

Abstract
Bacteriophage P22 is a prototypical member of the Podoviridae superfamily. Since its discovery in 1952, P22 has become a paradigm for phage transduction and a model for icosahedral viral capsid assembly. Here, we describe the complete architecture of the P22 tail apparatus (gp1, gp4, gp10, gp9, and gp26) and the potential location and organization of P22 ejection proteins (gp7, gp20, and gp16), determined using cryo-EM localized reconstruction, genetic knockouts, and biochemical analysis. We found that the tail apparatus exists in two equivalent conformations, rotated by ~6° relative to the capsid. Portal protomers make unique contacts with coat subunits in both conformations, explaining the 12:5 symmetry mismatch. The tail assembles around the hexameric tail hub (gp10), which folds into an interrupted β-propeller characterized by an apical insertion domain. The tail hub connects proximally to the dodecameric portal protein and head-to-tail adapter (gp4), distally to the trimeric tail needle (gp26), and laterally to six trimeric tailspikes (gp9) that attach asymmetrically to gp10 insertion domain. Cryo-EM analysis of P22 mutants lacking the ejection proteins gp7 or gp20 and biochemical analysis of purified recombinant proteins suggest that gp7 and gp20 form a molecular complex associated with the tail apparatus via the portal protein barrel. We identified a putative signal transduction pathway from the tailspike to the tail needle, mediated by three flexible loops in the tail hub, that explains how lipopolysaccharide (LPS) is sufficient to trigger the ejection of the P22 DNA in vitro.
Keywords: bacteriophage P22, Salmonella-phages, viral genome ejection, ejection proteins, DNA-ejectosome, cryo-EM, portal protein, asymmetric reconstruction
Graphical Abstract
INTRODUCTION
Salmonella enterica Serovar typhimurium infects 100,000 people annually and causes Salmonellosis, which can be potentially fatal due to its dehydrating effects, among other symptoms like fever and diarrhea. An agent of Salmonella enterica Serovar typhimurium eradication, bacteriophage P22, has been studied over the years as a model system phage. P22 is a generalized transducing bacteriophage of the Podoviridae superfamily and the prototype of the P22-like genus [1]. P22 capsid is assembled with 415 copies of coat protein (gp5), a triangulation number of 7, and an overall diameter of ~630 Å in the mature virion (MV). P22-like phages possess a short and noncontractile tail apparatus, too short to span the host cell envelope [2]. The P22 tail is a multi-subunit molecular machine [3, 4] that provides all structural determinants for host attachment, adsorption, cell envelope penetration, and DNA delivery. Asymmetric cryo-EM reconstructions of the P22 MV proved invaluable in deciphering the organization of the capsid and tail apparatus [5–9]. The tail emanates at a unique 5-fold vertex, interrupting the icosahedral capsid symmetry [10]. Cryo-EM reconstructions of the isolated tail apparatus extracted from the infectious P22 virion [11, 12] together with crystal structures of P22 tail factors (e.g., the portal protein, gp1 [13, 14]; portal protein bound to the head-to-tail adapter protein, gp4 [13]; the tail needle, gp26 [15, 16]; the tailspike, gp9 [17–20]) aided in elucidating the tail structure and assembly (Table S1). In vitro studies using purified P22 proteins revealed how the portal protein initiates procapsid assembly and tail factors assemble hierarchically at the end of genome packaging. The scaffolding protein regulates the polymerization of the P22 coat protein (gp5) [21] and facilitates the incorporation of portal protein monomers into nascent procapsids by triggering portal ring formation [22, 23]. Cryo-EM analysis of P22 precursor capsid (or procapsid) provided evidence that the scaffolding protein interacts with the N-terminus of the coat protein and the portal protein [8]. Biochemical studies found that in addition to interaction with portal protein, the scaffolding protein C-terminal helix-turn-helix motif interacts with the coat protein N-terminus. Scaffolding protein is also critical for incorporating the ejection proteins gp7, gp20, and gp16 [24, 25]. P22 procapsids package concatemeric DNA until the head is full by the action of a powerful motor formed by the large (TerL) [26] and small (TerS) [27] terminase subunits, possibly assembled into a complex [28]. After packaging is complete, the TerL nuclease domain cleaves off the concatemer, releasing the packaging motor and allowing tail factors to assemble onto the portal protein. The gp4 head-to-tail adaptor oligomerizes upon binding to a portal dodecamer [29–31], providing an assembly platform for the tail hub gp10, which is monomeric in solution but assembles onto gp4 [32], generating a 6-fold channel that is sealed by gp26 [33].
The three ejection proteins, gp7, gp20, and gp16, are essential for infectivity and required for genome ejection into the host [34–37]. Unlike tail components, identifying and characterizing P22 ejection proteins has been challenging [34]. These proteins are encapsulated into procapsids during morphogenesis and ejected into the host at infection [36]. They play a crucial role in the early stage of transferring P22 DNA into the host [38], as supported by in vitro liposome experiments [39]. Cryo-electron tomography (cryo-ET) analysis of P22 virions infecting Salmonella minicells revealed that the ejection proteins form a trans-envelope channel in the host cell envelope, involved in genome ejection [40]. However, unlike T7, where the ejection proteins stack symmetrically onto the portal protein [41], or phage ΦcrAss001 that contains cargo proteins poised for ejection around the portal perimeter [42], P22 ejection proteins reside inside the virion, loosely associated with the portal barrel, as suggested by bubblegram studies [43]. When the ejection proteins are genetically deleted from the virion, the P22 genome-packaging machinery packages a longer than average DNA molecule, suggesting the ejection proteins occupy volume within the virion that can be filled by packaged DNA when they are absent [44]. Finally, in vitro studies revealed that P22 ejection proteins are not ejected in Salmonella unless both lipopolysaccharide (LPS) and the membrane protein OmpA are present [38]. In contrast, slow ejection of P22 DNA without release of the ejection proteins can be triggered by the addition of LPS only [45].
This paper describes the high-resolution structure of the phage P22 tail machine before genome delivery that we characterized using cryo-EM, genetic, and biochemical analysis. Our work sheds light on the components in the tail and inside the virion responsible for host attachment and genome ejection.
RESULTS
High-resolution structure of P22 noncontractile tail machine
We used localized reconstruction to decipher the entire molecular architecture of the P22 tail machinery (Table 1). After identifying a 3D class average containing the phage tail, this class was selected and used to generate a twelve-fold symmetric (C12) and a six-fold symmetric (C6) map at a Fourier Shell Correlation resolution of 2.8 Å and 2.9 Å, respectively (Figure S1, S2, 1A). Symmetrized maps revealed atomic details of individual tail factors (Figure S3). Specifically, the C12 map was used to build the dodecameric portal protein bound to 12 copies of the head-to-tail factor; the C6 map revealed the atomic structures of six tail hubs (res. 2–472), each associated with the tailspike N-terminal domain (NTD) (res. 5–122). In total, 13,350 amino acids built in the cryo-EM maps (Figure 1B) were real space refined to a final Correlation Coefficient (CC) greater than 0.82 (Table 2). Due to their flexibility, the tailspike body (res. 126–623) and tail needle (res. 56–233) were poorly visible in the symmetrized maps but could be resolved in a lower-resolution asymmetric (C1) map (Figure S1, S2, Table 1). The full-length tailspike containing the symmetric core and asymmetric NTD was refined to a final CC 0.77 at 3.2 Å resolution (Table 2). Figure 1B shows a composite atomic model of the P22 tail apparatus, including all structural components visualized in C12, C6, and C1 maps. The tail is 2.9 MDa in mass, 360 Å in length, with an internal channel formed by the portal, head-to-tail adaptor, and tail hub that spans ~270 Å in length with a diameter between 48 – 68 Å (Figure 1C). The entire P22 tail lumen, from below the portal protein barrel to the top of the tail needle N-terminus, has an approximate volume of 2.1 × 106 Å3 (Figure 1C). It is large enough to encapsulate ~1.1 MDa of globular proteins [46] and does not have a density that could be ascribed to DNA. The electrostatic surface potential of the P22 channel is primarily neutral with two charged regions: a positively charged surface in the portal barrel proximal to the interior of the capsid and an acidic/negative surface in the portal vestibule close to the tail hub (Figure 1C). The lack of physical restrictions in the tail channel suggests that the tail needle is the only physical plug for genome retention [33, 47, 48].
Table 1.
Cryo-EM data collection statistics
| Data Collection | P22 WT | P22 gp7− | P22 gp20− | P22 gp9−/gp26− | P22 gp9−/gp26− + gp16/gp20 |
|---|---|---|---|---|---|
| Facility/Microscope | PNCC/Krios | in house/Glacios | in house/Glacios | in house/Glacios | PNCC/Krios |
| Camera | K3 | Falcon 4 | Falcon 4 | Falcon 4 | K3 |
| Voltage (kV) | 300 | 200 | 200 | 200 | 300 |
| Magnification | 29,000 X | 52,000 X | 52,000 X | 92,000 X | 18,000 X |
| Electron exposure (e−/Å2) | 30 | 25 | 25 | 25 | 30 |
| Defocus range (μm) | −0.8 to −2.1 | −1.0 to −2.4 | −1.0 to −2.4 | −1.0 to −1.5 | −0.8 to −2.0 |
| Pixel Size (Å) | 0.80 | 0.95 | 0.95 | 1.50 | 1.28 |
| Total movies (frames/movie) | 5,331 (30) | 7,669 (40) | 6,343 (40) | 4,440 (40) | 3,819 (20) |
| Initial particle no. | 41,597 | 386,885 | 205,120 | 20,717 | 136,921 |
| Final particle no. (symmetry) | 38,151 (C12) | 51,156 (C6) | 50,464 (C6) | 8,159 (C6) | 31,640 (C6) |
Figure 1. High-resolution view of P22 tail apparatus using localized reconstruction.
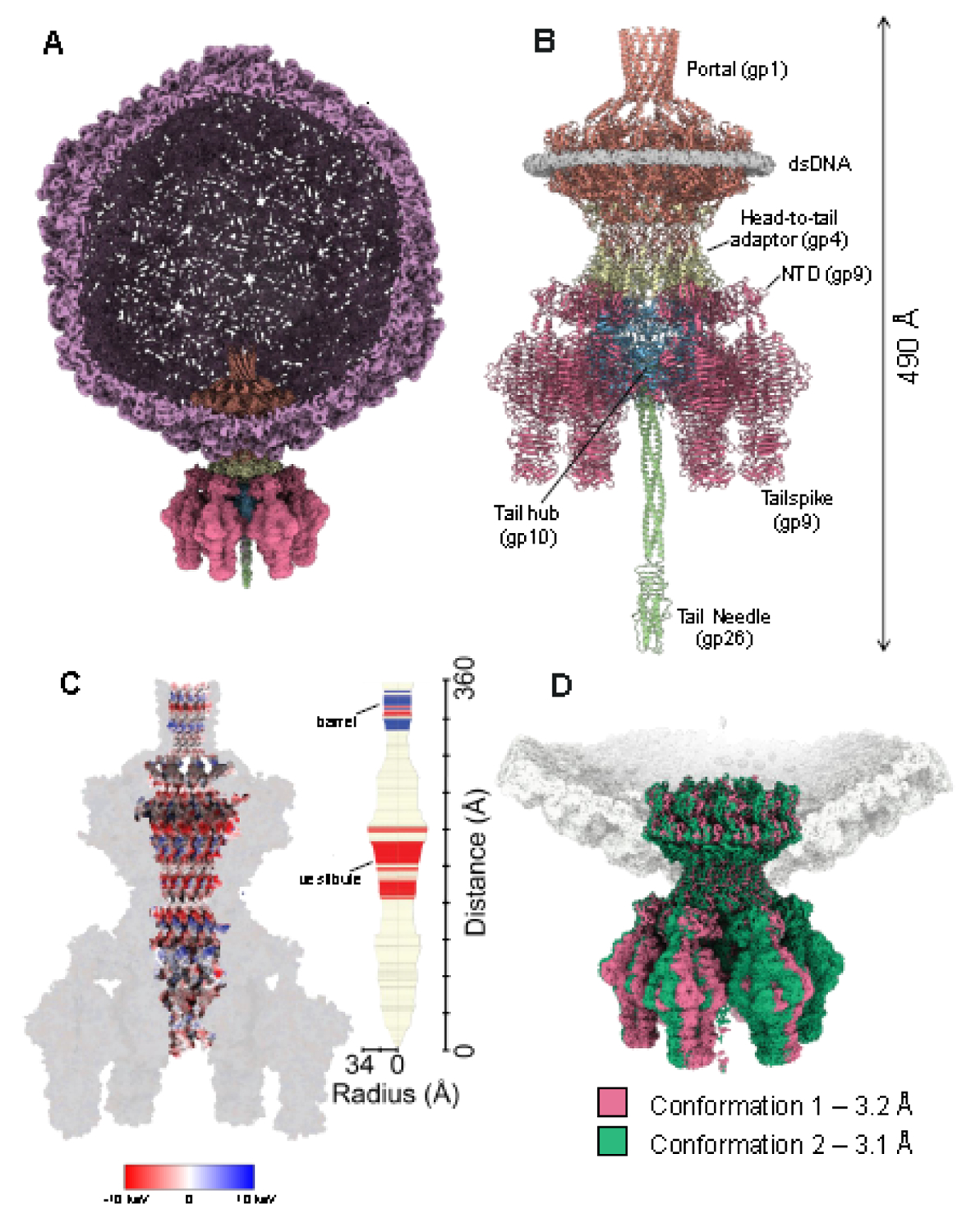
(A) A section through the P22 virion visualized at a high contour (~6.0 σ). The C6 localized reconstruction of the tail apparatus is overlaid to the unique vertex. (B) Cartoon representation of the P22 tail, including portal protein (light red), head-to-tail adaptor (light yellow), tail hub (light cyan blue), tailspike (pink), and tail needle (light green). The density for dsDNA surrounding the portal perimeter is displayed at 2.0 σ and colored in light gray. (C) Cross section of an electrostatic surface representation of the P22 tail highlights the surface inside the channel. Red and blue represent negative and positive charges near the surface, respectively, in keV. The regions in white have close to neutral surface charge. (D) Asymmetric (C1) reconstructions of the P22 tail aligned around the Z-axis, revealing two conformations of the tail apparatus rotated by 6 degrees.
