

Summary
Genetic studies have identified numerous loci associated with type 2 diabetes (T2D), but the functional roles of many loci remain unexplored. Here, we engineered isogenic knockout human embryonic stem cell lines for 20 genes associated with type 2 diabetes risk. We examined the impacts of each knockout on β-cell differentiation, functions and survival, and generated gene expression and chromatin accessibility profiles on β cells derived from each knockout line. Analyses of T2D-association signals overlapping HNF4A-dependent ATAC peaks identified a likely causal variant at the FAIM2 T2D-association signal. Additionally, the integrative association analyses identified four genes (CP, RNASE1, PCSK1N and GSTA2) associated with insulin production, and two genes (TAGLN3 and DHRS2) associated with β-cell sensitivity to lipotoxicity. Finally, we leveraged deep ATAC-seq read coverage to assess allele-specific imbalance at variants heterozygous in the parental line and identified a single likely functional variant at each of 23 T2D-association signals.
Graphical Abstract
In brief
Xue et al. engineered and characterized isogenic knockout human embryonic stem cell lines for 20 genes associated with type 2 diabetes risk. Integration of the genomic alterations and subsequent cellular assays in 20 hESC-β cells identifies genes affecting β-cell functionality and putative causal variants for type 2 diabetes.
Introduction
Type 2 diabetes (T2D) is a major contributor to the global burden of disease.1 It is characterized by impaired insulin secretion in pancreatic islet β cells and reduced insulin response in insulin-sensitive tissues.2 Despite success in identifying T2D-associated genetic effects in recent large-scale genetic studies2-5, the challenge of understanding the molecular and cellular mechanisms driving these associations remains difficult.6 In those instances where a candidate effector gene is known, few of these genes have been investigated through detailed functional studies in model systems of disease relevant tissues.7,8 As the catalog of effector genes underlying T2D genetic association9-14 grows, the T2D research community needs efficient model systems to probe the molecular and cellular consequences of perturbations of these genes.
Given the pathophysiology of T2D, much current genetic evidence supports the central role of pancreatic β-cell development and dysfunction in T2D disease progression.9,15 Robust protocols to differentiate human pluripotent stem cells (hPSCs) into insulin producing β-like cells have enabled in vitro model systems to study β-cell development.16-18 Coupled with the advent of flexible gene-editing technologies19, genetically engineered hPSCs promise to be an effective toolkit to investigate the effect of T2D-implicated genes on β-cell dysfunction. Indeed, recent studies have begun to implement this model system to generate isogenic hPSC-derived pancreatic β-like cells and characterize the effect of T2D-implicated genes on β-cell differentiation, function, and survival.20-29 However, these efforts have thus far studied only a limited set of genes, with most studies focusing on one or a few genes at one time. The polygenic nature of T2D demands larger studies to probe candidate effector genes more comprehensively.
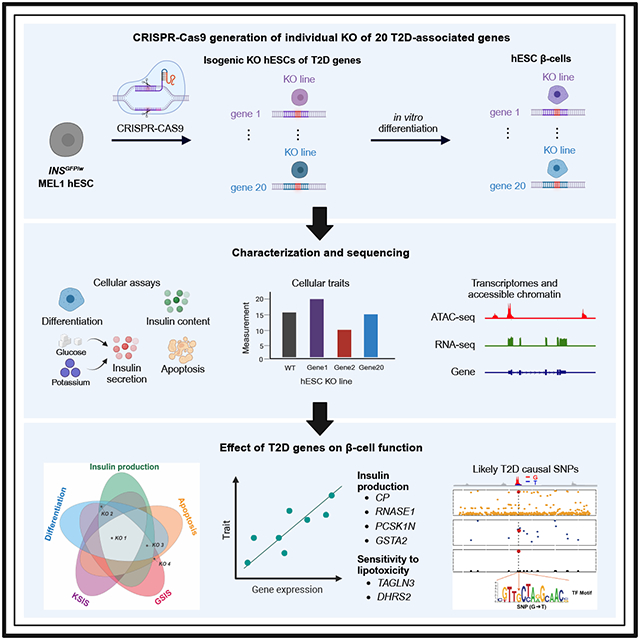
In this study, we employ an efficient CRISPR-based platform to generate isogenic knockout (KO) human embryonic stem cells (hESCs) across 20 T2D risk genes. We differentiate KO hESCs as well as two wildtype (WT) control hESCs into insulin-producing β-like cells (Figure 1A, Table 1). We then assess the effect of each KO across five different cellular phenotypes, including β-cell differentiation efficiency, insulin production and secretion, and β-cell survival after lipotoxic exposure. To understand the molecular mechanisms driving these differences, we generate gene expression and chromatin accessibility profiles of purified insulin-expressing β-like cells and characterize the transcriptional and epigenetic alterations caused by the loss of expression of each of these T2D-associated genes. Integrating functional traits and genomic alterations in 22 hESC-β and WT cells helps pinpoint putative likely causal variants and genes affecting β-cell functionality, providing valuable insights to the genetic architecture of T2D.
Figure 1. Isogenic hESC lines to evaluate the impact of loss of T2D-associated genes in β-cell generation, function, and survival.

(A) Schematic illustration of the experimental design. (B) Representative images of differentiated cells derived from WT and isogenic KO hESCs. Scale bar = 200 μm. (C) Quantification of the percentage of INS-GFP+ cells in the differentiated cells. (D) ELISA analysis of total intracellular insulin content of the purified β-like cells. (E and F) Static GSIS (E) and KSIS (F) of hESC-islet cells derived from WT and isogenic KO hESCs. The percent of insulin content under different stimulation conditions was shown in Figure S3A. (G and H) Representative flow cytometry analysis (G) and the quantification of the percentage of Annexin V+DAPI− cells (H) in INS-GFP+ cells after palmitate treatment. The gating strategy is shown in Figure S3B. (I) Summary of the impact of loss of T2D-associated genes in five cellular traits of hESC-β cells. The dot indicates the gene KO exhibited impairment effects on its overlapping cellular trait. For panels 1C-1F and 1H, data are shown as mean ± SD for two independent clones (#1 and #2) of each hESC line. The number of biological replicates is listed in Table S2. P-values were calculated by one-way ANOVA followed by Dunnet’s test. The n.s. indicates a non-significant difference and * symbol illustrates the significant difference of each KO line compared to the WT line. * P < 0.05, ** P < 0.01, ***P < 0.001, ****P < 0.0001.
Table 1.
20 T2D-associated genes selected for creation of isogenic KO hESCs.
| Gene | Gene description | T2D evidence | Expression (TPM) |
|
|---|---|---|---|---|
| hESC-β cells |
human islet β cells |
|||
| ABCC8 | ATP-binding cassette subfamily C member 8 | T2D knowledge portal effector gene30-33;T2D association from exome-sequencing14 | 53.75 | 31.27 |
| APOE | Apolipoprotein E | T2D knowledge portal effector gene31,30 | 667.30 | 0.62 |
| CDC123 | Cell division cycle protein 123 homolog | T2D knowledge portal effector3,33; T2D association from exome-sequencing14 | 69.85 | 12.08 |
| CDKAL1 | CDK5 Regulatory Subunit Associated Protein 1 Like 1 | T2D knowledge portal effector gene30,33 | 79.59 | 2.14 |
| COBLL1 | Cordon-bleu WH2 repeat protein like 1 | T2D knowledge portal effector gene30,33;T2D association from exome-sequencing14 | 9.39 | 1.07 |
| GCKR | Glucokinase regulatory protein | T2D knowledge portal effector gene31,30,33; T2D association from exome-sequencing14 | 51.29 | 0.01 |
| GIPR | Gastric inhibitory polypeptide receptor | T2D knowledge portal effector gene31,30,33 | 90.23 | 3.23 |
| HNF1A | Hepatocyte nuclear factor 1-alpha | T2D knowledge portal effector gene30-33 | 48.05 | 0.31 |
| HNF4A | Hepatocyte nuclear factor 4-alpha | T2D knowledge portal effector gene30-33 | 102.97 | 0.66 |
| HTT | Huntingtin | T2D knowledge portal effector gene32 | 28.73 | 0.66 |
| IGF2BP2 | Insulin-like growth factor 2 mRNA-binding protein 2 | T2D knowledge portal effector gene3,30-33 | 103.04 | 1.22 |
| KCNJ11 | ATP-sensitive inward rectifier potassium channel 11; Kir6.2 | T2D knowledge portal effector gene3,30-33; T2D association from exome-sequencing14 | 51.03 | 2.50 |
| SLC16A11 | Monocarboxylate transporter 11 | T2D knowledge portal effector gene30-32;T2D association from exome-sequencing14 | 29.52 | 0.67 |
| SLC30A8 | Zinc transporter 8 | T2D knowledge portal effector gene30-33; T2D association from exome-sequencing14 | 73.95 | 71.32 |
| TCF7L2 | transcription factor 7 like 2 | T2D knowledge portal effector gene3,30-32 | 18.96 | 3.42 |
| TGFB1 | Transforming growth factor β-1 | T2D association from exome-sequencing14 | 58.31 | 5.49 |
| TLE4 | Transducin-like enhancer protein 4 | Nearest gene to T2D association6 | 10.26 | 1.96 |
| TMCC2 | Transmembrane and coiled-coil domains protein 2 | T2D knowledge portal effector gene32; T2D association from exome-sequencing14 | 7.93 | 0.89 |
| WDR13 | WD repeat-containing protein 13 | T2D association from exome-sequencing14 | 32.36 | 12.38 |
| WFS1 | Wolframin | T2D knowledge portal effector gene3,32,33; T2D association from exome-sequencing14 | 163.18 | 7.38 |
| PDX1 | Pancreatic and duodenal homeobox 1 | Reference β-cell gene | 167.28 | 17.36 |
Results
Generation and functional characterization of isogenic T2D-KO hESC lines
We selected candidate T2D effector genes with various degrees of evidence in recent T2D genetic studies3,6,14,30-33, and prioritized 20 genes which showed detective expression in both primary human islet β cells and hESC-derived β cells (Table 1). We generated isogenic knockout lines using an INSGFP/w MEL1 hESC reporter line34 that enables the isolation of insulin-expressing cells by fluorescence-activated cell sorting (FACS). Two isogenic clones (labeled as #1 and #2), carrying either homozygous or compound heterozygous frameshift mutations (Figure S1), were identified for each T2D gene. For all target genes, we documented loss of function (LoF) mutations on both copies of the chromosome—except for CDC123, which was heterozygous LoF (CDC123 is a cell cycle protein; homozygous LoF would impair cell division35,36). As controls for subsequent analyses, we selected two wild type (WT) clones—one of which was exposed to Cas9 without the targeting sgRNA, the other of which was the unexposed INSGFP/w MEL1 cells. We confirmed that each clone retained typical hESC colony morphology and expressed pluripotency markers, including OCT4, SSEA4, NANOG, and TRA-1-81 (Figure S2A).
We differentiated the 42 hESC lines (2 WT lines, 20 KO lines with 2 biological replicate clones of each KO line; Table S2) into pancreatic β-like cells (hESC-β cells). We performed live-cell imaging and observed variable representation of INS-GFP+ cells in KO lines (Figure 1B), suggesting that some of the T2D-associated genes affect differentiation. We further quantified the percent of GFP+ cells using flow cytometry and found that the COBLL1−/−, GIPR−/−, HNF4A−/−, TCF7L2−/−, TGFB1−/−, and TLE4−/− lines with impaired differentiation efficiency (P-value<0.05; Figure 1C, Figure S2B). Notably, for the TCF7L2−/− line, the effect on differentiation efficiency was so severe that we dropped this line from some of the subsequent functional experiments that required many β-like cells (e.g., insulin secretion assays and apoptosis assays). To rule out off-target effects, we performed whole genome sequencing of two TCF7L2−/− clones and two WT controls and did not detect any clonal mutations in 250 bp flanking regions of 2,513 predicted CRISPR-Cas9 off-target cleavage sites. Meanwhile, we further examined the stepwise differentiation of TCF7L2−/− hESCs and found that loss of TCF7L2 damages definitive endoderm development (Figure S2C-E). The compromised differentiation of the definitive endoderm in TCF7L2−/− cells would inevitably hinder the subsequent differentiation toward pancreatic β cells.
Next, we measured three insulin-related cellular traits: (i) insulin production; (ii) glucose stimulated insulin secretion (GSIS), and (iii) KCl stimulated insulin secretion (KSIS) on β cells derived from WT and KO hESCs. First, we measured the total intracellular insulin content in purified INS-GFP+ hESC-β cells, and detected decreased total intracellular insulin content in ABCC8−/−, APOE−/−, CDKAL1−/−, COBLL1−/−, GIPR−/−, HNF1A−/−, HNF4A−/−, HTT−/−, IGF2BP2−/−, SLC16A11−/−, TCF7L2−/−, TGFB1−/−, WDR13−/−, and WFS1−/− cells (P-value<0.05; Figure 1D). Next, we differentiated all of the lines apart from TCF7L2−/− into islet-like organoids (hESC-islets) and assessed insulin secretion index after stimulation with 20 mM glucose or with 30 mM KCl (Figure S3A). Multiple KO hESC-islets exhibited impaired response to high glucose (P-value<0.05; Figure 1E) while only ABCC8−/−, HNF1A−/−, HNF4A−/−, HTT−/−, KCNJ11−/− and WDR13−/− hESC-islets showed defective insulin secretion in response to KCl stimulation (P-value<0.05; Figure 1F).
As a final cellular phenotype, we evaluated the apoptotic rate of WT and mutant hESC-β by quantifying the population of AnnexinV+DAPI− cells in INS-GFP+ cells (Figure S3B). Under regular cell culture conditions, we did not observe differences in β-cell survival between WT and KO hESC-β cells (Figure S3C). Given the importance of pancreatic β-cell death induced by lipid accumulation and stress in the context of T2D,37,38 we then assayed the apoptotic rate of WT and mutant hESC-β cells after exposing cells to 1 mM palmitate for 3 days. Comparing the KO lines to WT lines, we observed increased palmitate-induced β-cell apoptosis in ABCC8−/−, APOE−/−, CDC123+/−, CDKAL1−/−, COBLL1−/−, GIPR−/−, HNF1A−/−, HNF4A−/−, HTT−/−, IGF2BP2−/−, SLC16A11−/−, TLE4−/−, TMCC2−/−, WDR13−/−, and WFS1−/− hESC-β cells while detected a decreased apoptotic rate in SLC30A8−/− hESC-β cells (PP-value<0.05; Figure 1G-H). Overall, 19 out of the 20 T2D-associated genes showed an impaired effect in at least one of the five cellular assays considered, supporting our hypothesis that these 20 genes may affect T2D risk in part by perturbing the generation, function, and survival of pancreatic β cells. Strikingly, the loss of HNF4A affected all the five cellular traits, suggesting a particularly prominent role of HNF4A in the development and function of pancreatic β cells (Figure 1I).
Knockout of T2D genes results in large-scale transcriptomic and chromatin accessibility changes
To profile the transcriptomic and chromatin accessibility changes of each KO compared with WT, we purified INS-GFP+ hESC-β cells derived from one KO clone for each T2D gene along with the two WT lines and performed RNA-seq as well as ATAC-seq (Table S2). Focusing on the differentially expressed genes (DEGs; false discovery rate [FDR]<5% and ∣fold change [FC]∣>1.5), the range of the number of DEGs varied widely, from 295 genes in SLC30A8−/− hESC-β cells to 5,969 genes in HNF4A−/− hESC-β cells (Figure 2A). We found 171 out of 257 genes previously reported as T2D effector genes3,30-33 were DEGs in at least one line (Figure S4A), including PPARG, PAX4, and NEUROG3. In addition, we estimated the enrichment of DEGs in genes binned by their expression specificity in primary islet β cells compared to other islet cell types39. We observed that genes with expression profiles highly specific to β cells were enriched (FDR<5%) in DEGs for 17/20 of the KO lines (Figure 2B), underscoring the relevance of hESC-β cells as a model for primary islet β cells. These β-cell specific genes (Figure S4B and S4C) also included many well-characterized T2D genes, such as G6PC240 and NKX6-141. In addition, some disallowed genes that are typically repressed in mature adult β cells42, such as SLC16A143 and HSD11B144, were found to be upregulated in some KO lines (Figure S4D). This suggests alterations of gene regulatory networks and potential functional defects in those mutant β cells.
