

Abstract
Aging is marked by the gradual accumulation of deleterious changes that disrupt organ function, creating an altered physiological state that is permissive for the onset of prevalent human diseases. While the exact mechanisms governing aging remain a subject of ongoing research, there are several cellular and molecular hallmarks that contribute to this biological process. This review focuses on two factors, namely telomere dysfunction and inflammation, which have emerged as crucial contributors to the aging process. We aim to discuss the mechanistic connections between these two distinct hallmarks and provide compelling evidence highlighting the loss of telomere protection as a driver of pro-inflammatory states associated with aging. By reevaluating the interplay between telomeres, innate immunity, and inflammation, we present novel perspectives on the etiology of aging and its associated diseases.
Keywords: Telomeres, Innate immunity, Inflammation, Aging, Cancer, Genome stability
1. Protective properties of telomeres
The linear ends of eukaryotic chromosomes are constantly at risk of being mistaken for DNA breaks, creating a continuous threat to genome integrity and cell viability. Telomeres solve this end protection problem by employing diverse mechanisms capable of shielding chromosome termini from DNA damage surveillance and repair pathways [1–3]. Several factors, however, can compromise the functionality of this protective mechanism. First, telomeres in human somatic cells undergo gradual shortening due to the unidirectional nature of DNA polymerase and nucleolytic processing events during each round of cell cycle division [4–8]. In addition, replication forks are subject to stalling and collapsing as they navigate through telomeric sequences, resulting in incomplete replication and DNA sequences loss [9–12]. Last, the distinctive composition of telomeres, characterized by G-rich repeat sequences, renders them susceptible to oxidative DNA damage and buildup of persistent lesions [13–15], a process that has been recently termed TelOxidation [16]. This vulnerability extends to post-mitotic cells that do not undergo cell division, including cardiomyocytes, neurons, and osteoblasts [17] among others. The cumulative impact of these events can compromise telomere protection, resulting in premature aging syndromes and elevated susceptibility to cancer.
In humans, telomeres consist of repeated hexanucleotide sequences (5′-TTAGGG-3′) spanning several kilobases and terminating with a single-stranded (ss) 3′ G-overhang [18,19]. Telomeric DNA protects chromosome ends through its interaction with the six-subunit shelterin complex: TRF1 and TRF2 form homodimers and bind to double-stranded DNA [20,21], while POT1 specifically binds to ssDNA [22,23]. TIN2 and TPP1 act as connectors between the DNA-binding proteins [24–27], and RAP1 associates with TRF2 [28]. Among the shelterin proteins, TRF2 exhibits a remarkable capability to wrap DNA around itself [29,30] and promote Holliday junction formation [31–33]. These unique properties enable TRF2 to effectively remodel telomeric DNA into a closed t-loop configuration, where the 3′ overhang invades the adjacent double-stranded region [34]. This arrangement, first observed through electron microscopy [35] and later confirmed using super-resolution light microscopy [36], provides a first layer of protection by hiding the telomere terminus from DNA damage response factors that rely on exposed DNA ends. Other protective mechanisms include POT1 blocking RPA access to ssDNA to prevent ATR kinase activation [37–39] (Fig. 1A) and TRF1 preventing replication fork stalling during telomere replication [23,40]. In the absence of shelterin, telomeres are exposed to DNA damage surveillance and chromosome ends become inappropriately processed by three distinct and cell cycle-dependent repair pathways: classical non-homologous end joining (c-NHEJ), alternative end joining (alt-EJ) and homology-directed repair (HDR) [2] (Fig. 1). These repair activities set in motion a cascade of deleterious events ranging from chromosomal rearrangements, decrease of cellular fitness, to innate immune response and inflammation, each contributing to the characteristic features of aging.
Fig. 1. DNA repair pathways at deprotected telomeres.
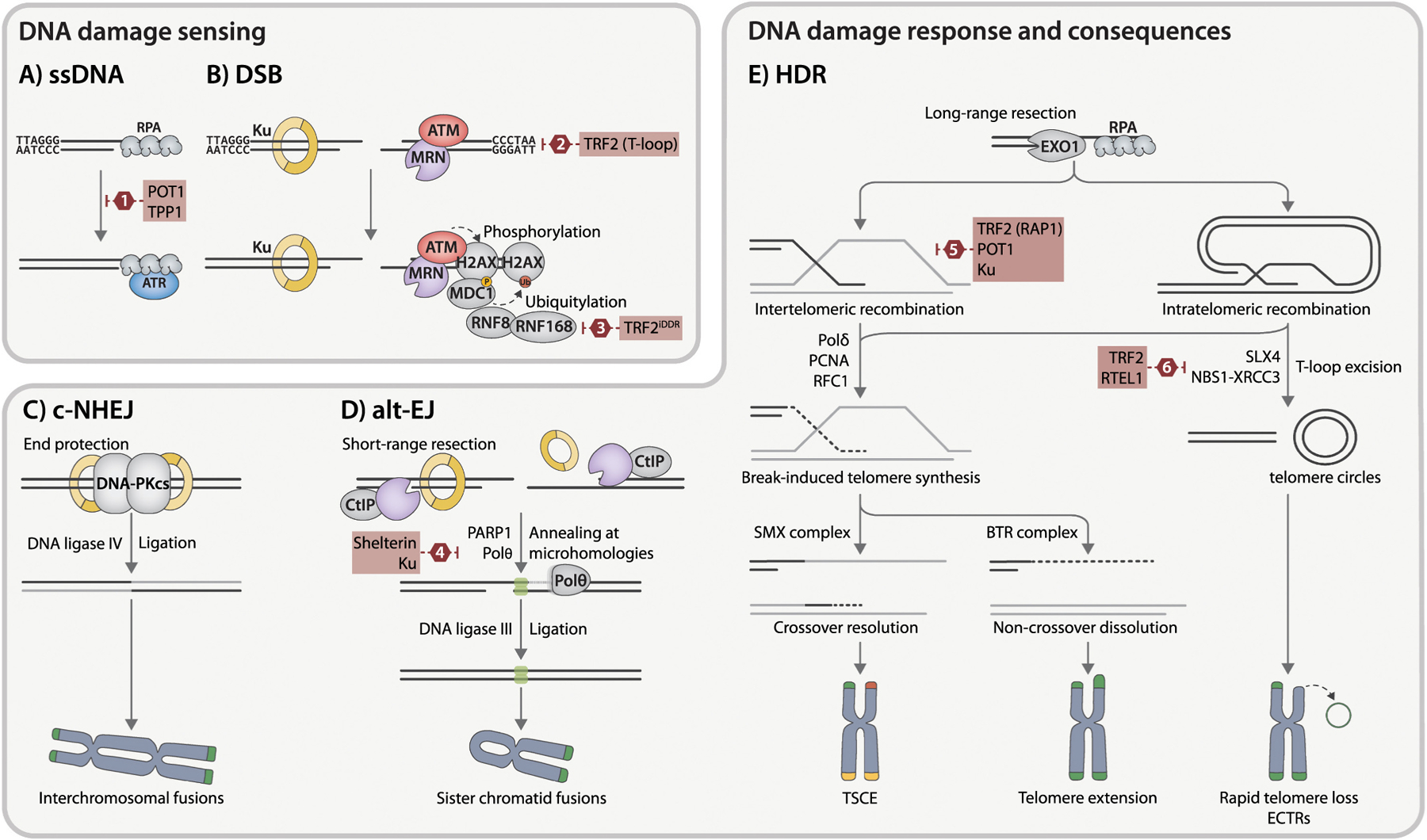
A) ssDNA activates the ATR kinase pathway. B) DSBs are recognized either by Ku heterodimer or MRN complex and activate the ATM kinase pathway. This triggers local phosphorylation of multiple ATM substrates in the surrounding chromatin, most notably the histone variant H2AX and the recruitment of additional DDR mediators, such as 53BP1 and MDC1, at the break site. These modifications give rise to TIF, macroscopic structures that mark sites of deprotected telomeres. This signaling switches from phosphorylation to ubiquitination through the recruitment of E3 ligases, including RNF8 and RNF168, resulting in the ubiquitination of several proteins, including H2A/H2AX. C) During c-NHEJ, broken ends are rapidly bound by Ku, followed by the recruitment and activation of the DNA-PKcs. Broken DNA ends are joined by DNA ligase IV. Depletion of TRF2 provokes undesired c-NHEJ activity at telomeres, potentially resulting in chromosomal end-to-end fusions. D) MRN/CtIP-mediated short-range resection disrupts the c-NHEJ pathway by displacing Ku from DNA ends and exposes short microhomologies of 2–20 bp (green boxes) required for alt-EJ. This repair process is contingent on PARP1 and Polθ and is finalized by DNA ligase III. E) Extensive resection is executed by EXO1 or combined action of BLM helicase and DNA2 nuclease. Generated 3’ ssDNA overhangs, rapidly coated with RPA heterotrimer, facilitate either inter- or intratelomeric strand invasion and recombination. This invasion results in break-induced telomere synthesis via a noncanonical Polδ-PCNA-RFC1 replisome. Formed D-loop can be resolved by the SMX endonuclease complex, causing crossover events without telomere elongation (telomeric sister chromatid exchange (T-SCE), indicated by yellow telomeres), or dissolved by the BTR complex, leading to telomere extension without crossover. Aberrant excision of T-loops formed by intratelomeric recombination via SLX4 or XRCC3 and NBS1 repair proteins can result in accumulation of ECTRs and rapid telomere shortening. Shelterin proteins and auxiliary factors, which inhibit specific stages of DNA repair mechanisms at deprotected telomeres, are highlighted with red boxes. (1) TPP1-POT1 complex represses ATR signaling by blocking RPA association with telomeric overhang. (2) TRF2 aids in forming the protective t-loop structure, shielding chromosome ends from DNA sensors. In addition, TRF2 inhibits Ku heterodimerization, thereby preventing c-NHEJ activation. (3) TRF2 through its iDDR domain impedes the recruitment of RNF168, limiting downstream signaling events. (4) Shelterin and the Ku heterodimer act in coordinated fashion to suppress alt-EJ. (5) components TRF2, RAP1, and POT1, in conjunction with Ku, impede sequence exchange between sister chromatid telomeres. (6) During the S phase, TRF2 recruits RTEL1 to unwind the t-loop, shielding it from cleavage by SLX4. Furthermore, TRF2 inhibits t-loop excision by suppressing NBS1–XRCC3 activity.
