

Abstract
The mRNA transcript of the human STMN2 gene, encoding for stathmin-2 protein (also called SCG10), is profoundly impacted by TDP-43 loss-of-function. The latter is a hallmark of several neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS). Using a combination of approaches, including transient antisense oligonucleotide (ASO)-mediated suppression, sustained shRNA-induced depletion in aging mice, and germline deletion, we show that stathmin-2 plays an important role in the establishment and maintenance of neurofilament-dependent axoplasmic organization that is critical for preserving the caliber and conduction velocity of myelinated large-diameter axons. Persistent stathmin-2 loss in adult mice results in pathologies found in ALS, including reduced inter-neurofilament spacing, axonal caliber collapse that drives tearing within outer myelin layers, diminished conduction velocity, progressive motor and sensory deficits, and muscle denervation. These findings reinforce restoration of stathmin-2 as an attractive therapeutic approach for ALS and other TDP-43-dependent neurodegenerative diseases.
Introduction
Amyotrophic lateral sclerosis (ALS) is characterized by selective degeneration of upper and lower motor neurons that results in muscle denervation, paralysis, and eventually death typically from respiratory failure within 2–5 years after diagnosis1. The loss of neuromuscular junctions (NMJs) is well established as one of the earliest pathological events in both familial and sporadic forms of ALS2–4. NMJs are vital synapses formed between motor neuron terminals and muscle cells, and their disruption occurs prior to disease onset and motor neuron degeneration in ALS3–5. Importantly, efforts focusing on the maintenance of motor neuron survival have failed to prevent muscle denervation or delay disease onset and progression in mice expressing ALS-causing mutations6–8, suggesting that pathological mechanisms involved in muscle denervation occur independently from the death of the motor neuron.
While initial axonal elongation of lower motor neurons can proceed independently of neurofilaments9, the formation of synapses at NMJs triggers enhanced synthesis and accumulation of neurofilaments10,11, which mediate a >10-fold growth in axonal diameter (and >100-fold increase in axonal volume)10,12. Neurofilaments become the most abundant structural element within mature, myelinated axons, outnumbering the microtubules by a factor of at least 3013–15. Importantly, neurofilament pathologies including intracellular inclusions containing neurofilaments have been reported in many neurodegenerative disorders, including ALS16–19. Transgenic mice expressing mutated20 or altered ratios of neurofilament subunits21 develop age-dependent motor neuron disease resembling aspects of human ALS20,22, further emphasizing the importance of neurofilaments in maintaining axonal function. Neurofilament sequence variants have been proposed as contributory risk factors23,24, although mutations in genes encoding neurofilament subunits have not been found as causative of ALS25.
The discovery of cytoplasmic mislocalization of the RNA binding protein TDP-43 in affected neurons in >95% of ALS and at least 50% of frontotemporal dementia (FTD) profoundly changed the direction of research in ALS and FTD26,27. TDP-43 is known to affect the levels or splicing patterns of mRNAs from more than 1500 genes28,29. Aside from TDP-43 aggregation, nuclear clearance of TDP-43 is observed in affected neurons in sporadic ALS and FTD27 and in the most frequent genetic cause of ALS and FTD, a GGGGCC repeat expansion in the C9orf72 gene30–32. Overwhelming evidence supports that TDP-43 loss of function (and by extension, errors of pre-RNA maturation) is a key aspect of disease mechanism underlying ALS and FTD.
Recently, we and others identified that the stathmin-2 mRNA (encoded by the STMN2 gene) is the transcript most affected by reduction in TDP-43 function33,34. TDP-43 suppression in human cells drives use of cryptic splice and polyadenylation sites in the STMN2 pre-mRNA, producing a truncated, non-functional mRNA that includes a new exon 2a that accumulates in brain and spinal cord of patients with TDP-43 pathology33–35. Importantly, the corresponding loss of stathmin-2 protein (also known as SCG10) results in an impaired ability of human motor neurons to recover after axonal injury33,34. Stathmin-2 mRNAs are among the top 25 most abundant transcripts in human33,36 and mouse37 motor neurons. Notably, TDP-43 dependent regulation of STMN2 pre-mRNAs is not conserved in rodents, as the three GU-rich TDP-43 binding motifs and the alternative polyadenylation site in intron 1 of the human STMN2 gene are not present in the corresponding murine Stmn2 pre-mRNA33. Correspondingly, TDP-43 nuclear clearance or impaired function does not drive loss of stathmin-2 in mice.
Stathmin-2 has been proposed as an important axonal-maintenance factor38 and an essential protein for motor axon regeneration33. Indeed, after axotomy of sensory axons, stathmin-2 synthesis is upregulated, with the protein accumulating in growth cones39. Chronic elimination of stathmin-2 starting from earliest development has been reported to yield mild sensory and motor deficits in young adult mice40,41. Despite an extensive literature on axonal development42 and impaired axonal regeneration in injured sensory neurons38,39,43,44, it remains unknown whether, and if so how, stathmin-2 contributes to maintenance of motor and sensory axons during aging. Here, we determine that sustained stathmin-2 loss from an otherwise mature adult nervous system provokes shrinkage of neurofilament spacing that defines axonal caliber, axonal collapse with tearing of outer myelin layers, progressive motor and sensory deficits, reduced conduction velocity, and muscle denervation. Remarkably, these pathological alterations derived from selectively suppressing stathmin-2 phenocopy disease events occurring in TDP-43 proteinopathies, including sporadic ALS.
Results
Denervation and slowed conduction after Stmn2 reduction
Evaluation of accumulated stathmin-2 RNA (determined by in situ hybridization – Extended Data Fig. 1a) and protein (determined by immunofluorescence – Extended Data Fig. 1b) within the normal adult nervous system revealed stathmin-2 expression in the soma of virtually every spinal neuron, with the highest accumulation in α-motor neurons followed by interneurons and γ-motor neurons. Stathmin-2 protein also accumulated at the presynaptic side of mature NMJs of gastrocnemius muscle from wild type (WT) mice (Extended Data Fig. 1c) and in the outer laminae of the dorsal horn that contains sensory neuron terminals projecting from the adjacent dorsal root ganglion (DRG) (Extended Data Fig. 1b), evidence suggesting roles for stathmin-2 within mature presynaptic motor and sensory neuronal synapses.
Intracerebroventricular (ICV) delivery of a Stmn2 mRNA-targeting antisense oligonucleotide (ASO) was used to test the consequence(s) of transient suppression of stathmin-2 synthesis in an otherwise normal adult murine nervous system (Fig. 1a). As early as 2 weeks after ASO injection, using either of two different Stmn2-targeting ASO variants, into 3-month-old adult mice, Stmn2 mRNA and protein were suppressed throughout the central nervous system, including cortex (Extended Data Fig. 1d,e) and spinal cord (Fig. 1b, Extended Data Fig. 1f). Suppression of Stmn2 RNAs and protein were sustained from 2 to 8 weeks post injection in spinal cord (Fig. 1c, Extended Data Fig. 1g) and cortex (Extended Data Fig. 1h,i). Although no significant clinical manifestations developed within 8 weeks of lowering stathmin-2, nerve conduction velocity (NCV, the speed at which electrical signals travel along axons) was significantly reduced within the sciatic nerve (Fig. 1d) without effect on compound muscle action potential (CMAP) (Extended Data Fig. 1j). Importantly, reduction in stathmin-2 for 8 weeks led to compromised integrity of neuromuscular synapses (Fig. 1e), including full denervation of 18% of NMJs and partial denervation of another 29% (Fig. 1f).
Figure 1: ASO-mediated transient stathmin-2 suppression reduces nerve conduction velocity and triggers muscle denervation.
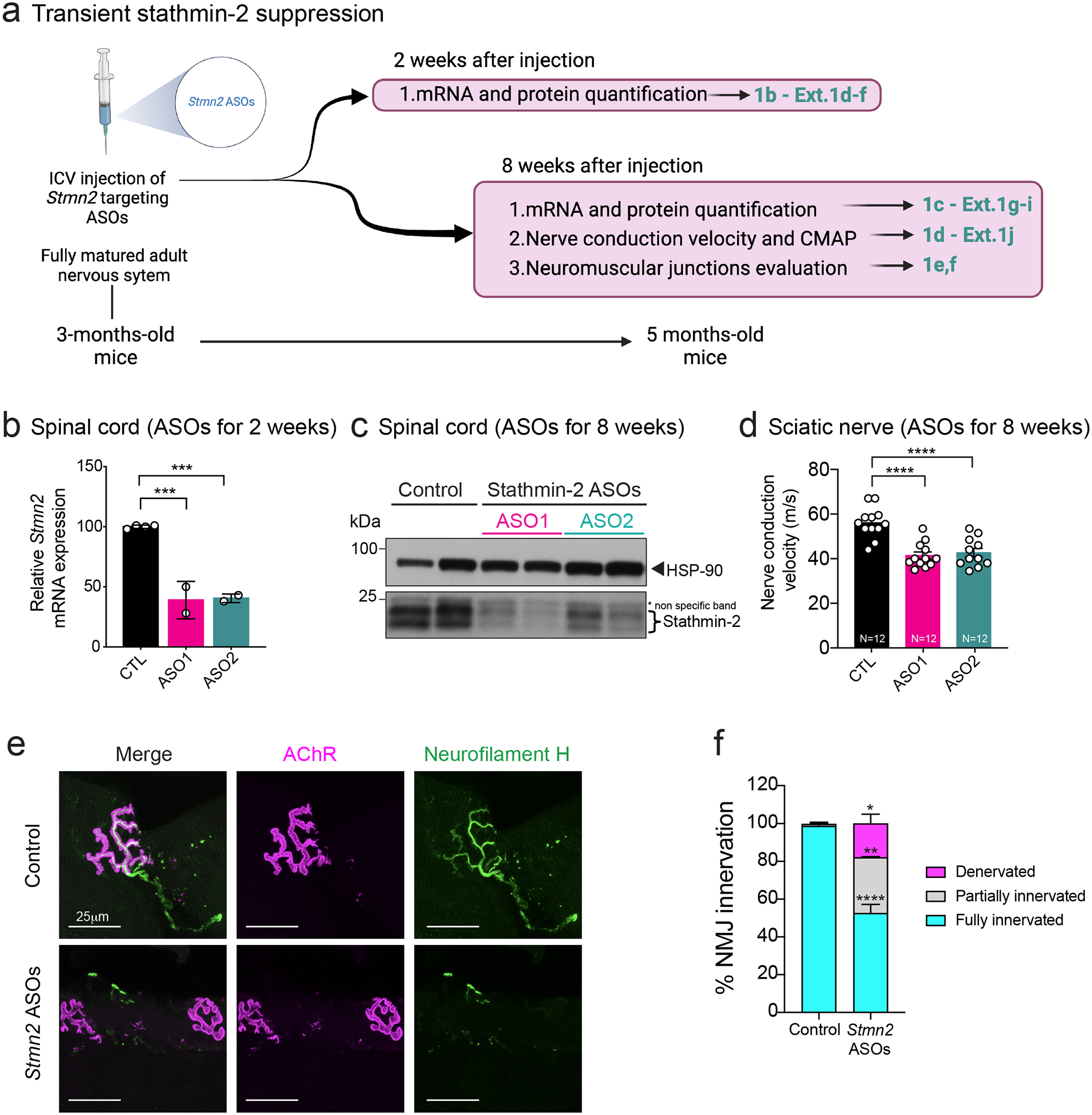
(a) Schematic representation of intraventricular (ICV) administration of control or Stmn2 targeting ASOs (ASO1 and ASO2) in 3-month-old wild-type mice. Figure created using Biorender. (b) Quantification of Stmn2 mRNA levels by qRT-PCR 2 weeks after ICV injection. Statistical analysis by two-sided, one-way ANOVA with Dunnett’s multiple comparison test. P = 0.0004 for both pairs. (c) Immunoblot showing stathmin-2 protein levels in mice spinal cord extracts 8 weeks after the ICV injection. Heat shock protein 90 (HSP-90) was used as a loading control. Each lane was loaded with protein extracted from a different animal. (d) Nerve conduction velocity measurement in mice hindlimbs 8 weeks after ICV injection of ASOs. Statistical analysis by two-sided, one-way ANOVA Tukey’s multiple comparisons test. N=12 animals per condition. P < 0.0001 for both pairs. (e-f) Representative confocal images (e) and innervation rate quantification (f) of neuromuscular junctions (NMJs) in the gastrocnemius muscle 8 weeks after delivery of non-targeting or Stmn2 targeting ASOs. At least n=3 animals and 250 NMJs were analyzed per condition. Statistical analysis by two-sided, two-way ANOVA followed by Sidak’s multiple comparisons test. Fully innervated P <0.0001; partially innervated P = 0.0012; denervated P = 0.0144. All panels: Data points represent individual mice. Error bars plotted as SEM. ****, p <0.0001; ***, p < 0.001; **, p < 0.01; *, p <0.05; ns, p>0.05.
Progressive motor deficits from chronic Stmn2 suppression
The consequence(s) of sustained stathmin-2 loss, thereby mimicking its reduction in ALS, was determined using adeno-associated virus (AAV) to chronically express a microRNA-embedded shRNA45,46 to suppress murine Stmn2 RNAs in motor and sensory neurons of an otherwise normal adult nervous system (Fig. 2a). A reporter gene encoding mClover (a brighter monomeric GFP derivative) was used to mark transduced neurons, their axons, and their terminals innervating hindlimb muscles (Extended Data Fig. 2a). Viral vectors encoding shRNA against murine Stmn2 or an irrelevant, control gene (bacterial β-galactosidase) were introduced into the lumbar spinal cord of 1-year-old WT mice via a single subpial delivery (i.e., injection just beneath the inner-most pia layer46–50). Unlike intrathecal administration, subpial injection in adult animals enables highly effective and long-lasting transduction to deep gray matter neurons and glia of the spinal cord46–50 (Extended Data Fig. 2b). Additionally, local axonal or synaptic uptake followed by retrograde AAV delivery also transduces cortical motor neurons and sensory neurons of the DRGs. Importantly, lumbar subpial administration achieves efficient focal transduction without targeting cervical or thoracic spinal motor neurons that control forelimbs and respiratory muscles46.
Figure 2: Focal, chronic, and selective suppression of stathmin-2 by subpial delivery of AAV9 shStmn2 in lumbar spinal cord results in motor deficits.
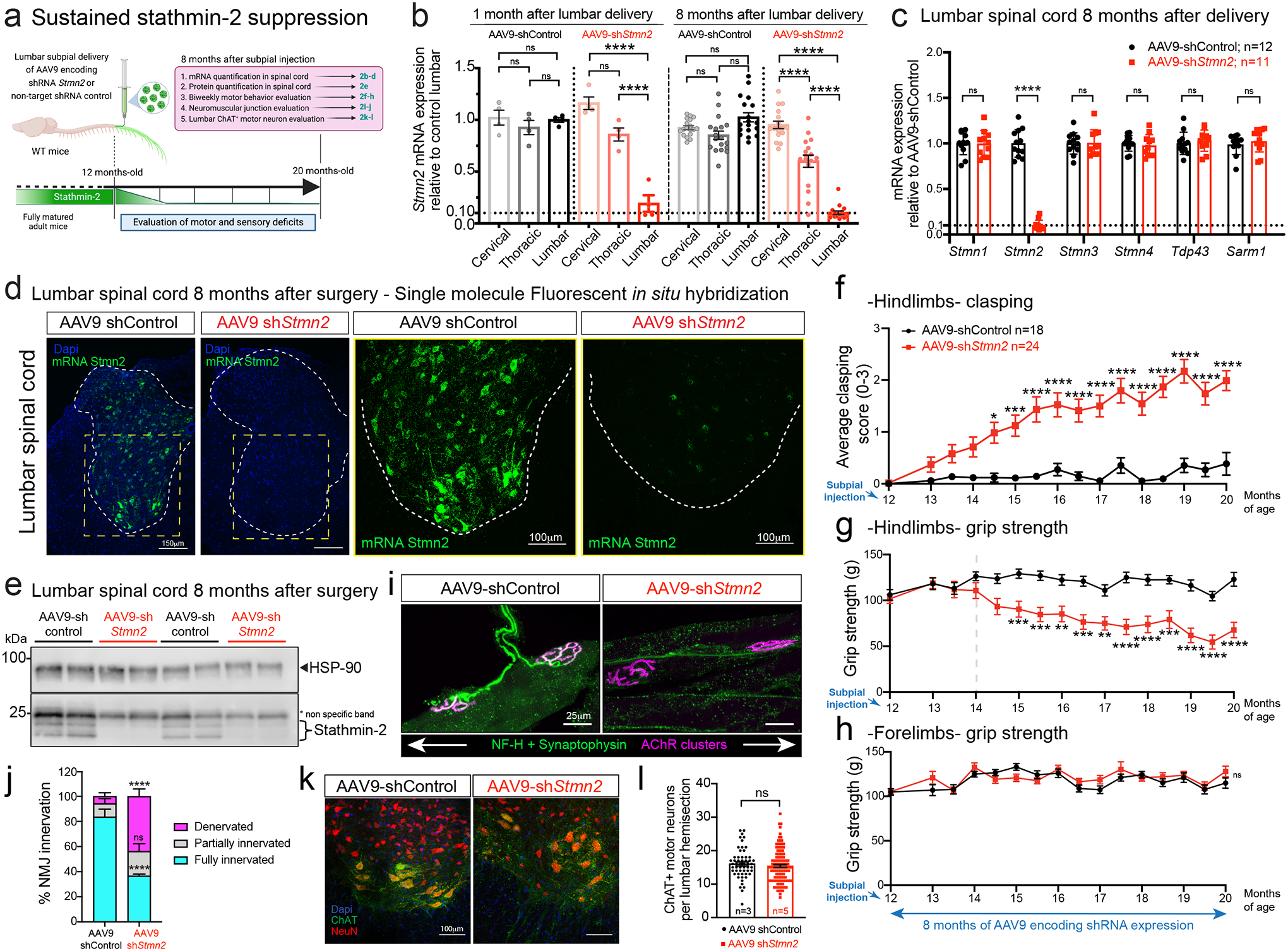
(a) Schematic of experiments evaluating sustained stathmin-2 depletion by AAV9-delivered shRNA subpially delivered into 12-month-old adult wildtype mice. Figure created using Biorender. (b) Measurement of Stmn2 mRNA expression by qRT-PCR in mice after subpial delivery of AAV9-shControl or AAV9-shStmn2 across cervical, thoracic, and lumbar segments at 1- and 8-months post-administration. Statistics by two-sided, one-way ANOVA with Tukey’s multiple comparisons correction. N=4 animals/condition at 1 month. N=18 animals with AAV9-shControl and n=17 animals with AAV9-shStmn2 at 8 months. P < 0.0001. (c) Measurement of mRNA expression for a panel of genes by qRT-PCR in lumbar spinal cord 8 months post-injection of AAV9-shRNAs. Statistics by two-sided, unpaired t-tests. P < 0.0001. (b,c) Gapdh used as an endogenous control gene. Each data point represents an individual mouse. (d) Stmn2 mRNA levels detected by single molecule FISH in mice lumbar spinal cord. (e) Immunoblots of stathmin-2 protein in mice lumbar spinal cord 8 months post-administration of AAV9-shControl (n= 4 animals) or Stmn2 shRNA (n=4 animals). HSP-90 was used as a loading control. *Indicates non-specific band. Each lane was loaded with a different animal. (f-h) Longitudinal analysis of hindlimb clasping (f), grip strength (g), and forelimb grip strength (h) of AAV9-shControl (n=18) or AAV9-shStmn2 (n=24); Statistics by two-sided, two-way ANOVA and Sidak’s multiple comparisons test. p values range from * P = 0.0141 to P < 0.0001. (i) Representative confocal images of gastrocnemius muscle immunostained with synaptophysin and neurofilament-H antibodies for axon terminals (green) and α-bungarotoxin for muscle endplates (magenta), 8 months post-injection of AAV9-shControl or AAV9-shStmn2. (j) Quantification of the neuromuscular innervation status. Statistics by two-sided, two-way ANOVA and Sidak’s multiple comparisons test. N=5 animals/condition. P <0.0001. (k,l) Representative images (k) and quantification (l) of ChAT positive motor neurons at the lumbar spinal cord of mice 8 months post-administration of AAV9-shControl (n=3 animals) or AAV9-shStmn2 (n=5 animals). Statistics by Mann-Whitney’s test. All panels: Error bars plotted as SEM. ****, p <0.0001; ***, p < 0.001; **, p < 0.01; *, p <0.05; ns, p>0.05.
