

Abstract
One potential advantage of live attenuated influenza vaccines (LAIVs) is their ability to establish both virus-specific antibody and tissue-resident memory T cells (TRM) in the respiratory mucosa. However, it is hypothesized that pre-existing immunity from past infections and/or immunizations prevents LAIV from boosting or generating de novo CD8+ T cell responses. To determine if we can overcome this limitation, we generated a series of drifted influenza A/PR8 LAIVs with successive mutations in the hemagglutinin (HA) protein, allowing for increasing levels of escape from pre-existing antibody. We also inserted a CD8+ T cell epitope from the Sendai virus nucleoprotein (SeV NP) to assess both generation of a de novo T cell response and boosting of pre-existing influenza-specific CD8+ T cells following LAIV immunization. Increasing the level of escape from antibody enabled boosting of pre-existing TRM, but we were unable to generate de novo SeV NP+ CD8+ TRM following LAIV immunization in PR8 influenza-immune mice, even with LAIV strains that can fully escape pre-existing antibody. As these data suggested a role for cell-mediated immunity in limiting LAIV efficacy, we investigated several scenarios to assess the impact of pre-existing LAIV-specific TRM in the upper and lower respiratory tract. Ultimately, we found that deletion of the immunodominant influenza NP366–374 epitope allowed for sufficient escape from cellular immunity to establish de novo CD8+ TRM. Combined, these studies demonstrate that both pre-existing humoral and cellular immunity can limit the effectiveness of LAIV, which is an important consideration for future design of vaccine vectors against respiratory pathogens.
Introduction
Despite the availability of a yearly vaccine, achieving effective protection against influenza infection remains a challenge. Current vaccination methods primarily focus on generating a strong antibody response against the viral surface proteins hemagglutinin (HA) and neuraminidase (NA). However, these proteins vary between influenza strains and are prone to mutation. For that reason, vaccine strategies that also generate a CD8+ T cell response, which is directed against epitopes within internal viral proteins such as nucleoprotein (NP) and matrix protein and are conserved across influenza strains, are likely to improve efficacy (1–4). Following clearance of influenza infection, tissue-resident memory CD8+ T cells (TRM) do not re-enter blood circulation and instead remain situated along the respiratory tract where they act as sentinels by rapidly responding to virus re-encounter. Although they alone cannot prevent infection with influenza virus, CD8+ TRM are important for limiting early viral replication and immunopathology and have been shown to play a vital role in mediating protection against a variety of respiratory pathogens in addition to influenza, such as RSV and SARS-CoV-2 (5–9). Given their demonstrated importance in the cell-mediated response to influenza infection, CD8+ TRM are prime targets (in addition to antibody) for designing vaccines that offer broad protection against a variety of influenza strains.
One influenza vaccine platform that can elicit both antibody and CD8+ T cell responses, including protective CD8+ TRM, is the live attenuated influenza vaccine or LAIV (10, 11). LAIV is administered as a nasal spray, thus mimicking a natural infection. However, due to its attenuation, it does not replicate effectively in the lower respiratory tract and generates only mild clinical symptoms (12). Unfortunately, despite its promising ability to engage both arms of the immune system, LAIV has historically performed poorly in adults and has not been recommended for use in recent years. Interestingly, LAIV did show marked efficacy in children when compared to adults (13–16). It is hypothesized that the reduced efficacy of LAIV immunization in adults is due to pre-existing influenza immunity from either prior infection and/or immunization. The implication that pre-existing immunity can impact the ability of a vaccine to “take” in individuals is an important consideration for future design of vaccine regimens, especially against respiratory viral infections. However, the relative contributions of pre-existing humoral and cellular immunity in limiting vaccine efficacy, specifically the resulting virus-specific T cell response, have not been thoroughly investigated.
In the present study, we use LAIV to investigate the impact of pre-existing humoral and cellular immunity on the dynamics of established and de novo CD8+ TRM in the respiratory tract. We find that vaccine escape from anti-HA antibodies on its own is not sufficient to generate a de novo antigen specific CD8+ lung TRM population in mice that have immunity from a prior influenza infection. However, we do find that LAIV immunization successfully boosts pre-existing influenza specific CD8+ lung TRM, with efficacy increasing as escape from anti-HA antibodies is achieved. Interestingly, LAIV immunization of mice with fewer pre-existing CD8+ lung TRM also failed to generate a de novo CD8+ TRM population, possibly demonstrating the important role for virus specific CD8+ TRM within the nasal cavity. Lastly, we show that pre-existing TRM specific for a single immunodominant CD8+ T cell epitope in LAIV are sufficient to prevent generation of de novo CD8+ TRM in the respiratory tract following immunization. Combined, these results show that although intranasal immunization with LAIV can boost already existing CD8+ lung TRM, its ability to generate a novel antigen specific CD8+ lung TRM population is hindered by these same pre-formed CD8+ memory T cells. These finding support a previously unrecognized role for T cell-mediated immunity in limiting LAIV efficacy and are important considerations for future design of vaccines against respiratory viral pathogens.
Materials & Methods
Mice
C57BL/6J mice (male and female) were bred in-house or purchased from Jackson Laboratory. All animals were housed at Emory University under specific pathogen-free conditions. Mice were between 8–12 weeks of age at time of infection, after which they were housed in specific animal biosafety level 2 conditions. All experiments were conducted in accordance with the Institutional Animal Care and Use Committee (IACUC) guidelines of Emory University.
Generation of live attenuated influenza virus
Influenza A viruses used in this study were derived from influenza A/Puerto Rico/8/34 (H1N1) virus (PR8). The wildtype genome sequence of this virus is available in GenBank with accession numbers AF389115-AF389122 (https://www.ncbi.nlm.nih.gov/nuccore). The following amino acid changes were introduced into this PR8 background to confer a live attenuated, cold-adapted phenotype: PB2 N265S, PB1 K391E, PB1 E581G, PB1 A661T and PB1 L319Q. The use of this genotype was based on published reports establishing the attenuating nature of the mutations in a murine model (17, 18). This strain is referred to herein as PR8 LAIV.
We further modified PR8 LAIV to introduce a foreign epitope from Sendai virus. Using commercial gene synthesis, the nucleotide sequence of the SeV nucleoprotein peptide FAPGNYPAL (5’ ttcgcacctggaaattaccctgcacta 3’) was inserted into the PR8 influenza A virus neuraminidase (NA) gene at the site corresponding to position 43–44 of the NA stalk domain. The modified NA gene was then cloned into the pDP reverse genetics vector (19). This was then combined with plasmids encoding the remaining seven gene segments of PR8 LAIV for reverse genetics-based recovery of PR8 LAIV SeV NP virus (referred to as LAIV-SeV NP in the main text).
A further set of virus variants was produced in the PR8 LAIV background, carrying the SeV NP epitope in NA and mutations in the viral hemagglutinin (HA) protein that disrupt known antibody epitopes. The antigenic changes introduced into HA are summarized in Table 1 and described by Das et al. (20). pDZ reverse genetics plasmids encoding svHA-3, svHA-6, svHA-10 and svHA-12 HA gene segments were kind gifts of Jonathan Yewdell and Christopher Brooke (21) and the HA mutations introduced in each variant are listed in Table 1. Finally, an additional amino acid change, N370Q, was introduced into the PR8 LAIV SeV NP svHA-12 virus to disrupt MHC-I loading of the native T cell epitope present in the NP protein of PR8 (svHA-12 N370Q).
Table 1.
Amino acid changes introduced into the PR8 HA sequential variant (sv) viruses
| Sequential Variant | Changes in HA, relative to PR8 wild type |
|---|---|
| svHA-WT | none |
| svHA-3 | G159S, N129D, R78G |
| svHA-6 | G159S, N129D, R78G, R224K, S145N, K163E |
| svHA-10 | G159S, N129D, R78G, R224K, S145N, K163E, E156K, E119K, G173R |
| svHA-12 | G159S, N129D, R78G, R224K, S145N, K163E, E156K, E119K, G173R, E70G, R48K, D225G |
Viruses were generated by reverse genetics as previously described (22, 23). In brief, 293T cells were transfected with reverse-genetics plasmids and then, after a 16–24 h incubation at 37°C, collected and injected into the allantoic cavity of 10–11 day old embryonated chicken’s eggs. Eggs were incubated at 33°C for 40–48 h and then chilled overnight prior to harvesting of allantoic fluid. Clarified allantoic fluid was aliquoted and stored at −80°C and virus therein was titrated by plaque assay on MDCK cells. The presence of introduced mutations was confirmed by Sanger sequencing of viral cDNA corresponding to the modified genome segments or by viral whole genome sequencing on an Illumina MiSeq platform.