Table 2.
Model Statistics
| Processing and refinement | ||||||
| PDB entry code | 8TVU | 8TVR | 8U10 | 8U11 | 8U1O | |
| Symmetry | C12 | C6 | C1 | C1 | C1 | |
| Model subunits | Portal: Head-to-Tail |
Tail hub: Tailspike N-term |
Coat: Portal: Head-to-Tail: Tail hub: Tailspike N-term Conf-1 |
Coat: Portal: Head-to-Tail: Tail hub: Tailspike N-term Conf-2 |
Full-length Tailspike |
|
| Map resolution (Å) | 3.0 Å | 2.8 Å | 3.2 Å | 3.1 Å | 3.3 Å | |
| Fourier Shell Correlation threshold | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | |
| Initial model used (PDB code) | de novo | de novo | 3LJ4; 1TYW | 3LJ4; 1TYW | 2BVE | |
| Correlation Coefficient (map-model) | 0.85 | 0.85 | 0.79 | 0.80 | 0.73 | |
| Model Composition | Number of Chains | 24 | 24 | 58 | 58 | 3 |
| Nonhydrogen Atoms | 72,840 | 38,532 | 138,421 | 138,416 | 15,096 | |
| Residues | 9,120 | 4,950 | 17,593 | 17,593 | 1,989 | |
| RMS deviations | Bond lengths (Å) | 0.004 | 0.004 | 0.003 | 0.006 | 0.005 |
| Bond angles (degrees) | 0.7 | 1.0 | 0.6 | 0.6 | 1.0 | |
| Validation | MolProbity Score | 2.1 | 2.0 | 1.8 | 1.9 | 2.1 |
| Clash Score | 8.4 | 7.8 | 8.9 | 10.1 | 12.3 | |
| Rotamer Outliers | 3.4 | 1.7 | 0.4 | 0.5 | 0.5 | |
| Ramachandran plot | Favored | 96.7 | 95.1 | 95.1 | 94.0 | 92.2 |
| Allowed | 3.3 | 4.9 | 4.8 | 5.9 | 7.6 | |
| Outliers | 0.0 | 0.0 | 0.1 | 0.1 | 0.3 | |
After determining the structures of individual tail components, we symmetry expanded the 2.9 Å C6 map and subjected the expanded pool of 38,308 particles to a new round of 3D classification (Figure S1). This yielded two predominant 3D class averages, containing 47.1 % (18,054) and 47.0 % (17,994) of the total particles, respectively. The two classes represent distinct tail conformations of the tail apparatus that we termed Conf-1 (3.2 Å) and Conf-2 (3.1 Å). Next, we docked the symmetrized atomic models of the P22 tail (Figure 1B) and modeled the coat protein into the asymmetric maps. The composite models for Conf-1 and Conf-2 were then real-space refined to a final CC of 0.79 and 0.80 respectively (Table 2). The two tail conformers are structurally identical but rotated approximately 6 degrees around the capsid 5-fold vertex (Figure 1D), as recently reported for the P22-like Shigella phage Sf6 tail [49].
Tail plasticity at the five-fold vertex
The portal protomer (Figure 2A) consists of four regions found in all portal proteins [50], the clip (res. 343–398), stem (res. 317–342; 399–420), wing (res. 1–316; 445–521), and crown (res. 522–605) [50], and also includes a C-terminal barrel (res. 600–725) [7]. No density was observed for two loops: residues 421–444 (hammer loop), which is ordered in the procapsid [14], and residues 481–490 (channel loop) that line the tail channel, potentially making direct contact with dsDNA during genome packaging (Figure 2A) [51]. Strong density for DNA surrounds the portal perimeter, contacting the portal wing domain through a basic and hydrophobic patch of residues consistent with this ring of DNA acting as a structural element to signal the end of headful packaging [5]. Superimposing the 12 portal subunits revealed structural similarity in Conf-1 (RMSD = 0.57 Å) and Conf-2 (RMSD = 0.60 Å), with some noticeable differences only in the coat-interacting region (res. 41–52, 200–217) (Figure 2A).
Figure 2. P22 portal protein and head-to-tail assembly at the five-fold vertex.
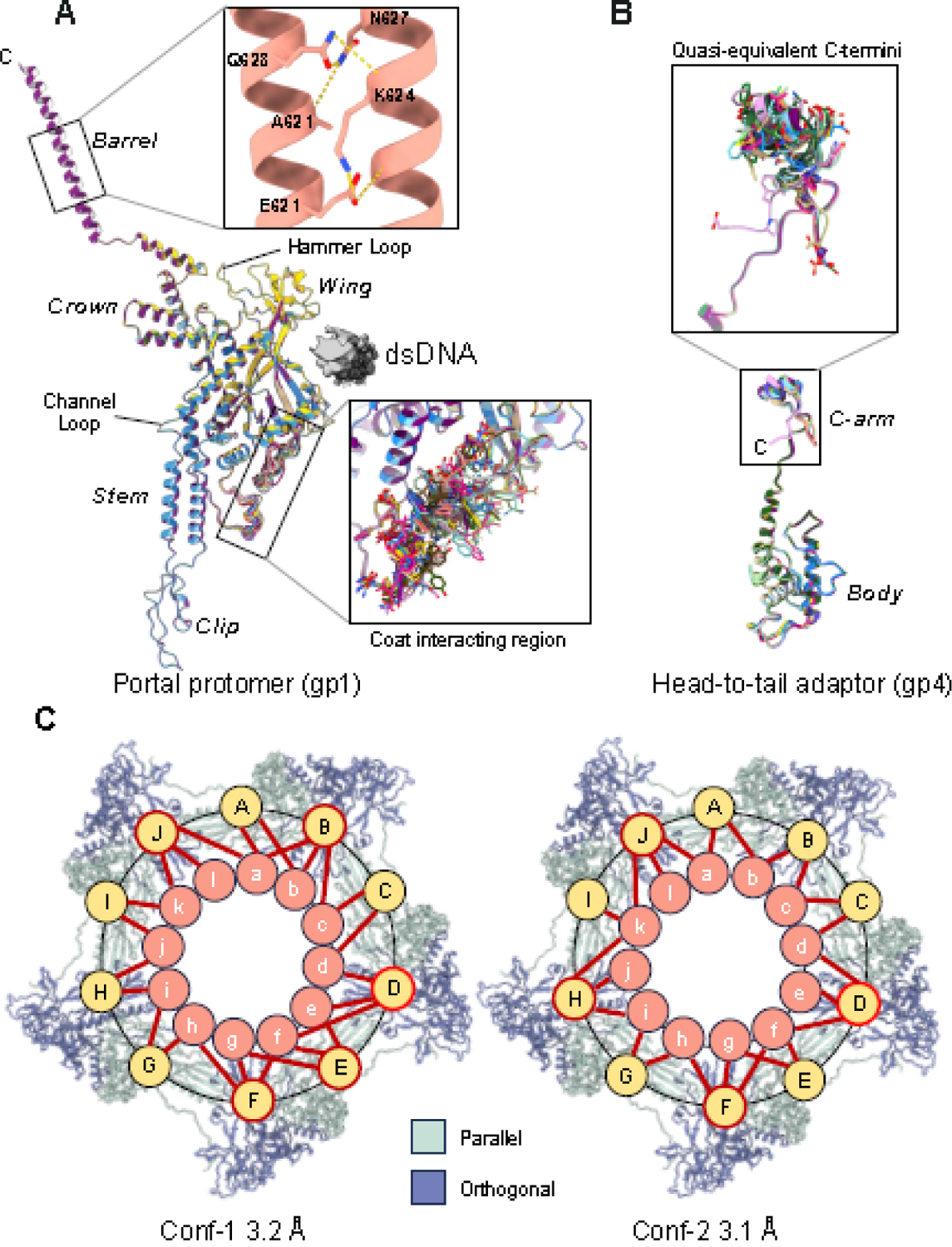
(A) Superimposition of twelve P22 portal subunits refined against the C1 map reveals significant structural variability in the coat interacting region of the wing (shown in the lower magnified panel). The top zoom-in panel shows sparse bonds stabilizing the barrel helical interface. (B) Superimposition of twelve head-to-tail adaptors refined against the C1 map reveals deviations in the C-terminal arm (shown in the magnified panel). (C) Cartoon schematic of the P22 portal subunits contacts with coat proteins at the five-fold vertex observed in Conf-1 and Conf-2. Coat proteins are shown as light and dark blue ribbons. Portal and coat subunits are schematized as circles and numbered ‘a-l’ and ‘A-J,’ respectively. Contacts between coat and portal subunits are shown with a red stick. Coat subunits D, F and J (circled in red) contact three portal subunits in both Conf-1 and Conf-2.
An intriguing feature of our high-resolution reconstruction, missing in previous X-ray structures [13] and cryo-EM reconstructions [5, 11], is that we could resolve side chains in the portal barrel. The C12 and C1 maps had strong density for the first ~70 amino acids of the barrel (res. 600–647) that we modeled and refined (Table 2). In contrast, the density for barrel residues 648–725 was very weak; thus, these residues were not modeled. We analyzed the inter-helical bonds keeping the barrel folded between residues 600–647 and found sparse molecular contacts stabilizing adjacent barrel helices, including five hydrogen bonds between residues 621–627 and mostly 20 van der Waals contacts (Figure 2A). The helix-helix interface buries 941.4 Å of solvent-accessible surface area, and the ΔG of association estimated by PISA (−18.5 kcal/mol) suggests the interface is likely unstable in solution. The modest binding interface supports experimental evidence that the isolated barrel helices (res. 605–725) do not form a stable quaternary structure in solution [14] or in procapsid [8] but assemble only in the MV surrounded by DNA [14] and ejection proteins [43]. Notably, the barrel appears to flower outward toward the C-terminus (Figure 1B), which reduces inter-helical bonding further, explaining the gradual disappearance of density for residues 648–725.
Next, we focused on the head-to-tail adapter gp4, which contains two domains: a globular body (res. 1–121) and a C-terminal arm (C-arm; res. 122–152), forming the portal protein interface (Figure 2B). Each portal subunit contacts four head-to-tail adaptors with a binding interface stabilized by eight hydrogen bonds, four salt bridges, and 46 van der Waals contacts (Figure 1B). Solution studies demonstrated that gp4 is mainly monomeric in solution but oligomerizes exclusively upon binding to the portal dodecamer [13]. Gp4 C-arm interacts with the portal interface while the α-helical body contacts the tail hub and tailspike N-termini (see below). Unlike phage Sf6 [49], the electron density of every copy of the head-to-tail adaptor ended at residue 151 in both conformations, lacking density for gp4 C-terminal residues 152–156. Head-to-tail subunits are very similar in Conf-1 and Conf-2, with RMSD = 0.65 Å and RMSD = 0.89 Å, respectively, with the only significant variations between C-terminal residues 136–152 (Figure 2B).