The sequence-independent c-NHEJ pathway is directed by the Ku70-Ku80 heterodimer (Ku) and engages the catalytic subunit of DNA-PKcs to coordinate end processing and produce blunt ended substrates for the DNA ligase IV complex [41] (Fig. 1C). On the other hand, alt-EJ requires microhomologies exposed by MRN-CtIP-mediated resection [42] and depends on PARP1, DNA polymerase theta, and DNA ligase III (Fig. 1D). Unlike intra-chromosomal breaks, the prevention of end-joining pathways at telomeres is essential. This is primarily achieved through TRF2-mediated t-loop formation while suppressing ATM activation [43,44], as well as the iDDR region of TRF2 inhibiting the recruitment of E3 ubiquitin ligase RNF168 [45] (Fig. 1B). Furthermore, the interaction between TRF2 and the α-helix 5 domain of Ku70—which mediates the heterotetramerization of Ku—prevents the synapsing of chromosome ends [46]. The unique sequence composition of telomeres makes minor resections highly favorable for alt-EJ, but research in mice reveals that this repair pathway only reaches full activity when both Ku and all shelterin components are missing [47]. During end-joining activities at telomeres, inter-chromosomal fusions are primarily driven by LIG4-dependent c-NHEJ [48,49] while intra-chromosomal fusions between sister chromatids rely on LIG3-mediated alt-EJ [50,51] (Fig. 1C, D). The subsequent formation of fused chromosomes, along with lagging chromosomes and acentric fragments that fail to incorporate into daughter nuclei during mitosis, leads to aneuploidy and multiple features of genome instability.
As cells progress through S and G2 phase, the HDR pathway becomes the primary strategy for handling DSBs and replication stress (Fig. 1E). This is facilitated by extensive long-range resection, mediated either by EXO1 or the BLM-DNA2 complex [52]. The resulting extended 3’ ssDNA tails provide a platform for the BRCA2-PALB2-BRCA1 complex to initiate the formation of RAD51 presynaptic filaments, required for strand invasion and homology search [53]. At deprotected telomeres, the invading strand can originate from the same (intra-chromosomal) or a different (inter-chromosomal) chromosome, and in both settings, it participates in break-induced telomere synthesis (BITS), a process contributing to the alternative lengthening of telomeres (ALT). Recombination intermediates can be either resolved by the BTR complex, promoting telomere extension without exchange, or directly cleaved by structure-specific nucleases like the SMX complex, resulting in telomeres with exchanged sequences [54]. The coordinated action of POT1, TRF2, and the Ku complex protects telomeres from unequal chromatid exchanges [38,55,56], with POT1 binding to ssDNA hindering RPA access to the telomeric overhang and preventing ATR signaling [57](Fig. 1A). A compromised TRF2 or Ku complex leads to aberrant HDR at telomeres, resulting in t-loop excision mediated by XRCC3, NBS1, and SLX4, accelerated telomere loss, and extrachromosomal telomeric repeats (ECTRs) [58,59]. The RTEL1 protein, an accessory component of the shelterin complex, mitigates this process by unwinding the t-loops, thus adding an additional layer of regulation in maintaining telomere integrity [60]. While HDR activities at telomeres may appear less harmful than end-joining, they can give rise to telomere sister chromatid exchanges (T-SCEs) [61], aberrant t-loop excisions leading to ECTRs, and recombination events that affect telomere length dynamics and jeopardize cellular viability (Fig. 1E).
2. Telomere-associated molecular patterns
Recent research has revealed that telomere deprotection serves as an intrinsic stimulus for innate immune sensing and signaling pathways, causing so-called sterile inflammation in the absence of pathogen infection. The connection between telomeres and inflammation is further supported by studies utilizing telomerase-null zebrafish [62] and mouse models [63–65], which exhibit accelerated telomere shortening, premature aging, and heightened inflammation. In humans, individuals with short telomere syndromes demonstrate premature aging and inflammation [66–68], while those with chronic inflammatory conditions often display compromised telomere protective mechanisms [69–72]. This section explores the molecular pathways that interconnect telomere biology and innate immunity, illuminating their dynamic interplay within the context of chronic age-related conditions.
2.1. Cytosolic DNA-sensing through cGAS
Innate immunity forms the initial line of defense against invading pathogens, priming the subsequent induction of an adaptive immune response [73]. This pathway relies on specialized sensors called pattern-recognition receptors (PRRs) capable of detecting a variety of pathogen-specific molecules, including lipids, sugars, and nucleic acids (NA), commonly referred to as danger-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) [74]. Self-NA resulting from cellular processes such as replication stress [75], genomic or mitochondrial DNA damage [76,77], or endogenous retroelement (ERE) activation [78] have been also identified as molecular patterns capable of triggering immune surveillance sensors and contributing to sterile inflammation.
Sensing cytosolic DNA is a fundamental aspect of innate immunity, with major sources including those acquired from the extracellular microenvironment or pathogens such as DNA viruses, retroviruses, bacteria, and parasites, as well as self-DNA leaking from the mitochondria and nucleus [79]. The nuclear envelope (NE) acts as a crucial regulatory barrier to prevent an aberrant innate immune response to host DNA molecules. However, during mitosis, cell migration, or other circumstances, the disintegration of the NE can lead to the mislocalization of DNA to the cytosol, triggering its subsequent detection by innate immune surveillance mechanisms. A key outcome of activating this signaling pathway is the generation of soluble mediators such as inflammatory cytokines and type I interferon (IFNs), which stimulate the production of IFN-stimulated genes (ISGs) with diverse antiviral, antitumor, and immunoregulatory properties [80].
Upon loss of telomere protection, DNA ends become susceptible to c-NHEJ and alt-EJ repair pathways, leading to the formation of three distinct types of fused chromosomes that fail to properly segregate during anaphase [50,81–84] (Fig. 2A). Genomic fusions take place between telomeres with non-telomeric loci, while inter-chromosomal fusions result in dicentric chromosomes, each retaining a functional centromere. Intra-chromosomal fusions occur between sister chromatids and generate pseudo-dicentric chromosomes, where the sister centromeres are correctly attached to opposing spindle poles during mitosis but remain linked at the fused end (Fig. 1C, D) [85].
