

SUMMARY
The integrated stress response (ISR) comprises the eIF2α kinases PERK, GCN2, HRI, and PKR, which induce translational and transcriptional signaling in response to diverse insults. Deficiencies in PERK signaling lead to mitochondrial dysfunction and contribute to the pathogenesis of numerous diseases. We define the potential for pharmacologic activation of compensatory eIF2α kinases to rescue ISR signaling and promote mitochondrial adaptation in PERK-deficient cells. We show that the HRI activator BtdCPU and GCN2 activator halofuginone promote ISR signaling and rescue ER stress sensitivity in PERK-deficient cells. However, BtdCPU induces mitochondrial depolarization, leading to mitochondrial fragmentation and activation of the OMA1-DELE1-HRI signaling axis. In contrast, halofuginone promotes mitochondrial elongation and adaptive mitochondrial respiration, mimicking regulation induced by PERK. This shows halofuginone can compensate for deficiencies in PERK signaling and promote adaptive mitochondrial remodeling, highlighting the potential for pharmacologic ISR activation to mitigate mitochondrial dysfunction and motivating the pursuit of highly-selective ISR activators.
Keywords: Unfolded protein response (UPR), integrated stress response (ISR), stress-responsive signaling pathway, pharmacologic activator
eTOC Blurb
Perea, Baron et al. demonstrate that pharmacologic activation of compensatory integrated stress response kinases mitigates cellular and mitochondrial dysfunction associated with deficiencies in Perk signaling. This work highlights the potential for highly-selective integrated stress response kinase activating compounds to reduce pathologies linked to PERK deficiency in human diseases.
Graphical Abstract
INTRODUCTION
Endoplasmic reticulum (ER) stress and mitochondrial dysfunction are inextricably linked in the onset and pathogenesis of etiologically diverse diseases including cancer, diabetes, and many neurodegenerative disorders. This has led to considerable interest in defining the biological mechanisms responsible for regulating mitochondria during ER stress. The PERK arm of the unfolded protein response (UPR) has emerged as an important stress-responsive signaling pathway for adapting mitochondria in response to pathologic ER insults.1,2 PERK is an ER-localized kinase that is activated in response to ER stress through a mechanism involving autophosphorylation and dimerization.3–5 Once activated, PERK primarily functions through selective phosphorylation of the α subunit of eukaryotic initiation factor 2 (eIF2α). This leads to both a reduction in new protein synthesis and the selective activation of stress-responsive transcription factors such as ATF4 that regulate expression of genes involved in many adaptive pathways including cellular redox, amino acid biosynthesis, and cellular proteostasis.6–8
PERK localizes to ER-mitochondrial contact sites, positioning this ER stress sensor to coordinate ER and mitochondria function in response to pathologic insults.9,10 Consistent with this, PERK-regulated transcriptional and translational signaling are both implicated in the adaptive remodeling of mitochondrial morphology and function.1,2 PERK signaling promotes both protective mitochondrial elongation and mitochondrial cristae formation in response to ER stress through mechanisms including adaptive remodeling of mitochondrial phospholipids and regulated import of the MICOS subunit MIC19.11–14 This organellar and ultrastructural remodeling functions to regulate mitochondrial bioenergetics and prevent premature mitochondrial fragmentation during ER stress. Similarly, ATF4-dependent upregulation of SCAF1 downstream of PERK increases assembly of respiratory chain supercomplexes to further adapt mitochondrial bioenergetics during ER stress.15 PERK signaling also regulates mitochondrial proteostasis through multiple mechanisms including ATF4-dependent expression of mitochondrial proteostasis factors (e.g., HSPA9, LON) and reductions in the core TIM23 import subunit TIM17A downstream of translation attenuation.8,16–18 Apart from these adaptive functions, PERK signaling regulates mitochondria-derived apoptotic signaling following prolonged, severe ER stress through multiple mechanisms primarily involving sustained upregulation of the transcription factor CHOP.19 Thus, PERK serves a central role in dictating mitochondrial adaptation and cellular survival in response to varying levels of ER stress.
Deficiencies in PERK activity induced by genetic, environmental, or aging-related factors are implicated in the onset and pathogenesis of numerous diseases.1 Loss-of-function mutations in EIF2AK3, the gene that encodes PERK, are causatively associated with Wolcott-Rallison syndrome – a rare autosomal-recessive disorder that involves multi-organ failures including prominent neonatal or early-childhood insulin-dependent diabetes, kidney and liver dysfunction, and cardiac abnormalities.20–22 Hypomorphic EIF2AK3 alleles are also implicated in neurodegenerative diseases including the tauopathy progressive supranuclear palsy (PSP).23–25 Further, deficiencies in PERK signaling have been implicated in the pathogenesis of many diseases including PSP and Huntington’s disease (HD).1,26–28 Intriguingly, the pathogenesis of these diseases all involves mitochondrial dysfunction, suggesting that impaired PERK signaling could contribute to disease pathology through deficient regulation of mitochondria.
The above results suggest that pharmacologic enhancement of PERK signaling offers a potential opportunity to mitigate pathologic cellular and mitochondrial dysfunction caused by PERK deficiency. Multiple compounds have been identified to activate PERK signaling including CCT020312 and MK28.27,29,30 Both of these compounds have been shown to be beneficial in cellular and mouse models of neurodegenerative diseases including PSP and HD.26,27 However, the mechanism by which these compounds activate PERK, their selectivity for PERK signaling over other stress-responsive signaling pathways, and the specific dependence of the observed protection on PERK activity remain poorly defined. Further, PERK activating compounds are likely to be limited in their ability to promote protective PERK-dependent signaling in cells expressing inactive or hypomorphic PERK variants. Thus, different strategies are likely required to fully access the therapeutic potential for enhancing adaptive PERK signaling in the context of disease.
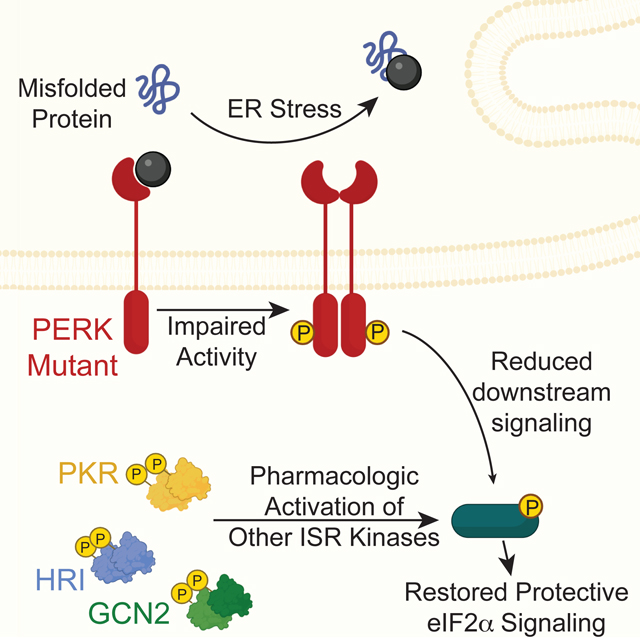
An alternative strategy to promote cellular and mitochondria remodeling in PERK-deficient cells is to activate compensatory kinases that induce similar signaling to that regulated by PERK. The integrated stress response (ISR) comprises four stress-activated eIF2α kinases activated in response to diverse pathologic insults (Fig. 1A).31,32 Apart from PERK, these include GCN2 (activated by amino acid deprivation), HRI (activated by heme deficiency, oxidative stress, and mitochondrial dysfunction), and PKR (activated by double stranded RNA). The activation of these kinases leads to eIF2α phosphorylation and similar translational and transcriptional signaling to that observed upon PERK activation. This suggests that pharmacologic activation of these other ISR kinases could compensate for deficiencies in PERK signaling and rescue pathologic cellular and mitochondrial dysfunction induced by reduced PERK activity. Consistent with this idea, HRI is activated in Perk-deficient neurons, but not Perk-deficient astrocytes, to promote translational attenuation and ATF4 activation downstream of eIF2α during ER stress.33 While this may reflect mitochondrial stress-dependent activation of HRI signaling induced by ER stress in neurons lacking Perk34,35, these results support the potential for pharmacologically enhancing alternative eIF2α kinases to promote ISR signaling in Perk-deficient cells.
Figure 1. Pharmacologic activation of integrated stress response (ISR) kinases.

A. Illustration showing the integrated stress response (ISR) comprising the stress-responsive eIF2α kinases PERK, GCN2, HRI, and PKR. Specific cellular insults that activate each individual kinase are shown. B. Structures of putative small molecule activators of PERK, HRI, or GCN2 used in this study. C. Graphs showing dose-responsive activation of an ATF4-FLuc ISR reporter (Fig. S1A) stably expressed in HEK293T cells treated for 8 h with the indicated ISR activator. The EC50 of activation is shown. Error bars show SEM for n=12 replicates. D. Graphs showing activation of an ATF4-mApple ISR reporter (Fig. S1A) stably expressed in HEK293T cells CRISPRi-depleted of the indicated ISR kinase and treated for 8 h with BtdCPU (10 μM), halofuginone (100 nM), Erlotinib (25 μM), or Sunitinib (10 μM). Error bars show SEM for n=3 replicates. ***p<0.005 for one-way ANOVA. See also Fig. S1.
Numerous small molecules have been identified to activate these other ISR kinases. BtdCPU and related N,N’-diarylureas are activators of HRI signaling, although the mechanism by which these compounds activate HRI are poorly defined.36,37 Alternatively, GCN2 is activated by the glutamyl-prolyl tRNA synthetase inhibitor halofuginone through a mechanism involving the accumulation of uncharged proline tRNA.38 Tyrosine kinase inhibitors such as erlotinib and sunitinib were also shown to activate GCN2 through a mechanism potentially involving direct binding to this ISR kinase.39 ATP competitive inhibitors of GCN2 have also been shown to activate GCN2 at lower concentrations.40 Similarly, PERK kinase inhibitors can also activate GCN2 signaling at concentrations higher than those observed for PERK inhibition.41 Like PERK activators, these other ISR kinase activators, most notably BtdCPU and halofuginone, have been shown to be protective in cellular and in vivo models of numerous disorders, further highlighting the potential for pharmacologic ISR activation in disease.37,38,42–44 However, the capacity for these compounds to promote adaptive mitochondrial remodeling in wild-type cells or cells deficient in PERK activity is currently unknown.
Here, we define the potential for pharmacologic, stress independent activation of ISR kinases to promote adaptive cellular and mitochondrial remodeling in wild-type and Perk-deficient cells. We identify two prioritized ISR activators, BtdCPU and halofuginone, that restores ISR signaling and ER stress sensitivity in Perk-deficient cells. However, we find that these compounds differentially impact mitochondria. We show that BtdCPU induces mitochondrial uncoupling, which in turn leads to mitochondrial fragmentation and ISR activation through the OMA1-DELE1-HRI mitochondrial stress signaling axis.34,35 In contrast, halofuginone induces ISR-dependent mitochondrial elongation and adaptive remodeling of mitochondrial respiratory chain activity, mimicking mitochondrial adaptations induced by PERK signaling. These results demonstrate the potential for pharmacologic, stress-independent activation of compensatory ISR kinases to promote adaptive mitochondrial remodeling in wild-type and PERK-deficient cells, motivating the development of highly-selective ISR kinase activating compounds for the treatment of diseases associated with PERK deficiency and/or mitochondrial dysfunction.
RESULTS
Activity profiling of ISR activating compounds.