Resolving the portal:coat symmetry mismatch
To rationalize the symmetry mismatch between the tail and icosahedral capsid, we inspected the interface between the 5-fold coat protein and the 12-fold portal/head-to-tail complex in both Conf-1 and -2 asymmetric reconstructions (Figure 2C). We focused on residues in bonding distance (<4 Å) between the 12 portal subunits and ten coat proteins surrounding the portal vertex that can be orthogonal or parallel to the portal wing (colored blue and light green, respectively, in Figure 2C). Parallel coat protein interacts with the portal with a greater surface area, mainly via the P-domain (Figure 2C). We found portal protomers make non-equivalent contacts with the coat subunits, which deviate from dodecameric symmetry and differ in the two conformations (Figure 2C). The coat-interacting region of each portal subunit (Figure 2A) contacts three parts of the P22 coat P-domain spanning residues 32–42, 96–105, and 364–392. The extent of interfacing residues within these three areas of the P-domain varies depending on how they bind relative to portal protein (orthogonal vs. parallel) (Figure 2C). Specifically, in Conf-1, portal contacts coat protein through 88 hydrogen bonds, 13 salt bridges, and 404 van der Waals interactions while having similar contacts in Conf-2 (85 hydrogen bonds, 14 salt bridges, 381 van der Waals interactions). In both conformations, each portal subunit contacts at least one coat subunit and vice-versa (Figure 2A). However, the portal:coat binding pattern is conformation-dependent (Figure 2C). In Conf-1, five of the ten coat subunits bond three different portal proteins, while the remaining coat subunits contact at least two different portal subunits. Meanwhile in Conf-2, only four of the ten coat subunits contact three portal subunits, five coat proteins contact two portal subunits and one coat is left bound to only one portal (Figure 2C). In both conformations, coat proteins D, F, and J which are all positioned orthogonal to portal, kept their triple binding interface to the same portal subunits (Figure 2C). Thus, non-equivalent bonds between the coat-interacting region of portal subunits (Figure 2A) and coat subunits cement the dodecamer portal at the icosahedral 5-fold vertex: these bonds are non-identical in the two portal conformers.
The tail hub is the P22 tail building block
Despite being the most conserved factor in P22-like phages [52], the structural details of the P22 gp10 tail hub have not been elucidated. Our C6 localized map revealed the atomic structure gp10 (res. 2–427), which consists of an interrupted β-propeller similar to phage Sf6 gp8 [49]. Gp10 comprises a β-propeller core (res. 50–362) and an insertion domain (res. 1–49; 363–471) (Figure 3A). The β-propeller core is formed by seven antiparallel β-sheets called blades, with each β-sheet containing four antiparallel β-strands. The insertion domain emanates from blades #1 and #7 and contains the protein N- and C- termini. Its quaternary structure is like the lamin B1 fold of nucleoporins forming the nuclear pore complex [53] (Figure 3A). Six gp10 subunits assemble as a hexamer onto the gp4 α-helical body (Figure 3B), while laterally, the tail hub serves as the docking point for binding six tailspike trimers (see next section). At the tail hub, the P22 tail changes symmetry from 12-fold symmetric to 6-fold symmetric, where one gp10 subunit contacts two head-to-tail adaptor subunits (Figure 3B).
Figure 3. Structure and assembly of the tail hub gp10.
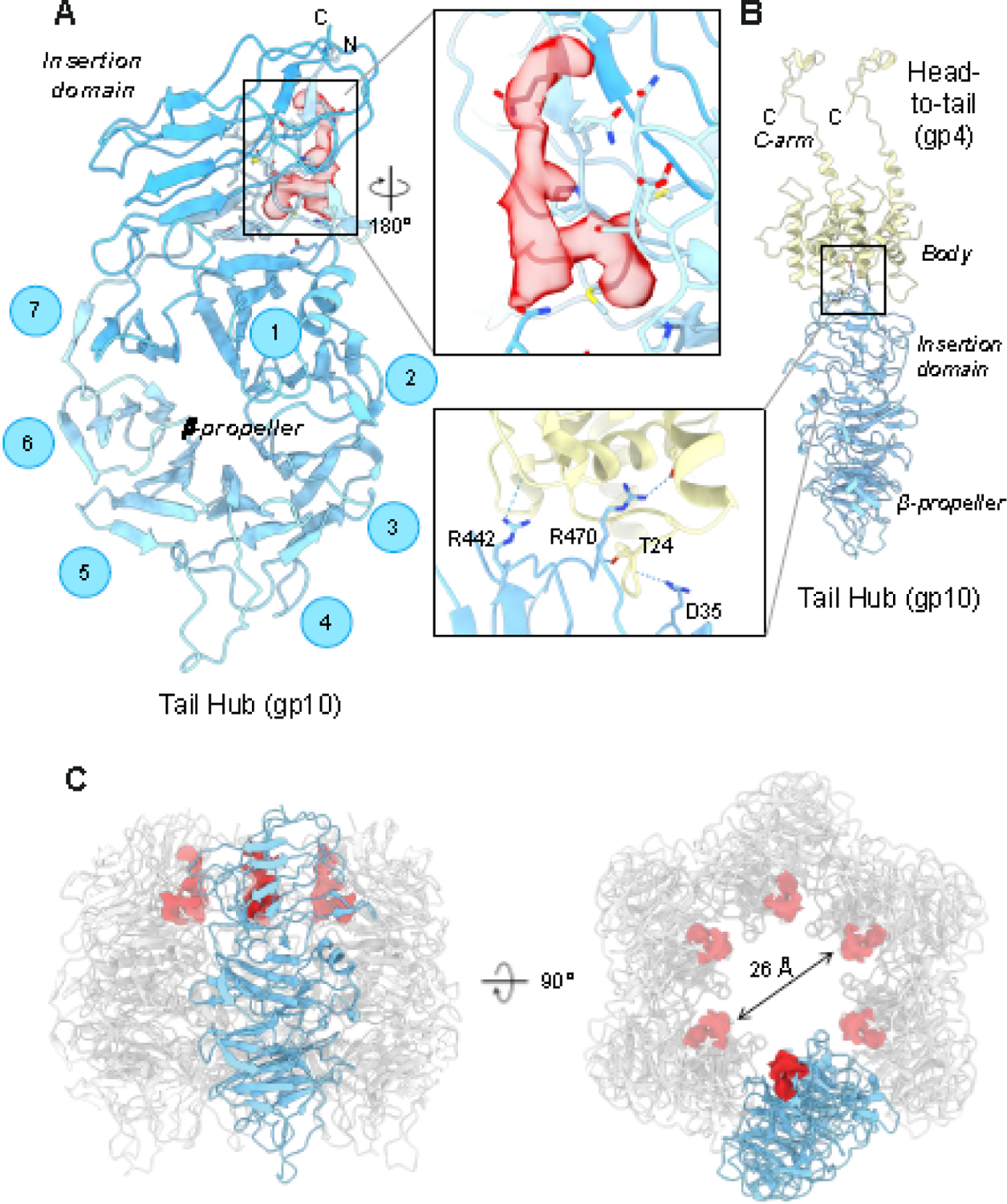
(A) P22 tail-hub tertiary structure reveals an interrupted β-propellor domain (res. 50–362) that contains an apical insertion domain (res. 1–49; 363–471). An elongated density, contour at ~3.5 σ (indicated in red), was identified as bonding with gp10 residues 5,7,9,31, and 41 (zoom-in panel). (B) Each gp10 monomer contacts two head-to-tail adaptor proteins. The inset shows hydrogen bonds between the tail hub and a pair of head-to-tail subunits. (C) Tail hub quaternary structure: one protomer is colored in light cyan-blue while the other five are in light gray. The unassigned density lining the tail channel is shown in red. The right panel is rotated along the y-axis by 90 degrees (right).
After fitting gp10 in the symmetrized and asymmetric maps, we identified an additional L-shaped density in the C6 map, clearly visible at 3σ contour (Figure 3A). The same density was also present in the C3 and C1 maps, albeit at a lower contour, and appears to be proteinaceous, with blobby main chain features and a bulky side chain density sandwiched between gp10 residues M9 and P33. The unknown density is located channel-side, facing the inside of the tail hub lumen, where it fills part of the positively charged collar lining a ~25 Å lumen (Figure 3C). At the current resolution, we cannot assign the density in the gp10 channel to a specific P22 open reading frame or host cell protein.
Asymmetric quaternary structure of the tailspike
P22 contains six trimeric tailspikes, each containing three endo-rhamnosidase active sites that cleave Salmonella O-antigen [17]. The tailspike is divided into two distinct domains (Figure 4A): the NTD (res. 5–122) that was built de novo in the C6 map and is intimately bound to gp10, and the C-terminal body (res.123–677), which consists of a prototypical triple β-helix made by identical protomers [18, 20]. Both domains are symmetric in the crystal structure of the full-length protein [19], whereas the NTD is profoundly asymmetric in our cryo-EM reconstruction. In complex with gp10, the tailspike NTD bends with respect to the body, with the linking α-helix (res. 113–122) serving as a molecular hinge (Figure 4A). The three tailspike protomers have RMSD 7.2 Å between residues 1–124, whereas the rest of the protein (res. 125–677) is relatively identical (RMSD 1.6 Å). The α-helical hinge between the tailspike body and NTD is positioned distal to the central axis rather than at the central axis, as in the homosymmetric crystal structure of P22 gp9 (PDB: 1LKT) [20] (Figure 4B).
Figure 4. The asymmetric structure of the P22 tailspike.
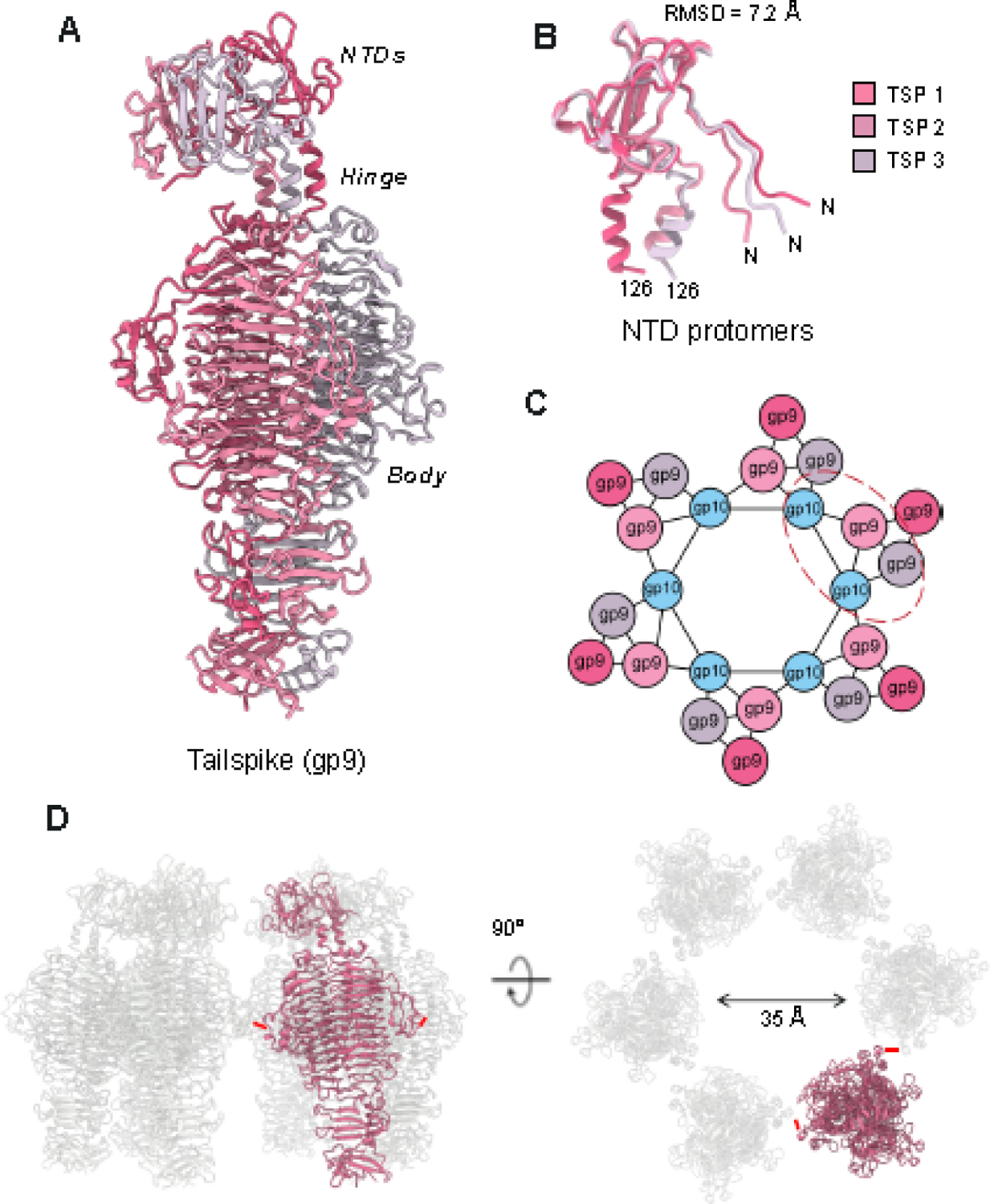
(A) The structure of the full-length P22 tailspike reveals an asymmetric NTD leaning to the left, connected to a triple β-helix body. (B) Superimposition of the tailspike N-termini (res 1–126). (C) Schematic diagram of the symmetry mismatch between the tail hub and trimer tailspikes. The red dashed line circles the pseudo-2:1 binding interface. (D) Quaternary structure of P22 tailspikes observed in the MV. Six tailspike (one subunit in red and five in light gray) are shown in a side view (left panel) and rotated along the y-axis by 90 degrees (right panel). The red dashes denote the smallest distance between adjacent tailspike subunits.
