

Abstract
Woodhouse–Sakati syndrome consists of hypogonadism, diabetes mellitus, alopecia, ECG abnormalities, and dystonia. This condition is caused by the loss of function of the DCAF17 gene. Most of the patients have been reported from Greater Middle Eastern countries. We report a 38 male from southern India who presented with syncope and massive hemoptysis due to ruptured bronchopulmonary collaterals. He also had alopecia, cataracts, recently diagnosed diabetes and hypogonadism. Whole exome sequencing showed a novel homozygous truncating variant in the DCAF17 gene. Despite embolization of the aortopulmonary collaterals, the patient died of recurrent hemoptysis.
Keywords: cardiac, Indian, Woodhouse–Sakati syndrome
1 |. INTRODUCTION
Woodhouse–Sakati (WS) syndrome consists of hypogonadism, diabetes, alopecia, dystonia, and incidentally detected ECG abnormalities (Kohil et al., 2023). It is a very rare condition, with less than 200 patients reported so far (Kohil et al., 2023). A few patients present with predominantly neurological features like dystonia and intellectual disabilities. Magnetic resonance imaging can show iron deposition in basal ganglia, and hence, WS syndrome is also classified as one of the neurodegeneration with brain iron accumulation syndromes (Abusrair et al., 2018).
Cardiac manifestations are limited to incidentally detected changes in the ST segment and T wave (Woodhouse & Sakati, 1983). Here, we report massive hemoptysis as a presenting feature of WS syndrome in an adult with a novel variant in the DCAF17 gene.