Figure 2. Loss of T2D associated genes results in large scale transcriptomic and epigenetic changes in hESC-β cells.

(A) Summary of differential gene expression (blue) and differential chromatin accessibility (red) in β-like cells. (B) Enrichment of DEGs in β-cell specific genes. (C) Correlation of DEGs and DARs. (D) Enrichment of DEGs around DARs in varying sizes of windows. (E) Distribution of accessible chromatin regions associated with nearby gene expression. For panels 2B and 2D, we applied the Benjamini-Hochberg procedure to correct for multiple hypotheses testing across all KO lines and highlighted the enrichment at FDR< 0.05 with triangles.
By comparing ATAC-seq data from KO lines to WT lines, we also identified differentially accessible chromatin regions (DARs; FDR<5% and ∣FC∣>1.5; Figure 2A). As with the DEG results, HNF4A−/− exhibited the greatest number of DARs (39,013; Figure 2A). Indeed, across all KO lines, the proportion of DARs identified was strongly correlated with the proportion of DEGs identified (Pearson’s r=0.71, P-value=4.1x10−4; Figure 2C). We found that DEGs were enriched near DARs up to 100 kb away (FDR<5%) in all but two KO lines, SLC30A8−/− and WDR13−/− (Figure 2D). At closer distances (<=25 kb), all lines showed substantial enrichment (FDR<5%). Finally, we considered the distance between each DAR and the nearest transcription start site (TSS) and observed that a large proportion of DARs (>21.7%) occur within 25 kb of a TSS (Figure 2E). We tested for an enrichment of HNF4A−/− suppressed DARs with HNF4A TF footprints around 25kb flanking regions of TSSs of DEGs and showed an abundance of overlap (P-value=1.01x10−5; Figure S4E).
To identify potential regulatory elements in hESC-β cells, we fit a regression to link accessible chromatin regions to nearby genes (<50kb) by jointly modeling ATAC-seq and RNA-seq signals across all 22 lines (KO+WT). We identified 1,150 associations (FDR<5%) spanning 726 genes and 1,035 accessible chromatin regions. While most genes were associated with a single chromatin region, we found that a few genes were associated with as many as 10 open chromatin peaks (Figure S4F). The same trend held true for accessible chromatin regions (Figure S4G). Notably, we identify chromatin regions associated with established T2D effector genes (e.g., TMEM176A/B; Figure S4H) and genes important in β-cell identity (e.g., NKX6-1; Figure S4H).
HNF4A regulates diabetes-relevant genes and HNF4A binding sites are perturbed by genetic variants associated with T2D
Across all KO lines, HNF4A−/− affected all five functional readouts compared to WT lines (Figure 1I), induced the greatest number of transcriptional and epigenomic changes compared to the WT lines (Figure 2A, 3A, 3B), and resulted in DEGs with the greatest enrichment in β-cell specific expression patterns (Figure 2B). We therefore further explored the role of HNF4A in the context of (i) hESC-β cell gene regulation and (ii) the genetics of T2D. First, we found that genes down-regulated in HNF4A−/− compared to WT were enriched (FDR<5%) in genes related to glucose metabolism and maturity onset diabetes of the young (MODY; Figure 3C), while up-regulated genes were enriched in processes not clearly relevant to diabetes; Figure S5A). Next, we expanded our characterization of the regulatory patterns of HNF4A−/− DARs. We observed that 58% of the 39,013 DARs in the line were suppressed while 42% were activated (Figure 3B). Among all the KO lines, HNF4A−/− showed the largest percentage of DEGs around DARs, with 79% of DEGs having a DAR within 50kb (P-value=3.1x10−10; Figure 2D). We hypothesized that such results may indicate that HNF4A−/− DARs occur in regulatory elements that may drive the observed changes in gene expression. We observed strong enrichment (FDR<0.05) for suppressed DARs in HNF4A−/− KO in islet enhancers and active promoters45 (Figure 3D)—notably islet stretch enhancers, which generally regulate tissue/cell-type specific gene expression46 - while we did not find such an enrichment for activated DARs. We scanned the HNF4A−/− DARs using binding site motifs for 677 transcription factors expressed in WT hESC-β cells and found that the suppressed DARs were most enriched in the HNF4A binding motif (FDR<5%; Figure 3E), while the activated DARs were most enriched in the FOXA1 binding motif (FDR<5%; Figure S5B). For the suppressed DARs the HNF4A binding motif most often occurred at the center of the region while for activated DARs there was no such trend (Figure 3F and Figure S5C). These results suggest that many of the suppressed DARs reflect direct changes due to binding of HNF4A, while many activated DARs reflect indirect effects resulting from the HNF4A knockout.
Figure 3. Fine mapping analysis of transcriptomic and epigenomic alterations in HNF4A−/− hESC-β cells prioritize a causal variant rs7132908 at a T2D risk locus.

(A) DEGs of the HNF4A−/− versus WT INS-GFP+ cells. Genes associated at FDR<0.05 and ∣FC∣ > 1.5 are highlighted with blue (down regulated) or red (up regulated). (B) DARs of the purified HNF4A−/− versus WT INS-GFP+ cells. Chromatin accessible regions associated at FDR<0.05 and ∣FC∣>1.5 are highlighted with blue (lost accessibility) or red (gained accessibility). (C) KEGG pathways enriched with down-regulated genes in HNF4A−/− versus WT INS-GFP+ cells (FDR<0.05). (D) Overlap of DARs in the HNF4A−/− versus WT INS-GFP+ cells with islet regulatory features defined by ChromHMM45. Counts of overlapping DARs were adjusted by total number of respective regulatory regions (number of DARs overlapping a regulatory feature x 10,000/total number of regulatory regions). (E) Enrichment of transcription factor binding site motifs in suppressed DARs of the HNF4A−/− hESC-β cells. Right panel shows the top 20 most enriched TFBSs. (F) Relative distance of HNF4A TFBSs from the center of suppressed DARs in the HNF4A−/− versus WT hESC-β cells. TFBS motif abundance was generated by scanning 150bp flanking regions around centers of all suppressed DARs. (G) T2D credible set of SNPs at a locus on chromosome 12 near FAIM2. rs7132908 overlaps a DAR and the A allele disrupts an HNF4A binding site. Top panel shows ATAC-seq (red) and RNA-seq (blue) read pileups in WT and HNF4A−/− hESC-β cells. “T2D_credible” shows two T2D credible set SNPs (height of the bar represents PPA). (H) Luciferase analysis to assess the functionality of the two credible set SNPs and an empty vector in EndoC-βH1 cell. Data was shown as mean ± SD. There are 3 biological replicates for each experimental group and 4 biological replicates for the empty vector control group. Unpaired Student’s t-test: ** P < 0.01.
Given the wide-spread diabetes-relevant effects of HNF4A−/− on hESC-β cells, we investigated if predicted HNF4A binding sites may be perturbed by candidate causal variants within 99% credible sets for genetic associations with T2D3. We focused on the suppressed DARs in HNF4A−/− compared to the WT lines and observed 64 out of 22,710 suppressed DARs overlap 90 credible SNPs, representing 57 T2D association signals. Two of these SNPs (rs7132908 and rs34033101), in two different T2D signals, are predicted to affect HNF4A TF footprints (Table S3). We selected the T2D genetic association signal at FAIM23 for experimental follow-up. The FAIM2 GWAS signal contained two credible SNPs: rs7132908 (MAF=0.25; posterior probability of association [PPA]=0.92) and rs3205718 (MAF=0.25; PPA=0.07). Of these two SNPs, only rs7132908 overlaps an HNF4A footprint, where the T2D risk allele, “A”, is predicted to disrupt HNF4A binding (Figure 3G). This HNF4A footprint occurred in a suppressed DAR in HNF4A−/− and was not associated with expression of FAIM2 or any other nearby gene (FDR>5%), making the effector gene unknown at this signal. We performed allele-specific luciferase assays for both variants in EndoC-βH1 cells, a human pancreatic β-cell line47. We observed a differential change in luciferase activity of the alleles for rs7132908 (P-value=0.002) —the “A” allele of rs7132908 was associated with increased luciferase activity—but not for rs3205718 (P-value=0.618; Figure 3H). Combined, these data suggest that rs7132908 is likely the causal variant at this T2D signal and that the “A” allele increases T2D risk by decreasing HNF4A binding and increasing the strength of an enhancer. In this situation, it thus appears that HNF4A is acting as a repressor.
Association between gene expression and cellular traits identifies genes controlling insulin production and β-cell survival
By comparing WT lines to KO lines spanning 20 genes, we identified downstream effects of T2D-relevant genes on β-cell cellular traits (Figure 1C-H), gene expression (Figure 2A), and chromatin accessibility (Figure 2A). In addition, the availability of cellular traits paired with -omics measurements across gene perturbations created a dataset where one could begin to map regulatory networks for cellular traits. Across all cell lines with paired cellular trait and -omics data, we jointly modeled gene expression and chromatin accessibility with each cellular trait. We identified 21 genes associated with insulin content and 35 genes associated with β-cell apoptotic rate after palmitate exposure (FDR<5% and ∣effect size∣>1.5, Table S4).
Focusing on the 21 genes associated with total insulin content (Figure 4A, Table S4), we selected five protein-coding genes (Figure 4B-F) to test for a causal relationship with insulin content based on the effect size of the association (∣effect size∣>1.5) and the gene’s expression in human islets (TPM>5) and hESC-β cells (TPM>5). Using EndoC-βH1 cells, we perturbed the expression of these candidate genes by inhibiting the expression of genes positively correlated with insulin content—CP and FOSB—through CRISPR interference (CRISPRi) and activating the expression of genes negatively correlated—RNASE1, PCSK1N, and GSTA2—through CRISPR activation (CRISPRa). Prior to testing for effects on insulin content, we confirmed the reduced or activated expression of the five genes in correspondingly perturbed EndoC-βH1 cells (Figure S6A). For 4/5 of the selected genes, we observed the predicted effect on total insulin content, where the inhibition of CP and the activation of RNASE1, PCSK1N, and GSTA2 decreased total insulin content (P-value<0.05; Figure 4G-H). For FOSB inhibition, there was no notable impact observed on the total insulin content of EndoC-βH1 cells (P-value=0.32). This suggests that the correlation between FOSB and total insulin content, as observed in hESC-β cells, may not stem from a causal relationship, or that EndoC-βH1 cells may not be an ideal model for detecting this effect.
Figure 4. Cellular trait association analysis identifies potential genes controlling insulin content.

(A) Identification of genes associated with total insulin content in hESC-β cells. Genes associated at FDR<0.05 and ∣effect size∣>1.5 are colored (negative: blue, positive: red). (B-F) Linear regression analysis of total insulin content in WT or KO INS-GFP+ cells with RNA expression of candidate gene CP (B), FOSB (C), PCSK1N (D), GSTA2 (E) and RNASE1 (F). The solid line and gray area indicate the regression line and 95% confidence interval (CI), respectively. (G) Total insulin content of EndoC-βH1 cells with transcriptional inhibition of CP or FOSB. N=3 biological replicates. (H) Total insulin content of EndoC-βH1 cells with transcriptional activation of RNASE1, PCSK1N, or GSTA2. N=3 biological replicates. (I) Relative expression of INS mRNA in EndoC-βH1 cells with transcriptional inhibition of CP. N=3 biological replicates. (J) Relative expression of INS mRNA in EndoC-βH1 cells with transcriptional activation of RNASE1, PCSK1N, or GSTA2. N=3 biological replicates. (K) Relative luciferase intensity of EndoC- βH1-luc cells with transcriptional inhibition of CP. N=3 biological replicates. (L) Relative luciferase intensity of EndoC- βH1-luc cells with transcriptional activation of RNASE1, PCSK1N, or GSTA2. Nano-luc intensity indicates the c-peptide content. N=3 biological replicates. For panels 4G-4L, data are shown as mean ± SD. P-values were calculated by unpaired Student’s t-test. The n.s. indicates a non-significant difference and * symbol illustrates the significant difference of each genetic perturbation line compared to the control line. * P < 0.05, ** P < 0.01, ***P < 0.001, ****P < 0.0001.
To better understand the molecular mechanisms underlying the observed effects of CP, RNASE1, PCSK1N, and GSTA2 on total insulin content, we conducted similar CRISPR perturbation experiments and measured (i) INS transcription and (ii) insulin protein translation/processing in EndoC-βH1-luc cells. For INS transcription, we used qRT-PCR in EndoC-βH1 cells, revealing that decreased CP expression and increased RNASE1 expression resulted in lower INS expression (P-value<0.05) (Figure 4I, 4J). To assess insulin protein translation/processing, we used EndoC-βH1-luc cells, in which Nano-Glo luciferase (Nano-luc) could be released via endogenous proinsulin convertase enzymes and its intensity could be used as a readout to track the change of proinsulin transgene translation and processing48. We first confirmed the reduced expression of CP and activated expression of RNASE1, PCSK1N, and GSTA2 in EndoC-βH1-luc cells using qRT-PCR (Figure S6B). Then Nano-luc assay showed that inhibition of CP had a mild effect on insulin protein translation/processing, reducing luciferase intensity by only ~10% (P-value<0.05; Figure 4K), while activation of PCSK1N, GSTA2, and RNASE1 greatly reduced intracellular Nano-luc production (P-value<0.05; Figure 4L). Collectively, these experiments indicate that PCSK1N and GSTA2 influence insulin production primarily by modulating insulin protein translation or processing, whereas CP and RNASE1 have implications for both INS transcription and downstream translation/processing processes.
In addition, from the 35 genes associated with palmitate-induced β-cell apoptotic rate (Figure 5A, Table S4), we selected five protein-coding genes to test for a causal relationship with palmitate-induced apoptotic rate based on the effect size of the association (∣effectsize∣>1.5) and the gene’s expression in human islets (TPM>5) and hESC-β cells (TPM>5): TAGLN3, ADCYAP1, DHRS2, CP, and SYNPO (Figure 5B-F). Similar to the assessment of insulin content, we used CRISPRa to activate the expression of positively correlated genes—TAGLN3, ADCYAP1, and DHRS2— and CRISPRi to inhibit the expression of negatively correlated genes—CP and SYNPO—in EndoC-βH1 cells. We confirmed that the expression of all five genes was reduced or activated in perturbed cells (Figure S6C). After stressed EndoC-βH1 cells with 1 mM of palmitate for 3 days, we detected an increased apoptotic rate in those cells with activation of TAGLN3 and DHRS2 (P-value<0.05; Figure 5G-H). Through immunofluorescence staining, we confirmed that activation of TAGLN3 and DHRS2 led to an increased percentage of cleaved caspase3+ cells (Figure 5K-L), supporting the role of TAGLN3 and DHRS2 in regulating β-cell survival. However, the other three correlated genes—CP, ADCYAP1 and SYNPO—exhibited no effects on palmitate-induced β-cell apoptosis (P-value>0.05; Figure 5G-J), indicating that they may only be involved in the innate β-cell survival response but do not exert a direct role to regulate β-cell apoptosis.
Figure 5. Cellular trait association analysis identifies genes controlling β-cell survival.

(A) Identification of genes correlated with palmitate-induced apoptotic rate in hESC-β cells. Genes associated at FDR<0.05 and ∣effect size∣>1.5 are colored (negative: blue, positive: red). (B-F) Linear regression analysis of apoptotic levels in each WT or KO line with RNA expression of candidate genes TAGLN3 (B), ADCYAP1 (C), DHRS2 (D), CP (E) and SYNPO (f). The solid line and gray area indicate the regression line and 95% CI, respectively. (G-J) Representative flow cytometry analysis (G and I) and the percentage of AnnexinV+DAPI− cells (H and J) in genetic perturbed EndoC-βH1 cells after palmitate treatment. Gating strategy is shown in Figure S6D. N=6 biological replicates. (K and L) Representative Immunofluorescent staining images (K), and the percentage of cleaved-caspase3+Insulin+ cells (L), in EndoC-βH1 cells carrying sgRNA to activate TAGLN3 or DHRS2. N=3 biological replicates. Scale bar = 200 μm. For panels 5H, 5J and 5L, data are shown as mean ± SD. P-values were calculated by unpaired Student’s t-test. The n.s. indicates a non-significant difference and * symbol illustrates the difference of each genetic perturbation line compared to the control line. * P < 0.05, ** P < 0.01, ***P < 0.001, ****P < 0.0001.