We generated a complete model of gp9 by solving the tailspike in two different maps. For the tailspike body, the crystal structure of the P22 tailspike (PDB: 1TSP) [18] was placed into the asymmetric maps as these maps had a stronger signal for this region than the C6 symmetrized map. Notably, the tail hub binds the tailspike with quasi-2-fold symmetric binding interfaces where one tailspike subunit binds one tail hub unit while the adjacent tailspike within the tailspike trimer binds in between two gp10 tail hub subunits (Figure 4C). The tailspike subunit facing outward from the tail does not contact gp10. Placing six tailspikes into the C1 density gave us the quaternary structure arrangement of the P22 tailspike in the MV (Figure 4D). The tailspikes do not make intermolecular contact with each other, and the closest distance between two strands (res. 244–257) of adjacent tailspikes (res. 197–260) is about 12.7 Å (red lines in Figure 4D).
P22 ejection proteins may reside inside the tail apparatus
We purified two P22 mutant phages lacking gp7 or gp20, termed P22 gp7— and P22 gp20— and determined their cryo-EM structures using the same single particle analysis (SPA) cryo-EM workflow described for the MV (Figure S1, S2). We obtained two medium-resolution reconstructions with resolved general tail features, including tailspike, tail hub, head-to-tail adaptor, and portal (Figure 5B, C). The maps of the P22 mutants were compared to the P22 MV (Figure 5A), and all the maps were low pass filtered to medium resolution ~5.0 Å and displayed at identical contours to ensure an objective comparison. Interestingly, the cross-sectional views for all three maps revealed three significant differences in the density corresponding to the portal protein C-terminus and the end of the tail needle gp26. First, P22 mutants of either deleted ejection protein had significantly less barrel density and, inside the barrel, less density for dsDNA, which is mostly similar in appearance to the density of the DNA ring surrounding the portal protein (Figure 1B). The barrel and DNA density disappearance can be attributed to the increased mobility of the barrel C-terminus without each ejection protein, suggesting a molecular complex of ejection proteins is formed in the WT phage, consistent with previous results [43]. Second, mutants lacking gp7 or gp20 had enhanced density for the tail needle gp26 distal tip. This flexible region of the needle tip [16], called the lasso domain, is blurred in the P22 virions lacking either ejection protein, suggesting increased tail needle mobility when all ejection proteins are present. Third, a large density was observed in the gp10 channel of P22 phages lacking gp7 (Figure 5C, D). To test whether the density inside the tail belongs to ejection proteins [40], AlphaFold2 [54] was used to predict the tertiary and quaternary structures of P22 gp16 and gp20. The observed density was too small to fit the predicted structure of gp16, while it could potentially accommodate the predicted model of gp20 (Figure 5D).
Figure 5. Cryo-EM reconstructions of P22 virion mutants.
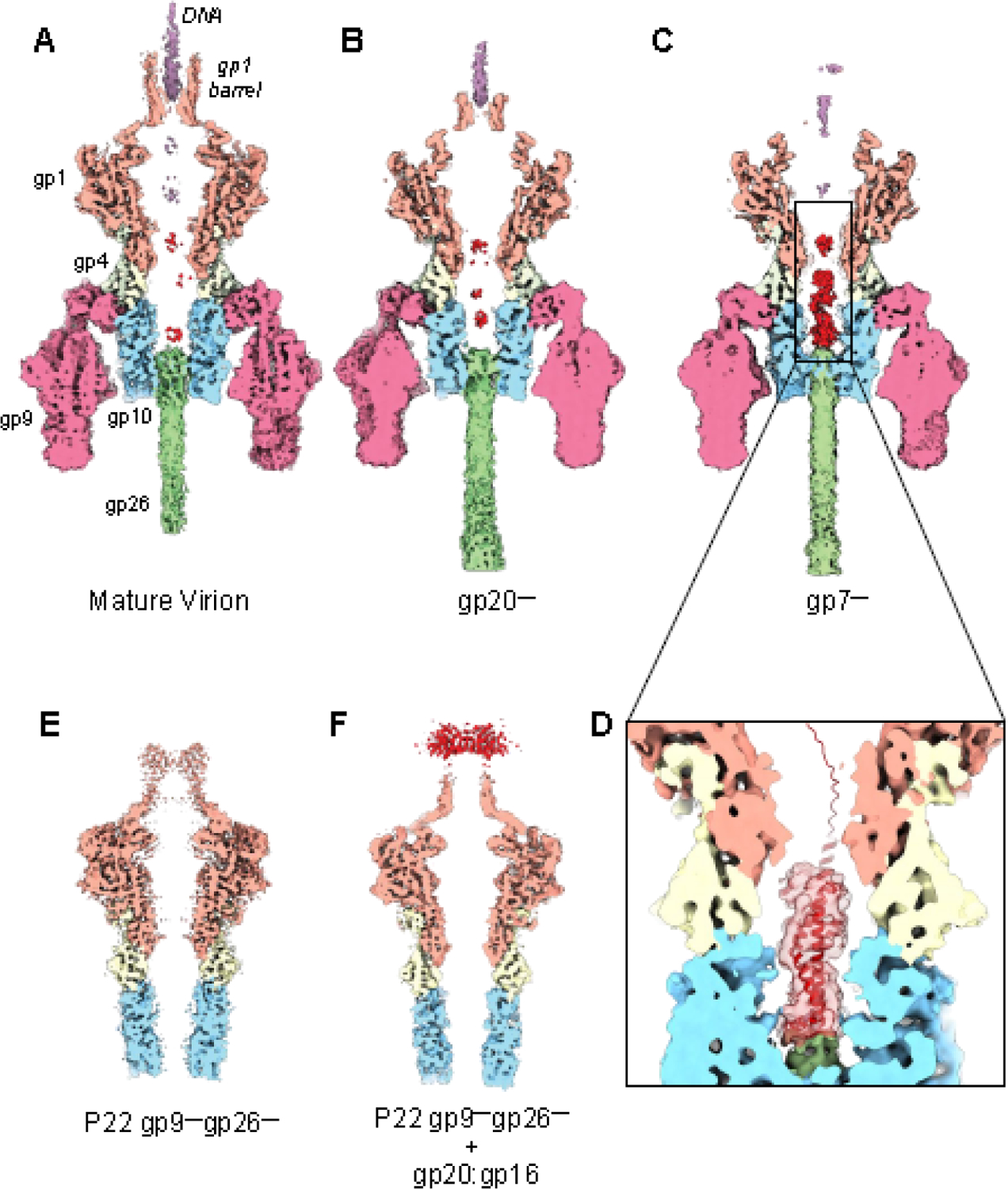
Cross-sectional views of P22 tail asymmetric reconstructions of the (A) P22 MV and mutants lacking one ejection protein, (B) P22 gp20— and (C) P22 gp7—. (D) A magnified view of the density in the channel (in light red) overlaid to an AlphaFold model of gp20. (E) Cross-sectional view of a P22 mutant lacking the tailspike and tail needle (P22 gp9—/gp26—). (F) Cross-sectional view of P22 gp9—/gp26— incubated with an excess of recombinant gp20:gp16 complex purified from a soluble bacterial lysate. All the maps shown in this figure were sharpened, low pass filtered to 5.2 Å, and are displayed at 2.0 σ.
We also reconstructed P22 mutants with both the tail needle and tailspikes deleted, which lack DNA inside the capsid (Figure 5E). We found that the structure of the gp10 tail hub was identical in the absence of a binding partner, suggesting that gp10 is rigid and unlikely to undergo significant structural rearrangements. This agrees with a previous study by cryo-ET and subtomogram averaging [40] that found the orientation and overall conformation of P22 gp10 are indistinguishable in free virions versus virions absorbed to Salmonella minicells. Accordingly, hexameric gp10 determined using single particle analysis fits well in the 28 Å subtomogram averaged map (Figure S5): gp10 overall structure appears unchanged after ejection of gp7, gp20, and gp16, suggesting the protein forms a rigid hub. Interestingly, a diffused density surrounding the barrel was observed when recombinant ejection proteins gp16 (609 aa, M.W. 64.4 kDa) and gp20 (471 aa, M.W. 49.9 kDa) were incubated with P22 gp9—/gp26— mutants (Figure 5F). For this experiment, histidine-tagged gp20 and (untagged) gp16 were co-expressed in bacteria, where the two proteins formed a soluble complex that we purified by metal-chelate affinity chromatography and size exclusion chromatography (SEC) (Figure S6). Although gp16 was reported to partition into the membrane fraction [39], the native protein is soluble in bacterial lysates [55], and recombinant gp16 was also partially soluble in our hands, underscoring an amphipathic nature, similar to T7 gp16 [56, 57]. In our experiments, the recombinant gp20:gp16 complex copurified as a single species but did not assemble into a higher order structure, unlike T7 homologous ejection proteins gp15 and gp16 that form tubes when co-expressed in bacteria or mixed in vitro [57]. We speculate that P22 gp20 and gp16 remain partially unfolded or adopt an elongated structure lacking a hydrophobic core like T7 gp15 [56], capable of entering the capsid from the tail hub and forming a complex with the barrel helices (Figure 5F) [43].
P22 ejection protein gp7 is a membrane protein that interacts with gp20
We attempted to purify the P22 ejection protein gp7 (229 aa, M.W. 23.4 kDa) but found that the recombinant protein is entirely insoluble. We extracted insoluble gp7 expressed in bacteria with sarcosine that was gradually exchanged to dodecyl β-D-maltoside (DDM), yielding a soluble species running as a monodisperse peak by SEC (Figure 6A). Gp7 was successfully incorporated in lipid nanodiscs (Figure 6A), confirming that the protein is a genuine membrane protein, like T7 gp14 [57]. This finding disagrees with a previous report that purified P22 gp7 is water soluble [39]. Gp7 is proteolytically cleaved at the N-terminus to remove the first 20 amino acids [58]. We also generated a construct lacking the N-terminal 20 residues but found no difference in the protein solubility (data not shown). When mixed, gp7 and gp20 in the absence of nanodiscs, the two proteins formed a molecular complex, migrating as an early-eluting peak by SEC (Figure 6B). However, cryo-EM analysis suggested that the in vitro assembled gp7:gp20 complex forms a partially oligomeric tube, possibly consistent with a highly flexible intermediate or a partially assembled DNA-ejectosome (Figure 6C). Notably, phage Sf6 gp12 (P22 gp20 homolog) also assembles into a tubular structure characterized by high flexibility in solution, as assessed by small-angle X-ray scattering [59]. In contrast, all efforts to form a complex of recombinant gp20 and gp16 (with or without gp7) were unsuccessful (data not shown), suggesting that the P22 DNA-ejectosome assembles only under physiological conditions and may require additional factors and a sequential order of assembly not recapitulated in vitro.