2 |. CASE REPORT
A 38-year-old unmarried male presented with recurrent syncope for 1 month. He belonged to Tamil ethnicity from southern India. He was diagnosed with diabetes 1 year ago. He was born of non consanguineous marriage, and his siblings did not have similar complaints. On examination, he had a total loss of scalp and body hair (alopecia totalis), which was from birth, a triangular face (Figure 1), bilateral cataracts, and testicular atrophy. He did not have any neurological deficit or deafness. CT brain showed white matter hyperdensities with basal ganglia calcification (Figure 2b). ECG showed delta waves suggestive of Wolff–Parkinson–White syndrome (Figure 2a), and he underwent radiofrequency ablation of the left lateral accessory pathway. A week later, he developed a sudden massive hemoptysis. CT angiogram showed large tortuous aorto pulmonary collaterals (Figure 2c,d). He underwent coil embolization of the arteries but had another bout of massive hemoptysis, which was fatal.
FIGURE 1.
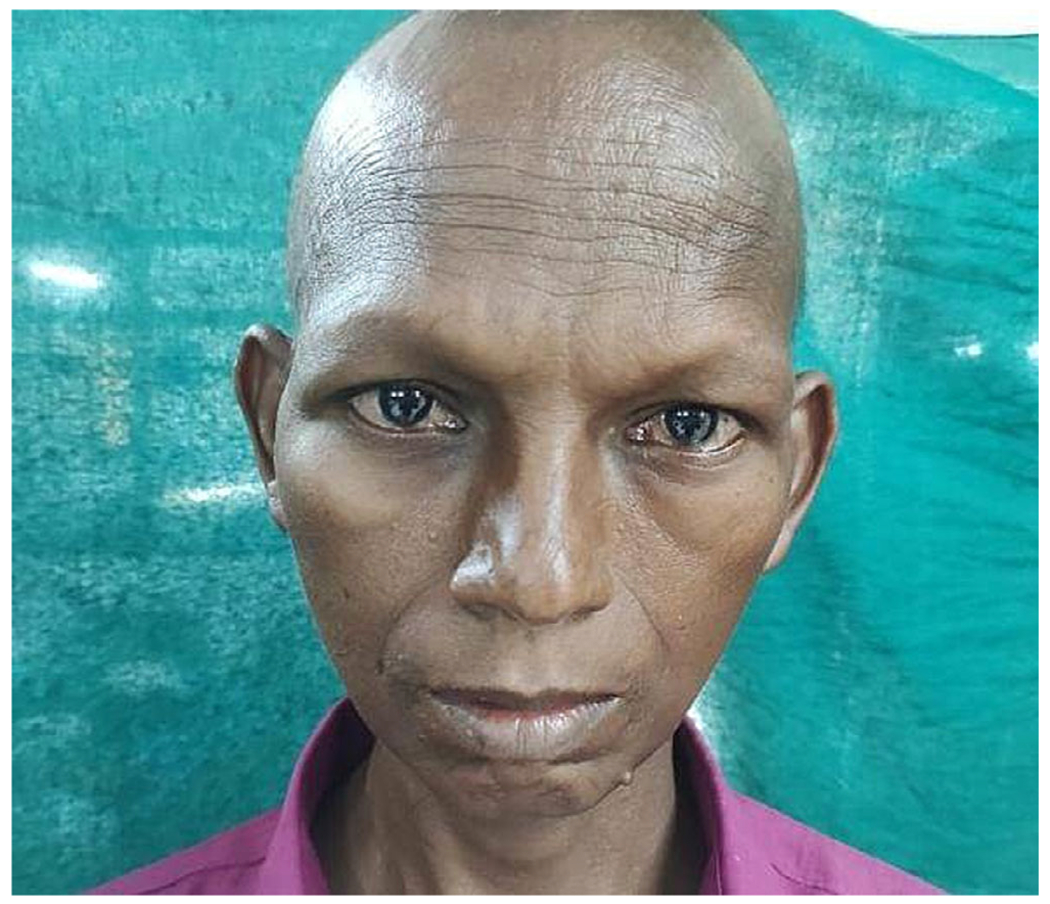
Triangular face with narrow chin, broad nasal bridge, and lack of scalp and facial hair.
FIGURE 2.
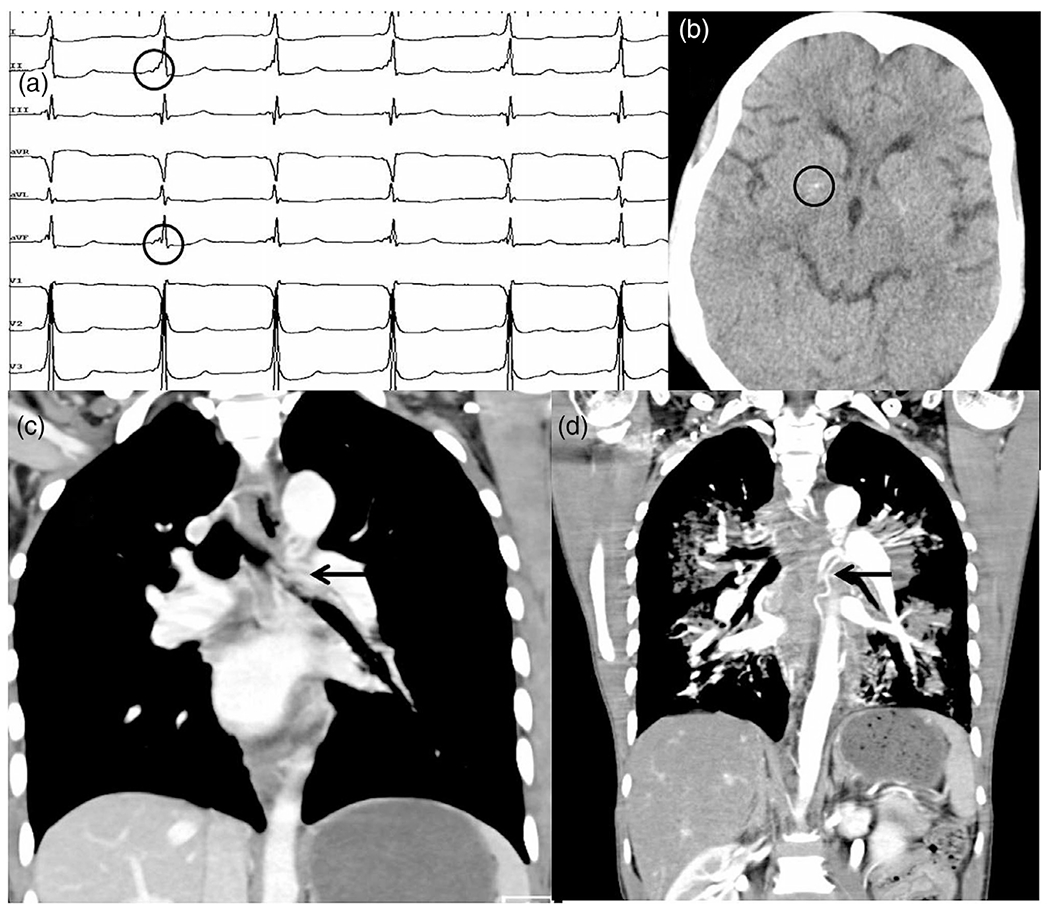
(a) ECG showing delta wave (circle). (b) CT brain showing calcification in basal ganglia (circle). (c, d) CT angiogram showing tortuous arteries from the aorta (arrow).