Analysis of allelic imbalance in accessible chromatin regions identifies a single candidate causal variant at 23 T2D genetic associations
A less obvious benefit of the inclusion of ATAC-seq in the experimental design was the chance to infer functional information about non-coding regions of the genome that are relevant to hESC-β cells, and to connect those to T2D genetic risk factors identified by GWAS. Statistical methods seek to reduce these multi-SNP signals to a “99% credible set”, including multiple tightly linked variants. But this “lumpy” architecture of genomic variation means that discerning the actual causative SNP at GWAS signals presents a major challenge for common disease genomics. We hypothesized that the causative variant is more likely to lie in an area of open chromatin in β cells. More than that, we hypothesized that the causative SNP should show evidence of differential chromatin accessibility between the risk and the non-risk alleles. ATAC-seq is capable of capturing instances where one allele is preferentially accessible (e.g., preferentially bound by a TF49,50). Such events can be quantified by measuring the difference in allele counts at heterozygous variants. Since both alleles occur within the same cell and have been exposed to the same experimental conditions, the intra-sample nature of this metric greatly reduces noise and maximizes signal.
In this study, the two WT lines and the 20 KO β-cell lines are all derived from the INSGFP/w MEL1 parental line. Therefore, for any SNP that happens to be heterozygous in this line, there is a large amount of ATAC-seq data available to examine for allelic imbalance. Furthermore, if one SNP in a credible set is heterozygous in INSGFP/w MEL1, linkage disequilibrium makes it likely that all of the SNPs in that credible set will be heterozygous. Using the chromatin accessibility data generated across the 20 KO lines and two WT lines, we quantified allelic imbalance across SNPs in 99% credible sets for T2D genetic associations.3 We identified 26 T2D association signals with ≥1 SNP that showed allelic imbalance (FDR<5%; Table S5; Figure 6A). At 18 of those signals, the INSGFP/w MEL1 cell line was heterozygous at all SNPs in the credible set and only one SNP showed allelic imbalance, which we conclude is likely to be the causal SNP at the T2D genetic association (Figure 6A).
Figure 6. ATAC-seq allelic imbalance analysis nominates functional candidates.

(A) Refinement of T2D GWAS signals using allelic imbalance analysis (binomial test from the common effect analysis). The INSGFP/w MEL1 hESC line is heterozygous at all credible set SNPs for 80/338 T2D association signals3. Within this group of 80 signals, we identified at least one SNP with allelic imbalance (FDR<5%) for 26 signals. At 18/26 signals, we identified a single SNP with allelic imbalance, thus likely to be the causative SNPs driving each association signal. (B) Candidate causal SNP at the ADCY5 locus. Top panel: UCSC browser of ATAC-seq (red) and RNA-seq (blue) reads around the credible set of SNPs in INSGFP/w MEL1 hESC-β cells. Next panels: −log10(P-values) from T2D genetic association; PPA from statistical analysis of genetic data on the credible set; −log10(P-value) of ATAC-seq allelic imbalance at each of the credible set SNPs. Dashed vertical blue line represents the candidate functional SNP and corresponds with the position of the disruption (G to A change) in the predicted TFBS motif (orange arrow). (C) An example of candidate functional SNP at the SEC16B locus. Order of panels is as in (B). (D) Nominating the likely functional SNP at the RALY locus. Order of panels is as in (B). (E) Association of ATAC reads imbalance at rs2284379 with total insulin content. The point size represents the total number of ATAC-seq reads covering the SNP position for the line.
As an example, we highlight a T2D association near ADCY5 (Figure 6B). Within the 99% credible set there are three SNPs, all of which are heterozygous in the INSGFP/w MEL1 cells. In our data, we found that rs11708067 (MAF=0.15) lies within an ATAC peak and exhibits allelic imbalance, where the “G” allele, associated with reduced T2D risk, shows increased accessibility. These results comport with a previous study that reports increased H3K27ac ChIP-seq reads from the “G” allele in human islets and increased luciferase activity of the “G” allele in a mouse β-cell line47, are in line with the deleterious effects of silencing ADCY5 in human islets51. We performed TF footprint analysis and discovered that the rs11708067 overlapped a E2F2 footprint where the “G” allele is predicted to have increased binding. We looked for, but did not find, an association between the chromatin accessibility of the region overlapping rs11708067 and the expression of nearby genes (FDR>5%), making the candidate effector gene at this signal an open question. Nonetheless, these results suggest that the T2D risk allele “A” may contribute to T2D risk by disrupting E2F2 binding.
As another example, we examined the complete credible set of 16 SNPs at a T2D association ~25kb downstream of SEC16B, all of which were heterozygous in the INSGFP/w MEL1 cells. Of the credible set SNPs, only rs574367 (MAF= 0.15) showed allelic imbalance (FDR<5%), with an increased proportion of reads from the non-risk “G” allele in 21 lines (Figure 6C). We performed TF footprint analysis and found that rs574367 strongly disrupts a predicted binding site for the RFX TF family, previously reported as an important T2D-relevant regulator of islet gene expression.45 We tested for an association between chromatin accessibility of the region overlapping rs574367 and the expression of nearby genes, but found no association (FDR>5%).
We also considered the possibility that some causal SNPs may not manifest an effect across all of the KO lines, since the knockouts have changed cellular phenotype and that may affect chromatin structure. Therefore, we also tested for allelic imbalance within each line individually. At an FDR<5%, we found 5 signals where a single SNP was not identified when considering common allelic imbalance effects across all lines, but the imbalance was significant in a subset of lines (Figure S7). Assuming that the SNP(s) driving the T2D genetic association manifest an effect in hESC-β cells, the 23 SNPs identified from these combined analyses represent strong candidates for being the causal SNP at these 23 T2D association signals.
As a third example, one signal located at chr20:32674967, all 95 SNPs in the 99% credible set were heterozygous in the parental INSGFP/w MEL1 line. SNP rs2284379, located in the first intron of the RALY locus, showed evidence of allelic imbalance (Figure 6D). But this association was particularly remarkable in that the allelic association favored the T allele in the majority of lines, but the C allele in the four lines with the lowest insulin content (see below; Figure 6E). The T2D risk allele of rs2284379, “C”, is predicted to better match the binding site motifs at footprints of RFX3, ZNF737, and MTF1. Given the location of this SNP, we tested for an association between chromatin accessibility of the region overlapping this SNP and RALY expression but did not find an association (FDR>5%).
Finally, we were also able to assess whether allelic imbalance at a particular SNP showed association with a phenotype of interest (e.g., a cellular trait ).52-54 To identify such effects, we jointly modeled allelic imbalance across all 22 lines, testing for an association with each cellular trait at all T2D 99% credible set SNPs. We identified two associations (FDR<5%), both with insulin content: rs2284379 (MAF=0.80) at the chr20:32674967 T2D association signal (RALY, Figure 6E,) and rs1800900 (MAF=0.60) at the chr20:57387352 T2D association signal (Table S5).
Discussion
We developed an isogenic KO hESC-derived β-cell platform to assess the molecular and cellular changes of human β cells carrying LoF mutations of 20 T2D-associated genes. The most fundamental detrimental effect, impaired β-cell differentiation, was observed for COBLL1, GIPR, HNF4A, TCF7L2, TGFB1, and TLE4. The defective differentiation in HNF4A−/− and TCF7L2−/− hESC lines is consistent with previous murine studies that the knockout of Hnf4a resulted in embryonic lethality55 and homozygous Tcf7l2 null mice experienced postnatal mortality56. Similarly, the impaired differentiation phenotype of TGFB1−/− hESCs also aligns with previous reports that dysregulation of TGFB1 signaling influences β-cell development.57 Interestingly, our finding of that GIPR KO impaired β-cell differentiation in hESCs, seems to diverge from the phenotype observed in Gipr null mice which display a twofold increase of β-cell area58, suggesting that the role of GIPR in β-cell differentiation may exhibit species-specific nuances. Of note, those Gipr−/− mice have a 40% reduction in pancreatic insulin content and gene expression despite the increased β-cell mass, indicating a functional abnormality of Gipr-deficiency β-cells58. The mechanisms underlying the relationship between T2D and COBLL1 or TLE4 remain unclear.
We observed multiple genes of diverse classes affecting a similar set of cellular traits. For example, loss of ABCC8, HNF1A, WDR13, and HTT does not affect β-cell generation in this system but impairs four cellular traits. Mutations in the genes ABCC859, HNF1A60, and WDR1361 have been associated with impaired β-cell function and insulin secretion. Recent studies suggest that mutations in ABCC8 and HNF1A may also contribute to β-cell dedifferentiation/transdifferentiation.60,62,63 Consistently, we found that ABCC8−/−, HNF1A−/−, and WDR13−/− hESC-β cells exhibit decreased total insulin content along with decreased expression of β-cell genes(Figure S3D and S4C). Meanwhile, for ABCC8−/−, HNF1A−/−, and WDR13−/− hESC-β cells, we also observed an upregulation of marker genes associated with other pancreatic endocrine cells (α, δ, ε and PP cells), ductal cells and acinar cells, further confirming the impaired β-cell identity in those KO cells (Figure S3D).
Among 20 T2D-associated genes studied, only loss of HNF4A caused effects in all five cellular traits (Figure 1I). HNF4A is an important transcription factor affecting liver64 and islet development and function65-67, Our analysis suggested that HNF4A might directly bind to regulatory elements that contribute to T2D risk, as we have shown for the FAIM2 signal. In contrast, SLC30A8−/− hESCs showed no impairments in any of five cellular traits. On the contrary, we found that homozygous LoF of SLC30A8 can actually enhance human β-cell survival from lipotoxicity, which is consistent with previous studies68-70 that LoF mutations of SLC30A8 showed a protective role in β-cell survival.
Notably, RNA-seq and ATAC-seq analysis suggested that knockout of KCNJ11 and ABCC8 results in large-scale transcriptomic and chromatin accessibility changes. One possible mechanism might operate through intracellular calcium. KATP channel closure due to LoF mutations in KCNJ11/ABCC8 can elevate intracellular Ca2+ levels in β cells,71 which disrupts Ca2+-regulated genes, leading to alterations in β-cell identity and islet morphology62.
By comparing across all 22 lines, we were able to identify genes whose expression correlated with insulin content, and with palmitate-induced apoptosis. Follow-up functional assays confirmed the regulatory roles of CP, RNASE1, PCSK1N, and GSTA2 in controlling insulin production, and identified TAGLN3 and DHRS2 as regulators of β-cell survival. CP, encoding for ceruloplasmin72, and GSTA2, encoding for glutathione S-transferase A273, are both enzymes involved in regulating oxidative stress in cells. Of particular interest, prior studies have reported elevated levels of serum ceruloplasmin in individuals with T2D.74,75 Additionally, reduced expression of GSTA2 has been observed in human islets treated with palmitate and high glucose.76 PCSK1N is an inhibitor of PCSK1, the key enzyme controlling the conversion of proinsulin to insulin.77 Thus, upregulation of PCSK1N might directly affect insulin content by inhibiting the proinsulin to insulin conversion. RNASE1 encodes an endonuclease that cleaves internal phosphodiester RNA bonds on the 3'-side of pyrimidine bases.78 Therefore, RNASE1 may indirectly regulate insulin production by affecting the stability of INS mRNA. Regarding genes associated with apoptosis, DHRS2 overexpression has been shown to induce apoptosis in certain cancer cells.79,80 TAGLN3 encodes transgelin 3, which has been shown to be involved in astrocyte inflammation.81 However, how transgelin 3 regulates β-cell survival is still not clear. Here our study suggests that TAGLN3 and DHRS2 may have a pro-apoptotic role in β cells, but further research is needed to fully elucidate their function and potential therapeutic implications.
Additional insights were derived by close inspection of allele-specific imbalance in ATAC-seq data at sites where the INSGFP/w MEL1 parental line was heterozygous. With 20 isogenic KO and two WT lines, the depth of coverage at an ATAC-seq peak was large enough to detect situations where one allele contributed substantially more than 50% of the reads. With this approach, we were able to pinpoint a single likely functional variant at each of 23 loci. The success of this effort, demonstrating that deep ATAC-seq coverage can discover biologically interesting allele-specific imbalance in a disease-relevant tissue, suggests that future work like this could be usefully done with a larger collection of iPSCs of diverse genotypes.
In summary, we developed an isogenic hESC platform to examine the impact of knocking out 20 T2D-associated genes on human β-cell generation, insulin content, glucose and KCl stimulated insulin secretion, and β-cell survival. Previously unknown insights were derived about each of the individual knockout lines, and the molecular comparison also revealed pathways involved in insulin production and apoptosis that would have been difficult to discern by other means. Future work to expand the panel to many more genes relevant to T2D, while maintaining the same standards for cellular phenotyping, is likely to be revealing. One can also readily imagine extrapolating this same platform to the analysis of any other polygenic disorder where relevant tissues can be differentiated from hPSCs and studied by integrative analysis methods.
Limitations of study
While this study represented a significant throughput advance by characterizing a total of 20 T2D gene knockouts in hESC-derived β cells, there are potentially dozens more effector genes that could not be included here. We observed that 15 out of 20 mutant β-like cell lines exhibited increased susceptibility to cell death under the in vitro conditions of palmitate-induced lipotoxicity. Though the 1 mM concentration of palmitate that we used falls within the normal range in human plasma, it is possible that the particular in vitro culture conditions, which naturally differ from what is experienced in vivo by pancreatic islets, might have heightened the sensitivity of hPSC-derived β cells towards palmitate-induced apoptosis. Finally, the ability to globally identify the likely functional basis for T2D GWAS risk loci by linking those up to hESC β-cell ATAC-seq peaks data was necessarily limited by the use of a single hESC line, the reality that many such risk loci reflect actions of other tissues, different differentiation states of β cells, or environmental influences that are unmeasured in this study.