Figure 6: Reconstitution of P22 ejection proteins gp7 and gp20.
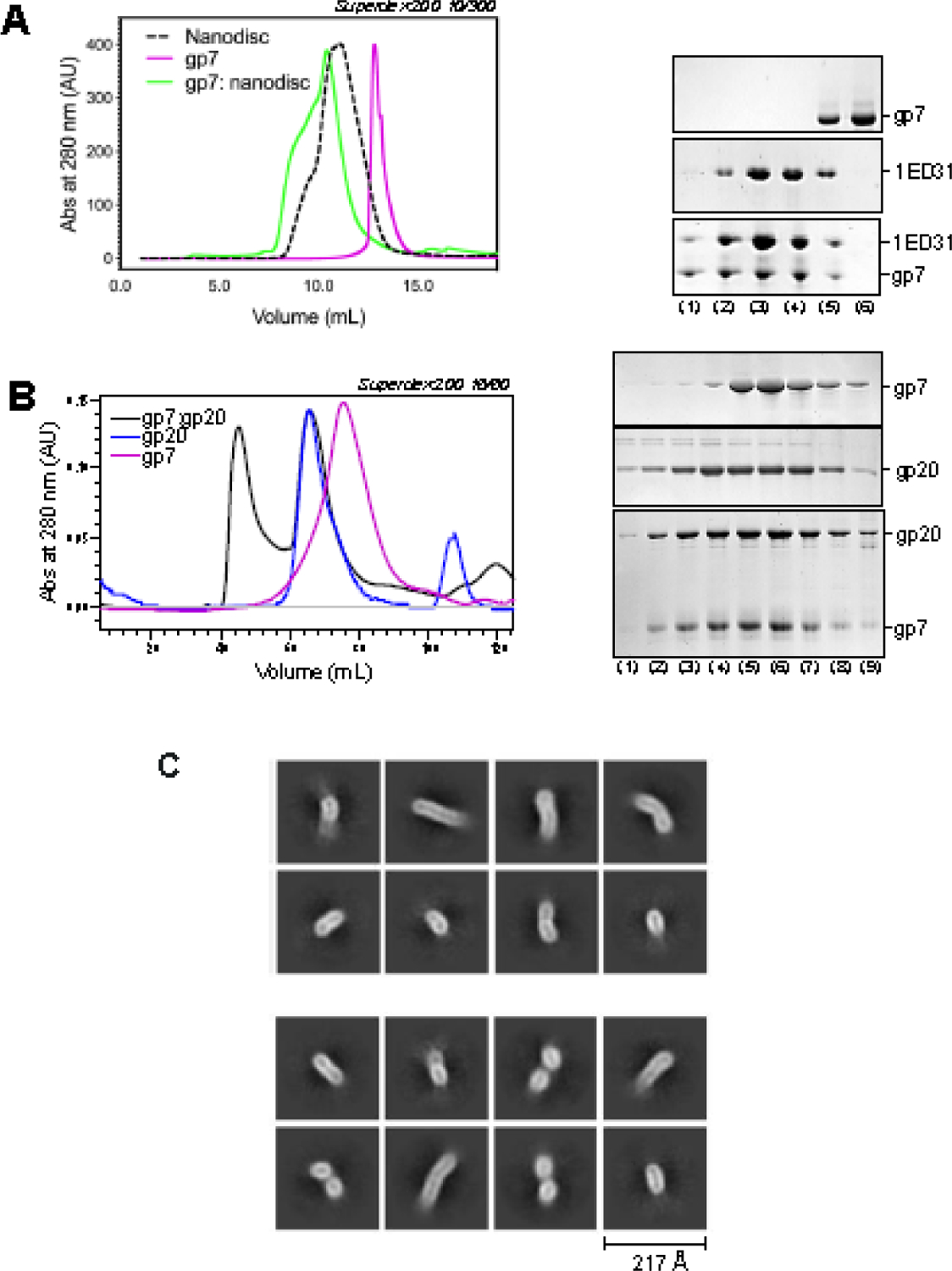
(A) SEC analysis (left panel) of solubilized gp7, scaffolding protein 1ED31 assembled into nanodiscs and gp7:nanodiscs. Fractions eluted from SEC were analyzed by SDS-PAGE (right panel). (B) SEC analysis (left panel) of solubilized gp7, gp20, and gp7:gp20 complex. Fractions eluted from SEC were analyzed by SDS-PAGE (right panel). (C) Representative 2D-class averages of vitrified gp7:gp20 complex.
DISCUSSION
In this paper, we described the structure of the phage P22 tail apparatus and the organization of the ejection proteins, determined using hybrid structural methods. We built accurate atomic models of all structural proteins, including the tail hub gp10, which was previously unknown, and the full-length tailspike that adopts a surprisingly asymmetric conformation assembled to the tail hub. The overall structural organization of the P22 tail apparatus is similar to the Shigella phage Sf6 [49]. However, P22 tail factors make significantly more bonds in their oligomeric structures and between different tail components than in the Sf6 tail (Table S2). For instance, the P22 gp10:gp10 binding interface is stabilized by 16 salt bridges, 32 hydrogen bonds, and 169 van der Waals interactions versus just nine salt bridges, nine hydrogen bonds, and 119 van der Waals, respectively, in Sf6. Even more so, the P22 tailspike gp9:gp9 interface has between four and six salt bridges in P22 versus just one in Sf6, indicating the P22 tail apparatus is more stable than in Sf6 and its tailspikes significantly more rigid, as previously observed [60]. Our structure-based observation of a super-stable tail reconciles the previous empirical finding that the P22 tail can be extracted from mature virions using prolonged heating at 60°C in a buffer containing 0.1 M EDTA, 0.1% (v/v) Triton X-100 and 2.0 M urea, which destroys the icosahedral shell liberating the tail as an intact macromolecular machine [12].
The tail’s building block, gp10, folds into an interrupted β-propeller, a fold seen throughout eukaryotes with various degrees of functions, including but not limited to ligand binding, signaling, or binding to other proteins [8]. Gp10 forms a hub around which the rest of the tail apparatus assembles, serving at least three functions: first, it reduces the C12 symmetry of the portal and head-to-tail from C12 to C3 for the tail needle that seals the tail channel; second, it generates a lumen for DNA passage that is filled with an unknown density; third, it provides asymmetric attachment surfaces laterally for the six trimeric tailspikes and distally to the tail needle. Thus, the tail hub, the most conserved tail factor in P22-like phages [52], is the keystone of this podovirus short noncontractile tail.
Previous work has shown that tailspikes binding to O-antigen and LPS hydrolysis is sufficient for the slow ejection of P22 DNA in vitro [45, 61]. However, the mechanisms of LPS-triggered signal transduction from the tailspike to the ejection proteins are unknown. P22 tailspikes are less flexible than in phage Sf6 [49, 60], making it difficult to explain how LPS could trigger the release of the tail needle and genome ejection in vitro [45, 61]. Our cryo-EM analysis of P22 MV and gp9—/gp26— found the tail hub is rigid and identical with or without tailspikes and tail needle (Figures 7E and F), suggesting the gp10 tube is unlikely to undergo major conformational changes sufficient to squeeze out the tail needle upon LPS binding to tailspikes. Wang et al. proposed that most of the interactions for P22 genome ejection require LPS hydrolysis by the tailspikes and the subsequent reorientation of the tail on the bacterial outer membrane [40]. However, our high-resolution structures reveal that P22 LPS binding sites in the three tailspike subunits are not identical and differ relative to their orientation to the tail hub. Only one of three subunits per trimer makes a unique contact with the tail hub loops, while the other two subunits have their dorsal fin domains facing away from the tail hub. We propose that first, LPS hydrolysis of the externally located tailspike subunits helps orient the P22 phage as suggested by Wang et al. [40] and that these binding and hydrolysis events also lead to the reorientation of the less populated, interior-facing tailspike subunits. These subunits would be positioned at an angle away from LPS molecules at this initial point of contact due to the orientation of the phage relative to the bacterial membrane. Then, the reorientation of the P22 tail would reorient the tail hub, allowing the internal facing tailspike subunit to bind and hydrolyze LPS to destabilize the contacts between the tail hub and the needle. How is the signal for tail needle ejection propagated in the tail upon LPS association with and hydrolysis by tailspikes? To answer this question, we zoomed in on the building blocks of the P22 tail: one trimer of gp9, a protomer of gp10, and the trimeric tail needle gp26 (Figure 7A). The tail hub is sealed by the N-terminus of the tail needle, which, like a plug, prevents premature DNA leakage from the phage [48] and ensures successful infection by only dissociating upon piercing the bacterial outer membrane (OM). Deuterium exchange mass spectrometry studies found that the gp26 N-terminal arm (res. 1–26) is the tail needle’s most dynamic part, forming many hydrogen bonds at its binding interface with gp10 [62]. This region of the tail needle contacts the first of three loops in the gp10 β-propeller domain (Figure 7B). We named the three loops, from the tail needle to the tailspike exterior, according to their arrangement: Loop I (res. 211–222) connecting gp10 blades 5 and 6, Loop II (res. 164–177), an extension of the 4th blade, and Loop III (res. 124–140) facing the tailspike dorsal fin loop, which is a region of ~24 aa protruding from the repetitive triple-β helix fold. Notably, the tailspike fin and Loop III are ~7.5 Å away, not in direct bonding distance but sufficiently close to engage in contacts if the tailspike undergoes conformational changes due to LPS binding and hydrolysis (Figure 7C). Residues 401–406 are not resolved in the crystal structure of tailspike protein [18] but are observed in the cryo-EM reconstruction of the tailspike dorsal fin loop, proximal to tail hub Loop III. We hypothesize that LPS-hydrolysis sends the signal from the fin loop extension, which pulls Loop III closer to the tailspike, pulling all other loops away from the tail needle N-terminal moiety. We propose such a conformational change may propagate through the three loops in gp10, leading to a stronger association between tail hub Loop III and tailspike dorsal fin loop, thereby disrupting the hydrophobic core between Loop II and Loop III, which ultimately frees the tail hub Loop I from bonding the tail needle. However, LPS alone is necessary but insufficient for releasing the ejection proteins gp7, gp20, and gp16 that leave the capsid before DNA only if the membrane protein OmpA and LPS are present [38]. This is likely a kinetic effect, consistent with the idea that the tail needle affects the rate of DNA delivery into bacteria [63].
Figure 7. A model for signal transduction.