Infections
Prior to all infections, mice were anesthetized using isoflurane (Patterson Veterinary). For primary A/Puerto Rico/8/34 (PR8) influenza infections, mice were infected either intranasally (i.n.) with 20 plaque forming units (PFU) in 30uL volume or via intraperitoneal (i.p.) injection with 106 PFU in 200uL volume. For adenovirus infections, mice were inoculated via both i.n. and footpad (s.c.) injections with 2×107 PFU of a replication-deficient adenovirus serotype 5 expressing influenza (A/Puerto Rico/8/34) nucleoprotein (AdNP) as described previously (24, 25). For LAIV immunizations, mice were given 20,000 –500,000 PFU i.n. in 30uL volume. For protection studies, mice were challenged with either 300,000 EID50 (50% egg infectious dose) x31 H3N2 influenza virus or 3,000 EID50 Sendai parainfluenza virus i.n. in 30uL volume. For EdU incorporation experiments, mice were dosed i.p. with 1mg of 5-ethynyl-2’-deoxyuridine (EdU, Cayman Chemical) in 200uL volume of sterile 1X PBS.
Single cell isolation
To distinguish tissue-resident cells from those in circulation, mice were intravenously (i.v.) labeled via tail vein injection of fluorescent anti-CD3e (1.5 ug) or anti-CD45.2 antibody (4 ug) in 200uL 1X PBS. Mice were euthanized 5 minutes later by intraperitoneal (i.p.) injection with Avertin (2,2,2-tribromoethanol) followed by brachial exsanguination. Spleen, lungs, bronchoalveolar lavage (BAL), and nasal cavity (NC) were then harvested. Lungs were enzymatically digested in Collagenase D (5g/L, Roche) and DNase (2×106 U/L, Sigma) for 30 minutes at 37C, with occasional mechanical dissociation. To enrich for lymphocytes, lung samples were centrifuged in a 40%/80% Percoll gradient. Nasal cavities were digested in a mixture of Collagenase D (5g/L), DNase (2×106 U/L), and Dispase (15U/mL, Sigma) at 37C with mechanical dissociation for a total of 30 minutes. Nasal cavities were then passed through a 70um filter prior to centrifugation. Spleens were mechanically dissociated and then RBC lysed.
Cell staining and flow cytometry
Single cell suspensions were first FC blocked using murine 2.4G2 antibody. Samples were then stained for 1 hour at room temperature with tetramers against influenza NP366–374Db, PA224–233Db, and Sendai parainfluenza NP324–332Kb (provided by the National Institutes of Health (NIH) Tetramer Core Facility at Emory University). Extracellular staining was then performed for 30 minutes. Cell viability was determined using Zombie fixable viability dye (BioLegend). For experiments utilizing EdU, samples were additionally stained using the Click-iT Plus EdU Flow Cytometry kit (Invitrogen) per the kit’s standard protocol. All samples were run on either a Fortessa X20 or a Symphony A3 (BD Biosciences) flow cytometer. FluNP366+ or Sendai NP+ CD8+ TRM were gated as: singlets, lymphocytes, live cells, CD4−CD8α+, i.v. label-, CD44hi, tetramer+. TRM were additionally gated on CD69 and CD103. For splenic TEM, the i.v. label gate was omitted and all live CD4−CD8α+ T cells were gated for CD44hi tetramer+. Flow cytometry data were analyzed using FlowJo v.10 software. For experiments evaluating LAIV immunization of PR8-immune or AdNP-immunized mice, a limit of detection was set at ≤ 10 cells within the final SeV NP+ gate.
Plaque assays
Following x31 influenza or Sendai parainfluenza challenge of LAIV-immunized mice, lung viral titers were determined as previously described (26).
Statistical analysis
Cell counts were determined either manually using a hemocytometer or with a LUNA-II automatic cell counter (Logos Biosystems). Statistical analyses were performed using GraphPad Prism Software. Unless otherwise indicated, data points represent individual mice or group mean (all biological replicates). Significance for all data was determined using a Mann-Whitney test and is displayed as mean ± SEM. p values are as follows: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Results
Live attenuated influenza vaccine elicits protective antigen specific CD8+ lung TRM
We first generated an LAIV strain by modifying wildtype (WT) influenza A/PR8 (H1N1) virus to contain a set of known attenuating point mutations within the polymerase proteins PB1 and PB2 (17, 27). To identify de novo CD8+ memory T cells following immunization, we further modified our LAIV to include the immunodominant epitope from the Sendai parainfluenza nucleoprotein (SeV NP), resulting in our final LAIV-SeV NP strain. We confirmed successful attenuation and generation of epitope-specific CD8+ splenic effector memory T cells (TEM) and lung and airway CD8+ TRM following intranasal LAIV-SeV NP immunization in vivo (Fig 1A–C). Importantly, SeV NP+ CD8+ TRM, identified using a combination of intravital labeling and surface staining for the canonical markers CD69 and CD103, were present in the lungs and airways for up to at least 90 days post-immunization with LAIV-SeV NP (Fig 1D–F).
Figure 1. Live attenuated influenza vaccine elicits protective antigen specific CD8+ lung TRM.
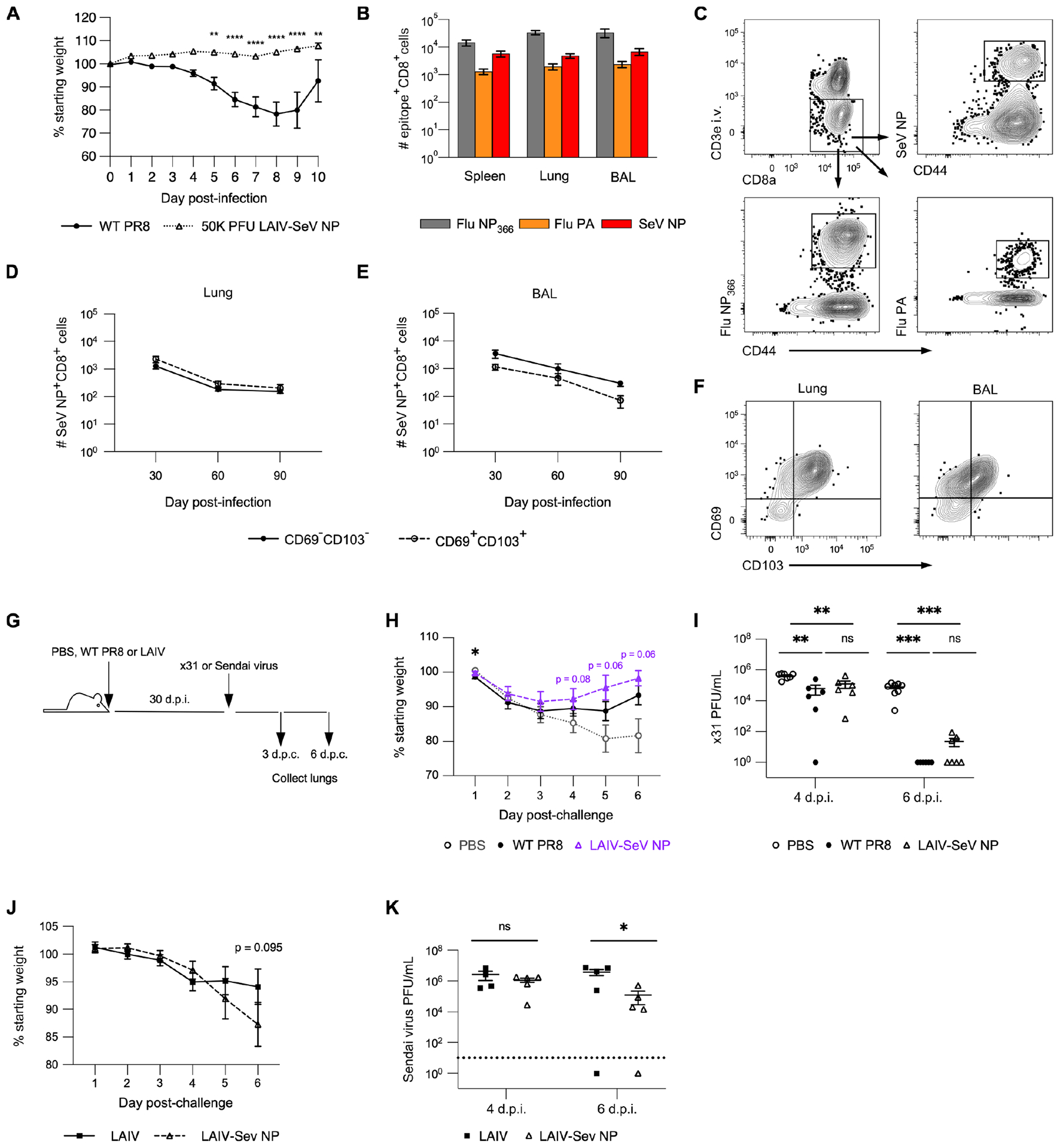