He was initially thought to have late-onset progeria (Werner syndrome). Whole exome sequencing, using HiSeq 2500 (Illumina) sequencer, showed a novel homozygous variant, c.153G>A (p.Trp51*), in exon 2 of DCAF17 gene leading to premature termination, resulting in a truncated or absent protein. This variant is absent in the Genome Aggregation Database and Indian database—IndiGenomes. Hence, this variant was classified as pathogenic (ACMG evidence codes used: PVS1, PM3_supporting, PM2_supporting). Other family members did not undergo genetic testing.
3 |. DISCUSSION
WS syndrome is a very rare condition with an estimated prevalence of <1/1,000,000 of the population (Kohil et al., 2023). About 187 genetically confirmed patients (98 families) have been reported, with almost all from the Greater Middle Eastern (GME) countries (168 patients, 89 families), while the rest of the patients are from Japan, Slovenia, Italy, Portugal (1 family each) and 2 families from China and India (Ali et al., 2022; Kohil et al., 2023). The first report from India was by Koshy et al. (2008), who had reported a 20-year-old lady who had immune-mediated thrombocytopenia and was incidentally detected to have diabetes, sparse scalp hair, hypogonadism, and mild ECG changes, and these findings were also present in both her siblings. Abdulla et al. (2015) reported a 30-year-old Indian lady with primary amenorrhea and was subsequently noted to have diabetes, sparse scalp hair, and mild intellectual disability. Similar findings were present in both of her siblings.
The causative gene has been identified as a nucleolar protein—DCAF17 (DDB1 and CUL4 associated factor 17, previously called C2orf37) (Alazami et al., 2008). Cullin 4 is a ubiquitin ligase that transfers ubiquitin to target proteins, which then undergo degradation by the proteasome. Also, the ubiquitination of histone proteins can lead to chromatin remodeling. DCAF brings together the substrate protein and Cullin, thereby facilitating the transfer of ubiquitin to the substrate protein (Fouad et al., 2019).
So far, 14 disease-causing variants have been described, with c.436delC (p.Ala147Hisfs*9) being the most common among the GME countries, suggesting a founder mutation (Ali et al., 2022; Kohil et al., 2023). The family reported by Koshy et al. had a homozygous splice site variant (c.1422+5G>T), while the second family had compound heterozygous variants (c.459-7499del, c.1238delA) (Abdulla et al., 2015; Ali et al., 2022). Our patient had c.153G>A (p.Trp51*) in exon 2 of DCAF17, which has not been reported so far.
Alopecia and hypogonadism are seen in all patients, while most have diabetes and intellectual disabilities and rarely dystonia (absent in our patient). Cardiac involvement in WS syndrome is usually incidentally detected ST segment and T-wave changes in the ECG (Woodhouse & Sakati, 1983). Ventricular septal defect and myocardial infarction with complete heart block have also been reported in two patients (Sendur et al., 2019; Steindl et al., 2010). However, pre-excitation syndrome in ECG and large aortopulmonary collaterals leading to fatal hemoptysis has not been reported earlier. Patient consent was taken for publishing his photograph.