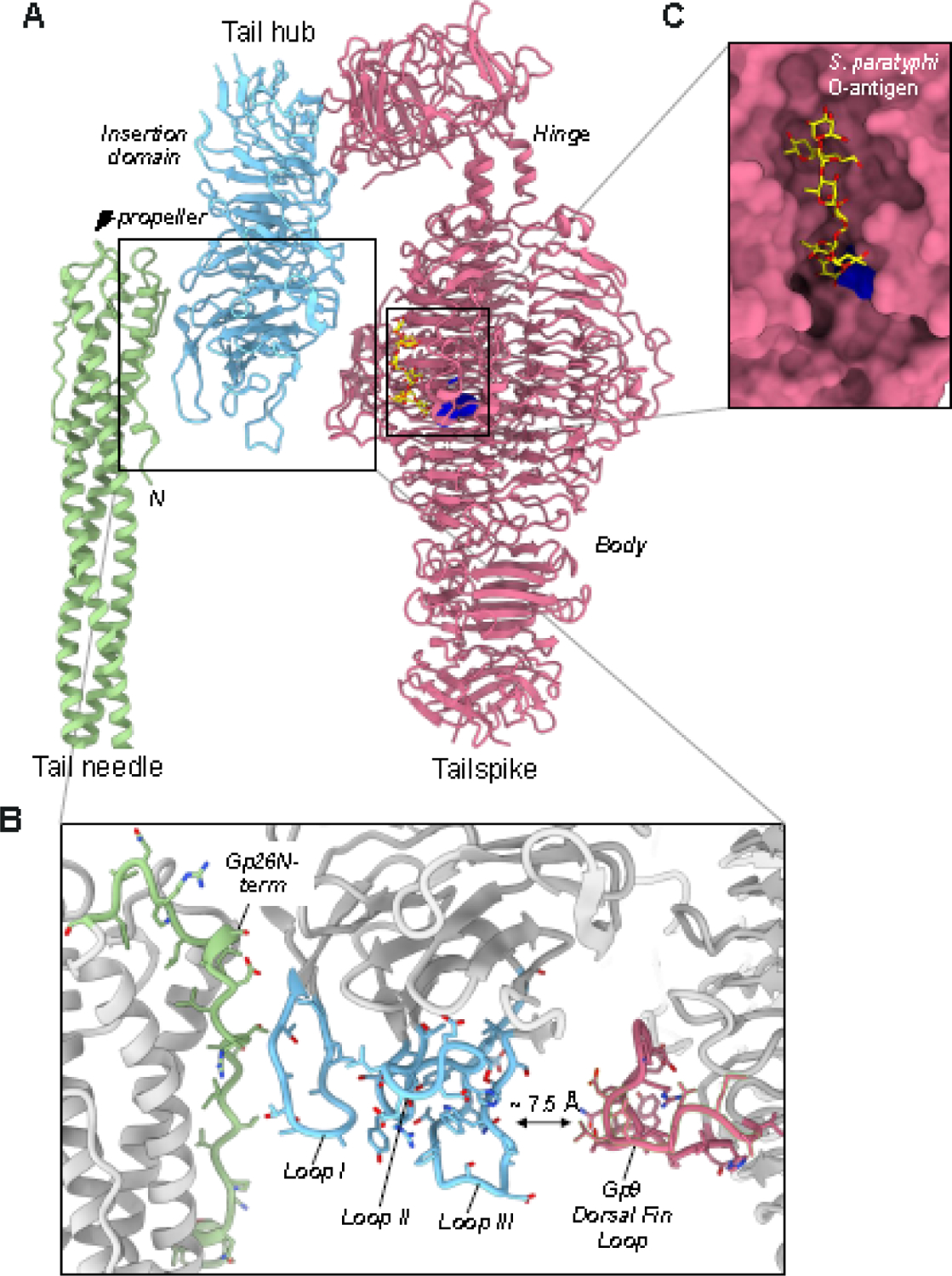
(A) Cartoon representation of a tailspike trimer shaded in pink, contacting one copy of the P22 tail hub (light cyan blue) and the tail needle (light green). (B) Zoom-in view of the P22 tailspike active sites colored as yellow spheres (res. 216–255, 358–387, 391–422). (C) Magnified view of gp10 loops mediating the putative crosstalk between the tailspike and tail needle.
We envision that releasing the tail needle opens the gp10 channel, providing an exit for the ejection proteins. These proteins were not explicitly visible in asymmetric reconstructions of the P22 MV or tail determined thus far [5–9]. Bubblegram studies suggested that the ejection proteins reside inside the virion, loosely interacting with the portal protein barrel [43]. In contrast, cryo-ET studies suggested that ejection proteins may be in the tail channel [40]. However, to challenge this idea, our reconstruction of the MVs revealed that the tail channel has limited room and could accommodate a maximum of ~1.1 MDa of proteinaceous mass, equal to ~3 copies of each ejection protein. This is less than the number of ejection proteins gp7, gp20, and gp16 estimated to exist inside each P22 MV that has been suggested to be 12, 12, and 30, respectively, which amounts to a total mass of ~3.0 MDa [43]. The disappearance of the barrel density in our P22 mutant virions lacking gp7 or gp20 suggests that the ejection proteins form a complex with the portal barrel that stabilizes the barrel. Thus, these proteins reside above the tail lumen and not inside the tail. Interestingly, we detected a density, possibly consistent with one equivalent of a single gp20, in the P22 mutant lacking gp7. Our biochemical data showed that gp7 and gp20 form a molecular complex that we hypothesize retains these two proteins at the barrel in the pre-ejection state. We also purified P22 ejection proteins and attempted to assemble a complex in vitro. We found that gp7 is a membrane protein, similar to T7 gp14 [34], that is thought to insert in the host OM, while gp20 is a bona fide periplasmic tunnel connecting to the inner membrane (IM) where we hypothesize gp16 resides. Notably, recombinant gp7 and gp20 assemble into a tube-like structure, likely consistent with the post-ejection state of these proteins, that was too flexible for meaningful single-particle analysis (Figure 6C). However, unlike T7, we were not able to assemble a higher-order complex of P22 gp20:gp16 complex in vitro, which remained monomeric at high concentrations or when incubated at room temperature to promote oligomerization. We hypothesize that the assembly of the P22 DNA-ejectosome is either assisted by Salmonella-specific chaperones or the ejection proteins made in E. coli adopt a non-native conformation incompatible with the assembly of an ejectosome.
In conclusion, the present study expands our understanding of the model system P22, providing a structural framework to probe the signal transduction pathway initiated by LPS interaction with tailspikes that culminates with the expulsion of the ejection proteins and assembly of a transenvelope channel into the bacterial host envelope.
MATERIALS AND METHODS
Purification of recombinant gp7, gp20, and gp16
The genes encoding gp7, gp20, and gp16 were cloned by PCR from a P22 genomic library in an engineered pET-28a (+) (Novagen), pETDuet-1 (Novagen), and an engineered pMAL-c6P (New England BioLabs), respectively. The modified plasmids contain the HRV3C protease cleavage site. The constructs were expressed in LOBSTR E. coli Expression Strain (Kerafast) supplemented with either 30 μg/mL kanamycin for gp7-pET28a or 50 μg/mL ampicillin for gp16-pETDuet-1 and gp20-pMAL-c6P. Bacterial cultures were grown in L.B. medium at 37 °C until A600 = ~0.3 when the temperature was dropped to 28 °C until an A600 = ~0.6 and were induced with 0.5 mM IPTG for 4 h.
For gp7, cell pellets were lysed by sonication in Lysis buffer (20 mM Tris-HCl pH 8.0, 300 mM NaCl, 4 mM MgCl2, 1% glycerol, 2 mM EDTA, 0.1% Triton X-100, 1 mM PMSF, 20 μg/mL DNase). As the gp7 was in the insoluble fraction, we retained the pellet after centrifugation at 18,000 rpm for 30 min, 4 °C. The resulting pellet was solubilized with rotation in Extraction buffer (20 mM Tris-HCl pH 8.0, 200 mM NaCl, 0.25–0.35% N-lauroylsarcosine, 1% glycerol, 20 μg/mL DNase) at room temperature for 1.5 – 2 h. The solubilization was followed by centrifugation at 18,000 rpm for 30 min, 4 °C. The resulting supernatant was incubated with Nickel Agarose beads (GoldBio) for 2 h with rotation at 4 °C. The beads were washed with Wash buffer (20 mM Tris-HCl pH 8.0, 200 mM NaCl, 0.025% DDM, 2 mM MgCl2, 1% glycerol, 1 mM PMSF, 5 mM imidazole) and eluted with Wash buffer containing 20, 40, 80, 160, 320 mM imidazole. The protein fractions were dialyzed against Dialysis buffer (20 mM Tris-HCl pH 8.0, 100 mM NaCl, 0.025% DDM, 2 mM MgCl2, 1% glycerol, 1 mM PMSF). The gp7 was further purified by SEC using Superdex 200 16/60 column equilibrated with Gel Filtration Buffer (20 mM Tris-HCl pH 8.0, 100 mM NaCl, 0.025% DDM, 2 mM MgCl2, 1% glycerol, 1 mM PMSF). The membrane scaffolding protein 2N2 (MSP2N2) that we expressed in BL21 (DE3) E. coli cells (Novagen) and purified as previously described [64]. Incorporation of DDM-solubilized gp7 in nanodiscs was done using the same procedure described for phage T7 ejection protein gp14 [57].
Individual ejection proteins gp20 and gp16 were expressed in bacteria as described for gp7. The cell pellets expressing gp20 were lysed by sonication in Lysis buffer (20 mM Tris-HCl pH 8.0, 300 mM NaCl, 4 mM MgCl2, 1% glycerol, 0.1% Triton X-100, 1 mM PMSF, 20 μg/mL DNase). The soluble portion, after centrifugation at 18,000 rpm for 30 min, 4 °C was incubated with Nickel Agarose beads (GoldBio) for 2 h with rotation at 4 °C. The beads were washed with Wash buffer (20 mM Tris-HCl pH 8.0, 300 mM NaCl, 2 mM MgCl2, 1% glycerol, 1 mM PMSF, 5 mM imidazole) and eluted with Wash buffer containing 20, 40, 80, 160, 320 mM imidazole. The relevant fractions containing gp20 were further purified by SEC using Superdex 200 16/60 column equilibrated with Gel Filtration Buffer (20 mM Tris-HCl pH 8.0, 100 mM NaCl, 2 mM MgCl2, 1% glycerol, 1 mM PMSF). The gp7:gp20 complex was formed by incubating a 2x molar excess of purified gp7 with gp20. The mixture was incubated overnight at 4 °C. The mixture was centrifuged at 13,000 rpm for 10 min, 4 °C, and purified using a Superose 12 preparative column.
To form the gp20:gp16 complex, we co-expressed his-gp20 (plasmid pETDuet-1-gp20) and untagged gp16 (plasmid pMAL-c6P-gp16) in BL21-AI E. coli Expression Strain (New England Biolabs) supplemented with 30 μg/mL kanamycin and 100 μg/mL ampicillin. Bacterial cultures were grown in L.B. medium at 37 °C until A600 = ~0.3 when the temperature was dropped to 28 °C until an A600 = ~0.6 and were induced with 0.1% arabinose for 2 h. The complex was purified from a soluble lysate using metal-chelate affinity chromatography (as described for gp20 alone) and SEC on a BioRad ENrich SEC 650 column.
Purification of P22 virions and virion-like particles
WT P22 phages were grown on Salmonella enterica strain DB7136 (leuA am414, hisC am525, sup0) and purified from a lysate, as described below. P22 gp20— and gp7— virus-like particles were prepared by induction of the prophages in the following strains (see also [43]): S. enterica LT2 galK∷TetRA strain UB-2285 that carries the gp20— prophage P22 c1-7, 20−Δ−1, 13−amH101, sieAΔ−1, orf25∷CamR-EG1 and UB-2289 that carries the gp7— prophage P22 c1-7, 7−Δ−1, 13−amH101, sieAΔ−1, orf25∷CamR-EG1 (these gene 7 and 20 deletions removed the entire gene except for the 3’−60 bp). Strain UB-2519 carries P22 c1-7, 26−amH204, 9−∷galK, 13−amH101, sieAD-1, orf25∷CamR-EG1 (where the galK gene replaces the first 100 bp of gene 9). Three liters of LB were inoculated with UB-2285, UB-2289, or UB-2519 overnight cultures and grown at 37 °C to an O.D. of ~0.6. Prophage expression in the lysogens was induced by adding 1.5 mg/ml carbadox and were allowed to grow for another 4 hours at 37 °C, harvested, resuspended in dilution fluid (20 mM Tris pH 7.5, 100 mM MgCl2), and stored at −20 °C. Cells were lysed in the presence of DNase and RNase (1 mg/ml each, final concentration), followed by a centrifugation step to spin out cellular debris. The supernatants were spun at 200,000 g to pellet the phages. Samples were then resuspended by gently shaking overnight in dilution fluid at 4 °C. The resulting 20— and 7— phage suspensions were further purified using cesium chloride gradients, followed by dialysis and differential centrifugation (72,000 g for 15 minutes) to separate phages from ribosomes. Samples were checked for purity using SDS-PAGE and transmission electron microscopy (data not shown).
Vitrification and data collection
2.5 μL of P22 virions, typically at 1 × 1014 phages/mL was applied to a 200-mesh copper Quantifoil R 2/1 holey carbon grid (EMS) previously glow-discharged for 60 sec at 15 mA using an easiGlow (PELCO). The grid was blotted for 7.5 sec at blot force 2 and vitrified immediately in liquid ethane using a Vitrobot Mark IV (Thermo Scientific). Cryo-grids were screened on 200 kV Glacios (Thermo Scientific) equipped with a Falcon4 detector (Thermo Scientific) at Thomas Jefferson University. EPU software (Thermo Scientific) was used for data collection using accurate positioning mode. For high-resolution data collection of the P22 MV, micrographs were collected on a Titan Krios (Thermo Scientific) microscope operated at 300 kV and equipped with a K3 direct electron detector camera (Gatan) at the National Cryo-EM Facility at the Pacific Northwest Cryo-EM Center, (PNCC). The high-resolution data collection of the P22 gp7— and P22 gp20— micrographs were collected on the in-house Glacios (Thermo Scientific).