(A) Percent original weight following i.n. infection with WT PR8 or immunization with LAIV-SeV NP. n = 5 mice per group from 2 experiments. (B) Number of FluNP366+, FluPA+, or SeV NP+ CD8+ T cells in the spleen, lung, and BAL 30 d.p.i. with LAIV-SeV NP. n = 2 independent experiments with 5 mice per experiment. (C) Example flow staining for epitope-specific lung TRM in mice 30 d.p.i. with LAIV-SeV NP. (D, E) Number of CD69− CD103− or CD69+ CD103+ SeV NP+ CD8+ TRM in the lung (D) and BAL (E) on indicated d.p.i. with LAIV-SeV NP. n = 2 independent experiments with 5 mice per timepoint per experiment. (F) Example flow staining for CD69 and CD103 amongst SeV NP+ CD8+ TRM in lung and BAL. (G) Experimental design. (H) Percent original weight following challenge of mock infected (PBS), WT PR8-infected, or LAIV-SeV NP immunized mice with x31 influenza. (I) Lung viral titers on days 4 and 6 post-challenge (d.p.c.) with x31 influenza in mice initially mock infected or given WT PR8 or LAIV-SeV NP. For H and I, n = 2 independent experiments with 6–8 mice per challenge group per timepoint. (J) Percent original weight following challenge of mice immunized with LAIV +/− SeV NP with Sendai parainfluenza virus. (K) Lung viral titers in mice immunized with LAIV +/− SeV NP and then challenged with Sendai parainfluenza virus. For J and K, n = 2 experiments with 5 mice per challenge group per timepoint.
To determine whether immunization with LAIV-SeV NP confers protection against heterosubtypic influenza challenge, we first infected animals WT PR8 (H1N1) or immunized with LAIV-SeV NP and then challenged them with x31 (H3N2) influenza (Fig 1G). Compared to mock immunized mice (PBS), mice that were infected with WT PR8 or immunized with LAIV-SeV NP experienced minimal weight loss following x31 influenza challenge (Fig 1H). Mice infected with WT PR8 or immunized with LAIV-SeV NP also had significantly lower lung viral titers by day 4 post-challenge (4 d.p.c.) (when compared to mock immunized mice) and had cleared most virus from the lung tissue within 6 d.p.c. (Fig 1I). Furthermore, we observed no significant difference in the protection conferred by infection with WT PR8 or immunization with LAIV-SeV NP (Fig 1H, I). To assess the protectiveness of SeV NP+ CD8+ memory T cells formed following immunization, we compared weight loss and lung viral titers following Sendai parainfluenza challenge between groups of mice immunized with either LAIV-SeV NP or a version of our LAIV that lacks the SeV NP epitope (Fig 1G). At 4 d.p.c., we observed no significant difference in morbidity or lung viral titers (Fig 1J, K). However, mice that had been immunized with LAIV-SeV NP did have significantly lower viral lung titers at 6 d.p.c. compared to those that were immunized with an LAIV that does not contain the SeV NP epitope (Fig 1K). Combined, these results show that our LAIV-SeV NP is successfully attenuated and generates protective epitope-specific CD8+ TRM in the lungs and airways following i.n. administration.
Immunization with LAIV-SeV NP strains capable of escaping established antibody fails to generate a de novo antigen specific CD8+ TRM population in mice with pre-existing immunity
To assess the impact of established anti-HA antibody on the efficacy of LAIV-SeV NP immunization, we generated a series of five additional LAIV-SeV NP strains that incorporate previously identified HA mutations that allow for each strain to have increasing ability to escape polyclonal anti-PR8 HA antibodies (Fig 2A). We termed these new LAIV-SeV NP “switch variant” strains based on their original publication: svHA-WT, svHA-3, svHA-6, svHA-10, and svHA-12 (20). Antigenically, svHA-WT is the same as LAIV-SeV NP and has an HA sequence identical to WT PR8 influenza, differing only by incorporation of the attenuating point mutations in PB1 and PB2, and the SeV NP epitope incorporated into the neuraminidase stalk. In contrast, svHA-12 is the most antigenically distinct from WT PR8 and is fully capable of escaping anti-PR8 HA antibodies (Fig 2A). We first used this series of switch variants to immunize mice that had been previously infected i.n. with WT PR8 influenza. As expected, svHA-WT immunization generated SeV NP+ CD8+ lung TRM in naïve animals but not in PR8-immune mice, with most samples in PR8-immune mice falling below the level of detection (Fig 2B–D). Surprisingly, immunization of PR8-immune mice with svHA-12 also failed to generate de novo SeV NP+ CD8+ lung TRM (Fig 2E–G). Both immunizations also failed to generate de novo CD8+ TRM in the airways (Supplemental Fig. 1A–F). We were also unable to identify de novo CD8+ TRM within the mediastinal lymph node of PR8-immune mice and excluded this tissue from further analyses (data not shown). In fact, none of our switch variant LAIV-SeV NP strains successfully generated SeV NP+ CD8+ TRM in the lungs or airways of mice that had established immunity to WT PR8 (Supplemental Fig. 2A–F). All together, these results demonstrate that immunization with LAIV-SeV NP is incapable of generating a de novo CD8+ TRM population in mice with pre-existing immunity, even when the established humoral response is evaded.
Figure 2. Immunization with LAIV-SeV NP strains capable of escaping pre-formed antibody fails to generate a de novo antigen specific CD8+ TRM population in mice with pre-existing immunity.