The ISR comprises four eIF2α kinases – PERK, HRI, GCN2, and PKR – that are activated in response to diverse stimuli (Fig. 1A).31,32 We initially probed the activity of previously reported compounds predicted to activate specific ISR kinases. These include the PERK activators CCT020312 and MK2827,29, the HRI activator BtdCPU37, and the GCN2 activators halofuginone, erlotinib, and sunitinib (Fig. 1B).38,39 Initially, we tested the ability of these compounds to activate a luciferase-based reporter of ATF4 translation (ATF4-FLuc) as an indicator of ISR activity in HEK293T cells (Fig. S1A).45 We confirmed that thapsigargin (Tg), a SERCA inhibitor that activates the ER stress responsive PERK ISR kinase, robustly increases ATF4-FLuc activity (Fig. S1B). All tested ISR activating compounds activated the ATF4-FLuc reporter with varying potency and efficacy that was consistent with previous reports (Fig. 1C).27,29,37–39 Next, to determine the specific kinase responsible for compound-dependent ISR activation, we used HEK293T cells expressing an ATF4-mApple translational reporter of ISR activation (Fig. S1A) and CRISPRi-depleted of individual ISR kinases.35 We confirmed the effectiveness of this approach by showing that Tg-dependent ATF4-mApple activation was inhibited in cells deficient in PERK, but not cells deficient in other ISR kinases, confirming that ER stress-dependent ATF4 activation is mediated through PERK (Fig. S1C). Treatment with CCT020312 or MK28 for 8 h increases ATF4-mApple fluorescence in all cell lines, indicating that, under these conditions, these two compounds activate the ISR reporter through a mechanism not solely dependent on a single kinase (Fig. S1C). HRI-depletion selectively blocked BtdCPU-dependent ATF4mApple fluorescence, demonstrating this compound activates the ISR through HRI (Fig. 1D). Similarly, GCN2-depletion selectively inhibited ATF4-mApple fluorescence induced by halofuginone, erlotinib, and sunitinib, confirming that these compounds activated ISR signaling through GCN2 (Fig. 1D). These results show that BtdCPU, halofuginone, erlotinib, and sunitinib activate the ISR through the activity of specific ISR kinases, confirming previously published results.37–39
Compensatory ISR kinase activation restores ISR signaling in Perk-deficient cells
We next sought to identify compounds that could restore ISR signaling and mitigate pathologic phenotypes linked to deficiencies in PERK activity. Initially, we treated Perk+/+ and Perk−/− MEFs with ISR activating compounds for 3 h and monitored expression of the ISR target protein ATF4. Tg-dependent increases of ATF4 were blocked in Perk−/− MEFs, further confirming that ER stress induces ATF4 through a PERK-regulated mechanism (Fig. 2A, B). In contrast, BtdCPU, halofuginone, erlotinib, sunitinib, CCT020312, and MK28 increased ATF4 in both Perk+/+ and Perk−/− MEFs, although Perk-deficient cells did show lower levels of ATF4 induction for many compounds, as compared to wild-type cells (Fig. 2A, B). Considering other activities of erlotinib (e.g., EGFR inhibition) and sunitinib (e.g., receptor tyrosine kinase inhibition) and the promiscuity for ISR kinase activation observed for CCT020312 and MK28 (Fig. S1C), we prioritized BtdCPU and halofuginone for further study. We showed that BtdCPU and halofuginone did not increase ATF4 in knockin MEFs expressing the non-phosphorylatable eIF2α mutant S51A (MEFA/A)46, confirming that the observed increase in ATF4 afforded by these compounds could be attributed to ISR signaling (Fig. S2A).
Figure 2. Pharmacologic ISR activators restore ISR signaling in Perk-deficient MEFs.

A,B. Representative immunoblots and quantification of ATF4 in Perk+/+ and Perk−/− MEFs treated for 3 h with thapsigargin (Tg, 500 nM), BtdCPU (10 μM), halofuginone (100 nM), erlotinib (25 μM), sunitinib (10 μM), MK28 (20 μM), or CCT020312 (10 μM). Error bars show SEM for n=3 replicates. C. Expression, measured by RNAseq, of genesets comprised of 16 ISR target genes in Perk+/+ and Perk−/− MEFs treated for 6 h with thapsigargin (Tg; 500 nM), BtdCPU (10 μM), or halofuginone (HF, 100 nM), as indicated. The expression of individual ISR target genes was normalized to their expression in Tg-treated Perk+/+ MEFs. ISR target genes are described in Table S2. *p<0.05, ***p<0.005 for one-way ANOVA relative to Tg-treated Perk−/− MEFS. D. Heat map showing expression, measured by RNAseq, of the UPR and ISR target genes in Perk+/+ and Perk−/− MEFs treated for 6 h with thapsigargin (Tg; 500 nM), BtdCPU (10 μM), or halofuginone (HF, 100 nM), as indicated. The expression of individual target genes was normalized to their expression in Tg-treated Perk+/+ MEFs. Data are included in Table S2. E,F. Gene set enrichment analysis (GSEA) for hallmark genesets of RNAseq data from Perk+/+ MEFs treated for 6 h with BtdCPU (10 μM; E) or halofuginone (HF, 100 nM; F). Full GSEA results are included in Table S3. See also Fig. S2 and Tables S1–S4.
To further probe the potential for BtdCPU and halofuginone to induce ISR signaling, we monitored gene expression by RNAseq in Perk+/+ and Perk−/− MEFs treated with these compounds in the presence or absence of Tg (Table S1). Initially, we compared the expression of 16 established ISR target genes47 to that observed in Tg-treated Perk+/+ cells across all conditions (Fig. 2C,D, Table S2). As expected, Perk-deficient cells treated with Tg showed no significant induction of ISR target genes. However, ER stress-responsive genes regulated by the IRE1/XBP1s and ATF6 arms of the UPR are efficiently induced in Tg-treated, Perk-deficient cells (Fig. 2D, Table S2). Treatment with BtdCPU or halofuginone increased ISR target gene expression in both Perk+/+ and Perk−/− MEFs in the presence or absence of Tg (Fig. 2C, D). We confirmed these results for the ISR target genes Ddit3/Chop and Chac1 by qPCR (Fig. S2B, C) These results indicate that BtdCPU and halofuginone both induce ISR signaling in wild-type cells and restore ISR signaling in Perk-deficient cells during conditions of ER stress.
Analysis of the transcriptome-wide changes indicate that BtdCPU is a more selective activator of the ISR than halofuginone. Gene set enrichment analysis (GSEA) showed that treatment with BtdCPU (Table S3) significantly induced expression of genes related to the unfolded protein response in both Perk+/+ and Perk−/− MEFs (Fig. 2E, Fig. S2D). This reflects the increased expression of ISR target genes such as Atf3, Chac1, Asns and Atf4 induced by this compound, as markers of other UPR pathways (i.e., IRE1/XBP1s or ATF6) are not significantly induced upon BtdCPU treatment in these cells (Fig. 2D). GSEA also identified increased inflammatory and hypoxic signaling in BtdCPU-treated cells (Fig. 2E, Fig. S2D). However, this appears to be primarily driven by the expression of established ISR target genes such as Atf3 and Ppp1r15a, which are included in all these genesets (Table S3). By profiling selective transcriptional targets of these and other stress-responsive signaling pathways47, we found that BtdCPU did not broadly induce activation of other stress-responsive signaling pathways including the NRF2-regulated oxidative stress response (OSR) (Fig. S2E, Table S4). Thus, our results indicate that BtdCPU is a preferential activator of ISR signaling in these cells.
In contrast, numerous pathways are impacted by halofuginone treatment in Perk+/+ and Perk−/− MEFs (Fig. 2F, Fig. S2F). These include the UPR (reflecting ISR activation) and NFκB-mediated inflammatory signaling. Consistent with this, we found that genesets comprised of ISR and NFκB targets are increased by halofuginone treatment in both genotypes (Fig. S2E, Table S4). However, halofuginone did not induce significant expression of genes regulated by other stress-responsive signaling pathways such as the IRE1/XBP1s or ATF6 arms of the UPR, heat shock response (HSR), or the NRF2-regulated OSR. Thus, while halofuginone robustly induces stress-responsive signaling through the ISR in both Perk+/+ and Perk−/− MEFs, our results indicate that this activity is not selective transcriptome-wide.
BtdCPU and halofuginone reduce ER stress sensitivity of Perk-deficient cells
Perk-deficiency leads to increased cellular sensitivity to ER stress.7 Consistent with this Perk−/− MEFs show reduced proliferation following Tg treatment, as compared to Perk+/+ MEFs, when measured by crystal violet staining (Fig. S3A). Re-overexpression of PERKWT rescues the increased ER stress sensitivity of Perk−/− MEFs, confirming this effect can be attributed to PERK. Co-treatment with BtdCPU or halofuginone improved proliferation of Perk-deficient cells treated with Tg (Fig. 3A, Fig. S3B). Other GCN2 activators including erlotinib and sunitinib also showed improved proliferation in Tg-treated Perk−/− MEFs (Fig. S3B). In contrast, the putative PERK activator CCT020312 did not increase viability in Tg-treated cells deficient in Perk. Further, MK28 showed toxicity in both Perk+/+ and Perk−/− MEFs, likely reflecting a PERK-independent, off-target activity of this compound.
Figure 3. BtdCPU and halofuginone reestablishes ER stress sensitivity in Perk-deficient MEFs.

A. Representative crystal violet staining of Perk+/+ and Perk−/− MEFs treated for 6 h with thapsigargin (Tg; 500 nM), BtdCPU (10 μM), and/or halofuginone (100 nM) and then replated and allowed to proliferate in 6-well plates. Crystal violet staining was performed 72 h after replating. B. Caspase 3/7 activity, measured by Caspase-Glo 3/7, in Perk+/+ and Perk−/− MEFs treated for 24 h with Tg (500 nM), BtdCPU (10 μM), and/or halofuginone (100 nM). Cells treated with staurosporine (1 μM) (STS) for 24 hr are shown as a control. Error bars show SEM for n=9 replicates. ***p<0.005 for one-way ANOVA. C-E. Representative images (C) and quantification of active caspase 3 (D) or cleaved PARP (E) in Perk+/+ and Perk−/− MEFs treated for 24 h with thapsigargin (Tg; 500 nM), BtdCPU (10 μM), and/or halofuginone (100 nM). Error bars show SEM for n=3. *p<0.05, **p<0.01, ***p<0.005 for one-way ANOVA. See also Fig. S3.
We next monitored the activity of the pro-apoptotic caspases 3 and 7 in Perk+/+ and Perk−/− MEFs co-treated with Tg and the ISR activators BtdCPU or halofuginone using Caspase-Glo 3/7. Tg-treated Perk−/− MEFs showed higher caspase 3/7 activity, as compared to Tg-treated Perk+/+ MEFs (Fig. 3B). Treatment with BtdCPU or halofuginone, on their own, did not significantly influence caspase 3/7 activity in either Perk+/+ or Perk−/− MEFs. However, co-treatment with Tg and either BtdCPU or halofuginone reduced Tg-dependent caspase activity in both cell types (Fig. 3B). Similar results were observed when caspase activity was monitored by immunoblotting for active, cleaved caspase 3 or cleaved PARP, a caspase substrate, although the BtdCPU-dependent reduction observed for cleaved caspase 3 in Tg-treated Perk-deficient cells did not quite reach significance (Fig. 3C–E). These results indicate that pharmacologic ISR activators such as BtdCPU or halofuginone restores ER stress sensitivity in Perk-deficient cells.
BtdCPU and halofuginone differentially impact mitochondria.