Fig. 2. Telomere-associated molecular patterns.
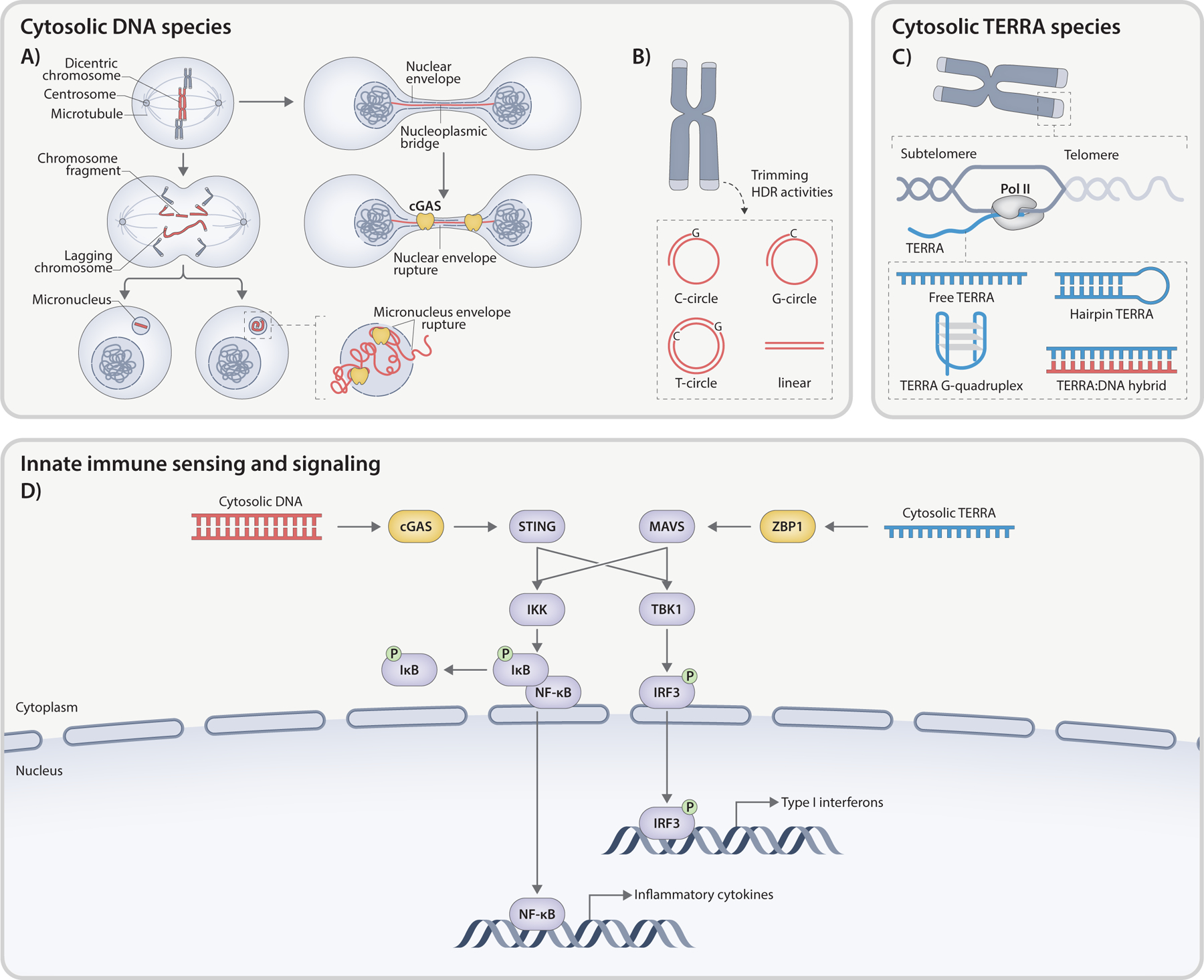
Telomere-driven innate immunity involves the activation of interconnected DNA- and RNA-sensing pathways. A) Deprotected telomeres result in chromosome fusion and formation of dicentric chromosomes. Tension across the centromeres of a dicentric chromosome can lead to chromosomal bridge formation, which is often resolved via fragmentation. Broken chromosome fragments lacking centromere may be mis-segregated, resulting in formation of one or more MN. MN can also emerge from lagging chromosomes that detach from both centromeres and are left behind at anaphase. These MN, characterized by an unstable nuclear envelope provoke cGAS activation upon envelope rupture. DNA bridges may endure past anaphase forming a link between two newly assembled daughter nuclei. These NPBs share similar nuclear envelope defects with micronuclei, leading to cGAS-dependent innate immune response. B) ECTRs, products of telomere trimming and HDR activity, may exhibit linear or circular configurations. The latter includes t-circles (both strands), C-circles (entire C-rich circle with a G-rich primer), or G-circles (complete G-rich circle with a C-rich primer). When released into cytoplasm, ECTRs can activate cGAS pathway. C) TERRA is a long non-coding RNA transcribed by RNA Polymerase II from subtelomeric regions toward chromosome ends, using the C-rich strand as a template. The cytosolic TERRA molecules can adopt multiple structures, such as ssRNA, G-quadruplex configurations, or RNA:DNA hybrids excised from telomeric R-loops. All TERRA species have the potential to provoke an innate immune response. D) In the cytosol, telomere-derived dsDNA species are recognized and bound by cGAS, activating STING. On the other hand, ZBP1 interacts with cytosolic TERRA, triggering MAVS-dependent signaling. Both pathways induce the transcription of interferon genes and proinflammatory cytokines via the TBK1-IRF3 and IKK-NFκB signaling cascades.
During anaphase, dicentric chromatids are pulled apart, creating a DNA bridge that can break by the spindle pulling forces, only to later rejoin over multiple breakage-fusion-bridge (BFB) cycles [86]. This catastrophic mutational process drives rapid genome evolution during tumorigenesis and is often accompanied by the generation of acentric fragments, forming one or more micronuclei (MN) at the end of mitosis [87,88] (Fig. 2A). It has also been described that, instead of breaking, dicentric chromosomes involved in anaphase bridges are sometimes detached from the two centrosomes, left behind at anaphase, and sequestered into MN [89] (Fig. 2A). Alternatively, DNA bridges can persist and extend beyond cytokinesis, carrying into the subsequent G1 phase [90]. The chromatin bridges linking the newly forming nuclei experience stretching due to cell migration and can reach hundreds of microns in length before eventually breaking (Fig. 2A). Transient accumulation of actin and myosin occurs at the bridge, and defects in myosin activation or actin assembly delay chromatin bridge breakage, suggesting that the tension generated by actomyosin force during cell division is required to break chromatin bridges [91]. In addition to DNA rupture through mechanical force, the TREX1 exonuclease is implicated in severing chromatin bridges [90]. This process involves a temporary rupture of the NE and loss of the nuclear permeability barrier, enabling cytoplasmic nucleases such as TREX1 to access chromatin and generate ssDNA fragments, which may facilitate breaks as DNA becomes increasingly stretched and fragile [90]. In both settings, the rupture of the chromatin bridge could also lead to the generation of acentric chromosome fragments distributed into MN.
Fused telomeres engage innate immunity through a mechanism involving the loss of cellular compartmentalization and the exposure of genomic DNA to cytoplasmic components including cGAS, an innate immune sensor that recognizes a wide range of cytoplasmic double-stranded DNA (dsDNA) species of both foreign and self-origin in a length-dependent but sequence-independent manner [92] (Fig. 2A). MN arising at mitotic exit, due to the incomplete partitioning of acentric chromosomal fragments or lagging chromosomes into daughter cells, frequently experience structural defects in the underlying lamina and nuclear pore complexes, collectively leading to the spontaneous collapse of the NE [93,94]. These disruptions not only impact crucial processes within MN, including DNA replication and repair, but also facilitate the access of cGAS to micronuclear DNA [95] (Fig. 2A). Further, persistent chromatin bridges during interphase induce a transient NE rupture during interphase, mixing nuclear and cytoplasmic components, and granting access to cGAS to the bridge DNA [90] (Fig. 2A). The precise connection between NE rupture and cGAS sensing remains unclear. Recent research indicates that a fraction of cGAS is located within the nucleus, where it is unable to oligomerize when tethered to nucleosomes [94,96–102]. The role of chromatin in restricting cGAS enzymatic activity suggests that NE rupture alone may not be sufficient to trigger cGAS activation, and additional factors such as DNA damage caused by exposure to cytoplasmic nucleases could alter chromatin state and facilitate cGAS activation. In line with this idea, genetic abnormalities in the histone pre-mRNA processing complex result in chromatin without linker histones, leading to cGAS stimulation and subsequent emergence of an IFN response [102].
