

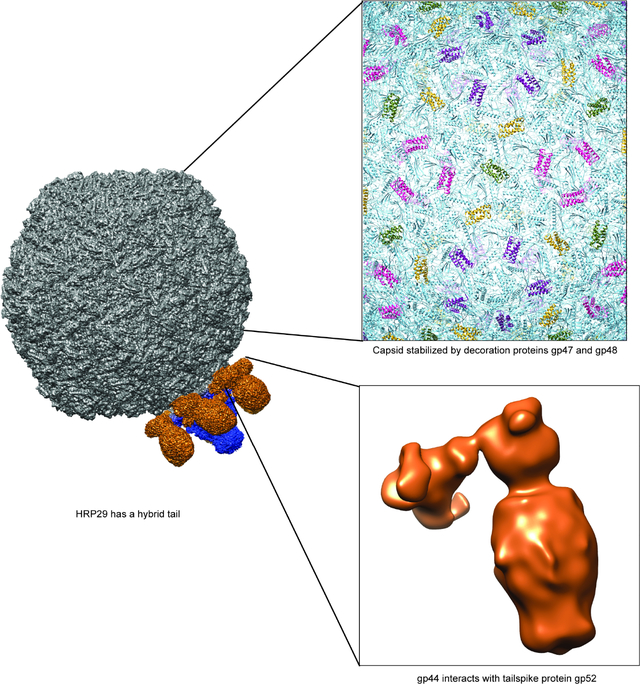
Summary
There is a paucity of high-resolution structures of phages infecting Shigella, a human pathogen and a serious threat to global health. HRP29 is a Shigella podophage belonging to the Autographivirinae family, and has very low sequence identity to other known phages. Here, we resolved the structure of the entire HRP29 virion by cryo-EM. Phage HRP29 has a highly unusual tail that is a fusion of a T7-like tail tube and P22-like tailspikes mediated by interactions from a novel tailspike adaptor protein. Understanding phage tail structures is critical as they mediate hosts interactions. Furthermore, we show that the HRP29 capsid is stabilized by two novel, and essential decoration proteins, gp47 and gp48. Only one high resolution structure is currently available for Shigella podophages. The presence of a hybrid tail and an adapter protein suggests that it may be a product of horizontal gene transfer, and may be prevalent in other phages.
Blurb
Shigella flexneri is a bacterial pathogen of concern and bacteriophage therapy to combat bacterial infections is gaining importance. To date there is only one high-resolution structure of a bacteriophage infecting S. flexneri known and here Subramanian et al. present the cryo-EM structure of the bacteriophage HRP29.
Grapchical Abstract
Introduction
An estimated 450,000 infections occur per year due to Shigella alone in the United States. Recently the development of extensively drug-resistant Shigella places this pathogen in the “serous threat” category according to the CDC 1. A rise in antibiotic resistance has increased the interest in using bacteriophages (phages) as treatment alternatives, or supplements for antibiotics. Phages infecting Shigella have been isolated and used for treatment as early as 1917 by Félix d’Herelle 2, but until recently only a limited collection of phages infecting Shigella were available 3,4. Most of the phages isolated against Shigella belong to the order Caudovirales and have capsids with dsDNA and a tail attached to the capsid that is crucial for the infection and propagation of the phage. Phages from Caudovirales can be morphologically classified into three groups based on their tail structures, Myoviridae (long, contractile), Siphoviridae (long, non-contractile) and Podoviridae (short, non-contractile) 5. Most phages isolated from the environment against Shigella flexneri belong to the Myoviridae family, with only two Podoviridae, observed to date 3,4,6. Both podophages, HRP29 and Moo19, were isolated by high school students in Lincoln, Nebraska. Surprisingly, we have not yet been successful in isolating podophages from environmental samples in Michigan. These podophages have distinct differences in physiology and structure compared to commonly studied podophages that infect enteric pathogens such as Salmonella and Escherichia species, including differences in host receptor interactions via the tails. Therefore, we were interested in characterizing these phages in greater detail.
HRP29 belongs to the Autographivirinae family and is a new species of Drulisvirus distantly related to phiKDA1 and phiKT. HRP29 has only 10% ANI to any known phage genome and further examination showed some potentially interesting features in the structural genes 6. For example, HRP29 has multiple genes encoding for tail proteins similar to a P22-like tailspike protein, as well as two T7-like tail protein genes. Various biochemical studies and previously-resolved structures of podophage T7, P22 and Sf6 virions, showed that T7 has a tail tube with a nozzle to which the tail fibers are attached to, whereas P22 and Sf6 have a tail needle and tailspike attached to the tail hub 7–9. The absence of structural information for close relatives of HRP29 and the genetic suggestion for the presence of a hybrid tail motivated us to understand the global architecture of the HRP29 phage, with an emphasis on determining the tail structure. Apart from the high-resolution structure of Sf6 8 there are no published structures for any podophage infecting Shigella.
We used single particle cryo-EM and image reconstruction methods to generate high-resolution structures of HRP29 allowing us to visualize the tail, and other features in greater detail. From the structure, the presence of two novel decoration proteins was identified and their role in stability was probed using CRISPR knockdown and particle stability tests. A focused reconstruction of the tail revealed the structure of portal, adaptor, and nozzle proteins as well as the N-terminus of the tailspike adaptor. Co-purification of the tailspike and the putative tailspike adaptor showed that they form a complex of trimers. Procapsids were also generated using a CRISPR knockdown of the large terminase (TerL) with the resulting structure indicating the scaffolding protein location. Our work has generated a high resolution structure and corresponding atomic model of the entire HRP29 virion, and reveals novel protein:protein interactions that form a hybrid structure between the T7-like and P22-like phages.
Results & Discussion
Asymmetric reconstruction of HRP29 reveals a hybrid tail
Plating experiments were performed to understand some of the basic requirements for HRP29 infection in Shigella flexneri and we compared these observations to the well-studied Shigella phage Sf6. In order for a successful infection, Sf6 requires initial interaction with the primary receptor, lipopolysaccharide (LPS), interactions with a secondary receptor, an outer membrane protein (either OmpA or OmpC), and interaction with an inner membrane protein, YajC 10,11. S. flexneri have a variety of serotypes like Y, 2a, 3a, 4a, 5a, X, 7a etc. that show variations in the LPS structure based on the presence or absence of glucosyl group, O-acetyl group and/or phosphoethanolamine group 12. Sf6 can infect only serotype Y, which is the simplest serotype as it lacks any of the additional sugar groups on the LPS. HRP29 also infects serotype Y and is partially dependent on outer membrane proteins OmpA and OmpC for infection (Supplementary table: 1). However, HRP29 does not require the inner membrane protein YajC for infection. This observation along with the presence of genes for both T7-like tail tube proteins and a Sf6-like tailspike proteins led us to investigate the structural details of the tail further. We generated an asymmetric reconstruction of HRP29 at 4.1 Å that showed that it has both a central tail tube structure similar to T7 along with six tailspikes similar to Sf6 (Fig. 1). The central tail tube was resolved better when compared to the tailspikes and the central section through the reconstruction showed the possible presence of inner core (Fig. S1).
Figure 1: HRP29 has a hybrid tail.
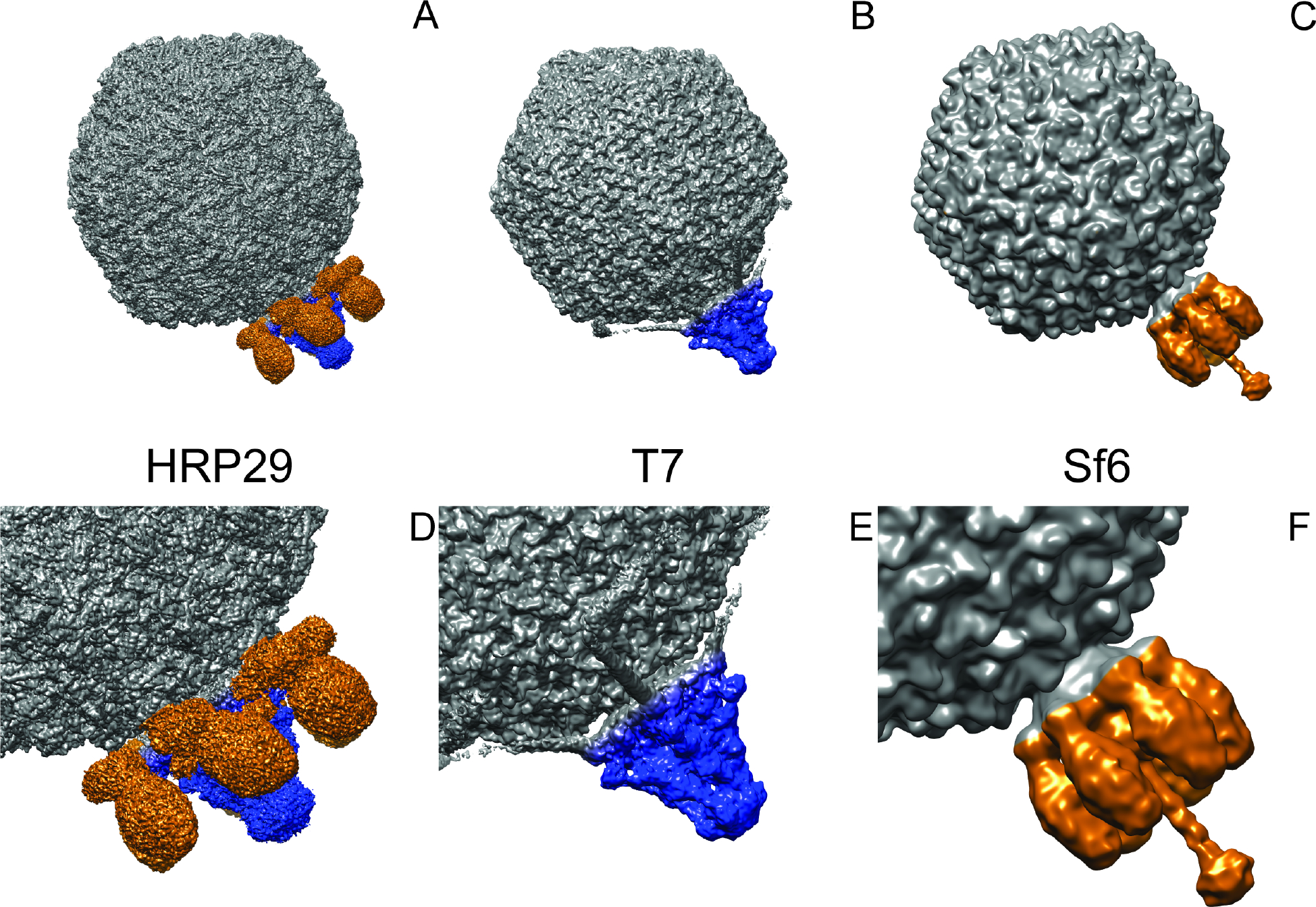
(A) Asymmetric reconstruction of HRP29 (EMD – 28227) with the central tail tube colored in blue and the tailspikes colored in orange (4.1 Å resolution). (B) Symmetry mismatch reconstruction of T7 (EMD – 31315) with the central tail tube colored in blue (7.0 Å resolution). (C) Asymmetric reconstruction of Sf6 (EMD – 5730) with the tail colored in orange highlighting the tailspike and tail needle (16.0 Å resolution). (D-F) Enlarged views of the tails of HRP29, T7 and Sf6.