PERK signaling promotes the ATF4-dependent induction of mitochondrial proteostasis factors including Lon (a mitochondrial AAA+ quality control protease), Dnaja3 (a mitochondrial HSP40 co-chaperone), and Hspa9 (the mitochondrial HSP70 chaperone).8,16 Thus, we monitored the expression of these mitochondrial proteostasis genes in our RNAseq data from wild-type and Perk-deficient cells treated with thapsigargin (Tg), BtdCPU, or halofuginone (HF). As expected, Tg treatment increased expression of Lon, Hspa9, and Dnaja3 in wild-type MEFs, but not Perk-deficient MEFs (Fig. 4A, Fig. S4A). This confirms that ER stress-dependent induction of these genes can be attributed to activation of the PERK ISR kinase. In contrast to Tg, BtdCPU and halofuginone induced expression of Lon, Dnaja3, and Hspa9 in both wild-type and Perk-deficient cells, with halofuginone showing the most robust induction (Fig. 4A, Fig. S4A). BtdCPU and halofuginone also increased expression of these mitochondrial proteostasis factors in Perk-deficient cells co-treated with Tg. This shows that treatment with BtdCPU or halofuginone mimic the PERK-dependent, ATF4-mediated regulation of these mitochondrial proteostasis factors in cells deficient in PERK both in the absence and presence of ER stress.
Figure 4. BtdCPU and halofuginone differentially impact mitochondrial morphology.

A. Expression, measured by qPCR, of the ATF4-regulated mitochondrial proteostasis factors LonP1 and Dnaja3 in Perk+/+ and Perk−/− MEFs treated for 6 h with thapsigargin (Tg; 0.5 μM), BtdCPU (10 μM), or halofuginone (HF; 100 nM). Error bars show SEM for n=3 replicates. *p<0.05, ***p<0.005 for one-way ANOVA. B,C., Representative images and quantification of mitochondrial morphology in Perk+/+ or Perk−/− MEFs expressing mitochondrial targeted GFP (mtGFP) and treated for 3 h with thapsigargin (Tg; 500 nM), BtdCPU (10 μM), halofuginone (HF; 100 nM), and/or ISRIB (200 nM). The inset shows 2-fold magnification of the image centered on the white asterisk. Scale bars, 5 μm. Error bars show SEM for n=3 replicates. *p<0.05, **p<0.01, ***p<0.005 for 2-way ANOVA (red indicates comparison between fragmented mitochondria fractions; green indicates comparisons between elongated mitochondria fractions). See also Fig. S4.
PERK-dependent ISR activation also promotes protective mitochondrial elongation in response to ER stress through an ATF4-independent mechanism downstream of PERK-regulated translation attenuation.11,14 Genetic depletion of Perk increases mitochondrial fragmentation and ablates ER stress-dependent increases in elongated mitochondria.11,14 Thus, we sought to determine whether compensatory ISR kinase activation could rescue mitochondrial elongation in Perk-deficient cells. We monitored mitochondrial morphology in Perk+/+ and Perk−/− MEFs transiently expressing mitochondrial-targeted GFP (mtGFP) treated with Tg, BtdCPU, or halofuginone. As reported previously11,14, Perk−/− MEFs showed increased basal mitochondrial fragmentation and were refractory to Tg-induced mitochondrial elongation. (Fig. 4B, C). Further, the increase in elongated mitochondria observed in Tg-treated Perk+/+ MEFs was inhibited by co-treatment with ISRIB – an ISR inhibitor that binds to eIF2B and desensitizes cells to eIF2α phosphorylation.48–51 These results further confirm that the mitochondria elongation observed in Tg-treated cells is attributed to PERK-dependent ISR signaling.
Treatment with BtdCPU did not increase mitochondria elongation (Fig. 4B, C). Instead, BtdCPU increased populations of fragmented mitochondria in both Perk+/+ and Perk−/− MEFs (Fig. 4B, C). Similar results were observed in MEF cells stably expressing mtGFP (MEFmtGFP; Fig. S4B). This increase in fragmentation was not inhibited by co-treatment with ISRIB (Fig. 4B, C, Fig. S4B). Similar results were observed in MEF cells expressing the non-phosphorylatable S51A eIF2α mutant (MEFA/A) 46 (Fig. S4C, D). These results suggest that BtdCPU promotes mitochondrial fragmentation through an ISR-independent mechanism.
In contrast, halofuginone increased mitochondrial elongation in both Perk+/+ and Perk−/− MEFs to levels similar to that observed in Tg-treated Perk+/+ cells (Fig. 4B, C). Halofuginone-dependent increases in elongated mitochondria were also observed in MEFmtGFP cells (Fig. S4E, F). This indicates that halofuginone can rescue mitochondrial elongation in Perk-deficient cells. Higher doses of halofuginone can induce translational attenuation independent of the ISR52, and ISR-independent translation attenuation afforded by compounds such as the ribosomal inhibitor cycloheximide induce mitochondrial elongation.11,14,53 Thus, to determine the dependence of halofuginone-dependent mitochondrial elongation on the ISR, we co-treated Perk+/+ and Perk−/− cells expressing mtGFP with halofuginone and ISRIB and monitored mitochondrial morphology. Co-treatment with ISRIB inhibited halofuginone-dependent increases in mitochondrial elongation in both genotypes (Fig. 4B, C). Halofuginone treatment also demonstrated impaired mitochondrial elongation in ISR-deficient MEFA/A cells (Fig. S4C, D). Further, we showed that halofuginone, like PERK activation11, increased mitochondrial elongation in Atf4-deficient MEFs(Fig. S4G).17 These results indicate that halofuginone induces mitochondrial elongation in wild-type and Perk-deficient cells through an ISR-dependent, but ATF4-independent, mechanism, mimicking the mitochondrial elongation induced by PERK activation.11,14
BtdCPU induces mitochondrial depolarization to promote OMA1-DELE1-HRI signaling
Mitochondrial fragmentation is a marker of mitochondrial dysfunction. Thus, the increased fragmentation observed in cells treated with BtdCPU suggests that this compound disrupts mitochondrial activity. To probe this, we monitored mitochondrial respiratory chain activity in Perk+/+ MEFs treated with BtdCPU using Seahorse (Fig. 5A). Treatment with BtdCPU did not impact basal respiration, but reduced ATP-linked respiration (Fig. 5A, B). This corresponded with an increase of proton leakage, suggesting that BtdCPU impacted mitochondrial membrane potential. Consistent with this, we observed reductions of mitochondrial membrane potential in Perk+/+ MEFs treated with BtdCPU, as measured by TMRE staining (Fig. 5C). Similar results were observed in other cell types including HEK293T and SHSY5Y cells (Fig. S5A, B). Co-treatment with ISRIB did not influence BtdCPU-dependent mitochondrial depolarization (Fig. S5B). This indicates that BtdCPU promotes mitochondrial depolarization independent of ISR activation.
Figure 5. BtdCPU promotes mitochondrial uncoupling.

A. Mitochondrial respiration, measured by Seahorse mitochondrial stress test, of Perk+/+ MEFs treated for 3 h with vehicle (Veh; black) or BtdCPU (10 μM; blue). Error bars show SEM for n=5 replicates. B. Basal respiration, ATP-linked respiration, and proton leak measured from the mitochondrial stress test shown in (A). Error bars show SEM for n=5 replicates. *p<0.05, **p<0.01, ***p<0.005 from unpaired t-test. C. TMRE fluorescence in Perk+/+ MEFs treated for 3 h with vehicle, BtdCPU (10 μM), or CCCP (25 μM). Error bars show SEM for n=3 replicates. ***p<0.005 from one-way ANOVA. D,E. Representative immunoblot (D) and quantification (E) of ATF4 in wild-type (WT) HEK293T cells or HEK293T cells CRISPR-deleted of HRI or DELE1 treated for the indicated time with BtdCPU (10 μM) or CCCP (5 μM). Actin is showed as a loading control. Error bars show SEM for n=3 replicates. ***p<0.005 for 2-way ANOVA. F. Immunoblot of lysates prepared from wild-type HEK293T cells, HEK293T cells CRISPR-deleted of OMA1, or HEK293T cells CRISPR-deleted of OMA1 overexpressing OMA1 treated for 3 h with vehicle, BtdCPU (10 μM), or CCCP (25 μM), as indicated. G,H. Representative immunoblot and quantification of long DELE1-mClover (DELE1L) cleavage to short DELE1-mClover (DELE1S) in HEK293T cells expressing DELE1L-mClover treated for the indicated time with BtdCPU (10 μM) or CCCP (5 μM). The cleavage of DELE1L (DELE1L / total DELE1) is shown normalized to untreated cells. Error bars show SEM for n=2 replicates. **p<0.01, ***p<0.005 for one-way ANOVA. I. Immunoblot of lysates prepared from wild-type MEFs or MEFs expressing the non-phosphorylatable S51A eIF2α mutant (MEFA/A) treated for 3 h with BtdCPU (10 μM) or CCCP (25 μM). See also Fig. S5.
Mitochondrial depolarization can activate ISR signaling through a mechanism involving stress-induced activation of the mitochondrial protease OMA1, cytosolic accumulation of full-length or a C-terminal cleavage products of DELE1, and subsequent DELE1-dependent HRI activation.34,35,54 Our results showing that BtdCPU depolarizes mitochondria suggests that this compound could activate ISR signaling through this OMA1-DELE1-HRI signaling axis. Consistent with this, deletion of HRI or DELE1 reduces BtdCPU-dependent ATF4 expression in HEK293T cells, mirroring results observed with the mitochondrial uncoupler CCCP (Fig. 5D, E). OMA1-deletion also inhibited BtdCPU-dependent ATF4 expression (Fig. 5F). Re-overexpression of OMA1 restored ATF4 induction in BtdCPU-treated OMA1-deficient cells, confirming that this effect can be attributed to OMA1.
OMA1 is a stress-activated protease localized to the inner mitochondrial membrane that is activated in response to mitochondrial stressors such as membrane depolarization.55 Once activated, OMA1 induces proteolytic processing of substrates including DELE1 and the inner membrane GTPase OPA1, which induces HRI activation and mitochondrial fission, respectively.34,35,56 We found that BtdCPU increased proteolytic processing of full-length DELE1-mClover expressed in HEK293T cells (Fig. 5G,H). Further, OMA1-dependent processing of OPA1 from long to short isoforms was increased in BtdCPU-treated HEK293T cells (Fig. 5F). Similar results were observed in MEFA/A cells expressing the non-phosphorylatable S51A eIF2α mutant46, indicating that this increase of OMA1-dependent OPA1 processing is independent of ISR activity (Fig. 5I). We also found that OMA1-deletion did not influence BtdCPU-dependent mitochondria depolarization, indicating that OMA1 activation was downstream of mitochondrial uncoupling (Fig. S5C). Collectively, these results indicate that BtdCPU-dependent mitochondria depolarization activates OMA1 proteolytic activity to promote mitochondrial fragmentation and ISR activation through a mechanism involving the OMA1-DELE1-HRI signaling pathway, revealing insights into the mechanism of action for ISR activation afforded by this compound.
Halofuginone promotes adaptive remodeling of mitochondria respiration in Perk-deficient cells.
Mitochondrial elongation is an adaptive mechanism that regulates mitochondrial bioenergetics during conditions of stress 57,58. Thus, our results that show PERK signaling impacts mitochondrial morphology both in the absence and presence of acute ER stress suggest an important role for PERK in regulating mitochondrial energy production. To test this, we monitored mitochondrial respiration in Perk-deficient MEFs using Seahorse. Cells deficient in Perk show increased basal respiration, ATP-linked respiration, spare respiratory capacity, and proton leak, as compared to Perk+/+ MEFs (Fig 6A, B). This is in contrast to experiments showing Perk-depletion in other cell types including HEK293, myeloid leukemia, and brown adipocyte cells reduce basal respiration.59–61 However, we demonstrate that re-overexpression of PERKWT in Perk−/− MEFs restores basal respiratory chain activity to the same levels observed in Perk+/+ MEFs, indicating the observed changes in Perk-deficient MEFs can be attributed to the presence of PERK. This likely highlights cell-type specific compensatory regulation of mitochondrial respiration associated with Perk-deficiency.
Figure 6. Halofuginone promotes adaptive remodeling of mitochondria respiration in PERK-deficient cells.

