

Abstract
The Y-family DNA polymerases – Pol ι, Pol η, Pol κ and Rev1 – are most well-known for their roles in the DNA damage tolerance pathway of translesion synthesis (TLS). They function to overcome replication barriers by bypassing DNA damage lesions that cannot be normally replicated, allowing replication forks to continue without stalling. In this work, we demonstrate a novel interaction between each Y-family polymerase and the nucleotide excision repair (NER) proteins, RAD23A and RAD23B. We initially focus on the interaction between RAD23A and Pol ι, and through a series of biochemical, cell-based, and structural assays, find that the RAD23A ubiquitin-binding domains (UBA1 and UBA2) interact with separate sites within the Pol ι catalytic domain. While this interaction involves the ubiquitin-binding cleft of UBA2, Pol ι interacts with a distinct surface on UBA1. We further find that mutating or deleting either UBA domain disrupts the RAD23A-Pol ι interaction, demonstrating that both interactions are necessary for stable binding. We also provide evidence that both RAD23 proteins interact with Pol ι in a similar manner, as well as with each of the Y-family polymerases. These results shed light on the interplay between the different functions of the RAD23 proteins and reveal novel binding partners for the Y-family TLS polymerases.
Keywords: UV excision repair protein RAD23, ubiquitin-associated (UBA) domains, hHR23, translesion polymerases
Introduction
Efficient and accurate DNA replication is essential for maintaining the integrity of the genetic code. DNA polymerases are central to this process, as they catalyze the template-directed synthesis of new DNA from individual nucleotides. The bulk of DNA replication during synthesis (S) phase of the cell cycle is performed by the replicative B-family DNA polymerases α, δ and ε.1 These polymerases synthesize new DNA with high accuracy due in part to their closed active sites, that contain binding pockets that accommodate only the four canonical Watson-Crick base pairs.2 While high nucleotide selectivity prevents the misincorporation of incorrect nucleotides, it also prevents the replicative polymerases from synthesizing new DNA from templates that contain damaged nucleotides. Replication is therefore vulnerable to DNA damaging events as they can disrupt the progress of replication forks.3 One way to overcome such disruptions is through the specialized replication pathway of translesion synthesis (TLS). This pathway employs DNA polymerases with open active sites that can accommodate, and bypass, a wide range of DNA adducts and is exemplified by the Y-family polymerases – Pol ι, Pol η, Pol κ and Rev1.4,5 Although these proteins are important for overcoming replication barriers, their open active sites render them inherently error prone.2 Translesion synthesis must therefore be tightly regulated, as its inappropriate use could result in unnecessary and detrimental DNA mutagenesis. Despite notable examples of TLS regulation – e.g. by the recruitment of TLS polymerases by mono-ubiquitinated PCNA6 – a complete understanding of how these proteins are regulated, and the proteins they interact with, remains unclear.7
RAD23A and RAD23B are important scaffold proteins with roles in the regulation of protein degradation and the initial stages of global genome nucleotide excision repair (GG-NER).8 In the former, the RAD23 proteins function to shuttle ubiquitinated substrates to the proteasome.9 This is facilitated by an N-terminal ubiquitin-like (UBL) domain that interacts with proteasomal subunit S5a,10 and two ubiquitin associated domains (UBA1 and UBA2) that bind ubiquitin.11,12 Despite associating with the proteasome, the RAD23 proteins are not readily degraded. One reason for this is the lack of a strong degradation signal within the RAD23 proteins.13 In addition, an intermolecular interaction between the UBL and UBA domains limits the ability of the RAD23 proteins to interact with the proteasome when not bound to ubiquitin.14,15 This interaction is highly dynamic and occurs through an interface on the UBL domain that over-laps with the S5a-binding site.16 The UBA domains also interact with UBL via their ubiquitin-binding surfaces, ensuring competition between intermolecular and ubiquitin binding.16,17
In contrast to their proteasomal roles, the RAD23 proteins function in GG-NER as stabilizers of the Xeroderma pigmentosum, complementation group C (XPC) protein. GG-NER is a major DNA repair pathway that removes a wide range of structurally unrelated DNA lesions from the genome.18 XPC functions in the earliest steps of NER, where it senses and binds to helix-distorting DNA lesions,19 and recruits other NER proteins including XPA and general transcription factor IIH (TFIIH).20,21 Here, the RAD23 proteins have functionally redundant roles, where they interact with XPC through a domain located between UBA1 and UBA2.22,23 This interaction is thought to protect XPC from proteasomal degradation,24 as well as enhance XPC binding to damaged DNA.22,25,26 Interestingly, while the vast majority of XPC in human whole cell extracts co-purifies with RAD23A or RAD23B, only a small portion of the RAD23 proteins co-purify with XPC.25,27 This suggests that the RAD23 proteins have numerous roles outside of XPC-binding.
In the current work, we demonstrate that the RAD23A and RAD23B proteins interact directly with the Y-family DNA polymerases. By focusing on the RAD23A-Pol ι interaction, we identify surfaces on the RAD23 UBA domains that are required for interacting with the Y-family polymerase catalytic domains. These findings thereby reveal a novel means through which RAD23A and RAD23B interact with DNA damage response proteins.
Results
Pol ι interacts with RAD23A and RAD23B
To expand our knowledge of the Y-family polymerase interactome, we first used a protein microarray to detect putative Pol ι protein interactions in vitro. For this assay, we isolated FLAG-tagged Pol ι from HEK293T cells and incubated this protein with microarrays containing 9480 immobilized recombinant human proteins. We then detected binding using Pol ι primary antibodies and Alexa647-conjugated secondary antibodies and compared the signal from Pol ι-incubated and control plates. This allowed us to calculate statistically significant differences in signal intensity for 2211 of the immobilized proteins (Dataset 1). Notably, a ubiquitin-fusion protein (RPS27A) was one of the top hits from our screen. As Pol ι is known to interact strongly with ubiquitin,28 the detection of this fusion protein supported the validity of our approach.
One of the proteins for which we observed the greatest positive difference in signal intensity was the nucleotide excision repair protein, RAD23 homolog A (RAD23A) (Figure 1a). RAD23A and its paralog, RAD23B (which was not included in the microarray), are closely related ubiquitin-binding proteins that function during the initial stages of global genome nucleotide excision repair (GG-NER).29 Given that Pol ι is also a ubiquitinated protein that is recruited to UV damage-induced repair sites,30,31 we considered whether RAD23A and RAD23B might also be genuine interactors of Pol ι.
Figure 1.
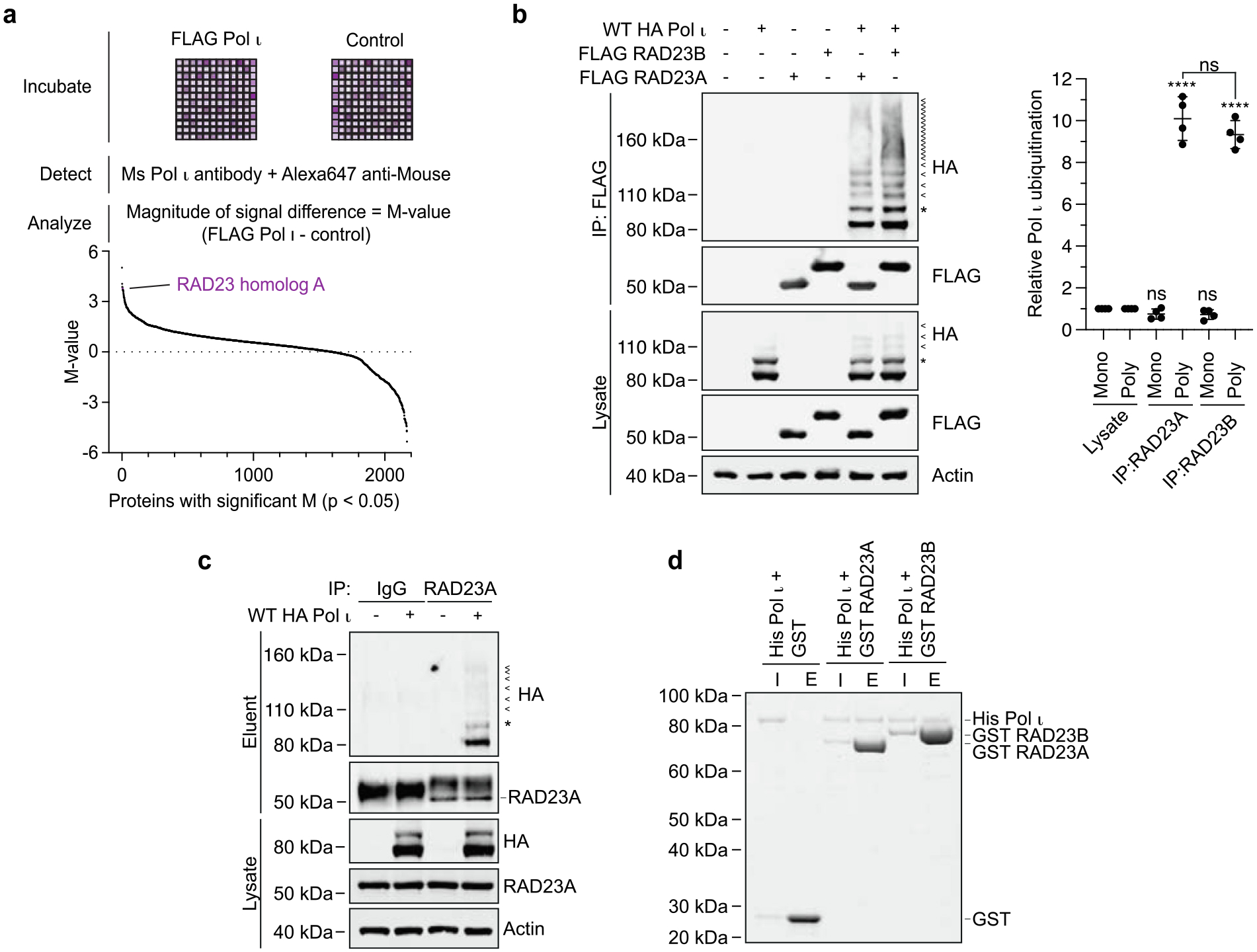
Pol ι interacts with RAD23A and RAD23B. (a) Schematic representing the protein microarray used to detect Pol ι protein interactions in vitro. The graph below represents the 2211 immobilized proteins for which statistically significant differences in signal intensity were detected between Pol ι-incubated and control plates. (b) Immunoprecipitation of WT FLAG-tagged RAD23A or RAD23B from cells co-expressing WT HA-Pol ι. Eluent and lysate (input) were immunoblotted as indicated. * = mono-ubiquitinated Pol ι, < = poly-ubiquitinated Pol ι. The bar graph represents the relative proportion of mono and poly-ubiquitinated Pol ι in the eluent, normalized to the proportion of Pol ι ubiquitination detected in the lysate. (c) Immunoprecipitation of endogenous RAD23A from 293 T cells co-expressing WT HA-Pol ι. Eluent and lysate (input) were immunoblotted as indicated. (d) Recombinant His-Pol ι (1.5 μg) was incubated with equimolar quantities of GST (0.5 μg) or GST-tagged RAD23A (2.5 μg) and GST-tagged RAD23B (2.7 μg). GST was captured on glutathione sepharose beads, washed, and eluted. Proteins were separated by gel electrophoresis and proteins stained with Coomassie blue (shown in greyscale). I = 10% input, E = 50% eluent.