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
REFERENCES
- Abdulla MC, Alazami AM, Alungal J, Koya JM, & Musambil M (2015). Novel compound heterozygous frameshift mutations of C2orf37 in a familial Indian case of Woodhouse-Sakati syndrome. Journal of Genetics, 94(3), 489–492. [DOI] [PubMed] [Google Scholar]
- Abusrair AH, Bohlega S, Al-Semari A, Al-Ajlan FS, Al-Ahmadi K, Mohamed B, & AlDakheel A (2018). Brain MR imaging findings in Woodhouse-Sakati syndrome. American Journal of Neuroradiology, 39(12), 2256–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alazami AM, Al-Saif A, Al-Semari A, Bohlega S, Zlitni S, Alzahrani F, Bavi P, Kaya N, Colak D, Khalak H, Baltus A, Peterlin B, Danda S, Bhatia KP, Schneider SA, Sakati N, Walsh CA, Al-Mohanna F, Meyer B, & Alkuraya FS (2008). Mutations in C2orf37, encoding a nucleolar protein, cause hypogonadism, alopecia, diabetes mellitus, mental retardation, and extrapyramidal syndrome. American Journal of Human Genetics, 83(6), 684–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali R, Al-Dewik N, Mohammed S, Elfituri M, Agouba S, Musa S, Mahmoud L, Almulla M, El-Akouri K, Mohd H, Bux R, Almulla H, Othman A, Al-Mesaifri F, Shahbeck N, Al-Muriekhi M, Khalifa A, Al-Sulaiman R, & Ben-Omran T (2022). Expanding on the phenotypic spectrum of Woodhouse-Sakati syndrome due to founder pathogenic variant in DCAF17: Report of 58 additional patients from Qatar and literature review. American Journal of Medical Genetics: Part A, 188(1), 116–129. [DOI] [PubMed] [Google Scholar]
- Fouad S, Wells OS, Hill MA, & D’Angiolella V (2019). Cullin ring ubiquitin ligases (CRLs) in cancer: Responses to ionizing radiation (IR) treatment. Frontiers in Physiology, 10, 1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohil A, Abdallah AM, Hussain K, & Al-Shafai M (2023). Genetic epidemiology of Woodhouse-Sakati syndrome in the greater Middle East region and beyond: A systematic review. Orphanet Journal of Rare Diseases, 18(1), 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshy G, Danda S, Thomas N, Mathews V, & Viswanathan V (2008). Three siblings with Woodhouse-Sakati syndrome in an Indian family. Clinical Dysmorphology, 17(1), 57–60. [DOI] [PubMed] [Google Scholar]
- Sendur SN, Oguz S, Utine GE, Dagdelen S, Oguz KK, Erbas T, & Alikasifoglu M (2019). A case of Woodhouse-Sakati syndrome with pituitary iron deposition, cardiac and intestinal anomalies, with a novel mutation in DCAF17. European Journal of Medical Genetics, 62(8), 103687. [DOI] [PubMed] [Google Scholar]
- Steindl K, Alazami AM, Bhatia KP, Wuerfel JT, Petersen D, Cartolari R, Neri G, Klein C, Mongiardo B, Alkuraya FS, & Schneider SA (2010). A novel C2orf37 mutation causes the first Italian cases of Woodhouse Sakati syndrome. Clinical Genetics, 78(6), 594–597. [DOI] [PubMed] [Google Scholar]
- Woodhouse NJ, & Sakati NA (1983). A syndrome of hypogonadism, alopecia, diabetes mellitus, mental retardation, deafness, and ECG abnormalities. Journal of Medical Genetics, 20(3), 216–219. [DOI] [PMC free article] [PubMed] [Google Scholar]