A. Mitochondrial respiration, measured by Seahorse mitochondrial stress test, of Perk+/+, Perk−/− MEFs, and Perk−/− MEFs overexpressing wild-type PERK. Error bars show SEM for n=5 replicates. B. Basal respiration, ATP-linked respiration, spare respiratory capacity and proton leak measured from the mitochondrial stress test shown in (A). Error bars show SEM for n=5 replicates. *p<0.05, ***p<0.005 for one-way ANOVA C. Mitochondrial respiration measured by Seahorse mitochondrial stress test, of Perk+/+ and Perk−/− MEFs treated for 3 h with thapsigargin (Tg; 500 nM), or halofuginone (HF; 100 nM). Error bars show SEM for n=5 replicates. D. Basal respiration and ATP-linked respiration measured from the mitochondrial stress test shown in (C). Error bars show SEM for n=5 replicates. ***p<0.005 for one-way ANOVA. See also Fig. S6.
Regardless, we show that Perk-deficient MEFs show impaired regulation of respiratory chain activity in response to acute ER stress. In Perk+/+ MEFs, treatment with Tg for 3 h reduces both basal mitochondrial respiration and ATP-linked respiration, while enhancing spare respiratory capacity (Fig. 6C, D; Fig. S6A). However, these changes were not observed in Perk-deficient cells. These results highlight an important role for PERK signaling in regulating mitochondrial respiration in the presence of acute ER stress.
Since halofuginone increases mitochondrial elongation in both Perk+/+ and Perk−/− MEFs (Fig. 4A,B) and mitochondrial elongation is linked to adaptive regulation of mitochondrial respiration57,58, we anticipated that treatment with this compound should promote similar changes to mitochondrial respiration to those observed in wild-type cells treated with PERK-activating ER stressors (e.g., Tg). Consistent with this, treatment of Perk+/+ MEFs with halofuginone induced identical changes of mitochondrial respiration to those observed in Tg-treated cells (Fig. 6C). This includes reductions in basal respiration and ATP-linked respiration, and increases in spare respiratory capacity (Fig. 6D, Fig. S6A, B). Halofuginone also reduced basal respiration and ATP-linked respiration in Perk−/− MEFs, restoring these parameters to levels similar to those observed in wild-type cells (Fig. 6C, D). However, halofuginone did not alter spare respiratory capacity or proton leakage in Perk-deficient cells (Fig. S6A, B), likely reflecting a chronic consequence of deficient PERK activity on mitochondrial function that cannot be rescued by this short treatment. Halofuginone-dependent alterations to mitochondrial respiration were inhibited in MEFA/A cells expressing the non-phosphorylatable S51A eIF2α mutant, indicating that these changes can be attributed to ISR activation (Fig. S6C). However, halofuginone reduced basal and ATP-linked respiration in Atf4-deficient cells (Fig. S6D, E). This suggests that these alterations to mitochondrial respiration are likely linked to ISR-dependent translational attenuation, but not ATF4-regulated transcriptional signaling – the same mechanism associated with ISR-dependent mitochondrial elongation.11,14 Consistent with this, treatment with the translation inhibitor cycloheximide (CHX) similarly reduced basal and ATP-linked respiration (Fig. S6D, E). Collectively, our results indicate that halofuginone can induce adaptive, ISR-regulated remodeling of mitochondrial respiration in both wild-type and Perk-deficient cells.
DISCUSSION
Pharmacologic ISR activation has emerged as a promising strategy to mitigate pathologies implicated in etiologically-diverse diseases including many types of cancers, ischemic diseases, and neurodegenerative disorders such as PSP.1,27,37,62–64 Here, we sought to further define the potential for pharmacologic ISR activation to promote adaptive mitochondrial remodeling and prevent pathologic mitochondrial dysfunction induced under conditions such as Perk-deficiency.1,25,65 By probing the activity of established ISR activating compounds, we demonstrate that treatment with halofuginone restores ER stress sensitivity and promotes adaptive mitochondrial remodeling in both wild-type and Perk-deficient cells. These results underscore the potential for pharmacologic ISR kinase activation to mitigate mitochondrial dysfunction associated with diverse disorders.
Numerous compounds have been reported to selectively activate ISR kinases independent of cellular stress; however, each of these compounds has liabilities that limit their ability to probe the biological and therapeutic potential for ISR signaling in cellular and in vivo models of disease. The PERK activator CCT020312 has been used to activate PERK signaling in multiple models, including mouse models of PSP where CCT020312 was shown to promote clearance of tau aggregates.26 However, recent results suggest this increased tau clearance reflects an off-target activity of this compound that increases autophagy independent of the ISR, questioning the dependence of this effect on PERK activation.66 Similarly, the PERK activator MK28 reduces toxic huntingtin aggregation in mouse models of HD.27 However, we found this compound to be cytotoxic in Perk+/+ and Perk−/− MEFs (Fig. S3B), likely reflecting a PERK-independent, off-target activity. Further, the mechanism of both CCT020312- and MK28-dependent PERK activation is currently poorly understood. While elucidation of the mechanism of PERK activation for these compounds could enable the establishment of next generation compounds with improved selectivity and potency, the use of CCT020312 and MK28 as PERK activators should be approached with care and include use of both pharmacologic or genetic controls to confirm that specific phenotypes can be attributed to PERK activation, as opposed to off-target activities.
BtdCPU and related N,N’-diarylureas have largely been developed to increase apoptotic signaling downstream of HRI for the treatment of hematologic cancers including multiple myeloma and acute leukemia.67,68 However, the mechanism of BtdCPU-dependent HRI activation was previously undefined. We demonstrate that BtdCPU disrupts the mitochondrial membrane potential to activate the stress-activated mitochondrial protease OMA1. This leads to ISR activation through the OMA1-DELE1-HRI mitochondrial stress-signaling axis and mitochondrial fragmentation through OMA1-dependent proteolytic processing of OPA1. Since mitochondrial uncoupling is associated many pathologic conditions, this mechanism of action limits the application of BtdCPU and related compounds to promote protective ISR signaling and mitochondrial adaptation in context of other diseases.
Unlike other ISR activator compounds discussed above, the mechanism of halofuginone-dependent ISR activation is known to be attributed to its inhibition of the glutamyl-prolyl tRNA synthetase.38 This leads to accumulation of uncharged proline tRNAs and subsequent GCN2 activation. Halofuginone has been shown to be protective in models of etiologically-diverse diseases including ischemic disorders, cardiovascular disease, and many cancers.42,44,69 However, apart from ISR activation, halofuginone can also promote protection through other mechanisms including inhibition of TGFβ signaling.69 In addition, our RNAseq transcriptional profiling indicates that halofuginone does not show transcriptome-wide selectivity for the ISR, as it also induces expression of genes regulated by other pathways including NFκB-mediated inflammatory signaling. Further, higher doses of halofuginone can inhibit global translation independent of the ISR, owing to its glutamyl-prolyl tRNA synthetase inhibition.38,52 These effects may limit the translational potential for halofuginone-dependent ISR activation as a strategy to mitigate dysfunction in diseases involving deficiencies of specific ISR kinases.
Regardless of these limitations, we show that halofuginone-dependent ISR activation both promotes ER stress sensitivity and adaptive mitochondrial remodeling in wild-type and Perk-deficient cells. This demonstrates the potential for pharmacologic activation of compensatory ISR kinases to promote adaptive mitochondria remodeling, even in cells deficient in PERK. However, our results also demonstrate the need for continued development of highly selective ISR kinase activators that can be used to further probe the potential for this approach to mitigate cellular and mitochondrial dysfunction in disease. While pharmacologic ISR activation offers unique opportunities to promote protective signaling through this pathway, a challenge for the translational development of pharmacologic ISR kinase activators is the pro-apoptotic signaling that could result from chronic ISR activation.19 Previous results have shown that optimization of compound PK/PD can allow transient, in vivo activation of similar stress-responsive signaling pathways such as the IRE1/XBP1s signaling arm of the UPR to induce protective, adaptive signaling without the pathologic consequences of chronic pathway activation.70 Similar strategies could be applied for ISR kinase activating compounds to allow protective, adaptive ISR signaling without inducing pro-apoptotic signaling associated with chronic ISR activity. As we, and others, continue identifying ISR kinase activating compounds, the therapeutic potential and optimized activity of this class of compounds will continue to be defined, revealing insights into the translational potential of pharmacologic ISR kinase activation to mitigate mitochondrial dysfunction for diverse disorders.
LIMITATIONS OF THE STUDY.
We would like to address some limitations of our study. We sought to define the potential for pharmacologic activation of ISR kinases to compensate for cellular and mitochondrial dysfunction induced by deficiencies in the ISR kinase PERK. For these efforts, we relied on the use of established ISR kinase activating compounds. While we identified compounds that selectively activated the GCN2 and HRI kinases (halofuginone and BtdCPU, respectively), we found that putative PERK kinase activators did not selectively induce ISR signaling through PERK. We also did not identify a suitable PKR activator for use in our study. Further, we found that the HRI activator BtdCPU induced mitochondrial dysfunction independent of the ISR, limiting its application for the mitochondrial aspects of our project. Ultimately, we demonstrated that pharmacologic GCN2 activation with halofuginone rescued cellular and mitochondrial phenotypes in Perk-deficient cells; however, we are unable to determine whether pharmacologic activation of other ISR kinases could similarly rescue these phenotypes.
Further, as pointed out above in our discussion, halofuginone does have some potential liabilities that could limit is application as a selective GCN2 activator. Halofuginone can induce genes regulated by other stress-responsive pathways including the NFκB-mediated inflammatory response. Further, halofuginone can inhibit other signaling pathways such as those regulated by TGFβ.69 Finally, halofuginone can induce global translational attenuation at higher concentrations.38,52 These other activities highlight the importance for using GCN2 or ISR inhibitors when probing the specific contributions of halofuginone-dependent ISR activation in different models.
Finally, we would like to point out that our experiments were solely performed in cell culture models, primarily mouse embryonic fibroblasts (MEFs). Our results suggest that pharmacologic activation of compensatory ISR kinases can rescue deficiencies linked to reduced PERK activity; however, it is important to note that similar effects may not be observed in other cell lines, primary cell culture models, and/or animal models. Thus, while we are encouraged about the potential for pharmacologic activation of compensatory ISR kinases to improve outcomes in the context of diseases linked to Perk-deficiency, it is important to remain cautious when extrapolating the results described in this manuscript. As we and others continue developing highly-selective ISR kinase activators and implementing these compounds in disease-relevant models, the therapeutic potential for this approach will become more clear.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact.
Further information and request for resources should be directed to and will be fulfilled by the lead contact, R.
Luke Wiseman (wiseman@scripps.edu).
Materials availability
All unique reagents and resources described in this manuscript are available from the lead contact, R. Luke Wiseman (wiseman@scripps.edu) upon reasonable request.
Data and code availability.