To test whether Pol ι might associate with the RAD23 paralogs in cell lines, we immunoprecipitated FLAG-tagged RAD23A or RAD23B from HEK293T cells and probed for HA-tagged Pol ι by western blotting (Figure 1b). This revealed an association between RAD23A and RAD23B with non-ubiquitinated Pol ι, as well as with several higher molecular weight species. We interpreted these higher molecular weight bands as being ubiquitinated forms of Pol ι, based on their migration pattern, as well as ours and others previous studies of Pol ι mono- and poly-ubiquitination.28,31,32 While we detected a similar proportion of mono-ubiquitinated to non-ubiquitinated Pol ι in the whole cell lysate and in the protein co-immunoprecipitated with RAD23A and RAD23B, poly-ubiquitinated Pol ι was enriched by ~10-fold in the eluted samples. We also detected an association between HA-Pol ι and immunoprecipitated endogenous RAD23A (Figure 1c).
The co-elution of poly-ubiquitinated Pol ι with RAD23A and RAD23B is consistent with the well-described role of the RAD23 paralogs in binding to K48-linked poly-ubiquitin chains.11,33 The co-elution of non-ubiquitinated Pol ι however led us to consider that a direct interaction may also exist between Pol ι and the RAD23 proteins independently of ubiquitination. To test this, we purified Pol ι and both RAD23 proteins from E. coli and tested whether they could interact with Pol ι in an in vitro pull-down assay (Figure 1d). Indeed, Pol ι co-eluted with GST-tagged RAD23A and RAD23B, although not with GST alone, from glutathione agarose beads. This suggests a direct interaction between the unmodified forms of either protein.
The Pol ι catalytic domain binds UBA1 and UBA2 of the RAD23 proteins
The RAD23 proteins associate with XPC via ubiquitin-independent interactions, mediated by their XPC-binding domains.22 To determine how the RAD23 proteins might associate with Pol ι, we created expression constructs where we individually deleted each domain of RAD23A – the N-terminal ubiquitin-like (UBL) domain, UBA1, the XPC-binding domain or UBA2 – and tested whether Pol ι co-immunoprecipitates with these mutants (Figure 2a). While deleting the UBL or UBA2 domain of RAD23A led to destabilization of the protein, we were nevertheless able to assess relative binding based on the proportion of Pol ι to RAD23A in the eluted sample. In these assays, deleting UBA2 caused a near-complete loss of binding to both ubiquitinated and non-ubiquitinated Pol ι, while deleting UBA1 reduced binding by ~75–85%. Removing the XPC-binding domain, or the unstructured regions between each domain, however had only minor effects on Pol ι-binding (Figure S1a). We obtained similar results using mutants of RAD23B that lacked either UBA domain (Figure S1b). This suggested to us that RAD23A and RAD23B binding to both non- and ubiquitinated-Pol ι is likely mediated by the UBA domains. Interestingly, for both RAD23A and RAD23B, we observed higher levels of Pol ι co-immunoprecipitating with mutants lacking the N-terminal UBL domain. This might be due to increased availability of the UBA domains, which are normally partially sequestered via intra-molecular interactions with the UBL domain.16,17
Figure 2.
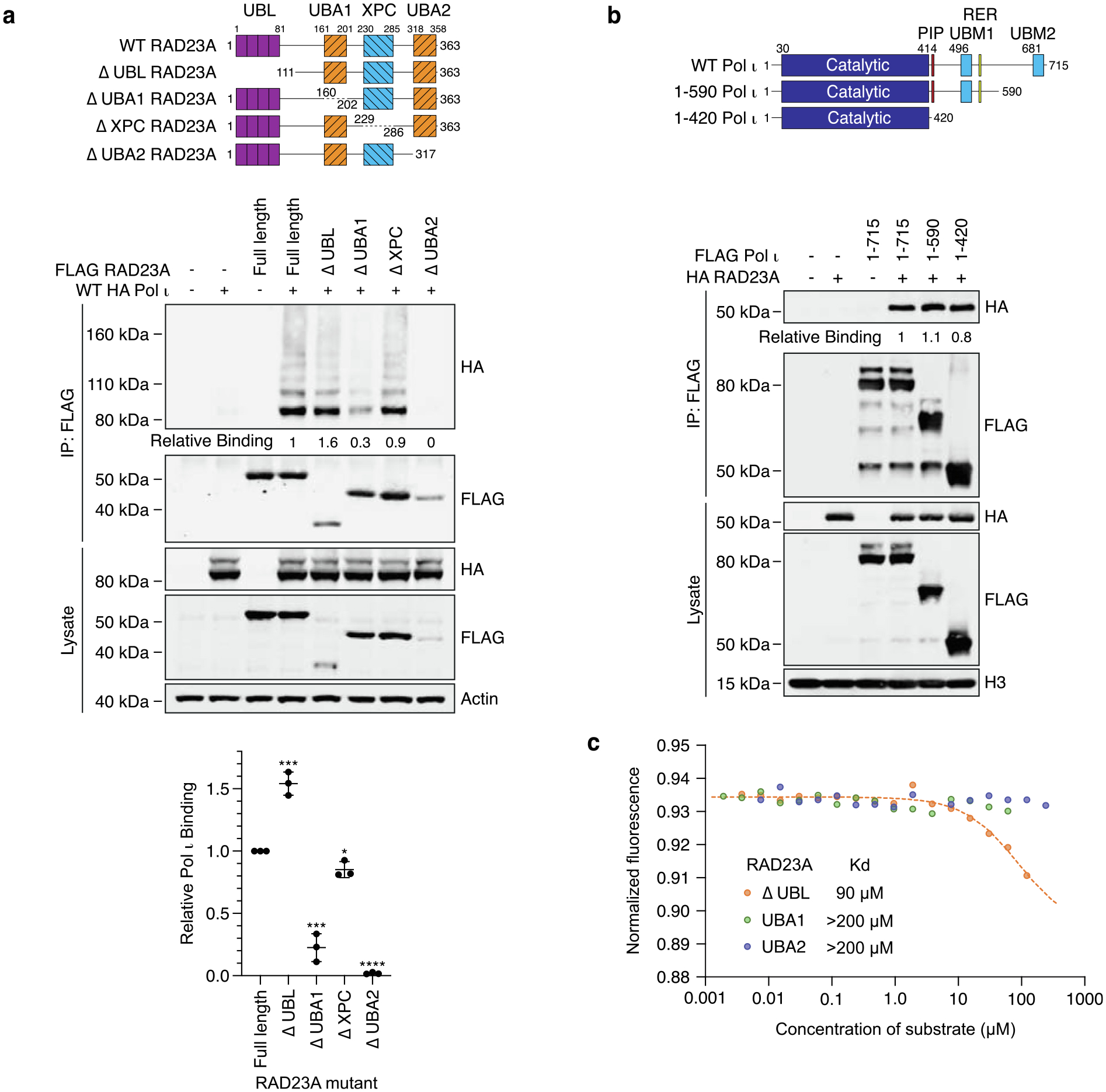
The Pol ι catalytic domain binds UBA1 and UBA2 of RAD23A. (a) Immunoprecipitation of the RAD23A mutants illustrated in the schematic, from 293 T cells co-expressing WT HA Pol ι. Eluent and WCL (input) were immunoblotted as indicated. Relative binding was calculated based on the ratio of HA to FLAG proteins in the eluent. The bar graph represents quantification of relative binding from three repeats. Error bars represent standard deviation. (b) Immunoprecipitation of the indicated Pol ι mutants from HEK293T cells co-expressing WT HA RAD23A. Eluent and WCL (input) were immunoblotted as indicated. Relative binding was calculated based on the ratio of HA to FLAG proteins in the eluent. (c) Microscale thermophoresis analysis between the catalytic domain of Pol ι and the indicated fragments of RAD23A.
We also sought to determine the region of Pol ι that the RAD23 proteins might interact with. To do so, we tested whether RAD23A could immunoprecipitate with Pol ι truncation mutants where we removed either half or the entire C-terminus34 (Figure 2b and S1c). Unexpectedly, while almost all other characterized Pol ι protein interactions occur via the C-terminus,35 RAD23A readily associated with the Pol ι catalytic domain. We also observed an association between the Pol ι catalytic domain and RAD23B (Figure S1d).
The RAD23 UBA domains have mostly been characterized for their roles in binding ubiquitin. The RAD23A UBA2 domain has however also been found to have an additional protein-binding role in interacting with the HIV-1 Vpr protein.36 Although our results suggested that non-ubiquitinated Pol ι might also bind the RAD23 UBA domains, we sought further proof of this interaction in a system that is devoid of ubiquitin. We therefore expressed and purified the Pol ι catalytic domain, the RAD23A C-terminus (containing UBA1 and UBA2), and both individual RAD23A UBA domains from E. coli and used microscale thermophoresis (MST) to measure their binding in vitro (Figure 2c). While we readily detected an association between the Pol ι catalytic domain and the RAD23A C-terminus (Kd = 90 μM), we detected only weak binding to either UBA domain, with a Kd that was unmeasurable in this assay. This, we reasoned, might reflect our observation from Figure 2a, that suggests full binding to Pol ι requires both RAD23 UBA domains.