Analysis of RNAs extracted after subpial administration of AAV9-shStmn2 revealed rapid and sustained reduction (80% at 1 month; 90% at 8 months) of Stmn2 mRNA selectively within lumbar spinal cord, with only mild or no reduction, respectively, in the thoracic and cervical segments (Fig. 2b). There were no compensatory changes in RNAs encoding the other three stathmin genes (stathmin-1, stathmin-3, and stathmin-4) or other potentially ALS-related genes including TDP-43 and SARM1 or genes whose mRNAs have similar nucleotide sequences that could potentially have been targeted by the shRNA (Fig. 2c, Extended Data Fig. 2c). Sustained suppression of Stmn2 RNAs and protein for 8 months was confirmed with single molecule fluorescence in situ hybridization (FISH) (Fig. 2d), immunoblotting (Fig. 2e, Extended Data Fig. 2d), and immunostaining (Extended Data Fig. 2e). Although body weight over an 8-month course remained unaffected (Extended Data Fig. 2f), within 2 months of reduced stathmin-2 synthesis, motor performance was impaired, including induction of hindlimb clasping (Fig. 2f) and reduced hindlimb grip strength (Fig. 2g), accompanied by loss of muscle volume as observed in the gastrocnemius muscle (Extended Data Fig. 2g). Biweekly analyses revealed that progressive lumbar motor deficiencies developed up to 20 months of age (Fig. 2f,g) without effect(s) on cervical spinal cord motor function (Fig. 2h).
Sustained stathmin-2 suppression resulted in disruption of more than half of the NMJs of lumbar motor neurons, with 43% of total (70% of affected) NMJs fully denervated after 8 months of stathmin-2 reduction (Fig. 2i,j). Despite this, there was no reduction in the number of lumbar ChAT+ motor neurons (Figures 2k,l) and no sign of neuroinflammation measured by changes in number or morphologies of astrocytes or microglia in the lumbar spinal cord (Extended Data Fig. 2h–j). Thus, sustained loss of stathmin-2 in motor neurons of an otherwise fully mature, adult nervous system drives muscle denervation without motor neuron perikaryon loss.
Collapse of motor axons in ALS and in mice upon Stmn2 loss
Continued suppression of stathmin-2 synthesis within lumbar spinal motor neurons of otherwise normal adult WT mice yielded a 35% reduction in conduction velocity of the sciatic nerve (Fig. 3a), similar to the conduction velocity reduction reported in ALS patients51–53. Conduction velocity is thought to be determined by two primary factors: the degree of myelination and axonal diameter, with bigger diameters driving faster conduction velocity54,55. Recognizing this, measurement of axonal diameters following 8-month suppression of stathmin-2 synthesis revealed a significant shrinkage in cross sectional area of the axons of the α-motor neurons (with diameters > 3μm) responsible for muscle contraction and a corresponding shrinkage in γ-motor axons (with diameters < 3μm). The average α-motor axonal diameter was reduced from 7.5μm to 5.5μm (Fig. 3b, Extended Data Fig. 3a), corresponding to a ~50% reduction in cross sectional area and producing the expected overall shrinkage (by 29%) of lumbar motor axon roots (Extended Data Fig. 3b), while the number of γ- and α-motor axons remained unchanged (Extended Data Fig. 3c).
Figure 3: Sustained loss of stathmin-2 reduces nerve conduction velocity and provokes axonal collapse by decreasing the spacing between axonal neurofilaments.
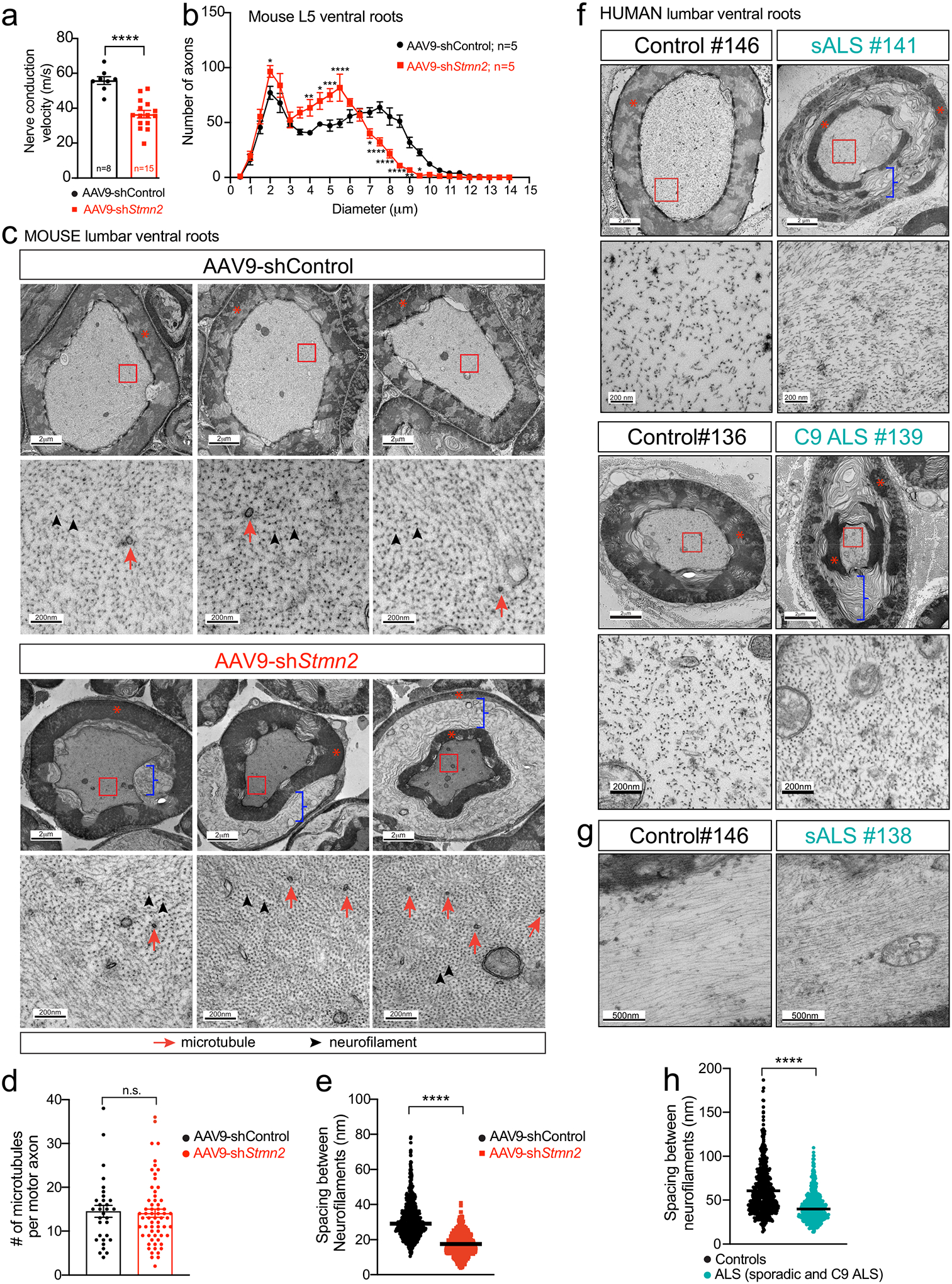
(a) Nerve conduction velocity on 20-month-old WT mice 8-months after lumbar subpial delivery of control or Stmn2 shRNAs. Each data point represents an individual mouse. Statistics by two-sided, unpaired t-tests. P <0.0001. (b) Size distribution of motor axon diameters in L5 motor roots 8-months post-injection of AAV9-shControl or AAV9-shStmn2. Statistics by two-sided, two-way ANOVA and Sidak’s multiple comparisons test. p values range from * P = 0.0426 to P < 0.0001. (c) (Top) Representative electron microscopy images of large caliber axons in ventral motor roots after subpial AAV9-shStmn2 injection reveal tearing within myelin layers. Higher magnification images (bottom) reveal altered spacing between neurofilament filaments in WT mice 8-months post-injection of AAV9-shStmn2 when compared to AAV9-shControl. Neurofilaments (highlighted by black arrowheads) outnumber microtubules (red arrows) in axoplasm of myelinated ventral axons. (d) Quantification of intra-axonal microtubules per ventral motor axon. Each data point represents an individual axon. 12–17 axons quantified per animal on n=2 AAV9-shControl and n=4 AAV9-shStmn2. Statistics by two-sided Mann Whitney t-test. (e) Quantification of intra-axonal distance between neurofilaments. At least 10 different axons were analyzed per group. N=2 animals on AAV9-shControl; n=4 animals on AAV9-shStmn2. Statistics by two-sided Mann Whitney t-test. P <0.0001 (f-g) Representative electron microscopy images of cross sectional (f) and longitudinal (g) sections of large caliber axons in the motor roots of postmortem human samples from non-neurological controls, sporadic ALS (sALS) and C9 ALS patients. Teared myelin layers are present in sALS and C9 ALS patients’ axons. Increased magnification micrographs of the axoplasm showing altered spacing between neurofilament filaments. (h) Quantification of the distance between neurofilaments in the axoplasm of human ventral roots. 8 different axons per condition derived from n=2 non-neurological controls, n=2 sALS and n=1 C9 ALS patient. Statistics by two-sided Mann Whitney t-test. P <0.0001. (c,f) Red asterisks indicate compacted myelin, blue brackets indicate ripped myelin. All panels: Error bars plotted as SEM. ****, p <0.0001; ***, p < 0.001; **, p < 0.01; *, p <0.05; ns, p>0.05.
Sustained reduction in stathmin-2 also produced dramatic changes in axoplasm and the interaction of the axon with its myelinating Schwann cells (Fig. 3c). Electron microscopic imaging of α-motor axons revealed axonal shrinkage, with the innermost myelin layers remaining attached to the axon, but with tearing within the outermost myelin layers, thereby at least partially disrupting connection of the Schwan cell body to its encapsulated axon. Indeed, we observed a separation between the inner and outer wrapping of myelin that could also be described as “delamination” of the (normally tightly wrapped) layers of the myelin sheath. Axoplasm of the shrunken axons appeared collapsed with compartments of highly compacted neurofilaments and higher microtubule density (Fig. 3c) although the overall microtubule number was unchanged (Fig. 3d). Neurofilament organization was highly abnormal, with average interfilament spacing reduced by nearly half (from median value of 29.02 nm to 16.86 nm – Fig. 3e).
Remarkably, evaluation of postmortem human ventral roots from lumbar spinal cord from 5 sporadic ALS patients, 2 C9-ALS patients and 2 healthy controls revealed similar axonal collapse and myelin tearing as in adult WT mice with sustained stathmin-2 suppression (Fig. 3f,g and Extended Data Figure 4). In ALS tissues, inter-neurofilament spacing was also reduced in the axoplasm of large caliber motor axons by nearly 30% (from median value of 54.42 nm to 39.90 nm – Fig. 3f–h and Extended Data Fig. 4). These results provide compelling evidence that TDP-43-dependent misprocessing of stathmin-2 mRNAs and subsequent stathmin-2 protein loss in sALS and C9-ALS patients contribute to motor axon defects and muscle denervation, two early pathological events in ALS.
Neurofilaments are responsible for the acquisition of normal axonal caliber13,56–59 and are assembled as obligate heteropolymers60 of three highly conserved polypeptide subunits: neurofilament light (NF-L), medium (NF-M), and heavy (NF-H). In mice, the long tails of NF-M and NF-H contain 7 or 51 lysine-serine-proline (KSP) phosphorylation sites, respectively, that extend from the filament surface to determine mean interfilament spacing59 and interactions with other cytoskeletal components and organelles59. A 10–20-fold increase in axonal caliber, corresponding to a 100–400 increase in axonal volume, initiates concomitantly with myelination that occurs within the first three postnatal weeks. An initial burst in caliber occurs in the first two to three months and is dependent on an “outside-in” signaling cascade from the myelinating cell to the underlying axon that requires the tail domain of NF-M59.
While levels of the three neurofilament subunits remained unchanged following sustained stathmin-2 suppression within the spinal cord (Extended Data Fig. 3d–h), levels of phosphorylated NF-H and NF-M were reduced (Extended Data Fig. 3i–k), the latter of which has been shown in mice to be required for establishing normal axonal caliber59. These results reveal a crucial role of stathmin-2 in the maintenance of axoplasm through orchestrating the “outside-in” phosphorylation of the larger two neurofilament subunits that act to mediate their nearest neighbor distances and the assembly of a three-dimensional, space-filling, interlinked neurofilament array that determines axonal caliber.
Reduced stathmin-2 levels impair tactile perception
Retrograde delivery of an AAV9 encoding GFP was used to validate that subpial injection yielded transduction of the majority of sensory neuron soma residing in the adjacent DRGs (Fig. 4a, Extended Data Fig. 5a), consistent with prior reports46,49. Virtually all normal DRG neurons express Stmn2 mRNAs detectable by FISH, albeit at diverse levels (Fig. 4b,c). Even though sensory alterations are not a major hallmark in ALS, an increasing number of studies have reported combined motor and sensory deficits in a significant group of ALS patients61. Interestingly, neuronopathies including facial-onset sensory and motor neuronopathy syndrome (FOSMN) present TDP-43 proteinopathy in DRG neurons with accumulation of TDP-43 aggregates in the cytoplasm and TDP-43 nuclear loss62–64. Thus, taking advantage of the technical features of subpial delivery and acknowledging the clinical relevance of the potential loss of stathmin-2 in sensory neurons, we explored the consequences of stathmin-2 loss in the mouse sensory system. Eight months after subpial administration of AAV9-shStmn2, Stmn2 mRNAs (detected by single molecule FISH – Fig. 4b) and stathmin-2 protein (detected by indirect immunofluorescence – Fig. 4d) were reduced in the majority of DRG neurons, with more than half expressing murine Stmn2 mRNA at ~30% of initial level (Fig. 4b,c) and a corresponding reduction in detectable stathmin-2 protein (Fig. 4d). These results were consistent with an overall 53% reduction of the initial Stmn2 mRNA level (measured by qRT-PCR in mRNA extracted from lumbar DRGs – Fig. 4e).
Figure 4: Reduced stathmin-2 levels in lumbar dorsal root ganglia impair hindlimb sensory system.
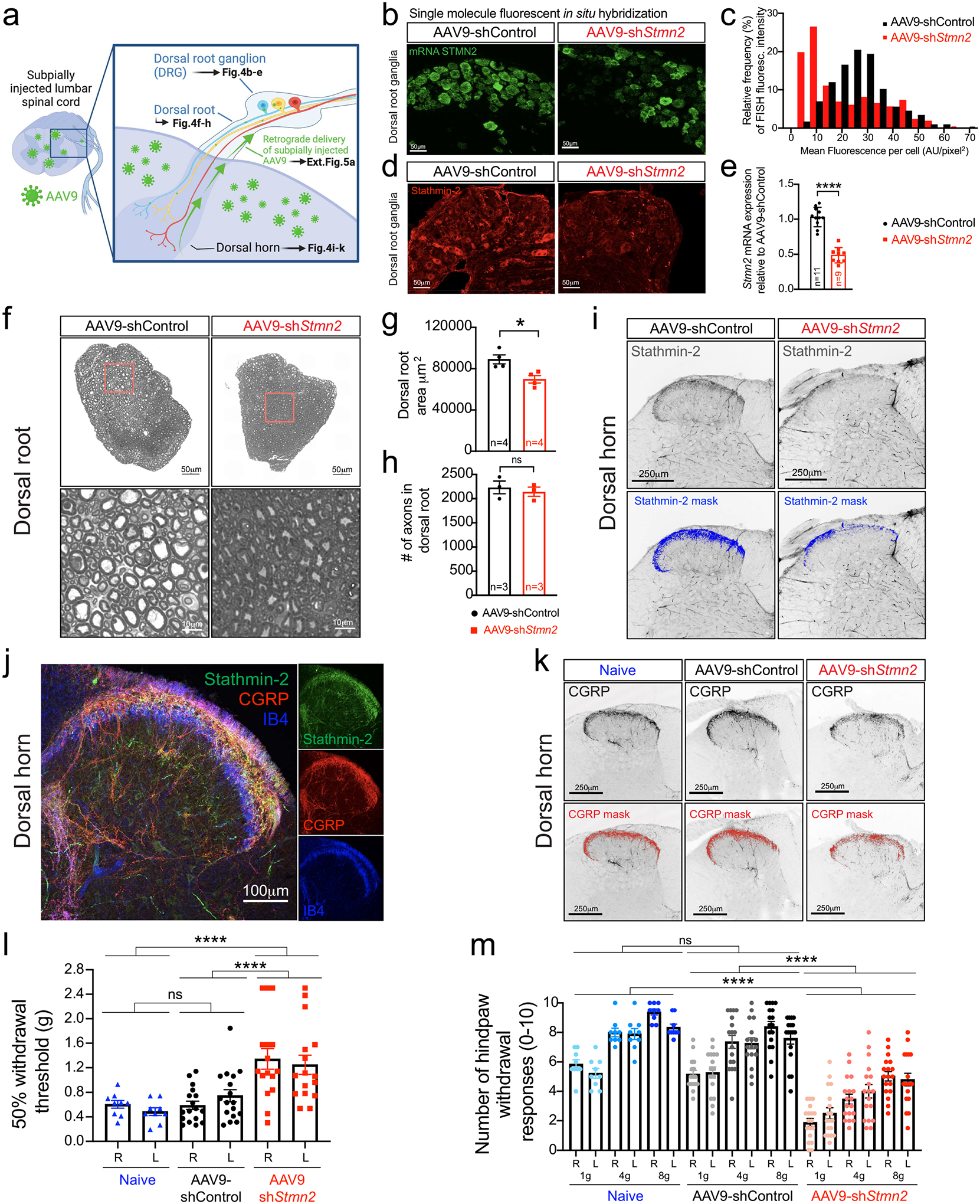
(a) Schematic of strategy to determine the impact of subpially injected AAV9-encoded shRNAs on the neurons in the dorsal root ganglion (DRG) innervating the dorsal spinal cord. Figure created using Biorender. (b-d) Representative confocal images from at least 3 animals per condition showing Stmn2 mRNA levels by single-molecule FISH (green, b) and its fluorescence distribution (c), and stathmin-2 protein (red, d) in the lumbar DRGs 8-months post-administration of AAV9-shRNAs. (e) Quantification of Stmn2 mRNA levels in DRG at 8-months post-injection, normalized to Gapdh. Statistics by two-sided, unpaired t-tests (P < 0.0001). (f-h) Representative images of cross-sectioned dorsal roots and higher magnification images showing axonal morphology and diameter size (f), quantification of area (g), and total number of sensory axons (h), of WT mice 8 months post-injection. Statistics by two-sided, unpaired t-tests (P = 0.0142). (i) Representative dorsal horns of lumbar spinal cord sections immunolabeled with stathmin-2, highlighted in blue, 8 months post-injection. N=2 animals on AAV9-shControl and n=4 animals with AAV9-shStmn2 were imaged with similar results. (j) Representative confocal micrograph of WT dorsal horn immunostained with stathmin-2 (green), CGRP (red), and IB4 (blue). N=3 wildtype animals were immunostained with similar results. (k) Representative lumbar spinal cord dorsal horn areas immunolabeled with CGRP, highlighted in red. N=5 non-injected (naïve) 20-month-old mice, or n=4 mice 8-months after subpial administration of AAV9-shRNAs were imaged with similar results. (l-m) Quantification of the 50% withdrawal threshold upon von Frey filament-based mechanical stimuli on mice hindlimbs (m) Quantification of hind paw response to increasing von Frey filament force stimuli on mice hindlimbs (l-m) Assays performed at 20 months-of-age when non-injected (naïve; n=9), or 8-months post-administration of AAV9-shControl (n=17) and AAV9-shStmn2 (n=16). Statistics by two-sided, one-way ANOVA Kruskal-Wallis with Dunn’s multiple comparisons test. P < 0.0001. For p values between specific conditions please see Source Data for Figure 4. All panels: Each data point represents an individual mouse. Error bars plotted as SEM. ****, P <0.0001; ***, P < 0.001; **, P < 0.01; *, P <0.05; ns, P >0.05.