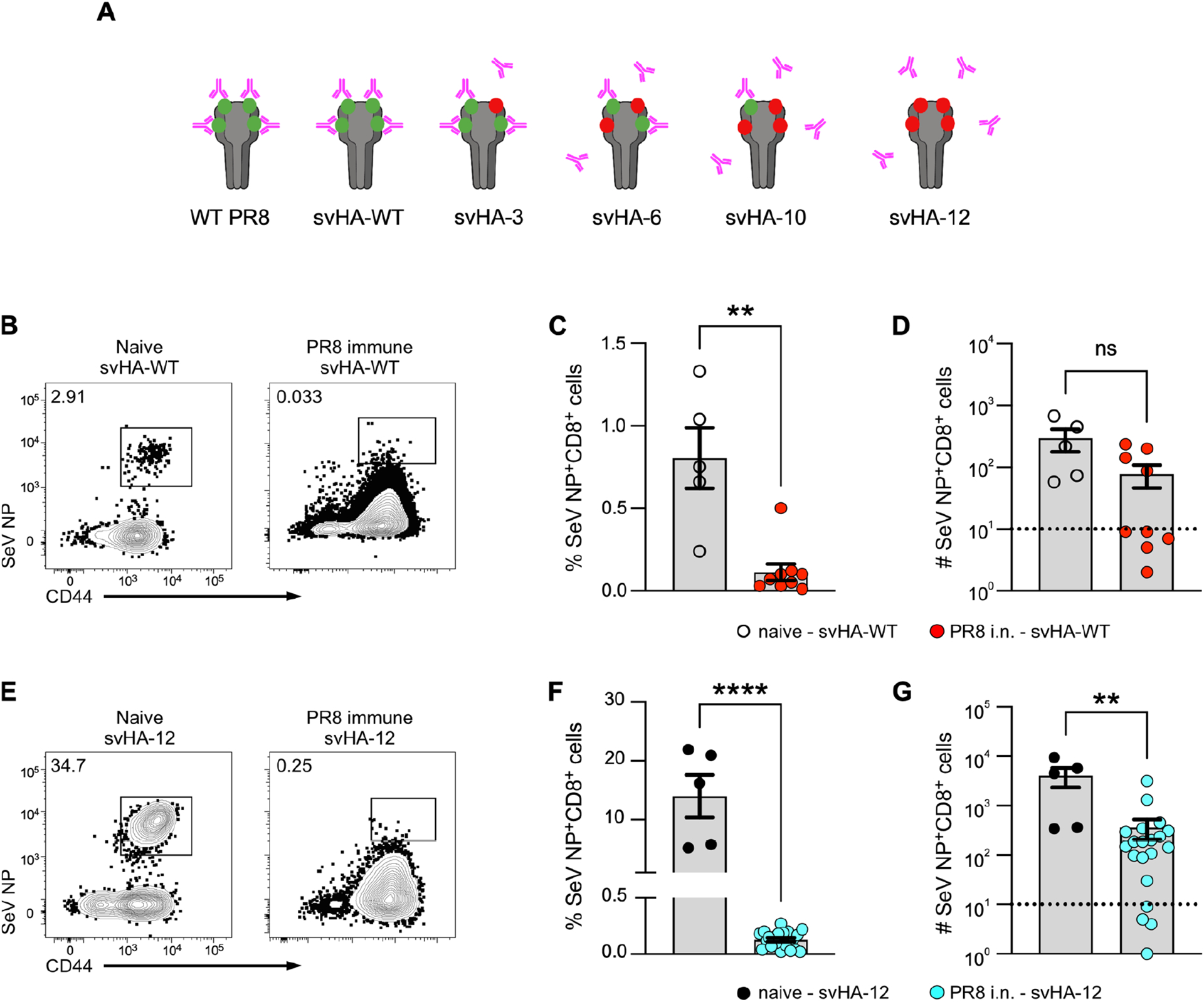
(A) Design of switch variant (sv) drifted LAIV-SeV NP strains that have either a WT HA sequence (svHA-WT) or a mutated HA sequence (svHA-3, svHA-6, svHA-10, svHA-12) with point mutations that allow for increasing escape from pre-existing anti-HA antibodies. (B) Example flow staining for SeV NP+ CD8+ TRM in the lung of naïve (left plot) or PR8-immune (right plot) mice subsequently immunized with LAIV svHA-WT. (C, D) Frequency (C) and number (D) of SeV NP+ CD8+ lung TRM 30 d.p.i. of naïve or PR8-immune mice with LAIV svHA-WT. (E-G) Same as B-D except with LAIV svHA-12 immunization. For naïve animals, n = 5 mice from 2 experiments. For PR8-immune animals for each immunization strain, n = 2 independent experiments with 5–11 mice per experiment. Limit of detection (LOD) indicated with dotted line at 101.
Pre-existing CD8+ TRM in the respiratory tract undergo expansion upon immunization with LAIV-SeV NP
Following the discovery that LAIV-SeV NP immunization of PR8-immune mice fails to generate a novel antigen-specific CD8+ lung TRM response, we sought to determine whether the pre-existing FluNP366+ CD8+ memory T cells responded to immunization and whether this was influenced by escape from pre-existing antibody. To accomplish this, we examined incorporation of EdU by FluNP366+ CD8+ TRM in PR8-immune mice following mock immunization (PBS) or immunization with svHA-WT, svHA-6, or svHA-12 (Fig 3A). At 7 days post-immunization, EdU+ FluNP366+ CD8+ TRM were clearly identifiable in both the lung interstitium and airways (Fig 3B). Within the lung, a significantly higher frequency and number of FluNP366+ CD8+ TRM incorporated EdU following immunization with svHA-WT, svHA-6 or svHA-12 when compared to samples from mock immunized mice (Fig 3C, D). Importantly, significantly more FluNP366+ CD8+ lung TRM in mice immunized with sv6 or svHA-12 incorporated EdU when compared to those from mice immunized with svHA-WT, indicating that the ability of LAIV-SeV NP immunization to induce T cell responses increases as anti-HA antibody is avoided (Fig 3C, D). Furthermore, the overall frequency and number of FluNP366+ CD8+ lung TRM increased upon immunization with svHA-6 and svHA-12 (Fig 3E, F). Immunization with svHA-6 or svHA-12 also resulted in significantly increased numbers of FluNP366+ CD8+ TRM as well as incorporation of EdU by these cells within the airways (Fig 3G–J). These results confirm that although immunization of PR8-immune mice with LAIV-SeV NP fails to generate de novo CD8+ TRM within the respiratory tract, it does succeed in boosting pre-existing lung CD8+ TRM populations more effectively when it escapes the anti-HA response.
Figure 3. Pre-existing CD8+ TRM in the respiratory tract undergo expansion upon immunization with LAIV-SeV NP.
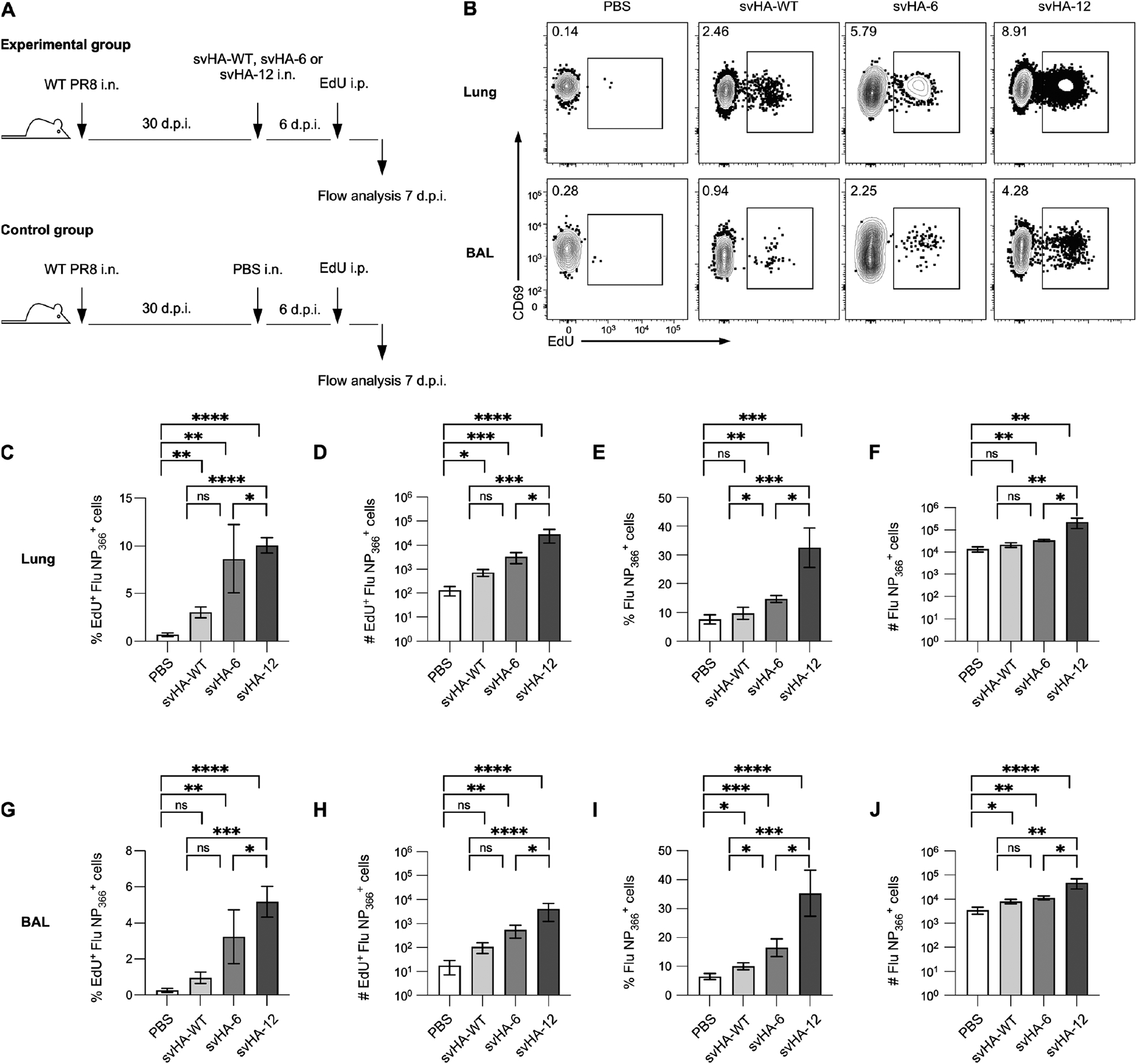
(A) Experimental design. (B) Example EdU staining amongst FluNP366+ CD8+ TRM in the lung and BAL following immunization of PR8-immune mice with PBS, svHA-WT, svHA-6, or svHA-12. (C, D) Frequency (C) and number (D) of EdU+ FluNP366+ CD8+ lung TRM following svHA-WT, svHA-6 or svHA-12 immunization. (E, F) Total frequency (E) and number (F) of FluNP366+ CD8+ lung TRM following the indicated immunization. (G-J) Same as C-F but in BAL. For each immunization strain, n = 2 independent experiments with 4–5 mice per experiment.