HRP29 portal protein is similar to T7
A C6 symmetry-imposed focused reconstruction of the HRP29 tail resolved atomic details of the portal protein (gp35), adapter protein (gp39), and the nozzle protein (gp40). Initial models for gp35, gp39 and gp40 were generated using Alphafold2 and then docked into the map followed by model building and refinement (Fig. 2A). A DALI search of HRP29 gp35 showed that it is similar to portal protein gp8 from the T7 from mature virion (PDB: 7EY8) with a RMSD of 2.7 Å and a Z-score of 33.4 (Fig. S2A). Portal protein gp35 had a similar domain structures including the stem, the clip, the wing and the crown domains when compared to T7 gp8 13. When compared to the portal structure from mature T7 virion, there were variations in the wing domain, the C-terminal residues (488 – 517) were not modeled in HRP29 and the channel valve also could not be modeled (Fig. S2A). The conformation of the channel valve, which holds the DNA in the capsid is likely similar to that of T7 7,13. Both P22 and Sf6 have a long helical barrel at the C-terminal end and this elongated structure is thought to form only in the mature virion as it might be stabilized by packaged DNA and ejection proteins 8,14. When compared to the Sf6 portal structure from the mature virion, HRP29 does not have a long helical barrel in the C-terminal end. HRP29 lacks this feature as the length of the HRP29 portal protein is 517 amino acids, and is considerably shorter than the Sf6 portal protein with 708 amino acids.
Figure 2: HRP29 has a central tail tube similar to T7.
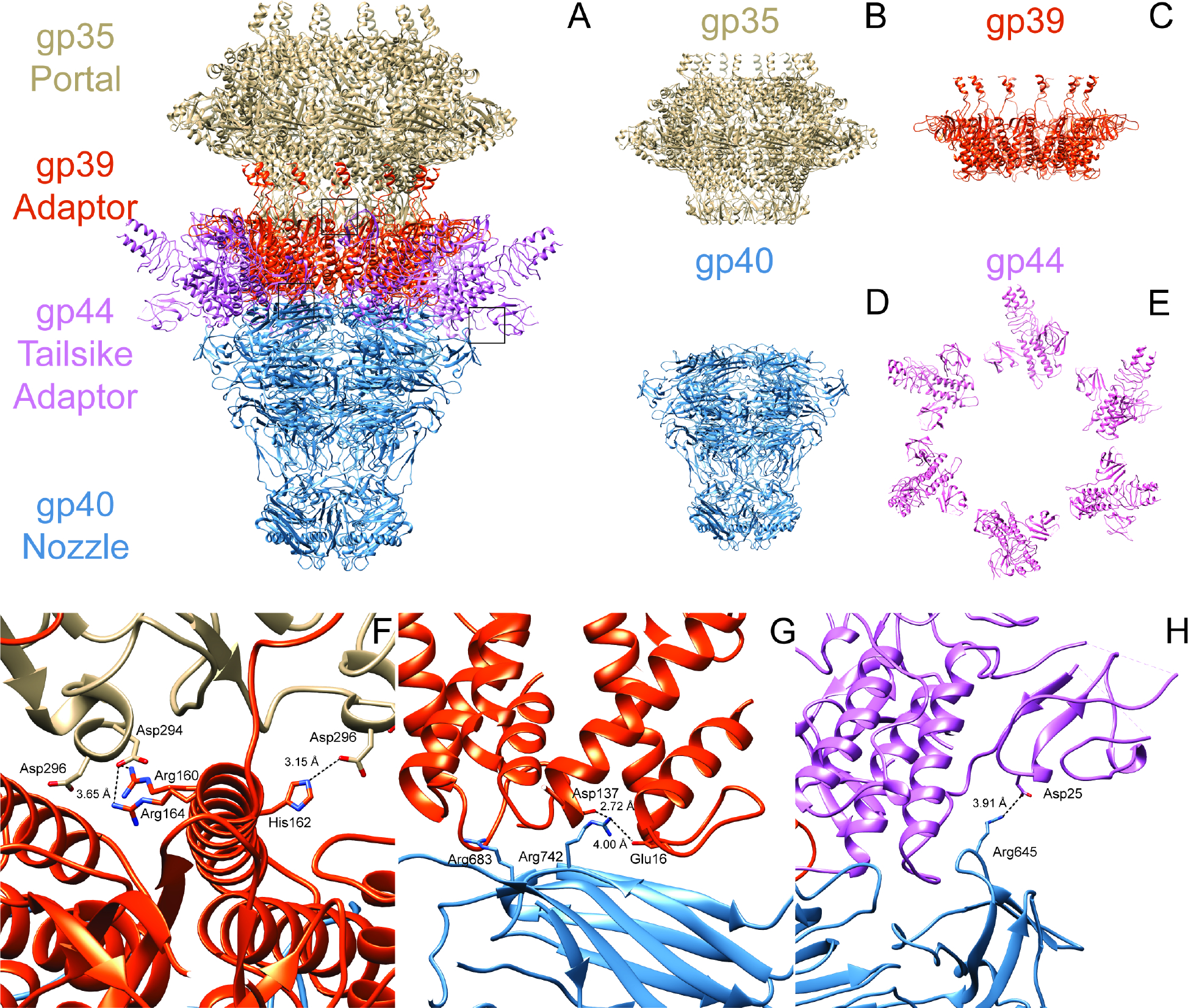
(A) Model of the tail generated from the focused C6 reconstruction of the tail with the portal gp35 colored in brown, adaptor gp39 in orange, nozzle gp40 in blue and tailspike adaptor gp44 in pink. (B-D) Side views of the tail tube proteins gp35, gp39 and gp40. (E) Top view of the tailspike adaptor gp44. (F) Interaction between gp35 (brown) and gp39 (orange) mediated by negatively charged amino acids on gp35 and positively charged amino acids on gp39. This interaction is flipped in terms of charge when compared to T7. (G) Negatively charged amino acids from two gp39 monomers interact with one monomer of gp40 at two distinct sites with positively charged amino acids. (H) Interaction between nozzle gp40 and tailspike adaptor gp44 mediated by Arg645 in gp40 and Asp25 in gp44. This interaction is also flipped in terms of charge when compared to T7.
The HRP29 tail complex is composed of gp39, gp40 and gp44
Analysis of the HRP29 tail complex revealed structures of three major components gp39, gp40 and gp44. A DALI search of HRP29 gp39 showed it is similar to gp11 of T7 from mature virion (PDB: 7EY9) with a RMSD of 2.5 Å and a Z-score of struct 18.2 (fig. S2B). Similar to T7, HRP29 adapter protein gp39 has an a-helix bundle, a fiber dock domain and a C-terminal embracing helix domain (Fig. S2B)13. In comparison to the homolog T7 gp11, HRP29 gp39 lacks the disulfide bond stabilizing the α-helix bundle. Instead, HRP29 gp39 interacts with gp35 through electrostatic interactions mediated by residues Arg160, Arg164 and His162 of gp39 and Asp294 and Asp296 of gp35 (Fig. 2B). This electrostatic interaction is opposite those in T7, as negatively charged residues in adapter protein gp11 interact with positively charged residues of gp8 in T7. The C-terminal end of HRP29 gp39 could not be modeled completely when compared to T7 gp11. The C-terminal end sequence of gp39 is similar to gp11, and thus might form a similar electrostatic interaction with the portal protein gp35. Next, a DALI search of HRP29 gp40 showed that it is similar to gp12 of T7 from the mature virion (PDB: 7EY9) with an RMSD of 2.9 Å and a Z-score of 29.1 (Fig. S2C). Similar to T7, HRP29 nozzle protein, gp40, has a β-propeller domain, platform domain, fiber dock domain and a nozzle tip domain, with differences in the fiber dock domain (Fig. S2C) 13. The β-propeller in gp40 lacks the disulfide bond and the hydrophobic interaction at the base of the propeller that are found in T7. In addition, positively charged residues of gp40 at different locations interact with negatively charged residues from two gp39 subunits (Fig. 2C). The nozzle of HRP29 also has gates similar to T7 to restrict and secure the DNA with in the mature virion with the largest differences being in the gate 1 region.
Apart from gp35, gp39 and gp40, there were six densities with protruding trimeric α-helices (Fig. 2A). An alphafold prediction of the gp44 tailspike adapter protein trimer was docked into the map and then refined. This density was similar to the initial parts of a trimer of gp17 from the phage T7 tail, and the first 147 of 260 amino acids were modeled. A DALI search of HRP29 gp44 showed that it is similar to gp17 of T7 from the mature virion (PDB: 7EY9) with a RMSD of 3.8 Å and a Z-score of 13.1, (Fig. S2D). In T7, the first 143 amino acids form the fiberdock domain and the long flexible tail is formed by the following region 7,15. The flexibility of the T7 tail fiber likely comes from the region following the fiberdock domain, as these residues were not able to be modeled in the T7 structure. Similarly, in HRP29, the region after the first 147 amino acids may also be flexible thereby preventing modeling. HRP29 gp44 forms a trimer that appears to interact with the tail adapter gp39 and the tail nozzle gp40. Approximately the first 69 amino acids of gp44 form a β-bundle structure followed by a long loop that connects to α-helix which forms the initial part of the trimerization domain. Residues 105 to 138 are unstructured intertwine with the same region from the other monomers in the trimer. The core of the unstructured trimer region is hydrophobic and electrostatic interactions between the monomers on the exterior stabilize this region. Two of the three β-bundles from the gp44 interact with the fiberdock domain from nearby gp39. The third β-bundle from the gp44 trimer is nearby the fiberdock domain of gp44 and Asp25 from gp44 interacts with Arg645 of gp40 (Fig. 2D). This electrostatic interaction is opposite in comparison to T7 as negatively charged residues in the nozzle gp12 interact with positively charged residues of gp17 in T7. The base of the gp44 trimer formed by the loop connecting the β-bundle and α-helix, interacts with the platform domain of gp40 and the helix domain of gp39.