Sensory neurons extend an axon that bifurcates, with one branch directed towards the periphery to sense environmental stimuli, while the other branch enters the dorsal root to innervate neurons of the dorsal spinal cord (Fig. 4a)65. As observed in ventral roots (Fig. 3b,c), the size distribution of axons in the dorsal root shifted towards smaller diameters upon sustained reduction of stathmin-2 (Extended Data Fig. 5b,c), producing an overall 22% shrinkage of cross-sectional area of the affected dorsal root (Fig. 4f,g) without any axonal loss (Fig. 4h). Reduction of stathmin-2 was also observed in the sensory neuron terminals innervating the dorsal horn of the lumbar spinal cord of AAV9-shStmn2 mice (Fig. 4i, Extended Data Fig. 5d), with the area occupied by stathmin-2 positive terminals diminished by two-thirds within 8-months of sustained suppression (Extended Data Fig. 5d). Interestingly, the stathmin-2-containing sensory terminal area in the dorsal horn colocalized with calcitonin gene-related peptide (CGRP)-positive fibers (Fig. 4j) corresponding to unmyelinated, slowly conducting peptidergic C fibers (Lamina I) and thinly myelinated more rapidly conducting Aγ fibers (outer Lamina II)65–67. By contrast, the inner lamina II mainly comprised of unmyelinated, slowly conducting non-peptidergic C fibers (identifiable by isolectin B4 (IB4) staining) barely overlapped with stathmin-2 positive terminals (Fig. 4j), indicating specificity of stathmin-2 role in peptidergic terminals of pain transmitting sensory neurons.
Consistently, loss of stathmin-2 in the sensory terminals was accompanied by a reduction of CGRP positive fibers innervating the dorsal spinal cord (Fig. 4k, Extended Data Fig. 5e), evidence of the importance of stathmin-2 in terminal maintenance of peptidergic sensory neurons. These molecular alterations in sensory neurons altered somatosensory behavior. Mice with sustained reduction of stathmin-2 had suppressed tactile evoked responses (i.e., increased paw withdrawal thresholds) when compared to mice injected with non-targeting AAV9-shControl (Fig. 4l) or age-matched non-injected, naïve animals. Response to tactile stimuli was also impaired in response to mechanical nociceptive stimuli (pain). Indeed, while naïve and AAV9-shControl animals behaved indistinguishably, responses to nociceptive mechanical stimuli, determined by hindpaw withdrawal frequency upon increasing force, were reduced in mice with diminished stathmin-2 (Fig. 4m).
Increased perinatal lethality from absence of stathmin-2
CRISPR/Cas9 genome editing was used to inactivate one endogenous Stmn2 allele in C57BL/6J mice by deletion of a 1028bp segment including exon 3 (Extended Data Fig. 6a). The deletion was predicted to produce an unstable RNA that is a substrate for nonsense-mediated decay as a consequence of a frameshift mutation at codon 38 (Extended Data Fig. 6a). Although mice were born in expected Mendelian ratios from breeding Stmn2+/− animals (Extended Data Fig. 6b), 80% of Stmn2−/− pups died shortly after birth (median survival of only 1.5 days), indicating one or more roles for stathmin-2 in early development. However, in Stmn2−/− mice that survived to weaning age, no further accelerated lethality was observed (Fig. 5a). As expected, Stmn2 mRNAs were reduced or undetectable, respectively, in cortical (Fig. 5b) or spinal cord (Fig. 5c) from Stmn2+/− or Stmn2−/− adult mice. Stathmin-2 protein was also reduced by 50% in heterozygous Stmn2+/− mice and was undetectable in Stmn2−/− mice in lumbar spinal cord protein extracts (Fig. 5d, Extended Data Fig. 6c) and with immunostaining of lumbar spinal cord sections (Fig. 5e). Reduction or complete absence of stathmin-2 did not produce compensatory changes in expression of the other stathmin family members (Extended Data Fig. 6d,e).
Figure 5: Stathmin-2 has an important role early after birth, rescued by FVB genetic background but not by Sarm1 ablation.
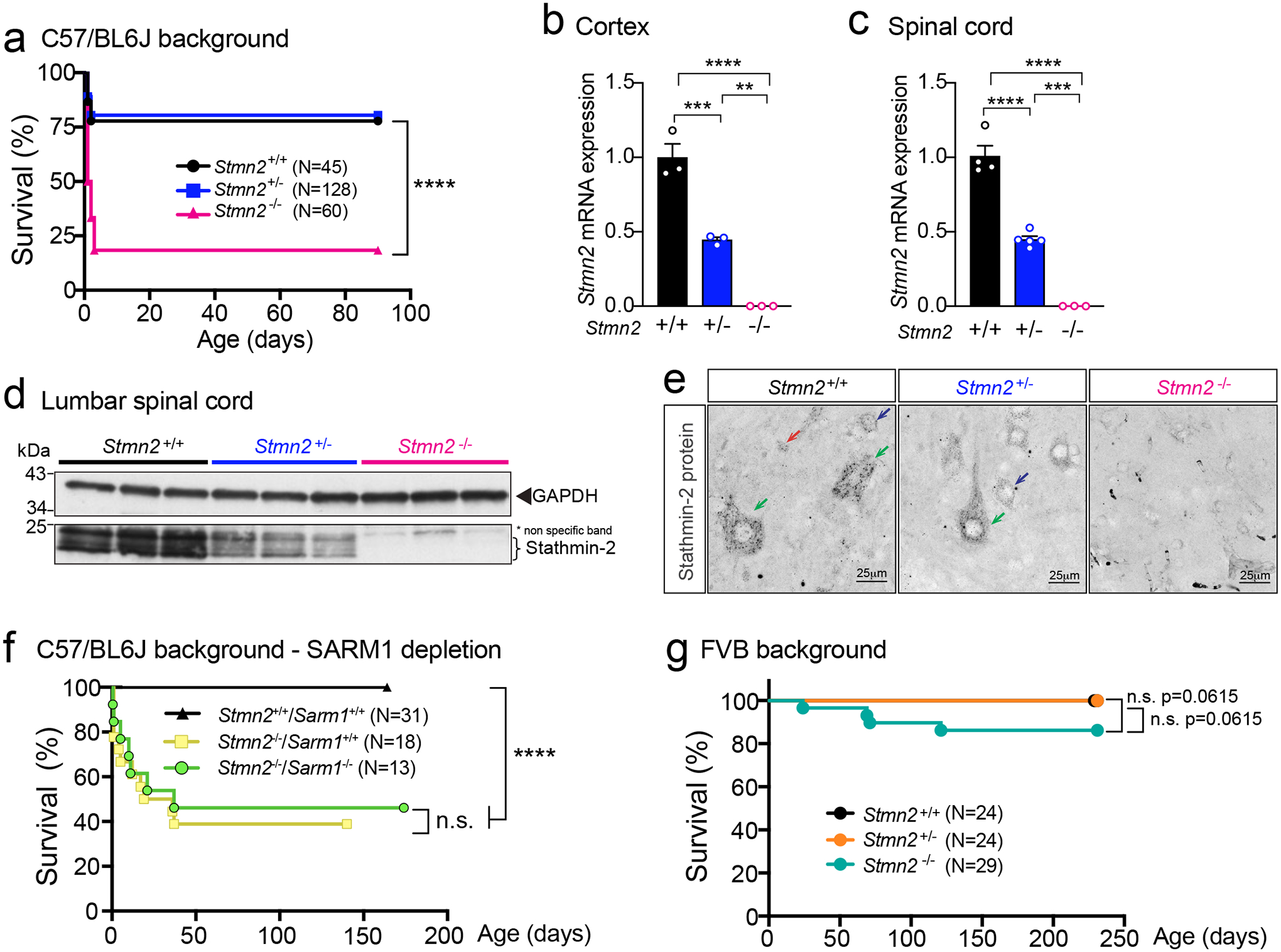
(a) Survival curve of Stmn2+/+, Stmn2+/− and Stmn2−/− mice in a C57/BL6J background. Statistical analysis by Log-rank Mantel-Cox test (P < 0.0001) (b-c) Measurement of murine Stmn2 mRNA levels extracted from cortex (n=3 animals per genotype) (b), and spinal cord (c) of Stmn2+/+, Stmn2+/− and Stmn2−/− mice. Each data point represents an individual mouse. Gapdh was used as an endogenous control gene. Statistical analysis by two-sided, one-way ANOVA post hoc Tukey’s multiple comparisons test. For spinal cords, n=4 animals for Stmn2+/+; n=5 animals for Stmn2+/−; and n=3 animals for Stmn2−/− were used. Range of p values from P = 0.0027 to P < 0.0001 (b) and P = 0.0002 to P < 0.0001 (c). (d) Immunoblot showing levels of the ~21 kDa mouse stathmin-2 protein from n = 3 different animals per genotype. GAPDH was used as a loading control. (e) Confocal micrographs of stathmin-2 immunolabeling at the ventral spinal cord of 12-month-old Stmn2+/+, Stmn2+/− and Stmn2−/− mice. Green arrows: α-motor neurons; blue arrows: γ-motor neurons; red arrows: interneurons. At least n = 3 different animals per genotype were imaged with similar results. (f) Survival curve of Stmn2+/+/Sarm1+/+, Stmn2−/−/Sarm1+/+ and Stmn2−/−/Sarm1−/− mice in a C57/BL6J background. Statistical analysis by Log-rank Mantel-Cox test (P < 0.0001). (g) Survival curve of Stmn2+/+, Stmn2+/− and Stmn2−/− mice in FVB background. Statistical analysis by Log-rank Mantel-Cox Test. All panels: Error bars plotted as SEM. ****, P <0.0001; ***, P < 0.001; **, P < 0.01; *, P <0.05; ns, P >0.05.
The threshold of stathmin-2 levels needed to rescue perinatal lethality in Stmn2−/− mice was determined by breeding with two newly generated BAC transgenic lines harboring a complete human STMN2 gene. Expression of human stathmin-2 at 10% of the endogenous protein level (Extended Data Fig. 6f – corresponding to BAC mouse line 9446 with 1 copy of the transgene – Extended Data Fig. 6g) was not enough to counteract the lethality after birth (8% of progeny that survived to p10 were Stmn2−/−, BAC9446, only 1/3rd of the 25% expected). However, lethality was fully rescued (24% of the progeny survived at p10 from 25% expected) by accumulation of BAC-encoded human stathmin-2 to 50% of the endogenous protein level (Extended Data Fig. 6h – corresponding to BAC mouse line 9439 with 3 copies of the transgene – Extended Data Fig. 6g).
Developmental requirement for STMN2 is SARM1 independent
Both stathmin-2 and NMNAT2 (nicotinamide mononucleotide adenylyltransferase 2 which functions in NAD+ biosynthesis) have been proposed as axonal survival factors68,69 that act to oppose the toll-like receptor adapter sterile alpha and TIR motif containing 1 (SARM1). SARM1 is implicated as a central executioner of axonal degeneration (referred to as Wallerian degeneration) and its suppression has been proposed as a therapeutic strategy for multiple neurodegenerative diseases, including ALS68,69. While loss of Nmnat2 induces perinatal lethality that is restored by ablation of Sarm170, perinatal death driven by stathmin-2 loss was unaffected by reduction in or absence of Sarm1 in cohorts of Stmn2−/− mice bred to have neither or both Sarm1 alleles inactivated (Fig. 5f). Thus, postnatal death from Stmn2 loss is independent of SARM1 pro-degeneration activity.
Lethality from Stmn2 absence is mouse strain dependent
Inbred genetic backgrounds can significantly influence expression of phenotypes associated with known genetic perturbations71 and can underlie variation in disease severity between individuals with the same mutation(s). To test if this was true for stathmin-2, we backcrossed the B6-Stmn2+/− mice into an alternative genetic background, the Friend leukemia virus B (FVB) mouse strain. In contrast with the widespread perinatal lethality of Stmn2−/− mice in the C57BL/6J background, FVB:B6 mice deficient for Stmn2 were born in the expected Mendelian ratios without significant perinatal lethality (Fig. 5g).
Absence of Stmn2 results in motor deficits and denervation
The effects of homozygous Stmn2 loss from earliest development were determined in C57BL/6J and FVB mice that survived to weaning age. Body weight in males (Fig. 6a,b) and females (Fig. 6c,d) was reduced compared to age-matched Stmn2+/− and WT littermates. Similar to the motor impairment developed following chronic AAV9-shStmn2 suppression in the normal adult nervous system, motor performance evaluated by hindlimb clasping (Fig. 6e,f), grip strength (Fig. 6g,h), and rotarod (Fig. 6i,j) was significantly impaired at 3 months of age in both Stmn2−/− mice strains compared to WT mice. Once developed, initial motor deficits did not progress in Stmn2−/− mice that were monitored up to 12 months of age. Motor performance of Stmn2+/− mice was indistinguishable from WT littermates.
Figure 6: Absence of stathmin-2 results in motor deficits and muscle denervation without motor neuron loss.
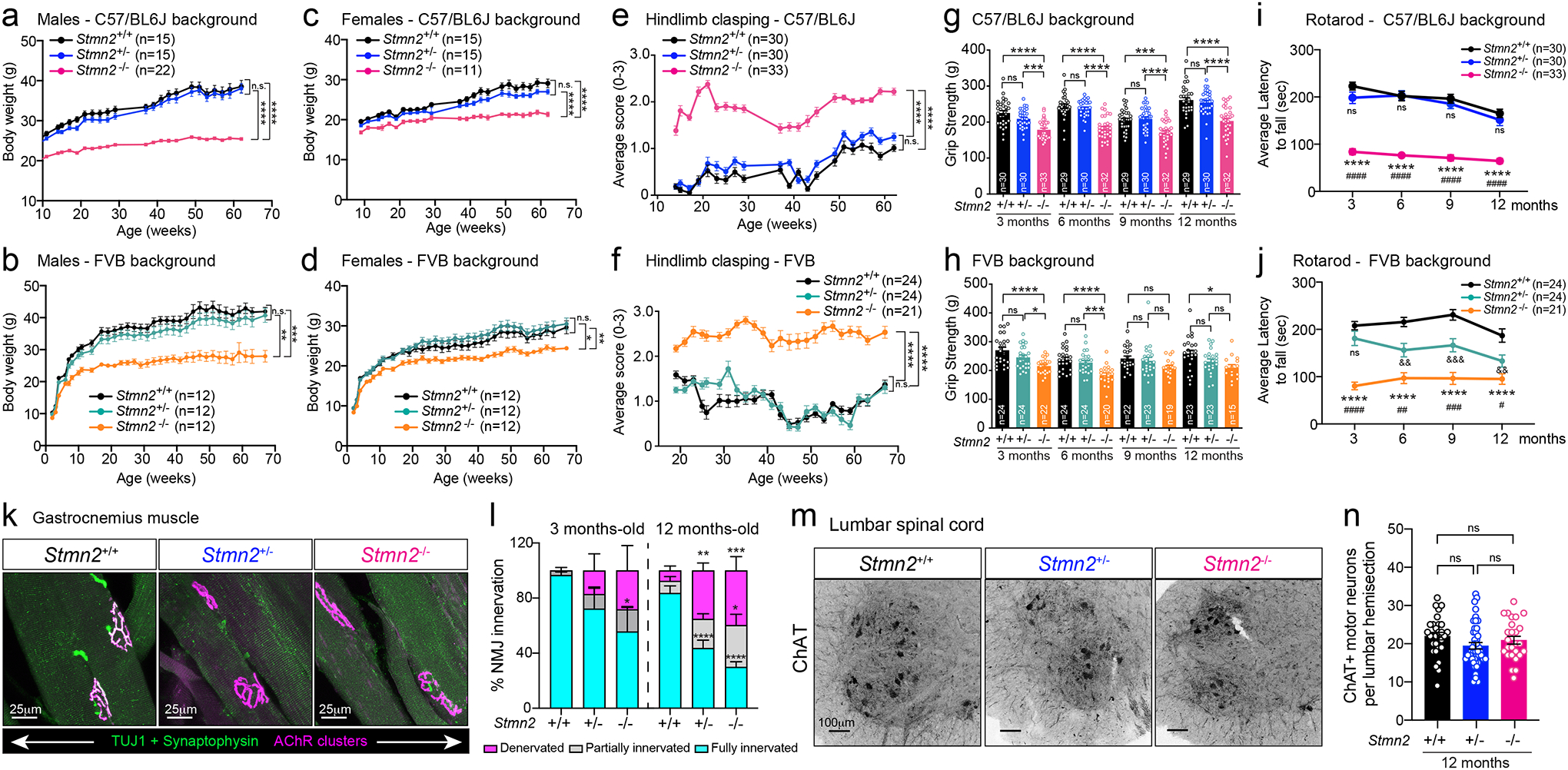
(a-b) Body weight from Stmn2+/+, Stmn2+/− and Stmn2−/− male mice in C57/BL6J (a) and FVB backgrounds (b), and (c-d) female mice in C57/BL6J (c) and FVB (d) backgrounds. (e-f) Hindlimb clasping measurements of Stmn2+/+, Stmn2+/− and Stmn2−/− mice in C57/BL6J (e) and FVB (f) backgrounds. (a-f) Each dot represents the mean value per genotype in each time-point. Statistics by two-sided, two-way ANOVA post hoc Tukey’s multiple comparisons test. (g-h) Hindlimb grip strength measurements of Stmn2+/+, Stmn2+/− and Stmn2−/− mice at 3, 6, 9, and 12 months-old in C57/BL6J (g) or FVB (h) backgrounds. Each data point represents an individual mouse. Statistics by two-sided, one-way ANOVA post hoc Tukey’s multiple comparisons. (a-h) P value range from P = 0.0496 to P < 0.0001. Specific p values for each time-point are specified in Source Data for Figure 6. (i-j) Rotarod performance of Stmn2+/+, Stmn2+/− and Stmn2−/− mice at 3, 6, 9, and 12 months in C57/BL6J (i) or FVB (j) backgrounds. Each dot represents mean values per genotype in each time-point. Statistics by two-sided, two-way ANOVA post hoc Tukey’s multiple comparisons. *, #, and & represent statistical tests performed between Stmn2+/+ and −/−; Stmn2+/− and −/−; and Stmn2+/+ and +/−, respectively. (k) Gastrocnemius muscle from 12-month-old Stmn2+/+, Stmn2+/− and Stmn2−/− mice immunolabelled with synaptophysin and β3-tubulin (Tuj1) (green) for axon terminals, and α-bungarotoxin (magenta) for muscle endplates. (l) Innervation frequency quantified using confocal microscopy at 3- and 12-months. Statistics by two-sided, two-way ANOVA post hoc Tukey’s multiple comparisons test. N=3 animals/genotype at 3 months; n=4–5 animals/genotype at 12 months. P value range from P = 0.0396 to P < 0.0001. (m) Representative lumbar spinal cord ventral sections of 12-month-old Stmn2+/+, Stmn2+/− and Stmn2−/− mice immunolabelled with ChAT. (n) Quantification of ChAT positive motor neurons per hemisections. N= 5 animals/genotype. Statistical analysis by one-way ANOVA post hoc Tukey’s multiple comparisons. All panels: Error bars plotted as SEM. ****, P <0.0001; ***, P < 0.001; **, P < 0.01; *, P <0.05; ns, P >0.05.