RNAseq data has been deposited at GEO and is publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-ATF4 antibody (D4B8) | Cell Signaling | Cat # 11815 |
| Mouse anti-tubulin antibody [B-5–1-2] | Sigma | Cat # T6704 |
| Mouse anti-HSP60 antibody (LK1) | ThermoFisher | Cat # MA5–45109 |
| Rabbit anti-PERK (C33E10) | Cell Signaling | Cat # 3192 |
| Mouse anti-HA [Clone: 16B12] | Biolegend | Cat # 901501 |
| Mouse anti-GFP (B2) | Santa Cruz | Cat # Sc-9996 |
| Mouse anti-OPA1 | BD Bioscience | Cat # 612606 |
| Rabbit anti-OMA1 | Cell Signaling | Cat # 95473 |
| Rabbit anti-cleaved Caspase 3 (D175) | Cell Signaling | Cat # 96611 |
| Rabbit anti-PARP | Cell Signaling | Cat # 9542 |
| Mouse anti-GAPDH | Genetex | Cat # GTX627408 |
| Mouse anti-beta-actin | Cell Signaling | Cat # 3700 |
| Chemicals, peptides, and recombinant proteins | ||
| Thapsigargin | Fisher Scientifics | Cat # 50–464-295 |
| ISRIB | Sigma | Cat # SML0843 |
| CCCP | Sigma | Cat # C2759 |
| BtdCPU | Fisher | Cat # 32489210MG |
| Halofuginone | Sigma | Cat # 50–576-300001 |
| Erlotinib | Selleckchem | Cat # S7786 |
| Sunitinib | Selleckchem | Cat # S7781 |
| MK28 | MedChemExpress | Cat # HY-137207 |
| CCT020312 | MedChemExpress | Cat # HY-119240 |
| Staurosporine | Fisher | Cat # NC1407148 |
| MEF Avalanche Transfection Reagent | EZ Biosystems | Cat # EZT-MEFS-1 |
| Critical commercial assays | ||
| Quick RNA mini-prep kit | Zymo Research | Cat # R1055 |
| High Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat # 4368702 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | Cat # 4367659 |
| Caspase 3/7 Glo Assay Kit | Promega | Cat # G8090 |
| Data Deposited | ||
| RNAseq data generated from Perk+/+ and Perk−/− treated with thapsigargin, halofuginone, and/or BtdCPU were deposited to the Gene Expression Omnibus (GEO). | This manuscript. | GEO: GSE227134 |
| Experimental models: Cell lines | ||
| Perk+/+ and Perk−/− MEFs | Harding et al (2000) Mol Cell7 | David Ron’s lab |
| Atf4+/+ and Atf4−/− MEFs | Harding et al (2003) Mol Cell17 | Established by David Ron’s lab; Provided by Jonathan Lin’s lab |
| MEFA/A MEFs | Scheuner et al (2001) Mol Cell46 | Randal Kaufman’s lab |
| HEK293T cells | ATCC | CRL-3216 |
| OMA1-deficient HEK293T cells | Guo et al (2020) Nature35 | Martin Kampmann’s lab |
| DELE1-deficient HEK293T cells | Guo et al (2020) Nature35 |
Martin Kampmann’s lab |
| HRI-deficient HEK293T cells | Guo et al (2020) Nature35 | Martin Kampmann’s lab |
| HEK293T cells stably expressing ATF4-FLuc | Yang et al (2023) Nat Struct Mol Biol45 | Martin Kampmann’s lab |
| HEK293T cells stably expressing ATF4-mAPPLE and CRISPRi-depleted of specific ISR kinases | Guo et al (2020) Nature35 | Martin Kampmann’s lab |
| MEFmtGFP cells | Wang et al (2012) Angew Chem Int Ed Engl71 |
Peter Schultz’s lab |
| SHSY5Y cells | ATCC | Cat # CRL-2266 |
| Oligonucleotides | ||
|
M. musculus Chop (forward qPCR primer):
CCTAGCTTGGCTGACAGAGG |
Integrated DNA Technologies | N/A |
|
M. musculus Chop (reverse qPCR primer):
CTGCTCCTTCTCCTTCATGC |
Integrated DNA Technologies | N/A |
|
M. musculus Chac1 (forward qPCR primer):
TGACCCTCCTTGAAGACCGTGA |
Integrated DNA Technologies | N/A |
|
M. musculus Chac1 (reverse qPCR primer):
AGTGTCATAGCCACCAAGCACG |
Integrated DNA Technologies | N/A |
|
M. musculus RiboP (forward qPCR primer):
TGTCATCGCTCAGGGTGTTG |
Integrated DNA Technologies | N/A |
|
M. musculus RiboP (reverse qPCR primer):
AAGCCAAATCCCATGTCGTC |
Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| hsPerkWT.pcDNA3.1 | Yuan et al (2018) Hum Mol Gen23 | Jonathan Lin’s lab |
| mtGFP.pDEST40 | Lebeau et al (2018) Cell
Reports11 |
Luke Wiseman’s lab |
| DELE1L-mCLOVER plasmid | Guo et al (2020) Nature35 | Martin Kampmann’s lab |
| Software and algorithms | ||
| FlowJo™ Software | BD Biosciences | N/A |
| PRISM 10.0.2 | Graphpad | N/A |
| LICOR Imaging Studio | Licor Biosciences | N/A |
| DNAstar Lasergene SeqManPro | DNAStar Inc. | N/A |
| ArrayStar 12.2 with Qseq | DNAStar Inc. | N/A |
| DESeq 2 v. 1.34 | Bioconductor | N/A |
| BiomaRt v. 2.50.3 | Bioconductor | N/A |
| Other | ||
| Infinite F200 Pro Plate Reader | Tecan | N/A |
| Bio-Rad ZE5 Cell Analyzer | Bio-Rad | N/A |
| Olympus IX71 microscope with 60x oil objective | Olympus | N/A |
| Hamamatsu C8484 camera | Hamamatsu Photonics | N/A |
| Zeiss LSM 880 Airyscan confocal laser scanning microscope | Zeiss | N/A |
| Odyssey Infrared Imaging System | Licor Biosciences | N/A |
| ABI 7900HT Fast Real Time PCR machine | Applied Biosciences | N/A |
| XF96 Extracellular Flux Analyzer | Seahorse Biosciences | N/A |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mammalian Cell Culture
Perk+/+ and Perk−/− MEFs (kind gifts from David Ron, Cambridge)7, Atf4+/+ and Atf4−/− MEFs (kind gifts from Jonathan Lin, Stanford), and MEFA/A cells (kind gifts from Randal Kaufman; Sanford-Burnham-Prebys)46 were cultured as previously described at 37°C and 5% CO2 in DMEM (Corning-Cellgro) supplemented with 10% fetal bovine serum (FBS; Omega Scientific), 2 mM L-glutamine (GIBCO), 100 Units/mL penicillin, 100 mg/mL streptomycin (GIBCO), non-essential amino acids (GIBCO), and 2-mercaptoethanol (ThermoFisher). HEK293T cells (purchased from ATCC) and SHSY5Y cells (purchased from ATCC) were cultured at 37°C and 5% CO2 in DMEM (Corning-Cellgro) supplemented with 10% fetal bovine serum (FBS; Omega Scientific), 2 mM L-glutamine (GIBCO), 100 Units/mL penicillin, and 100 mg/mL streptomycin (GIBCO). OMA1-deficient HEK293T cells, DELE1-deficient HEK293T cells, and HRI-deficient HEK293T cells were described previously 35 and cultured at 37°C and 5% CO2 in DMEM (Corning-Cellgro) supplemented with 10% fetal bovine serum (FBS; Omega Scientific), 2 mM L-glutamine (GIBCO), 100 Units/mL penicillin, and 100 mg/mL streptomycin (GIBCO). HEK293T cells stably expressing ATF4-FLuc or ATF4-mAPPLE CRISPRi-depleted of HRI, GCN2, PERK, or PKR were described previously 35,45 and cultured at 37°C and 5% CO2 in DMEM (Corning-Cellgro) supplemented with 10% fetal bovine serum (FBS; Omega Scientific), 2 mM L-glutamine (GIBCO), 100 Units/mL penicillin, and 100 mg/mL streptomycin (GIBCO). MEFmtGFP cells (kind gift from Peter Schultz, TSRI)71 were cultured at 37°C and 5% CO2 in DMEM (Corning-Cellgro) supplemented with 10% fetal bovine serum (FBS; Omega Scientific), 2 mM L-glutamine (GIBCO), 100 Units/mL penicillin, and 100 mg/mL streptomycin (GIBCO).
METHOD DETAILS
Plasmids, compounds, and reagents
All compounds used in this study were purchased: thapsigargin (Tg; 50–464-295, Fisher Scientific), ISRIB (SML0843, Sigma), CCCP (C2759, Sigma), BtdCPU (32–489-210MG, Fisher), halofuginone (50–576-30001, Sigma), erlotinib (S7786, Selleckchem), sunitinib (SU11248, Selleckchem), MK-28 (HY-137207, MedChemExpress), CCT020312 (HY-119240, Fisher), staurosporine (S1421, Selleckchem). PerkWT overexpression plasmid was a kind gift from Jonathan Lin (Stanford) 23. The mitochondrial-targeted GFP plasmid 11 and the DELE1L-mClover plasmid 35 were described previously. MEF cells were transfected with MEF Avalanche Transfection Reagent (EZ Biosystems) according to the manufacturer’s protocol.
Measurements of ISR activation in ATF4-reporter cell lines
ATF4-FLuc reporter cells were seeded at a density of 15,000 cells per well in Greiner Bio-One CELLSTAR flat 384-well white plates with clear bottoms. The following day, cells were treated with the indicated compound in triplicate 10-point dose response format for eight hours. After treatment, an equal volume of Promega Bright-Glo substrate was added to the wells and allowed to incubate at room temperature for 10 minutes. Luminescence was then measured in an Infinite F200 PRO plate reader (Tecan) with an integration time of 1000 ms.
ATF4-mApple reporter cells were seeded at a density of 300,000 cells per well in 6-well TC-treated flat bottom plates (Genesee Scientific). The following day, cells were treated for eight hours with the indicated concentrations. Following treatment, cells were washed twice with phosphate-buffered saline (PBS) and dissociated using TrypLE Express (ThermoFisher). The enzymatic reaction was neutralized through addition of flow buffer containing PBS and five percent fetal bovine serum (FBS). Flow cytometry was performed on a Bio-Rad ZE5 Cell Analyzer. mApple (568/592 nm) was measured using the 561 nm green-yellow laser in combination with the 577/15 filter. Analysis was performed using FlowJo™ Software (BD Biosciences).
Fluorescence Microscopy
MEF or HeLa cells transiently transfected with mtGFP or MEFmtGFP cells were seeded at a density of 100,000 cells/well on glass-bottom dishes (MatTek) coated with poly-D-lysine (Sigma) or rat tail collagen 1 (GIBCO). Cells were then treated as indicated and images were recorded with an Olympus IX71 microscope with 60x oil objective (Olympus), a Hamamatsu C8484 camera (Hamamatsu Photonics), and HCI image software (Hamamatsu Photonics). The images for the Atf4+/+ and Atf4−/− MEFs shown in Fig. S4G were recorded on a Zeiss LSM 880 Airyscan confocal laser scanning microscope. Quantification was performed by blinding the images and then scoring cells based on the presence of primarily fragmented, tubular, or elongated mitochondria, as before 11. At least three different researchers scored each set of images and these scores were averaged for each individual experiment and all quantifications shown were performed for at least 3 independent experiments quantifying a total of >60 cells/condition across all experiments. The data were then analyzed in PRISM (GraphPad, San Diego, CA) and plotted on a stacked bar plot to show the average morphology and standard error of the mean across all experiments. Statistical comparisons were performed using a 2-way ANOVA in PRISM, comparing the relative amounts of fragmented, tubular, or elongated mitochondria across different conditions.
Immunoblotting and Antibodies
Whole cells were lysed on ice in HEPES lysis buffer (20 mM HEPES pH 7.4, 100 mM NaCl, 1 mM EDTA, 1% Triton X100) supplemented with 1x Pierce protease inhibitor (ThermoFisher). Total protein concentrations of lysates were then normalized using the Bio-Rad protein assay and lysates were combined with 1x Laemmli buffer supplemented with 100 mM DTT and boiled for 5 min. Samples (100 μg) were then separated by SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad). Membranes were blocked with 5% milk in tris-buffered saline (TBS) and then incubated overnight at 4°C with the indicated primary antibody. The next day, membranes were washed in TBS supplemented with Tween, incubated with the species appropriate secondary antibody conjugated to IR-Dye (LICOR Biosciences), and then imaged using an Odyssey Infrared Imaging System (LICOR Biosciences). Quantification was then carried out using the LICOR Imaging Studio software. Primary antibodies were acquired from commercial sources and used in the indicated dilutions in antibody buffer (50mM Tris [pH 7.5], 150mM NaCl supplemented with 5% BSA (w/v) and 0.1% NaN3 (w/v)): ATF4 (Cell Signaling, 1:500), Tubulin [B-5–1-2] (Sigma, 1:5000), HSP60 [LK1] (ThermoFisher, 1:1000), PERK (C33E10) (Cell Signaling, 1:1000), HA [Clone: 16B12] (Biolegend, 1: 1000), GFP (B2) (Santa Cruz, 1:1000), OPA1 (BD Bioscience, 1:2000), OMA1 (Cell Signaling, 1:1000), beta-actin (Cell Signaling 1:5,000), cleaved Caspase 3 (Cell Signaling, 1:1000), PARP (Cell Signaling, 1:1000), GAPDH (GeneTex, 1:1000).