Tailspike adapter gp44 interacts with gp52 tailspike to form a hybrid tail
Although the lower resolution asymmetric reconstruction of the entire virion showed the entire tail machinery, a higher resolution, focused reconstruction of the HRP29 tail resolved only the N-terminal part of gp44 and did not show connection to the tailspike protein, gp52 likely due to inherent flexibility. The HRP29 tailspike (gp52) is similar to the Sf6 tailspike (gp14) and the active site residues Glutamate 366 and Aspartate 399 are conserved between both (Fig. S3). The only significant difference is in the first 40 residues. HRP29 and Sf6 both infect the serotype Y strain of S. flexneri and in the case of Sf6 the N-terminus of the tailspike (gp14) directly interacts with the tail hub. In HRP29, gp44 and gp52 may interact directly with each other to form the tail or another protein could mediate this interaction. Since the resolution of the map was limited, we used protein co-purification to test this hypothesis. We co-expressed gp44 containing a N-terminal 6x His tag along with gp52 without the addition of any tags and the complex was purified from crude lysate using a Ni-NTA coloumn. Size exclusion chromatography showed that gp44 and gp52 co-elute and form a stable complex. Furthermore, the density of each band from SDS-PAGE showed that there were similar amounts of gp44 and gp52 with a ~1:1 stoichiometry of each protein (Fig. S4A). Therefore, a trimer of gp44 likely interacts with a trimer of gp52. We used electron microscopy of negatively stained co-eluted complexes and performed image analysis (Fig. 3A, S4B). A low-resolution 3D reconstruction showed a L-shaped structure formed by the gp44-gp52 complex (Fig. 3B). Alphafold2 could not predict the complex formation between gp44-gp52 accurately and only a trimer of gp52 could be reliably fit into the density (Fig. 3C, S4C). The interaction interface between the C-terminal region of gp44 and the N-terminal region of gp52 could not be resolved clearly. The gp44 N-terminal region was likely not clearly resolved due to flexibility, especially in the absence of any interactions with the tail. We attempted to solve the structure of this complex by cryo-EM studies, and were met with several challenges, including severe orientation bias and inherent flexibility of some regions (see Supplementary Fig. S4D, S4E). Even though the interaction between gp44 and gp52 could not be visualized in detail, the ability of gp44 and gp52 to be co-purified and the low-resolution negative stain model of the complex support the hypothesis that gp44 and gp52 interact with each other. In order to determine if both tail proteins were essential for infection by HRP29, we designed CRISPR knockdowns using CRISPR-dCas9. Knockdowns of gp44 and gp52 had mild effects: 10-fold and 100-fold reduction in plaquing efficiency, respectively (Fig. 5A). Therefore, we conclude that a combination of both proteins does indeed confer an advantage to phage HRP29.
Figure 3: gp52 interacts with the tail through gp44.
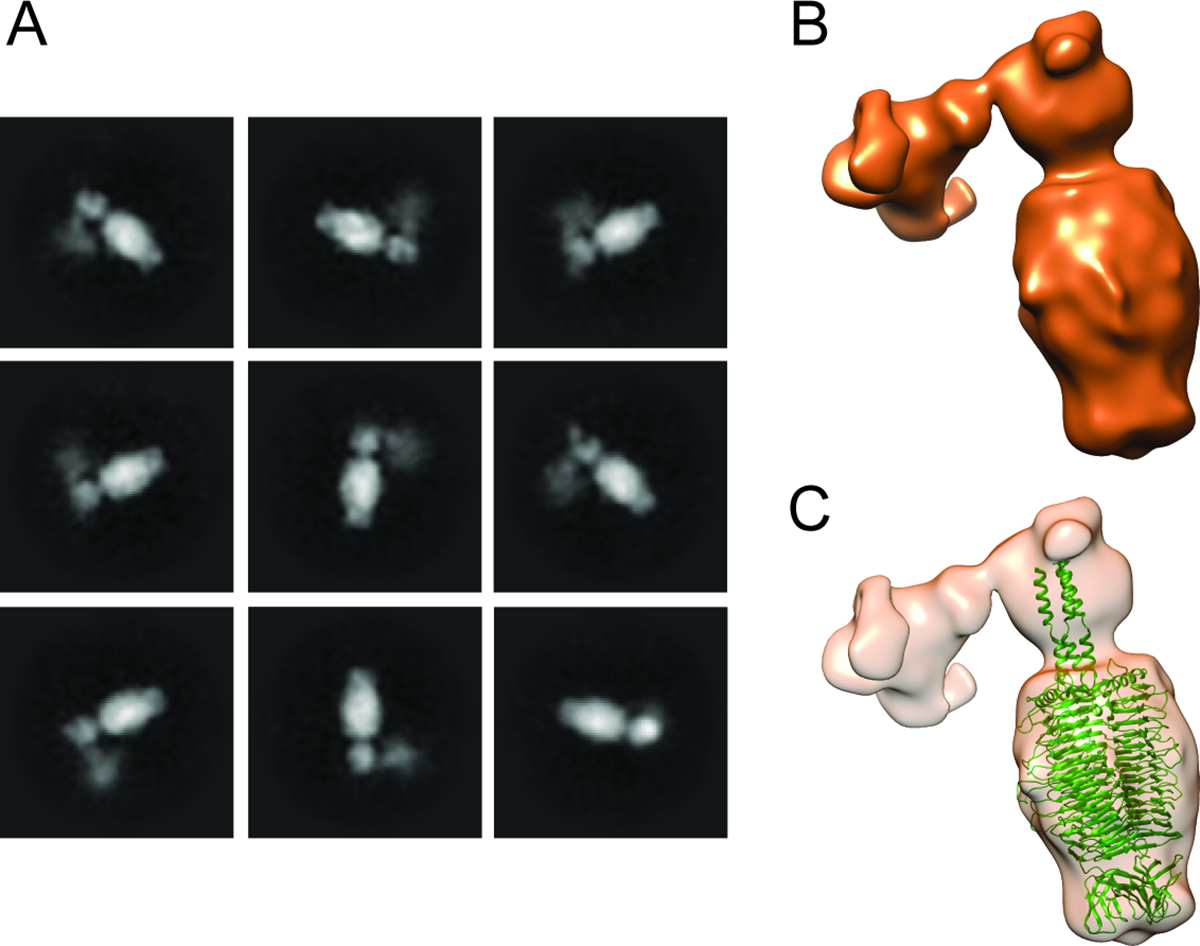
(A) Representative 2D class averages of negatively stained gp44-gp52 complex purified from E. coli, shows a strong signal for tailspike protein gp52, whereas the signal for gp44 is more diffuse and noisy. (B) 3D reconstruction of gp44 and gp52 complex from a total of 8,583 particles show that the complex has an elbow shape. (C) Alphafold prediction of gp52 trimer (green) docked into the 3D reconstruction of gp44-gp52 complex; the alphafold prediction of gp44 trimer could not be reliably fit into the model.
Figure 5: CRISPR knockdown of gp44, gp47, gp48, gp52 and thermal stability assay.
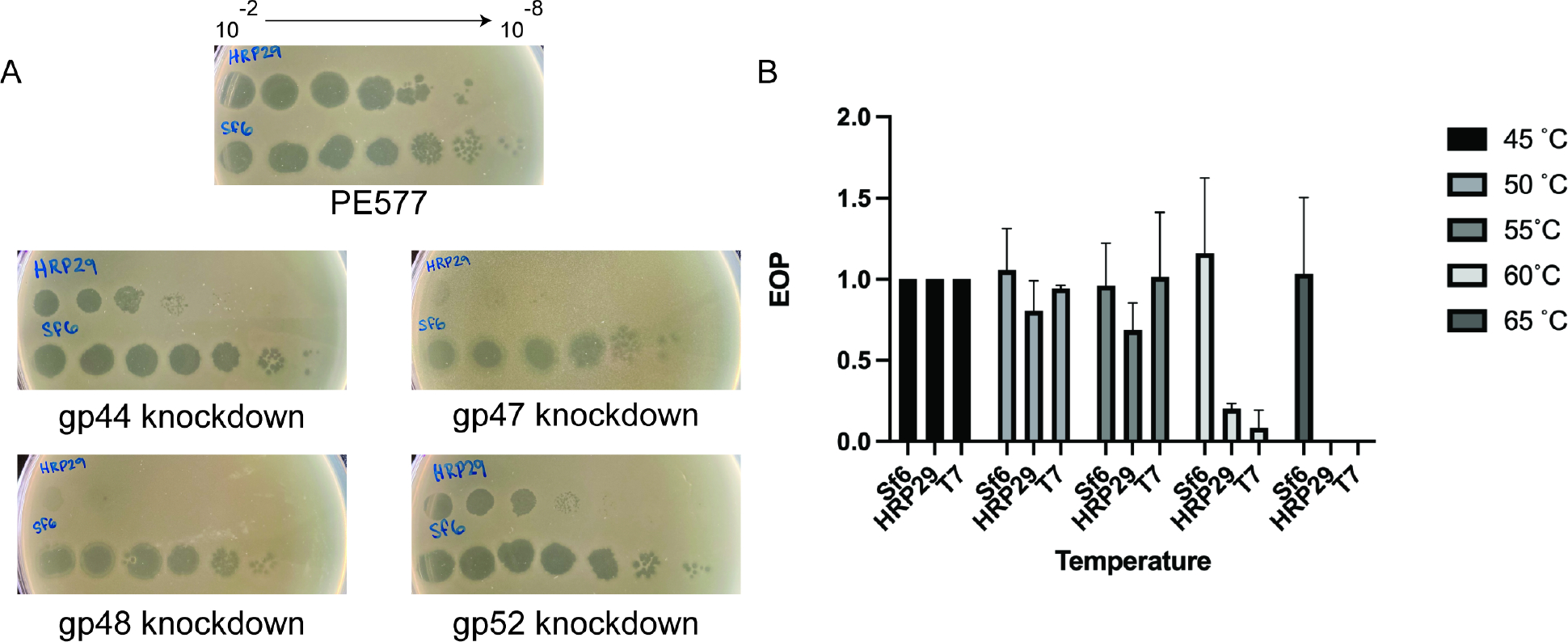
(A) Spot assays of HRP29 and Sf6 at various dilutions on PE577, cells expressing a dead cas9 nuclease along with a gRNA targeting either gp44, gp47, gp48 or gp52. (B) Quantitative plaque assays of Sf6, HRP29 and T7 after incubation for 10 minutes at the indicated temperature. The efficiency of plating was determined as the titer of phage at the experimental temperature, divided by the titer of phage at the permissive temperature of 45 °C. Error bars reflect the standard deviation from at least three biological replicates.