Loss of stathmin-2 from early embryogenesis induced progressive NMJ denervation of adult hindlimb muscles as observed at 3- and 12 months, the latter being comparable to that following 8-months of AAV9-shStmn2-mediated reduction of stathmin-2 within a fully matured motor neuron (Fig. 6k,l), accompanied by mildly reduced CMAP when compared to aged-matched WT and Stmn2+/− mice (Extended Data Fig. 7a). Denervation was not accompanied by a reduction in the number of ChAT positive spinal motor neurons even at 12 months of age (Fig. 6m,n), nor by increased accumulation in serum of NF-L or pNF-H, biomarkers widely used as an indicator of neurodegeneration (Extended Data Fig. 7b–e). Once again, presence or absence of a functional SARM1-dependent degenerative pathway did not affect development of motor deficits upon loss of stathmin-2 (Extended Data Fig. 7f). Thus, even when stathmin-2 loss is initiated in earliest embryogenesis, the resultant alterations in distal axons that trigger reduced conduction velocity and motor deficits are independent of SARM1 activity and are insufficient to drive adult motor neuron death. Examination of Stmn2−/− mice confirmed reduced nociception relative to Stmn2+/+ mice in both genetic backgrounds (Extended Data Fig. 7g,h, respectively), accompanied by reduced accumulation of CGRP (Extended Data Fig. 7i), findings reinforcing the importance of stathmin-2 in adult sensory systems.
Stmn2 loss reduces axonal caliber and conduction velocity
Complete absence of stathmin-2 yielded reduced conduction velocity (in comparison to WT littermates) in the sciatic nerves of 3- and 6-month-old C57BL/6J Stmn2−/− mice (Fig. 7a). Measurement of axonal diameters at 3 months of age in L5 ventral motor axons of Stmn2−/− mice revealed reduced diameters (Fig. 7b,c) without any axonal loss (Fig. 7d), demonstrating inhibited or delayed axonal caliber acquisition, a feature that is normally completed in mice by 3 months of age72. Levels of all three neurofilament subunits and their KSP phosphorylated forms were comparable in Stmn2−/− and Stmn2+/+ mice at 3 months of age (Extended Data Fig. 8a–f). By 12 months of age, however, chronic absence of stathmin-2 yielded markedly diminished levels of NF-H and its phosphorylated form (pNF-H, detected with the SMI-31 monoclonal antibody) (Fig. 7e,f, quantified in Fig. 7g,h). Levels of NF-M (Fig. 7i; quantified in Fig. 7j) and its phosphorylated form (pNF-M, also detected with SMI-31 antibody) were also reduced (Fig. 7e, quantified in Fig. 7l). No changes were observed in NF-L levels among the genotypes at the different timepoints tested (Fig. 7k,m and Extended Data Fig. 8a,e). Thus, absence of stathmin-2 impaired accumulation of NF-M and NF-H, as well as their phosphorylation, which together provide explanations for reduced acquisition or maintenance of axonal calibers59,73–75 and reduced nerve conduction velocity76.
Figure 7: Absence of stathmin-2 reduces nerve conduction velocity and alters axonal radial growth, and neurofilament composition.
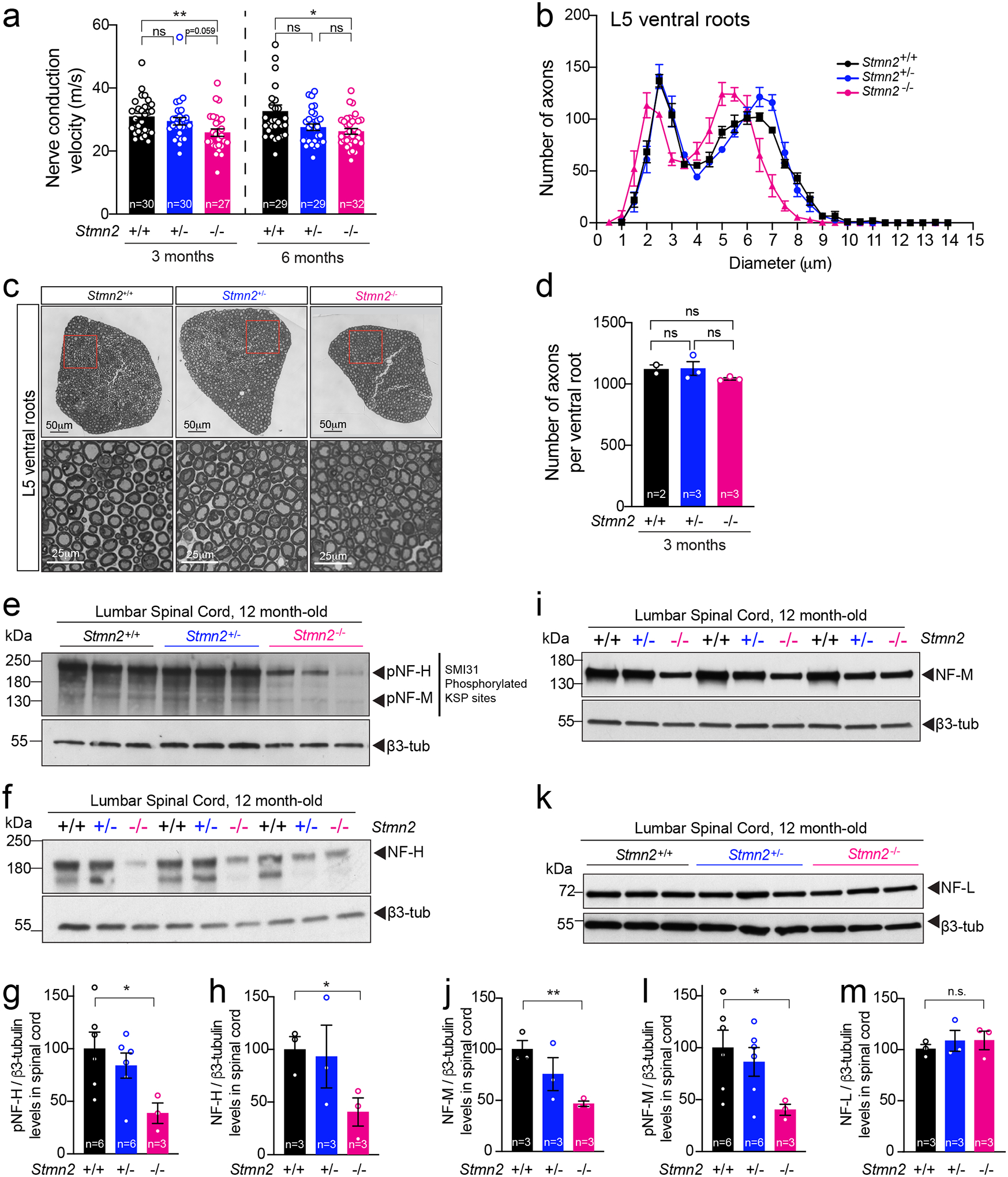
(a) Nerve conduction velocity of Stmn2+/+, Stmn2+/− and Stmn2−/− at 3 and 6 month of age in the C57/BL6J background. Each data point represents an individual mouse. Corresponding n number specified in the graph. Statistics by two-sided, Kruskal-Wallis post hoc Dunn’s multiple comparisons test. P = 0.0033 and P = 0.0162 at 3 and 6 months, respectively. (b) Size distribution of motor axons in the L5 ventral motor roots of 3-month-old Stmn2+/+, Stmn2+/− and Stmn2−/− mice in the C57/BL6J background. N= 2 for Stmn2+/+; n= 3 animals/genotype for Stmn2+/− and Stmn2−/−. (c) Representative motor roots micrographs and higher magnification images from 3-month-old Stmn2+/+, Stmn2+/− and Stmn2−/− mice in the C57/BL6J background showing reduced axon diameter in Stmn2−/−. (d) Number of total axons per L5 ventral root quantified in Stmn2+/+ (n=2 animals), Stmn2+/− (n=3 animals), and Stmn2−/− (n=3 animals). Statistics by two-sided, Kruskal-Wallis post hoc Dunn’s multiple comparisons test. (e,f) Immunoblotting for phosphorylated forms of neurofilament heavy (pNF-H) and neurofilament medium (pNF-M) (e) and total neurofilament heavy (NF-H) (f) in spinal cord protein extracts of 12 months-old Stmn2+/+, Stmn2+/− and Stmn2−/− mice. (g-h) Quantifications of pNF-H, (P = 0.0381) (g), NF-H, P = 0.0307 (h), and pNF-M, (P = 0.0486) (l), normalized to the amount of β3-tubulin in (e) and (f) respectively. β3-tubulin used as loading control since it remained unchanged upon the same amount of protein loading. Each data point represents an individual mouse. Corresponding n numbers specified in the graphs. Statistics by two-sided, unpaired t-test. (i-k) Immunoblotting for neurofilament medium (NF-M) (i) and neurofilament light (NF-L) (k) in spinal cord protein extracts of 12 months-old Stmn2+/+, Stmn2+/− and Stmn2−/− mice. Quantification for NF-M, (P = 0.0039) (j), and NF-L (m) normalized to the amount of β3-tubulin are shown. Each data point represents an individual mouse. Corresponding n numbers specified in the graphs. Statistics by two-sided, unpaired t-test. All panels: Error bars plotted as SEM. ****, P <0.0001; ***, P < 0.001; **, P < 0.01; *, P <0.05; ns, P >0.05.
Discussion
Using transient or sustained suppression, we have established the importance of stathmin-2 in acquisition and maintenance of proper caliber of motor axons, in neurofilament-dependent structuring of axoplasm, in acquisition and retention of normal conduction velocity, and in maintenance of mature NMJs and CGRP+ sensory terminals. Sustained reduction of stathmin-2 in an otherwise mature nervous system induced collapse of axonal caliber and tearing of outer myelin layers, thereby at least partially disconnecting the “outside-in” signaling cascade from myelinating cell to the ensheathed axon59. Correspondingly, neurofilament phosphorylation is inhibited, resulting in the collapse of the inter-neurofilament spacing that is required to produce the three-dimensional space filling array that mediates axonal caliber59. This in turn yields neuronal dysfunction that includes reduced conduction velocity, progressive motor and sensory deficits (including diminished tactile and nociceptive somatosensory responses), and NMJ denervation. Most importantly, even transient reduction of stathmin-2 in the adult nervous system is sufficient to provoke denervation of NMJs. Remarkably, postmortem evaluation of ventral roots from ALS patients using electron microscopy revealed similar reduction of inter-neurofilament spacing accompanied by axonal collapse and myelin tearing of large caliber motor axons. This axonal defect is a consistent (5 out of 5 sALS, and 2 out of 2 C9orf72 inherited ALS) feature of TDP-43-associated ALS, reflecting the relevance of stathmin-2 loss in the physiopathology of ALS. Chronic reduction of stathmin-2 alone initiated distal motor axonal degeneration independent of the SARM1 prodegenerative pathway, including failure of synaptic maintenance and axoplasmic collapse into compartments of highly disorganized, closely spaced neurofilaments and tearing of the layers of close-packed myelin.
Although stathmin-2 has been previously proposed to regulate the rapid growth and shrinkage of microtubules (referred to as dynamic instability)77–80 through its direct binding to α/β tubulin heterodimers42 in a phosphorylation-dependent manner81, our data establish that reduction in stathmin-2 in a normal adult nervous system provokes strikingly altered neurofilament organization without affecting microtubule number. Reduced level and phosphorylation of NF-M and NF-H upon stathmin-2 loss reflect disruption of the “outside-in” signaling pathway from myelinating cell to the underlying axoplasm that is required to mediate and maintain axonal diameter59.
Perhaps most importantly, chronic reduction of stathmin-2 in motor neurons in an otherwise normal adult nervous system is itself sufficient to drive the initial steps of an ALS-like motor phenotype, a finding that strongly supports a mechanistic contribution to disease initiation and progression from the established stathmin-2 loss in sporadic and familial ALS pathogenesis36. Although TDP-43 loss of function affects the levels or splicing of more than 1500 RNAs28,29, the mRNA encoding stathmin-2 is the transcript most affected in human neurons and its restoration alone is sufficient to rescue compromised axonal regeneration after injury of TDP-43 depleted motor neurons33,34. In fact, our group has recently demonstrated the use of ASOs to block the usage of the cryptic splice and polyadenylation sites in the first intron of the stathmin-2 pre-mRNA as a way to prevent the truncation of the stathmin-2 mRNA and restore the normal levels of stathmin-2 expression82. Our effort has also identified a significant role of stathmin-2 in tactile and nociceptive pain transmission and expands restoration or maintenance of stathmin-2 as a therapeutic target for sensory neuropathies, such as FOSMN62–64, that include TDP-43 mislocalization in sensory neurons.
In addition to its requirement for axoplasmic and synaptic maintenance in the adult, our evidence establishes that absence of stathmin-2 in mice can compromise perinatal survival (in the C57/BL6J background) independent of the SARM1 prodegenerative pathway, consistent with a prior study41. The near absence of early lethality in a different genetic background (FVB) implicates contribution(s) of yet unidentified genetic variants to perinatal survival in the absence of stathmin-2, implicating a role of stathmin-2 in early development.
Finally, we have demonstrated that not only does stathmin-2 accumulate at the axon terminals of mature motor and sensory neurons in the adult nervous system (e.g., NMJs and dorsal horn lamina I and outer laminal II, respectively), but also that there is a continuing requirement for stathmin-2 in the maintenance of motor and sensory neuron synapses. Upon sustained reduction in stathmin-2, synapses are disrupted, triggering a “dying back” degenerative process similar to that recognized as an early event in ALS3. However, reduction in stathmin-2 does not compromise motor neuron survival in mice, at least within the 8-month timeframe we have analyzed. This might be due to additional mRNA alterations occurring in TDP-43 proteinopathies, including the loss of UNC13A83,84, that may compound the pathology driven by loss of stathmin-2 alone. Nevertheless, our data strongly support stathmin-2 loss as an early contributor to ALS disease initiation and progression and highlight the attractiveness of restoring stathmin-2 as a therapeutic approach for TDP-43-dependent neurodegenerative diseases82.
Methods
Animals
All animals were Mus musculus species, with C57BL/6N mice (female and male, ~12 months of age) sourced from the Cleveland Laboratory (University of California San Diego, UCSD) and Sarm1 deficient mice (B6.129X1-Sarm1tm1Aidi/J, Jax catalog number 18069) obtained from The Jackson Laboratory. Mice (3–4 per cage) were housed in individually high efficiency particulate air (HEPA)-filtered polysulfonate cages, under controlled conditions of 12 h light/dark cycles (6 am to 6 pm), room temperature of 22 ± 4°C, and 50 ± 15% humidity, with 15 air exchanges per hour. Animals had access to acidified tap water (pH 2.5 to 3.0), and normal rodent chow ad libitum. Animal health and body weight were monitored on a daily basis. All experiments followed NIH Guidelines and were approved by the Institutional Animal Care and Use Committee (IACUC) at UCSD (Protocol S00225) and The Jackson Laboratory (Protocol 20029–1).
• Generation of Stmn2 deficient mice
C57BL/6J-Stmn2 deficient mice were created via CRISPR/Cas9 technology. Two RNA guides were used to target upstream (Stmn2_up_crRNA1:TGCGCAGACTCCATCAGACT; Stmn2_up_crRNA2: ATTTTACACTCTGCTCTATG) and downstream (Stmn2_down_crRNA1: ATCCTACTGTAGAGAATTGA; Stmn2_down_crRNA2: ACTATGGACATTAAGACTGG) of Stmn2-exon 3. A founder mouse carrying a 1028 nucleotide deletion spanning Stmn2-exon 3 was backcrossed twice to C57BL/6J, establishing the stock C57BL/6J-Stmn2em2Lutzy/Mmjax, (here B6-Stmn2+/−, Jax catalog number 33740). For survival studies, 60 breeding pairs B6-Stmn2(+/−) were stablished. All pups were genotyped at birth and included in the survival study. For mixed genetic background experiments, B6-Stmn2−/+ mice were outcrossed with FVB/NJ (Jax catalog number 1800) for one generation. Twenty-five heterozygous pairs were intercrossed to generate F2 FVB;B6-Stmn2−/− homozygous mice. All F2 progeny was genotyped at birth and enrolled in the survival study. For longitudinal phenotyping a minimum cohort of n=11–15 mice by sex and genotype were enrolled from both backgrounds. All the cohorts were randomly selected from age matched litters. Survival was monitored weekly. All animals were tested at 3, 6, 9, and 12 months for rotarod, grip strength, clasping score, von Frey test, and electrophysiology (CMAP, and NCV), with technicians blinded to genotypes. To assess the role of Sarm1 in neonatal lethality, Sarm1tm1Aidi/ deficient mice were crossed with B6-Stmn2−/+ mice. From the F1 progeny, twenty (Stmn2−/+, Sarm1−/+) heterozygous mice were intercrossed to generate F2 homozygous mice for both Sarm1 and Stmn2 null alleles. Survival studies included: double homozygous (Stmn2−/−, Sarm1−/−), single Sarm1-KO (Stmn2+/+, Sarm1−/−), and wild type (Stmn2+/+, Sarm1+/+). Kaplan-Meyer survival curves were reported. Inclusion/exclusion criteria was as follows: every mouse found dead or requiring euthanasia due to reduced body conditioning and more than 15 % loss of maximum body weight were scored as 1. Mice requiring euthanasia for unrelated conditions like paraphimosis, severe fight wounds, or dermatitis were censored and scored as 0.
• Generation of human BAC STMN2 transgenic lines
We generated a C57BL/6J BAC transgenic mouse by microinjecting the BAC clone RP11–761J24, a 202,106bp DNA fragment carrying the complete human STMN2 gene, into C57BL/6J zygotes. Two transgenic founder lines, BAC-line 9439 and 9446, were identified. For genetic rescue experiments, B6-Stmn2−/+ were crossed with both B6-Tg(STMN2) transgenic lines. F1 heterozygous Stmn2−/+ and hemizygous Tg(STMN2) mice were crossed with B6-Stmn2−/+ to generate B6-Stmn2−/−Tg(STMN2) (Jax catalog number 36033 for BAC-line 9439, and Jax catalog number 36034 for BAC-line 9446), homozygous for the mouse Stmn2-null allele and hemizygous for the human BAC-transgene. At least 150 progeny per strain were analyzed, and genotype rates and survival were determined to evaluate rescue of perinatal lethality associated with murine Stmn2 loss on C57BL/6J background.
All genotyping primers are detailed in Supplementary Table 1.