RNA sequencing and analysis
Perk+/+ and Perk−/− MEFs cells were treated for 6 h with respective compounds as noted. Cells were rinsed with DPBS, lysed, and total RNA was collected using the QuickRNA mini kit (Zymo) according to the manufacturer′s instructions. Transcriptional profiling using whole transcriptome RNA sequencing was conducted via BGI Americas on the DNBseq platform with three biological replicates for each condition. All samples were sequenced to a minimum depth of 20 M PE 150 bp stranded reads. Alignment of reads was performed using DNAstar Lasergene SeqManPro to the mouse genome GRCm39 assembly. Aligned reads were imported into ArrayStar 12.2 with Qseq (DNAStar Inc.) to quantify the gene expression levels. Differential expression analysis and statistical significance comparisons were assessed using DESeq 2 v. 1.34 in R compared to vehicle-treated Perk+/+ cells. Mouse annotations were converted to human orthologs using BiomaRt v. 2.50.3 prior to functional gene set enrichment with the R package fast gene set enrichment (fgsea) v. 1.20.0 using the Hallmark Pathways Geneset v7.5.1 from MsigDB. Code can be provided upon request. The complete RNAseq data is deposited in gene expression omnibus (GEO) as GSE227134.
Quantitative Polymerase Chain Reaction (qPCR)
The relative mRNA expression of target genes was measured using quantitative RT-PCR. Cells were treated as indicated and then washed with phosphate buffered saline (PBD; Gibco). RNA was extracted using Quick-RNA MiniPrepKit (Zymo Research) according to the manufacturers protocol. RNA (500 ng) was then converted to cDNA using the High Capacity Reverse Transcription Kit (Applied Biosystems). qPCR reactions were prepared using Power SYBR Green PCR Master Mix (Applied Biosystems), and primers (below) were obtained from Integrated DNA Technologies. Amplification reactions were run in an ABI 7900HT Fast Real Time PCR machine with an initial melting period of 95 °C for 5 min and then 45 cycles of 10 s at 95 °C, 30 s at 60 °C. qPCR Primers
| Forward | Reverse | |
|---|---|---|
| M. musculus Chop | CCTAGCTTGGCTGACAGAGG | CTGCTCCTTCTCCTTCATGC |
| M. musculus Chac1 | TGACCCTCCTTGAAGACCGTGA | AGTGTCATAGCCACCAAGCACG |
| M. musculus RiboP | TGTCATCGCTCAGGGTGTTG | AAGCCAAATCCCATGTCGTC |
Measurement of Mitochondrial Membrane Potential
Cells were seeded at a density of 200,000 cells per well in a 6-well plate and treated with 500 nM Tg for 3h prior to collection. CCCP (10 μM) was added 50 min before collection, followed by 200 nM TMRE (ThermoFisher) 20 min before collection. Following treatments, cells were washed twice with phosphate-buffered saline (PBS) and dissociated using TrypLE Express (ThermoFisher). The enzymatic reaction was neutralized through addition of flow buffer containing PBS and five percent fetal bovine serum (FBS). Fluorescence intensity of TMRE (552/574 nm) for 20,000 cells/condition was measured on a Bio-Rad ZE5 Cell Analyzer using the 561 nm green-yellow laser in combination with the 577/15 filter. Analysis was performed using FlowJo™ Software (BD Biosciences). Data are presented as geometric mean of the fluorescence intensity from three experiments normalized to vehicle-treated cells.
Cell Proliferation and Apoptosis Assays
Perk+/+ and Perk−/− MEFs were treated for 6 h with either vehicle (DMSO) or the indicated drugs. Cells were washed with room temperature PBS and then trypsinized. Fresh media was added and cells were counted using a Countess Automated Cell Counter (ThermoFisher). Cells were seeded in a 6-well plate at three different dilutions: 30,000, 15,000 and 7,500 and then allowed to proliferate for five days while incubated at 37°C. At five days, media was aspirated off and cells were rinsed with PBS. Cells were then fixed with crystal violet staining solution (0.5% crystal violet (w/v), 20% methanol) for 10 minutes in room temperature. The crystal violet staining solution was then removed and cells were washed 3x with PBS and allowed to dry overnight. Images were then taken of the stained plates. Other parameters of ER Stress induced cell death were measured through immunoblotting or with the Caspase 3/7 Glo Assay Kit (Promega) according to the manufacturer’s protocol. In brief, cells were plated at 10k/well in a white 96-well plate. The next day, cells were treated with the indicated drugs for 24 h (n=5 for each condition). After treatment, an equal volume of Caspase 3/7 substrate was added to the wells and allowed to incubate at room temperature for 1 h at 37°C and 5% CO2. Luminescence was then measured in an Infinite F200 PRO plate reader (Tecan) with an integration time of 1000 ms.
Mitochondrial Respiration
Mitochondrial respiration parameters were measured using a Mito Stress Test Kit and XF96 Extracellular Flux Analyzer (Seahorse Bioscience) according to the manufacturer’s protocol. In brief, 15,000 cells/well were plated on a 96-well plate on Cell-Tak (Corning) coated wells in their standard growing media and cultured overnight. The next day, cells were treated with drugs and incubated at 37°C, 5% CO2 for the indicated treatment time. Seahorse XF assay media (Agilent) was used to wash and remove the standard growing media from the cell plate and the sensor plate was prepped with standard Mito Stress Test drugs. After calibration of the sensor plate on the XF96 Extracellular Flux Analyzer, the cell plate was inserted into the machine and each parameter was calculated with 4 measurements separated by 5 min intervals following injections of drugs. Basal respiration measurement was followed by injections of oligomycin (2 μM), FCCP (0.5 μM) and Rotenone/Antimycin (1μM each), respectively. Individual respiratory parameters were then calculated according to the manufacturer’s protocol.
QUANTIFICATION AND STATISTICAL ANALYSIS.
Statistics were calculated in PRISM 9 (GraphPad, San Diego, CA). Data are presented as mean ± SEM and were analyzed by two-way ANOVA with Tukey’s multiple correction test, one-way ANOVA, or the appropriate Student’s t-tests, as indicated in the accompanying figure legends. Indications of nonsignificant interactions from ANOVA were generally omitted for clarity.
Supplementary Material
Figure S1. Pharmacologic activation of integrated stress response (ISR) kinases, related to Fig. 1.
Figure S2. Pharmacologic ISR activators restore ISR signaling in Perk-deficient MEFs, related to Fig. 2.
Figure S3. BtdCPU and halofuginone reestablish ER stress sensitivity in Perk-deficient MEFs, related to Fig. 3.
Figure S4. BtdCPU and halofuginone differentially impact mitochondrial morphology, related to Fig. 4.
Figure S5. BtdCPU promotes mitochondrial uncoupling, related to Fig. 5.
Figure S6. Halofuginone promotes adaptive remodeling of mitochondria respiration in PERK-deficient cells, related to Fig. 6.
Table S1. Pharmacologic ISR activators restore ISR signaling in Perk-deficient MEFs, related to Fig. 2 and Fig. S2.
Table S2. Pharmacologic ISR activators restore ISR signaling in Perk-deficient MEFs, related to Fig. 2 and Fig. S2.
Table S3. Pharmacologic ISR activators restore ISR signaling in Perk-deficient MEFs, , related to Fig. 2 and Fig. S2.
Table S4. Pharmacologic ISR activators restore ISR signaling in Perk-deficient MEFs, related to Fig. 2 and Fig. S2.
HIGHLIGHTS.
Halofuginone and BtdCPU activate the integrated stress response via GCN2 and HRI
Pharmacologic GCN2 or HRI activation rescues ER stress resilience in Perk−/− cells
The HRI activator BtdCPU depolarizes mitochondria to induce OMA1-DELE1-HRI signaling
Halofuginone restores mitochondrial adaptation in Perk-deficient cells
SIGNIFICANCE.
The integrated stress response (ISR) consists of four eIF2α kinases that are activated in response to diverse types of cellular stress. Activation of these ISR kinases induces downstream translational and transcriptional signaling that promotes adaptive remodeling of cellular physiology to help cells recover from an acute cellular stress. Deficiencies in the ISR kinase PERK leads to cellular and mitochondrial dysfunction and contributes to the pathogenesis of numerous diseases. Here, we use established chemical activators of the ISR to probe the potential for activating compensatory eIF2α kinases to mitigate cellular and mitochondrial dysfunction in Perk-deficient cells. We show that halofuginone and BtdCPU, compounds that activate the ISR kinases GCN2 and HRI, respectively, robustly induce ISR signaling and rescue ER stress resilience in cells deficient in Perk. However, we find that BtdCPU induces mitochondrial depolarization, leading to both mitochondrial fragmentation and activation of the OMA1-DELE1-HRI mitochondrial stress signaling pathway. In contrast, halofuginone-dependent ISR activation promotes adaptive remodeling of mitochondria in both wild-type and Perk-deficient cells, mimicking the mitochondrial adaptation regulated by PERK signaling. These results indicate that pharmacologic activation of compensatory ISR kinases using compounds like halpofuginone can mitigate cellular and mitochondrial dysfunction associated with diseases involved reduced PERK activity and motivate the development of highly selective ISR kinase activating compounds for the treatment of these disorders.
ACKNOWLEDGEMENTS.
We thank Evan Powers for critical reading of this manuscript. This work was funded by the National Institutes of Health (NS095892, NS125674 to RLW), a National Science Foundation predoctoral fellowship (VP), and the George E. Hewitt Postdoctoral fellowship (VD).
APPENDIX
INCLUSION AND DIVERSITY
One or more authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS.