Additionally, we hypothesize that since Sf6 (has a tailspike and a tail needle) requires yajC, an inner membrane for infection, and T7 (uses a nozzle instead) does not need an inner membrane protein, that this hybrid assembly joining a tailspike with a nozzle allows this Shigella podophage to bypass this requirement. The presence of a T7-like tail tube and functional tailspikes instead of tail fibers has been previously observed in closely related bacteriophages phages K1–5, K1E and SP6, yet these have two sets of tailspikes instead of the one set of tailspike that HRP29 possesses 16,17. K1–5 bacteriophage can infect two strains of E. coli K1 and E. coli K5 that have distinct capsule structures. The central tail tube is connected to a total of six gp37 adapters and each gp37 has binding sites for two types of tailspikes, gp46 and gp47. The N-terminal region of the adapter protein gp37 is similar to T7 gp17. K1–5 gp46 is similar to P22 tailspike and gp47 is a endosialidase enabling K1–5 to infect strains with distinct capsule structures. Bacteriophage K1E is similar to K1–5 but can infect only E. coli K1 as it lacks a functional tailspike gp46 as only the first 111 amino acids of gp46 are present in K1E. Bacteriophage SP6 can infect both the S. enterica subsp. enterica serovar Typhimurium and S. enterica subsp. enterica serovar Newport. Similar to K1–5, SP6 also has an adapter protein gp37 that is connected to two tailspikes gp46 and gp47. SP6 gp46 is similar to P22 tailspike and gp47 is also supposed to form a trimeric structure but with less similarity to P22 tailspike. Only one of the tailspikes is engaged on the cell surface when infecting a particular serotype and the two tailspikes can rotate with respect to each other to engage with the O-antigen. A common theme utilized by K1–5, K1E, SP6 and also HRP29 is the tailspike adapter. This adapter has an N-terminal region similar to gp17 of T7 connecting the tailspikes to the central T7 like tail and a C-terminal region that has evolved to bind tailspikes. HRP29 tailspike gp52 is similar to the Sf6 tailspike gp14 but gp52 is significantly shorter (508 amino acids) when compared to gp14 (623 amino acids). The N-terminal region of HRP29 gp52 encompassing the first 41 amino acids is however dissimilar to Sf6 gp14 as it may have evolved to bind to the tailspike adapter gp44. The active site residues in Sf6 tailspike gp14 Glu366 and Asp399 (numbering does not include the N-terminal methionine) 18 are also present in HRP29 gp52 at positions 367 and 400, indicating that gp52 may employ a similar catalytic mechanism to degrade the O-antigen. To our knowledge HRP29 is the first phage with a T7 like tail tube attached to only one functional tailspike (gp52) via a tailspike adapter (gp44).
Casjens and Thuman-Commike 19 examined evolutionary relationships of 57 mosaically related P22-like phages and made several observations regarding the tailspike: 1) the tailspike gene is often separated by the other morphogenetic genes by several kbp, 2) the N-terminal domain is the capsid binding region, and this is highly conserved to maintain productive protein:protein interactions with the capsids, 3) where the C-terminal domain is highly diverse and dependent on receptor binding protein interactions. They reasoned that this is indicative of horizontal exchange among phage types, that the mosaic boundaries do not necessarily coincide with gene boundaries, and that there is strong evolutionary pressure to maintain a consistent gene order. While some of these principles hold true in HRP29-related phages such as consistent gene order, it seems that exchange along gene boundaries is more commonly found. In phages HRP29, K1–5, K1E, and SP6 it appears an additional gene is needed to serve as an adapter, as these phages are completely missing the tailspike head-binding domains. Instead, HRP29-like phages have taken advantage of a structural motif similar to the T7 fiber in order to allow for a productive tailspike attachment to the capsid, instead of a conserved N-terminal domain.
HRP29 internal proteins gp41, gp42 and gp43
Podophages like T7 and P22 form a channel bridging the inner and outer membranes of the host to enable ejection of DNA into the cytoplasm 20,21. The channel is formed by ejection proteins gp14, gp15 and gp16 in T7, gp7, gp16 and gp20 in P22 and gp11. HRP29 has three annotated internal virion proteins gp41, gp42 and gp43, and the presence of a core inside the virion can be visualized in the central slices of the asymmetric reconstruction (Supplemental Figure S1). The internal proteins have no significant sequence similarity to T7 ejection proteins (gp14, gp15 and gp16) nor Sf6 ejection proteins (gp11, gp12 and gp13). We were not able to obtain a high-resolution reconstruction of internal proteins even with the focused asymmetric reconstruction of the HRP29 tail. The lack of obvious similarity of HRP29 gp41 and gp42 to other phage proteins can be due to either the prediction not being accurate, or they may have a significantly different fold. Additional data will be needed to confirm that these proteins are indeed ejection proteins.
The portal (12 monomers) is attached to the capsid at one of the five-fold vertices and thereby creates a non-divisible symmetry mismatch. Interactions between portal and capsid proteins have been resolved for T4, P23–45 and Sf6 8,22,23. In the case of T4 and P23–45, the portal accommodates interactions with each of the capsid subunits at the five-fold vertex by undergoing localized conformation changes. However, in the case of Sf6 the portal protein remains fixed and instead forms symmetry matching interactions with the capsid. Attempts to identify the interactions between the capsid and the portal to understand whether portal undergoes localized conformational changes to interact with the capsid or form symmetrical interactions with the capsid using focused asymmetric reconstruction failed. The failure of focused asymmetric reconstruction may be due to a variety of factors including but not limited to lower number of tail particles, capsid density dominating alignment and/or flexibility within the tail.
HRP29 has two decoration proteins: gp47 and gp48
In addition to the hybrid tail, our asymmetric reconstruction of the virion revealed interesting surface features. Specifically, the surface of the capsid showed bumpy features around hexamers and pentamers, yet the asymmetric reconstruction was too low in resolution to accurately determine the details on the capsid surface. Phage capsids hold packaged genomic DNA, and as a result the internal pressure is extremely high. Capsids such as for phage HK97 are stabilized by both covalent (chain mail interlocking disulfide bridges), and non-covalent interactions between the subunits of the capsid 24,25. In addition, phage capsids can also be stabilized by cementing or decoration proteins 26. We used an icosahedral reconstruction to improve the resolution and examine the capsid surface in more detail. The 3.1 Å density map clearly showed distinct α-helical structures interacting with the major capsid protein subunits.
A homology model of HRP29 was generated using Phyre2. The T7 major capsid protein was one of the top hits, which was then used as the initial model to build and refine HRP29 gp37 (Fig. 4A). Additionally, a DALI search of gp37 showed that HRP29 gp37 is most similar to gp10A of T7 from the mature virion (PDB: 3J7X) with a RMSD of 2.9 Å and a Z-score of 23.1. The overall fold of the HRP29 capsid monomer resembles T7 with a RMSD of 1.299 Å. HRP29 gp37 has a long unstructured surface-exposed N-terminal arm, along with a surface-exposed C-terminus similar to T7 27. The T7 capsid is largely stabilized by intracapsomere interactions between Asp103 and Arg262 in the A-loop, and an intracapsomere charge-based interaction between the N-terminal residues (14–19) and the A-pocket (151–156) of an adjacent subunit. However, these intercapsomere and and intracapsomere stabilizing interactions are absent in HRP29. The absence of the intracapsomere stabilizing forces in HRP29 is due to the difference in the A-loop conformations and the absence of a similar pair of oppositely charged residues in the region. The N-terminal loop in HRP29 also adopts a slightly different conformation which puts it farther away from the A-pocket.
Figure 4: HRP29 mature virion has two decoration proteins gp47 and gp48.
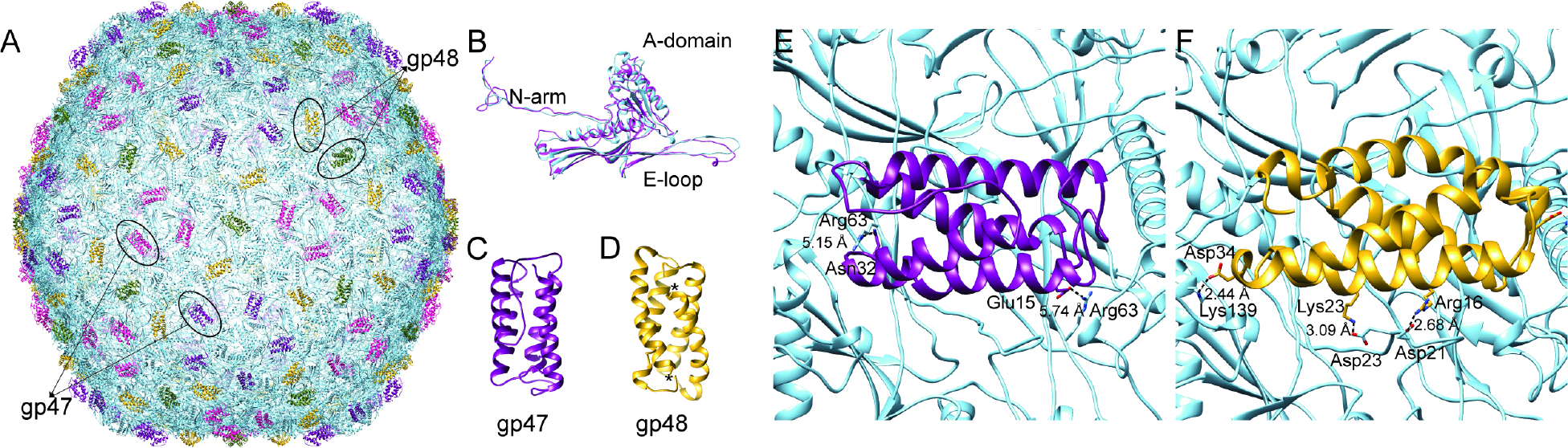
(A) Model of HRP29 capsid generated form an icosahedral reconstruction with the capsid protein (gp37) colored in blue and the decoration protein gp47 colored in purple and magenta and the other decoration protein gp48 colored in yellow and green. (B) One of the monomers from the hexamers of the asymmetric unit from HRP29 and T7 were aligned against each other with a RMSD of 1.299 Å using match maker in chimera. (C) Monomer of gp47 from the penton-hexon junction. (D) Dimer of gp48 with the asterisk mark representing the two C-terminus. (E) gp47 interaction with the capsid proteins is stabilized by positively charged amino acid Arg63 from two capsid monomers. (F) gp48 interaction with the capsid is stabilized by a series of electrostatic interactions from each of the gp48 monomer mediated by residues Arg16, Lys23 and Asp34 from gp48 and Asp21, Asp23 and Lys139 of the capsid protein.