The recombinant gp7: gp20 protein complex was applied to a 200-mesh UltrAuFoil R 1.2/1.3 holey gold grids (EMS) previously-glow-discharged for 60 sec at 15 mA using an easiGlow (PELCO). The grid was blotted and vitrified using the procedures used for the other samples. A high-resolution dataset was collected using EPU software (Thermo Scientific) on a 200 kV Glacios (Thermo Scientific) equipped with a Falcon4 detector (Thermo Scientific) at Thomas Jefferson University.
Cryo-EM single particle analysis of P22 particles
All P22 datasets were motion-corrected with MotionCorr2 [65]. RELION’s implementation of motion correction was applied to the micrographs with options of dose-weighted averaged micrographs and the sum of non-dose weighted power spectra every 4 e−/Å2. CTF (Contrast Transfer Function) was estimated using CTFFIND4 [66]. After initial reference picking and 2D classification, particles were subjected to a reference-free low-resolution reconstruction without imposing symmetry. The particles were then 3D classified into four classes with I4 symmetry imposed. Of the four classes, the best class was chosen and was subjected to 3D auto-refinement to align the particles finely. The particles were then expanded according to I4 symmetry using RELION’s relion_particle_symmetry_expand function to obtain 60 times the initial particles. A cylindrical mask (r = 200 Å) was generated using SCIPION 3.0 [67] and then resampled onto a reference map covering the five-fold vertex in Chimera [68]. The cylindrical mask was then used for non-sampling 3D classification without imposing symmetry to search for the tail. Locally aligned particles were then combined, and duplicate particles were removed. The initial localized reference map was reconstructed directly from one of the classes using RELION’s ab initio 3D Initial Model. Selected 3D classes were auto-refined using C5 symmetry, followed by 5-fold particle expansion. The expanded particles were subjected to a third 3D classification, and the map was symmetrized by imposing C12 and C6, which gave the best density for the portal protein/head-to-tail adaptor and tail hub/tailspike, respectively. After the fourth 3D classification using C6 symmetry expanded particles, local maps with two tail apparatus conformations were obtained. All steps of SPA, including 2D/3D classification, 3D refinement, CTF refinement, particle polishing, post-processing, and local resolution calculation, were carried out using RELION 3.1.2 [69, 70]. The final densities were sharpened using phenix.autosharpen [71]. RELION_postprocess [69, 70] was used for local resolution estimation. All cryo-EM data collection statistics are in Table 1. A pipeline of SPA is shown in Figure S1.
De novo model building, oligomer generation, and refinement
The 3.0 Å C12-averaged localized reconstruction was used to build a model of dodecameric portal protein bound to twelve copies of the head-to-tail adaptor. P22 portal barrel was modeled between 606–647, while C-terminal residues 647–725 had no discernable density. Similarly, the head-to-tail adaptor C-terminal residues 153–166 were not visible in the C12 map. Using the 2.8 Å C6 localized reconstruction, we built a complete model of the hexameric tail hub (res. 2–472) bound to six copies of the trimeric tailspike N-termini (res. 5–123). The crystal structure of the gp9 tailspike body (PDB: 2VBE; res. 124–623) was placed and real-space refined in the Conf-1 asymmetric (C1) map. All de novo atomic models (except the tail needle gp26 and tailspike gp9 body) shown in this paper were built manually using Coot [72] or Chimera [68] and refined using several rounds of rigid-body, real-space and B-factor refinement using phenix.real_space_refinement [73]. The final models of the portal: head-to-tail adaptor (24 chains) and tail hub: tailspike-N (24 chains) yielded a final Correlation Coefficient (CC) of 0.85 and 0.82, respectively, and excellent stereochemistry (Table 2). After obtaining accurate atomic models from the C12/C6 symmetrized local reconstruction, the portal, head-to-tail adaptor, tail hub, and tailspike models were placed in the C1 maps, revealing different densities for the C-terminal residues 151–160 for the five head-to-tail adaptors. The models were extended by hand in Coot [72], and all tail components corresponding to ~21,977 and 21,891 residues for Conf-1 and Conf-2, respectively, were subjected to rigid-body and real-space refinement, yielding a final CC = 0.77 and 0.74, respectively (Table 2). The full-length tailspike was modeled by combining N-terminal residues 4–123 (build de novo in the C6 map) and residues 124–623 in the tailspike body, which are like the crystal structure (PDB: 2VBE) [74]. Finally, the tail needle knob (PDB: 3RWN) [75] and a model of the full-length P22 tail needle (PDB: 3C9I) [33] were placed in the C1 density refined using the fit-into-map command in Chimera [68]. All final models were validated using MolProbity [76] (Table 2).
Structural analysis
All ribbon and surface representations were generated using ChimeraX [77] and PyMol [78]. Drawings of electron density maps and local resolution maps were generated using ChimeraX [77]. Structural neighbors and flexible regions were identified using the DALI server [79]. Binding interfaces were analyzed using PISA [80] and PDBsum [81] to determine bonding interactions, interatomic distances, and types of bonds. The tail volume was analyzed using the 3v website [82]. The Coulombic Electrostatic Potential was calculated and displayed with surface coloring using ChimeraX [77].
Supplementary Material
Highlights.
P22 tail apparatus exists in two equivalent conformations rotated by ~6°
The conserved tail hub gp10 folds into an interrupted β-propeller
The tailspike N-terminal domains attach asymmetrically to the tail hub insertion domain
Ejection proteins gp7 and gp20 form a molecular complex
Proposed model for signal transduction from the tailspike to the tail needle
ACKNOWLEDGMENTS
We thank the staff at PNCC for their assistance with remote data collection. This work was supported by National Institutes of Health grants R01 GM100888, R35 GM140733, and S10 OD030457 to G.C.; R01 GM076661 to C.M.T., and R01 GM114817 to S.R.C. Research in this publication includes work carried out at the Sidney Kimmel Cancer Center X-ray Crystallography and Molecular Interaction Facility at Thomas Jefferson, partly supported by National Cancer Institute Cancer Center Support Grant P30 CA56036. A portion of this research was supported by NIH grant U24 GM129547 and performed at the PNCC at OHSU and accessed through EMSL (grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research. Atomic coordinates for P22 Portal:Head-to-Tail (C12), Tail hub:Tailspike-N (C6), Portal:Head-to-Tail:Coat:Tail hub:Tailspike N-term Conf-1 (C1), Portal:Head-to-Tail:Coat:Tail hub:Tailspike N-term Conf-2 (C1), full-length tailspike (C3) complexes have been deposited in the Protein Data Bank with accession codes 8TVU, 8TVR, 8U10, 8U11, and 8U1O. The cryo-EM density maps have been deposited in the Electron Microscopy Data Bank with accession codes EMD-41651, EMD-41649, EMD-41791, EMD-41792, and EMD-41819.
Abbreviations used:
- cryo-EM
cryogenic electron microscopy
- FL-portal
full-length portal protein
- M.V.
mature virion
- OM
outer membrane
- IM
inner membrane
- SEC
size exclusion chromatography
- SPA
single-particle analysis
- CC
correlation coefficient
- M.W.
molecular weight
- RMSD
room-mean square deviation
- dsDNA
double-stranded DNA
- SSM
secondary structure superimposition
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- DDM
dodecyl β-D-maltoside
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing interests.
CRediT author statement:
Stephano M. Iglesias: Software, Validation, Formal Analysis, Data Curation, Visualization, Writing – Original Draft, Writing – Review & Editing, Resources, Investigation. Ravi K Lokareddy: Software, Validation, Formal Analysis, Data Curation, Visualization, Writing – Review & Editing. Ruoyu Yang: Software, Validation, Formal Analysis, Data Curation, Visualization. Fenglin Li: Software, Validation, Formal Analysis, Data Curation, Visualization. Daniel P. Yeggoni: Resources, Investigation. Chun-Feng David Hou: Software, Validation, Formal Analysis, Data Curation, Visualization, Writing– Review & Editing. Makayla N. Leroux: Methodology, Investigation, Resources. JulianaR. Cortines: Methodology, Investigation, Resources, Writing – Review & Editing. JustinC. Leavitt: Investigation, Resources. Mary Bird: Investigation, Resources. Sherwood R.Casjens: Funding acquisition, Resources. Simon White: Resources, Investigation.Carolyn M. Teschke: Funding acquisition, Methodology, Investigation, Resources,Writing – Review & Editing. Gino Cingolani: Software, Validation, Formal Analysis, Data Curation, Visualization, Supervision, Funding acquisition, Project administration, Writing -Original Draft, Writing – Review & Editing, Resources, Methodology.