STAR Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Oct-4A (C30A3) Rabbit mAb | Cell Signaling Technologies | #2840 |
| Nanog (D73G4) XP® Rabbit mAb | Cell Signaling Technologies | #4903 |
| SSEA4 (MC813) Mouse mAb | Cell Signaling Technologies | #4755 |
| TRA-1-81 Mouse mAb | Cell Signaling Technologies | #4745 |
| Polyclonal Guinea Pig Anti-Insulin | Dako | #A0564 |
| Human SOX17 Antibody | R&D | AF1924 |
| Anti-HNF3β/FOXA2 Antibody | Millipore | 07-633 |
| Purified Rabbit Anti- Active Caspase-3 | BD bioscience | #559565 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | A-11055 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 647 | Thermo Fisher Scientific | A-214477 |
| Alexa Fluor 488 AffiniPure Donkey Anti-Guinea Pig IgG (H+L) | Jackson ImmunoResearch Labs | #706-545-148 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 488 | Thermo Fisher Scientific | #A32790 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | #A-21203 |
| Donkey anti-Rabbit IgG (H+L) Secondary Antibody, Alexa Fluor 594 conjugate | Thermo Fisher Scientific | #A-21207 |
| Chemicals, peptides, and recombinant proteins | ||
| Normocin | Invivogen | #ant-nr-2 |
| Y-27632 | MedchemExpress | #HY-10583 |
| Activin A | R&D Systems | #338-AC-500/CF |
| Recombinant Human KGF (FGF-7) Protein | Peprotech | #100-19-500UG |
| CHIR99021 | Cayman Chemical | #13122 |
| SANT-1 | Sigma Aldrich | #S4572-25MG |
| Retinoic acid | Sigma Aldrich | #R2625-500MG |
| LDN 193189 hydrochloride | Axon Medchem | #Axon 1509 |
| TPPB | Tocris Bioscience | #5343 |
| T3 hormone | Sigma Aldrich | #T6397-100MG |
| Zinc sulfate heptahydrate | Sigma Aldrich | #Z0251-100G |
| Heparin sodium salt | Sigma Aldrich | #H3149-1MU |
| γ-Secretase Inhibitor XX | Millipore | #565789-1MG |
| ALK5 Inhibitor II | Cayman Chemical | #14794 |
| L-Ascorbic acid | Sigma Aldrich | #A4544-100G |
| R428 | MedchemExpress | #HY-15150 |
| N-acetyl-L-cysteine | Sigma Aldrich | #A9165-5G |
| Trolox | Millipore | #648471 |
| Matrigel | Corning | #354234 |
| TWEEN® 20 | Sigma-Aldrich | #P9416 |
| APC Annexin V | BD Biosciences | # 550475 |
| Paraformaldehyde, 4% in PBS | Thermo Fisher Scientific | #J61899.AK |
| Penicillin and Streptomycin | Thermo Fisher Scientific | #10378016 |
| Fetal Bovine Serum, qualified, heat inactivated | Thermo Fisher Scientific | #10438026 |
| GlutaMAX | Thermo Fisher Scientific | #35050-061 |
| D-Glucose | Sigma-Aldrich | #G8769 |
| β-mercaptoethanol | Thermo Fisher Scientific | #21985023 |
| Nicotinamide | Sigma-Aldrich | #72340 |
| Sodium selenite | Sigma-Aldrich | #S9133 |
| Transferrin | Sigma-Aldrich | #T8158 |
| Accutase | Innovative Cell Technologies | #MSPP-AT104 |
| ReLeSR | STEMCELL Technologies | #05872 |
| ITS-X | Thermo Fisher Scientific | #51500056 |
| NaHCO3 | Thermo Fisher Scientific | #S6267 |
| Lipofectamine™ 2000 Transfection Reagent | Thermo Fisher Scientific | #11668027 |
| Opti-MEM | Thermo Fisher Scientific | #331985070 |
| Triton X-100 | Sigma-Aldrich | #X100 |
| Igepal CA-630 | Sigma-Aldrich | #I8896 |
| Digitonin | Promega | #G9441 |
| L-Ascorbic acid (vitamin C) | Sigma-Aldrich | #4544 |
| RIPA buffer | Sigma-Aldrich | #R0278 |
| BSA, Fatty Acid Free, Fraction V | Lampire | #7500804 |
| Bovine Serum Albumin, low endotoxin | Sigma-Aldrich | #A1470 |
| Lenti-X Concentrator | Takara | #631232 |
| Blasticidin | Thermo Fisher Scientific | #R21001 |
| Puromycin Dihydrochloride | Thermo Fisher Scientific | #A1113803 |
| EDTA | Thermo Fisher Scientific | #15575020 |
| DAPI | Sigma Aldrich | #D9542 |
| Critical commercial assays | ||
| Dual-Luciferase® Reporter Assay System | Promega | #E1980 |
| Nano-Glo® Luciferase Assay System | Promega | #N1130 |
| STELLUX Chemi Human Insulin ELISA Jumbo | Alpco | #80-INSHU-CH10 |
| High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor | Thermo Fisher Scientific | #4374966 |
| SYBR™ Green PCR Master Mix | Roche | #4309155 |
| Absolutely RNA Microprep Kit | Agilent Technologies | #400805 |
| RNeasy Plus Mini Kit | Qiagen | #74136 |
| Zymo DNA Clean & Concentrator Kits | Zymo Research | #D4003 |
| TruSeq Stranded mRNA LP (48 Spl) | Illumina | #20020594 |
| IDT for Illumina – TruSeq RNA UD Indexes (96 Indexes, 96 Samples) | Illumina | #20022371 |
| Illumina Tagment DNA Enzyme and Buffer Large Kit | Illumina | #20034198 |
| Deposited data | ||
| RNA-seq data | This paper | GSE228665 |
| ATAC-seq data | This paper | GSE228665 |
| SNP array genotyping data | This paper | GSE228665 |
| Whole genome sequence data | This paper | GSE228665 |
| Source data for manuscript figures | This paper | Table S1 |
| Experimental models: Cell lines | ||
| hESC line MEL-1 | Monash University | #CVCL_XA16 |
| EndoC-βH1 | INSERM | #CVCL_L909 |
| HEK293T cells | ATCC | #CRL-11268 |
| Oligonucleotides | ||
| Primers used for DNA sequencing | Integrated DNA Technologies | Sequences in Table S1 |
| Primers used for qRT-PCR | Integrated DNA Technologies | Sequences in Table S1 and Table S6 |
| Primers used for constructing luciferase vectors | Integrated DNA Technologies | Sequences in Table S6 |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-Puro (PX459) V2.0 | Ran et al. 201319 | Addgene plasmid # 62988; RRID: Addgene_62988 |
| pCC_12 - hU6-BsmBI-sgRNA(E+F)-barcode-EFS-KRAB-dxCas9NG-NLS-2A-Puro-WPRE | Legut et al. 202082 | Addgene plasmid # 139097; RRID: Addgene_139097 |
| pCC_05 - hU6-BsmBI-sgRNA(E+F)-barcode-EFS-dCas9-NLS-VPR-2A-Puro-WPRE | Legut et al. 202082 | Addgene plasmid # 139090; RRID: Addgene_139090 |
| psPAX2 | Didier Trono83 | Addgene plasmid # 12260; RRID: Addgene_12260 |
| pMD2.G | Didier Trono84 | Addgene plasmid # 12259; RRID: Addgene_12259 |
| Proinsulin-NanoLuc in pLX304 | Burns et al. 201548 | Addgene plasmid # 62057; RRID: Addgene_62057 |
| pGL4.23[luc2/minP] vector | Promega | #E8411 |
| phRL-SV40 Vector | Promega | #E6261 |
| Software and algorithms | ||
| FlowJo | FLOWJO LLC | https://www.flowjo.com/ |
| GraphPad Prism | GraphPad Software | http://www.graphpad.com/scientific-software/prism |
| Adobe illustrator | Adobe | https://www.adobe.com/products/illustrator.html |
| MetaMorph® image analysis software | Molecular Devices | https://www.moleculardevices.com/products/cellular-imaging-systems/acquisition-and-analysis-software/metamorph-microscopy |
| ZEN | ZEISS | https://www.zeiss.com/microscopy/en/products/software/zeiss-zen-lite.html |
| novoalign v2.07.11 | http://www.novocraft.com/products/novoalign | |
| Michigan TOPmed Server (Minimac v4) | Das et al. 201685 | https://imputation.biodatacatalyst.nhlbi.nih.gov/#! |
| STAR v2.73a | Dobin et al. 201386 | https://github.com/alexdobin/STAR |
| QoRTs (v1.3.6,87 | Hartley & Mullikin 201587 | https://hartleys.github.io/QoRTs/ |
| GATK v4.1.9.0 | Auwera & O’Connor 202088 | https://gatk.broadinstitute.org/hc/en-us/articles/360041320571--How-to-Install-all-software-packages-required-to-follow-the-GATK-Best-Practices |
| Samtools v1.9 | Danecek et al. 202189,90 | https://github.com/samtools/samtools |
| verifybamID v1.1.191 | Jun et al. 201291 | https://github.com/statgen/verifyBamID/releases |
| BWA-MEM v0.7.17-r1194 | Li & Durbin 200992 | https://github.com/lh3/bwa |
| bedtools v2.26.0 | Quinlan and Hall 201093 | https://bedtools.readthedocs.io/en/latest/content/installation.html |
| MACS2 v2.2.7.194 | Zhang et al. 200894 | https://github.com/macs3-project/MACS/wiki/Install-macs2 |
| DESeq2 v1.32.095 | Love et al. 201495 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| MetaVolcano v1.10.096 | Cesar Prada 201996 | https://www.bioconductor.org/packages/release/bioc/html/MetaVolcanoR.html |
| fgsea v1.20.0 | Korotkevich et al. 201697 | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| msigdbr v7.5.1 | Liberzon et al. 201198 | https://igordot.github.io/msigdbreak/ |
| UCSC Genome Browser | Nassar et al. 202399 | https://genome.ucsc.edu/ |
| LIMIX v1.0.17 | Lippert et al. 2014100 | https://github.com/limix/limix/blob/master/doc/qtl.rst |
| CELLEX v1.2.2 | Timshel et al. 2020101 | https://github.com/perslab/CELLEX/blob/master/tutorials/demo_mousebrain_vascular_cells.ipynb |
| CTA: C++ implementation of Buenrostro adapter trimming | John Hensley 2017John Hensley 2017 | https://github.com/ParkerLab/cta |
| FIMO v5.4.1 | Grant et al. 2011102 | https://meme-suite.org/meme/doc/download.html |
| SEA v5.4.1 | McLeay & Bailey 2010103 | https://meme-suite.org/meme/doc/download.html |
| CENTIPEDE v1.2 | Pique-Regi et al. 2011104 | https://rdrr.io/rforge/CENTIPEDE/man/CENTIPEDE-package.html |
| WASP v0.3.4 | van de Geijn et al. 2015105 | https://github.com/bmvdgeijn/WASP |
| Annotate_variation.pl (v2019-10-24) | Wang et al. 2010106 | https://annovar.openbioinformatics.o |
| Other | ||
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Dr. Francis S. Collins (francis.collins@nih.gov).
Materials availability
Cell lines generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement.
Data and code availability
The RNA-seq, ATAC-seq, whole genome sequence data, and SNP array genotyping data generated during this study are available at GEO under accession no. GSE228665. Source data used to generate the graphs in the paper can be found in the file Data S1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell lines and culture conditions
We obtained INSGFP/w MEL-1 (RRID: CVCL_XA16, male) human embryonic stem cell (hESC) stocks from Dr. Ed Stanley at Monash University. All hESC studies were approved by the Tri-Institutional Embryonic Stem Cell Research Committee (ESCRO). To culture and maintain both wildtype (WT) and isogenic (see Generation of isogenic hESC lines) hESCs, we followed a previously described protocol.107 We grew hESCs on Matrigel-coated plates in StemFlex medium (Thermo Fisher Scientific) supplemented with 50 μg/mL Normocin (InvivoGen), with medium changed daily and cultures passaged at 1:6-1:10 with ReLeSR (Stem Cell Technologies). We obtained EndoC-βH1 cells (RRID: CVCL_L909, female) from CNRS, and cultured them in DMEM containing 5.6 mM glucose, 2% BSA (Sigma-Aldrich), 50 μM 2-mercaptoethanol (Thermo Fisher Scientific), 10 mM nicotinamide (Sigma-Aldrich), 5.5 μg/ml transferrin (Sigma-Aldrich), 6.7 ng/ml selenite (Sigma-Aldrich), 100 U/ml penicillin and 100 μg/ml streptomycin. HEK293T cells (purchased from ATCC, CRL-11268, female) were cultured in DMEM supplemented with 10% FBS (Thermo Fisher Scientific). All cell lines were cultured at 37 °C with 5% CO2 and were tested for mycoplasma contamination every six months using MycoAlertTM PLUS Mycoplasma Detection Kit (Lonza).
METHOD DETAILS
All studies were performed in a blinded manner without inclusion and exclusion applied. The sample size and statistical analysis method of each experiment have been provided in the figure legends.
Selection of T2D associated genes
For the isogenic knockout lines, we selected 20 genes that are enriched with T2D effector genes defined by the Accelerating Medicines Partnership (AMP)12 (https://t2d.hugeamp.org) (Table 1). Briefly, we considered two pools of genes for the candidate selection: 1) The genes implicated to be causal by having T2D-associated coding variant(s) in a large-scale exome sequencing study14; OR 2) closest genes to a 99% credible set of SNP with a high posterior probability of association (PPA>0.7)6. Next, we prioritized the ones that are expressed in human pancreatic β cells (TPM≥0.01). We finally chose 20 genes with relatively high expression in hESC-β cells (TPM ≥7.50) for subsequent knock-out assays. It includes 17 predicted T2D effector genes (AMP) and 3 genes that might have implications in islet β cells based on published literature57,61,108.
Generation of isogenic hESC lines
To create the isogenic KO hESC lines, we designed short guide RNAs (sgRNAs) targeting exons of 20 genes with evidence for T2D (Table 1; Table S1) using the web resources available at http://chopchop.cbu.uib.no/. We cloned them into the pSpCas9(BB)-2A-Puro (PX459) V2.0 vector (Addgene #62988) according to the instructions described in our previous publication.109 All KO lines were generated from INSGFP/w MEL-1. Briefly, MEL-1 cells were dissociated using Accutase (Innovative Cell Technologies) and electroporated (5× 105 cells per sample) with 4 μg sgRNA-construct plasmids using Human Stem Cell Nucleofector™ solution (Lonza) following manufacturer’s instructions. The cells were then seeded into 2 wells of 24-well plates and cultured in StemFlexTM medium with 10 μM Y-27632. They were switched to StemFlexTM medium with 0.5mg/ml puromycin on the next day and maintained for 2 days. After puromycin selection, hESCs were dissociated into single cells with Accutase and re-plated at a density of 5 cells/well in 96-well plates. 10 μM Y-27632 was added for the first 2 days. 10 days later, individual colonies were picked and re-plated into two wells of 96-well plates. When hESCs reached ~90% confluence, one well of each clone was analyzed to confirm the indel information of each clone by Sanger sequencing a ~500 bp window around the Cas9-sgRNA recognition site (Figure S1). For biallelic frameshift mutants, we expanded two clones (clones #1 and #2) with either homozygous indel mutations or compound heterozygous indel mutations in each target gene to perform cellular assays. We also expanded two WT clonal lines as WT controls to account for potential non-specific effects associated with the gene-targeting process.
Assessment of knockout off-target effects
We performed an in-silico scanning of potential off-target sites for the TCF7L2 sgRNA using CRISPRitz110 across the entire genome. By allowing up to 4 mismatches in the sgRNA sequence, we identified 2,513 cleavage sites that could be affected by the sgRNA. We extracted DNAs of two WT and two TCF7L2−/− lines with the DNeasy® Blood & Tissue Kit (QIAGEN) according to the manufacturer’s protocol. We performed whole genome sequencing of the cells with an average depth of >26x across the genome per clonal library. We aligned the reads to the GRCh38 genome assembly using BWA-MEM v0.7.17-r1194 with the -M option.92 After removing duplicate reads and filtering for properly paired reads with mapping quality ≥30 with samtools v1.9,89,90 we retained remaining uniquely aligned primary reads per library for downstream analyses. We identified all somatic variants (including single nucleotide polymorphisms (SNVs) and indels) in 250bp flanking regions of the predicted cleavage sites that are present in only TCF7L2−/− lines but not in the WTs using GATK v4.0.5.1 Mutect288 (pair-wse comparisons) with the “--min-base-quality-score 30” option. As expected, we only observed homozygous on-target indels but no other variants with allele frequency >15% in the KO lines. As these allele frequencies fall well below the heterozygous level, we conclude that they are sequencing errors or mosaics arising during cellular expansion. Furthermore, as the variants are different in each of the two KO lines, we conclude that they cannot be responsible for the cellular phenotype.