While building the capsid asymmetric unit it became evident that there were two different densities not accounted for by the major capsid protein. The two densities were segmented and submitted individually to the DeepTracer for identification. DeepTracer identified the two densities as a monomer of gp47 and a dimer of gp48. The presence of gp47 and gp48 in the mature virion was also confirmed by mass spectrometry analysis (data not shown). Individual CRISPR knockdowns of gp47 and gp48 were used to understand if they are essential proteins, or dispensable. Spot assays of HRP29 on cells expressing gRNA targeting either gp47 or gp48 along with an inactive cas9 nuclease showed that HRP29 produced significantly fewer viable phages (~10^6 fold reduction) when compared to cells without the CRISPR knockdown system (Fig. 5A). The severely reduced ability of HR29 to produce viable phages in the absence of either gp47 or gp48 indicate that they are indeed both essential. On the capsid, gp47 monomer binds at the pentamer – hexamer and hexamer – hexamer junction whereas a dimer of gp48 binds at the hexamer-hexamer junction surrounding pentamers (Fig. 4A). The binding pockets of gp47 and gp48 are similar to each other but the stabilizing interactions formed by gp47monomer and gp48 dimers are unique. In the case of gp47 there is a single hydrogen bond with the gp37 capsid monomer mediated by residue Glu15 at each of the pentamer - hexamer and hexamer – hexamer junction (Fig. 4E). Each monomer of gp48 forms hydrogen bonds with two gp37 capsomeres mediated by residues Lys23 and Asp34; a total of four hydrogen bonds are present at each of the hexamer – hexamer junctions (Fig. 4F). A DALI search of gp47 and gp48 showed no similarity to known published phage proteins.
The lack of viable phages when production of gp47 and gp48 was inhibited indicates that gp47 and gp48 might provide stability to the HRP29 capsid. Thermal stability assays were performed to test the overall stability of HRP29 in comparison to Sf6 and T7 (Fig. 5B). HRP29 was slightly more stable against heating than T7, but not as stable as Sf6 (Fig 5B). Both gp47 and gp48 can be considered essential decoration/cementing proteins as they bind to the surface of the capsid and seem to provide some stability. Decoration proteins such as P30 (phage PRD1), gpD (phage λ), Soc (phage T4), Dec (phage L), and Psu (phageP4) bind to defined positions on the capsid such as the 2-fold, 3-fold, quasi 2-fold, quasi 3-fold and the quasi 6-fold symmetry axes and stabilize particular stress points 28–32. By contrast, gp47 and gp48 bind and stabilize all the individual monomers in the capsid by binding at the interfaces where two monomers meet. Instead of binding and stabilizing particular stress points on the capsid, gp47 and gp48 seem to form stabilizing interactions throughout the entire capsid.
Decoration proteins from bacteriophages can adopt various structures with well-defined features like the β-tulip motif, OB-fold, β-tadpole fold, Ig-like domain and a knotted α-helical fold 26. Adenovirus uses decoration proteins that adopt the 4-helix bundle, triskelion structures adopted by the decoration proteins of adenovirus 33. When compared to known decoration protein structures, the folds adopted by gp47 and gp48 along with their short length are quite unique. gp47 adopts a four α-helix bundle structure where in two of the four α- helices lie on top of each other and the helices on the bottom interact with the capsid. Whereas, gp48 is present as a dimer on the capsid and each monomer consists of two α-helices that are stacked. Oligomerization of gp48 may occur after binding to the capsid like other decoration proteins such as λ’s gpD, or phageL Dec protein or, alternatively, gp48 might exist as a dimer before binding to the capsid. The two helices on the bottom of the gp48 dimer interact with the capsid in a location that is similar to the gp47 interaction site. Decoration proteins like Hoc (phage T4) 34,35, pb10 (phage T5) 36 can assist in host attachment and whether gp47 and gp48 play a role in attachment is not clear. Since these proteins are essential for phage viability, we cannot directly test their roles in host attachment.
The HRP29 procapsid has one scaffolding per capsomere, and no decoration proteins
Most phages assemble from a precursor procapsid, which is a spherical capsid structure with the major capsid protein organized with icosahedral symmetry. Maturation involves packaging of the genome through the portal using a complex of two terminase proteins. Packaging induces capsid expansion into a larger, more angular capsid shell, and phage assembly then concludes with the addition of the tail proteins. In order to understand whether the decoration proteins are incorporated early during assembly into the procapsids, or onto mature heads, we examined the HRP29 procapsid. Procapsids were isolated from cells expressing a dead cas9 nuclease and a guide RNA targeting the large terminase to stop DNA packaging and arrest the assembly process. Procapsids were then further purified using a cesium chloride gradient and particles were imaged by single-particle cryo-EM. An icosahedral reconstruction of the procapsid showed that the overall domain architecture is similar to dsDNA phages that have the HK97-like fold (Fig. 6A) 37. The first 27 amino acids of the major capsid protein were not modeled as the density was missing in the cryo-EM map; the rest of the protein was modeled and refined. Similar to the mature virion, a DALI search of gp37 showed that HRP29 gp37 in the procapsid is most similar to gp10A of T7 (PDB: 3J7V) with a RMSD of 3.0 Å and a Z-score of 20.8. The P-loops and the E-loops from three capsomeres come together at the three-fold and quasi three-fold axes to form a two layered interaction to stabilize the procapsid similar to T7 27. Density for the putative decoration proteins gp47 and gp48 were not visualized in the procapsid structure and mass spectrometry confirmed these proteins were not present in the purified sample (data not shown). Therefore, we conclude that they are added after competition of the procapsid, but cannot determine if the addition is concurrent with packaging, or after maturation.
Figure 6: HRP29 procapsid has scaffolding protein density just beneath the N-terminus.
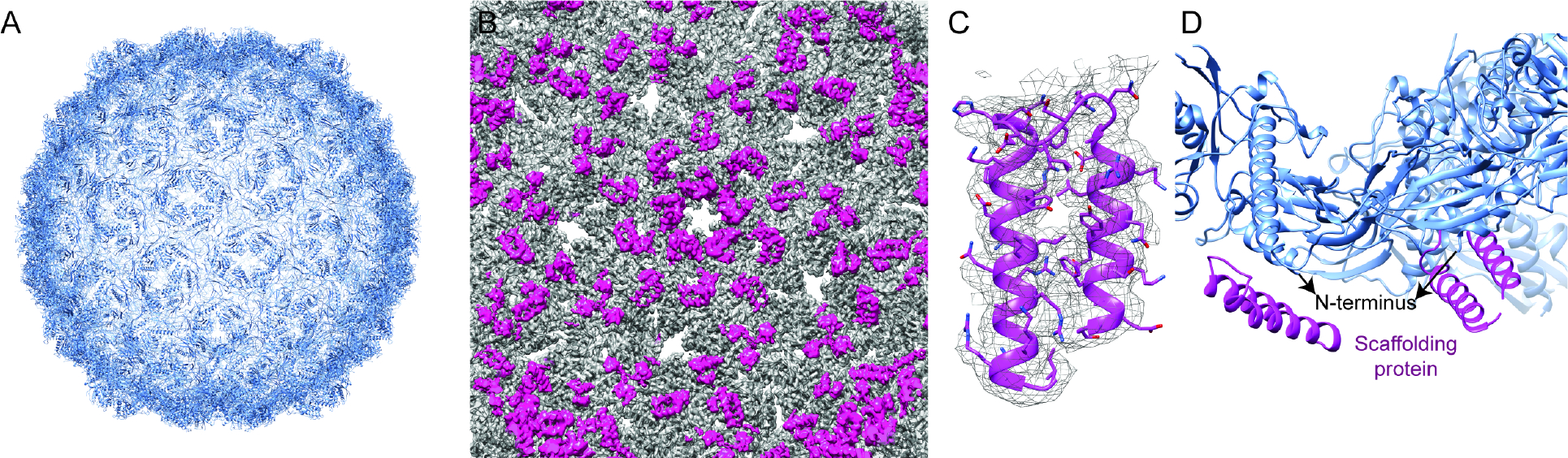
(A) Icosahedral model of the procapsid show a characteristic bumpy surface along with holes in the center of the pentons and hexons. (B) Interior view of the procapsid with the capsid density colored in grey and the scaffolding density from the difference map colored in magenta (resolution = 3.5 Å at FSC0.143). The difference map was calculated by subtracting a map generated from the icosahedral model using the molmap command in chimera from the original cryo-EM density map. (C) Fit of scaffolding helix turn helix region shown in mesh representation and good fit was observed for the bulky amino acids. (D) The helix turn helix region of the C-terminal part of the scaffolding protein sits right under the modeled N-terminus which lacks the first 27 amino acids and the spine helix. The capsid protein is in blue and the scaffolding protein is colored in magenta.