Route of prior infection does not alter the ability of LAIV to establish de novo CD8+ TRM
Given that our results suggest the cellular immune response on its own is enough to prevent LAIV-SeV NP immunization from generating a novel CD8+ TRM population, we repeated our immunization experiment, but infected mice with WT PR8 via i.p. injection rather than the i.n. route. Previous studies have shown that a non-pulmonary infection route results in significantly fewer CD8+ TRM in the lungs and airways, while generating similar numbers of circulating CD8+ TEM (28). However, neither svHA-WT nor svHA-12 immunization generated de novo SeV NP+ CD8+ TRM in the lung (Fig 4A–F). The immunizations did, however, boost the pre-existing FluNP366+ CD8+ T cells in the lung (Fig 4G–J). We also confirmed boosting of the pre-existing FluNP366+ CD8+ T cells in the airways, spleen, and nasal cavity (Supplemental Fig. 3A–C). Apart from the frequency of EdU+ FluNP366+ TRM in the lung, immunization with svHA-12 had significantly higher efficacy when compared to immunization with svHA-WT (Fig 4G–J). Though this result was somewhat surprising, we hypothesize that it can be explained by the presence of CD8+ TRM within the nasal cavity, which is the primary site of LAIV replication. The fold change decrease of CD8+ TRM in the lungs and airways is much greater than that seen in the nasal cavity when comparing i.n. versus i.p. infection routes (Fig 4K).
Figure 4. Route of prior infection does not alter the ability of LAIV to establish de novo CD8+ TRM.
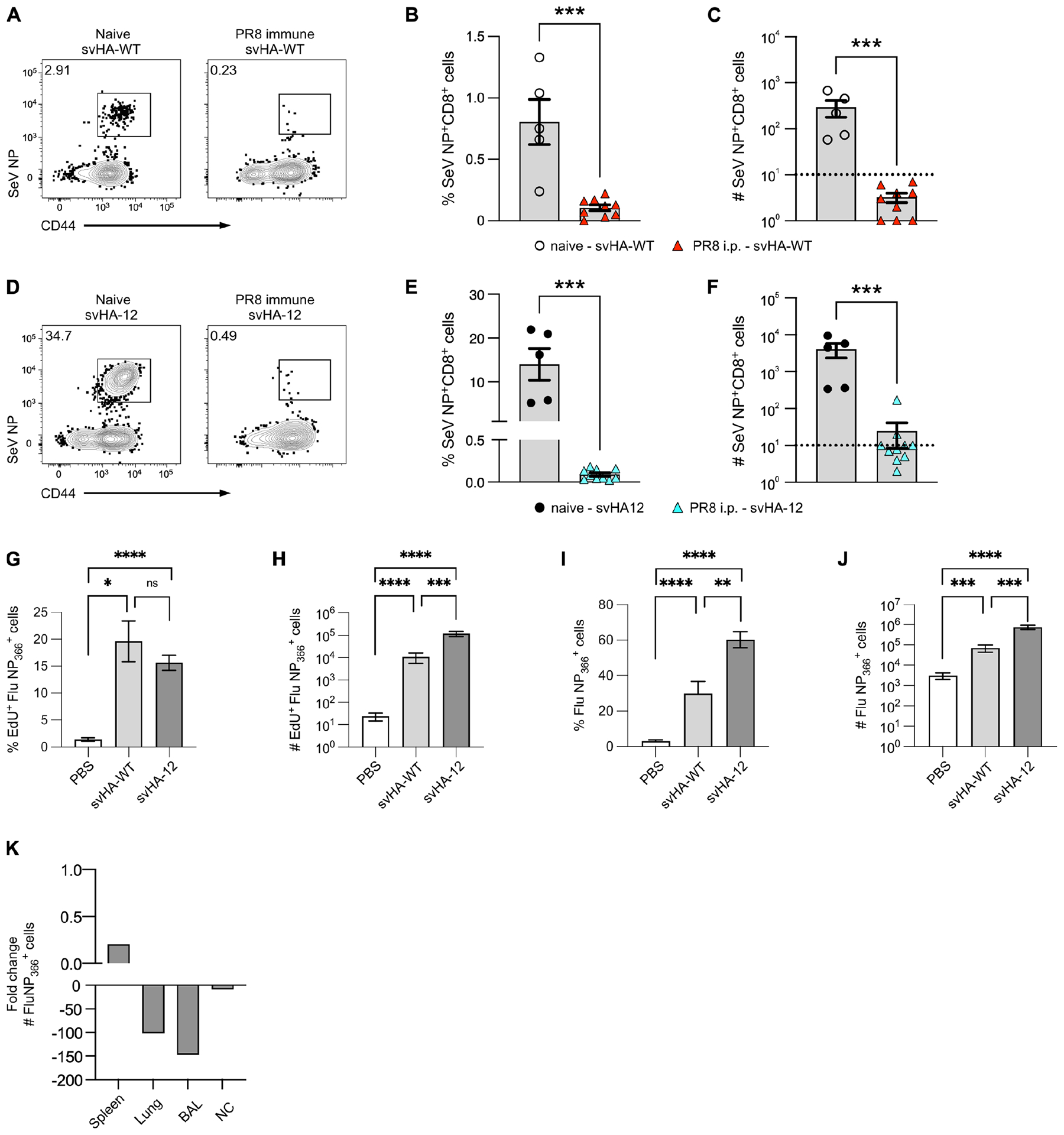
(A-C) Example staining (A), frequency (B) and number (C) of SeV NP+ CD8+ lung TRM following svHA-WT immunization (30 d.p.i.) of naïve mice or mice infected via i.p. injection with WT PR8. (D-F) Same as A-C but with svHA-12. LOD indicated with dotted line at 101. For naïve animals, n = 5 mice from 2 experiments. For PR8-immune animals, n= 2 independent experiments with 4–5 mice per experiment. (G, H) Frequency (G) and number (H) of EdU+ FluNP366+ CD8+ lung TRM following indicated immunization. (I, J) Total frequency (I) and number (J) of FluNP366+ CD8+ lung TRM following indicated immunization. For G-J, n = 2 independent experiments with 5 mice per experiment for each immunization strain. (K) Fold change in the number of FluNP366+ CD8+ T cells in indicated tissues when comparing PR8 infection via i.n. or i.p. route.