We declare no conflicts related to the work described in this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Almeida LM, Pinho BR, Duchen MR, and Oliveira JMA (2022). The PERKs of mitochondria protection during stress: insights for PERK modulation in neurodegenerative and metabolic diseases. Biol Rev Camb Philos Soc 97, 1737–1748. 10.1111/brv.12860. [DOI] [PubMed] [Google Scholar]
- 2.Rainbolt TK, Saunders JM, and Wiseman RL (2014). Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol Metab 25, 528–537. 10.1016/j.tem.2014.06.007. [DOI] [PubMed] [Google Scholar]
- 3.Gardner BM, Pincus D, Gotthardt K, Gallagher CM, and Walter P. (2013). Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol 5, a013169. 10.1101/cshperspect.a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walter P, and Ron D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 5.Hetz C, Zhang K, and Kaufman RJ (2020). Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol 21, 421–438. 10.1038/s41580-020-0250-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wek RC, and Cavener DR (2007). Translational control and the unfolded protein response. Antioxid Redox Signal 9, 2357–2371. 10.1089/ars.2007.1764. [DOI] [PubMed] [Google Scholar]
- 7.Harding HP, Zhang Y, Bertolotti A, Zeng H, and Ron D. (2000). Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 5, 897–904. 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 8.Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, et al. (2013). ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15, 481–490. 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A, and Agostinis P. (2012). PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ 19, 1880–1891. 10.1038/cdd.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munoz JP, Ivanova S, Sanchez-Wandelmer J, Martinez-Cristobal P, Noguera E, Sancho A, Diaz-Ramos A, Hernandez-Alvarez MI, Sebastian D, Mauvezin C, et al. (2013). Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J 32, 2348–2361. 10.1038/emboj.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lebeau J, Saunders JM, Moraes VWR, Madhavan A, Madrazo N, Anthony MC, and Wiseman RL (2018). The PERK Arm of the Unfolded Protein Response Regulates Mitochondrial Morphology during Acute Endoplasmic Reticulum Stress. Cell Rep 22, 2827–2836. 10.1016/j.celrep.2018.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Latorre-Muro P, O’Malley KE, Bennett CF, Perry EA, Balsa E, Tavares CDJ, Jedrychowski M, Gygi SP, and Puigserver P. (2021). A cold-stress-inducible PERK/OGT axis controls TOM70-assisted mitochondrial protein import and cristae formation. Cell Metab 33, 598–614 e597. 10.1016/j.cmet.2021.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barad BA, Medina M, Fuentes D, Wiseman RL, and Grotjahn DA (2022). A surface morphometrics toolkit to quantify organellar membrane ultrastructure using cryo-electron tomography. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perea V, Cole C, Lebeau J, Dolina V, Baron KR, Madhavan A, Kelly JW, Grotjahn DA, and Wiseman RL (2023). PERK signaling promotes mitochondrial elongation by remodeling membrane phosphatidic acid. EMBO J 42, e113908. 10.15252/embj.2023113908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balsa E, Soustek MS, Thomas A, Cogliati S, Garcia-Poyatos C, Martin-Garcia E, Jedrychowski M, Gygi SP, Enriquez JA, and Puigserver P. (2019). ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes through the PERK-eIF2alpha Axis. Mol Cell 74, 877–890 e876. 10.1016/j.molcel.2019.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hori O, Ichinoda F, Tamatani T, Yamaguchi A, Sato N, Ozawa K, Kitao Y, Miyazaki M, Harding HP, Ron D, et al. (2002). Transmission of cell stress from endoplasmic reticulum to mitochondria: enhanced expression of Lon protease. J Cell Biol 157, 1151–1160. 10.1083/jcb.200108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11, 619–633. 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 18.Rainbolt TK, Atanassova N, Genereux JC, and Wiseman RL (2013). Stress-regulated translational attenuation adapts mitochondrial protein import through Tim17A degradation. Cell Metab 18, 908–919. 10.1016/j.cmet.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hetz C, and Papa FR (2018). The Unfolded Protein Response and Cell Fate Control. Mol Cell 69, 169–181. 10.1016/j.molcel.2017.06.017. [DOI] [PubMed] [Google Scholar]
- 20.Julier C, and Nicolino M. (2010). Wolcott-Rallison syndrome. Orphanet J Rare Dis 5, 29. 10.1186/1750-1172-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mann JP, Lenz D, Stamataki Z, and Kelly D. (2022). Common mechanisms in pediatric acute liver failure. Trends Mol Med. 10.1016/j.molmed.2022.11.006. [DOI] [PubMed] [Google Scholar]
- 22.Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, and Julier C. (2000). EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet 25, 406–409. 10.1038/78085. [DOI] [PubMed] [Google Scholar]
- 23.Yuan SH, Hiramatsu N, Liu Q, Sun XV, Lenh D, Chan P, Chiang K, Koo EH, Kao AW, Litvan I, and Lin JH (2018). Tauopathy-associated PERK alleles are functional hypomorphs that increase neuronal vulnerability to ER stress. Hum Mol Genet 27, 3951–3963. 10.1093/hmg/ddy297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park G, Xu K, Chea L, Kim K, Safarta L, Song KH, Wu J, Park S, Min H, Hiramatsu N, et al. (2022). Neurodegeneration risk factor, EIF2AK3 (PERK), influences tau protein aggregation. J Biol Chem 299, 102821. 10.1016/j.jbc.2022.102821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, Rademakers R, de Silva R, Litvan I, Riley DE, et al. (2011). Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet 43, 699–705. 10.1038/ng.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruch J, Xu H, Rosler TW, De Andrade A, Kuhn PH, Lichtenthaler SF, Arzberger T, Winklhofer KF, Muller U, and Hoglinger GU (2017). PERK activation mitigates tau pathology in vitro and in vivo. EMBO Mol Med 9, 371–384. 10.15252/emmm.201606664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ganz J, Shacham T, Kramer M, Shenkman M, Eiger H, Weinberg N, Iancovici O, Roy S, Simhaev L, Da’adoosh B, et al. (2020). A novel specific PERK activator reduces toxicity and extends survival in Huntington’s disease models. Sci Rep 10, 6875. 10.1038/s41598-020-63899-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shacham T, Patel C, and Lederkremer GZ (2021). PERK Pathway and Neurodegenerative Disease: To Inhibit or to Activate? Biomolecules 11. 10.3390/biom11030354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stockwell SR, Platt G, Barrie SE, Zoumpoulidou G, Te Poele RH, Aherne GW, Wilson SC, Sheldrake P, McDonald E, Venet M, et al. (2012). Mechanism-based screen for G1/S checkpoint activators identifies a selective activator of EIF2AK3/PERK signalling. PLoS One 7, e28568. 10.1371/journal.pone.0028568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grandjean JMD, and Wiseman RL (2020). Small molecule strategies to harness the unfolded protein response: where do we go from here? J Biol Chem 295, 15692–15711. 10.1074/jbc.REV120.010218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, and Gorman AM (2016). The integrated stress response. EMBO Rep 17, 1374–1395. 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Costa-Mattioli M, and Walter P. (2020). The integrated stress response: From mechanism to disease. Science 368. 10.1126/science.aat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wolzak K, Nolle A, Farina M, Abbink TE, van der Knaap MS, Verhage M, and Scheper W. (2022). Neuron-specific translational control shift ensures proteostatic resilience during ER stress. EMBO J 41, e110501. 10.15252/embj.2021110501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fessler E, Eckl EM, Schmitt S, Mancilla IA, Meyer-Bender MF, Hanf M, Philippou-Massier J, Krebs S, Zischka H, and Jae LT (2020). A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 579, 433–437. 10.1038/s41586-020-2076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin YT, Wiita AP, Xu K, Correia MA, and Kampmann M. (2020). Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 579, 427–432. 10.1038/s41586-020-2078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Q, Du R, Reis Monteiro Dos Santos GR, Yefidoff-Freedman R, Bohm A, Halperin J, Chorev M, and Aktas BH (2020). New activators of eIF2alpha Kinase Heme-Regulated Inhibitor (HRI) with improved biophysical properties. Eur J Med Chem 187, 111973. 10.1016/j.ejmech.2019.111973. [DOI] [PubMed] [Google Scholar]
- 37.Chen T, Ozel D, Qiao Y, Harbinski F, Chen L, Denoyelle S, He X, Zvereva N, Supko JG, Chorev M, et al. (2011). Chemical genetics identify eIF2alpha kinase heme-regulated inhibitor as an anticancer target. Nat Chem Biol 7, 610–616. 10.1038/nchembio.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keller TL, Zocco D, Sundrud MS, Hendrick M, Edenius M, Yum J, Kim YJ, Lee HK, Cortese JF, Wirth DF, et al. (2012). Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat Chem Biol 8, 311–317. 10.1038/nchembio.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang CP, Clark O, Ferrarone JR, Campos C, Lalani AS, Chodera JD, Intlekofer AM, Elemento O, and Mellinghoff IK (2022). GCN2 kinase activation by ATP-competitive kinase inhibitors. Nat Chem Biol 18, 207–215. 10.1038/s41589-021-00947-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carlson KR, Georgiadis MM, Tameire F, Staschke KA, and Wek RC (2023). Activation of Gcn2 by small molecules designed to be inhibitors. J Biol Chem 299, 104595. 10.1016/j.jbc.2023.104595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szaruga M, Janssen DA, de Miguel C, Hodgson G, Fatalska A, Pitera AP, Andreeva A, and Bertolotti A. (2023). Activation of the integrated stress response by inhibitors of its kinases. Nat Commun 14, 5535. 10.1038/s41467-023-40823-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Juarez P, Mohammad KS, Yin JJ, Fournier PG, McKenna RC, Davis HW, Peng XH, Niewolna M, Javelaud D, Chirgwin JM, et al. (2012). Halofuginone inhibits the establishment and progression of melanoma bone metastases. Cancer Res 72, 6247–6256. 10.1158/0008-5472.CAN-12-1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tian X, Zhang S, Zhou L, Seyhan AA, Hernandez Borrero L, Zhang Y, and El-Deiry WS (2021). Targeting the Integrated Stress Response in Cancer Therapy. Front Pharmacol 12, 747837. 10.3389/fphar.2021.747837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ishii H, Choudhuri R, Mathias A, Sowers AL, Flanders KC, Cook JA, and Mitchell JB (2009). Halofuginone mediated protection against radiation-induced leg contracture. Int J Oncol 35, 315–319. [PMC free article] [PubMed] [Google Scholar]