The procapsid reconstruction also revealed density on the inside of the structure that could not be accounted for by the major capsid protein. Scaffolding proteins help in the nucleation of capsid assembly by binding to capsid proteins and starting the assembly process around the portal. The stoichiometry of scaffolding proteins to capsid proteins differ among various phages but their function is the same. The scaffolding proteins either leave the procapsid intact via intracapsomere holes during maturation, or they are proteolytically digested. The location of scaffolding proteins inside the procapsids have been visualized previously in other phages like T7 and P22, but only partial structures of the protein were visualized due to the inherent flexibility of scaffolding proteins 27,38. A helix-turn-helix (HTH) density was present in the interior of the HRP29 procapsid under the N-terminal helix and the spine helix (Fig. 6B, 6D). In phages T7, Phi29 and P22 similar densities were also observed in the interior of their procapsids, which were identified as the C-terminal part of the scaffolding protein. Alphafold2 prediction of HRP29 scaffolding protein showed that it also has a similar HTH motif in the C-terminus that fit well into the density in the interior of the procapsid (Fig. 6C). HRP29 has strong HTH density at each capsomere of the pentons and the capsomeres of the hexons surrounding the pentons. Apart from these locations the HTH density is also visible beneath capsomers around the icosahedral threefold axis. There were weaker densities for scaffolding protein at other hexamers too indicating that there may be a scaffolding protein for every major capsid protein, but these sites may bind with lower affinity (Fig S5A, S5B). Two different populations of binding sites for scaffolding proteins have been identified previously, such as in phage P22 39. The HRP29 scaffolding protein interacts with the N-helix of the capsomere from one subunit and the loop following the N-helix of the neighboring capsid subunit. In T7, scaffolding proteins are not found beneath the pentons and the data suggests a proposed ratio of two scaffolding proteins per hexon, whereas in P22 each capsomere is associated with an individual scaffolding protein. In the case of T7 the presence of the inner stack on top of the portal was suggested to be a factor in influencing the 1:3 ratio of scaffold proteins to capsomeres. The exact ratio of scaffold protein to a capsomere in the case of HRP29 is not clear but it appears to differ from T7 due to the presence of the scaffolding protein density at the pentons.
The structure of the portal when assembled in the procapsid state has been determined previously for P22 14. Recently a high-resolution structure of the portal from the procapsid of thermophilic phage P23–45 has also been determined using the local reconstruction approach developed by Ilca et. Al 23,40. All the five-fold vertices from the procapsid map were computationally isolated using the local-rec plugin in Scipion 41. Sub-particles centered around the five-fold vertex were extracted utilizing the parameters from the icosahedral reconstruction and subjected to alignment free 3D-classification. Despite our best efforts we were not able to computationally isolate the portal vertex and resolve a structure of the portal from the HRP29 procapsid.
In summary, the procapsid of HRP29 has a scaffolding protein present for every capsid protein like P22, but the overall fold of the major capsid protein in both the procapsid state and mature virion is most similar to T7. The mature capsid of HRP29 is stabilized by two decoration proteins, gp47 and gp48, to likely compensate for the stabilizing forces that are lacking in comparison to T7. The decoration proteins are small and have a helical architecture when compared to others, and are absent in the procapsid, indicating that they are likely assembled directly onto the mature capsid. HRP29 possesses a tail that’s a hybrid of the T7-like tail tube and an Sf6/P22-like tailspike. This hybrid interaction is possible with the help of the tailspike adaptor protein that has evolved to have a N-terminal domain similar to T7 gp17 that can dock with the tail tube and a C-terminal domain that can interact with the tailspike. The infection process of T7 and P22 have been studied by cryo-electron tomography and the various steps involved in host-recognition and DNA ejection have been elucidated 20,21. Future studies using mini cells of Shigella flexneri serotype Y strain and HRP29 will be necessary to elucidate the exact mechanism of infection of HRP29 and to test the hypothesis of whether the mechanism of infection is a hybrid mechanism combining T7 and P22.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Kristin Parent (kparent@msu.edu).
Materials Availability
All plasmids used in this study will be made available upon request. The study did not generate other new or unique reagents.
Data and code availability
The cryo-EM density maps have been deposited in the Electron Microscopy Data Bank with accession codes EMD-28226, EMD-28227, EMD-28238, and EMD-28562.
Atomic coordinates for capsid, C6 reconstruction of tail and procapsid have been deposited in the PDB with accession codes 8ELD, 8ES4 and 8EM6.
The paper does not report code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Study Participant Details
The following strains were used in this study:
Method details
Bacterial strains and plasmids
The strain of bacteria used as host for HRP29, “PE577” is an avirulent derivative of S. flexneri and has been described previously 12,42,43. A yajC knockout of PE577 was generated by lambda red recombineering and kanamycin cassette recombination using overlapping arms from the yajC Keio knockout strain. The kanamycin cassette along with the overlapping arms were amplified using primers SPK049 & SPK050 and used for transformation into strain PE577. All the other knockouts used in this study have been described previously 10. Plasmids pdCas9 (CamR) 44 and pGRB 45 (AmpR) were obtained from Addgene. Plasmid pRSFDuet (KanR) was obtained from Novagen.
Efficiency of plating assay
Serial dilutions of high titer HRP29 phage stock (~1×1011 phage/mL) were plated on various strains using the double agar overlay method. Efficiency of plating was calculated as the titer on the test bacteria divided by the titer on the permissive strain, S. flexneri PE577, grown overnight at 37 °C. Three biological replicates of each host strain were used for each experiment.
HRP29 virion purification for cryoEM
PE577 cells were grown to a OD 600nm of 0.2 then infected with HRP29 at a multiplicity of infection (MOI) of 0.1 phage/cell at 37 °C. After 6 hours of growth remaining cells were lysed with chloroform and pelleted by centrifugation at 4000×g for 10 minutes at 4°C. The supernatant was then spun at 27,000×g for 90 min at 4°C and the phage pellet was resuspended in phage buffer (10 mM Tris, pH=7.5, 10 mM MgCl2). The sample was further purified using a cesium chloride gradient as described 43, the band containing the virions was extracted with a syringe, and the sample was buffer exchanged with phage buffer overnight at 4 °C. Finally, the phage was concentrated using a 100-kDa MWCO Amicon spin concentrator to ~1013 phage/mL.
HRP29 Procapsid purification
A guide RNA targeting the large terminase subunit (Supplementary Table 2) was designed and cloned into the pGRB vector digested with SpeI. The cloned vector was then introduced into PE577 cells also containing a pdCas9 vector. Cells containing both the pdCas9 vector and the pGRB vector expressing the guide RNA targeting the large terminase subunit were grown to a OD 600nm of 0.2 then infected with HRP29 at a MOI of 0.1 phage/cell. After 6 hours of growth the cells were lysed with chloroform and pelleted by centrifugation at 4000×g for 10 minutes. The supernatant was then spun at 12,000×g for 18 hours and the phage pellet was resuspended in phage buffer by nutation. The sample was further purified using a cesium chloride gradient (see above). Finally, procapsids were concentrated using a 100-kDa MWCO Amicon spin concentrator prior to cryo-EM imaging. In addition, purified procapsids were run on a agarose gel stained with ethidium bromide, and then with Coomassie blue to confirm the absence of DNA (data not shown). Procapsids were also TCA precipitated and run on and SDS-PAGE gel for mass spectrometry analysis to identify the proteins present (data not shown).
CRISPR knockdown of gp47 and gp48
A guide RNA targeting gp47 (or) gp48 (Supplementary Table 2) was designed and cloned into the pGRB vector digested with SpeI. The cloned vector was then introduced into PE577 cells also containing the pdCas9 vector. Cells containing both the pdCas9 vector and the pGRB vector were then used spot titer assays. Briefly, 5 uL of serial dilutions of a high titer phage stock were spotted on plates with cells prepared using the double agar overlay method.
Thermal stability tests
Small aliquots (50 μL) of phage stocks of T746, Sf6, and HRP29 at 1 ×1010 phage/mL were incubated at 5 degree intervals ranging 45 – 65 °C for 10 minutes each using a BioRad T100 thermal cycler. After incubation at the desired temperature, quantitative plaque assays were performed and the efficiency of plating was determined as the titer of phage at the experimental temperature, divided by the titer of phage at the permissive temperature of 45 °C. The experiment was performed using three biological replicates, and the error bars reflect the standard deviation.
CryoEM Sample preparation and data acquisition
5 μL aliquots of purified HRP29 virions or procapsids were applied to R2/2 Quantifoil grids (Electron Microscopy Solutions) that had been glow discharged for 45 seconds in a Pelco Easiglow glow discharging unit. The samples were plunge frozen in liquid ethane using a Vitrobot Mark IV operated at 4°C and 100% humidity, with a blot force of 1 and 5 seconds of blotting time per grid.
Cryo-EM data were collected at Purdue Cryo-EM facility using a Titan Krios equipped with a K3 direct electron detector, and operating at 300 keV with a post-column GIF (20 eV slit width) under low dose conditions. For HRP29 virions, micrographs (Fig S6A) were collected at 53,000X nominal magnification (0.816 Å/pixel) by recording 40 frames over 4.4 sec for a total dose of 33 e−/Å2. For HRP29 procapsids, micrographs (Fig S6B) were collected at 64,000X nominal magnification (0.66 Å/pixel) by recording 43 frames over 3.4 sec for a total dose of 38 e−/Å2. See Supplementary Table 3 for full parameters.
Icosahedral and asymmetric image reconstructions of the HRP29 virion
Data processing was carried out using Relion 3.1.1 47. Briefly, the dose-fractionated movies were subjected to motion correction using Relion’s own implementation of MotionCor2. CTF estimation of the resulting images were estimated using CTFFIND-4.1 48 and particles were picked using the Autopick option. 4X binned particles were then extracted and subjected to 2D classification. Full cryoEM image processing statistics are listed in Supplementary Table 4. For the icosahedral reconstruction of the virion capsid, 38,666 particles were used for 3D refinement, with an ab-initio model serving as the initial model. For the asymmetric reconstruction of the virion, a total of 33,127 particles were used for 3D refinement, with the Sf6 virion map (EMD:5730, scaled by a factor of 1.27 in EMAN2 49, symmetrized using Relion 3.0.8) serving as an initial model. Refined particles were extracted again with 2X binning and subjected to 3D refinement and CTF refinement. The overall resolution was estimated based on the gold-standard Fourier shell correlation (FSC0.143) (Fig S6C, S6D). The final maps were deposited into EMDB (accession number EMD - 28226 & EMD - 28227).
Localized reconstruction of the HRP29 tail
Sub-particle extraction was carried out on 33,127 particles from HRP29 for an asymmetric reconstruction using the localrec plugin in Scipion3 40,41,50. 33,119 extracted particles were then imported in Relion 3.1.1 and subjected to further processing. 3D refinement was carried out imposing C6 symmetry with an initial model generated from the sub-particles. Particle subtraction was carried out to remove capsid density using a mask encompassing the tail generated with the segger tool in chimera51. Alignment free 3D classification was then carried out which resulted in 24,391 selected particles and subjected to further 3D refinement and CTF refinement. The overall resolution was estimated based on the gold-standard Fourier shell correlation (FSC0.143) (Fig S6E). The final map was deposited into the EMDB (accession number EMD - 28562).