Pre-existing CD8+ TRM are sufficient to prevent the generation of de novo CD8+ TRM following LAIV immunization
To investigate whether evasion of the influenza-specific CD8+ T cell response enables formation of a novel CD8+ lung TRM population, we generated a new version of svHA-12 that includes a point mutation, N370Q, within the nucleoprotein sequence that prevents presentation of NP antigen on MHC class I (29) (Fig 5A). Inclusion of this point mutation thereby eliminates responses to the immunodominant NP366–374 CD8+ T cell epitope from our vaccination strain and allows the immunization to circumvent the pre-existing FluNP366-specific CD8+ T cell response. To test whether the presence of TRM specific for a single immunodominant CD8+ T cell epitope alone is sufficient to prevent LAIV immunization from generating a de novo T cell response, we first infected mice using a replication-deficient recombinant adenovirus vector that expresses PR8 NP (AdNP). These mice do not generate any antibodies against influenza HA and will only generate influenza-specific CD8+ memory T cells to the FluNP366 epitope. We then immunized these mice with either svHA-12 or svHA-12 N370Q (Fig 5B) and looked one month later for SeV NP+ CD8+ TRM in the respiratory tract (Fig 5C). Like our results in PR8-immune mice, immunization of AdNP-immune mice with svHA-12 largely failed to generate SeV NP+ CD8+ TRM. However, we did observe a significantly higher frequency and number of SeV NP+ CD8+ TRM in the lung, BAL, and nasal cavity when AdNP-infected mice were immunized with svHA-12 N370Q (Fig 5D, E). We also confirmed that these SeV NP+ CD8+ TRM express both CD69 and CD103 (Fig 5F). Combined, these results demonstrate that CD8+ TRM on their own can prevent LAIV-SeV NP immunization from generating de novo antigen-specific CD8+ TRM.
Figure 5. Pre-existing CD8+ TRM are sufficient to prevent the generation of de novo CD8+ TRM following LAIV immunization.
(A) Schematic illustrating the design of LAIV strain svHA-12 N370Q. (B) Experimental design. (C) Example staining for SeV NP+ CD8+ T cells in indicated tissues in AdNP-immunized mice 30 d.p.i. with svHA-12 or svHA-12 N370Q. (D, E) Frequency (D) and number (E) of SeV NP+ CD8+ T cells in indicated tissues 30 d.p.i. with svHA-12 or svHA-12 N370Q. (F) Number of CD69+ CD103+ cells amongst SeV NP+ CD8+ T cells. Samples with the parent gate falling below the LOD were excluded. Data represent 3 independent experiments with n = 5 mice per group per experiment. LOD indicated with dotted line at 101. (G) Experimental design. (H-K) Same as C-F except in mice first immunized with LAIV lacking SeV NP. Data represent 3 independent experiments with n = 5 mice per group per experiment. LOD indicated with dotted line at 101.
Given this finding, we next established pre-existing immunity by immunizing animals with LAIV lacking SeV NP and then immunized with svHA-12 or svHA-12 N370Q (Fig 5G). Like our results in AdNP immunized mice, svHA-12 N370Q immunization resulted in generation of significantly more SeV NP+ CD8+ T cells in all tissues compared with svHA-12 immunization, although the number of SeV NP+ CD8+ T cells was increased to a lesser degree than observed in AdNP immunized animals (Fig 5H–J). However, we did not observe any significant difference in the number of CD69+ CD103+ SeV NP+ CD8+ T cells generated following immunization with either svHA-12 or svHA-12 N370Q (Fig 5K). Lastly, svHA-12 N370Q immunization of LAIV-immune mice also resulted in a significantly larger population of Flu PA+ CD8+ T cells compared to svHA-12 immunization, confirming that immunization with a drifted LAIV-SeV NP strain lacking the immunodominant Flu NP366 epitope is also capable of boosting pre-existing T cells to other epitopes (Supplemental Fig. 4A–D). Overall, these data show that both humoral and cellular immunity can limit the ability of LAIV to promote virus-specific CD8+ T cell responses.
Discussion:
Despite ongoing research efforts, achieving long-term protection against many respiratory pathogens through vaccination remains a challenge. In the case of influenza, the virus’s capacity to rapidly mutate its surface proteins results in its ability to evade the antibodies generated during a primary immune response. Influenza-specific CD8+ memory T cells, however, target conserved internal viral proteins that are shared across different subtypes of influenza virus, thereby making them crucial for cross-reactive protection (1–4). CD8+ TRM, a specialized subset of memory T cells that remain embedded along the respiratory tract and do no re-enter the circulation, have been shown to be particularly important for mediating protection against influenza virus challenge (7, 9, 30). Several vaccination strategies developed against influenza virus have been shown to be effective at inducing a CD8+ T cell response, including live attenuated influenza vaccine (LAIV). However, while LAIV has been effective in children, it has shown minimal efficacy and variable T cell responses in adults (13–16, 31). We therefore hypothesized that LAIV’s ability to “take” is impacted by pre-existing influenza immunity due to prior infections and/or immunizations.
In the present study, we assess the impact of pre-existing humoral and cellular immunity on the ability of LAIV to generate de novo antigen-specific CD8+ TRM in the respiratory tract. Not surprisingly, we observed that LAIV-SeV NP immunization can boost pre-existing influenza NP-specific CD8+ TRM with increasing efficacy as escape from anti-HA antibodies increased. However, LAIV-SeV NP immunization failed to generate de novo SeV NP CD8+ TRM even when the immunization strain is capable of fully escaping anti-HA antibodies. It is unlikely that this is due competition for MHC-I binding between T cell epitopes as Sendai parainfluenza nucleoprotein does not share homology with influenza virus. Furthermore, the SeV NP324–332 epitope inserted within our LAIV strains is Kb-restricted, whereas the immunodominant PR8 influenza NP366–374 epitope is Db-restricted. It has been shown that influenza NP366-specific memory CD8+ T cells are preferentially activated and expanded during secondary infection due to differential antigen processing; perhaps explaining why NP366-specific CD8+ TRM were successfully boosted upon immunization (32).
Intriguingly, immunization with an LAIV-SeV NP strain fully capable of escaping antibodies (svHA-12) still failed to generate de novo CD8+ TRM when fewer pre-existing virus-specific CD8+ TRM were present in the lung and airways. This finding is particularly interesting to us, as it suggests that CD8+ TRM within the nasal cavity are sufficient at preventing LAIV-SeV NP immunization from stimulating formation of CD8+ TRM against novel antigens. In contrast to lung TRM, nasal cavity TRM are formed independently of local cognate antigen recognition and are relatively stable over time (30). This explains why intraperitoneal infection, which does not deliver antigen to the lungs, results in similar numbers of TRM in the nasal cavity when compared to intranasal administration. Importantly, CD8+ TRM within the nasal cavity can prevent viral transmission down to the lung by controlling pathogen replication within the nasal epithelium. This, in turn, could limit the amount and duration antigen available for CD8+ T cell priming and expansion, especially in the lower respiratory tract. Given that LAIV is temperature sensitive and predominantly replicates in the upper respiratory tract, and that optimal TRM formation within the lung requires recognition of local antigen, this is a plausible explanation for why LAIV-SeV NP immunization failed to generate de novo lung TRM in PR8-immune mice, even when significantly fewer influenza-specific TRM were present in the lower respiratory tract (27). It is possible that less related pathogens would be capable of evading nasal cavity CD8+ TRM; however, our finding with AdNP-immunized animals discussed below indicates that limited epitope similarity is enough to prevent immunization from “taking.” Additional experiments are needed to confirm these hypotheses.
Lastly, immunization with an LAIV-SeV NP strain that fully escapes antibodies and does not present the immunodominant NP366 epitope (svHA-12 N370Q) did succeed in generating de novo CD8+ TRM in the lungs and BAL of mice that only have pre-existing NP366+ TRM. Immunization with svHA-12 N370Q also generated SeV NP+ CD8+ T cells in mice that were first immunized with an LAIV lacking the SeV NP epitope. However, the numbers of SeV NP+ T cells were less than those observed in AdNP immunized animals, suggesting that the other CD8+ T cell epitopes present in the LAIV are still impacting the formation of de novo TRM. It is important to note that we also cannot fully rule out a role for NP-specific B cells in our model. However, given our finding in AdNP mice, we suspect NP-specific antibodies to play a minimal role. Taken together, our findings suggest that escape from both the pre-existing humoral and cellular responses is necessary to generate a novel epitope-specific CD8+ lung TRM population following immunization with LAIV.
There has been longstanding interest in broadening our understanding of how pre-existing immunity impacts susceptibility to influenza infection (33). Original antigenic sin, a phenomenon whereby the immune system preferentially recalls existing memory cells made in response to a previous antigen encounter, is a well described impediment to generating de novo protection against influenza virus (34). The process has been best described in relation to antibody responses and is typically not observed during subsequent exposures to distantly related or entirely unrelated antigens (35). It has been proposed that altered antigen trafficking and prevention of LAIV replication in the nasal epithelia post-vaccination are potential mechanisms by which pre-existing antibodies reduce the overall effectiveness of LAIV, where effectiveness was measured as strong serum antibody levels and protection against homologous and heterologous strains (36). Here, our data suggests CD8+ TRM within the nasal epithelia may also work in a similar way to limit LAIV efficacy.