The RAD23 UBA1 and UBA2 domains bind distinct surfaces on the Pol ι catalytic domain
To determine how the UBA domains bind to Pol ι, and to provide a rationale for why both interactions might be required, we sought to characterize the interfaces that mediate these interactions. As binding to either UBA domain is relatively weak on their own, we opted to conduct these experiments using solution nuclear magnetic resonance (NMR) spectroscopy, as this technique is uniquely suited to studying interactions of this nature.37
Here, we isotopically labeled the RAD23A UBA1 or UBA2 domains, incubated either protein with increasing concentrations of the unlabeled Pol ι catalytic domain, and monitored the resulting peak intensity changes in the 15N-TROSY HSQC spectrum at each point of the titration (Figure 3a–b and S2). We then quantified and mapped the resulting loss of intensity on the known structures of the UBA1 and UBA2 domains (1IFY and 1OQY, respectively). This revealed continuous binding interfaces on the surfaces of either domain (Figure 3c–d).
Figure 3.
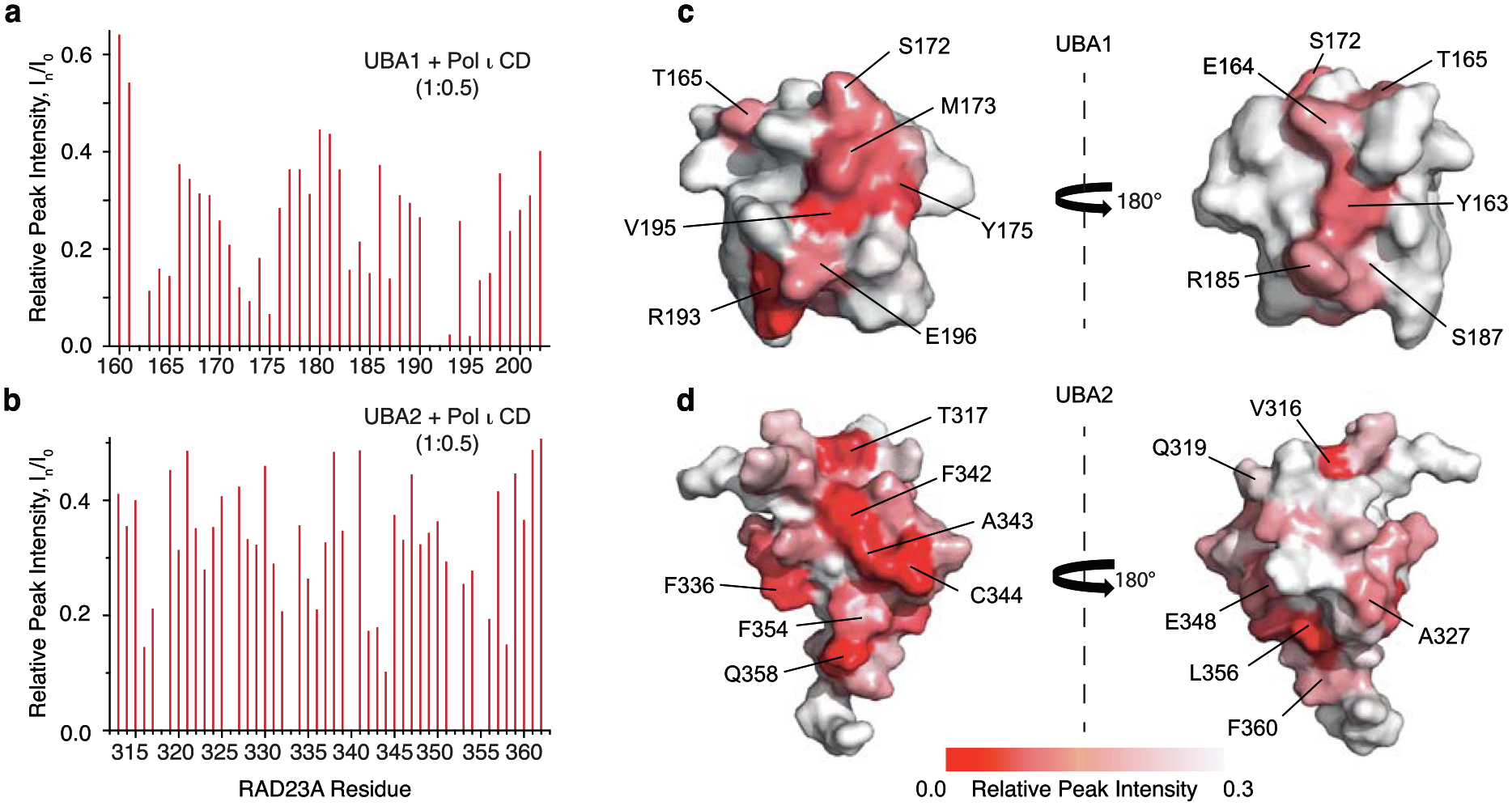
UBA1 and UBA2 directly interact with the Pol ι catalytic domain. A bar graph of per-residue NMR peak intensity loss in 15N-TROSY-HSQC spectrum of 15N-labeled RAD23A UBA1 (a) and UBA2 (b) caused by the addition of unlabeled Pol ι (1–419). (c-d) Pol ι binding site mapped on the surface of RAD23A UBA domains. Relative NMR peak intensities, In/I0, are color-coded from smallest (red) to largest (white) and mapped on the structures of UBA1 (PDB: 1IFY) (c) and UBA2 (PDB: 1OQY) (d) Amino acid residues displaying the largest peak intensity perturbations are labeled. The structures are shown in two orientations with a 180° rotation.
To confirm that we had identified the correct binding sites, we next made point mutations in UBA1 (S172A, M173A, V195A) and UBA2 (C344A, F354A) of RAD23A at specific residues that had the greatest loss of peak intensity in our above assay. Importantly, only surface-exposed residues were chosen for mutations to avoid potential structural impairment of the UBA fold. As with the complete deletion of UBA1 or UBA2 in Figure 2b, these mutations led to a substantial decrease in Pol ι-binding in co-IP assays (Figure 4a), supporting their involvement in these interactions. We also observed a similar disruption of Pol ι-binding following the mutation of corresponding amino acid residues in UBA1 and UBA2 of RAD23B (Figure S3).
Figure 4.
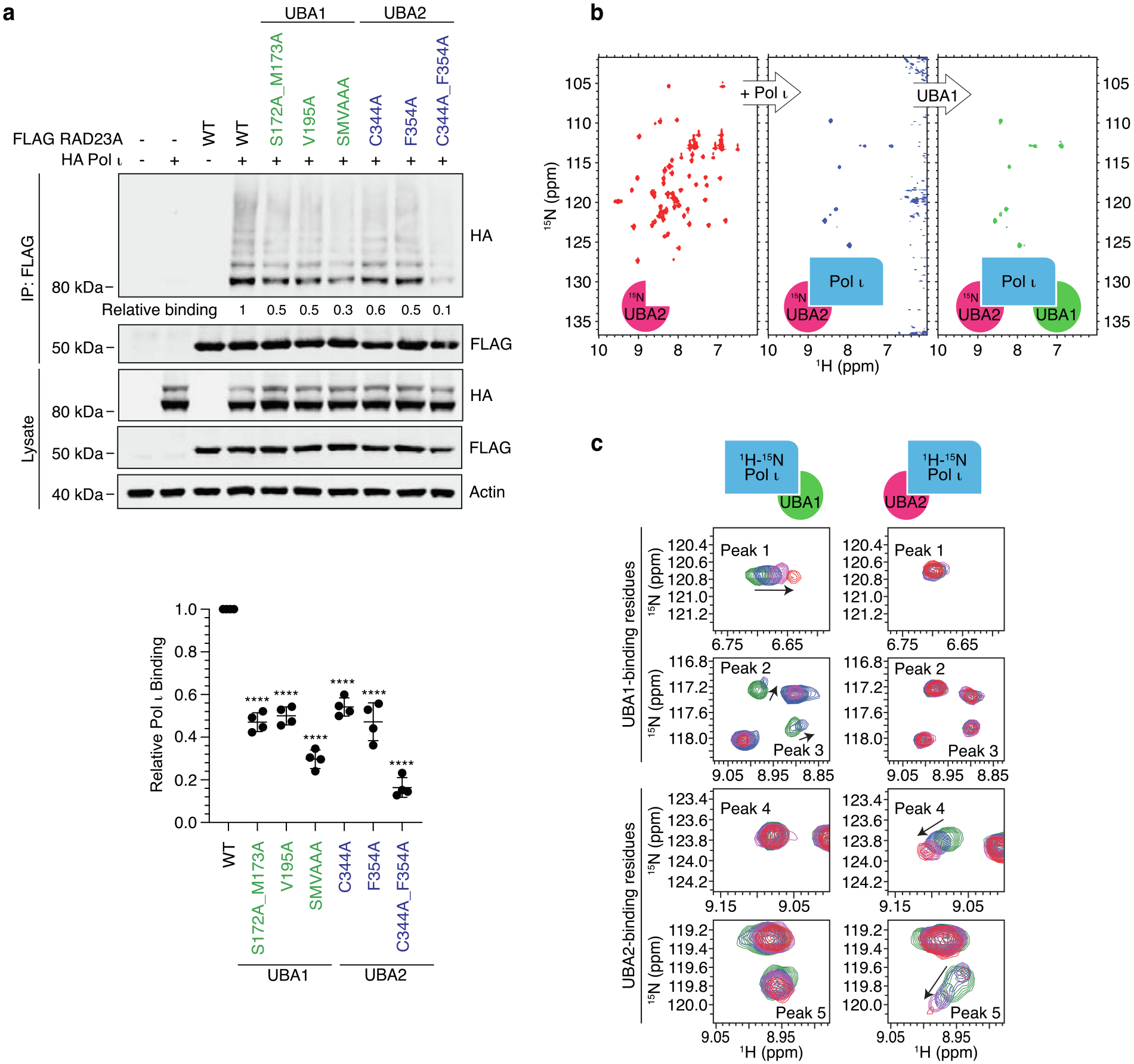
UBA1 and UBA2 of RAD23A make distinct interactions with the Pol ι catalytic domain. (a) Immunoprecipitation of the indicated RAD23A truncations from 293 T cells co-expressing WT HA Pol ι. Eluent and WCL (input) were immunoblotted as indicated. Relative binding was calculated based on the ratio of HA to FLAG proteins in the eluent. The bar graph represents the quantification of relative binding from three repeats. Error bars represent standard deviation. Unpaired t-tests were used to assess differences in binding of Pol ι with WT vs mutant RAD23A.**** = p < 0.001. (b) UBA1 and UBA2 domains of RAD23A simultaneously bind to the catalytic domain of Pol ι. 15N-TROSY HSQC spectra of the 15N/2H-labeled UBA2 domain alone (red), after the addition of 1.5 molar excess of unlabeled Pol ι (blue) and after further addition of 5-molar excess of the unlabeled UBA1 domain (green). (c) Comparison of Pol ι regions involved in binding to either UBA1 or UBA2 domains of RAD23A. Representative backbone amide peaks in the 15N-TROSY HSQC spectrum of Pol ι alone (green) and in the presence of either UBA domain at 1:1 (blue), 1:3 (magenta) and 1:6 (red) molar ratios. Identical spectral regions are shown for UBA1 and UBA2 titrations.