Directed differentiation of hESC to β cells
We differentiated hESCs into pancreatic β-like cells using a modified protocol from previous studies.16,17,25 Briefly, on day 0, we exposed cells to basal medium RPMI 1640 (Corning) supplemented with 1× Glutamax (Thermo Fisher), 50 μg/mL Normocin, 100 ng/mL Activin A (R&D systems), and 3 μM of CHIR99021 (Cayman Chemical) for 24 hours. On day 1, we changed the medium to basal RPMI 1640 medium supplemented with 1× Glutamax, 50 μg/mL Normocin, 0.2% FBS (Thermo Fisher Scientific), 100 ng/mL Activin A for 2 days, producing definitive endoderm cells. On day 3, we cultured the definitive endoderm cells in basal MCDB131 supplemented with 1× Glutamax (Thermo Fisher Scientific), 1.5 g/L sodium bicarbonate (Sigma-Aldrich), 2% bovine serum albumin (BSA, Lampire), 10 mM glucose (Sigma Aldrich), 50 ng/mL FGF7 (Peprotech) and 0.25 mM L-ascorbic acid (Sigma Aldrich) for 2 days to acquire primitive gut tube. On day 5, we induced the cells to differentiate to posterior foregut in basal medium MCDB 131 supplemented with 2% BSA, 2.5 g/L sodium bicarbonate, 1× Glutamax, 10 mM glucose, 0.25 mM L-ascorbic acid, 50 ng/mL FGF-7, 2 μM Retinoic acid (RA; Sigma Aldrich), 100 nM LDN193189 (LDN, Axon Medchem), 1:200 ITS-X (Thermo Fisher Scientific), 200 nM TPPB (Tocris Bioscience) and 0.25 μM SANT-1 (Sigma Aldrich) for 2 days. On day 7, we induced the cells to differentiate to pancreatic endoderm in MCDB 131 medium supplemented with 2% BSA, 2.5 g/L sodium bicarbonate, 1× Glutamax, 10 mM glucose, 0.25 mM L-ascorbic acid, 2 ng/mL of FGF-7, 0.2 μM RA, 200 nM LDN193189, 1:200 ITS-X, 100 nM TPPB and 0.25 μM SANT-1 for 3 days. On day 10 the cells were induced to differentiate to pancreatic endocrine precursors in MCDB 131 medium supplemented with 1.5 g/L sodium bicarbonate, 1×Glutamax, 20 mM glucose at final concentration, 2% BSA, 0.1 μM RA, 100 nM LDN193189, 1:200 ITS-X, 0.25 mM SANT-1, 1 μM T3 hormone (Sigma Aldrich), 10 μM ALK5 inhibitor II (Cayman Chemical), 10 μM zinc sulfate heptahydrate (Sigma Aldrich) and 10 μg/mL of heparin (Sigma Aldrich) for 3 days. On day 13, we exposed cells to MCDB 131 medium supplemented with 1.5 g/L sodium bicarbonate, 1× Glutamax, 20 mM glucose at final concentration, 2% BSA, 100 nM LDN193189, 1:200 ITS-X, 1 μM T3, 10 μM zinc sulfate, 10 μg/mL of heparin, 100 nM gamma secretase inhibitor XX (Millipore) for the 7 days. On day 21, cells were exposed to MCDB 131 medium supplemented with 1.5 g/L sodium bicarbonate, 1× Glutamax, 20 mM glucose, 2% BSA, 1:200 ITS-X, 1 μM T3, 10 μM ALK5 inhibitor II, 10 μM zinc sulfate, 10 μg/mL of heparin, 1 mM N-acetyl cysteine (Sigma Aldrich), 10 μM Trolox (Millipore), 2 μM R428 (MedchemExpress) for 7-15 days. We refreshed the medium every day. Specially, for GSIS and KSIS assay, we dissociated cells at stage 6 using Accutase, and seeded them into 96-well U-bottom low attachment plates as described in Static GSIS and KSIS Assays. We presented the actual number of biological replicates (n) for each downstream assay in Table S2.
Immunofluorescence staining and confocal microscopy
We fixed cells in 4% paraformaldehyde solution (Thermo Fisher Scientific) for 20 minutes, washed them three times in PBS with 5 minutes incubation for each wash, and blocked and permeabilized cells in a PBS solution containing 5% horse serum and 0.3% Triton X-100 (Sigma Aldrich) for 1 hour at room temperature. Then, we incubated the cells with primary antibodies overnight at 4°C and washed them in PBS with a 5-minute incubation three times. After a 1-hour incubation with fluorescence-conjugated secondary antibodies (Alexafluor, Thermo Fisher Scientific) at room temperature, we washed the cells with PBS three times. The detailed antibody information has been included as Table S6. Images in Figure S2A and S2E were taken by Inverted Microscope/Apotome (Zeiss). Images in Figure 5K were taken by LSM 800 confocal microscope (Zeiss) and scored using MetaMorph® image analysis software (Molecular Devices). We calculated mean ± SD for each assay using 3 independent biological replicates, and we present those data in Figure 5L.
Fluorescence-activated cell sorting
We dissociated hESC-derived cells at day 24 into single cells using Accutase and resuspended in PBS supplemented with 0.5% BSA, 300 nM DAPI, and 2 mM EDTA. The Flow Cytometry Core Facility in Weill Cornell Medicine helped conduct the sorting experiments and collect GFP+DAPI-cells by BD FACS Melody™ Cell Sorter. All experiments were performed with >=3 independent replicates. For RNA-seq, we collected 500,000 cells for each replicate. For ATAC-seq or ELISA assay, we collected 50,000 cells per replicate. We present the actual number of biological replicates (n) in Table S2.
Flow cytometry analysis
We dissociated hESC-derived cells or EndoC-βH1 cells using Accutase. To analyze GFP expression, we resuspended the hESC-derived cells in PBS and used them directly for analysis. The gating strategy for the analysis of GFP+ cells is shown in Figure S3B. For Annexin V cellular apoptosis analysis, we stained hESC-derived or EndoC-βH1 cells with the APC/Annexin V apoptosis detection Kit (BD Bioscience) and DAPI according to manufacturer’s instructions and analyzed cells using Attune NxT Flow Cytometer (Thermo Fisher Scientific) within 30 minutes. The gating strategy for the analysis of apoptotic rate in hESC-derived β cells and EndoC-βH1 cells is shown in Figure S3B and Figure S6D, respectively. All experiments were performed with >=3 independent replicates. We present the actual number of biological replicates in Table S2 and the legends of Figure 5H and 5J.
Static GSIS and KSIS Assays
We dissociated cells at stage 6 using Accutase and resuspended them in S6 medium supplemented with 10 μM Y-27632 at a final concentration of 300 cells/μl. Using a multichannel pipette and trough, we filled 96-well U-bottom low attachment plates with 100 cell suspensions in each well and spun at 300g for 5 minutes. Cells were aggregated into clusters by incubating for at least 24 hours at 37°C with 5% CO2 and then fed every 48 hours until at least the 8th day at Stage 7. Before the static GSIS/KSIS assays, 6-8 islet-like clusters were combined into one well as one replicate and starved in S7 medium but with 5 mM glucose for 12 hours. Subsequently we removed the medium and washed cell clusters with fresh KRBH Buffer. We then incubated the cells in LG KRBH (with 0.1% BSA and 2 mM glucose) for 1 hour in an air incubator at 37°C. We aspirated the media and replaced it with 200 μL LG KRBH buffer or LG KRBH buffer with combinations of 20 mM glucose, or 30 mM KCl to each well and incubated at 37°C for 1 hour. Plates were spun and the top 120 μl supernatants were collected. The residual medium was removed, and cell clusters of each well were lysed by RIPA buffer (Sigma Aldrich) supplemented with Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). We measured insulin content in both supernatant and cell lysis using STELLUX Chemi Human Insulin ELISA Jumbo kit (Alpco). Stimulation index represents the fold change of the percent of insulin secreted upon 20 mM glucose or 30 mM KCl stimulation divided by the percent of insulin secreted upon 2 mM glucose stimulation. We calculated mean ± SD for each assay using at least 6 independent biological replicates, and we present the actual number of biological replicates in Table S2.
Luciferase Reporter Assay
We based the construction of all luciferase vectors on the pGL4.23[luc2/minP] vector (Promega) which contains a firefly luciferase gene luc2 under regulation of a TATA-box minimal promoter (minP). From genomic DNA of EndoC-βH1 cells, we cloned the DNA region (723bp, from chr12:49868906 to chr12:49869637) at the locus of T2D_fmap. FAIM2.chr12:50263148. This construct included the “A” allele at rs7132908. We then subcloned this into pGL4.23[luc2/minP] vector (Promega) between the XhoI and Bgl II restriction sites. Using PCR amplification with mutated primers, followed by DpnI digestion and nick ligation in E. coli111, we performed site-directed mutation of the plasmid to produce the same vector with the “G” allele at rs7132908. Constructs of all plasmids were confirmed by Sanger sequencing. The sequences of primers used to construct and validate each vector are shown in Table S6. For luciferase assays, we seeded EndoC-βH1 cells into 12-well plates at a density of 5.0 × 105 cells/well, cultured those for 48 hours, and then transfected with firefly luciferase reporter vectors. We used a Renilla luciferase vector carrying the SV40 promoter, phRL-SV40 (Promega) as an internal control. We co-transfected cells with firefly luciferase reporters (1 μg/well) and phRL-SV40 (20 ng/well), using Lipofectamine 2000 (Thermo Fisher Scientific), following the manufacturer’s instructions. Transfections were performed in triplicate for experimental group using constructed vectors and in quadruplicate for control group using empty vector. We harvested cells at 48 hours after transfection and lysed them in the passive lysis buffer (Promega). We measured luciferase activity of the lysates with the Dual-Luciferase® Reporter Assay System (Promega) according to the manufacturer’s protocols. We calculated the ratio of firefly/Renilla luciferase activity for each tested enhancer candidate vector and normalized that to the empty vector pGL4.23[luc2/minP] as the final relative luciferase intensity. We calculated mean ± SD for each assay using 3-4 independent biological replicates and we present those data in Figure 3H.
CRISPR perturbation experiments
To perturb the transcriptional expression of candidate genes, we designed two different sgRNAs for each candidate gene, using the web resources available at http://chopchop.cbu.uib.no/. We cloned sgRNAs targeting RNASE1, PCSK1N, GSTA2, TAGLN3, ADCYAP1 and DHRS2 (sequences of sgRNA targeting regions are listed in Table S1) into dSpCas9-VPR vector (Addgene #139090) for gene activation. We cloned sgRNAs targeting CP, FOSB, and SYNPO into the dCas9-KRAB vector (Addgene #139097) according to the previously described instructions82. We produced lentivirus expressing each CRISPRa or CRISPRi system in HEK293T cells, using a second-generation viral packaging system, and used the virus to infect EndoC-βH1 cells or EndoC-luc cells as previously described48. At 48 hours post transduction, we treated cells with 2 μg/mL puromycin for one week to select for infected cells, which were then used for downstream functional assays.
Generation of EndoC-βH1-luc cells and Nano-luc reporter assay
We produced lentivirus expressing proinsulin-luciferase fusion protein in 10-cm diameter dishes from 80% confluent HEK293T cells, transfected with lentiviral packaging plasmid psPAX2 (Addgene #12260), envelope plasmid pMD2.G (Addgene #12259) and Proinsulin-NanoLuc plasmid (Addgene #62057). We pooled viral supernatant harvested at 48h and 72h post-transfection and concentrated it using Lenti-X Concentrator (Takara) according to the instructions. We added the virus prep to EndoC-βH1 cells in fresh culture medium (see Cell lines and culture conditions) with 8 μg/ml Polybrene (Sigma-Aldrich), and spun the cells at 800 x g for 1 hour at 30 °C. After 24 hours in the presence of virus, we placed cells in fresh growth media. Subsequently, we treated the infected EndoC-βH1 cells with 5 μg/mL blasticidin (Thermo Fisher Scientific) for one week to produce the stable EndoC-luc lines. To test if CP, RNASE1, PCSK1N and GSTA2 have effects on insulin translation/processing, we conducted CRISPR perturbation experiments in EndoC--βH1-luc cells (see CRISPR perturbation experiments). We dissociated EndoC--1-luc cells into single cells and counted them by a Countess II Cell Counter (Thermo Fisher Scientific). 10,000 EndoC-βH1-luc cells were lysed in 100 μl passive lysis buffer (Promega) and we then measured intracellular Nano-luc intensity of lysate with the Nano-Glo® Luciferase Assay System (Promega) according to the manufacturer’s protocols. We calculated mean ± SD for each assay using 3 independent biological replicates and we present those data in Figure 4K-4L.
qRT-PCR
We isolated total RNA from EndoC-βH1 cells or EndoC-luc cells using the RNeasy Plus Mini Kit (QIAGEN), quantified RNA with a NanoDrop spectrophotometer (Thermo Fisher Scientific), and synthesized cDNA with a high-capacity reverse transcription kit (Thermo Fisher Scientific). We performed real-time qPCR with a LightCycler 480 (Roche) instrument with LightCycler DNA master SYBR Green I reagents (Roche). Primer sequences specific to INS, candidate genes being tested, and the reference gene (GAPDH) are listed in Table S1 and S6. We determined Delta-delta-cycle threshold (DDCT) relative to the GAPDH and control samples. We calculated mean ± SD for each assay using 3 independent biological replicates, and we present those data in Figure 4I-J and Figure S6A-C.
INSGFP/w MEL1 genotyping, quality control, and imputation
We genotyped the parental INSGFP/w MEL1 hESC line used for generating the isogenic hESC lines using the Infinium Omni2.5Exome-8 BeadChip array v1.3 (Illumina, San Diego, CA) at the NHGRI Genomics Core facility, resulting in a call rate of 99.7% (out of 2,612,357 SNPs). Using novoalign v2.07.11 (http://www.novocraft.com/products/novoalign), we mapped the array probe sequences to the GRCh37 (hg19) genome assembly and filtered variants with ambiguous probe alignments as previously described in Currin et al..112 We combined the INSGFP/w MEL1 genotypes with 15 samples genotyped on the same chip and 2,504 samples from 1000G project phase 3 release113. We removed variants not in the 1000G Phase 3 release panel, with missing genotypes in >1 of the 16 genotyped samples, that are likely palindromic variants with MAF>0.4 in the 16 genotyped samples, or with a genotype distribution that deviates from Hardy-Weinberg equilibrium (P-value<1x10−4). After filtering the genotypes, we used the remaining 1,589,371 SNPs for genotype imputation on the Michigan TOPmed Server (Minimac v485). In total, we generated imputed genotypes of all SNPs (r2>0.3) included in the TOPmed panel for the analysis described in ATAC-seq allelic imbalance analysis.
RNA isolation, sequencing, and processing
For the 20 KO and 2 WT hESC lines described in Generation of isogenic hESC lines, we selected and expanded clones, differentiated hESCs into hESC-β cells (see β-cell differentiation protocol of hESCs), and generated RNA-seq data on the purified hESC-β cells (Table S2). We selected a single clone for the KO lines and two clones for the WT lines (Table S2). For each clone, we performed the differentiation and RNA-seq experiment in 3-4 replicates (Table S2). For each replicate, we extracted and purified total RNA from the FACS-sorted hESCs-β cells (see Table S2) using the Absolutely RNA Nanoprep kit (Agilent Technologies), quantified with a NanoDrop spectrophotometer (Thermo Fisher Scientific). We used the Weill Cornell Genomics Core to sequence the purified RNA. Briefly, we evaluated the quality of RNA samples using the Agilent bioanalyzer (Agilent Technologies), generated cDNA libraries using TruSeq RNA Sample Preparation (Illumina) and sequenced the cDNA libraries using an Illumina NovaSeq 6000 with 2x51 bp cycles (Illumina). We aligned the processed reads to the GRCh38 genome assembly using STAR v2.73a86 with default parameters and quantified expression levels of Gencode v19 genes (Ensembl release 103) using GoRTs (v1.3.687; Table S7). Finally, we generated a raw read count matrix of gene by library and a normalized mRNA expression matrix of transcripts per million (TPM). On average, we generated 36,586,780 (13,273,689-123,518,245) paired-end reads per library, of which 84.36% were uniquely aligned to the genome. Out of the aligned reads, 79.42% were unambiguously assigned to unique genes, emphasizing the quality of these data. For sequencing statistics, see Table S7.
RNA-seq quality control
To assess the reproducibility of RNA-seq data from replicate libraries, we normalized the gene expression using log2(counts per million total reads [CPM]) and calculated Pearson correlation of pair-wise replicate libraries. We did not identify any outlier (minimum Pearson's r≥0.95) and used the resulting data for downstream analysis.