A recent study demonstrated that pre-existing humoral responses to LAIV prevent formation of CD8+ lung TRM and that this limitation could be overcome by increasing the vaccine dose (37). Although B cell responses to LAIV were not a focus of this manuscript, our data complement these findings by showing that pre-existing memory T cells in the upper respiratory tract also contribute to limiting LAIV efficacy. The ability of TRM to rapidly eliminate virus-infected cells could limit the absolute amount and duration of viral antigen production, further biasing the antibody response toward pre-existing memory B cells. Additional studies are needed to determine the impact of pre-existing cellular immunity on LAIV’s ability to stimulate de novo B cell responses.
Overall, our results provide critical insight to the roles that both pre-existing humoral and cellular immunity play in restricting the ability of LAIV immunization to promote antigen-specific T cell responses, and in particular the ability of pre-existing TRM to limit the development of de novo T cell responses. Recently, it’s been shown that prior immunity impacts the immune response to vaccination against SARS-CoV-2. While individuals that recovered from SARS-CoV-2 infection tend to experience a broader and stronger antibody response after booster vaccinations, a larger proportion of naïve T cells was found to be required for superior quality CD4+ T cell responses post-vaccination (38). In addition, LAIVs and other viral vectors, such as recombinant adenoviruses and orthopoxviruses, have been proposed as delivery platforms for vaccine antigens or for gene therapy (25, 39–43). Our data suggest that vector-specific memory T cell responses may also limit the efficacy of these platforms and should be considered in their design. In the case of recombinant adenovirus vaccines, vectors built from serotypes of non-human species are being evaluated that may circumvent this issue (44). While pre-existing antibodies and memory T cells are known to shape the establishment of novel antigen responses, their impact can vary depending on the specific pathogen and vaccination platform used, and additional research is needed to better inform vaccine design.
Supplementary Material
Key points:
LAIV fails to elicit de novo TRM in the face of pre-existing antibodies
Escape from cellular immunity allows for generation of T cell memory following LAIV
Acknowledgements:
We thank Dr. Johnathan Yewdell and Dr. Christopher Brooke or providing the influenza A/PR8 HA switch variant plasmids. We also thank the NIH Tetramer Core Facility (Contract 75N93020D00005) for providing class I tetramers. This research project was supported by the Emory University School of Medicine Flow Cytometry Core and the Pediatric/Winship Flow Cytometry Core. Portions of figures were made using BioRender.com.
This work was supported by National Institutes of Health (NIH) grants R35HL150803 and U01HL139483. J.L.L. was supported by NIH grant F31HL156639. J.L.L., A.C.L., and J.E.K. designed the study. S.D. generated all virus strains. K.E.H. validated virus sequences. J.L.L. performed experiments and data analysis with input from A.C.L. and J.E.K. J.L.L. wrote the manuscript and S.D., K.E.H., A.C.L., and J.E.K. edited the manuscript.
References:
- 1.Liang S, Mozdzanowska K, Palladino G, and Gerhard W. 1994. Heterosubtypic immunity to influenza type A virus in mice. Effector mechanisms and their longevity. J Immunol 152: 1653–1661. [PubMed] [Google Scholar]
- 2.Taylor PM, and Askonas BA. 1986. Influenza nucleoprotein-specific cytotoxic T-cell clones are protective in vivo. Immunology 58: 417–420. [PMC free article] [PubMed] [Google Scholar]
- 3.Terajima M, Cruz J, Leporati AM, Orphin L, Babon JA, Co MD, Pazoles P, Jameson J, and Ennis FA. 2008. Influenza A virus matrix protein 1-specific human CD8+ T-cell response induced in trivalent inactivated vaccine recipients. J Virol 82: 9283–9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu T, Guan J, Handel A, Tscharke DC, Sidney J, Sette A, Wakim LM, Sng XYX, Thomas PG, Croft NP, Purcell AW, and La Gruta NL. 2019. Quantification of epitope abundance reveals the effect of direct and cross-presentation on influenza CTL responses. Nat Commun 10: 2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grau-Exposito J, Sanchez-Gaona N, Massana N, Suppi M, Astorga-Gamaza A, Perea D, Rosado J, Falco A, Kirkegaard C, Torrella A, Planas B, Navarro J, Suanzes P, Alvarez-Sierra D, Ayora A, Sansano I, Esperalba J, Andres C, Anton A, Ramon YCS, Almirante B, Pujol-Borrell R, Falco V, Burgos J, Buzon MJ, and Genesca M. 2021. Peripheral and lung resident memory T cell responses against SARS-CoV-2. Nat Commun 12: 3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jozwik A, Habibi MS, Paras A, Zhu J, Guvenel A, Dhariwal J, Almond M, Wong EHC, Sykes A, Maybeno M, Del Rosario J, Trujillo-Torralbo MB, Mallia P, Sidney J, Peters B, Kon OM, Sette A, Johnston SL, Openshaw PJ, and Chiu C. 2015. RSV-specific airway resident memory CD8+ T cells and differential disease severity after experimental human infection. Nat Commun 6: 10224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McMaster SR, Wilson JJ, Wang H, and Kohlmeier JE. 2015. Airway-Resident Memory CD8 T Cells Provide Antigen-Specific Protection against Respiratory Virus Challenge through Rapid IFN-gamma Production. J Immunol 195: 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poon MML, Rybkina K, Kato Y, Kubota M, Matsumoto R, Bloom NI, Zhang Z, Hastie KM, Grifoni A, Weiskopf D, Wells SB, Ural BB, Lam N, Szabo PA, Dogra P, Lee YS, Gray JI, Bradley MC, Brusko MA, Brusko TM, Saphire EO, Connors TJ, Sette A, Crotty S, and Farber DL. 2021. SARS-CoV-2 infection generates tissue-localized immunological memory in humans. Sci Immunol 6: eabl9105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu T, Hu Y, Lee YT, Bouchard KR, Benechet A, Khanna K, and Cauley LS. 2014. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J Leukoc Biol 95: 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zens KD, Chen JK, and Farber DL. 2016. Vaccine-generated lung tissue-resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zens KD, Chen JK, Guyer RS, Wu FL, Cvetkovski F, Miron M, and Farber DL. 2017. Reduced generation of lung tissue-resident memory T cells during infancy. J Exp Med 214: 2915–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fiore AE, Uyeki TM, Broder K, Finelli L, Euler GL, Singleton JA, Iskander JK, Wortley PM, Shay DK, Bresee JS, Cox NJ, C. Centers for Disease, and Prevention. 2010. Prevention and control of influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Recomm Rep 59: 1–62. [PubMed] [Google Scholar]
- 13.Belshe RB, Edwards KM, Vesikari T, Black SV, Walker RE, Hultquist M, Kemble G, Connor EM, and C.-T. C. E. S. Group. 2007. Live attenuated versus inactivated influenza vaccine in infants and young children. N Engl J Med 356: 685–696. [DOI] [PubMed] [Google Scholar]