Human STMN2 copy number analysis on BAC-Tg lines
Transgene copy number was determined by droplet digital PCR. Genomic DNA (gDNA) was extracted from tissues of hemizygous BAC-Tg(STMN2) mice (8 weeks old, N=3 by sex and genotype), and 20 ng were used by reaction. A FAM-labeled probe was used to target human STMN2 gene (dHsaCNS772300147, Bio-Rad) and a Hex-labeled probe was used to target mouse ApoB (dMmuCNS407594696, Bio-Rad) as two copies reference gene. Copy number was determined using QuantaSoft™ Analysis Pro v1.0.596.0525 from Bio-Rad, and reported as copies/mouse genome.
RNAi AAV vector development
One hundred Psm2 97-mer hairpin oligos were designed against the 3’UTR of the murine Stmn2 mRNA using the “Hannon Lab RNAi Central siRNA design tool” (http://katahdin.mssm.edu/siRNA/RNAi.cgi?type=shRNA). Top candidate hairpin sequences were hand-picked using internal sequence content criteria and bioinformatic filtering to eliminate potential off-targets. The top five hairpin design sequences were produced as ssDNA oligonucleotides (IDT) and amplified as previously described45 for plasmid integration into a miR30a backbone vector by restriction digest and ligation. A control hairpin was similarly produced targeting the bacterial beta-galactosidase gene (with no predicted human or mouse target). The resulting microRNA-embedded RNAi elements were subcloned into a custom AAV transfer vector encoding an RNA Polymerase II driven human Ubiquitin C promoter and Clover fluorescent protein coding sequence, with RNAi elements inserted between Clover stop codon and an SV40 polyadenylation signal sequence. All bacterial plasmids were grown in One Shot Stbl3 chemically competent E. coli (Thermo) at 30°C to prevent recombination of hairpin sequence or inverted terminal repeat (ITR) elements. Candidate and control vectors were tested for activity by transient transfection with Trans-IT LT1 transfection reagent (Mirus) into murine neuron-like N2A cells followed by Trizol RNA isolation (Thermo) and qRT-PCR quantification with primers amplifying murine Stmn2 and Gapdh mRNAs. The vector with the top activity against murine Stmn2 was produced at high volume alongside the control vector for viral packaging (>400μg plasmid per transfer vector), followed by complete sanger sequencing vector verification in 1% DMSO containing reactions. AAV9 rep/cap and helper plasmids were produced in parallel, and virus was generated at the UCSD viral core by 3-plasmid transfection into 293T cells, with virions purified as previously described47. Viral genome titers were measured by limiting dilution series and qPCR using primers to amplify the Clover coding sequence and using the transfer plasmid dilution series as a quantification reference. Hairpin oligonucleotide sequences are listed in Supplementary Table 2.
Subpial injections
Subpial vector delivery in mice was performed as previously described50. In anesthetized mice with Isofluorane 2–3%, a skin incision was made at lumbar (Th8-L1) vertebra level. Using a dissecting microscope, a dorsal laminectomy of Th12 vertebra was performed to expose and cut open with a 30G needle the dura matter membrane overlying the L1-L2 spinal segment. Exposed pia matter was punctured with a 36 G penetrating needle, immediately after, a delivery needle (blunt 36 G needle) loaded with the scAAV9 vector was inserted. Precise placement of both, pia-penetrating and subpial delivery needle was accomplished using a fine XYZ manipulator (SMM 100B; Narishige, Tokyo, Japan). The viral vector scAAV9-UbC-Clover-miR30-ShRNA-Control (1.0X1013gc/ml) or scAAV9-UbC-Clover-miR30-ShRNA-STMN2 (1.0X1013gc/ml) was then delivered into lumbar subpial space. The vector solution was diluted 1:2 with PBS1X just before the injection and a total of 10μl was delivered (5μl bilateral over a 5 min each) using a 50 μl Hamilton syringe and a manual infusion pump (Stoelting, cat# 51222). Animals received subcutaneous fluids (Ringer lactate solution) antibiotics (cefazolin, 10mg/kg) and pain medication (Buprenorphine sustain release, 0.05mg/Kg).
Hindlimb clasping test
The hindlimb clasping test was performed as described previously for other mouse models of neurodegeneration85,86. Depending on the hindlimb placement and the duration of the position the animal received a score from 0 to 386.The average score of three consecutive liftings was used for each mouse.
Grip strength test
Forelimbs or hindlimbs paws were independently tested using a grip strength meter (Columbus Instruments, 04219). Animals were hold allowing them to grasp a bar with only the forelimbs or the hindlimbs paws and gently pulled back with steady force until both paws released the bar. Peak tension in gram was recorded for 5 consecutive trials. For Sarm1 and Stmn2-deficient mice cohorts and their matching controls, grip strength was assessed using a grid attachment to the instrument, recording the front paws and all four paws strength, the average strength in grams of three consecutive trials was recorded.
Rotarod test
Motor coordination was evaluated using an Ugo-Basile accelerating rotarod model 47600. Animals were acclimated to the testing room for 60min and subjected to a “training session” on day one and a “test” session on day 2. Mice were placed on a rotating rod at 4rpm which accelerates up to 40rpm over the course of 300-secs. Each mouse was subjected to four consecutive trials with a 45-sec resting interval. The time in seconds when the mouse falls from the rod was recorded. Average latency to fall of the last three trials was reported.
Mechanical sensitivity assays
Mechanical threshold was assessed on right and left glabrous hindpaw skin of the animals using a method adapted from previous studies87–89. Mice were placed into individual testing cages on a wire mesh bottom to acclimate for at least 30min before any testing. Calibrated von Frey filaments (1.65–4.31) were applied to determine the 50% paw withdrawal using the Up-Down method90–92. Frequency of withdrawal to supra-threshold mechanical stimuli was evaluated as a mechanical sensitivity measurement. Measurements were recorded by applying bending forces of 1, 4 and 8 g filaments to the base of the third toe on the plantar surface of both paws 10 times during each testing period to determine the response frequency for each filament. Chosen von Frey filaments warranted a supra-threshold stimulus within the noxious mechanical range93–96.
Compound muscle action potential
Compound action potential (CMAP) analysis was conducted as previously described97,98. CMAP responses were recorded from the proximal hindlimb using two recording electrodes. The active electrode was positioned over the tibialis anterior (TA) muscle. The reference electrode was positioned at the metatarsal region of the foot on the same limb. Supramaximal stimulation of the sciatic nerve was elicited via two needle electrodes placed subcutaneously over the sacrum (anode) and the sciatic notch (cathode). Stimulations were delivered through a Stimulus Isolator (FE180, AD Instruments). CMAP amplitude was recorded and the peak-to-peak amplitude of the CMAPs was measured. For mice with stathmin-2 knock-down either by ASOs or subpially delivered AAV9, responses were recorded from the TA using 30G platinum transcutaneous needle electrodes (distance between recording electrodes ~1cm; Grass Technologies, Astro-Med). Recording electrodes were connected to an active headstage (3110W; Warner Instruments). Amplified signal was acquired by the PowerLab 8/30 data acquisition system (ADInstruments) at a sampling frequency of 20kHz, digitized and analyzed.
Nerve conduction velocity
Nerve conduction velocity at the sciatic nerve was determined as previously described99 by measuring differences in compound muscle action potential (CMAP) latency in the muscles of the hind paw following sciatic nerve stimulation at two points, hip and ankle. For recording, the active needle electrode was inserted in the center of the paw and a reference electrode was placed in the skin between the first and second digits. The distance between points of stimulation and the recorded latencies was used in the calculation of velocity.
Tissue collection
Animals were euthanized with pentobarbital (100mg/kg) and perfused with 20 ml of ice-cold PBS 1X. Mice tissues were collected as follows: spinal cords were divided in cervical, thoracic, and lumbar segments. Each segment was then divided in 3 equal size pieces. The rostral pieces were post-fixed for 24 h in freshly prepared 4% paraformaldehyde (PFA) (4% PFA dissolved in 0.15 M sodium phosphate buffer, pH 7.4) at 4°C and the other 2 pieces flash frozen in dry ice. Left and right muscles were dissected and post-fixed for 5 days in 4% PFA at 4°C. Right and left dorsal root ganglion L5 (DRG L5) were dissected and post-fixed for 24 h in 4% PFA at 4°C. L3,4,6 lumbar DRG’s were pooled and flash frozen in dry ice. Samples for immunofluorescence and FISH were cryo-protected for 72 hours in 30% sucrose buffer after their post-fixation period and stored at 4°C. Animals used for electron microscopy were euthanized with pentobarbital and the whole body perfused with 20 ml of ice-cold PBS 1x followed by 20 ml freshly prepared fixative (2 % PFA, 2 % glutaraldehyde, 0.1 M cacodylate buffer pH=7.2). After perfusion, L5 dorsal and ventral roots, and sciatic nerves were dissected, and preserved in the same fixative solution at 4°C until processing.
Immunofluorescence
Cryoprotected tissues were embedded in Optimal Cutting Temperature (OCT) matrix compound (Tissue-Tek, Sakura Finetek), frozen with dry ice and mounted in the cryostat. Sections were cut from the spinal cords (30-μm thickness), DRGs (15-μm thickness) or muscles (40-μm thickness). Free-floating sections were washed three times in wash-buffer (PBS 1X with 0.3% Triton-X100) followed by a blocking step with 5% donkey serum in wash-buffer for 1h. Sections were incubated in primary antibodies (in blocking solution) overnight at 4°C, then washed three times with wash-buffer and incubated with secondary antibodies diluted in blocking solution for 1h at room temperature. Muscle endplates were labeled with 555-α-bungarotoxin (1:500) for 1h at room temperature. Sections were mounted on slides, dried at room temperature and cover slipped in ProLong Gold antifade mounting medium, with DAPI (Invitrogen). Primary and secondary antibodies and dilutions used are described on Supplementary Table 3. Antigen retrieval was performed to detect stathmin-2 protein in spinal cords and DRGs. 30-μm sections were mounted on slides and let dried overnight. Sections were washed 3 times with wash-buffer and immersed in pre-heated (90–100°C) 1X antigen retrieval solution (Dako, cat # S1699) for 25 min, and washed with PBS 1X. At least 2 animals per comparing conditions were processed, stained, and imaged in every round of experiments. Image acquisition was done on LEICA SP8 high resolution confocal microscope. Fluorescence intensity from unsaturated images were captured with identical confocal settings. Image processing was done using Fiji Software100 and comparing samples were treated identically.
Fluorescent In situ Hybridization - FISH
Spinal cord and DRGs were cryo-sectioned (thickness of 15 μm), directly mounted on Superfrost Plus slides (Thermo Fisher) and dried overnight at room temperature. RNA FISH was performed using RNAscope Multiplex Fluorescent v.2 (no. 323100), following fixed frozen tissue protocol according to manufacturer’s instructions. A mouse-Stmn2-specific RNAscope probe (498391-C2; NM_025285.2, 20 pairs, 898 −1849 nucleotides, ACD) was used (Supplementary Table 4). Hybridization signals were detected by a chromogenic reaction using red chromogen dilution 1:3000 (PerkinElmer TSA Plus Cyanine 3 System). RNA quality was evaluated for each sample using RNAscope 3-plex Positive Control Probe containing the housekeeping gene cyclophilin B, also known as peptidylprolyl isomerase B (PPIB), RNA polymerase subunit IIA (PolR2A) and ubiquitin C (UBC) specific for mice. Negative control background staining was evaluated using a probe specific to bacterial dapB gene (RNAscope 3-plex Negative Control Probe).
Motor neuron counting
Lumbar ChAT positive ventral horn motor neurons were counted from 10–15 lumbar spinal cord cryosections (per animal) spaced 300μm apart and expressed as total motor neurons counted per spinal cord hemi section.
Neuromuscular junction innervation evaluation
Innervation analysis of neuromuscular junctions (NMJs) in gastrocnemius muscles was performed on images obtained on SP8 Leica confocal microscope. Individual NMJs were considered as innervated when the presynaptic axon, (labelled either by neurofilament-H+synaptophysin or β3-tubulin+synaptophysin) staining covered at least 50% of the area of α-bungarotoxin staining. Partially denervated was considered when the area of α-bungarotoxin staining was covered between 10 to 50% by the axon terminal staining, while when no axon terminal labelling was detected in the α-bungarotoxin positive area, the NMJ was considered denervated.
CGRP and IB4 Quantification in the spinal cord
Maximum projections were analyzed using Fiji Software100. A region of interest was created to selectively analyze CGRP and IB4 projections arriving to the dorsal horn of the spinal cord. To enhance detection of the borders, images were blurred with a gaussian blur. The area of CGRP and IB4 staining was determined by counting the pixels with intensity higher than a pre-set threshold (constant for all analyzed images) inside the respective regions of interest.
Morphometric Analysis of Axons
L5 roots were embedded in Epon-Araldite as described in the electron microscopy section, thin sections (0.75μm) were prepared and stained for light microscopy with toluidine blue. Cross sections of L5 motor axons were analyzed at each experimental group. Diameters of all axons in the ventral and dorsal roots were measured and determined using Fiji Software100.
RNA extraction for qRT-PCR quantification
Tissue sections snap frozen at the time of collection were mechanically homogenized in 1ml Trizol (Thermo) with a rotor stator homogenizer (Omni International). RNA was extracted using chloroform and quantified on a Nanodrop spectrophotometer (Thermo). 1μg (or 200ng for the DRGs) was used for first-strand cDNA synthesis with SuperScript-III reverse transcriptase (Thermo) and oligo dT priming. cDNA was diluted to 1ng/μL, and 4ng loaded per 10μL SYBR Green reaction (BioRad), with three technical replicates loaded per biological sample, run on a C1000 qPCR thermocycler (BioRad) using primers detailed in Supplementary Table 2. All melt-curves showed a single distinct amplification product. Murine Gapdh and Rps9 genes were used as endogenous controls and showed equivalent results. The data normalized with Gapdh was graphed for the figures. Relative expression for each gene was calculated from delta-Cq data.
Tissue Protein Extraction and Immunoblotting
Mouse spinal cords were homogenized in cold RIPA Buffer supplemented with protease and phosphatase inhibitors and extracts were clarified by cold centrifugation for 20min at 20,000g. Equal protein amounts were separated by SDS-PAGE, transferred to nitrocellulose membranes, and probed with the indicated antibodies diluted in 5% nonfat dry milk in Tris-buffered saline (TBS)-Tween 0.1%, followed by horseradish peroxidase-conjugates (HRP) Sheep Anti-Mouse IgG or Donkey Anti-Rabbit IgG secondary antibodies (Prometheus, 1:5000–1:15000 depending on the primary antibody). Immunoblots were developed on film through enhanced chemiluminescence (ECL).
Human postmortem tissues
Human tissues were obtained using a short postmortem interval acquisition protocol that followed HIPAA-compliant informed consent procedures and were approved by Institutional Review Board (Benaroya Research Institute, Seattle, WA IRB# 10058 and UCSD, San Diego, CA IRB# 120056). Tissue samples were obtained from patients who met the modified El Escorial criteria for definite ALS. Nervous systems used as control samples were obtained from non-neurological patients either after life support was withdrawn or from patients on hospice care. Autopsies were performed within 8 hours of death, with an average post-mortem interval of 5 hours for the cohort of patients used in this study.
Human roots processing and morphometric analysis of axons
Human tissues were fixed using neutral buffered formalin. Roots used for electron microscopy were transferred into freshly prepared fixative solution (2% PFA, 2% glutaraldehyde, 0.1 M cacodylate buffer pH=7.2) and kept at 4°C until embedding in Epon-Araldite. For morphometric analysis of axons, 0.75μm sections were stained with toluidine blue.
Statistical analysis and reproducibility
All mice were randomly allocated into age-matched experimental groups. The data were analyzed for statistical significance and graphed using GraphPad Prism 9 Version 9.5.1. Whether data followed a normal distribution was determined. Whenever the data did not follow a normal distribution, nonparametric tests were used. P < 0.05 was considered significant. Statistical tests and number of biological replicates used throughout the manuscript are depicted in the Figure Legends and Source Data files. Investigators were blinded during data collection and image analysis, and the order of images analyzed was randomized. Specific number of animals or other biological replicates processed and quantified for immunofluorescence and electron microscopy are annotated in the figure legends.
Data and materials availability:
All mouse strains are publicly available through The Jackson Laboratory. Other materials are available under materials transfer agreement for noncommercial replication or extension of this work, upon request to corresponding authors. All other information is available in the manuscript or the supplementary materials. Figures 1a,2a and 4a were created with BioRender.com.
Extended Data
Extended Data Figure 1: Intraventricular ASO delivery efficiently reduces stathmin-2 expression in mouse cortex and spinal cord.
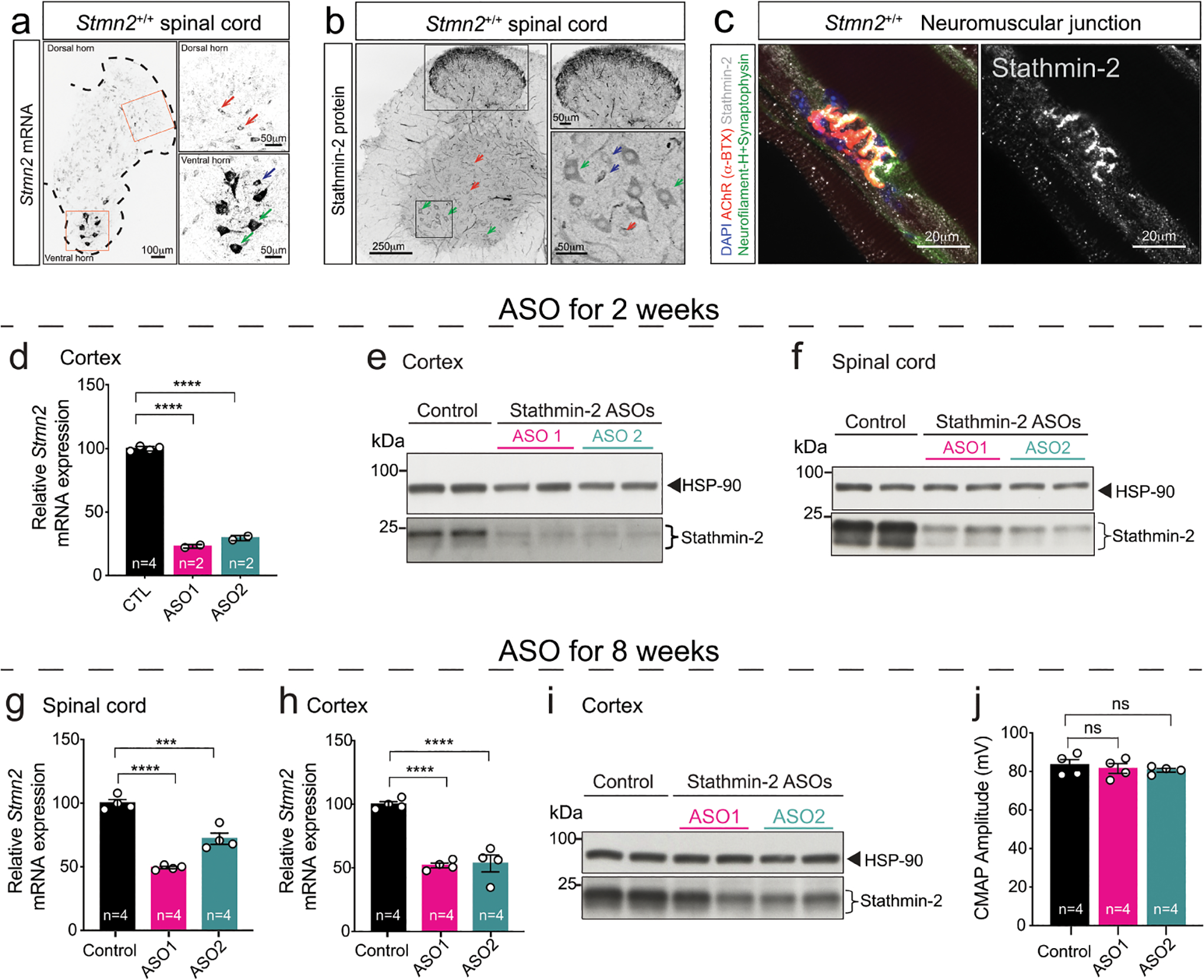
(a-b) Stmn2 mRNA levels detected by FISH (a), and immunofluorescence confocal image immunolabeled for stathmin-2 protein (b), from 12-month-old WT mice spinal cord hemisections. Green arrows: α-motor neurons; blue arrows: γ-motor neurons; red arrows: interneurons. (c) Immunofluorescence confocal image of 12-month-old WT mice gastrocnemius muscle revealing stathmin-2 presence at the neuromuscular junction. (a-c) At least n=3 animals per condition were imaged with similar results. (d,e) Quantification of Stmn2 mRNA levels by qPCR (d) and immunoblots (e) showing stathmin-2 protein levels in mice cortex 2 weeks after the ICV injection of non-targeting (n=4 animals) or Stmn2 targeting ASOs (n=2 animals/per ASO). HSP-90 was used as a loading control in the immunoblotting. Statistics by two-sided, one-way ANOVA Dunnett’s multiple comparison test (P < 0.0001). (f) Immunoblots showing stathmin-2 protein levels in mouse spinal cord 2 weeks after the ICV injection of non-targeting or Stmn2 targeting ASOs. HSP-90 was used as a loading control. *Indicates non-specific band. (g) Quantification of Stmn2 mRNA levels by qPCR in mouse spinal cord 8 weeks after the ICV injection of non-targeting (n=4 animals) or Stmn2 targeting ASOs (n=4 animals/per ASO). Gapdh was used as an endogenous control gene. *** P = 0.0002 and **** P < 0.0001 (h,i) Quantification of Stmn2 mRNA levels by qPCR, (P < 0.0001) (h), and immunoblots (i) showing stathmin-2 protein levels in mice cortex 8 weeks after the ICV injection of non-targeting (n=4 animals) or Stmn2 targeting ASOs (n=4 animals/per ASO). HSP-90 was used as a loading control in the immunoblotting. Gapdh was used as an endogenous control gene. (j) Compound muscle action potential (CMAP) measurements in muscles of WT mice treated with non-targeting (n=4 animals) or Stmn2 targeting ASOs (n=4 animals/per ASO) for 8 weeks. (g,h,j) Statistics by two-sided, one-way ANOVA Dunnett’s multiple comparison test. All panels: Each data point represents an individual mouse. Error bars plotted as SEM. ****, P <0.0001; ***, P < 0.001; **, P < 0.01; *, P <0.05; ns, P >0.05.