In addition to the exposed DNA at the chromatin bridge and MN, ECTRs released into the cytoplasm can be also recognized by cGAS [103] (Fig. 2B). ECTRs manifest in both linear and circular configurations [58, 104–106] and are frequently observed in a subset of cancer cell lines, particularly those utilizing the ALT pathway to maintain telomere length without relying on telomerase activity [58,105]. Telomeric (t)-circles, a type of ECTR, are predominantly described as double-stranded circles with nicks or gaps, resulting from the trimming of elongated telomeres [58,105,107] (Fig. 2B). They are observed in various scenarios, including cells experiencing telomere elongation like telomerase+ cancer cell lines [107], and mitogen-stimulated T-lymphocytes [108]. Several factors have been shown to regulate t-circle elaboration in cancer cells, including NBS1, XRCC3, ORC2, and TZAP [106,109–112]. Telomeric (C)- and (G)-circles are distinguished by the presence of intact C-rich and G-rich telomeric DNA, respectively, with partial double-stranded structures [113] (Fig. 2B). Their production is regulated by factors associated with telomere replication stress, with C-circles being more abundant in ALT+ cells. SLX4 and FANCM have been identified as suppressors, while BLM and other members of the BTR complex have been found to promote C-circle formation, in coordination to their impact on ALT-telomere synthesis [54,114–116]. Additionally, MRE11 and RAD51 act as suppressors of C-circle formation in ALT+ cells through a RAD52-independent pathway [117]. The recognition of cytosolic ECTRs by cGAS initiates an innate immune response marked by the production of IFNB1 and defects in cell proliferation [103]. This discovery has prompted the proposal of selectively inducing the cGAS pathway in ALT+ cancer cells with a high abundance of ECTRs as a promising therapeutic approach.
cGAS is activated through direct and high-affinity binding to longer dsDNA fragments (>45 bp), promoting dimerization and facilitating the formation of more stable complexes through a 2:2 DNA/cGAS interaction [118,119]. The formation of phase-separated liquid droplets driven by non-specific interactions results in the coalescence of hundreds of cGAS molecules with immunostimulatory DNA [119,120], enhancing activity and synthesis of the second messenger 2’,3’ cGAMP [92]. The latter binds to STING, located at the endoplasmic reticulum (ER), triggering conformational changes, oligomerization, and translocation of STING to the ER-Golgi intermediate compartment and Golgi apparatus [121,122]. At these sites, STING recruits TBK1 and/or IKKε kinases, along with the transcription factor IRF3, to form a functional signalosome. The STING-TBK1-IRF3 complex signifies the canonical activation of STING signaling, characterized by various regulatory proteins and post-translational modifications on STING, such as Ser 366 phosphorylation and palmitoylation [123]. Once phosphorylated by TBK1, IRF3 undergoes dimerization and translocates into the nucleus, initiating the transcription of IFNB1 that acts through autocrine and paracrine signaling to activate the IFNAR1 and IFNAR2 heterodimeric receptor complex and inducing the transcription of numerous ISGs [124] (Fig. 2D). NF-κB activation, on the other hand, relies on the phosphorylation of its inhibitor IκB by the IκB kinase (IKK) complex. Following phosphorylation, IκB undergoes polyubiquitination and subsequent proteasomal degradation, liberating NF-κB to translocate to the nucleus and regulate downstream proinflammatory cytokines and chemokines [125–127] (Fig. 2D). However, the host has developed mechanisms to regulate the availability of ligands and maintain immunological balance. This is achieved through the degradation activities of DNases, such as extracellular DNases (DNase I and DNase IL3), phagolysosomal DNase (DNase II), and cytosolic DNase (TREX1 or DNase III) [103]. Dysregulation of this pathway can contribute to the development of type I interferonopathies, such as Aicardi-Goutières syndrome (AGS) [128] and systemic lupus erythematosus (SLE) [129], characterized by a chronic activation of cGAS-STING and excessive IFN production.
2.2. Cytosolic RNA-sensing through ZBP1
Telomere-driven innate immunity involves the recognition of cytosolic DNA by the cGAS-STING machinery, as well as the activation of a parallel pathway that relies on sensing Telomeric Repeat-containing RNA (TERRA) transcripts [130–134]. TERRA is synthesized by RNA polymerase II using CpG-rich promoter sequences located in the subtelomeric regions of a majority of human chromosomes, utilizing the C-rich telomeric strand as template [135] (Fig. 2C). Consequently, TERRA molecules consist of chromosome-specific subtelomeric sequences followed by a variable number of telomeric UUAGGG repeats [130,136,137]. The regulation of TERRA promoters involves epigenetic modifications, including DNA methylation mediated by DNMT1 and DNMT3B [137], as well as chromatin alterations facilitated by the homodimerization domain of TRF2 [138]. Depletion of TRF2 or expression of the dominant-negative mutant TRF2ΔBΔM compromises telomere protection and stimulates TERRA synthesis, resembling the altered telomere state observed during an anti-proliferative barrier known as replicative crisis [138–140].
Polyadenylated TERRA transcripts reside in the nucleoplasm, while non-polyadenylated transcripts are found in the nucleoplasm as well as associated with telomeric chromatin [136,141]. The interaction of TERRA with telomeres is facilitated by its telomeric 3’ end, allowing for binding to telomere-associated proteins [142,143] and the formation of RNA:DNA hybrids (R-loops) [114,136,144,145]. Additionally, TERRA molecules have been found in the cytosol [140] and can be secreted within exosome vesicles [146], yet their shuttling dynamics between the nucleus, cytoplasm, and extracellular environment are poorly understood. The biological functions of TERRA at telomeres include regulating telomere replication [147], promoting heterochromatin formation [148], facilitating telomerase recruitment [149–152], and enabling HDR among telomeres in ALT cancer cells [114,142,153–155], while its broader functions at non-telomeric compartments remain mostly elusive.
Our recent understanding of immunostimulatory RNA species has expanded to encompass host-derived RNA ligands and include those transcribed from endogenous EREs, in addition to viral genome and replication intermediates. Upregulation, mislocalization, or misprocessing of these self-RNA species confers them with immunostimulatory properties, potentially contributing to sterile inflammation observed in autoimmune or autoinflammatory disorders [156,157]. TERRA molecules serve as a prime example of this phenomenon. TERRA interacts with the cytosolic NA sensor human ZBP1 (also known as DAI or DLM-1) and triggers innate immune responses and IFN signaling [140]. Originally identified as a cytosolic receptor for DNA [158], ZBP1 has emerged as a versatile sensor capable of detecting Z-form NA, with a particular affinity for RNA molecules [159–166]. This recognition is mediated by its N-terminal Zα1 and Zα2 domains, which can bind to Z-NA in a structure-specific manner, independent of specific sequences or individual base contacts [167–169]. ZBP1 contains two receptor-interacting protein homotypic interaction motif (RHIM) domains. The latter enable interactions with RIPK1 and RIPK3, which are involved in cell death signaling, innate immunity, and inflammation [170,171], as evidenced by studies in mouse models infected with influenza A virus (IAV), vaccinia virus (VACV), and certain herpesviruses [172].