- 14.Forrest BD, Pride MW, Dunning AJ, Capeding MR, Chotpitayasunondh T, Tam JS, Rappaport R, Eldridge JH, and Gruber WC. 2008. Correlation of cellular immune responses with protection against culture-confirmed influenza virus in young children. Clin Vaccine Immunol 15: 1042–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He XS, Holmes TH, Zhang C, Mahmood K, Kemble GW, Lewis DB, Dekker CL, Greenberg HB, and Arvin AM. 2006. Cellular immune responses in children and adults receiving inactivated or live attenuated influenza vaccines. J Virol 80: 11756–11766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoft DF, Babusis E, Worku S, Spencer CT, Lottenbach K, Truscott SM, Abate G, Sakala IG, Edwards KM, Creech CB, Gerber MA, Bernstein DI, Newman F, Graham I, Anderson EL, and Belshe RB. 2011. Live and inactivated influenza vaccines induce similar humoral responses, but only live vaccines induce diverse T-cell responses in young children. J Infect Dis 204: 845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cox A, and Dewhurst S. 2015. A Single Mutation at PB1 Residue 319 Dramatically Increases the Safety of PR8 Live Attenuated Influenza Vaccine in a Murine Model without Compromising Vaccine Efficacy. J Virol 90: 2702–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cox A, Schmierer J, D’Angelo J, Smith A, Levenson D, Treanor J, Kim B, and Dewhurst S. 2020. A Mutated PB1 Residue 319 Synergizes with the PB2 N265S Mutation of the Live Attenuated Influenza Vaccine to Convey Temperature Sensitivity. Viruses 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perez DR, Angel M, Gonzalez-Reiche AS, Santos J, Obadan A, and Martinez-Sobrido L. 2017. Plasmid-Based Reverse Genetics of Influenza A Virus. Methods Mol Biol 1602: 251–273. [DOI] [PubMed] [Google Scholar]
- 20.Das SR, Hensley SE, Ince WL, Brooke CB, Subba A, Delboy MG, Russ G, Gibbs JS, Bennink JR, and Yewdell JW. 2013. Defining influenza A virus hemagglutinin antigenic drift by sequential monoclonal antibody selection. Cell Host Microbe 13: 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quinlivan M, Zamarin D, Garcia-Sastre A, Cullinane A, Chambers T, and Palese P. 2005. Attenuation of equine influenza viruses through truncations of the NS1 protein. J Virol 79: 8431–8439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fodor E, Devenish L, Engelhardt OG, Palese P, Brownlee GG, and Garcia-Sastre A. 1999. Rescue of influenza A virus from recombinant DNA. J Virol 73: 9679–9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoffmann E, Neumann G, Hobom G, Webster RG, and Kawaoka Y. 2000. “Ambisense” approach for the generation of influenza A virus: vRNA and mRNA synthesis from one template. Virology 267: 310–317. [DOI] [PubMed] [Google Scholar]
- 24.Becker TC, Noel RJ, Coats WS, Gomez-Foix AM, Alam T, Gerard RD, and Newgard CB. 1994. Use of recombinant adenovirus for metabolic engineering of mammalian cells. Methods Cell Biol 43 Pt A: 161–189. [DOI] [PubMed] [Google Scholar]
- 25.Uddback IE, Pedersen LM, Pedersen SR, Steffensen MA, Holst PJ, Thomsen AR, and Christensen JP. 2016. Combined local and systemic immunization is essential for durable T-cell mediated heterosubtypic immunity against influenza A virus. Sci Rep 6: 20137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kohlmeier JE, Miller SC, Smith J, Lu B, Gerard C, Cookenham T, Roberts AD, and Woodland DL. 2008. The chemokine receptor CCR5 plays a key role in the early memory CD8+ T cell response to respiratory virus infections. Immunity 29: 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cox A, Baker SF, Nogales A, Martinez-Sobrido L, and Dewhurst S. 2015. Development of a mouse-adapted live attenuated influenza virus that permits in vivo analysis of enhancements to the safety of live attenuated influenza virus vaccine. J Virol 89: 3421–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McMaster SR, Wein AN, Dunbar PR, Hayward SL, Cartwright EK, Denning TL, and Kohlmeier JE. 2018. Pulmonary antigen encounter regulates the establishment of tissue-resident CD8 memory T cells in the lung airways and parenchyma. Mucosal Immunol 11: 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Webby RJ, Andreansky S, Stambas J, Rehg JE, Webster RG, Doherty PC, and Turner SJ. 2003. Protection and compensation in the influenza virus-specific CD8+ T cell response. Proc Natl Acad Sci U S A 100: 7235–7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pizzolla A, Nguyen THO, Smith JM, Brooks AG, Kedzieska K, Heath WR, Reading PC, and Wakim LM. 2017. Resident memory CD8(+) T cells in the upper respiratory tract prevent pulmonary influenza virus infection. Sci Immunol 2. [DOI] [PubMed] [Google Scholar]
- 31.Hoft DF, Lottenbach KR, Blazevic A, Turan A, Blevins TP, Pacatte TP, Yu Y, Mitchell MC, Hoft SG, and Belshe RB. 2017. Comparisons of the Humoral and Cellular Immune Responses Induced by Live Attenuated Influenza Vaccine and Inactivated Influenza Vaccine in Adults. Clin Vaccine Immunol 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crowe SR, Turner SJ, Miller SC, Roberts AD, Rappolo RA, Doherty PC, Ely KH, and Woodland DL. 2003. Differential antigen presentation regulates the changing patterns of CD8+ T cell immunodominance in primary and secondary influenza virus infections. J Exp Med 198: 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cobey S, and Hensley SE. 2017. Immune history and influenza virus susceptibility. Curr Opin Virol 22: 105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Francis T Jr. 1960. On the Doctrine of Original Antigenic Sin. Proceedings of the American Philosophical Society 104: 572–578. [Google Scholar]
- 35.Webster RG 1966. Original antigenic sin in ferrets: the response to sequential infections with influenza viruses. J Immunol 97: 177–183. [PubMed] [Google Scholar]
- 36.Roy S, Williams CM, Wijesundara DK, and Furuya Y. 2020. Impact of Pre-Existing Immunity to Influenza on Live-Attenuated Influenza Vaccine (LAIV) Immunogenicity. Vaccines (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng MZM, Fritzlar S, Wang Z, Tan TK, Kedzierska K, Townsend A, Reading PC, and Wakim LM. 2022. Cutting Edge: High-Dose Live Attenuated Influenza Vaccines Elicit Pulmonary Tissue-Resident Memory CD8+ T Cells in the Face of Pre-Existing Humoral Immunity. J Immunol 209: 1832–1836. [DOI] [PubMed] [Google Scholar]
- 38.Saggau C, Martini GR, Rosati E, Meise S, Messner B, Kamps AK, Bekel N, Gigla J, Rose R, Voss M, Geisen UM, Reid HM, Sumbul M, Tran F, Berner DK, Khodamoradi Y, Vehreschild M, Cornely O, Koehler P, Krumbholz A, Fickenscher H, Kreuzer O, Schreiber C, Franke A, Schreiber S, Hoyer B, Scheffold A, and Bacher P. 2022. The pre-exposure SARS-CoV-2-specific T cell repertoire determines the quality of the immune response to vaccination. Immunity 55: 1924–1939 e1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coughlan L, Sridhar S, Payne R, Edmans M, Milicic A, Venkatraman N, Lugonja B, Clifton L, Qi C, Folegatti PM, Lawrie AM, Roberts R, de Graaf H, Sukhtankar P, Faust SN, Lewis DJM, Lambe T, Hill A, and Gilbert SC. 2018. Heterologous Two-Dose Vaccination with Simian Adenovirus and Poxvirus Vectors Elicits Long-Lasting Cellular Immunity to Influenza Virus A in Healthy Adults. EBioMedicine 29: 146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lillie PJ, Berthoud TK, Powell TJ, Lambe T, Mullarkey C, Spencer AJ, Hamill M, Peng Y, Blais ME, Duncan CJ, Sheehy SH, Havelock T, Faust SN, Williams RL, Gilbert A, Oxford J, Dong T, Hill AV, and Gilbert SC. 2012. Preliminary assessment of the efficacy of a T-cell-based influenza vaccine, MVA-NP+M1, in humans. Clin Infect Dis 55: 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Powell TJ, Peng Y, Berthoud TK, Blais ME, Lillie PJ, Hill AV, Rowland-Jones SL, McMichael AJ, Gilbert SC, and Dong T. 2013. Examination of influenza specific T cell responses after influenza virus challenge in individuals vaccinated with MVA-NP+M1 vaccine. PLoS One 8: e62778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vitelli A, Quirion MR, Lo CY, Misplon JA, Grabowska AK, Pierantoni A, Ammendola V, Price GE, Soboleski MR, Cortese R, Colloca S, Nicosia A, and Epstein SL. 2013. Vaccination to conserved influenza antigens in mice using a novel Simian adenovirus vector, PanAd3, derived from the bonobo Pan paniscus. PLoS One 8: e55435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wold WS, and Toth K. 2013. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr Gene Ther 13: 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fausther-Bovendo H, and Kobinger GP. 2014. Pre-existing immunity against Ad vectors: humoral, cellular, and innate response, what’s important? Hum Vaccin Immunother 10: 2875–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.