The apparent requirement for both RAD23A/B UBA domains for proper binding to Pol ι, led us to reflect on the likelihood that these domains might bind to separate sites within the Pol ι catalytic domain. To assess if this is the case, we used NMR to test whether the UBA domains compete for binding to Pol ι. Firstly, we incubated isotopically labeled UBA2 from RAD23A (which we had found to bind the catalytic domain less readily than UBA1) with 1.5-fold excess of the unlabeled catalytic domain of Pol ι. As shown in Figure 3b, this caused a considerable reduction in peak intensity. We then added a 5-fold excess of unlabeled UBA1 (Figure 4b). Doing so however failed to restore the UBA2 peaks that were broadened due to its interaction with the Pol ι catalytic domain, suggesting that UBA1 does not compete with UBA2 for binding to Pol ι. This supports our findings, suggesting that both domains might simultaneously bind to the Pol ι catalytic domain and are likely required for a stable interaction.
We also tested directly whether UBA1 and UBA2 interact with distinct residues within the Pol ι catalytic domain. Unlike in Figure 4b, in these assays we isotopically labeled the Pol ι catalytic domain, titrated it with unlabeled UBA domains, and monitored changes occurring in the 15N-TROSY HSQC spectrum at each point of the titrations (Figure 4c). Remarkably, distinctly unique peaks within the Pol ι catalytic domain spectrum were affected by the addition of UBA1 and UBA2 domains. For example, peaks 1, 2 and 3 in Figure 4C move upon addition of UBA1 but not UBA2 domain, similarly, peaks 4 and 5 move upon UBA2 but not UBA1 binding. These data together suggest that the RAD23A UBA domains bind to Pol ι via distinct non-overlapping interfaces.
Pol ι and ubiquitin-binding occur on overlapping surfaces of UBA1, but distinct interfaces of UBA2
Most of the known roles for the RAD23 proteins rely on their ability to bind ubiquitin, mediated by their UBA domains. Previous work has revealed that each domain interacts with an almost identical surface on ubiquitin.15 Given that our data indicate that UBA1 and UBA2 bind separate surfaces on Pol ι, we were interested in determining whether these domains interact with Pol ι and ubiquitin via the same surfaces.
While the structure of RAD23A UBA1 in complex with ubiquitin was previously determined (PDB 5XBO,38 Figure 5a), the structure of the UBA2-ubiquitin complex remains unknown. To characterize the ubiquitin-binding interfaces on the UBA domains, we titrated either isotopically labeled domain with increasing amounts of unlabeled ubiquitin and monitored changes in the 15N-TROSY HSQC spectrum at each point of the titration. We then quantified and mapped the resulting chemical shift perturbations (CSPs) on the structures of the UBA domains to define the ubiquitin-binding interfaces (Figure S4). These data allowed us to use HADDOCK software to calculate data-based structural models of ubiquitin in complex with the UBA2 domain (Figure 5b). We then colored the Pol ι-binding surfaces within the UBA-ubiquitin complexes as per Figure 3c–d, allowing us to visually compare the binding surfaces for Pol ι and ubiquitin on UBA1 and UBA2.
Figure 5.
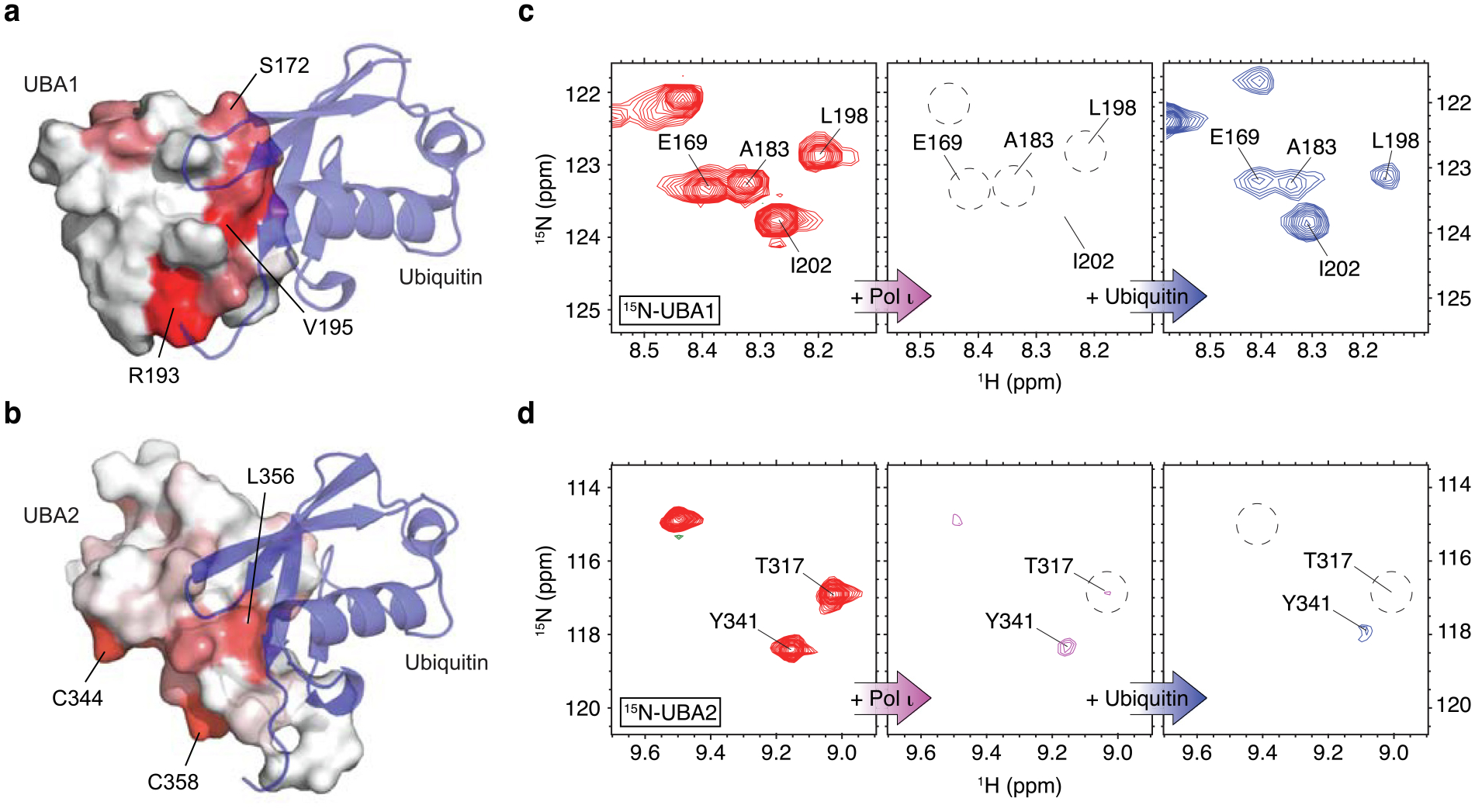
RAD23A UBA1 domain binding to ubiquitin interferes with its Pol ι interaction. (a) Structure of the RAD23A UBA1 domain in complex with ubiquitin (PDB: 5XBO). Ubiquitin is shown as a blue ribbon while the UBA1 domain is shown as a surface with Pol ι-binding site mapped in red. (b) HADDOCK model of the RAD23A UBA2 domain in complex with ubiquitin (blue). The Pol ι interacting residues of UBA2 are mapped in red. (c-d) 15N TROSY HSQC spectra of 15N/2H -labeled UBA1 (c) and UBA2 (d) domains alone (red), in complex with the unlabeled catalytic domain of Pol ι (magenta), and after further treatment with the excess of ubiquitin (blue).
These analyses revealed that ubiquitin and Pol ι bind UBA1 via a similar surface (Figure 5a). Indeed, both binding events involve a molecular surface that wraps around UBA1, centralized on proximally located residues (e.g. M171, S172, V195). By contrast, our modeling suggests that ubiquitin binds UBA2 on a surface on the opposite side of the domain from the key Pol ι-binding residues (e.g. C344, F354, Q358) (Figure 5b).
The results above suggested to us that UBA1 binding to ubiquitin- and Pol ι is likely to be mutually exclusive. The distinct ubiquitin- and Pol ι-binding surfaces on UBA2, however, could allow both interactions to occur simultaneously. To determine if this is the case, we used solution NMR to test whether ubiquitin can compete with Pol ι for binding to either UBA domain. Here, we incubated isotopically labeled RAD23A UBA1 or UBA2 with a 1:1.2 molar ratio of the unlabeled Pol ι catalytic domain. As in Figure S2, this led to a severe broadening of NMR peaks in the 15N-TROSY HSQC spectrum for each domain, due to formation of a large UBA-Pol ι complex (Figure 5c). We then added unlabeled ubiquitin to these solutions. For UBA1, this led to a return of peak intensities, which matched with the spectrum of the UBA-ubiquitin complex. These data suggest that ubiquitin could replace Pol ι in binding to UBA1, leading to formation of a final smaller UBA1-ubiquitin complex. For UBA2, however, the addition of unlabeled ubiquitin did not result in a return of peak intensities, indicating that UBA2 binds to Pol ι and ubiquitin simultaneously. These data support our hypothesis that ubiquitin- and Pol ι-binding to UBA2 is not competitive. Interestingly, the Pol ι-binding interface of UBA2 identified in this study has been previously shown to selectively recognize K48-linked di-ubiquitin.39 This presents an interesting possibility that the length and the linkage of the ubiquitin chain may affect the competitive dynamics of the UBA2 interactions.
RAD23A and RAD23B also bind to other human Y-family DNA polymerases
Pol ι and the other human Y-family DNA polymerases – Pol η, Pol κ and Rev1 – share a structurally similar catalytic domain, with regions of high sequence conservation40 (Figure 6a and S5a). We therefore wondered whether the other Y-family polymerases might also interact with the RAD23 proteins. To test this, we expressed the catalytic domains of Pol η, Pol κ and REV1 in cells and performed co-IP experiments with RAD23A (Figure 6a) and RAD23B (Figure S5b). This revealed that the RAD23 proteins can indeed bind to the catalytic domains of each polymerase.
