-Saez AJ, Garg AD, Garrido C, Gavathiotis E, Golstein P, Gottlieb E, Green DR, Greene LA, Gronemeyer H, Gross A, Hajnoczky G, Hardwick JM, Harris IS, Hengartner MO, Hetz C, Ichijo H, Jaattela M, Joseph B, Jost PJ, Juin PP, Kaiser WJ, Karin M, Kaufmann T, Kepp O, Kimchi A, Kitsis RN, Klionsky DJ, Knight RA, Kumar S, Lee SW, Lemasters JJ, Levine B, Linkermann A, Lipton SA, Lockshin RA, Lopez-Otin C, Lowe SW, Luedde T, Lugli E, MacFarlane M, Madeo F, Malewicz M, Malorni W, Manic G, Marine JC, Martin SJ, Martinou JC, Medema JP, Mehlen P, Meier P, Melino S, Miao EA, Molkentin JD, Moll UM, Munoz-Pinedo C, Nagata S, Nunez G, Oberst A, Oren M, Overholtzer M, Pagano M, Panaretakis T, Pasparakis M, Penninger JM, Pereira DM, Pervaiz S, Peter ME, Piacentini M, Pinton P, Prehn JHM, Puthalakath H, Rabinovich GA, Rehm M, Rizzuto R, Rodrigues CMP, Rubinsztein DC, Rudel T, Ryan KM, Sayan E, Scorrano L, Shao F, Shi Y, Silke J, Simon HU, Sistigu A, Stockwell BR, Strasser A, Szabadkai G, Tait SWG, Tang D, Tavernarakis N, Thorburn A, Tsujimoto Y, Turk B, Vanden Berghe T, Vandenabeele P, Vander Heiden MG, Villunger A, Virgin HW, Vousden KH, Vucic D, Wagner EF, Walczak H, Wallach D, Wang Y, Wells JA, Wood W, Yuan J, Zakeri Z, Zhivotovsky B, Zitvogel L, Melino G, Kroemer G, Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018, Cell Death Differ 25(3) (2018) 486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Inserte J, Cardona M, Poncelas-Nozal M, Hernando V, Vilardosa U, Aluja D, Parra VM, Sanchis D, Garcia-Dorado D, Studies on the role of apoptosis after transient myocardial ischemia: genetic deletion of the executioner caspases-3 and −7 does not limit infarct size and ventricular remodeling, Basic Res Cardiol 111(2) (2016) 18. [DOI] [PubMed] [Google Scholar]
- [54].Cho JH, Lee PY, Son WC, Chi SW, Park BC, Kim JH, Park SG, Identification of the novel substrates for caspase-6 in apoptosis using proteomic approaches, BMB Rep 46(12) (2013) 588–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Pop C, Salvesen GS, Human caspases: activation, specificity, and regulation, J Biol Chem 284(33) (2009) 21777–21781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sun Y, Myocardial repair/remodelling following infarction: roles of local factors, Cardiovasc Res 81(3) (2009) 482–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN, A mechanistic role for cardiac myocyte apoptosis in heart failure, J Clin Invest 111(10) (2003) 1497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ, Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death, Science 292(5517) (2001) 727–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ, Role of Bax and Bak in mitochondrial morphogenesis, Nature 443(7112) (2006) 658–62. [DOI] [PubMed] [Google Scholar]
- [60].Ramachandra CJA, Hernandez-Resendiz S, Crespo-Avilan GE, Lin YH, Hausenloy DJ, Mitochondria in acute myocardial infarction and cardioprotection, EBioMedicine 57 (2020) 102884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zhang Q, Wang L, Wang S, Cheng H, Xu L, Pei G, Wang Y, Fu C, Jiang Y, He C, Wei Q, Signaling pathways and targeted therapy for myocardial infarction, Signal Transduct Target Ther 7(1) (2022) 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Baseline mRNA and protein levels of BCL-2 family proteins in hearts of wild type and Bak−/− mice
A. Quantification of baseline levels of the indicated transcripts by qRT-PCR in hearts of wild type (black) and Bak−/− (red) mice (N = 5 male mice/group). B. Western blot and quantification of baseline levels of the indicated proteins in hearts of wild type (black) and Bak−/− (red) mice. (N=4 male mice/group in top immunoblot and 3 mice/group in bottom immunoblot). Statistic: Student’s t-test. ** P < 0.01, **** P < 0.0001.
Fig. S2. Left ventricular wall thicknesses in wild type and Bak−/− mice in MI/R study in Fig 1E.
Echocardiographically-determined interventricular septal thicknesses in diastole (IVSd) and systole (IVSs) and left ventricular posterior wall thicknesses in diastole (LVPWd) and systole (LVPWs) in wild type and Bak−/− mice at baseline and following sham operation or MI/R at the indicated time points (N=4 sham-operated for each genotype, 7 wild type MI/R, 10 Bak−/− MI/R male mice). Data – mean ± SEM. Statistic: ANOVA with Tukey’s post-test for ≥ 3 groups. * P < 0.05. Remainder of echocardiographic data are shown in Fig. 1E.
Fig. S3. Control immunostaining studies for troponin T and TUNEL
Micrograph of heart section showing negative staining for troponin T when IgG (3.5 μg/ml) substituted for troponin T primary antibody (top). Micrograph of heart section treated with DNase I (10 units/ml) showing positive TUNEL signal (bottom).
Fig. S4. Larger images of TUNEL shown in Fig 3A.
Micrographs of TUNEL-stained nuclei in infarct zone of wild type mice subjected to 45 min ischemia followed by 4 h or 24 h reperfusion to show cardiomyocyte (white arrows) and non-cardiomyocyte (yellow arrows) TUNEL. Note that TUNEL signal (green) overlaps with DAPI (blue) to give an aqua color on the merged image. This image is representative of hearts of N=5 males/group.
Fig. S5. Left ventricular wall thicknesses in wild type and Bak−/− mice in non-reperfused MI study in Fig 5E.
Echocardiographically-determined interventricular septal thicknesses in diastole (IVSd) and systole (IVSs) and left ventricular posterior wall thicknesses in diastole (LVPWd) and systole (LVPWs) in wild type and Bak−/− mice at baseline and following sham operation and non-reperfused MI at the indicated time points. Baseline: wild type (black) N=20 males; Bak−/− (red) N=22 males; 7 d: wild type N= 12 males; Bak−/− 13 males; 14 d, 21 d, and 28 d: wild type N=11 males; Bak−/− N=13 males. Data – mean ± SEM. Statistic: ANOVA with Tukey’s post-test for ≥ 3 groups. **** P < 0.0001. Remainder of echocardiographic data are shown in Fig. 5E.
Data Availability Statement
The datasets generated and analyzed in this study are available from the corresponding author upon reasonable request.