Extended Data Figure 2: Sustained stathmin-2 depletion induces axonal withdrawal from neuromuscular junctions without compromising motor neuron survival.
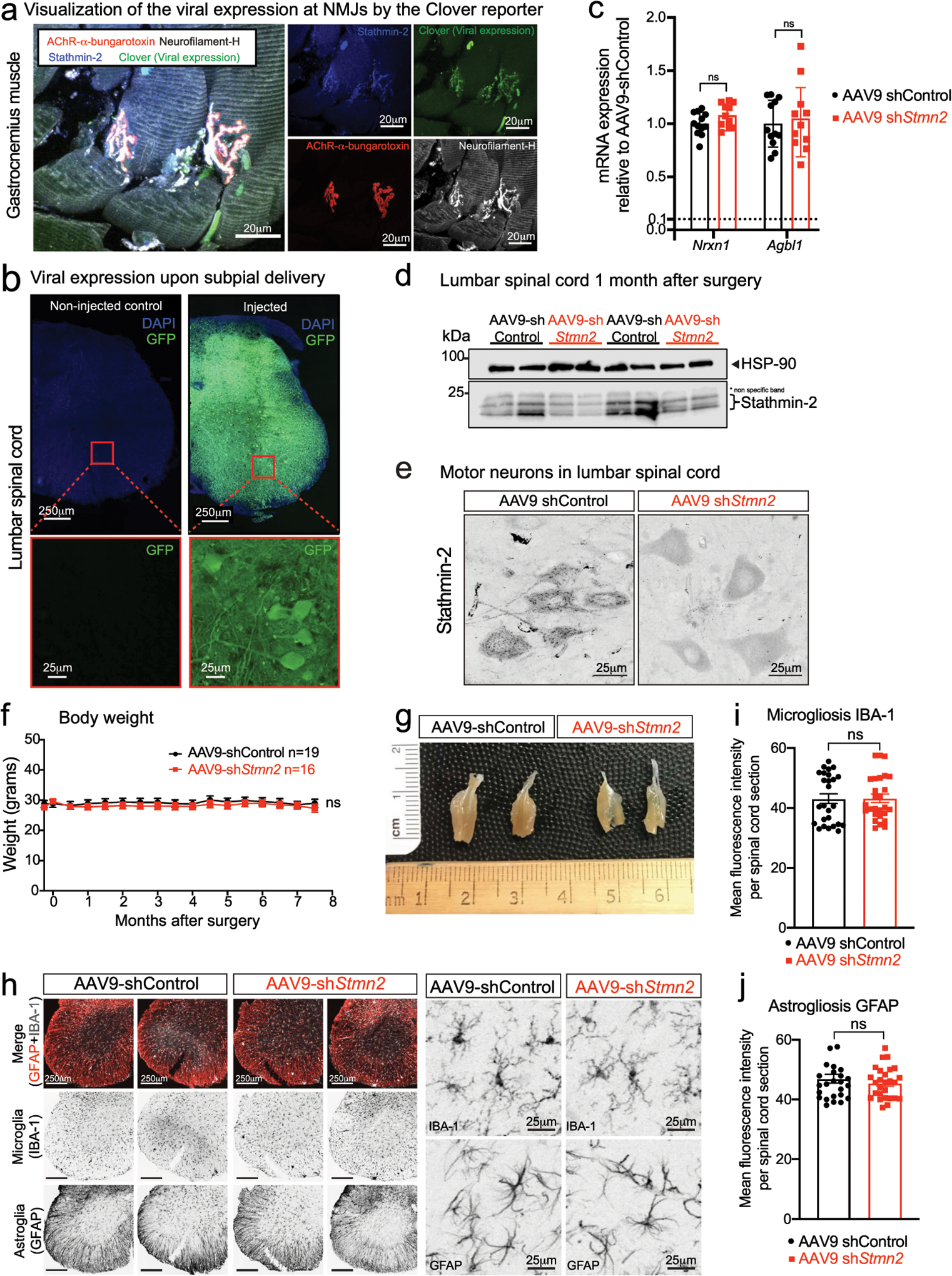
(a) Representative confocal image of gastrocnemius muscle stained for stathmin-2 (blue), muscle AChR clusters using α-bungarotoxin (red), direct imaging of clover in the 488-wavelength (green) representing viral expression, and neurofilament-H (white). At least n=3 animals were imaged with similar results. (b) Representative image of lumbar spinal cord of non-injected (left) and 2 months after subpial injection with AAV9 expressing green fluorescent protein (GFP) (right) with the respective high magnification images of the ventral spinal cord regions, below each panel. At least n=3 animals were imaged with similar results. (c) Measurement in lumbar spinal cord segments at 8-months post injection of control or Stmn2 targeting AAV9 of potential off-target genes by qRT-PCR. N=12 animals with AAV9-shControl and n=11 animals with AAV9-shStmn2. Gapdh was used as an endogenous control gene. Statistics by two-sided, unpaired t-tests. (d) Immunoblots to determine stathmin-2 protein level in mouse lumbar spinal cord 1-month after subpial injection of a Stmn2 reducing AAV9 or control shRNA. HSP-90 was used as a loading control. *Indicates non-specific band. (e) Mouse lumbar spinal cord immunofluorescence micrographs visualized with stathmin-2 antibody 8 months after subpial injection into the lumbar spinal cord of non-targeting control or Stmn2-reducing AAV9. (f) Bi-weekly measurements of mouse body weight after subpial injection of AAV9 encoding either non-targeting or Stmn2-targeting shRNA. Statistics by two-sided, two-way ANOVA and Sidak’s multiple comparison test. (g) Representative images of entire gastrocnemius muscles from mice 8 months after subpial delivery of AAV9 encoding either a non-targeting or an Stmn2-shRNA AAV9. (h-j) Representative immunofluorescence images of mouse lumbar spinal cord stained with the microglial and astrocytic markers IBA1 and GFAP (h) and quantification of microgliosis (i) and astrogliosis (j), 8 months after subpial delivery of a non-targeting control (n=4 animals) or Stmn2-targeting AAV9 shRNA (n=4 animals). Statistics by two-sided, unpaired t-tests. All panels: Each data point represents an individual mouse. Error bars plotted as SEM. ns, p>0.05.
Extended Data Figure 3: Decreased phosphorylation of NF-M and NF-H upon sustained Stmn2 suppression.
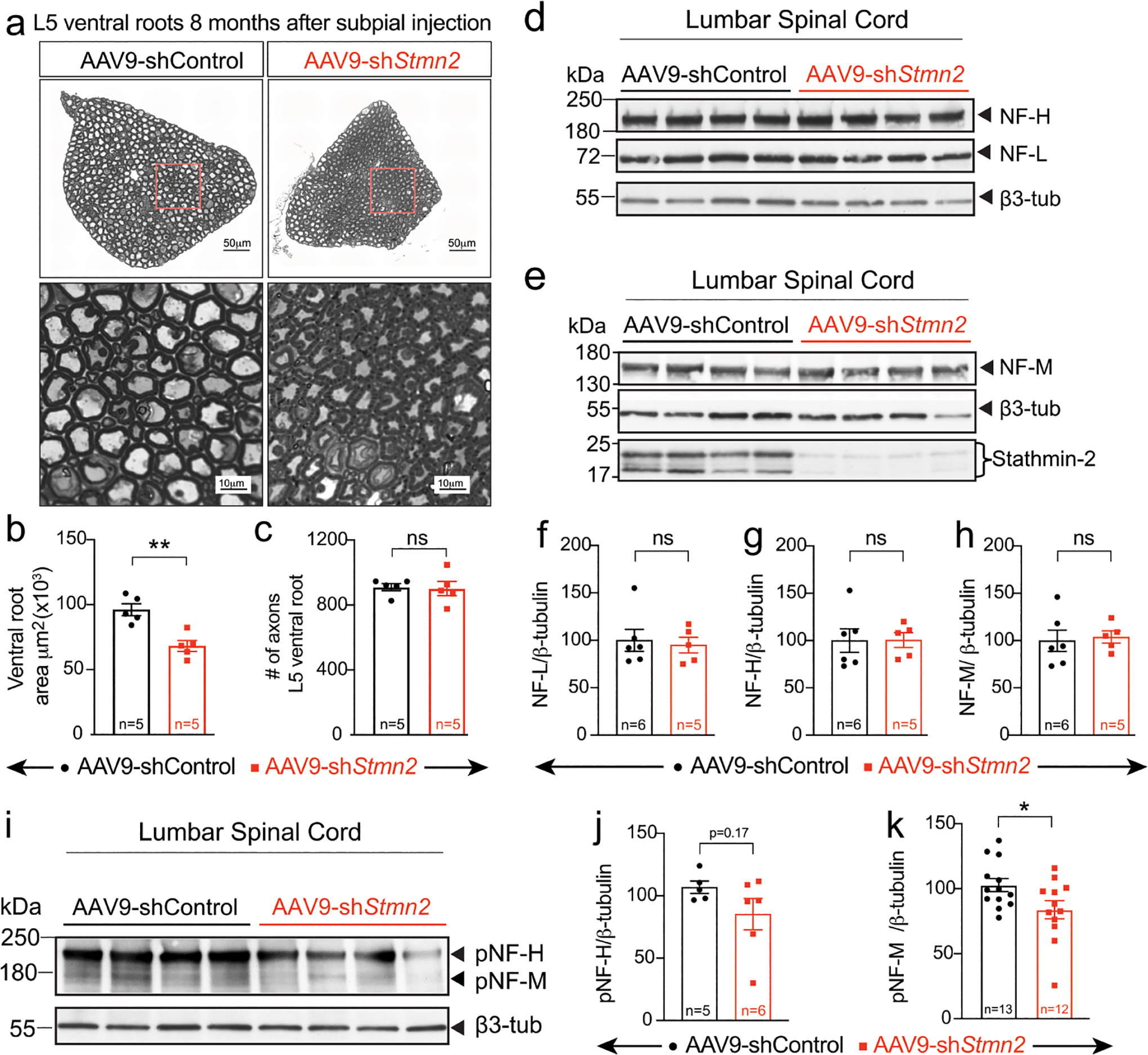
(a) Representative micrographs of motor roots and higher magnification images of ventral root motor axon morphology and diameters, 8 months after subpial injection of AAV9 encoding non-targeting or Stmn2 targeting shRNA. (b,c) Quantification of cross-sectional area (n=5 animals/condition) (b) and of the total number of axons per ventral root (n=5 animals/condition) (c). Statistical analysis by two-sided, Mann Whitney t-test, (P = 0.0079). (d,e) Levels of total neurofilament heavy (NF-H) and neurofilament light (NF-L) (f) and total neurofilament medium (NF-M) (g) analyzed by immunoblotting spinal cord extracts from WT mice 8 months after subpial injection of either AAV9 encoding a non-targeting (n=6 animals) or Stmn2 targeting shRNA (n=5 animals). β3-tubulin was used as loading control. AAV9-shRNA-mediated suppression of stathmin-2 protein levels was confirmed in all examined samples (e). (f-h) Quantification of the immunoblots in panels d,e. Statistics by two-sided, unpaired t-test. (i) Levels of phosphorylated neurofilament heavy (pNF-H) and medium (pNF-M) subunits analyzed by immunoblotting of spinal cord extracts from WT mice, 8 months after subpial AAV9 encoding a non-targeting or Stmn2 targeting shRNA. β3-tubulin was used as a loading control. (j,k) Quantification of the immunoblots from panel i. N=5 animals with AAV9-shControl and n=6 animals with AAV9-shStmn2 (j), and n=13 animals with AAV9-shControl and n=12 animals with AAV9-shStmn2, (P = 0.036) (k). Statistics by two-sided, unpaired t-test. All panels: Each data point represents an individual mouse. Error bars plotted as SEM. ****, P <0.0001; ***, P < 0.001; **, P < 0.01; *, P <0.05; ns, P >0.05.
Extended Data Figure 4: Axonal shrinkage, collapse of neurofilament spacing, and tearing of myelin in sporadic ALS and C9-ALS.
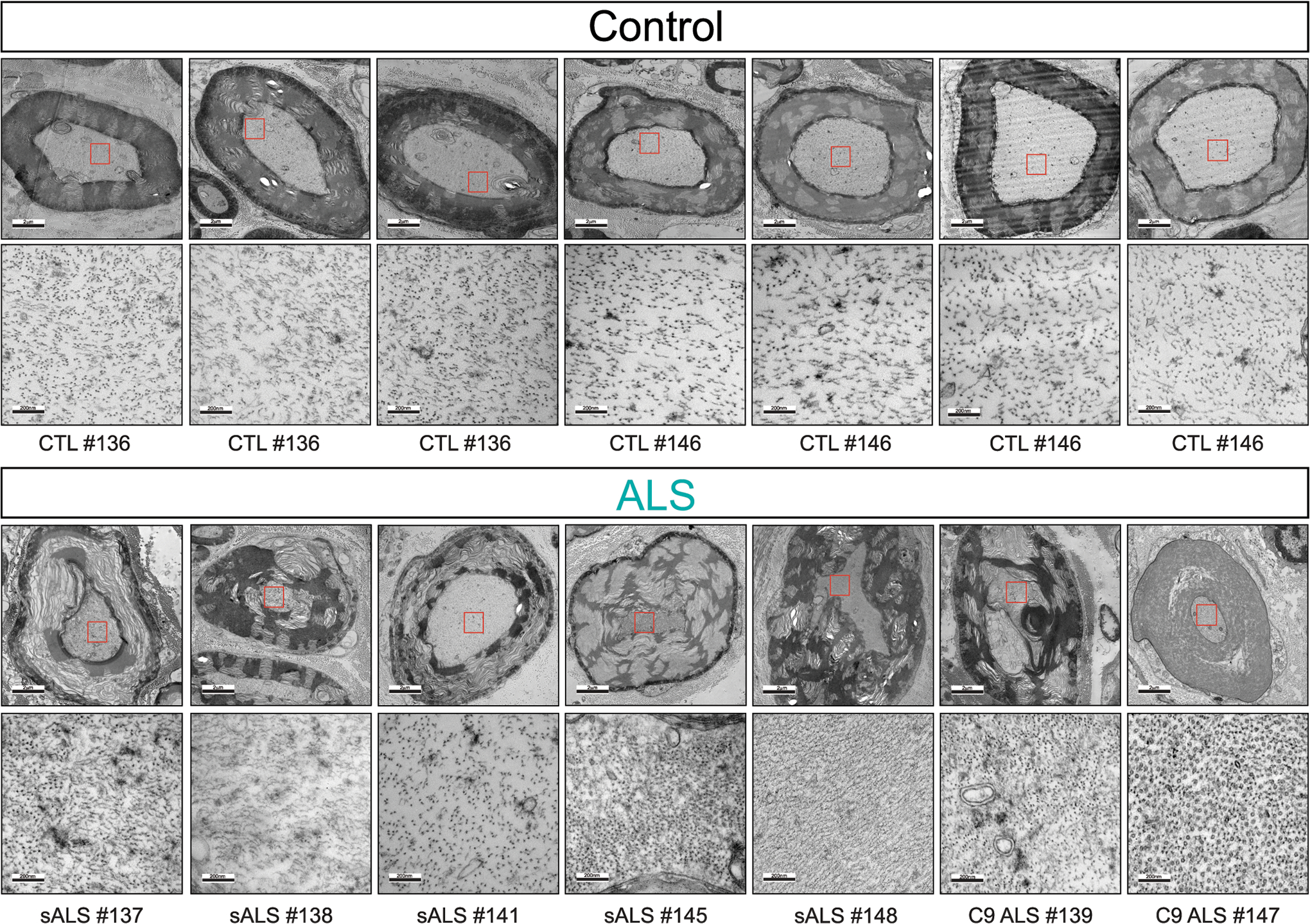
Representative electron microscopy images of large caliber axon cross sections in the motor roots of postmortem human samples from healthy controls n = 2 (upper panel) and sporadic ALS (sALS, n=5) and C9orf72 ALS patients (C9 ALS, n=2) (lower panel). Increased magnification micrographs of the axoplasm showing altered spacing between neurofilament filaments is shown.