The initial evidence of mouse ZBP1 recognizing viral RNA came from a study with the orthomyxoviruses influenza A and influenza B, which induced RIPK3-dependent cell death [173]. The second Zα domain (Zα2) appears to be responsible for activating cell death signaling as mutations in key Z-NA contact amino acids abolished cell death [160]. During infections with DNA viruses such as murine cytomegalovirus (MCMV) and herpes simplex virus-1, the inhibition of RNA synthesis effectively prevented the activation of ZBP1, suggesting that Z-RNAs, rather than Z-DNAs, are the primary activating ligands [161,162,174]. A recent study revealed that Z-RNAs produced by SARS-CoV-2, potentially from ORF1a and ORF1b genomic regions, activate ZBP1 [175]. It has been proposed that the formation of Z-RNA structures may occur due to torsional stress induced by negative supercoiling during viral replication, similar to what has been observed with dsDNA during cellular transcription [176,177]. Once activated, ZBP1 associates with RIPK3 to execute programmed cell death pathways such as necroptosis, apoptosis, and pyroptosis, depending on the cell type and caspase activity. ZBP1-mediated programmed cell death can even involve the simultaneous activation of these three modes of cell death, leading to the emergence of a cell death pathway termed PANoptosis [178].
In the absence of adenosine deaminase RNA specific 1 (ADAR1) activity, ZBP1 can be activated by EREs derived from mammalian genomes [179–182]. The IFN-inducible p150 isoform of ADAR1, which contains a Zα domain, can bind to Z-NA [183,184], thereby reducing their capacity to activate host RNA sensors. This reduction is achieved in part by introducing adenosine-to-inosine edits in their sequences [185]. Mice lacking ADAR1 p150 expression or carrying a mutation of its Zα domain, exhibit type I IFN-dependent pathology reminiscent of AGS [179–182]. Deletion of ZBP1 mitigates the pathology, reduces the IFN signature, and prolongs animal survival. Despite the technical difficulties in directly evaluating Z-RNA formation in living cells and tissues, the available data indicate that ADAR1 plays a major role in editing RNA transcripts derived from EREs, such as LINE-1 and SINEs, which have a propensity to form Z-RNA structures [181], thereby preventing aberrant activation of ZBP1. The precise mechanism by which ZBP1 triggers pathogenesis in the absence of ADAR1 remains uncertain, as it occurs independently of canonical pathways involving RIPK1, RIPK3, MLKL-mediated necroptosis, and caspase-8-dependent apoptosis, suggesting a novel mode of action [179–182].
Our understanding of ZBP1 signaling mechanisms is largely derived from mouse models, despite the limited similarity in protein sequences between human and murine ZBP1 [186]. The mRNA profiles also differ, with human ZBP1 showing heterogeneity due to alternative splicing and multiple transcription start sites [187]. Many predicted isoforms in humans are not expressed in mouse cells, including the short isoform (ZBP1-S) lacking exon 2, which encodes the first Zα domain [140,187, 188]. ZBP1-S displays distinct subcellular localizations, implying diverse biological functions compared to the full-length protein [168]. In line with this idea, our recent study implicated ZBP1-S in mediating the onset of replicative crisis and preventing the propagation of cells at risk of neoplastic transformation [140]. During crisis, cGAS–STING drives an initial transcriptional induction of ZBP1-S, a prototypical ISG, and primes cells to respond to aberrant accumulation of additional cytosolic immunostimulatory NA species. IFN-induced ZBP1-S is activated by sensing TERRA molecules derived from damaged telomeres, resulting in its redistribution to a distinct subset of mitochondria and the formation of filamentous structures (Fig. 2D). The assembly of ZBP1-S filaments at mitochondria is not dependent on interacting with mitochondrial DNA (mtDNA) or mitochondrial RNA (mtRNA). Disruption of the RHIM1 or Zα2 domains hinders the formation of ZBP1-S filaments and diminishes their involvement in the innate immune response. Interestingly, multimerization, as well as mitochondrial translocation to activate the adaptor protein MAVS [189–192] is shared between ZBP1 and the innate immune RNA sensors RIG-I [193] and MDA-5 [194]. However, these RNA sensors often form aggregated structures rather than filaments, suggesting an alternative oligomerization mechanism for ZBP1 [195].
MAVS, anchored to various subcellular compartments through its C-terminal transmembrane domain, elicits different antiviral signaling responses: The mitochondrial MAVS promotes type I IFN expression, while the peroxisomal MAVS signals for the induction of type III IFN [196–198]. MAVS contains an N-terminal caspase recruitment domain (CARD) that interacts with similar CARD domains present in the RNA sensors RIG-I and MDA5 [199–203]. Upon association with RIG-I and/or MDA5, MAVS forms prion-like polymers on the mitochondrial surface [204,205]. This active state facilitates the recruitment of downstream TRAF effector proteins to create the “MAVS signalosome” [206]. As a result, two molecular cascades become engaged: (a) the phosphorylation of the transcription factors IRF3 and IRF7 by TBK1 and IKKε to induce the expression of type I/III IFNs and (b) the phosphorylation of the transcription factor NF-κB by the IKKα/β/γ complex to upregulate the expression of inflammatory genes [189–191,207] (Fig. 2D).
During crisis, ZBP1-S triggers cell death through a MAVS-mediated IFN response and autophagy, with no evidence of ongoing PANoptosis under these circumstances [140]. Remarkably, cells with compromised ZBP1-MAVS-autophagy pathways gain resistance to cell death and persistently proliferate beyond the crisis barrier, despite their critically short and fused telomeres [140]. These findings firmly establish ZBP1-S signaling as a pivotal component of the crisis program, acting as a tumor suppressive mechanism for eliminating cells with dysfunctional telomeres predisposed to neoplastic transformation (Fig. 2D).
The discovery of a molecular connection between deprotected telomeres, mitochondrial signaling, and innate immune responses underscores a novel interplay among major hallmarks of aging. However, several aspects of this relationship remain poorly understood. One aspect involves the nature of the immunogenic TERRA ligands recognized by ZBP1-S. TERRA, characterized by G-rich repeated sequences, has the ability to form stable hairpins and non-canonical G-quadruplex structures (RG4s) in vitro [208–211] (Fig. 2D). The in vivo formation and functional significance of RG4 structures within TERRA are still uncertain; however, the conformation-specific sensing ability of ZBP1 suggests that RG4s may share similarities with the left-handed Z-RNA structures detected by ZBP1. An alternative possibility is the formation of R-loops resulting from the hybridization of TERRA with the C-rich telomeric strand. These R-loops may undergo processing and excision, leading to their abnormal accumulation in the cytoplasm and subsequent activation of ZBP1-driven innate immunity (Fig. 2C, D). The second unexplored aspect relates to the redistribution of ZBP1 to mitochondria that is essential for activating a MAVS-dependent IFN response. Unlike other RNA sensors that rely on CARD-CARD interactions with MAVS to translocate to mitochondria, ZBP1 lacks a CARD domain. ZBP1 is likely recruited to mitochondria via interactions with proteins at the mitochondria-ER contact sites, which are known to be involved in innate immune signaling [212].
3. Telomere-driven innate immunity and inflammation
Altered immune function and chronic inflammation have long been recognized as substantial contributors to the aging process and its associated diseases [213]. Contrary to previous assumptions [214], recent advancements in genetic studies and clinical observations indicate that the loss of telomere protection as a function of replicative age is not merely a consequence of inflammation but actively fuels the inflammatory process [215,216]. It is therefore essential to reevaluate our understanding of the synergy between telomeres and inflammation, offering valuable insights into the fundamental mechanisms of aging.