To assess the contamination of the total RNA isolated for RNA-seq, we combined the aligned reads across replicate libraries and flagged duplicate reads in the bam files using GATK v4.1.9.0 MarkDuplicates88 with default options. Using the “view” function from Samtools v1.989,90 with option “-F 3840”, we removed duplicate reads as well as those reads that mapped to supplementary/secondary alignments. Finally, using the INSGFP/w MEL1 genotypes (see INSGFP/w MEL1 genotyping, quality control, and imputation) as the reference panel, we used verifybamID v1.1.191 with options --ignoreRG --precise --self --maxDepth 100 to identify clones with RNA that was likely contaminated (FREEMIX>5%) or did not match reference genotypes (CHIPMIX>5%). We did not identify any problematic clones.
ATAC-seq library preparation, sequencing, and processing
We used the FACS-sorted hESC-β cells described in RNA isolation, sequencing, and processing to perform ATAC-seq and prepared samples according to Weill Cornell Medicine Epigenetics Core facility protocol114. Briefly, we sorted 50,000 INS-GFP+ cells, washed them with 1000 μl of ice-cold PBS, and resuspended the pellets in 25 μl of ice cold 1X ATAC Buffer [20mM Tris-HCl (pH 7.4), 20mM NaCl and 6mM MgCl2]. We incubated the samples for 5 minutes on ice, thoroughly mixed in 25 μl of ice cold ATAC-Detergent-buffer [20mM Tris-HCl (pH 7.4), 20 mM NaCl and 6 mM MgCl2, 0.2% Igepal CA-630 (Sigma Aldrich), 0.2% Tween 20 (Sigma Aldrich) and 0.02% Digitonin (Promega) and continued incubating the samples on ice for another 3 minutes. After incubation, we centrifuged the samples and collected the pellets. Next, we resuspended the pellets in the following transposase mixture: 25 μl 2X TD Buffer (Illumina), 2.5 μl TDE1 (Illumina), 16.5 ul PBS, 0.5 ul Digitonin (1%), 0.5 ul Tween-20 (10%), and 5 ul H2O. We incubated the suspended cells at 37°C for 30 minutes in a thermomixer (Benchmark) set to 500 rpm. We added 250 μl of Zymo DNA binding buffer to the suspension and purified the tagmented DNA with Zymo DNA clean and concentrator (Zymo research) according to manufacturer’s instructions. We submitted the samples to the Weill Cornell Medicine Epigenetics Core facility for library preparation according to a previously published method115 and NovaSeq SP (800M reads) 2x50 cycles (PE50) sequencing. We trimmed adaptor sequences using CTA (vO. 1.2) and aligned the trimmed reads to the GRCh38 genome assembly using BWA-MEM v0.7.17-r1194 with the - M option.92 On average, we generated 94,307,193 (70,146,700-111,259,636) reads per library, of which 96.84% aligned to the genome as primary alignments. After removing duplicate reads with GATK v4.1.9.0 MarkDuplicates and filtering for autosomal, properly paired reads with mapping quality ≥30 with samtools v1.989,90, we retained 63,255,116 uniquely aligned primary reads per library (minimum of 47,329,535) for downstream analyses (Table S7).
Using the filtered reads, we called ATAC peaks as described in Rai et al..116 Briefly, we converted the aligned BAM files to BED files using the bamtobed function from bedtools v2.26.093 and called peaks using MACS2 v2.2.7.194 with options “--nomodel --shift -100 --extsize 200 -B --keep-dup all”, removing candidate peaks that overlap with ENCODE blacklists117 and controlling for a false discovery rate (FDR) of 5%. For each hESC-β line (20 KO and 2 WT), we merged peaks across replicates and retained peaks present in ≥2 replicates. Next, we created a master set of peaks by merging peaks across all 22 lines, generating 208,945 peaks. Finally, we used this master set of peaks to quantify the number of reads mapping to the peaks within each library using the “multicov” function from bedtools v— with the option “-q 30” and the aligned BAM files, generating the accessible chromatin region count matrix.
ATAC-seq quality control
We used FastQC and MultiQC to generate and aggregate QC metrics across libraries. We evaluated the base quality scores (“per_base_sequence_quality_scores”) and sequence quality scores (“per_sequence_quality_scores”), and detected all samples passed MultiQC thresholds with “pass”. To assess reproducibility across hESC-β line replicates, we normalized the peak count matrix (see ATAC-seq library preparation, sequencing, and processing) using log2(CPM) and calculated the pairwise Pearson correlation of the normalized counts between replicates for each hESC-β line. We identified no outlier libraries (Pearson correlation coefficients ≥0.97). Finally, we identified potential contamination or sample swaps in the isogenic hESC lines by combining the filtered, aligned reads (see ATAC-seq library preparation, sequencing, and processing) across replicates (merge function from Samtools v1.989,90) and using verifybamID v1.1.191 with options “--ignoreRG --precise --self -maxDepth 100” on the merged reads and INSGFP/w MEL1 genotypes (as the reference panel). We identified no contamination (all lines with FREEMIX<5%) or sample swaps (all lines with CHIPMIX<3%).
Identification of differentially expressed genes and differentially accessible regions in KO hESC-β cells compared to WT cells
We tested for differential expression and accessibility in the KO hESC-β cells compared to the WT cells (see Table S2) using DESeq2 v1.32.0.95 For each set of comparisons, we retained features (e.g., genes or chromatin regions) with CPM≥0.5 in ≥50% of the replicate libraries across the KO and WT lines (>=3). To identify shared differences between the KO line and both WT lines, we used the Wald test implemented in DESeq2 with default options to compare the KO line against each WT line and meta-analyzed the results using the rem_mv function from MetaVolcano v1.10.096 with default parameters. We performed multiple hypothesis correction using the Benjamini-Hochberg procedure118 and considered features with ∣FC∣>1.5 and FDR<5% to be differentially expressed or accessible.
To test for differential expression, we used the gene expression matrix described in RNA isolation, sequencing, and processing. To test for differential chromatin accessibility, we used the accessible chromatin region count matrix described in ATAC-seq library preparation, sequencing, and processing.
T2D effector genes
We downloaded a list of 257 T2D predicted effector genes generated by integrating the results from three different approaches, namely, “Curated T2D effector gene prediction”, “Effector index predictions”, and “Integrated classifier predictions” (https://t2d.hugeamp.org/method.html?trait=t2d&dataset=egls, accessed October 1, 2022) and considered them as the “T2D effector genes”.
Enrichment of gene sets in KO hESC-β cells compared to WT cells
We tested for gene sets enriched in the KO hESC-β cells compared to WT cells using fgsea v1.20.0.97 Using the differential expression results from each KO line (see Identifying differentially expressed genes and differentially accessible regions in KO hESC-β cells compared to WT cells), we ranked genes by the meta-analysis log2(FC) in descending order. We performed gene set enrichment analysis using fgsea with default parameters, the ranked gene list, and gene sets from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (obtained from the R package msigdbr v7.5.198). We performed multiple hypothesis correction using the Benjamini-Hochberg procedure118 and considered gene sets with FDR<5% as enriched.
β-cell gene expression specificity scores
We used a single cell RNA-seq dataset of 12 libraries prepared from human pancreatic islets of one donor39 to assess islet cell type specific expression of genes. We collected sequence reads of 16,028 cells representing major endocrine and exocrine cell types. We normalized the raw read counts by library size and transcript length for each cell and generated a TPM matrix of cell barcode by gene. Next, we applied CELLEX v1.2.2101 to derive cell type specificity scores using the TPM matrix and cell type labels.
Enrichment of differentially expressed genes in β-cell specific genes
We binned protein coding genes into 10 groups with equal number of genes where we have β-cell expression specificity scores (>0) using the single cell RNA-seq data from 39 (see β-cell gene expression specificity scores). This approach defines the genes in bin 1 to be highly specific to β cells and those in bin 10 the least specific. We included genes that are expressed ubiquitously (specificity score = 0) into the bin 10. Next, we tested enrichment of differentially expressed genes in each bin per KO line using Fisher’s exact test (fisher.test function of R stats package; v4.1.2). We performed multiple hypothesis correction using the Benjamini-Hochberg procedure118 per KO line and considered gene sets with FDR<5% as enriched.
Association between chromatin accessibility/gene expression and cellular traits
To identify associations with insulin content, apoptotic rate, differentiation efficiency, glucose-induced insulin secretion (GSIS), and KCl-induced insulin secretion (KSIS), we performed differential chromatin accessibility and gene expression analysis across all samples. Since sequencing replicates and phenotypic assay replicates were not paired, we summed the feature (i.e., accessible chromatin regions or genes) reads and averaged the cellular assay results across replicates. We standardized the cellular trait values, removed features with low signal—keeping accessible regions and genes with CPM ≥0.5 in ≥50% of samples—and used DESeq2 v1.32.095 to test for an association between each feature and cellular trait. For each omics feature type and cellular trait pair, we removed tests where the regression was driven by an outlier(s) (minimum Cook's distance P-value<0.01119), used the Benjamini-Hochberg procedure118 to control for the number of tests, and considered tests with ∣FC∣>1.5 and FDR<5% to be associated.
Enrichment of differentially expressed genes at differentially accessible regions
For each KO line, we tested for enrichment of DEGs nearby DARs (see Identifying differentially expressed genes and differentially accessible regions in KO hESC-β cells compared to WT cells). Briefly, we performed a Fisher’s exact test (fisher.test function of R stats package; v4.1.2) to evaluate the enrichment of DARs out of all ATAC-seq peaks that overlap with DARs where the transcription start site (TSS) is within a specified window size. For TSSs, we used the genomic coordinates defined in the NCBI RefSeq release (NCBI RefSeq table; GRCh38 assembly) from UCSC Genome Browser (https://genome.ucsc.edu/)99. We tested using 5kb, 10kb, 25kb, 50kb, and 100kb window sizes. For each window size analysis, we applied the Benjamini-Hochberg procedure118 to correct for multiple hypotheses testing across all KO lines. We considered DARs to be enriched around DEGs at 5% FDR.
Association of gene expression and chromatin accessibility
Using the paired RNA-seq and ATAC-seq data for each hESC-β replicate, we tested for associations between gene expression and chromatin accessibility. We removed any gene or ATAC peak with CPM≤0.5 in ≥50% of all 67 libraries to focus on shared features across all KO lines and considered gene-peak pairs where the peak was within 50kb of either side of the gene TSS, which was derived from NCBI RefSeq release (NCBI RefSeq table; GRCh38 assembly) from UCSC Genome Browser (https://genome.ucsc.edu/).99 For each gene-peak pair, we used the qtl_test_lmm function from LIMIX v1.0.17100 to fit a linear regression to model the inverse-normalized peak counts as the dependent variable and the inverse-normalized gene expression as the independent variable. We controlled for the FDR across all gene-peak tests using the BH procedure.118 To explore the effects of different window sizes, we also tested for associations using 5kb, 25kb, and 100kb windows around the gene TSSs to identify gene-peak pairs. For the gene expression and chromatin accessibility data, we used the counts matrices described in Association between chromatin accessibility/gene expression and cellular trait.
Effects of T2D GWAS credible set of SNPs on transcription factor (TF) footprints
To assess the effect of SNPs on regulatory elements, we performed a TF footprint analysis in the merged ATAC-seq peaks for each cell line individually. We scanned the peak regions with position weight matrices (PWMs) of the directly determined TF motifs included in Cis-BP v2120 using “Find Individual Motif Occurrences” (FIMO) v5.4.1102 with default options. Next, we used CENTIPEDE v1.2104 to call footprints for each FIMO scan result in combination with the corresponding ATAC-seq aligned bam file. This approach allowed us to measure the number of transposase Tn5 integration events at a region ±100bp from each motif occurrence. We defined a motif occurrence to be bound by the respective TF if the CENTIPEDE posterior probability was ≥0.95 and its coordinates were fully contained within an ATAC-seq peak. We further considered any T2D 99% credible set SNP3 overlapping such a motif occurrence to be potentially disrupting the binding site of the respective TF.
Enrichment of differential ATAC-seq peaks in ChromHMM
To investigate the enrichment of DARs in islet regulatory regions, we analyzed the regulatory features defined by ChromHMM45. Using the intersect function of bedtools (v2.26.0), we compared DARs identified in "Identification of differentially expressed genes and differentially accessible regions in KO hESC-β cells compared to WT cells" with each ChromHMM feature, with a restriction of DARs that overlapped at least 50% with a feature of interest (using option "-f 0.5"). We conducted Fishers’ exact test (fisher.test function of R stats package; v4.1.2) to evaluate the enrichment of suppressed DARs among all DARs that overlap with a feature. We also performed the same test in the other way around - for the enrichment of activated DARs for the set of features. We applied the Benjamini-Hochberg procedure118 to correct for multiple hypotheses across all 14 ChromHMM features. We considered a feature of interest in a line to have enriched suppressed/activated DARs at 5% FDR.
Enrichment of TFBSs in differential ATAC-seq peaks
We tested for enrichment of TF binding site motifs for 677 TFs that are expressed in WT line (TPM>0) in the suppressed or activated ATAC-seq peaks in each KO and WT line (see Effects of T2D GWAS credible set of SNPs on transcription factor (TF) footprint and Comparison of chromatin accessibility in KO and WT lines) using “Simple Enrichment Analysis” (SEA) with default options (v5.4.1)103. We performed multiple hypothesis correction using the Benjamini-Hochberg procedure118 and considered motifs with FDR<5% to be enriched.
ATAC-seq allelic imbalance analysis
For each line, we filtered duplicate reads, reads identified as secondary alignments, or reads with poor mapping quality (<30) and merged the aligned, paired reads across replicates using the “merge” function from samtools v1.9.89,90 We applied WASP v0.3.4105 to quantify allele counts while controlling for mapping biases at heterozygous variants in the INSGFP/w MEL1 parental line, using only variants with an imputation quality r2>0.3. From the allelic counts generated by WASP, we selected T2D 99% credible sets SNPs from Mahajan et al.3 and performed a two-sided binomial test (binom.test function in R v4.2.2) in each line across all variants with >=1 total counts. To identify common effects across lines, we performed a meta-analysis using Stouffer’s Z-score method,121,122 weighting the Z-scores from each line by the total read counts overlapping the variant (sumz method from metap R package v1.8). For both line-specific and common effect analyses, we controlled for the number of tests using the Benjamini-Hochberg procedure118.
We performed gene-based annotation for the imbalanced SNPs using ANNOVAR106 (GRCh38 refSeq table).
Association between ATAC-seq allelic imbalance signals with cellular traits
Using the allelic counts generated by WASP (see ATAC-seq allelic imbalance analysis), we selected T2D 99% credible sets SNPs from Mahajan et al.3 and fit a binomial regression (sm.GLM function with family set to sm.families. Binomial() from the statsmodels Python package v0.13.2) across all lines, testing for an association between allelic imbalance and each cellular trait. For each variant considered for allelic imbalance, we standardized the cellular trait values prior to fitting the regression. We included all variants in the analysis with >=1 count. Finally, we removed associations driven by outliers, dropping those with a minimum Cook's distance P-value<0.01119, and controlled for the number of tests for each cellular trait using the Benjamini-Hochberg procedure118.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis for functional assay
All experiments were performed with >=3 independent replicates unless otherwise specified in the Figure legends. Unless otherwise noted in the rest of the method sections, for comparisons of functional assay results, we calculated mean ± SD for each assay using >=3 independent biological replicates. We included descriptions of each statistical test and the n and P values in each Figure legend and related experimental method sections.
Supplementary Material
Document S1. Figures S1-S7 and Tables S1-S4 and S7
Table S5. Summary of ATAC-seq allelic imbalance signatures at heterozygous SNPs in 99% credible sets for T2D association, related to Figure 6 and STAR Methods.
Table S7. Summary of RNA/ATAC-seq sequencing statistics, related to STAR Methods.
Highlight.
Deficiency of 20 T2D genes affects molecular and cellular phenotypes of β cells.
rs7132908 affects HNF4A binding and is likely a T2D causal variant.
Four genes are associated with insulin production and two with β-cell survival.
ATAC-seq allele imbalance analysis refines 23 GWAS signals at single-SNP resolution.