Extended Data Figure 5. Reduced stathmin-2 levels by subpial injection alters sensory marker in lumbar spinal cord:
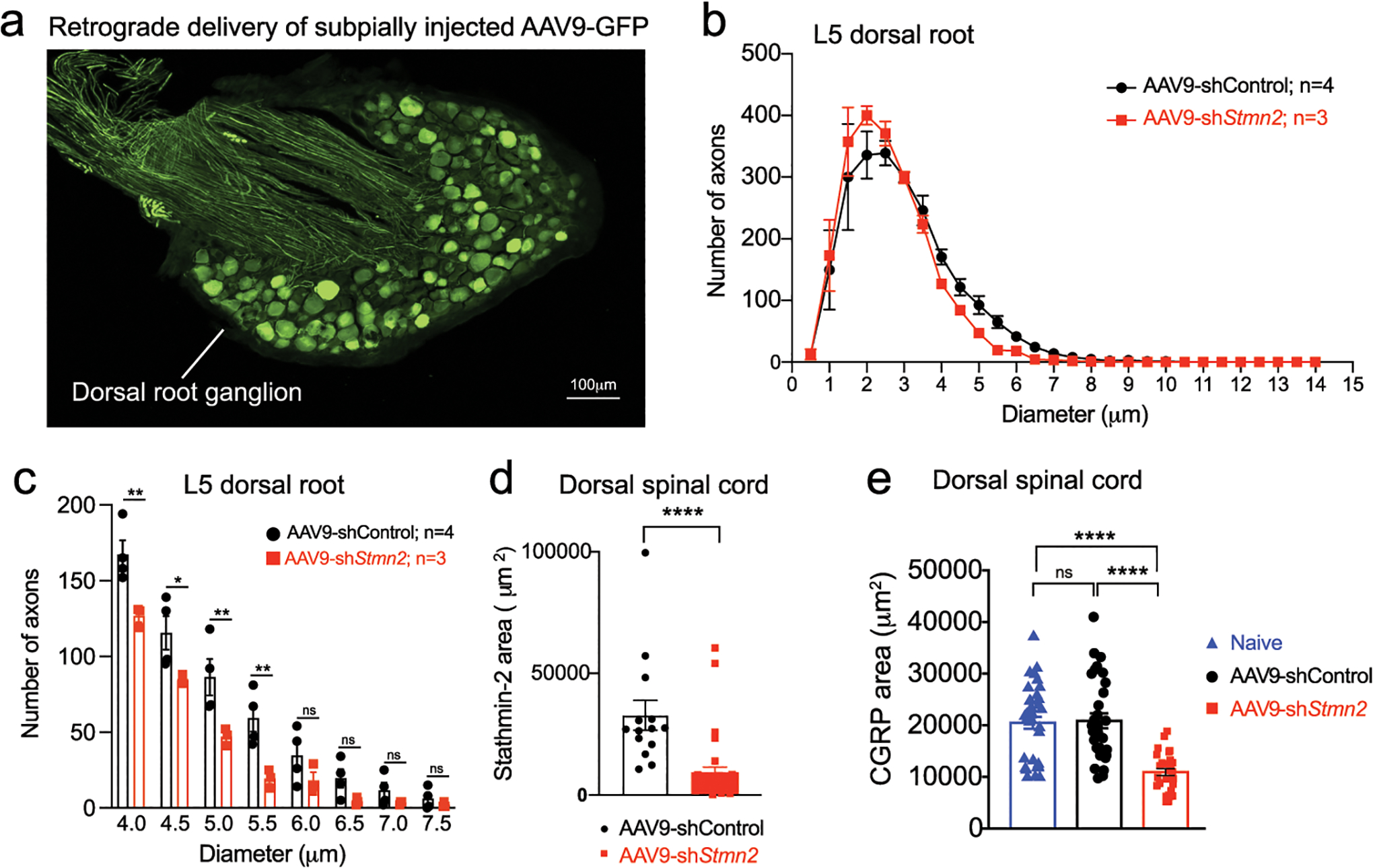
(a) Representative image of lumbar dorsal root ganglion 2 months after subpial injection into lumbar spinal cord and subsequent retrograde delivery of AAV9 expressing green fluorescent protein (GFP). (b-c) Size distribution of axonal diameter of sensory axons innervating the dorsal spinal cord (b), and axon numbers in the 4 μm to 8 μm diameter range in the L5 dorsal root (c). Statistical analysis by two-sided, two-way ANOVA and Sidak’s multiple comparison test. P values range from P = 0.0189 to P = 0.0028. N=4 animals with AAV9-shControl and n=3 animals with AAV9-shStmn2. (d-e) Quantification related to Figure 4i,k of positive area for stathmin-2 (n=2 animals injected with AAV9-shControl and n=4 animals injected with AAV9-shStmn2, (P <0.0001) (d), and CGRP (e) in the dorsal spinal cord of age-matched non-injected naïve animals (n=5) or 8 months after subpial injection of AAV9 encoding either non-targeting sequence (n=4) or Stmn2 shRNA (n=4). P <0.0001. Statistical analysis by two-sided, Mann Whitney test (d) and Kruskal-Wallis nonparametric tests (e). All panels: Each data point represents an individual mouse. Error bars plotted as SEM. ****, P <0.0001; ***, P < 0.001; **, P < 0.01; *, P <0.05; ns, P >0.05.
Extended Data Figure 6: Stathmin-2 related genes remain unchanged upon stathmin-2 loss.
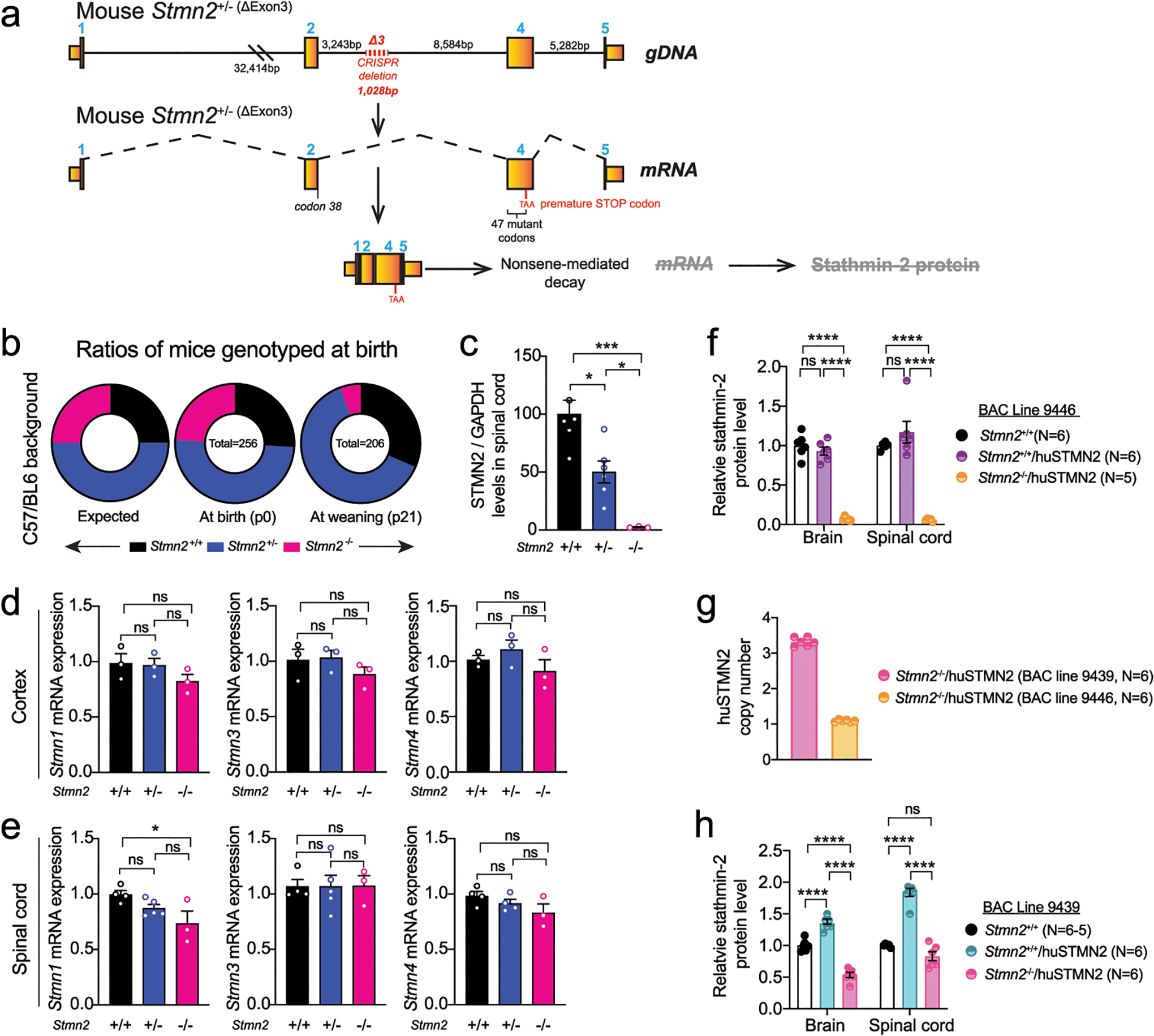
(a) Diagram of genome editing design by CRISPR-Cas9-mediated excision of mouse Stmn2 exon 3 that leads to complete absence of stathmin-2 protein. (b) Ratios of mice expected, genotyped at birth (p0) and alive at weaning age (p21) from Stmn2+/− to Stmn2+/− crossing in C57/BL6J background. (c) Stathmin-2 protein quantification from the immunoblots in Fig. 5d normalized by GAPDH level. N=6 animals per Stmn2+/+ and Stmn2+/−; n=3 animals per Stmn2−/−. Statistical analysis by two-sided, one-way ANOVA post hoc Tukey’s multiple comparison test. * P = 0.0103; * P = 0.041; *** P = 0.0003. (d,e) Measurement of mouse Stmn-1, −3 and −4 mRNA levels extracted from 12-month-old cortex (d) and spinal cord (e) of Stmn2+/+, Stmn2+/− Stmn2−/− mice. Gapdh was used as an endogenous control gene. N=3 animals per genotype (d), and n= 3–5 per genotype (e). Statistical analysis by two-sided one-way ANOVA post hoc Tukey’s multiple comparisons test. (f) Stathmin-2 protein quantification from brain and spinal cord extract of Stmn2+/+, Stmn2+/+/huSTMN2 and Stmn2−/−/huSTMN2 (BAC line 9439) by immunoblotting. N=5 animals per genotype. (g) huSTMN2 transgene copy numbers measured in BAC transgenic lines 9439 and 9446. N=6 animals per genotype. (h) Stathmin-2 protein quantification from brain and spinal cord extract of Stmn2+/+, Stmn2+/+/huSTMN2 and in Stmn2−/−/huSTMN2 (BAC line 9446) by immunoblotting. N=6 animals per genotype. (f,h) Statistical analysis by two-sided, two-way ANOVA post hoc Tukey’s multiple comparisons test, (P <0.0001). All panels: Each data point represents an individual mouse. Bar graphs represent mean values. Error bars plotted as SEM. ****, P <0.0001; ***, P < 0.001; **, P < 0.01; *, P <0.05; ns, P >0.05.
Extended Data Figure 7: Absence of stathmin-2 results in motor deficits regardless of the genetic background.
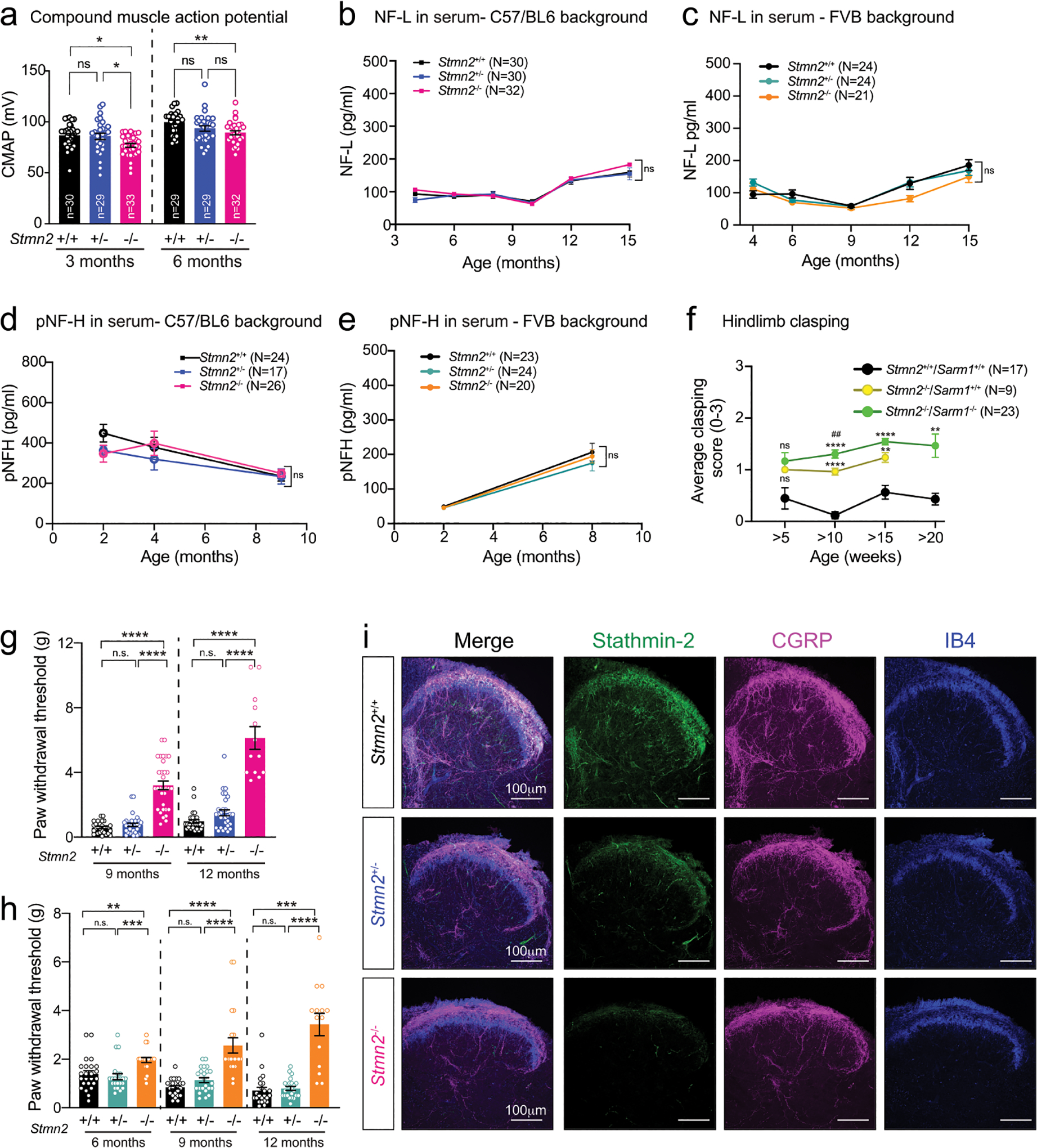
(a) Compound muscle action potential (CMAP) measurements in gastrocnemius muscle of mice. Statistics by two-sided, one-way ANOVA post hoc Tukey’s multiple comparisons test. P = 0.0131; P = 0.0252 at 3 months; P = 0.0045. (b-c) Neurofilament light (NF-L) levels in serum at different time-points in Stmn2+/+, Stmn2+/− and Stmn2−/− mice in the C57/BL6J (b) and FVB (c) backgrounds. (d-e) Phosphorylated neurofilament heavy (pNF-H) levels in serum at different time-points in Stmn2+/+, Stmn2+/− and Stmn2−/− mice in the C57/BL6J (d) and FVB (e) backgrounds. (b-e) Statistics by two-sided, two-way ANOVA post hoc Tukey’s multiple comparison test. (f) Hindlimb clasping test of Stmn2+/+/Sarm1+/+, Stmn2+/+/Sarm1−/− and Stmn2−/−/Sarm1−/− mice in C57/BL6J background for 20 weeks. Statistics by two-sided, two-way ANOVA post hoc Tukey’s multiple comparison test. **P = 0.0018; **** P <0.0001 when comparing to Stmn2+/+/Sarm1+/+. ## P = 0.0069 when comparing between Stmn2+/+/Sarm1−/− and Stmn2−/−/Sarm1−/−. (g-h) Von Frey analysis for the sensory response in hindlimbs of Stmn2+/+, Stmn2+/− and Stmn2−/− mice in C57/BL6J background at 9 and 12 month of age, (P <0.0001) (g), and in FVB background at 6, 9 and 12 month of age. P value range from P = 0.0037 to P <0.0001 (h). Statistics by two-sided, one-way ANOVA Kruskal-Wallis with Dunn’s multiple comparison test. Number of animals used were n=28 for Stmn2+/+; n=30 for Stmn2+/− and n=32 for Stmn2−/− at 9 months, and n=29 for Stmn2+/+; n=30 for Stmn2+/− and n=13 for Stmn2−/− at 12 months (g). Number of animals used were n=24 for Stmn2+/+; n=23 for Stmn2+/− and n=20 for Stmn2−/− at 6 months and 9 months; n=23 for Stmn2+/+; n=23 for Stmn2+/− and n=14 for Stmn2−/− at 12 months (h). (i) Lumbar spinal cord dorsal section of Stmn2+/+, Stmn2+/− and Stmn2−/− mice immunostained for stathmin-2 (green), CGRP (magenta) and isolectin B4 (IB4) in blue. N=3 animals/genotype were imaged. (a,g,h) Each data point represents an individual mouse. All panels: Error bars plotted as SEM. ****, P <0.0001; ***, P < 0.001; **, P < 0.01; *, P <0.05; ns, P >0.05.
Extended Data Figure 8: Absence of stathmin-2 alters neurofilament composition over time in mice spinal cords.
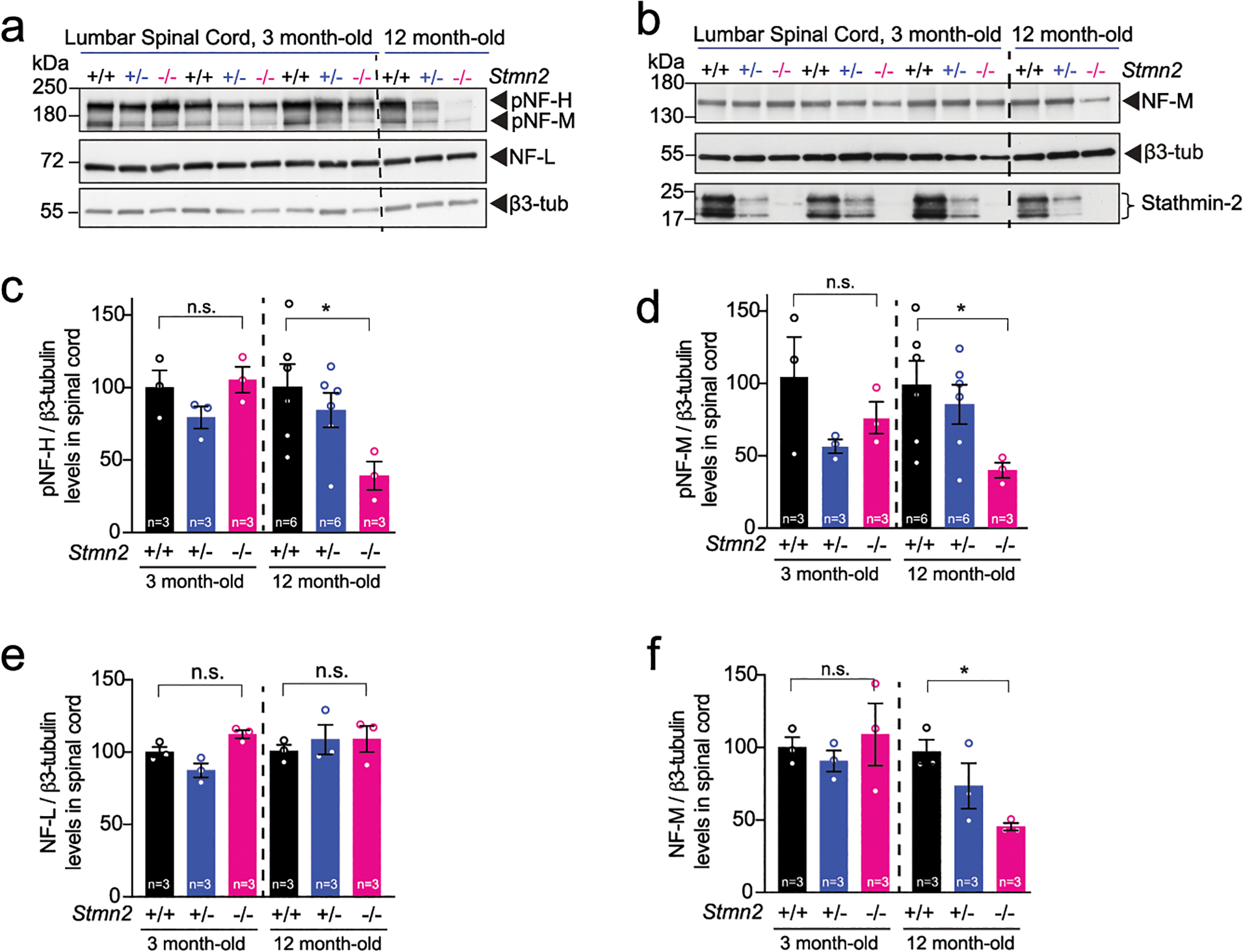
(a) Immunoblotting for phosphorylated neurofilament heavy (pNF-H), phosphorylated neurofilament medium (pNF-M), and neurofilament light (NF-L) analyzed on 3 and 12 months-old Stmn2+/+, Stmn2+/− and Stmn2−/− mice lumbar spinal cord protein extracts. (b) Immunoblotting for neurofilaments medium (NF-M) and stathmin-2 from Stmn2+/+, Stmn2+/− and Stmn2−/− mice lumbar spinal cord protein extracts at 3 and compared to 12 months-old. β3-tubulin was used as an endogenous protein loading control. Stathmin-2 levels of Stmn2+/+, Stmn2+/− and Stmn2−/− at ~21 kDa are also shown. (c-f) Quantifications from immunoblots for pNF-H (c), pNF-M (d), NF-L (e) and NF-M (f) are shown. N=3 animals per genotype were used for 3 months, and n=6 animals for Stmn2+/+ and Stmn2+/− and n=3 animals for Stmn2−/− were used for 12 months. β3-tubulin remained unchanged confirming amount of protein loading control. Statistical analysis by two-sided, unpaired t-test. Each data point represents an individual mouse. Error bars plotted as SEM. P = 0.0381 (c); P = 0.0486 (d); P = 0.0039 (f).