3.1. Telomere deprotection and aging
Telomere deprotection, attributed to factors such as the end replication problem, replication slippage, and oxidative damage, imposes replicative barriers that limit the proliferative potential of normal human somatic cells [217–221]. Replicative senescence is triggered upon the recognition of DNA ends as DNA breaks, leading to the activation of DDR pathways, formation of telomere-induced DNA damage foci (TIF), and subsequent cell cycle arrest mediated by the p53/p21WAF1/CIP1 and p16INK4A/pRB tumor suppressor pathways [44, 222–224]. Telomere length profiles in senescent cells are heterogeneous and only a few exhibit TIF, suggesting that individual DDR signaling events at telomeres contribute to the onset of senescence. In line with this idea, telomeres in senescence retain a sufficient level of shelterin to partially protect against DNA repair mechanisms while simultaneously triggering DDR signals necessary for an irreversible cell cycle arrest [44, 222,224–227]. In addition to the permanent cessation of cell division, senescence is characterized by morphological and metabolic alterations, resistance to cell death, and the accumulation of cytoplasmic chromatin fragments (CCFs) formed through the extrusion of nuclear chromatin into the cytoplasm due to the loss of nuclear membrane integrity [228]. MN, distinct from CCFs, exhibit a reduced propensity to accumulate in senescent cells. Distinguishing between CCFs and MN microscopically remains challenging, often leading to confusion between the two. While MN-dependent activation of cGAS typically leads to the induction of IFN genes, the activation of cGAS by CCFs in senescent cells is predominantly associated with an NF-κB-dependent cytokine response, contributing to the emergence of the senescence-associated secretory phenotype (SASP) [229–232]. Studies on aged tissues obtained from primate models, including mice, baboons, and humans, have provided evidence of the accumulation of TIF and other markers of replicative senescence during aging [233–240]. Based on these observations, senescent cells are proposed to contribute to tissue aging through two main mechanisms: First, they lose the ability to proliferate and replenish tissues with new cells [241,242]. Second, they secrete molecules that promote tissue dysfunction and inflammation [243], such as degradative enzymes capable of breaking down the extracellular matrix, growth factors, and inflammatory cytokines. Building upon this concept, the targeted elimination of senescent cells or suppressing SASP production using genetic and pharmacological approaches has demonstrated effectiveness in reducing age-related decline and extending the health-span in aged mice [244–246].
Oncogenic events that interfere with the functions of p53 and/or pRB enable cells to proliferate past the senescence barrier until their telomeres reach a critically shortened state, characterized by a near absence of shelterin subunits and activation of DNA repair mechanisms [217, 218,247]. At this stage, a second proliferative barrier known as replicative crisis emerges, marked by a substantial decline in viable cells and increased genome instability often including chromosome fusions, nucleoplasmic bridges (NPBs), MN, and aneuploidy [51,81,248–250]. During crisis, cell death is initiated by the telomere dysfunction-driven accumulation of self-NA species in the cytoplasm, recognized as danger signals that alert the immune system to cope with aberrant cellular activities. Two interconnected cytosolic NA-sensing pathways, namely cGAS-STING activated by cytosolic DNA fragments, and ZBP1-MAVS activated by cytoplasmic TERRA molecules synthesized from damaged telomeres, contribute to the clearance of cells undergoing crisis [140,248]. These pathways synergistically induce a robust type-I IFN responses and sustained inflammatory cascades, leading to the activation of an atypical form of autophagy that facilitates the elimination of cells, rather than promoting viability [140,248]. Autophagy-mediated cell death has been observed in various organisms during developmental [251–254] and pathophysiological [255–258] processes, as well as in cancer cell lines [259], particularly those with impaired apoptosis. While the precise mechanisms underlying the transition of autophagy from a survival pathway to a lethal one are not fully understood, two propositions have emerged: Excessive autophagy flux can lead to the detrimental breakdown of cellular organelles and disrupt cellular homeostasis, or autophagosome membranes can serve as platforms to assemble death-inducing signaling complexes [260].
The accumulation of dicentric chromosomes during crisis, primarily through alt-EJ instead of c-NHEJ [81,261–263], drives complex chromosomal rearrangements and BFB cycles observed in various human tumors, including acute lymphocytic leukemia, squamous cell cancers, and esophageal adenocarcinoma [264,265]. The observations suggest that, during carcinogenesis, replicative crisis occurs prior to the activation of telomere maintenance mechanisms, effectively eliminating pre-malignant cells and serving as a potent barrier against age-related cancer initiation. Further evidence comes from observations of shortened and fused telomeres during the transition from ductal hyperplasia to invasive cancer [266], the initiation of colorectal adenomas [267], and the early stages of chronic lymphocytic leukemia in humans [268]. Studies in mice lacking telomerase and p53 have revealed that telomere dysfunction is associated with the formation of non-reciprocal translocations, focal amplifications, and deletions, mirroring observations made in human cancers [63]. However, the impact of replicative crisis on the aging process, beyond its role in preventing age-related cancer, is not well understood, and the accumulation of crisis cells in aged tissues has not been explored due to the lack of reliable biomarkers, pointing at necessary future research to fill these knowledge gaps.
In addition to the progressive telomere shortening observed during repeated passages in vitro, most human tissues and organs undergo telomere shortening throughout the aging process. Telomere length is negatively associated with age in various cell types such as peripheral blood cells, dermal fibroblasts, intestinal epithelial cells, and mucosal keratinocytes [269–272]. TIF assays, employed to profile telomere deprotection in cells and tissues, have revealed an elevated occurrence of dysfunctional telomeres during aging in various mammalian tissues, including the brain, liver, and skin of baboons [233,234], proliferating and non-proliferating tissues in mice [237–240,273], as well as human skin melanocytes and CD8 + T cells [235,236]. The impact of telomere shortening on organismal aging has been further studied in animal models, particularly in mice and zebrafish, by genetically deleting telomerase component genes either the telomerase RNA component (Terc) or the telomerase reverse transcriptase (TERT) [63,274–276]. Both components function to synthesize telomere repeats on chromosome ends, thereby maintaining telomere length in germline tissues as well as in immortal and cancer cells. Telomerase-deficient zebrafish possess human-like short telomeres that decline with age, disrupting homeostasis in highly proliferative tissues and leading to various degenerative phenotypes and inflammation [275–277]. Late-generation telomerase-knockout mice revealed signs of premature aging, including shortened life expectancy, depletion of tissue stem cell reserves, organ atrophy, and inflammation [274,278–280]. In humans, germline mutations in genes affecting telomerase function or telomere maintenance result in a range of clinical conditions that are collectively termed telomeropathies, short-telomere syndromes, or telomere biology disorders. This wide spectrum of disorders, which include dyskeratosis congenita, pulmonary fibrosis, and aplastic anemia, is characterized by severely shortened telomeres, often resulting in hematopoietic stem cell failure in the most severe cases, inflammation, premature aging and shortened lifespan [66], highlighting the substantial impact of telomere shortening on overall organismal health and its pivotal role in the aging process.
3.2. Telomere deprotection at the core of inflammaging?
Epidemiological studies have established a connection between inflammation and numerous aging-related diseases, including obesity, type 2 diabetes, cardiovascular diseases, Alzheimer’s disease, and cancer [281]. This led to the concept of inflammaging—a low-grade systemic inflammation that accompanies aging in mammals and links normal aging with age-related diseases sharing an inflammatory foundation [213]. Despite this knowledge, the precise causes of inflammaging remain largely unknown, emphasizing the need for further research to uncover the underlying mechanisms.
The loss of telomere protection contributes to cellular and organismal aging, and disturbances in telomere homeostasis have been associated with several chronic inflammatory conditions. Telomerase-deficient zebrafish show heightened inflammation, increased expression of TNF1-α, and accelerated aging [62]. Late-generation telomerase null mice exhibit both impaired telomeres and age-associated degenerative pathology, including inflammation [63–65]. In the intestinal epithelium, telomere dysfunction results in the activation of the YAP1 transcriptional coactivator and up-regulation of pro-IL-18, a critical immune regulator in the pathogenesis of inflammatory bowel disease [279]. The role of telomere dysfunction as a driver of inflammatory diseases is evident from telomeropathy studies. These disorders arise from germline defects in telomere maintenance genes such as TERT, TERC, PARN, DKC1, NHP2, NOP10, NAF1, WRAP53, as well as mutations affecting the shelterin complex (TIN2, POT1, and TPP1) and telomere replication (CTC1 and RTEL1) [66]. Individuals exhibit impairments in telomere protective mechanisms, experience subclinical manifestations in multiple organs, and develop an increased incidence of idiopathic pulmonary fibrosis, liver cirrhosis, and kidney diseases, all of which relate to increased inflammation [66–68]. Moreover, telomere dysfunction has been recognized as major contributor to a range of inflammatory diseases in older individuals, independent of the presence of genetic polymorphisms in telomere-associated genes [69–72]. In such cases, the presence of dysfunctional telomeres in a subset of cells within affected tissues can trigger localized inflammation, leading to an increased production of reactive oxygen species and further telomere dysfunction in neighboring cells. This exacerbates the inflammatory response and contributes to the development of various age-related conditions like inflammatory bowel disease [282], pancreatitis [283], non-alcoholic fatty liver disease [284], and chronic obstructive pulmonary disease [285]. These findings have established connections between telomere dynamics and age-associated inflammation. However, the debate regarding whether telomere attrition or inflammation is the primary factor remains inconclusive, indicating a likely coexistence and interaction of both components forming a detrimental cycle.