Acknowledgments
This research was supported in part by the National Institutes of Health grant 1-ZIA-HG000024 (to F.S.C.), NIDDK grants (R01 DK124463, R01 DK116075-01A1, R01 DK119667-01A1, and 1U01DK127777-01 to S.C.), and the American Diabetes Association grant (9-22-PDFPM-06 to D.X.). The authors thank Kevin W. Currin and Cassie Robertson for helpful advice in the data analysis, Efsun Arda for her valuable feedback, Vivian Guo for her assistance in the molecular cloning work, and Tuo Zhang for helping with data transfer and submission. The authors also are grateful for support provided by the Genomics and Epigenomics Core Facility and Flow Cytometry Core Facility at Weill Cornell Medicine and acknowledge the NIAMS Genomic Technology Section for conducting whole genome sequencing work and the NIH HPC Biowulf cluster for providing computational resources.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests
S.C. is the co-founder of OncoBeat, LLC. and a consultant of Vesalius Therapeutics. The other authors declare no competing interest.
References
- 1.Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, Colagiuri S, Guariguata L, Motala AA, Ogurtsova K, et al. (2019). Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract 157, 107843. 10.1016/j.diabres.2019.107843. [DOI] [PubMed] [Google Scholar]
- 2.Galicia-Garcia U, Benito-Vicente A, Jebari S, Larrea-Sebal A, Siddiqi H, Uribe KB, Ostolaza H, and Martín C (2020). Pathophysiology of type 2 diabetes mellitus. Int. J. Mol. Sci 21. 10.3390/ijms21176275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mahajan A, Spracklen CN, Zhang W, Ng MCY, Petty LE, Kitajima H, Yu GZ, Rüeger S, Speidel L, Kim YJ, et al. (2022). Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat. Genet 54, 560–572. 10.1038/s41588-022-01058-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vujkovic M, Keaton JM, Lynch JA, Miller DR, Zhou J, Tcheandjieu C, Huffman JE, Assimes TL, Lorenz K, Zhu X, et al. (2020). Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet 52, 680–691. 10.1038/s41588-020-0637-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spracklen CN, Horikoshi M, Kim YJ, Lin K, Bragg F, Moon S, Suzuki K, Tam CH, Tabara Y, Kwak S-H, et al. (2019). Identification of type 2 diabetes loci in 433,540 East Asian individuals. BioRxiv. 10.1101/685172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, Payne AJ, Steinthorsdottir V, Scott RA, Grarup N, et al. (2018). Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet 50, 1505–1513. 10.1038/s41588-018-0241-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krentz NAJ, and Gloyn AL (2020). Insights into pancreatic islet cell dysfunction from type 2 diabetes mellitus genetics. Nat. Rev. Endocrinol 16, 202–212. 10.1038/s41574-020-0325-0. [DOI] [PubMed] [Google Scholar]
- 8.Mattis KK, and Gloyn AL (2020). From Genetic Association to Molecular Mechanisms for Islet-cell Dysfunction in Type 2 Diabetes. J. Mol. Biol 432, 1551–1578. 10.1016/j.jmb.2019.12.045. [DOI] [PubMed] [Google Scholar]
- 9.Viñuela A, Varshney A, van de Bunt M, Prasad RB, Asplund O, Bennett A, Boehnke M, Brown AA, Erdos MR, Fadista J, et al. (2020). Genetic variant effects on gene expression in human pancreatic islets and their implications for T2D. Nat. Commun 11, 4912. 10.1038/s41467-020-18581-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alonso L, Piron A, Morán I, Guindo-Martínez M, Bonàs-Guarch S, Atla G, Miguel-Escalada I, Royo R, Puiggròs M, Garcia-Hurtado X, et al. (2021). TIGER: The gene expression regulatory variation landscape of human pancreatic islets. Cell Rep. 37, 109807. 10.1016/j.celrep.2021.109807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weeks EM, Ulirsch JC, Cheng NY, Trippe BL, Fine RS, Miao J, Patwardhan TA, Kanai M, Nasser J, Fulco CP, et al. (2020). Leveraging polygenic enrichments of gene features to predict genes underlying complex traits and diseases. medRxiv. 10.1101/2020.09.08.20190561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Costanzo MC, von Grotthuss M, Massung J, Jang D, Caulkins L, Koesterer R, Gilbert C, Welch RP, Kudtarkar P, Hoang Q, et al. (2023). The Type 2 Diabetes Knowledge Portal: An open access genetic resource dedicated to type 2 diabetes and related traits. Cell Metab. 35, 695–710.e6. 10.1016/j.cmet.2023.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deaton AM, Parker MM, Ward LD, Flynn-Carroll AO, BonDurant L, Hinkle G, Akbari P, Lotta LA, Regeneron Genetics Center, DiscovEHR Collaboration, et al. (2021). Gene-level analysis of rare variants in 379,066 whole exome sequences identifies an association of GIGYF1 loss of function with type 2 diabetes. Sci. Rep 11, 21565. 10.1038/s41598-021-99091-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flannick J, Mercader JM, Fuchsberger C, Udler MS, Mahajan A, Wessel J, Teslovich TM, Caulkins L, Koesterer R, Barajas-Olmos F, et al. (2019). Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature 570, 71–76. 10.1038/s41586-019-1231-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Udler MS, Kim J, von Grotthuss M, Bonàs-Guarch S, Cole JB, Chiou J, Christopher D Anderson on behalf of METASTROKE and the ISGC, Boehnke M, Laakso M, Atzmon G, et al. (2018). Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: A soft clustering analysis. PLoS Med. 15, e1002654. 10.1371/journal.pmed.1002654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O’Dwyer S, Quiskamp N, Mojibian M, Albrecht T, et al. (2014). Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol 32, 1121–1133. 10.1038/nbt.3033. [DOI] [PubMed] [Google Scholar]
- 17.Davis JC, Alves TC, Helman A, Chen JC, Kenty JH, Cardone RL, Liu DR, Kibbey RG, and Melton DA (2020). Glucose Response by Stem Cell-Derived β Cells In Vitro Is Inhibited by a Bottleneck in Glycolysis. Cell Rep. 31, 107623. 10.1016/j.celrep.2020.107623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Domcke S, Hill AJ, Daza RM, Cao J, O’Day DR, Pliner HA, Aldinger KA, Pokholok D, Zhang F, Milbank JH, et al. (2020). A human cell atlas of fetal chromatin accessibility. Science 370, eaba7612. 10.1126/science.aba7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lithovius V, Saarimäki-Vire J, Balboa D, Ibrahim H, Montaser H, Barsby T, and Otonkoski T (2021). SUR1-mutant iPS cell-derived islets recapitulate the pathophysiology of congenital hyperinsulinism. Diabetologia 64, 630–640. 10.1007/s00125-020-05346-7. [DOI] [PubMed] [Google Scholar]
- 21.McGrath PS, Watson CL, Ingram C, Helmrath MA, and Wells JM (2015). The Basic Helix-Loop-Helix Transcription Factor NEUROG3 Is Required for Development of the Human Endocrine Pancreas. Diabetes 64, 2497–2505. 10.2337/db14-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, Sterr M, Ansarullah, Burtscher I, Böttcher A, Beckenbauer J, Siehler J, Meitinger T, Häring H-U, Staiger H, et al. (2019). Point mutations in the PDX1 transactivation domain impair human β-cell development and function. Mol. Metab 24, 80–97. 10.1016/j.molmet.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amin S, Cook B, Zhou T, Ghazizadeh Z, Lis R, Zhang T, Khalaj M, Crespo M, Perera M, Xiang JZ, et al. (2018). Discovery of a drug candidate for GLIS3-associated diabetes. Nat. Commun 9, 2681. 10.1038/s41467-018-04918-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gage BK, Asadi A, Baker RK, Webber TD, Wang R, Itoh M, Hayashi M, Miyata R, Akashi T, and Kieffer TJ (2015). The role of ARX in human pancreatic endocrine specification. PLoS ONE 10, e0144100. 10.1371/journal.pone.0144100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi Z-D, Lee K, Yang D, Amin S, Verma N, Li QV, Zhu Z, Soh C-L, Kumar R, Evans T, et al. (2017). Genome Editing in hPSCs Reveals GATA6 Haploinsufficiency and a Genetic Interaction with GATA4 in Human Pancreatic Development. Cell Stem Cell 20, 675–688.e6. 10.1016/j.stem.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tiyaboonchai A, Cardenas-Diaz FL, Ying L, Maguire JA, Sim X, Jobaliya C, Gagne AL, Kishore S, Stanescu DE, Hughes N, et al. (2017). GATA6 Plays an Important Role in the Induction of Human Definitive Endoderm, Development of the Pancreas, and Functionality of Pancreatic β Cells. Stem Cell Reports 8, 589–604. 10.1016/j.stemcr.2016.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Velazco-Cruz L, Goedegebuure MM, Maxwell KG, Augsornworawat P, Hogrebe NJ, and Millman JR (2020). SIX2 Regulates Human β Cell Differentiation from Stem Cells and Functional Maturation In Vitro. Cell Rep. 31, 107687. 10.1016/j.celrep.2020.107687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo M, Zhang T, Dong X, Xiang JZ, Lei M, Evans T, Graumann J, and Chen S (2017). Using hESCs to Probe the Interaction of the Diabetes-Associated Genes CDKAL1 and MT1E. Cell Rep. 19, 1512–1521. 10.1016/j.celrep.2017.04.070. [DOI] [PubMed] [Google Scholar]
- 29.Zeng H, Guo M, Zhou T, Tan L, Chong CN, Zhang T, Dong X, Xiang JZ, Yu AS, Yue L, et al. (2016). An Isogenic Human ESC Platform for Functional Evaluation of Genome-wide-Association-Study-Identified Diabetes Genes and Drug Discovery. Cell Stem Cell 19, 326–340. 10.1016/j.stem.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Human Genetics Knowledge Portal - Research Method. https://t2d.hugeamp.org/method.html?trait=t2d&dataset=egls.
- 31.Human Genetics Knowledge Portal - Research Method. https://t2d.hugeamp.org/method.html?trait=t2d&dataset=mccarthy.
- 32.Su C, Gao L, May CL, Pippin JA, Boehm K, Lee M, Liu C, Pahl MC, Golson ML, Naji A, et al. (2022). 3D chromatin maps of the human pancreas reveal lineage-specific regulatory architecture of T2D risk. Cell Metab. 34, 1394–1409.e4. 10.1016/j.cmet.2022.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forgetta V, Jiang L, Vulpescu NA, Hogan MS, Chen S, Morris JA, Grinek S, Benner C, Jang D-K, Hoang Q, et al. (2022). An effector index to predict target genes at GWAS loci. Hum. Genet 141, 1431–1447. 10.1007/s00439-022-02434-z. [DOI] [PubMed] [Google Scholar]
- 34.Micallef SJ, Li X, Schiesser JV, Hirst CE, Yu QC, Lim SM, Nostro MC, Elliott DA, Sarangi F, Harrison LC, et al. (2012). INS(GFP/w) human embryonic stem cells facilitate isolation of in vitro derived insulin-producing cells. Diabetologia 55, 694–706. 10.1007/s00125-011-2379-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bieganowski P, Shilinski K, Tsichlis PN, and Brenner C (2004). Cdc123 and checkpoint forkhead associated with RING proteins control the cell cycle by controlling elF2gamma abundance. J. Biol. Chem 279, 44656–44666. 10.1074/jbc.M406151200. [DOI] [PubMed] [Google Scholar]
- 36.Ohno K, Okuda A, Ohtsu M, and Kimura G (1984). Genetic analysis of control of proliferation in fibroblastic cells in culture. I. Isolation and characterization of mutants temperature-sensitive for proliferation or survival of untransformed diploid rat cell line 3Y1. Somat. Cell Mol. Genet 10, 17–28. 10.1007/BF01534469. [DOI] [PubMed] [Google Scholar]
- 37.Oh YS, Bae GD, Baek DJ, Park E-Y, and Jun H-S (2018). Fatty Acid-Induced Lipotoxicity in Pancreatic Beta-Cells During Development of Type 2 Diabetes. Front Endocrinol (Lausanne) 9, 384. 10.3389/fendo.2018.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dinić S, Arambašić Jovanović J, Uskoković A, Mihailović M, Grdović N, Tolić A, Rajić J, Ðorđević M, and Vidaković M (2022). Oxidative stress-mediated beta cell death and dysfunction as a target for diabetes management. Front Endocrinol (Lausanne) 13, 1006376. 10.3389/fendo.2022.1006376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bonnycastle LL, Gildea DE, Yan T, Narisu N, Swift AJ, Wolfsberg TG, Erdos MR, and Collins FS (2020). Single-cell transcriptomics from human pancreatic islets: sample preparation matters. Biol. Methods Protoc 5, bpz019. 10.1093/biomethods/bpz019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen W-M, Erdos MR, Jackson AU, Saxena R, Sanna S, Silver KD, Timpson NJ, Hansen T, Orrù M, Grazia Piras M, et al. (2008). Variations in the G6PC2/ABCB11 genomic region are associated with fasting glucose levels. J. Clin. Invest 118, 2620–2628. 10.1172/JCI34566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aigha II, and Abdelalim EM (2020). NKX6.1 transcription factor: a crucial regulator of pancreatic β cell development, identity, and proliferation. Stem Cell Res. Ther 11, 459. 10.1186/s13287-020-01977-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rutter GA, Georgiadou E, Martinez-Sanchez A, and Pullen TJ (2020). Metabolic and functional specialisations of the pancreatic beta cell: gene disallowance, mitochondrial metabolism and intercellular connectivity. Diabetologia 63, 1990–1998. 10.1007/s00125-020-05205-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pullen TJ, Sylow L, Sun G, Halestrap AP, Richter EA, and Rutter GA (2012). Overexpression of monocarboxylate transporter-1 (SLC16A1) in mouse pancreatic β-cells leads to relative hyperinsulinism during exercise. Diabetes 61, 1719–1725. 10.2337/db11-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pullen TJ, Huising MO, and Rutter GA (2017). Analysis of Purified Pancreatic Islet Beta and Alpha Cell Transcriptomes Reveals 11β-Hydroxysteroid Dehydrogenase (Hsd11b1) as a Novel Disallowed Gene. Front. Genet 8, 41. 10.3389/fgene.2017.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Varshney A, Scott LJ, Welch RP, Erdos MR, Chines PS, Narisu N, Albanus RD, Orchard P, Wolford BN, Kursawe R, et al. (2017). Genetic regulatory signatures underlying islet gene expression and type 2 diabetes. Proc Natl Acad Sci USA 114, 2301–2306. 10.1073/pnas.1621192114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parker SCJ, Stitzel ML, Taylor DL, Orozco JM, Erdos MR, Akiyama JA, van Bueren KL, Chines PS, Narisu N, NISC Comparative Sequencing Program, et al. (2013). Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci USA 110, 17921–17926. 10.1073/pnas.1317023110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsonkova VG, Sand FW, Wolf XA, Grunnet LG, Kirstine Ringgaard A, Ingvorsen C, Winkel L, Kalisz M, Dalgaard K, Bruun C, et al. (2018). The EndoC-βH1 cell line is a valid model of human beta cells and applicable for screenings to identify novel drug target candidates. Mol. Metab 8, 144–157. 10.1016/j.molmet.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burns SM, Vetere A, Walpita D, Dančík V, Khodier C, Perez J, Clemons PA, Wagner BK, and Altshuler D (2015). High-throughput luminescent reporter of insulin secretion for discovering regulators of pancreatic Beta-cell function. Cell Metab. 21, 126–137. 10.1016/j.cmet.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 49.Rozowsky J, Abyzov A, Wang J, Alves P, Raha D, Harmanci A, Leng J, Bjornson R, Kong Y, Kitabayashi N, et al. (2011). AlleleSeq: analysis of allele-specific expression and binding in a network framework. Mol. Syst. Biol 7, 522. 10.1038/msb.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reddy TE, Gertz J, Pauli F, Kucera KS, Varley KE, Newberry KM, Marinov GK, Mortazavi A, Williams BA, Song L, et al. (2012). Effects of sequence variation on differential allelic transcription factor occupancy and gene expression. Genome Res. 22, 860–869. 10.1101/gr.131201.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hodson DJ, Mitchell RK, Marselli L, Pullen TJ, Gimeno Brias S, Semplici F, Everett KL, Cooper DMF, Bugliani M, Marchetti P, et al. (2014). ADCY5 couples glucose to insulin secretion in human islets. Diabetes 63, 3009–3021. 10.2337/db13-1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moyerbrailean GA, Richards AL, Kurtz D, Kalita CA, Davis GO, Harvey CT, Alazizi A, Watza D, Sorokin Y, Hauff N, et al. (2016). High-throughput allele-specific expression across 250 environmental conditions. Genome Res. 26, 1627–1638. 10.1101/gr.209759.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knowles DA, Davis JR, Edgington H, Raj A, Favé M-J, Zhu X, Potash JB, Weissman MM, Shi J, Levinson DF, et al. (2017). Allele-specific expression reveals interactions between genetic variation and environment. Nat. Methods 14, 699–702. 10.1038/nmeth.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taylor DL, Knowles DA, Scott LJ, Ramirez AH, Casale FP, Wolford BN, Guan L, Varshney A, Albanus RD, Parker SCJ, et al. (2018). Interactions between genetic variation and cellular environment in skeletal muscle gene expression. PLoS ONE 13, e0195788. 10.1371/journal.pone.0195788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duncan SA, Nagy A, and Chan W (1997). Murine gastrulation requires HNF-4 regulated gene expression in the visceral endoderm: tetraploid rescue of Hnf-4(−/−) embryos. Development 124, 279–287. 10.1242/dev.124.2.279. [DOI] [PubMed] [Google Scholar]