- 45.Yang J, Baron KR, Pride DE, Schneemann A, Guo X, Chen W, Song AS, Aviles G, Kampmann M, Wiseman RL, and Lander GC (2022). DELE1 oligomerization promotes integrated stress response activation. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, and Kaufman RJ (2001). Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 7, 1165–1176. 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- 47.Grandjean JMD, Plate L, Morimoto RI, Bollong MJ, Powers ET, and Wiseman RL (2019). Deconvoluting Stress-Responsive Proteostasis Signaling Pathways for Pharmacologic Activation Using Targeted RNA Sequencing. ACS Chem Biol 14, 784–795. 10.1021/acschembio.9b00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KK, Wilson C, et al. (2013). Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2, e00498. 10.7554/eLife.00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zyryanova AF, Weis F, Faille A, Alard AA, Crespillo-Casado A, Sekine Y, Harding HP, Allen F, Parts L, Fromont C, et al. (2018). Binding of ISRIB reveals a regulatory site in the nucleotide exchange factor eIF2B. Science 359, 1533–1536. 10.1126/science.aar5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsai JC, Miller-Vedam LE, Anand AA, Jaishankar P, Nguyen HC, Renslo AR, Frost A, and Walter P. (2018). Structure of the nucleotide exchange factor eIF2B reveals mechanism of memory-enhancing molecule. Science 359. 10.1126/science.aaq0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schoof M, Boone M, Wang L, Lawrence R, Frost A, and Walter P. (2021). eIF2B conformation and assembly state regulate the integrated stress response. Elife 10. 10.7554/eLife.65703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pitera AP, Szaruga M, Peak-Chew SY, Wingett SW, and Bertolotti A. (2022). Cellular responses to halofuginone reveal a vulnerability of the GCN2 branch of the integrated stress response. EMBO J 41, e109985. 10.15252/embj.2021109985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, Da Cruz S, Clerc P, Raschke I, Merkwirth C, et al. (2009). SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J 28, 1589–1600. 10.1038/emboj.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fessler E, Krumwiede L, and Jae LT (2022). DELE1 tracks perturbed protein import and processing in human mitochondria. Nat Commun 13, 1853. 10.1038/s41467-022-29479-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang K, Li H, and Song Z. (2014). Membrane depolarization activates the mitochondrial protease OMA1 by stimulating self-cleavage. EMBO Rep 15, 576–585. 10.1002/embr.201338240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.MacVicar T, and Langer T. (2016). OPA1 processing in cell death and disease - the long and short of it. J Cell Sci 129, 2297–2306. 10.1242/jcs.159186. [DOI] [PubMed] [Google Scholar]
- 57.Gomes LC, and Scorrano L. (2011). Mitochondrial elongation during autophagy: a stereotypical response to survive in difficult times. Autophagy 7, 1251–1253. 10.4161/auto.7.10.16771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yao CH, Wang R, Wang Y, Kung CP, Weber JD, and Patti GJ (2019). Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation. Elife 8. 10.7554/eLife.41351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kato H, Okabe K, Miyake M, Hattori K, Fukaya T, Tanimoto K, Beini S, Mizuguchi M, Torii S, Arakawa S, et al. (2020). ER-resident sensor PERK is essential for mitochondrial thermogenesis in brown adipose tissue. Life Sci Alliance 3. 10.26508/lsa.201900576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grenier A, Poulain L, Mondesir J, Jacquel A, Bosc C, Stuani L, Mouche S, Larrue C, Sahal A, Birsen R, et al. (2022). AMPK-PERK axis represses oxidative metabolism and enhances apoptotic priming of mitochondria in acute myeloid leukemia. Cell Rep 38, 110197. 10.1016/j.celrep.2021.110197. [DOI] [PubMed] [Google Scholar]
- 61.Bassot A, Chen J, Takahashi-Yamashiro K, Yap MC, Gibhardt CS, Le GNT, Hario S, Nasu Y, Moore J, Gutierrez T, et al. (2023). The endoplasmic reticulum kinase PERK interacts with the oxidoreductase ERO1 to metabolically adapt mitochondria. Cell Rep 42, 111899. 10.1016/j.celrep.2022.111899. [DOI] [PubMed] [Google Scholar]
- 62.Zhang G, Wang X, Rothermel BA, Lavandero S, and Wang ZV (2022). The integrated stress response in ischemic diseases. Cell Death Differ 29, 750–757. 10.1038/s41418-021-00889-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rosarda JD, Baron KR, Nutsch K, Kline GM, Stanton C, Kelly JW, Bollong MJ, and Wiseman RL (2021). Metabolically Activated Proteostasis Regulators Protect against Glutamate Toxicity by Activating NRF2. ACS Chem Biol 16, 2852–2863. 10.1021/acschembio.1c00810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hughes D, and Mallucci GR (2019). The unfolded protein response in neurodegenerative disorders - therapeutic modulation of the PERK pathway. FEBS J 286, 342–355. 10.1111/febs.14422. [DOI] [PubMed] [Google Scholar]
- 65.Stutzbach LD, Xie SX, Naj AC, Albin R, Gilman S, Group PSPGS, Lee VM, Trojanowski JQ, Devlin B, and Schellenberg GD (2013). The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol Commun 1, 31. 10.1186/2051-5960-1-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yoon L, Botham RC, Verhelle A, Sanz-Martinez P, Xu J, Cole CM, Tan EP, Chou CC, Cuoco CA, Massey LA, et al. (2022). mTOR inhibitor-independent Autophagy Activator Ameliorates Tauopathy and Prionopathy Neurodegeneration Phenotypes. bioRxiv. [Google Scholar]
- 67.Burwick N, Zhang MY, de la Puente P, Azab AK, Hyun TS, Ruiz-Gutierrez M, Sanchez-Bonilla M, Nakamura T, Delrow JJ, MacKay VL, and Shimamura A. (2017). The eIF2-alpha kinase HRI is a novel therapeutic target in multiple myeloma. Leuk Res 55, 23–32. 10.1016/j.leukres.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Smith KH, Budhraja A, Lynch J, Roberts K, Panetta JC, Connelly JP, Turnis ME, Pruett-Miller SM, Schuetz JD, Mullighan CG, and Opferman JT (2021). The Heme-Regulated Inhibitor Pathway Modulates Susceptibility of Poor Prognosis B-Lineage Acute Leukemia to BH3-Mimetics. Mol Cancer Res 19, 636–650. 10.1158/1541-7786.MCR-20-0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pines M, and Spector I. (2015). Halofuginone - the multifaceted molecule. Molecules 20, 573–594. 10.3390/molecules20010573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Madhavan A, Kok BP, Rius B, Grandjean JMD, Alabi A, Albert V, Sukiasyan A, Powers ET, Galmozzi A, Saez E, and Wiseman RL (2022). Pharmacologic IRE1/XBP1s activation promotes systemic adaptive remodeling in obesity. Nat Commun 13, 608. 10.1038/s41467-022-28271-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang D, Wang J, Bonamy GM, Meeusen S, Brusch RG, Turk C, Yang P, and Schultz PG (2012). A small molecule promotes mitochondrial fusion in mammalian cells. Angew Chem Int Ed Engl 51, 9302–9305. 10.1002/anie.201204589. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Pharmacologic activation of integrated stress response (ISR) kinases, related to Fig. 1.
Figure S2. Pharmacologic ISR activators restore ISR signaling in Perk-deficient MEFs, related to Fig. 2.
Figure S3. BtdCPU and halofuginone reestablish ER stress sensitivity in Perk-deficient MEFs, related to Fig. 3.
Figure S4. BtdCPU and halofuginone differentially impact mitochondrial morphology, related to Fig. 4.
Figure S5. BtdCPU promotes mitochondrial uncoupling, related to Fig. 5.
Figure S6. Halofuginone promotes adaptive remodeling of mitochondria respiration in PERK-deficient cells, related to Fig. 6.
Table S1. Pharmacologic ISR activators restore ISR signaling in Perk-deficient MEFs, related to Fig. 2 and Fig. S2.
Table S2. Pharmacologic ISR activators restore ISR signaling in Perk-deficient MEFs, related to Fig. 2 and Fig. S2.
Table S3. Pharmacologic ISR activators restore ISR signaling in Perk-deficient MEFs, , related to Fig. 2 and Fig. S2.
Table S4. Pharmacologic ISR activators restore ISR signaling in Perk-deficient MEFs, related to Fig. 2 and Fig. S2.
Data Availability Statement
RNAseq data has been deposited at GEO and is publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-ATF4 antibody (D4B8) | Cell Signaling | Cat # 11815 |
| Mouse anti-tubulin antibody [B-5–1-2] | Sigma | Cat # T6704 |
| Mouse anti-HSP60 antibody (LK1) | ThermoFisher | Cat # MA5–45109 |
| Rabbit anti-PERK (C33E10) | Cell Signaling | Cat # 3192 |
| Mouse anti-HA [Clone: 16B12] | Biolegend | Cat # 901501 |
| Mouse anti-GFP (B2) | Santa Cruz | Cat # Sc-9996 |
| Mouse anti-OPA1 | BD Bioscience | Cat # 612606 |
| Rabbit anti-OMA1 | Cell Signaling | Cat # 95473 |
| Rabbit anti-cleaved Caspase 3 (D175) | Cell Signaling | Cat # 96611 |
| Rabbit anti-PARP | Cell Signaling | Cat # 9542 |
| Mouse anti-GAPDH | Genetex | Cat # GTX627408 |
| Mouse anti-beta-actin | Cell Signaling | Cat # 3700 |
| Chemicals, peptides, and recombinant proteins | ||
| Thapsigargin | Fisher Scientifics | Cat # 50–464-295 |
| ISRIB | Sigma | Cat # SML0843 |
| CCCP | Sigma | Cat # C2759 |
| BtdCPU | Fisher | Cat # 32489210MG |
| Halofuginone | Sigma | Cat # 50–576-300001 |
| Erlotinib | Selleckchem | Cat # S7786 |
| Sunitinib | Selleckchem | Cat # S7781 |
| MK28 | MedChemExpress | Cat # HY-137207 |
| CCT020312 | MedChemExpress | Cat # HY-119240 |
| Staurosporine | Fisher | Cat # NC1407148 |
| MEF Avalanche Transfection Reagent | EZ Biosystems | Cat # EZT-MEFS-1 |
| Critical commercial assays | ||
| Quick RNA mini-prep kit | Zymo Research | Cat # R1055 |
| High Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat # 4368702 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | Cat # 4367659 |
| Caspase 3/7 Glo Assay Kit | Promega | Cat # G8090 |
| Data Deposited | ||
| RNAseq data generated from Perk+/+ and Perk−/− treated with thapsigargin, halofuginone, and/or BtdCPU were deposited to the Gene Expression Omnibus (GEO). | This manuscript. | GEO: GSE227134 |
| Experimental models: Cell lines | ||
| Perk+/+ and Perk−/− MEFs | Harding et al (2000) Mol Cell7 | David Ron’s lab |
| Atf4+/+ and Atf4−/− MEFs | Harding et al (2003) Mol Cell17 | Established by David Ron’s lab; Provided by Jonathan Lin’s lab |
| MEFA/A MEFs | Scheuner et al (2001) Mol Cell46 | Randal Kaufman’s lab |
| HEK293T cells | ATCC | CRL-3216 |
| OMA1-deficient HEK293T cells | Guo et al (2020) Nature35 | Martin Kampmann’s lab |
| DELE1-deficient HEK293T cells | Guo et al (2020) Nature35 |
Martin Kampmann’s lab |
| HRI-deficient HEK293T cells | Guo et al (2020) Nature35 | Martin Kampmann’s lab |
| HEK293T cells stably expressing ATF4-FLuc | Yang et al (2023) Nat Struct Mol Biol45 | Martin Kampmann’s lab |
| HEK293T cells stably expressing ATF4-mAPPLE and CRISPRi-depleted of specific ISR kinases | Guo et al (2020) Nature35 | Martin Kampmann’s lab |
| MEFmtGFP cells | Wang et al (2012) Angew Chem Int Ed Engl71 |
Peter Schultz’s lab |
| SHSY5Y cells | ATCC | Cat # CRL-2266 |
| Oligonucleotides | ||
|
M. musculus Chop (forward qPCR primer):
CCTAGCTTGGCTGACAGAGG |
Integrated DNA Technologies | N/A |
|
M. musculus Chop (reverse qPCR primer):
CTGCTCCTTCTCCTTCATGC |
Integrated DNA Technologies | N/A |
|
M. musculus Chac1 (forward qPCR primer):
TGACCCTCCTTGAAGACCGTGA |
Integrated DNA Technologies | N/A |
|
M. musculus Chac1 (reverse qPCR primer):
AGTGTCATAGCCACCAAGCACG |
Integrated DNA Technologies | N/A |
|
M. musculus RiboP (forward qPCR primer):
TGTCATCGCTCAGGGTGTTG |
Integrated DNA Technologies | N/A |
|
M. musculus RiboP (reverse qPCR primer):
AAGCCAAATCCCATGTCGTC |
Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| hsPerkWT.pcDNA3.1 | Yuan et al (2018) Hum Mol Gen23 | Jonathan Lin’s lab |
| mtGFP.pDEST40 | Lebeau et al (2018) Cell
Reports11 |
Luke Wiseman’s lab |
| DELE1L-mCLOVER plasmid | Guo et al (2020) Nature35 | Martin Kampmann’s lab |
| Software and algorithms | ||
| FlowJo™ Software | BD Biosciences | N/A |
| PRISM 10.0.2 | Graphpad | N/A |
| LICOR Imaging Studio | Licor Biosciences | N/A |
| DNAstar Lasergene SeqManPro | DNAStar Inc. | N/A |
| ArrayStar 12.2 with Qseq | DNAStar Inc. | N/A |
| DESeq 2 v. 1.34 | Bioconductor | N/A |
| BiomaRt v. 2.50.3 | Bioconductor | N/A |
| Other | ||
| Infinite F200 Pro Plate Reader | Tecan | N/A |
| Bio-Rad ZE5 Cell Analyzer | Bio-Rad | N/A |
| Olympus IX71 microscope with 60x oil objective | Olympus | N/A |
| Hamamatsu C8484 camera | Hamamatsu Photonics | N/A |
| Zeiss LSM 880 Airyscan confocal laser scanning microscope | Zeiss | N/A |
| Odyssey Infrared Imaging System | Licor Biosciences | N/A |
| ABI 7900HT Fast Real Time PCR machine | Applied Biosciences | N/A |
| XF96 Extracellular Flux Analyzer | Seahorse Biosciences | N/A |