Our recent research suggests that telomere-associated molecular patterns may be the root cause of inflammaging [140] (Fig. 2). In normal cells with functional p53 and pRB pathways, telomere deprotection leads to replicative senescence and the emergence of SASP, characterized by increased secretion of soluble pro-inflammatory cytokines, chemokines, and other inflammatory molecules [286]. The SASP involves a series of poorly understood events that are interconnected with the DDR and NFκB signaling pathways [287,288]. Several groups have identified a consistent pattern of abnormal cGAS-STING activity in different types of senescent cells, including replicative senescence triggered by telomere shortening [231,239]. It has been observed that cGAS is enriched on CCFs that accumulate within senescent cells, contributing to the secretion of specific components of the SASP. In addition to CCFs, other sources of cytosolic DNA species, such as mitochondrial DNA and retrotransposon cDNA, have been observed to accumulate in senescent cells and contribute to the inflammatory SASP [79]. Despite the requirement of the cGAS-STING pathway for both inducing and maintaining the SASP program, its absence has no impact on the activation of ATM-dependent DDR pathway, p53 stabilization, and it does not prevent the onset of replicative senescence in human primary cells [229]. As senescent cells accumulate during aging in both model organisms and humans, the SASP may possess a long-term destructive potential by mediating tissue damage and chronic inflammation [289]. In cells with compromised cell cycle checkpoints, telomere deprotection triggers a distinct cellular program called replicative crisis [140,248,290]. During this phase, dysfunctional telomeres generate cell-intrinsic stimulatory products: Micronuclear DNA activates the cGAS-STING pathway, while cytosolic TERRA RNA molecules interact with the ZBP1-MAVS pathways [140]. These pathways collaborate to induce potent IFN and inflammatory responses, potentially contributing to the increased inflammation observed during the aging process (Fig. 2C). The pro-inflammatory state observed during replicative crisis is fundamentally different from the one associated with senescence, as it predominantly involves IFN signaling and ISGs, which are not actively involved in senescence and therefore not associated with the SASP [291]. To describe the inflammatory secretome produced by crisis cells, we propose the term CASP (Crisis-Associated Secretory Phenotype). The CASP appears to be required for crisis-associated cell death and the onset of this second replicative barrier [140]. Further research will be essential to comprehensively characterize this inflammatory secretome and its implications in cell death during crisis.
As individuals age, systemic chronic inflammation tends to increase, as evidenced by studies showing higher levels of cytokines and chemokines in older individuals [292,293]. Based on our recent research [140, 217,248], we argue that age-related telomere dysfunction is a central driver of inflammaging, with the major source of inflammatory stimuli arising from misplaced self-NA sensed by receptors of the innate immune system cGAS and ZBP1 (Fig. 2). In addition to telomere deprotection, other age-associated alterations can lead to innate immune activation [281]. One particular example is the release of mtDNA into the cytosol, which can be recognized by NLRP3 inflammasome [294], cGAS [295], as well as ZBP1 [296]. The presence of mtDNA circulating in plasma has also been reported during aging and in several specific age-associated diseases [79,297–299]. Although it is not yet known whether circulating cell-free mtDNA is causative in age-associated diseases, given its strong pro-inflammatory potential, it is likely that cytoplasmic and extracellular mtDNA contribute to some extent to chronic inflammation. Modulating innate immune pathways holds significant promise for mitigating chronic inflammation and age-related diseases. However, it is essential to carefully consider potential drawbacks, such as compromising immune surveillance and tumor suppression, and possible side effects on the immune microenvironment.
The study of inflammation and telomere deprotection in relation to aging offers valuable insights. However, their interdependent nature calls for a more synergistic approach to fully grasp the underlying mechanisms of aging and devise effective therapeutic interventions for the elderly. Considering their interconnected roles holds great potential in tackling inflammaging, ultimately leading to improved health and well-being in aging populations.
Acknowledgements
We apologize to our colleagues whose excellent work could not be cited due to space limitations. We thank Tobias T Schmidt and Samuel I Bloom for the insightful comments and suggestions.
J.N. is supported by the European Molecular Biology Organization (ALTF 213-216), the Hewitt Foundation, and the National Cancer Institute (K99CA252447 and R00CA252447).
S.P. has received support from the Swiss National Science Foundation Early Postdoc Mobility Fellowship (P2ZHP3_195173), and the Glenn Foundation for Biology of Aging Research.
J.K. receives support from The Salk Institute Cancer Center Core Grant (P30CA014195), the National Institutes of Health (RO1CA227934, RO1CA234047, RO1CA228211, RO1AG077324), the Donald and Darlene Shiley Chair, the Samuel Waxman Cancer Research Foundation, the Auen Foundation, and the American Heart Association (19PABHI34610000).
Abbreviations:
- 53BP1
Tumor Protein P53 Binding Protein 1
- AGS
Aicardi-goutières syndrome
- ALT
Alternative lengthening of telomeres
- alt-EJ
Alternative end-joining
- ATM
Ataxia telangiectasia mutate
- ATR
Ataxia telangiectasia and Rad3-related protein
- BFB
Breakage-fusion-bridge
- BITS
Break-induced telomere synthesis
- BLM
Bloom
- BRCA1
Breast cancer type 1 susceptibility protein
- BRCA2
Breast cancer type 2 susceptibility protein
- BTR complex
RecQ helicase BLM, topoisomerase IIIα, and RMI
- c-NHEJ
Canonical non-homologous end-joining
- CARD
Caspase recruitment domain
- CARDIF
CARD adapter inducing interferon beta
- CASP
Crisis-associated secretory phenotype
- CCFs
Cytoplasmic chromatin fragments
- cGAMP
Cyclic guanosine monophosphate–adenosine monophosphate
- cGAS
Cyclic GMP-AMP synthase
- CTC1
Conserved telomere maintenance component 1
- CtIP
CtBP-interacting protein
- DAI
DNA-dependent activator of IFN-regulatory factors
- DAMPs
Damage-associated molecular patterns
- DDR
DNA damage response
- DKC
Dyskeratosis congenital
- DLM-1
Tumor stroma and activated macrophage protein
- DNA-PKcs
DNA-dependent protein kinase catalytic subunit
- DNA2
DNA replication ATP-dependent helicase/nuclease DNA2
- DNMT1
DNA (cytosine-5)-methyltransferase 1
- dsDNA
Double-stranded DNA
- ECTR
Extrachromosomal telomere repeat
- ER
Endoplasmic reticulum
- ERE
Endogenous retroviral elements
- ERGIC
ER-Golgi intermediate compartment
- EXO1
Exonuclease 1
- HDR
Homology-directed repair
- IAV
Influenza A virus
- iDDR
Inhibitor of DDR
- IFN
Interferon
- IFNAR1
Interferon alpha/beta receptor 1
- IFNAR2
Interferon alpha/beta receptor 2
- IFNB1
Interferon beta
- IKK
IκB kinase complex
- IKK-β
Inhibitor of nuclear factor kappa-B kinase ε
- IL
Interleukin
- IPS-1
Interferon beta promoter stimulator protein 1
- IRF3
Interferon regulatory factor 3
- IRF7
Interferon regulatory factor 7
- ISG
Interferon-stimulated gene
- IκB
Inhibitor of nuclear factor kappa B
- Ku
complex Ku70-Ku80
- LIG3
Ligase 3
- LIG4
Ligase 4
- LINE-1
Long interspersed nuclear elements 1
- MAM
Mitochondria-associated endoplasmic reticulum membrane
- MAPK
Mitogen-activated protein kinase
- MAVS