- 56.Korinek V, Barker N, Moerer P, van Donselaar E, Huls G, Peters PJ, and Clevers H (1998). Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat. Genet 19, 379–383. 10.1038/1270. [DOI] [PubMed] [Google Scholar]
- 57.Wang H-L, Wang L, Zhao C-Y, and Lan H-Y (2022). Role of TGF-Beta Signaling in Beta Cell Proliferation and Function in Diabetes. Biomolecules 12. 10.3390/biom12030373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pamir N, Lynn FC, Buchan AMJ, Ehses J, Hinke SA, Pospisilik JA, Miyawaki K, Yamada Y, Seino Y, McIntosh CHS, et al. (2003). Glucose-dependent insulinotropic polypeptide receptor null mice exhibit compensatory changes in the enteroinsular axis. Am. J. Physiol. Endocrinol. Metab 284, E931–9. 10.1152/ajpendo.00270.2002. [DOI] [PubMed] [Google Scholar]
- 59.Flanagan SE, Clauin S, Bellanné-Chantelot C, de Lonlay P, Harries LW, Gloyn AL, and Ellard S (2009). Update of mutations in the genes encoding the pancreatic beta-cell K(ATP) channel subunits Kir6.2 (KCNJ11) and sulfonylurea receptor 1 (ABCC8) in diabetes mellitus and hyperinsulinism. Hum. Mutat 30, 170–180. 10.1002/humu.20838. [DOI] [PubMed] [Google Scholar]
- 60.Miyachi Y, Miyazawa T, and Ogawa Y (2022). HNF1A mutations and beta cell dysfunction in diabetes. Int. J. Mol. Sci 23. 10.3390/ijms23063222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Singh VP, Lakshmi BJ, Singh S, Shah V, Goel S, Sarathi DP, and Kumar S (2012). Lack of Wdr13 gene in mice leads to enhanced pancreatic beta cell proliferation, hyperinsulinemia and mild obesity. PLoS ONE 7, e38685. 10.1371/journal.pone.0038685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stancill JS, Cartailler J-P, Clayton HW, O’Connor JT, Dickerson MT, Dadi PK, Osipovich AB, Jacobson DA, and Magnuson MA (2017). Chronic β-Cell Depolarization Impairs β-Cell Identity by Disrupting a Network of Ca2+-Regulated Genes. Diabetes 66, 2175–2187. 10.2337/db16-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang W, and Zhang C (2021). Targeting β-cell dedifferentiation and transdifferentiation: opportunities and challenges. Endocr. Connect 10, R213–R228. 10.1530/EC-21-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.DeLaForest A, Nagaoka M, Si-Tayeb K, Noto FK, Konopka G, Battle MA, and Duncan SA (2011). HNF4A is essential for specification of hepatic progenitors from human pluripotent stem cells. Development 138, 4143–4153. 10.1242/dev.062547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boj SF, Petrov D, and Ferrer J (2010). Epistasis of transcriptomes reveals synergism between transcriptional activators Hnf1alpha and Hnf4alpha. PLoS Genet. 6, e1000970. 10.1371/journal.pgen.1000970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ng NHJ, Jasmen JB, Lim CS, Lau HH, Krishnan VG, Kadiwala J, Kulkarni RN, Ræder H, Vallier L, Hoon S, et al. (2019). HNF4A Haploinsufficiency in MODY1 Abrogates Liver and Pancreas Differentiation from Patient-Derived Induced Pluripotent Stem Cells. iScience 16, 192–205. 10.1016/j.isci.2019.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miura A, Yamagata K, Kakei M, Hatakeyama H, Takahashi N, Fukui K, Nammo T, Yoneda K, Inoue Y, Sladek FM, et al. (2006). Hepatocyte nuclear factor-4alpha is essential for glucose-stimulated insulin secretion by pancreatic beta-cells. J. Biol. Chem 281, 5246–5257. 10.1074/jbc.M507496200. [DOI] [PubMed] [Google Scholar]
- 68.Dwivedi OP, Lehtovirta M, Hastoy B, Chandra V, Krentz NAJ, Kleiner S, Jain D, Richard A-M, Abaitua F, Beer NL, et al. (2019). Loss of ZnT8 function protects against diabetes by enhanced insulin secretion. Nat. Genet 51, 1596–1606. 10.1038/s41588-019-0513-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ma Q, Xiao Y, Xu W, Wang M, Li S, Yang Z, Xu M, Zhang T, Zhang Z-N, Hu R, et al. (2022). ZnT8 loss-of-function accelerates functional maturation of hESC-derived β cells and resists metabolic stress in diabetes. Nat. Commun 13, 4142. 10.1038/s41467-022-31829-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Davidson HW, Wenzlau JM, and O’Brien RM (2014). Zinc transporter 8 (ZnT8) and β cell function. Trends Endocrinol. Metab 25, 415–424. 10.1016/j.tem.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Karunakaran V, Wei C, and Bano G (2020). Monogenic Diabetes due to ABCC8/KCNJ11 Mutation: Case Study and Review of Literature. Journal of Endocrinology and Metabolism Research 1, 1–16. [Google Scholar]
- 72.Liu Z, Wang M, Zhang C, Zhou S, and Ji G (2022). Molecular functions of ceruloplasmin in metabolic disease pathology. Diabetes Metab. Syndr. Obes 15, 695–711. 10.2147/DMSO.S346648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Choi YJ, Shin MJ, Youn GS, Park JH, Yeo HJ, Yeo EJ, Kwon HJ, Lee LR, Kim NY, Kwon SY, et al. (2023). Protective Effects of PEP-1-GSTA2 Protein in Hippocampal Neuronal Cell Damage Induced by Oxidative Stress. Int. J. Mol. Sci 24. 10.3390/ijms24032767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Skalnaya MG, Skalny AV, and Tinkov AA (2017). Serum copper, zinc, and iron levels, and markers of carbohydrate metabolism in postmenopausal women with prediabetes and type 2 diabetes mellitus. J. Trace Elem. Med. Biol 43, 46–51. 10.1016/j.jtemb.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 75.Hasan A, Ahmed S, and Ali D (2021). Study of serum ceruloplasmin and zinc in type 2 diabetes mellitus. ZJMS 25, 480–485. 10.15218/zjms.2021.010. [DOI] [Google Scholar]
- 76.Marselli L, Piron A, Suleiman M, Colli ML, Yi X, Khamis A, Carrat GR, Rutter GA, Bugliani M, Giusti L, et al. (2020). Persistent or Transient Human β Cell Dysfunction Induced by Metabolic Stress: Specific Signatures and Shared Gene Expression with Type 2 Diabetes. Cell Rep. 33, 108466. 10.1016/j.celrep.2020.108466. [DOI] [PubMed] [Google Scholar]
- 77.Liu T, Zhao Y, Tang N, Feng R, Yang X, Lu N, Wen J, and Li L (2012). Pax6 directly down-regulates Pcsk1n expression thereby regulating PC1/3 dependent proinsulin processing. PLoS ONE 7, e46934. 10.1371/journal.pone.0046934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pringle NP, Guthrie S, Lumsden A, and Richardson WD (1998). Dorsal spinal cord neuroepithelium generates astrocytes but not oligodendrocytes. Neuron 20, 883–893. 10.1016/s0896-6273(00)80470-5. [DOI] [PubMed] [Google Scholar]
- 79.Zhou Y, Wang L, Ban X, Zeng T, Zhu Y, Li M, Guan XY, and Li Y (2018). DHRS2 inhibits cell growth and motility in esophageal squamous cell carcinoma. Oncogene 37, 1086–1094. 10.1038/onc.2017.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li Z, Tan Y, Li X, Quan J, Bode AM, Cao Y, and Luo X (2022). DHRS2 inhibits cell growth and metastasis in ovarian cancer by downregulation of CHKα to disrupt choline metabolism. Cell Death Dis. 13, 845. 10.1038/s41419-022-05291-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Arnaud L, Benech P, Greetham L, Stephan D, Jimenez A, Jullien N, García-González L, Tsvetkov PO, Devred F, Sancho-Martinez I, et al. (2022). APOE4 drives inflammation in human astrocytes via TAGLN3 repression and NF-κB activation. Cell Rep. 40, 111200. 10.1016/j.celrep.2022.111200. [DOI] [PubMed] [Google Scholar]
- 82.Legut M, Daniloski Z, Xue X, McKenzie D, Guo X, Wessels H-H, and Sanjana NE (2020). High-Throughput Screens of PAM-Flexible Cas9 Variants for Gene Knockout and Transcriptional Modulation. Cell Rep. 30, 2859–2868.e5. 10.1016/j.celrep.2020.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Addgene: psPAX2. http://n2t.net/addgene:12260.
- 84.Addgene: pMD2.G. http://n2t.net/addgene:12259.
- 85.Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, et al. (2016). Next-generation genotype imputation service and methods. Nat. Genet 48, 1284–1287. 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hartley SW, and Mullikin JC (2015). GoRTs: a comprehensive toolset for quality control and data processing of RNA-Seq experiments. BMC Bioinformatics 16, 224. 10.1186/s12859-015-0670-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Auwera G der AV, and O’Connor BD (2020). Genomics in the Cloud: Using Docker, GATK, and WDL in Terra 1st ed. (O’Reilly Media; ). [Google Scholar]
- 89.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, et al. (2021). Twelve years of SAMtools and BCFtools. Gigascience 10. 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jun G, Flickinger M, Hetrick KN, Romm JM, Doheny KF, Abecasis GR, Boehnke M, and Kang HM (2012). Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am. J. Hum. Genet 91, 839–848. 10.1016/j.ajhg.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cesar Prada HN (2019). MetaVolcanoR. Bioconductor. 10.18129/b9.bioc.metavolcanor. [DOI] [Google Scholar]
- 97.Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, and Sergushichev A (2016). Fast gene set enrichment analysis. BioRxiv. 10.1101/060012. [DOI] [Google Scholar]
- 98.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nassar LR, Barber GP, Benet-Pagès A, Casper J, Clawson H, Diekhans M, Fischer C, Gonzalez JN, Hinrichs AS, Lee BT, et al. (2023). The UCSC Genome Browser database: 2023 update. Nucleic Acids Res. 51, D1188–D1195. 10.1093/nar/gkac1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lippert C, Casale FP, Rakitsch B, and Stegle O (2014). LIMIX: genetic analysis of multiple traits. BioRxiv. 10.1101/003905. [DOI] [PubMed] [Google Scholar]
- 101.Timshel PN, Thompson JJ, and Pers TH (2020). Genetic mapping of etiologic brain cell types for obesity. eLife 9. 10.7554/eLife.55851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.McLeay RC, and Bailey TL (2010). Motif Enrichment Analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics 11, 165. 10.1186/1471-2105-11-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pique-Regi R, Degner JF, Pai AA, Gaffney DJ, Gilad Y, and Pritchard JK (2011). Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res. 27, 447–455. 10.1101/gr.112623.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.van de Geijn B, McVicker G, Gilad Y, and Pritchard JK (2015). WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat. Methods 12, 1061–1063. 10.1038/nmeth.3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang K, Li M, and Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tang X, Uhl S, Zhang T, Xue D, Li B, Vandana JJ, Acklin JA, Bonnycastle LL, Narisu N, Erdos MR, et al. (2021). SARS-CoV-2 infection induces beta cell transdifferentiation. Cell Metab. 33, 1577–1591.e7. 10.1016/j.cmet.2021.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Theis A, Singer RA, Garofalo D, Paul A, Narayana A, and Sussel L (2021). Groucho co-repressor proteins regulate β cell development and proliferation by repressing Foxa1 in the developing mouse pancreas. Development 148. 10.1242/dev.192401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tang X, Xue D, Zhang T, Nilsson-Payant BE, Carrau L, Duan X, Gordillo M, Tan AY, Qiu Y, Xiang J, et al. (2023). A multi-organoid platform identifies CIART as a key factor for SARS-CoV-2 infection. Nat. Cell Biol 25, 381–389. 10.1038/s41556-023-01095-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cancellieri S, Canver MC, Bombieri N, Giugno R, and Pinello L (2020). CRISPRitz: rapid, high-throughput and variant-aware in silico off-target site identification for CRISPR genome editing. Bioinformatics 36, 2001–2008. 10.1093/bioinformatics/btz867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu H, and Naismith JH (2008). An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91. 10.1186/1472-6750-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Currin KW, Erdos MR, Narisu N, Rai V, Vadlamudi S, Perrin HJ, Idol JR, Yan T, Albanus RD, Broadaway KA, et al. (2021). Genetic effects on liver chromatin accessibility identify disease regulatory variants. Am. J. Hum. Genet 108, 1169–1189. 10.1016/j.ajhg.2021.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Epigenomics Core @ WCMC. https://epicore.med.Cornell.edu/services.php?option=atacseqdescription#seq.
- 115.Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rai V, Quang DX, Erdos MR, Cusanovich DA, Daza RM, Narisu N, Zou LS, Didion JP, Guan Y, Shendure J, et al. (2020). Single-cell ATAC-Seq in human pancreatic islets and deep learning upscaling of rare cells reveals cell-specific type 2 diabetes regulatory signatures. Mol. Metab 32, 109–121. 10.1016/j.molmet.2019.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Benjamini Y, and Hochberg Y (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society: Series B (Methodological) 57, 289–300. 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- 119.Cook RD (1977). Detection of Influential Observation in Linear Regression. Technometrics 19, 15. 10.2307/1268249. [DOI] [Google Scholar]
- 120.Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443. 10.1016/j.cell.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Samuel A,S, Edward A,S, Leland C,D, Shirley A,S, and Robin M,W Jr. (1949). The American Soldier: Adjustment During Army Life (Princeton University Press; ). [Google Scholar]
- 122.Lipták T. (1958). On the combination of independent tests. Magyar Tud Akad Mat Kutato Int Kozl 3, 171–197. Sciwheel inserting bibliography... [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Figures S1-S7 and Tables S1-S4 and S7
Table S5. Summary of ATAC-seq allelic imbalance signatures at heterozygous SNPs in 99% credible sets for T2D association, related to Figure 6 and STAR Methods.
Table S7. Summary of RNA/ATAC-seq sequencing statistics, related to STAR Methods.
Data Availability Statement
The RNA-seq, ATAC-seq, whole genome sequence data, and SNP array genotyping data generated during this study are available at GEO under accession no. GSE228665. Source data used to generate the graphs in the paper can be found in the file Data S1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.