Figure 6.
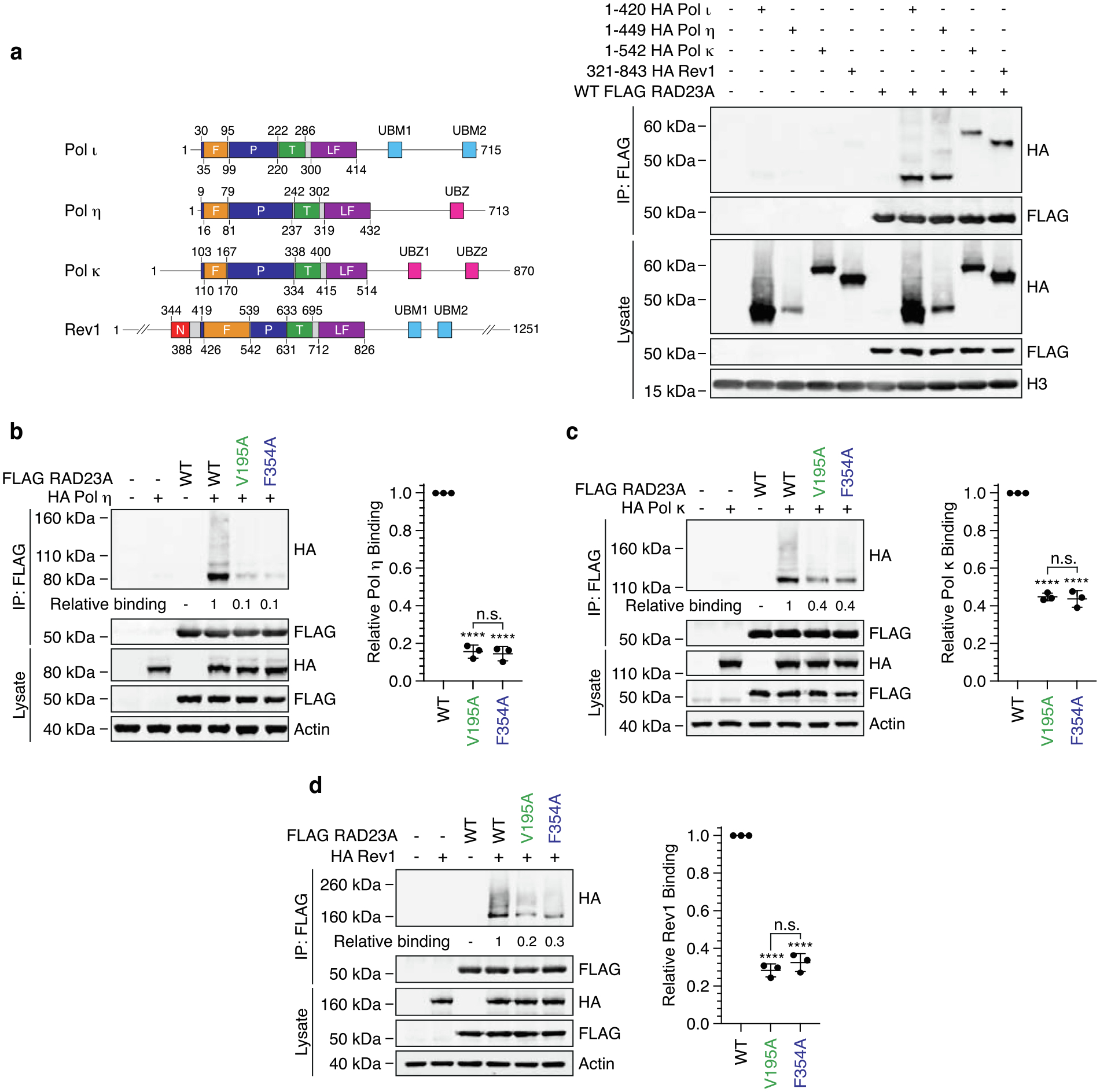
RAD23A associates with the catalytic domain of Pol η, Pol κ and REV1. (a) The schematic illustrates the domain structure of the human Y-family polymerases. F = fingers, P = palm, T = thumb, LF = little finger, UBM = ubiquitin-binding motif, UBZ = ubiquitin-binding zinc finger. WT FLAG-RAD23A was immunoprecipitated from cells co-expressing HA-tagged catalytic domains of Pol ι, Pol η, Pol κ and REV1. The eluent and lysate were immunoblotted as indicated. (b, c, d) Immunoprecipitation of WT, V195A and F354A FLAG-RAD23A from cells co-expressing HA-tagged Pol η (b), Pol κ (c) or REV1 (d). Eluent and lysate were immunoblotted as indicated and relative binding calculated based on the ratio of HA polymerase / FLAG-RAD23A in the eluent. Bar graphs illustrate relative binding from three replicates. Error bars indicate the standard deviation. ns = not significant, * = p < 0.5, ** = p < 0.1, *** = p < 0.01, **** = p < 0.001.
We also considered the probability that these interactions might be mediated by similar binding interfaces on UBA1 and UBA2 of the RAD23 proteins. We therefore performed co-IPs to compare the binding of each polymerase with WT RAD23A and variants containing point mutations in these domains. Here, mutation of V195 (UBA1) or F354 (UBA2) of RAD23A disrupted binding to Pol η (Figure 6b), Pol κ (Figure 6c) and Rev1 (Figure 6d). Furthermore, as with Pol ι, mutations in the RAD23A UBA domains disrupted binding to the predominant and higher molecular weight forms of each polymerase. These results suggested to us that RAD23A and RAD23B likely bind each Y-family polymerase via a common mechanism.
Discussion
The RAD23 paralogs have largely been characterized as scaffold proteins with dual functions in proteasomal degradation and nucleotide excision repair.8 Central to these roles is the ability to bind ubiquitin, mediated by their two ubiquitin-binding domains. UBA2 of RAD23A has also been found to mediate an interaction with the HIV-1 Vpr protein. This interaction is however relatively weak, and association between these proteins requires an additional interaction between Vpr and the RAD23A XPC-binding domain.36,41 In our work, we show that RAD23A UBA2 also interacts directly with the catalytic domain of Pol ι. In an arrangement that is analogous to the RAD23A-Vpr complex, binding between UBA2 and Pol ι is insufficient for a stable complex to form and must occur in addition to a second interaction. Unlike in the complex with Vpr however, this interaction is mediated not by the XPC-binding domain, but by a distinct interaction between the Pol ι catalytic domain and UBA1 of RAD23A. These data thereby reveal a new role for the RAD23A UBA domains in binding non-ubiquitin human proteins.
While we have largely focused on the interaction between RAD23A and Pol ι, our immunoprecipitation data suggest that RAD23A and RAD23B form interactions with each of the human Y-family polymerases. Although the precise location of the UBA1 and UBA2 binding sites within the polymerase catalytic domains remain unclear, these interactions are likely to occur through similar mechanisms. Indeed, mutation of the RAD23A UBA domains disrupts binding to each polymerase, suggesting each interacts with the same UBA surfaces. This said, we did observe some variation in the degree to which this disruption occurs. For instance, while single point mutations in RAD23A UBA1 or UBA2 disrupted binding to Pol ι and Pol κ by 40–60%, this increased to 70–80% for Rev1, and ~90% for Pol η. This variation likely reflects differences in the catalytic domains of these polymerases, which although architecturally similar, are sufficiently varied to exhibit differences in catalytic activity and substrate specificity.42
Although our work has focused on the interaction between RAD23A/B and the non-modified catalytic domains of the TLS polymerases, our analyses demonstrate that the RAD23 proteins also associate with higher molecular weight forms of these proteins. Furthermore, these species are enriched in samples co-immunoprecipitating with RAD23A/B. These bands most likely represent poly-ubiquitinated forms of the polymerases, and their association with the RAD23 proteins is consistent with the high affinity of the RAD23 UBA domains for K48-linked ubiquitin chains.11,33 Whether or not these interactions are part of a multi-step binding mechanism – e.g., occurring after binding to the polymerase catalytic domain – however remains unclear. We also cannot exclude the possibility that binding between the RAD23 proteins with poly-ubiquitinated forms of the Y-family polymerases may not be physiologically relevant, but rather an artefact resulting from transient overexpression of these proteins. Nevertheless, our finding that UBA1 of RAD23A binds the Pol ι catalytic domain and ubiquitin via the same interface, as well that both UBA1 and UBA2 are required to form a stable interaction with Pol ι, suggests that interactions between Y-family polymerases and the RAD23 proteins is likely to be affected by ubiquitin signalling.
An unexpected finding from our study was that binding with RAD23A and RAD23B is mediated by direct interactions with the Y-family polymerase catalytic domains. This was surprising, as most characterized interactions with these polymerases occur via their C-termini. Pol ι, for example, contains distinct C-terminal binding sites that interact with PCNA,43 Rev1,44 ubiquitin,28 and USP7.34 One exception we have previously identified is that the Pol ι catalytic domain is required for complex formation with the p300 acetyltransferase45; the precise mode of this association is however yet to be determined. UBA domains are well conserved amongst ubiquitin-binding proteins with both proteasomal and non-proteasomal functions.46 It is interesting to consider whether these domains in other proteins might also mediate interactions with catalytic domains of TLS polymerases. In any case, the finding that RAD23A and RAD23B makes two distinct and direct interactions with the Pol ι catalytic domain raises questions regarding interplay between the DNA-processing and protein-binding functions of the Y-family polymerases. Understanding such interplay will likely be a focus of future work.
In summary, our work here reveals a novel interaction between human Y-family polymerases and the RAD23 proteins. These interactions are mediated by the polymerase catalytic domains, and UBA1 and UBA2 of RAD23A/B. While UBA2 contains separate polymerase- and ubiquitin-binding sites, UBA1 forms both interactions via the same interface. These findings reveal new interactions for Y-family polymerases, as well as demonstrate a novel binding mode for the RAD23 UBA domains, that is competitive with ubiquitin binding.
Material and Methods
Expression vectors
All mammalian and bacterial expression vectors created for this work are available from Addgene (www.addgene.org/browse/article/28234221/). Plasmid files have also been submitted to Mendeley Data. All synthetic DNA fragments were chemically synthesized by GenScript (Piscataway, NJ).