Icosahedral reconstruction of the HRP29 procapsid
Data processing was carried out using Relion 3.1.1 47. Briefly, the dose-fractionated movies were subjected to motion correction using Relion’s own implementation of MotionCor2. CTF estimation of the resulting images was estimated using CTFFIND-4.1 48 and particles were picked using the Autopick option. 4X binned particles were then extracted and subjected to 2D classification. Full cryoEM image processing statistics are listed in the Supplementary Table 4. For an icosahedral reconstruction, 78,902 particles were used for 3D refinement, with an ab-initio model serving as the initial model. Refined particles were extracted with 2X binning and subjected to 3D refinement. Alignment free 3D classification was then carried which resulted in 60,962 particles and subjected to further 3D refinement and CTF refinement. The overall resolution was estimated based on the gold-standard Fourier shell correlation (FSC0.143). The final maps were deposited into EMDB (accession number EMD- 28238 ) (Fig S6F8D).
Model building and refinement
AlphaFold2 52,53 and Phyre2 54 were used to generate homology models for all modeled protein chains. Initial models were then docked into EM maps for further refinement. For the HRP29 virion capsid and procapsid maps, the asymmetric unit consisting of seven subunits of the major capsid protein was refined against a segmented map generated using the segger extension in chimera. Additionally, there are two gp47 and two gp48 molecules in the asymmetric unit and they were also refined against the segmented map. Initial models of tail subunit proteins were docked into the localized reconstruction map (C6 symmetry) using chimera and used for further refinement. Refinement was carried out using Phenix 55 and model adjustments were carried out in COOT 56. Model parameters were monitored using Molprobity 57 in Phenix and the values are listed in Supplementary Table 4 along with the respective PDB IDs.
Purification of gp44 bound to gp52
The gene encoding gp44 was amplified from HRP29 genomic DNA using primers (SPK146 & SPK147; Supplementary Table 2) and cloned into pRSFDuet vector (Novagen) digested with HindII and SacI using Gibson assembly which added a 6X-His tag to gp44. The gene encoding gp52 was amplified from HRP29 genomic DNA using primers (SPK148 & SPK149; Supplementary Table 2) and cloned into the same vector digested with NdeI and XhoI using Gibson assembly. Upon sequencing we discovered a mutation in gp52, A754G which resulted in amino acid change T252A. However, the change was also present in the HRP29 genomic DNA and we decided to proceed forward as the tailspike is known to have variation in the region upon repeated propagation of phage. The plasmid was transformed into BL21DE3 cells and a starter culture was grown in LB media at 37 °C with 100 μg/mL of kanamycin. The starter culture was used to inoculate 1L of LB media and grown at 37 °C with 100 μg/mL of kanamycin until OD 600nm of 0.5 and protein expression was induced with 1mM IPTG. After 3h of induction the cells were harvested by centrifugation at 4000×g for 10 minutes and resuspended in Buffer A (10 mM Tris pH 7.6, 100 mM NaCl, and 10 mM MgCl2) at 4 °C. The cells were again pelleted by centrifugation at 4000×g for 10 minutes and were flash frozen in liquid nitrogen.
Frozen cell pellets were thawed and resuspended in Buffer A supplemented with Roche Protease inhibitor cocktail tablet, 1 mg/mL lysozyme, 10 mM Cacl2 and 20 ug/mL of DNaseI. Cells were lysed by sonication and cell debris was removed by centrifugation at 20,000×g for 30 min at 4°C. The resulting supernatant was applied to Ni-NTA agarose resin and washed with lysis buffer followed by a wash with buffer A containing 25mM Immidazole. The Ni-NTA resin was subsequently washed with Buffer A containing 50mM Immidazole, 100mM Immidazole and 250mM Immidazole to elute the protein. The presence of both gp44 and gp52 in the various washes was confirmed by SDS-PAGE and washes containing pure gp44-gp52 complex were pooled and concentrated using a 100-kDa MWCO spin concentrator. The concentrated sample was then injected into a Superose 6 column equilibrated with Buffer A. Peak fractions corresponding to pure gp44-gp52 complex were pooled together and used for subsequent analysis by negative stain.
Negative staining of gp44 bound to gp52
A small portion (~3 μL) of the gp44-gp52 complex at a concentration of 0.01 mg/mL was applied to freshly glow discharged (PELCO easiGlow, 15 mA, 45 sec) continuous carbon support film grids (Ted Pella) for 60 seconds, followed by washing with distilled water and then staining with 1% aqueous Uranyl Acetate (Electron Microscopy Solutions). The samples were imaged at the RTSF Cryo-EM Core Facility at Michigan State University using a Talos Arctica operated at 200 keV. Micrographs were collected on a Ceta camera at a nominal magnification of 57,000x (1.80 Å/pixel) with an exposure time of 4 seconds and objective lens setting of 5 μm underfocus. 2D class averages of the gp44-gp52 complex were calculated from 8,583 particles using Cryosparc v3.31 58.
Quantification and Statistical Analysis
Statistical details of experiments:
FSC measurements were used to estimate resolution of cryo-EM maps as a bult-in feature of the software Relion 3.1.1 47. Details of this analysis can be found throughout the Results, Figure Legends, and Supplementary Materials. For this we applied the “gold standard approach”, where FSC is calculated from independently refined half reconstructions.
Phage Thermal Stability experiments were performed using three biological replicates (n = 3), and error bars reflect the standard deviation.
Supplementary Material
Highlights.
HRP29 is podophage with a hybrid tail
The hybrid tail is composed of a tail tube like T7 and tailspikes similar to Sf6.
Tailspike adapter (gp44) connects the tailspikes (gp52) to the tail tube.
HRP29 capsid is stabilized by two decoration proteins gp47 and gp48.
Acknowledgements
We would like to thank Dr. Ian Molineux at the University of Texas, Austin for helpful discussion. We would like to thank Dr. Pavel Plevka and Dominik Hrebik at CEITEC, Brno, Czech Republic for helpful suggestions on data processing. We would like to thank the MSU RTSF Cryo-EM Core Facility for use of the Talos Arctica. Additionally, we thank Dr. T. Klose at Purdue University’s Midwest Cryo-EM Consortium (NIH Consortium #U24GM116789-03). This work was supported by the National Institutes of Health GM110185, GM140803 and National Science Foundation CAREER Award 1750125 to K.N.P.
Inclusion and Diversity
One or more of the authors in this paper self-identifies as a gender minority in science. One or more of the authors in this paper identifies as a member of the LGBTQIA+ community. One or more of the authors in this paper received support from a program designed to increase minority representation in science.
Footnotes
Declaration of interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Qiu S, Liu K, Yang C, Xiang Y, Min K, Zhu K, Liu H, Du X, Yang M, Wang L, et al. (2022). A Shigella sonnei clone with extensive drug resistance associated with waterborne outbreaks in China. Nat Commun 13, 7365. 10.1038/s41467-022-35136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fruciano DE, and Bourne S (2007). Phage as an antimicrobial agent: d’Herelle’s heretical theories and their role in the decline of phage prophylaxis in the West. Can J Infect Dis Med Microbiol 18, 19–26. 10.1155/2007/976850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doore SM, Schrad JR, Dean WF, Dover JA, and Parent KN (2018). Shigella Phages Isolated during a Dysentery Outbreak Reveal Uncommon Structures and Broad Species Diversity. J Virol 92. 10.1128/JVI.02117-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Subramanian S, Parent KN, and Doore SM (2020). Ecology, Structure, and Evolution of Shigella Phages. Annu Rev Virol 7, 121–141. 10.1146/annurev-virology-010320-052547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ackermann HW (1998). Tailed bacteriophages: the order caudovirales. Adv Virus Res 51, 135–201. 10.1016/s0065-3527(08)60785-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doore SM, Schrad JR, Perrett HR, Schrad KP, Dean WF, and Parent KN (2019). A cornucopia of Shigella phages from the Cornhusker State. Virology 538, 45–52. 10.1016/j.virol.2019.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen W, Xiao H, Wang L, Wang X, Tan Z, Han Z, Li X, Yang F, Liu Z, Song J, et al. (2021). Structural changes in bacteriophage T7 upon receptor-induced genome ejection. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2102003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li F, Hou CD, Yang R, Whitehead R, Teschke CM, and Cingolani G (2022). High-resolution cryo-EM structure of the Shigella virus Sf6 genome delivery tail machine. Sci Adv 8, eadc9641. 10.1126/sciadv.adc9641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lander GC, Tang L, Casjens SR, Gilcrease EB, Prevelige P, Poliakov A, Potter CS, Carragher B, and Johnson JE (2006). The structure of an infectious P22 virion shows the signal for headful DNA packaging. Science 312, 1791–1795. 10.1126/science.1127981. [DOI] [PubMed] [Google Scholar]
- 10.Subramanian S, Dover JA, Parent KN, and Doore SM (2022). Host Range Expansion of Shigella Phage Sf6 Evolves through Point Mutations in the Tailspike. J Virol 96, e0092922. 10.1128/jvi.00929-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parent KN, Erb ML, Cardone G, Nguyen K, Gilcrease EB, Porcek NB, Pogliano J, Baker TS, and Casjens SR (2014). OmpA and OmpC are critical host factors for bacteriophage Sf6 entry in Shigella. Mol Microbiol 92, 47–60. 10.1111/mmi.12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muthuirulandi Sethuvel DP, Devanga Ragupathi NK, Anandan S, and Veeraraghavan B (2017). Update on: Shigella new serogroups/serotypes and their antimicrobial resistance. Lett Appl Microbiol 64, 8–18. 10.1111/lam.12690. [DOI] [PubMed] [Google Scholar]