Supplementary Material
Acknowledgements
We are grateful to the Muscular Dystrophy Association for supporting JL-E and ZM with MDA Development grants. We thank UCSD Genetics Training Program and the National Institute for General Medical Sciences, T32 GM008666 for supporting MWB and T32AG066596-01 for supporting MSB. A.R.A.A.Q is the recipient of a Postdoctoral Fellowship from the BrightFocus Foundation (Grant Number A2022002F). This work was supported by grants from ALS finding a Cure (to CL-T), NINDS/NIH: R01NS112503 to MM, CL-T and DWC, RF1NS124203 to CML, DWC and CL-T. The microscope core was supported by NINDS/NIH P30NS047101. CL-T is the recipient of the Araminta Broch-Healey Endowed Chair in ALS.
Footnotes
Competing Interests Statement
C.F.B., M.B.-H., K.L., P.J.-n., and F.R. are employees of Ionis Pharmaceuticals. D.W.C. is a consultant for Ionis Pharmaceuticals. Z.M. and D.W.C. have a relevant patent. C.L.-T serves on the scientific advisory board of SOLA Biosciences, Libra Therapeutics, Arbor Biotechnologies and Dewpoint Therapeutics and has received consultant fees from Mitsubishi Tanabe Pharma Holdings America and Applied Genetic Technologies Corporation. All other authors declare no competing interests.
References
- 1.Rowland LP & Shneider NA Amyotrophic lateral sclerosis. N Engl J Med 344, 1688–1700 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Cleveland DW & Rothstein JD From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci 2, 806–819 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Fischer LR, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol 185, 232–240 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Killian JM, Wilfong AA, Burnett L, Appel SH & Boland D Decremental motor responses to repetitive nerve stimulation in ALS. Muscle Nerve 17, 747–754 (1994). [DOI] [PubMed] [Google Scholar]
- 5.Dengler R, et al. Amyotrophic lateral sclerosis: macro-EMG and twitch forces of single motor units. Muscle Nerve 13, 545–550 (1990). [DOI] [PubMed] [Google Scholar]
- 6.Gould TW, et al. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci 26, 8774–8786 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parone PA, et al. Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J Neurosci 33, 4657–4671 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perez-Garcia MJ & Burden SJ Increasing MuSK activity delays denervation and improves motor function in ALS mice. Cell Rep 2, 497–502 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beaulieu JM & Julien JP Peripherin-mediated death of motor neurons rescued by overexpression of neurofilament NF-H proteins. J Neurochem 85, 248–256 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Lee MK & Cleveland DW Neuronal intermediate filaments. Annu Rev Neurosci 19, 187–217 (1996). [DOI] [PubMed] [Google Scholar]
- 11.Muma NA, Slunt HH & Hoffman PN Postnatal increases in neurofilament gene expression correlate with the radial growth of axons. J Neurocytol 20, 844–854 (1991). [DOI] [PubMed] [Google Scholar]
- 12.Hoffman PN & Cleveland DW Neurofilament and tubulin expression recapitulates the developmental program during axonal regeneration: induction of a specific beta-tubulin isotype. Proc Natl Acad Sci U S A 85, 4530–4533 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffman PN, et al. Neurofilament gene expression: a major determinant of axonal caliber. Proc Natl Acad Sci U S A 84, 3472–3476 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee MK & Cleveland DW Neurofilament function and dysfunction: involvement in axonal growth and neuronal disease. Curr Opin Cell Biol 6, 34–40 (1994). [DOI] [PubMed] [Google Scholar]
- 15.Hirokawa N Cross-linker system between neurofilaments, microtubules, and membranous organelles in frog axons revealed by the quick-freeze, deep-etching method. J Cell Biol 94, 129–142 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sobue G, et al. Phosphorylated high molecular weight neurofilament protein in lower motor neurons in amyotrophic lateral sclerosis and other neurodegenerative diseases involving ventral horn cells. Acta Neuropathol 79, 402–408 (1990). [DOI] [PubMed] [Google Scholar]
- 17.Cote F, Collard JF & Julien JP Progressive neuronopathy in transgenic mice expressing the human neurofilament heavy gene: a mouse model of amyotrophic lateral sclerosis. Cell 73, 35–46 (1993). [DOI] [PubMed] [Google Scholar]
- 18.Hirano A, Donnenfeld H, Sasaki S & Nakano I Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 43, 461–470 (1984). [DOI] [PubMed] [Google Scholar]
- 19.Hirano A, et al. Fine structural study of neurofibrillary changes in a family with amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 43, 471–480 (1984). [DOI] [PubMed] [Google Scholar]
- 20.Lee MK, Marszalek JR & Cleveland DW A mutant neurofilament subunit causes massive, selective motor neuron death: implications for the pathogenesis of human motor neuron disease. Neuron 13, 975–988 (1994). [DOI] [PubMed] [Google Scholar]
- 21.Xu Z, Cork LC, Griffin JW & Cleveland DW Increased expression of neurofilament subunit NF-L produces morphological alterations that resemble the pathology of human motor neuron disease. Cell 73, 23–33 (1993). [DOI] [PubMed] [Google Scholar]
- 22.Bruijn LI, Miller TM & Cleveland DW Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci 27, 723–749 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Vechio JD, Bruijn LI, Xu Z, Brown RH Jr. & Cleveland DW Sequence variants in human neurofilament proteins: absence of linkage to familial amyotrophic lateral sclerosis. Ann Neurol 40, 603–610 (1996). [DOI] [PubMed] [Google Scholar]
- 24.Xiao S, McLean J & Robertson J Neuronal intermediate filaments and ALS: a new look at an old question. Biochim Biophys Acta 1762, 1001–1012 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Garcia ML, et al. Mutations in neurofilament genes are not a significant primary cause of non-SOD1-mediated amyotrophic lateral sclerosis. Neurobiol Dis 21, 102–109 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Arai T, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351, 602–611 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Polymenidou M, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14, 459–468 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tollervey JR, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci 14, 452–458 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeJesus-Hernandez M, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Renton AE, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nana AL, et al. Neurons selectively targeted in frontotemporal dementia reveal early stage TDP-43 pathobiology. Acta Neuropathol 137, 27–46 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melamed Z, et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci 22, 180–190 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klim JR, et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci 22, 167–179 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prudencio M, et al. Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J Clin Invest 130, 6080–6092 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krach F, et al. Transcriptome-pathology correlation identifies interplay between TDP-43 and the expression of its kinase CK1E in sporadic ALS. Acta Neuropathol 136, 405–423 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun S, et al. Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc Natl Acad Sci U S A 112, E6993–7002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shin JE, et al. SCG10 is a JNK target in the axonal degeneration pathway. Proc Natl Acad Sci U S A 109, E3696–3705 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shin JE, Geisler S & DiAntonio A Dynamic regulation of SCG10 in regenerating axons after injury. Exp Neurol 252, 1–11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guerra San Juan I, et al. Loss of mouse Stmn2 function causes motor neuropathy. Neuron 110, 1671–1688 e1676 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krus KL, et al. Loss of Stathmin-2, a hallmark of TDP-43-associated ALS, causes motor neuropathy. Cell Rep 39, 111001 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chauvin S & Sobel A Neuronal stathmins: a family of phosphoproteins cooperating for neuronal development, plasticity and regeneration. Prog Neurobiol 126, 1–18 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Summers DW, Milbrandt J & DiAntonio A Palmitoylation enables MAPK-dependent proteostasis of axon survival factors. Proc Natl Acad Sci U S A 115, E8746–E8754 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Selvaraj BT, Frank N, Bender FL, Asan E & Sendtner M Local axonal function of STAT3 rescues axon degeneration in the pmn model of motoneuron disease. J Cell Biol 199, 437–451 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stegmeier F, Hu G, Rickles RJ, Hannon GJ & Elledge SJ A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci U S A 102, 13212–13217 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miyanohara A, et al. Potent spinal parenchymal AAV9-mediated gene delivery by subpial injection in adult rats and pigs. Mol Ther Methods Clin Dev 3, 16046 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bravo-Hernandez M, et al. Spinal subpial delivery of AAV9 enables widespread gene silencing and blocks motoneuron degeneration in ALS. Nat Med 26, 118–130 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bravo-Hernandez M, Tadokoro T & Marsala M Subpial AAV Delivery for Spinal Parenchymal Gene Regulation in Adult Mammals. Methods Mol Biol 1950, 209–233 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Tadokoro T, et al. Precision spinal gene delivery-induced functional switch in nociceptive neurons reverses neuropathic pain. Mol Ther (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tadokoro T, et al. Subpial Adeno-associated Virus 9 (AAV9) Vector Delivery in Adult Mice. J Vis Exp (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Joyce NC & Carter GT Electrodiagnosis in persons with amyotrophic lateral sclerosis. PM R 5, S89–95 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mills KR & Nithi KA Peripheral and central motor conduction in amyotrophic lateral sclerosis. J Neurol Sci 159, 82–87 (1998). [DOI] [PubMed] [Google Scholar]
- 53.de Carvalho M & Swash M Nerve conduction studies in amyotrophic lateral sclerosis. Muscle Nerve 23, 344–352 (2000). [DOI] [PubMed] [Google Scholar]
- 54.Hursh JB Conduction velocity and diameter of nerve fibers. American Journal of Physiology—Legacy Content. 127, 131–139 (1939). [Google Scholar]
- 55.de Waegh SM, Lee VM & Brady ST Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann cells. Cell 68, 451–463 (1992). [DOI] [PubMed] [Google Scholar]
- 56.Friede RL & Samorajski T Axon caliber related to neurofilaments and microtubules in sciatic nerve fibers of rats and mice. Anat Rec 167, 379–387 (1970). [DOI] [PubMed] [Google Scholar]
- 57.Ohara O, Gahara Y, Miyake T, Teraoka H & Kitamura T Neurofilament deficiency in quail caused by nonsense mutation in neurofilament-L gene. J Cell Biol 121, 387–395 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu Q, Couillard-Despres S & Julien JP Delayed maturation of regenerating myelinated axons in mice lacking neurofilaments. Exp Neurol 148, 299–316 (1997). [DOI] [PubMed] [Google Scholar]
- 59.Garcia ML, et al. NF-M is an essential target for the myelin-directed “outside-in” signaling cascade that mediates radial axonal growth. J Cell Biol 163, 1011–1020 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee MK, Xu Z, Wong PC & Cleveland DW Neurofilaments are obligate heteropolymers in vivo. J Cell Biol 122, 1337–1350 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tao QQ, Wei Q & Wu ZY Sensory nerve disturbance in amyotrophic lateral sclerosis. Life Sci 203, 242–245 (2018). [DOI] [PubMed] [Google Scholar]
- 62.Rossor AM, Jaunmuktane Z, Rossor MN, Hoti G & Reilly MM TDP43 pathology in the brain, spinal cord, and dorsal root ganglia of a patient with FOSMN. Neurology 92, e951–e956 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sonoda K, et al. TAR DNA-binding protein 43 pathology in a case clinically diagnosed with facial-onset sensory and motor neuronopathy syndrome: an autopsied case report and a review of the literature. J Neurol Sci 332, 148–153 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Camdessanche JP, et al. Sensory and motor neuronopathy in a patient with the A382P TDP-43 mutation. Orphanet J Rare Dis 6, 4 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Patapoutian A, Peier AM, Story GM & Viswanath V ThermoTRP channels and beyond: mechanisms of temperature sensation. Nat Rev Neurosci 4, 529–539 (2003). [DOI] [PubMed] [Google Scholar]
- 66.Julius D & Basbaum AI Molecular mechanisms of nociception. Nature 413, 203–210 (2001). [DOI] [PubMed] [Google Scholar]
- 67.Le Pichon CE & Chesler AT The functional and anatomical dissection of somatosensory subpopulations using mouse genetics. Front Neuroanat 8, 21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Figley MD & DiAntonio A The SARM1 axon degeneration pathway: control of the NAD(+) metabolome regulates axon survival in health and disease. Curr Opin Neurobiol 63, 59–66 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Coleman MP & Hoke A Programmed axon degeneration: from mouse to mechanism to medicine. Nat Rev Neurosci 21, 183–196 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gilley J, Orsomando G, Nascimento-Ferreira I & Coleman MP Absence of SARM1 rescues development and survival of NMNAT2-deficient axons. Cell Rep 10, 1974–1981 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Doetschman T Influence of genetic background on genetically engineered mouse phenotypes. Methods Mol Biol 530, 423–433 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hoffman PN, Griffin JW, Gold BG & Price DL Slowing of neurofilament transport and the radial growth of developing nerve fibers. J Neurosci 5, 2920–2929 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marszalek JR, et al. Neurofilament subunit NF-H modulates axonal diameter by selectively slowing neurofilament transport. J Cell Biol 135, 711–724 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wong PC, et al. Increasing neurofilament subunit NF-M expression reduces axonal NF-H, inhibits radial growth, and results in neurofilamentous accumulation in motor neurons. J Cell Biol 130, 1413–1422 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xu Z, et al. Subunit composition of neurofilaments specifies axonal diameter. J Cell Biol 133, 1061–1069 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rao MV, et al. Gene replacement in mice reveals that the heavily phosphorylated tail of neurofilament heavy subunit does not affect axonal caliber or the transit of cargoes in slow axonal transport. J Cell Biol 158, 681–693 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morii H, Shiraishi-Yamaguchi Y & Mori N SCG10, a microtubule destabilizing factor, stimulates the neurite outgrowth by modulating microtubule dynamics in rat hippocampal primary cultured neurons. J Neurobiol 66, 1101–1114 (2006). [DOI] [PubMed] [Google Scholar]
- 78.Riederer BM, et al. Regulation of microtubule dynamics by the neuronal growth-associated protein SCG10. Proc Natl Acad Sci U S A 94, 741–745 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jourdain L, Curmi P, Sobel A, Pantaloni D & Carlier MF Stathmin: a tubulin-sequestering protein which forms a ternary T2S complex with two tubulin molecules. Biochemistry 36, 10817–10821 (1997). [DOI] [PubMed] [Google Scholar]
- 80.Marklund U, Larsson N, Gradin HM, Brattsand G & Gullberg M Oncoprotein 18 is a phosphorylation-responsive regulator of microtubule dynamics. Embo J 15, 5290–5298 (1996). [PMC free article] [PubMed] [Google Scholar]
- 81.Charbaut E, et al. Stathmin family proteins display specific molecular and tubulin binding properties. J Biol Chem 276, 16146–16154 (2001). [DOI] [PubMed] [Google Scholar]
- 82.Baughn MW, et al. Mechanism of STMN2 cryptic splice-polyadenylation and its correction for TDP-43 proteinopathies. Science 379, 1140–1149 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ma XR, et al. TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature 603, 124–130 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brown AL, et al. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 603, 131–137 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chou AH, et al. Polyglutamine-expanded ataxin-3 causes cerebellar dysfunction of SCA3 transgenic mice by inducing transcriptional dysregulation. Neurobiol Dis 31, 89–101 (2008). [DOI] [PubMed] [Google Scholar]
- 86.Guyenet SJ, et al. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Laird JMA, Martinez-Caro L, Garcia-Nicas E & Cervero F A new model of visceral pain and referred hyperalgesia in the mouse. Pain 92, 335–342 (2001). [DOI] [PubMed] [Google Scholar]
- 88.Pitcher MH, Price TJ, Entrena JM & Cervero F Spinal NKCC1 blockade inhibits TRPV1-dependent referred allodynia. Mol Pain 3, 17 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Inyang KE, et al. The antidiabetic drug metformin prevents and reverses neuropathic pain and spinal cord microglial activation in male but not female mice. Pharmacol Res 139, 1–16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dixon WJ Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol 20, 441–462 (1980). [DOI] [PubMed] [Google Scholar]
- 91.Chaplan SR, Bach FW, Pogrel JW, Chung JM & Yaksh TL Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53, 55–63 (1994). [DOI] [PubMed] [Google Scholar]
- 92.Malmberg AB, Chen C, Tonegawa S & Basbaum AI Preserved acute pain and reduced neuropathic pain in mice lacking PKCgamma. Science 278, 279–283 (1997). [DOI] [PubMed] [Google Scholar]
- 93.Gilchrist LS, et al. Re-organization of P2X3 receptor localization on epidermal nerve fibers in a murine model of cancer pain. Brain Res 1044, 197–205 (2005). [DOI] [PubMed] [Google Scholar]
- 94.Wacnik PW, et al. Functional interactions between tumor and peripheral nerve: morphology, algogen identification, and behavioral characterization of a new murine model of cancer pain. J Neurosci 21, 9355–9366 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Garrison SR, Dietrich A & Stucky CL TRPC1 contributes to light-touch sensation and mechanical responses in low-threshold cutaneous sensory neurons. J Neurophysiol 107, 913–922 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ranade SS, et al. Piezo2 is the major transducer of mechanical forces for touch sensation in mice. Nature 516, 121–125 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bogdanik LP, et al. Systemic, postsymptomatic antisense oligonucleotide rescues motor unit maturation delay in a new mouse model for type II/III spinal muscular atrophy. Proc Natl Acad Sci U S A 112, E5863–5872 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ling KK, et al. Antisense-mediated reduction of EphA4 in the adult CNS does not improve the function of mice with amyotrophic lateral sclerosis. Neurobiol Dis 114, 174–183 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bogdanik LP, et al. Loss of the E3 ubiquitin ligase LRSAM1 sensitizes peripheral axons to degeneration in a mouse model of Charcot-Marie-Tooth disease. Dis Model Mech 6, 780–792 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.