Mammalian expression vectors:
The wild-type FLAG-Pol ι (pJRM46) and wild-type HA-Pol η (pJRM56) expression vectors has been described previously,32 as have the FLAG-Pol ι 1–590 and 1–420C-terminal truncations constructs.34 The wild-type HA-Pol ι expression plasmid was created by subcloning the Pol ι coding sequence from the wild-type FLAG-Pol ι vector into the AsiSI and MluI restriction sites of pCMV6-AN-HA (Origene). The coding sequences of Pol κ and Rev1 were synthesized and cloned into the AsiSI and HindIII restriction sites of pCMV6-AN-HA. Pol ι 1–420, Pol η 1–449 and Pol κ 1–542c-terminal truncation constructs were created by introducing STOP codons into the WT plasmids by site-directed mutagenesis. The coding sequence for the Rev1 catalytic domain (aa 321–843) was synthesized and cloned into pCMV6-AN-HA.
The coding sequences for RAD23A and RAD23B were synthesized and cloned into the AsiSI and HindIII restriction sites of pCMV6-AN-DDK (Origene) and pCMV6-AN-HA to create the N-terminal FLAG- and HA-RAD23A and RAD23B mammalian expression constructs, respectively. Truncation and point mutant constructs of the RAD23 proteins were constructed by synthesizing new gene fragments and subcloning into the wild-type plasmids using restriction sites flanking or contained within the coding sequences.
Bacterial expression vectors:
The WT His-Pol ι bacterial expression vector (pJM919) is a derivative of pJM86847 that has been modified to contain an NdeI restriction site at the N-terminus of the Pol ι coding sequence. The catalytic domain of Pol ι (aa 1–419) was codon optimized for E. coli expression, chemically synthesized, and cloned into the NdeI-XhoI restriction sites of pJM919. The GST-RAD23A and GST-RAD23B expression vectors were created by cloning coding-optimized synthetic sequences for either protein into the NdeI and BamHI sites of pGEX-6P-1. Coding sequences for UBA1 (aa 160–205) and UBA2 (aa 311–363) of RAD23A were synthesized and cloned into NdeI-BamHI of pET28b (+) and used for E. coli expression of either domain with an N-terminal His-tag. The ubiquitin expression plasmid was created by sub-cloning a ubiquitin-coding sequence into pET-15b (Sigma Aldrich) using NdeI-BamHI restriction sites.
Mammalian cell culture
Mammalian cell culture and transfection was performed as described previously.34
Immunoprecipitation, immunoblotting and antibodies
Immunoprecipitation and immunoblotting experiments were performed as previously described.34 Antibodies against HA (1:2000; clone C29F4, cat # 3724S), β-actin (1:5000; clone 13E5, cat # 4970S), H3 (1:4000; clone 1B1B2, cat # 14269S) and RAD23A (1:1000; clone D7U7Z, cat # 24555) were purchased from Cell Signaling Technology, as was a non-specific isotype control IgG (clone DA1E, cat # 3900S). The FLAG antibody was from Sigma-Aldrich (1:4000; clone M2, cat # F1804). The Pol ι antibody was from Abnova (1:1000; clone M01, cat # H00011201-M01). Primary antibodies were detected with fluorescent secondary antibodies, and immunoblots quantified, as described previously.34 For the microarray, the Pol ι antibody was detected with an Alexa Fluor 647-conjugated goat anti-mouse secondary antibody (Thermo Fisher Scientific cat #A-21240).
Protein microarray
The protein microarray experiment was performed as a custom service by LifeSensors, Inc. For this, WT FLAG-Pol ι was immunoprecipitated from HEK293T cells and sent to LifeSensors for challenging the ProtoArray Human Protein Microarray (ThermoFisher Scientific). Arrays were removed from −20 °C storage and placed at room temperature (RT) for 15 min before opening, to avoid formation of condensation. Arrays were then blocked for 1 h at RT in PBS containing 0.05% Tween-20, 20 mM reduced glutathione, 1 mM DTT, 1% BSA, and 25% glycerol. Arrays were washed in 3 changes of ~500 mL PBST before application of Pol ι or continued incubation in blocking buffer. After 1 h, both arrays were washed as before, then incubated with anti-Pol ι antibodies at 1:1000 dilution in PBST for 1 h RT. Arrays were again washed, then incubated with an Alexa647-conjugated anti-mouse secondary antibody. Arrays were then washed twice more with PBST, twice with 0.2 micron-filtered water, dried by centrifugation (1000 RPM for 5 min at RT) and scanned using a GenePix 4100A instrument (Molecular Devices).
Microarray images were gridded and quantitated using GenePix Pro (v7) software. Median intensities (features and local backgrounds) were utilized, and signal to noise ratio (SNR) calculated. Data was log(2) transformed, and values at or below zero set to a positive fraction (1.0 × 109). Magnitude change was calculated as (log(SNR) Pol-iota-treated -log(SNR)control). These values were then Loess transformed by print tip and location to remove technical sources of error,48 resulting in the final estimate of magnitude change (M-value). Duplicate features (representing identical proteins) were summarized by average (avg M) and standard deviation. T-test (paired, 2-tailed) was used to assess the statistical significance (p-value) of each estimate. A threshold of 95% confidence (p < 0.05) was employed to filter data. Positive M values graphically determined to be deviant from the trend were further selected.
Protein purification
Purification of GST RAD23A and RAD23B:
GST tagged-RAD23A and RAD23B were expressed and purified in a similar manner from BL21 (DE3) E. coli. Transformed cells were grown at 37 °C until OD600 of 0.5–0.6; protein expression was then induced with addition of 500 μM isopropyl 1-thio-β-D-galactopyranoside (IPTG) and cells grown overnight at 20 °C. Cells were harvested by centrifugation, resuspended in GST binding & Wash Buffer (50 mM Tris pH 8.0, 150 mM NaCl, 0.1 mM EDTA) and lysed by sonication (30 s sonication, 90 s rest: total sonication for 5 min). Samples were then clarified by centrifugation at 15,000g for 30 min, then at for 35,000 RPM for 45 min, both at 4 °C. The clarified supernatant was incubated with glutathione Sepharose 4 Fast Flow beads (Cytiva cat # 17513202) for 1 h at 4 °C, then poured onto a 10 mL chromatography column. Beads were washed with 5 × column volumes of GST binding and wash buffer and bound proteins eluted with 10 mM glutathione. The eluent was then diluted 1:3 with DEAE-R buffer (20 mM Tris, 50 mM NaCl, 0.1 mM EDTA, 10% glycerol) and incubated with Q Sepaharose Fast Flow (Cytiva cat # 17101401) for 1 h at 4 °C. Beads were poured onto a 10 mL chromatography column, washed with 1 column volume of DEAE-R buffer, and proteins eluted by incubation with increasing concentrations of NaCl (100, 200, 300, 400, 500 mM).
Purification of WT His-Pol ι.
The WT His-Pol ι bacterial expression vector (pJM919) was transformed into chemically competent RW644 E. coli49 the day before growth. The following day, colonies were used to inoculate flasks containing 50 mL of LB and 30 μg mL−1 kanamycin. These cultures were grown at 37 °C until OD600 of 0.4–0.6, then used to start two flasks containing 1 L of pre-warmed LB. These cultures were grown until OD600 of 1.2–1.5, then cells harvested by centrifugation and pellets frozen on dry ice prior to storage at −80 °C. Cell pellets were resuspended in 50 mL of lysis buffer (50 mM Tris pH 7.5, 300 mM NaCl, 20 mM imidazole, 10% glycerol, 10 mM β-mercaptoethanol) supplemented with 1 × protease inhibitor cocktail (Complete, EDTA free; Roche, Sigma-Aldrich) and lysed by sonication. The lysate was then clarified by centrifugation and incubated with Ni-NTA agarose beads (Qiagen cat # 30210, 8 mL of slurry) for 30 min at 4 °C. Beads were poured onto a 30 mL column, washed 3x with wash buffer 1 (20 mM Tris pH 7.5, 1 M NaCl, 20 mM imidazole, 10% glycerol, 10 mM β-mercaptoethanol), 3x with wash buffer 2 (10 mM 10 mM Na3PO4, 125 mM NaCl, 20 mM imidazole, 10% glycerol, 10 mM β-mercaptoethanol), then eluted with 2 mL fractions of buffer B (10 mM 10 mM Na3PO4, 125 mM NaCl, 200 mM imidazole, 10% glycerol, 10 mM β-mercaptoethanol). Fractions containing Pol ι were pooled and dialyzed overnight against 1 L of HPQ buffer A (10 mM 10 mM Na3PO4 pH 7.3, 100 mM NaCl, 10% glycerol, 10 mM β-mercaptoethanol), then incubated with 3 mL of packed Q sepharose agarose beads (Cytiva cat # 17101401). Beads were washed with buffer HPQ A and Pol ι eluted with a gradient of HPQ buffer A containing 150, 175, 200, 250, 500 mM NaCl. Fractions containing Pol ι were diluted at least 50% with HAP buffer (10 mM Na3PO4 pH 7.7, 300 mM NaCl, 10% glycerol, 10 mM β-mercaptoethanol) and applied to a column containing hydrated and packed hydroxyapatite resin (Bio-Rad Bio-Gel HTP, cat # 1300420; 1 g dry resin). Beads were washed with 2x column volumes of HAP buffer, then Pol ι eluted with increasing concentrations of Na3-PO4 (20, 30, 60, 100 mM).
Purification of the Pol ι catalytic domain and ubiquitin:
Ubiquitin and the catalytic domain of Pol ι were expressed and purified in a similar manner. Briefly, plasmids encoding His Pol ι aa 1–419 and full-length ubiquitin were transformed into E. coli BL21(DE3) cells. Unlabeled proteins used for NMR experiments were expressed in 1 L of Luria broth (LB) media. The 15N-labeled sample of Pol ι was obtained by expressing proteins in M9 minimal medium supplemented with 15N-ammonium chloride as a sole source of nitrogen. Transformed cells were grown at 37 °C until OD600 of 0.8–1.0. Protein expression was induced with 1 mM isopropyl 1-thio-β-ᴅ-galactopyranoside (IPTG) overnight at 20 °C. Cells were harvested by centrifugation, resuspended in a buffer containing 20 mM Tris pH 8.0, 200 mM NaCl, 10 mM imidazole and 1 mM PMSF, lysed by sonication, and centrifuged at 15,000 rpm for 30 min. The supernatant was filtered and applied to equilibrated HisPur™ Cobalt resin (Thermo Scientific). Proteins were eluted in a buffer containing 20 mM Tris pH 8.0, 200 mM NaCl and 250 mM imidazole and then subjected to size-exclusion chromatography using a HiLoad Superdex 75 column (GE Healthcare) in a buffer containing 20 mM Tris pH 6.5, 100 mM NaCl and 10 mM β-mercaptoethanol.