- 13.Cuervo A, Fàbrega-Ferrer M, Machón C, Conesa JJ, Fernández FJ, Pérez-Luque R, Pérez-Ruiz M, Pous J, Vega MC, Carrascosa JL, and Coll M (2019). Structures of T7 bacteriophage portal and tail suggest a viral DNA retention and ejection mechanism. Nat Commun 10, 3746. 10.1038/s41467-019-11705-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lokareddy RK, Sankhala RS, Roy A, Afonine PV, Motwani T, Teschke CM, Parent KN, and Cingolani G (2017). Portal protein functions akin to a DNA-sensor that couples genome-packaging to icosahedral capsid maturation. Nat Commun 8, 14310. 10.1038/ncomms14310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuervo A, Pulido-Cid M, Chagoyen M, Arranz R, González-García VA, Garcia-Doval C, Castón JR, Valpuesta JM, van Raaij MJ, Martín-Benito J, and Carrascosa JL (2013). Structural characterization of the bacteriophage T7 tail machinery. J Biol Chem 288, 26290–26299. 10.1074/jbc.M113.491209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leiman PG, Battisti AJ, Bowman VD, Stummeyer K, Mühlenhoff M, Gerardy-Schahn R, Scholl D, and Molineux IJ (2007). The structures of bacteriophages K1E and K1–5 explain processive degradation of polysaccharide capsules and evolution of new host specificities. J Mol Biol 371, 836–849. 10.1016/j.jmb.2007.05.083. [DOI] [PubMed] [Google Scholar]
- 17.Tu J, Park T, Morado DR, Hughes KT, Molineux IJ, and Liu J (2017). Dual host specificity of phage SP6 is facilitated by tailspike rotation. Virology 507, 206–215. 10.1016/j.virol.2017.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Müller JJ, Barbirz S, Heinle K, Freiberg A, Seckler R, and Heinemann U (2008). An intersubunit active site between supercoiled parallel beta helices in the trimeric tailspike endorhamnosidase of Shigella flexneri Phage Sf6. Structure 16, 766–775. 10.1016/j.str.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 19.Casjens SR, and Thuman-Commike PA (2011). Evolution of mosaically related tailed bacteriophage genomes seen through the lens of phage P22 virion assembly. Virology 411, 393–415. 10.1016/j.virol.2010.12.046. [DOI] [PubMed] [Google Scholar]
- 20.Wang C, Tu J, Liu J, and Molineux IJ (2019). Structural dynamics of bacteriophage P22 infection initiation revealed by cryo-electron tomography. Nat Microbiol 4, 1049–1056. 10.1038/s41564-019-0403-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu B, Margolin W, Molineux IJ, and Liu J (2013). The bacteriophage t7 virion undergoes extensive structural remodeling during infection. Science 339, 576–579. 10.1126/science.1231887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang Q, Tang WC, Tao P, Mahalingam M, Fokine A, Rossmann MG, and Rao VB (2020). Structural morphing in a symmetry-mismatched viral vertex. Nat Commun 11, 1713. 10.1038/s41467-020-15575-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bayfield OW, Steven AC, and Antson AA (2020). Cryo-EM structure in situ reveals a molecular switch that safeguards virus against genome loss. Elife 9. 10.7554/eLife.55517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Helgstrand C, Wikoff WR, Duda RL, Hendrix RW, Johnson JE, and Liljas L (2003). The refined structure of a protein catenane: the HK97 bacteriophage capsid at 3.44 A resolution. J Mol Biol 334, 885–899. 10.1016/j.jmb.2003.09.035. [DOI] [PubMed] [Google Scholar]
- 25.Popa MP, McKelvey TA, Hempel J, and Hendrix RW (1991). Bacteriophage HK97 structure: wholesale covalent cross-linking between the major head shell subunits. J Virol 65, 3227–3237. 10.1128/JVI.65.6.3227-3237.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dedeo CL, Teschke CM, and Alexandrescu AT (2020). Keeping It Together: Structures, Functions, and Applications of Viral Decoration Proteins. Viruses 12. 10.3390/v12101163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo F, Liu Z, Fang PA, Zhang Q, Wright ET, Wu W, Zhang C, Vago F, Ren Y, Jakana J, et al. (2014). Capsid expansion mechanism of bacteriophage T7 revealed by multistate atomic models derived from cryo-EM reconstructions. Proc Natl Acad Sci U S A 111, E4606–4614. 10.1073/pnas.1407020111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rydman PS, Bamford JK, and Bamford DH (2001). A minor capsid protein P30 is essential for bacteriophage PRD1 capsid assembly. J Mol Biol 313, 785–795. 10.1006/jmbi.2001.5068. [DOI] [PubMed] [Google Scholar]
- 29.Yang Q, Maluf NK, and Catalano CE (2008). Packaging of a unit-length viral genome: the role of nucleotides and the gpD decoration protein in stable nucleocapsid assembly in bacteriophage lambda. J Mol Biol 383, 1037–1048. 10.1016/j.jmb.2008.08.063. [DOI] [PubMed] [Google Scholar]
- 30.Qin L, Fokine A, O’Donnell E, Rao VB, and Rossmann MG (2010). Structure of the small outer capsid protein, Soc: a clamp for stabilizing capsids of T4-like phages. J Mol Biol 395, 728–741. 10.1016/j.jmb.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newcomer RL, Schrad JR, Gilcrease EB, Casjens SR, Feig M, Teschke CM, Alexandrescu AT, and Parent KN (2019). The phage L capsid decoration protein has a novel OB-fold and an unusual capsid binding strategy. Elife 8. 10.7554/eLife.45345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banerjee R, Nath S, Ranjan A, Khamrui S, Pani B, Sen R, and Sen U (2012). The first structure of polarity suppression protein, Psu from enterobacteria phage P4, reveals a novel fold and a knotted dimer. J Biol Chem 287, 44667–44675. 10.1074/jbc.M112.423202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu H, Jin L, Koh SB, Atanasov I, Schein S, Wu L, and Zhou ZH (2010). Atomic structure of human adenovirus by cryo-EM reveals interactions among protein networks. Science 329, 1038–1043. 10.1126/science.1187433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fokine A, Islam MZ, Zhang Z, Bowman VD, Rao VB, and Rossmann MG (2011). Structure of the three N-terminal immunoglobulin domains of the highly immunogenic outer capsid protein from a T4-like bacteriophage. J Virol 85, 8141–8148. 10.1128/JVI.00847-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sathaliyawala T, Islam MZ, Li Q, Fokine A, Rossmann MG, and Rao VB (2010). Functional analysis of the highly antigenic outer capsid protein, Hoc, a virus decoration protein from T4-like bacteriophages. Mol Microbiol 77, 444–455. 10.1111/j.1365-2958.2010.07219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vernhes E, Renouard M, Gilquin B, Cuniasse P, Durand D, England P, Hoos S, Huet A, Conway JF, Glukhov A, et al. (2017). High affinity anchoring of the decoration protein pb10 onto the bacteriophage T5 capsid. Sci Rep 7, 41662. 10.1038/srep41662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duda RL, and Teschke CM (2019). The amazing HK97 fold: versatile results of modest differences. Curr Opin Virol 36, 9–16. 10.1016/j.coviro.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen DH, Baker ML, Hryc CF, DiMaio F, Jakana J, Wu W, Dougherty M, Haase-Pettingell C, Schmid MF, Jiang W, et al. (2011). Structural basis for scaffolding-mediated assembly and maturation of a dsDNA virus. Proc Natl Acad Sci U S A 108, 1355–1360. 10.1073/pnas.1015739108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parker MH, Brouillette CG, and Prevelige PE (2001). Kinetic and calorimetric evidence for two distinct scaffolding protein binding populations within the bacteriophage P22 procapsid. Biochemistry 40, 8962–8970. 10.1021/bi0026167. [DOI] [PubMed] [Google Scholar]
- 40.Ilca SL, Kotecha A, Sun X, Poranen MM, Stuart DI, and Huiskonen JT (2015). Localized reconstruction of subunits from electron cryomicroscopy images of macromolecular complexes. Nat Commun 6, 8843. 10.1038/ncomms9843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abrishami V, Ilca SL, Gomez-Blanco J, Rissanen I, de la Rosa-Trevín JM, Reddy VS, Carazo JM, and Huiskonen JT (2021). Localized reconstruction in Scipion expedites the analysis of symmetry mismatches in cryo-EM data. Prog Biophys Mol Biol 160, 43–52. 10.1016/j.pbiomolbio.2020.05.004. [DOI] [PubMed] [Google Scholar]
- 42.Doore SM, Subramanian S, Tefft NM, Morona R, TerAvest MA, and Parent KN (2021). Large metabolic rewiring from small genomic changes between strains of. J Bacteriol 203. 10.1128/JB.00056-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Casjens S, Winn-Stapley DA, Gilcrease EB, Morona R, Kühlewein C, Chua JE, Manning PA, Inwood W, and Clark AJ (2004). The chromosome of Shigella flexneri bacteriophage Sf6: complete nucleotide sequence, genetic mosaicism, and DNA packaging. J Mol Biol 339, 379–394. 10.1016/j.jmb.2004.03.068. [DOI] [PubMed] [Google Scholar]
- 44.Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, and Marraffini LA (2013). Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res 41, 7429–7437. 10.1093/nar/gkt520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Y, Lin Z, Huang C, Zhang Y, Wang Z, Tang YJ, Chen T, and Zhao X (2015). Metabolic engineering of Escherichia coli using CRISPR-Cas9 meditated genome editing. Metab Eng 31, 13–21. 10.1016/j.ymben.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 46.Dunn JJ, and Studier FW (1983). Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J Mol Biol 166, 477–535. 10.1016/s0022-2836(83)80282-4. [DOI] [PubMed] [Google Scholar]
- 47.Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7. 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–221. 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, and Ludtke SJ (2007). EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157, 38–46. 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 50.de la Rosa-Trevín JM, Quintana A, Del Cano L, Zaldívar A, Foche I, Gutiérrez J, Gómez-Blanco J, Burguet-Castell J, Cuenca-Alba J, Abrishami V, et al. (2016). Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. J Struct Biol 195, 93–99. 10.1016/j.jsb.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 51.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 52.Holm L (2022). Dali server: structural unification of protein families. Nucleic Acids Res 50, W210–215. 10.1093/nar/gkac387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kelley LA, Mezulis S, Yates CM, Wass MN, and Sternberg MJ (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10, 845–858. 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, and Adams PD (2018). Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D Struct Biol 74, 531–544. 10.1107/S2059798318006551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 57.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, and Richardson DC (2007). MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35, W375–383. 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296. 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM density maps have been deposited in the Electron Microscopy Data Bank with accession codes EMD-28226, EMD-28227, EMD-28238, and EMD-28562.
Atomic coordinates for capsid, C6 reconstruction of tail and procapsid have been deposited in the PDB with accession codes 8ELD, 8ES4 and 8EM6.
The paper does not report code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.