REFERENCES
- [1].Teschke CM, Parent KN. ‘Let the phage do the work’: using the phage P22 coat protein structures as a framework to understand its folding and assembly mutants. Virology. 2010;401:119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hyman P, Abedon ST. Bacteriophage (overview). Encyclopedia of Microbiology (Third Edition), Academic Press. 2009:322–38. [Google Scholar]
- [3].Bhardwaj A, Olia AS, Cingolani G. Architecture of viral genome-delivery molecular machines. Curr Opin Struct Biol. 2014;25:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Casjens SR, Molineux IJ. Short noncontractile tail machines: adsorption and DNA delivery by podoviruses. Adv Exp Med Biol. 2012;726:143–79. [DOI] [PubMed] [Google Scholar]
- [5].Lander GC, Tang L, Casjens SR, Gilcrease EB, Prevelige P, Poliakov A, et al. The structure of an infectious P22 virion shows the signal for headful DNA packaging. Science. 2006;312:1791–5. [DOI] [PubMed] [Google Scholar]
- [6].Chang J, Weigele P, King J, Chiu W, Jiang W. Cryo-EM asymmetric reconstruction of bacteriophage P22 reveals organization of its DNA packaging and infecting machinery. Structure. 2006;14:1073–82. [DOI] [PubMed] [Google Scholar]
- [7].Tang J, Lander GC, Olia A, Li R, Casjens S, Prevelige P Jr., et al. Peering down the barrel of a bacteriophage portal: the genome packaging and release valve in p22. Structure. 2011;19:496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chen DH, Baker ML, Hryc CF, DiMaio F, Jakana J, Wu W, et al. Structural basis for scaffolding-mediated assembly and maturation of a dsDNA virus. Proc Natl Acad Sci U S A. 2011;108:1355–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Parent KN, Khayat R, Tu LH, Suhanovsky MM, Cortines JR, Teschke CM, et al. P22 coat protein structures reveal a novel mechanism for capsid maturation: stability without auxiliary proteins or chemical crosslinks. Structure. 2010;18:390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Parent KN, Schrad JR, Cingolani G. Breaking Symmetry in Viral Icosahedral Capsids as Seen through the Lenses of X-ray Crystallography and Cryo-Electron Microscopy. Viruses. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lander GC, Khayat R, Li R, Prevelige PE, Potter CS, Carragher B, et al. The P22 tail machine at subnanometer resolution reveals the architecture of an infection conduit. Structure. 2009;17:789–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tang L, Marion WR, Cingolani G, Prevelige PE, Johnson JE. Three-dimensional structure of the bacteriophage P22 tail machine. Embo J. 2005;24:2087–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Olia AS, Prevelige PE Jr., Johnson JE, Cingolani G. Three-dimensional structure of a viral genome-delivery portal vertex. Nat Struct Mol Biol. 2011;18:597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lokareddy RK, Sankhala RS, Roy A, Afonine PV, Motwani T, Teschke CM, et al. Portal protein functions akin to a DNA-sensor that couples genome-packaging to icosahedral capsid maturation. Nat Commun. 2017;8:14310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Olia AS, Casjens S, Cingolani G. Structural plasticity of the phage P22 tail needle gp26 probed with xenon gas. Protein Sci. 2009;18:537–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Olia AS, Casjens S, Cingolani G. Structure of phage P22 cell envelope-penetrating needle. Nat Struct Mol Biol. 2007;14 1221–6. [DOI] [PubMed] [Google Scholar]
- [17].Andres D, Gohlke U, Broeker NK, Schulze S, Rabsch W, Heinemann U, et al. An essential serotype recognition pocket on phage P22 tailspike protein forces Salmonella enterica serovar Paratyphi A O-antigen fragments to bind as nonsolution conformers. Glycobiology. 2013;23:486–94. [DOI] [PubMed] [Google Scholar]
- [18].Steinbacher S, Seckler R, Miller S, Steipe B, Huber R, Reinemer P. Crystal structure of P22 tailspike protein: interdigitated subunits in a thermostable trimer. Science. 1994;265:383–6. [DOI] [PubMed] [Google Scholar]
- [19].Seul A, Muller JJ, Andres D, Stettner E, Heinemann U, Seckler R. Bacteriophage P22 tailspike: structure of the complete protein and function of the interdomain linker. Acta Crystallogr D Biol Crystallogr. 2014;70:1336–45. [DOI] [PubMed] [Google Scholar]
- [20].Steinbacher S, Miller S, Baxa U, Budisa N, Weintraub A, Seckler R, et al. Phage P22 tailspike protein: crystal structure of the head-binding domain at 2.3 A, fully refined structure of the endorhamnosidase at 1.56 A resolution, and the molecular basis of O-antigen recognition and cleavage. J Mol Biol. 1997;267:865–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Prevelige PE Jr., Thomas D, King J. Scaffolding protein regulates the polymerization of P22 coat subunits into icosahedral shells in vitro. J Mol Biol. 1988;202:743–57. [DOI] [PubMed] [Google Scholar]
- [22].Motwani T, Lokareddy RK, Dunbar CA, Cortines JR, Jarrold MF, Cingolani G, et al. A viral scaffolding protein triggers portal ring oligomerization and incorporation during procapsid assembly. Sci Adv. 2017;3:e1700423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Thuman-Commike PA, Greene B, Malinski JA, Burbea M, McGough A, Chiu W, et al. Mechanism of scaffolding-directed virus assembly suggested by comparison of scaffolding-containing and scaffolding-lacking P22 procapsids. Biophys J. 1999;76:3267–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cortines JR, Motwani T, Vyas AA, Teschke CM. Highly specific salt bridges govern bacteriophage P22 icosahedral capsid assembly: identification of the site in coat protein responsible for interaction with scaffolding protein. J Virol. 2014;88:5287–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cortines JR, Weigele PR, Gilcrease EB, Casjens SR, Teschke CM. Decoding bacteriophage P22 assembly: identification of two charged residues in scaffolding protein responsible for coat protein interaction. Virology. 2011;421:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Roy A, Cingolani G. Structure of p22 headful packaging nuclease. J Biol Chem. 2012;287:28196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Roy A, Bhardwaj A, Datta P, Lander GC, Cingolani G. Small terminase couples viral DNA binding to genome-packaging ATPase activity. Structure. 2012;20:1403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].McNulty R, Lokareddy RK, Roy A, Yang Y, Lander GC, Heck AJ, et al. Architecture of the Complex Formed by Large and Small Terminase Subunits from Bacteriophage P22. J Mol Biol. 2015;427:3285–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Olia AS, Al-Bassam J, Winn-Stapley DA, Joss L, Casjens SR, Cingolani G. Binding-induced stabilization and assembly of the phage P22 tail accessory factor gp4. J Mol Biol. 2006;363:558–76. [DOI] [PubMed] [Google Scholar]
- [30].Lorenzen K, Olia AS, Uetrecht C, Cingolani G, Heck AJ. Determination of stoichiometry and conformational changes in the first step of the P22 tail assembly. J Mol Biol. 2008;379:385–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zheng H, Olia AS, Gonen M, Andrews S, Cingolani G, Gonen T. A conformational switch in bacteriophage p22 portal protein primes genome injection. Mol Cell. 2008;29:376–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Olia AS, Bhardwaj A, Joss L, Casjens S, Cingolani G. Role of gene 10 protein in the hierarchical assembly of the bacteriophage P22 portal vertex structure. Biochemistry. 2007;46:8776–84. [DOI] [PubMed] [Google Scholar]
- [33].Bhardwaj A, Olia AS, Walker-Kopp N, Cingolani G. Domain organization and polarity of tail needle GP26 in the portal vertex structure of bacteriophage P22. J Mol Biol. 2007;371:374–87. [DOI] [PubMed] [Google Scholar]
- [34].Swanson NA, Hou CD, Cingolani G. Viral Ejection Proteins: Mosaically Conserved, Conformational Gymnasts. Microorganisms. 2022;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Botstein D, Waddell CH, King J. Mechanism of head assembly and DNA encapsulation in Salmonella phage p22. I. Genes, proteins, structures and DNA maturation. J Mol Biol. 1973;80:669–95. [DOI] [PubMed] [Google Scholar]
- [36].Israel V. E proteins of bacteriophage P22. I. Identification and ejection from wild-type and defective particles. J Virol. 1977;23:91–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Poteete AR, King J. Functions of two new genes in Salmonella phage P22 assembly. Virology. 1977;76:725–39. [DOI] [PubMed] [Google Scholar]
- [38].Jin Y, Sdao SM, Dover JA, Porcek NB, Knobler CM, Gelbart WM, et al. Bacteriophage P22 ejects all of its internal proteins before its genome. Virology. 2015;485:128–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Perez GL, Huynh B, Slater M, Maloy S. Transport of phage P22 DNA across the cytoplasmic membrane. J Bacteriol. 2009;191:135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wang C, Tu J, Liu J, Molineux IJ. Structural dynamics of bacteriophage P22 infection initiation revealed by cryo-electron tomography. Nat Microbiol. 2019;4:1049–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chen W, Xiao H, Wang L, Wang X, Tan Z, Han Z, et al. Structural changes in bacteriophage T7 upon receptor-induced genome ejection. Proc Natl Acad Sci U S A. 2021;118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bayfield OW, Shkoporov AN, Yutin N, Khokhlova EV, Smith JLR, Hawkins D, et al. Structural atlas of a human gut crassvirus. Nature. 2023;617:409–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wu W, Leavitt JC, Cheng N, Gilcrease EB, Motwani T, Teschke CM, et al. Localization of the Houdinisome (Ejection Proteins) inside the Bacteriophage P22 Virion by Bubblegram Imaging. MBio. 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Leavitt JC, Gilcrease EB, Woodbury BM, Teschke CM, Casjens SR. Intravirion DNA Can Access the Space Occupied by the Bacteriophage P22 Ejection Proteins. Viruses. 2021;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Andres D, Hanke C, Baxa U, Seul A, Barbirz S, Seckler R. Tailspike interactions with lipopolysaccharide effect DNA ejection from phage P22 particles in vitro. J Biol Chem. 2010;285:36768–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sakikawa C, Taguchi H, Makino Y, Yoshida M. On the maximum size of proteins to stay and fold in the cavity of GroEL underneath GroES. J Biol Chem. 1999;274:21251–6. [DOI] [PubMed] [Google Scholar]
- [47].Bhardwaj A, Walker-Kopp N, Casjens SR, Cingolani G. An evolutionarily conserved family of virion tail needles related to bacteriophage P22 gp26: correlation between structural stability and length of the alpha-helical trimeric coiled coil. J Mol Biol. 2009;391:227–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Strauss H, King J. Steps in the stabilization of newly packaged DNA during phage P22 morphogenesis. J Mol Biol. 1984;172:523–43. [DOI] [PubMed] [Google Scholar]
- [49].Li F, Hou CD, Yang R, Whitehead R 3rd, Teschke CM, Cingolani G. High-resolution cryo-EM structure of the Shigella virus Sf6 genome delivery tail machine. Sci Adv. 2022;8:eadc9641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Dedeo CL, Cingolani G, Teschke CM. Portal Protein: The Orchestrator of Capsid Assembly for the dsDNA Tailed Bacteriophages and Herpesviruses. Annu Rev Virol. 2019;6:141–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lebedev AA, Krause MH, Isidro AL, Vagin AA, Orlova EV, Turner J, et al. Structural framework for DNA translocation via the viral portal protein. Embo J. 2007;26:1984–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Casjens SR, Thuman-Commike PA. Evolution of mosaically related tailed bacteriophage genomes seen through the lens of phage P22 virion assembly. Virology. 2011;411:393–415. [DOI] [PubMed] [Google Scholar]
- [53].Ruan J, Xu C, Bian C, Lam R, Wang JP, Kania J, et al. Crystal structures of the coil 2B fragment and the globular tail domain of human lamin B1. FEBS Lett. 2012;586:314–8. [DOI] [PubMed] [Google Scholar]
- [54].Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Thomas D, Prevelige P Jr. A pilot protein participates in the initiation of P22 procapsid assembly. Virology. 1991;182:673–81. [DOI] [PubMed] [Google Scholar]
- [56].Swanson NA, Lokareddy RK, Li F, Hou CD, Leptihn S, Pavlenok M, et al. Cryo-EM structure of the periplasmic tunnel of T7 DNA-ejectosome at 2.7 A resolution. Mol Cell. 2021;81:3145–59 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Swanson NA, Lokareddy RK, Li F, Hou CF, Pavlenok M, Niederweis M, et al. Expression and purification of phage T7 ejection proteins for cryo-EM analysis. STAR Protoc. 2021;2:100960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Conlin CA, Vimr ER, Miller CG. Oligopeptidase A is required for normal phage P22 development. J Bacteriol. 1992;174:5869–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhao H, Speir JA, Matsui T, Lin Z, Liang L, Lynn AY, et al. Structure of a Bacterial Virus DNA-Injection Protein Complex Reveals a Decameric Assembly with a Constricted Molecular Channel. PLoS One. 2016;11:e0149337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Parent KN, Gilcrease EB, Casjens SR, Baker TS. Structural evolution of the P22-like phages: comparison of Sf6 and P22 procapsid and virion architectures. Virology. 2012;427:177–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Baxa U, Weintraub A, Seckler R. Self-Competitive Inhibition of the Bacteriophage P22 Tailspike Endorhamnosidase by O-Antigen Oligosaccharides. Biochemistry. 2020;59:4845–55. [DOI] [PubMed] [Google Scholar]
- [62].Bhardwaj A, Sankhala RS, Olia AS, Brooke D, Casjens SR, Taylor DJ, et al. Structural Plasticity of the Protein Plug That Traps Newly Packaged Genomes in Podoviridae Virions. J Biol Chem. 2016;291:215–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Leavitt JC, Gogokhia L, Gilcrease EB, Bhardwaj A, Cingolani G, Casjens SR. The tip of the tail needle affects the rate of DNA delivery by bacteriophage p22. PLoS One. 2013;8:e70936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tan YZ, Zhang L, Rodrigues J, Zheng RB, Giacometti SI, Rosario AL, et al. Cryo-EM Structures and Regulation of Arabinofuranosyltransferase AftD from Mycobacteria. Mol Cell. 2020;78:683–99 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, Agard DA. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods. 2017;14:331–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Rohou A, Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol. 2015;192:216–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Conesa Mingo P, Gutierrez J, Quintana A, de la Rosa Trevin JM, Zaldivar-Peraza A, Cuenca Alba J, et al. Scipion web tools: Easy to use cryo-EM image processing over the web. Protein Sci. 2018;27:269–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. [DOI] [PubMed] [Google Scholar]
- [69].Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Terwilliger TC, Sobolev OV, Afonine PV, Adams PD. Automated map sharpening by maximization of detail and connectivity. Acta Crystallogr D Struct Biol. 2018;74:545–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. [DOI] [PubMed] [Google Scholar]
- [73].Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D Struct Biol. 2018;74:531–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Muller JJ, Barbirz S, Heinle K, Freiberg A, Seckler R, Heinemann U. An intersubunit active site between supercoiled parallel beta helices in the trimeric tailspike endorhamnosidase of Shigella flexneri Phage Sf6. Structure. 2008;16:766–75. [DOI] [PubMed] [Google Scholar]
- [75].Bhardwaj A, Molineux IJ, Casjens SR, Cingolani G. Atomic structure of bacteriophage Sf6 tail needle knob. J Biol Chem. 2011;286:30867–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].DeLano WL. The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC. 2002. [Google Scholar]
- [79].Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–97. [DOI] [PubMed] [Google Scholar]
- [81].Laskowski RA. PDBsum new things. Nucleic Acids Res. 2009;37:D355–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Voss NR, Gerstein M. 3V: cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 2010;38:W555–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.