Purification of the RAD23A UBA domains:
RAD23A UBA1 and UBA2 domains were expressed and purified in a similar manner. Briefly, UBA1 (160–205) and UBA2 (361–409) plasmids were transformed into Escherichia coli BL21(DE3) cells. 15N/2H-labeled proteins used in NMR titration experiments were expressed in deuterated M9 minimal media containing with 15NH4Cl as a sole source of nitrogen. Transformed cells were grown at 37 °C until OD600 of 0.8–1.0. Protein expression was induced with 1 mM isopropyl 1-thio-β-ᴅ-galactopyranoside (IPTG) overnight at 20 °C. Cells were harvested by centrifugation, resuspended in a buffer containing 20 mM Tris pH 7.5, 200 mM NaCl and 1 mM PMSF, lysed by sonication, and centrifuged at 15,000 rpm for 30 min. The supernatant was filtered and applied to equilibrated Glutathione resin. Proteins were eluted in a buffer containing 20 mM Tris pH 7.5, 200 mM NaCl and 32.5 mM reduced glutathione. PreScission protease was then added to samples overnight at room temperature to remove the GST tag. Proteins were then subjected to size-exclusion chromatography using a HiLoad Superdex 75 column (GE Healthcare) in a buffer containing 20 mM Tris pH 6.5, 100 mM NaCl and 10 mM β-mercaptoethanol.
Pull-down assay
Pull-down assays were performed between His Pol ι (3.2 μg) and equimolar quantities of GST (1 μg), GST RAD23A (2.5 μg), or GST RAD23B (2.7 μg), using a previously described methodology.34
NMR spectroscopy
All NMR experiments were collected at 15 °C on Bruker NEO 800 NMR spectrometer equipped with a 5 mm TCI HCN cryoprobe unless stated otherwise. All spectra were processed using Bruker Topspin 4.1 software and analyzed using CARA (https://wiki.cara.nmr.ch/). Previously reported NMR backbone assignment of UBA150 and UBA251 (BMRB 27978) were used in data analyses.
NMR chemical shift perturbation assays.
NMR chemical shift perturbations were used to identify Pol ι-binding interface of RAD23A. Each NMR sample contained 300 μM of a 15N/2H-labeled RAD23A UBA domain (either UBA1 or UBA2) dissolved in 20 mM Tris pH 6.5, 100 mM NaCl, 10 mM β-mercaptoethanol and 10% v/v D2O. Unlabeled Pol ι (1–419) was gradually added to UBA samples up to 1.2 molar excess and 15N-TROSY HSQC spectra were collected at every point of the titration. The resulting NMR peak broadening was quantified as relative peak intensities, In/I0, where I0 and In are peak intensities in the free UBA spectrum and the spectrum of UBA bound to Pol ι, respectively. The relative peak intensities at 1:0.5 molar ratio of UBA to Pol ι were mapped onto the structures of UBA1 (PDB: 1IFY) and UBA2 (PDB: 1OQY) domains.
Similarly, to map the ubiquitin-binding interface of RAD23A, 15N-TROSY HSQC spectra of 300 μM 15N/2H-labeled UBA domains (either UBA1 or UBA2) were compared to the spectra of each UBA domain saturated with unlabeled ubiquitin (up to 1:3 molar ratio). Ubiquitin-induced NMR chemical shift perturbations were quantified as , where ΔωN and ΔωH are changes in N and H frequencies in Hz, respectively, and reported in 1H ppm.
NMR titrations of the catalytic domain of Pol ι (1–419) with UBA domains of RAD23A were performed at 30 °C. 100 μM sample of 15N-labeled Pol ι dissolved in 20 mM Tris pH 6.5, 100 mM NaCl, 10 mM β-mercaptoethanol and 10% v/v D2O was gradually titrated with the unlabeled RAD23A UBA domains (ether UBA1 or UBA2) and 15N-TROSY HSQC spectra were recorded at their 1:0, 1:1, 1:3 and 1:6 molar ratios.
NMR competition experiments.
Pol ι and ubiquitin competition for UBA binding. NMR competition experiments were performed using 15N-TROSY HSQC experiments. All samples were prepared in 20 mM Tris pH 6.5, 100 mM NaCl, 10 mM β-mercaptoethanol and 10% v/v D2O. To test whether ubiquitin can displace UBA1 and UBA2 domains from UBA/Pol ι complexes, the spectra of 300 μM 15N-RAD23A-UBA domains alone, in complex with 1.2 molar excess of unlabeled Pol ι (1–419) and the UBA/Pol ι complexes further treated with > 2-molar excess of unlabeled ubiquitin were recorded and compared. The appearance of sharp NMR signals corresponding to UBA/ubiquitin complexes served as an indicator of successful displacement.
UBA1 and UBA2 competition for Pol ι binding. Competition experiments were performed at 15 °C on a Bruker NEO 600 NMR spectrometer equipped with an NCH cryoprobe. All samples were prepared in 20 mM Tris pH 6.5, 100 mM NaCl, 10 mM β-mercaptoethanol, and 10% v/v D2O. To test whether UBA1 can compete with UBA2 for Pol ι binding, 15N-TROSY HSQC spectra of 300 μM 15N-UBA2 domain alone, in complex with 1.5-molar excess of unlabeled Pol ι (1–419), and UBA2/Pol ι complex further treated with 5-fold excess of unlabeled UBA1 (1:1.5:5 molar ratio of UBA2:Pol ι:UBA1) were recorded.
Molecular docking.
The structural model of the RAD23A UBA2 domain in a complex with ubiquitin was generated with HADDOCK 2.2 (High Ambiguity Driven protein – protein Docking)52–54 using experimental NMR chemical shift perturbations. RAD23A residues 331, 332, 348, and 356 and ubiquitin residues 8, 44, and 70 which undergo the largest chemical shift perturbations upon reciprocal NMR titrations were used as “active” residues”. Previously reported structures of ubiquitin (PDB: 1UBQ) and the RAD23A residues 311–363 (PDB: 1OQY16) were used for docking. Docking yielded 118 structures in 11 clusters. The top cluster with the lowest z-score was chosen as the most reliable.
Microscale thermophoresis
The interactions between the catalytic domain of Pol ι (1–419) and RAD23A UBA1, UBA2 and ΔUBL fragments were monitored using microscale thermophoresis. All samples were prepared in 20 mM PBS buffer with 100 mM NaCl at pH 8.0. The recombinant 6xHis-tagged Pol ι protein was labeled using RED-tris-NTA dye (Monolith His-Tag labeling kit, NanoTemper technologies). Subsequently, 0.125 μM Pol ι was mixed with 16 serial dilutions of each UBA1 (0.002 to 62.5 μM), UBA2 (0.008 to 250 μM) and ΔUBL (0.008 to 250 μM). Samples were loaded into the premium glass capillaries (Monolith NT.115 series) and the fluorescence signal was measured using a Monolith NT.115 instrument (NanoTemper Technologies). Monolith™ NT analysis software was used to determine the dissociation constants (Kd).
Supplementary Material
Acknowledgement
We thank Mallory Smith (Laboratory of Genomic Integrity) for help and advice with purifying His-Pol ι, GST-RAD23A and GST-RAD23B.
Funding
This work was supported by funds from the National Institute of Child Health and Human Development (NICHD)/National Institutes of Health (NIH) Intramural Research Program [to RW], as well by NIH grants [R35 GM128864 to IB]. Funding for open access charge: NICHD/NIH Intramural Research Program.
Footnotes
CRediT authorship contribution statement
Nicholas W. Ashton: Conceptualization, Methodology, Investigation, Visualization, Formal analysis, Writing – original draft, Supervision. Nancy Jaiswal: Conceptualization, Methodology, Investigation, Visualization, Formal analysis, Writing – original draft. Natália Cestari Moreno: Investigation. Irina V. Semenova: Investigation. Dana A. D’Orlando: Investigation. Marcela Teatin Latancia: Investigation. Justyna McIntyre: Conceptualization. Roger Woodgate: Conceptualization, Writing – review & editing, Funding acquisition. Irina Bezsonova: Conceptualization, Writing – review & editing, Visualization, Supervision, Funding acquisition.
DECLARATION OF COMPETING INTEREST
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2023.168353.
DATA AVAILABILITY
Uncropped immunoblot and protein gel images, as well as raw microarray data, are available from Mendeley Data (https://data.mendeley.com/datasets/ht763x684h). All raw NMR data are available upon request.
References
- 1.Stillman B, (2008). DNA polymerases at the replication fork in eukaryotes. Mol. Cell 30, 259–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCulloch SD, Kunkel TA, (2008). The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res 18, 148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Branzei D, Foiani M, (2005). The DNA damage response during DNA replication. Curr. Opin. Cell Biol 17, 568–575. [DOI] [PubMed] [Google Scholar]
- 4.Yang W, Woodgate R, (2007). What a difference a decade makes: insights into translesion DNA synthesis. PNAS 104, 15591–15598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sale JE, Lehmann AR, Woodgate R, (2012). Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nature Rev. Mol. Cell Biol 13, 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waters LS, Minesinger BK, Wiltrout ME, D’Souza S, Woodruff RV, Walker GC, (2009). Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev 73, 134–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anand J, Chiou L, Sciandra C, Zhang X, Hong J, Wu D, et al. , (2023). Roles of trans-lesion synthesis (TLS) DNA polymerases in tumorigenesis and cancer therapy. NAR Cancer 5, zcad005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gronbaek-Thygesen M, Kampmeyer C, Hofmann K, Hartmann-Petersen R, (2023). The moonlighting of RAD23 in DNA repair and protein degradation. Biochim. Biophys. Acta 1866, 194925. [DOI] [PubMed] [Google Scholar]
- 9.Chen L, Madura K, (2002). Rad23 promotes the targeting of proteolytic substrates to the proteasome. Mol. Cell Biol 22, 4902–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hiyama H, Yokoi M, Masutani C, Sugasawa K, Maekawa T, Tanaka K, et al. , (1999). Interaction of hHR23 with S5a. The ubiquitin-like domain of hHR23 mediates interaction with S5a subunit of 26 S proteasome. J. Biol. Chem 274, 28019–28025. [DOI] [PubMed] [Google Scholar]
- 11.Chen L, Shinde U, Ortolan TG, Madura K, (2001). Ubiquitin-associated (UBA) domains in Rad23 bind ubiquitin and promote inhibition of multi-ubiquitin chain assembly. EMBO Rep 2, 933–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilkinson CR, Seeger M, Hartmann-Petersen R, Stone M, Wallace M, Semple C, Gordon C, (2001). Proteins containing the UBA domain are able to bind to multi-ubiquitin chains. Nature Cell Biol 3, 939–943. [DOI] [PubMed] [Google Scholar]
- 13.Fishbain S, Prakash S, Herrig A, Elsasser S, Matouschek A, (2011). Rad23 escapes degradation because it lacks a proteasome initiation region. Nature Commun 2, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heessen S, Masucci MG, Dantuma NP, (2005). The UBA2 domain functions as an intrinsic stabilization signal that protects Rad23 from proteasomal degradation. Mol. Cell 18, 225–235. [DOI] [PubMed] [Google Scholar]
- 15.Wang Q, Goh AM, Howley PM, Walters KJ, (2003). Ubiquitin recognition by the DNA repair protein hHR23a. Biochemistry 42, 13529–13535. [DOI] [PubMed] [Google Scholar]
- 16.Walters KJ, Lech PJ, Goh AM, Wang Q, Howley PM, (2003). DNA-repair protein hHR23a alters its protein structure upon binding proteasomal subunit S5a. PNAS 100, 12694–12699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryu KS, Lee KJ, Bae SH, Kim BK, Kim KA, Choi BS, (2003). Binding surface mapping of intra- and interdomain interactions among hHR23B, ubiquitin, and polyubiquitin binding site 2 of S5a. J. Biol. Chem 278, 36621–36627. [DOI] [PubMed] [Google Scholar]
- 18.Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH, (2014). Understanding nucleotide excision repair and its roles in cancer and ageing. Nature Rev. Mol. Cell Biol 15, 465–481. [DOI] [PubMed] [Google Scholar]
- 19.Sugasawa K, Ng JM, Masutani C, Iwai S, van der Spek PJ, Eker AP, et al. , (1998). Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol. Cell 2, 223–232. [DOI] [PubMed] [Google Scholar]
- 20.Bunick CG, Miller MR, Fuller BE, Fanning E, Chazin WJ, (2006). Biochemical and structural domain analysis of xeroderma pigmentosum complementation group C protein. Biochemistry 45, 14965–14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uchida A, Sugasawa K, Masutani C, Dohmae N, Araki M, Yokoi M, et al. , (2002). The carboxy-terminal domain of the XPC protein plays a crucial role in nucleotide excision repair through interactions with transcription factor IIH. DNA Repair (Amst) 1, 449–461. [DOI] [PubMed] [Google Scholar]
- 22.Masutani C, Araki M, Sugasawa K, van der Spek PJ, Yamada A, Uchida A, et al. , (1997). Identification and characterization of XPC-binding domain of hHR23B. Mol. Cell Biol 17, 6915–6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L, Lu X, Peterson C, Legerski R, (1997). XPC interacts with both HHR23B and HHR23A in vivo. Mutat. Res 383, 197–203. [DOI] [PubMed] [Google Scholar]
- 24.Ng JM, Vermeulen W, van der Horst GT, Bergink S, Sugasawa K, Vrieling H, Hoeijmakers JH, (2003). A novel regulation mechanism of DNA repair by damage-induced and RAD23-dependent stabilization of xeroderma pigmentosum group C protein. Genes Dev 17, 1630–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sugasawa K, Masutani C, Uchida A, Maekawa T, van der Spek PJ, Bootsma D, et al. , (1996). HHR23B, a human Rad23 homolog, stimulates XPC protein in nucleotide excision repair in vitro. Mol. Cell Biol 16, 4852–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergink S, Toussaint W, Luijsterburg MS, Dinant C, Alekseev S, Hoeijmakers JH, et al. , (2012). Recognition of DNA damage by XPC coincides with disruption of the XPC-RAD23 complex. J. Cell Biol 196, 681–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van der Spek PJ, Eker A, Rademakers S, Visser C, Sugasawa K, Masutani C, et al. , (1996). XPC and human homologs of RAD23: intracellular localization and relationship to other nucleotide excision repair complexes. Nucleic Acids Res 24, 2551–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bienko M, Green CM, Crosetto N, Rudolf F, Zapart G, Coull B, et al. , (2005). Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science 310, 1821–1824. [DOI] [PubMed] [Google Scholar]
- 29.Dantuma NP, Heinen C, Hoogstraten D, (2009). The ubiquitin receptor Rad23: at the crossroads of nucleotide excision repair and proteasomal degradation. DNA Repair (Amst) 8, 449–460. [DOI] [PubMed] [Google Scholar]
- 30.Kannouche P, Fernandez de Henestrosa AR, Coull B, Vidal AE, Gray C, Zicha D, et al. , (2003). Localization of DNA polymerases η and ι to the replication machinery is tightly co-ordinated in human cells. EMBO J 22, 1223–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McIntyre J, McLenigan MP, Frank EG, Dai X, Yang W, Wang Y, Woodgate R, (2015). Posttranslational regulation of human DNA polymerase iota. J. Biol. Chem 290, 27332–27344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McIntyre J, Vidal AE, McLenigan MP, Bomar MG, Curti E, McDonald JP, et al. , (2013). Ubiquitin mediates the physical and functional interaction between human DNA polymerases η and ι. Nucleic Acids Res 41, 1649–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raasi S, Pickart CM, (2003). Rad23 ubiquitin-associated domains (UBA) inhibit 26 S proteasome-catalyzed proteolysis by sequestering lysine 48-linked polyubiquitin chains. J. Biol. Chem 278, 8951–8959. [DOI] [PubMed] [Google Scholar]
- 34.Ashton NW, Valles GJ, Jaiswal N, Bezsonova I, Woodgate R, (2021). DNA polymerase ιinteracts with both the TRAF-like and UBL1–2 domains of USP7. J. Mol. Biol 433, 166733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McIntyre J, (2020). Polymerase iota - an odd sibling among Y family polymerases. DNA Repair (Amst) 86, 102753. [DOI] [PubMed] [Google Scholar]
- 36.Jung J, Byeon IJ, DeLucia M, Koharudin LM, Ahn J, Gronenborn AM, (2014). Binding of HIV-1 Vpr protein to the human homolog of the yeast DNA repair protein RAD23 (hHR23A) requires its xeroderma pigmentosum complementation group C binding (XPCB) domain as well as the ubiquitin-associated 2 (UBA2) domain. J. Biol. Chem 289, 2577–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vinogradova O, Qin J, (2012). NMR as a unique tool in assessment and complex determination of weak protein-protein interactions. Top. Curr. Chem 326, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang WX, Gu XH, Dong X, Tang C, (2017). Lanthanoid tagging via an unnatural amino acid for protein structure characterization. J. Biomol. NMR 67, 273–282. [DOI] [PubMed] [Google Scholar]
- 39.Varadan R, Assfalg M, Raasi S, Pickart C, Fushman D, (2005). Structural determinants for selective recognition of a Lys48-linked polyubiquitin chain by a UBA domain. Mol. Cell 18, 687–698. [DOI] [PubMed] [Google Scholar]
- 40.Pata JD, (2010). Structural diversity of the Y-family DNA polymerases. Biochim. Biophys. Acta 1804, 1124–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Byeon IL, Calero G, Wu Y, Byeon CH, Jung J, DeLucia M, et al. , (2021). Structure of HIV-1 Vpr in complex with the human nucleotide excision repair protein hHR23A. Nature Commun 12, 6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang W, (2014). An overview of Y-Family DNA polymerases and a case study of human DNA polymerase η. Biochemistry 53, 2793–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haracska L, Acharya N, Unk I, Johnson RE, Hurwitz J, Prakash L, Prakash S, (2005). A single domain in human DNA polymerase iota mediates interaction with PCNA: implications for translesion DNA synthesis. Mol. Cell Biol 25, 1183–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohashi E, Hanafusa T, Kamei K, Song I, Tomida J, Hashimoto H, et al. , (2009). Identification of a novel REV1-interacting motif necessary for DNA polymerase κ function. Genes Cells 14, 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McIntyre J, Sobolewska A, Fedorowicz M, McLenigan MP, Macias M, Woodgate R, Sledziewska-Gojska E, (2019). DNA polymerase ιis acetylated in response to S(N)2 alkylating agents. Sci. Rep 9, 4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Su V, Lau AF, (2009). Ubiquitin-like and ubiquitin-associated domain proteins: significance in proteasomal degradation. Cell. Mol. Life Sci 66, 2819–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frank EG, McDonald JP, Karata K, Huston D, Woodgate R, (2012). A strategy for the expression of recombinant proteins traditionally hard to purify. Anal. Biochem 429, 132–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smyth GK, Speed T, (2003). Normalization of cDNA microarray data. Methods 31, 265–273. [DOI] [PubMed] [Google Scholar]
- 49.Karata K, Vaisman A, Goodman MF, Woodgate R, (2012). Simple and efficient purification of Escherichia coli DNA polymerase V: cofactor requirements for optimal activity and processivity in vitro. DNA Repair (Amst) 11, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mueller TD, Feigon J, (2002). Solution structures of UBA domains reveal a conserved hydrophobic surface for protein-protein interactions. J. Mol. Biol 319, 1243–1255. [DOI] [PubMed] [Google Scholar]
- 51.Byeon IL, Jung J, Byeon CH, DeLucia M, Ahn J, Gronenborn AM, (2020). Complete (1)H, (13)C, (15)N resonance assignments and secondary structure of the Vpr binding region of hHR23A (residues 223–363). Biomol. NMR Assign 14, 13–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dominguez C, Boelens R, Bonvin AM, (2003). HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc 125, 1731–1737. [DOI] [PubMed] [Google Scholar]
- 53.van Zundert GCP, Rodrigues J, Trellet M, Schmitz C, Kastritis PL, Karaca E, et al. , (2016). The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol 428, 720–725. [DOI] [PubMed] [Google Scholar]
- 54.Wassenaar TA, van Dijk M, Loureiro-Ferreira N, van der Schot G, Sjoerd J, de Vries SJ, Schmitz C, et al. , (2012). WeNMR: structural biology on the grid. J. Grid Comput 10, 743–767. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Uncropped immunoblot and protein gel images, as well as raw microarray data, are available from Mendeley Data (https://data.mendeley.com/datasets/ht763x684h). All raw NMR data are available upon request.