Mitochondrial antiviral signaling protein
- MCMV
Murine cytomegalovirus
- MDA5
Melanoma differentiation-associated protein 5
- MDC1
Mediator of DNA damage checkpoint 1
- MLKL
Mixed lineage kinase domain like pseudokinase
- MN
Micronuclei
- MRN
MRE11, RAD50, and NBS1
- NAF1
Nuclear assembly factor 1 ribonucleoprotein
- NBS1
Nijmegen breakage syndrome 1
- NE
Nuclear envelope
- NF-κB
Nuclear factor kappa B
- NHP2
Nucleolar protein family A, member 2
- NOP10
Nucleolar protein 10
- NPB
Nucleoplasmic bridges
- OMM
Outer mitochondrial membrane
- ORC2
Origin recognition complex subunit 2
- ORF
Open reading frame
- PALB2
Partner and localizer of BRCA2
- PAMPs
Pathogen-associated molecular pattern molecules
- PARN
Poly(A)-specific ribonuclease
- PARP1
Poly(ADP-Ribose) polymerase 1
- PCNA
Proliferating Cell Nuclear Antigen
- PNPase
Polyribonucleotide Nucleotidyltransferase 1, Mitochondrial
- POT1
Protection of telomeres 1
- pRb
RB transcriptional corepressor 1
- PRRs
Pattern recognition receptors
- RFC1
Replication Factor C Subunit 1
- RG4s
RNA G-Quadruplexes
- RHIM
RIP homotypic interaction motif
- RIG-I
Retinoic Acid-Inducible Gene 1 Protein
- RIPK1
Receptor Interacting Serine/Threonine Kinase 1
- RIPK3
Receptor Interacting Serine/Threonine Kinase 3
- RMI
RecQ-mediated genome instability
- RNF168
Ring finger protein 168
- RPA
Replication Protein A
- RTEL1
Regulator of telomere elongation helicase 1
- SARS-CoV-2
Severe acute respiratory syndrome coronavirus 2
- SASP
Senescence-associated secretory phenotype
- SINEs
Short interspersed nuclear elements
- SLE
Systemic lupus erythematosus
- SLX4
Structure-specific endonuclease subunit
- SMX complex
SLX1-SLX4, MUS81-EME1, and XPF-ERCC1
- ssDNA
Single-stranded DNA
- STING
Stimulator of interferon genes protein
- SUV3
Suppressor of Var1, 3-Like 1
- t-loop
Telomere-loop
- T-SCE
Telomere sister chromatid exchange
- TBD
Telomere biology disorders
- TBK1
TANK binding kinase 1
- TCAB1
Telomerase cajal body protein 1
- TERC
Telomerase RNA component
- TERRA
Telomeric repeat-containing RNA
- TERT
Catalytic subunit telomerase reverse transcriptase
- TIF
Telomere dysfunction induced foci
- TIN2
TERF1 interacting nuclear factor 2
- TOP3A
DNA topoisomerase III alpha
- TPP1
Tripeptidyl peptidase 1
- TRAF
TNF receptor associated factor
- TREX1
Three prime repair exonuclease 1
- TRF1
Telomeric repeat binding factor 1
- TRF2
Telomeric repeat binding factor 2
- TZAP
Zinc finger and BTB domain containing 48
- VACV
Vaccinia related kinase 2
- VISA
Virus-induced signaling adaptor
- WRAP53
WD repeat containing antisense to TP53
- XRCC3
X-ray repair cross complementing 3
- YAP1
Yes1 associated transcriptional regulator
- Z-NAs
Z-nucleic acid
- ZBP1
Z-DNA binding protein 1
Footnotes
CRediT authorship contribution statement
JN, SP, JK: Conceptualization, Visualization, Writing: Initial draft, Writing: Final Manuscript, Rosponding to referees and editing.
Declaration of Competing Interest
The authors of the review entitled ‘Telomeres as hotspots for innate immunity and inflammation’ declare no conflict of interest.
Data Availability
No data was used for the research described in the article.
References
- [1].de Lange T, Shelterin: the protein complex that shapes and safeguards human telomeres, Genes Dev 19 (2005) 2100–2110. [DOI] [PubMed] [Google Scholar]
- [2].de Lange T, Shelterin-mediated telomere protection, Annu. Rev. Genet 52 (2018) 223–247. [DOI] [PubMed] [Google Scholar]
- [3].Palm W, de Lange T, How shelterin protects mammalian telomeres, Annu. Rev. Genet 42 (2008) 301–334. [DOI] [PubMed] [Google Scholar]
- [4].Chow TT, Zhao Y, Mak SS, Shay JW, Wright WE, Early and late steps in telomere overhang processing in normal human cells: the position of the final RNA primer drives telomere shortening, Genes Dev 26 (2012) 1167–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sfeir AJ, Chai W, Shay JW, Wright WE, Telomere-end processing the terminal nucleotides of human chromosomes, Mol. Cell 18 (2005) 131–138. [DOI] [PubMed] [Google Scholar]
- [6].Watson JD, Origin of concatemeric T7 DNA, Nat. N. Biol 239 (1972) 197–201. [DOI] [PubMed] [Google Scholar]
- [7].Olovnikov AM, A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon, J. Theor. Biol 41 (1973) 181–190. [DOI] [PubMed] [Google Scholar]
- [8].Harley CB, Futcher AB, Greider CW, Telomeres shorten during ageing of human fibroblasts, Nature 345 (1990) 458–460. [DOI] [PubMed] [Google Scholar]
- [9].Makovets S, Herskowitz I, Blackburn EH, Anatomy and dynamics of DNA replication fork movement in yeast telomeric regions, Mol. Cell. Biol 24 (2004) 4019–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Verdun RE, Karlseder J, The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres, Cell 127 (2006) 709–720. [DOI] [PubMed] [Google Scholar]
- [11].Miller KM, Rog O, Cooper JP, Semi-conservative DNA replication through telomeres requires Taz1, Nature 440 (2006) 824–828. [DOI] [PubMed] [Google Scholar]
- [12].Ivessa AS, Zhou J-Q, Schulz VP, Monson EK, Zakian VA, Saccharomyces Rrm3p, a 5′ to 3′ DNA helicase that promotes replication fork progression through telomeric and subtelomeric DNA, Genes Dev 16 (2002) 1383–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Barnes RP, et al. , Telomeric 8-oxo-guanine drives rapid premature senescence in the absence of telomere shortening, Nat. Struct. Mol. Biol 29 (2022) 639–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fouquerel E, et al. , Targeted and persistent 8-oxoguanine base damage at telomeres promotes telomere loss and crisis, e6, Mol. Cell 75 (2019) 117–130. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Opresko PL, Fan J, Danzy S, Wilson DM, Bohr VA, Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2, Nucleic Acids Res 33 (2005) 1230–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jacome Burbano MS, Cherfils-Vicini J, Gilson E, Neutrophils: mediating TelOxidation and senescence, EMBO J 40 (2021), e108164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F, Cellular senescence in ageing: from mechanisms to therapeutic opportunities, Nat. Rev. Mol. Cell Biol 22 (2021) 75–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Makarov VL, Hirose Y, Langmore JP, Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening, Cell 88 (1997) 657–666. [DOI] [PubMed] [Google Scholar]
- [19].McElligott R, Wellinger RJ, The terminal DNA structure of mammalian chromosomes, EMBO J 16 (1997) 3705–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Broccoli D, Smogorzewska A, Chong L, de Lange T, Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2, Nat. Genet 17 (1997) 231–235. [DOI] [PubMed] [Google Scholar]
- [21].Bilaud T, et al. , Telomeric localization of TRF2, a novel human telobox protein, Nat. Genet 17 (1997) 236–239. [DOI] [PubMed] [Google Scholar]
- [22].Baumann P, Cech TR, Pot1, the putative telomere end-binding protein in fission yeast and humans, Science 292 (2001) 1171–1175. [DOI] [PubMed] [Google Scholar]
- [23].Loayza D, De Lange T POT1 as a terminal transducer of TRF1 telomere length control, Nature 423 (2003) 1013–1018. [DOI] [PubMed] [Google Scholar]
- [24].Kim S, Kaminker P, Campisi J, TIN2, a new regulator of telomere length in human cells, Nat. Genet 23 (1999) 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].O’Connor MS, Safari A, Xin H, Liu D, Songyang Z, A critical role for TPP1 and TIN2 interaction in high-order telomeric complex assembly, Proc. Natl. Acad. Sci. USA 103 (2006) 11874–11879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ye JZ-S, et al. , POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex, Genes Dev 18 (2004) 1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Takai KK, Kibe T, Donigian JR, Frescas D, de Lange T, Telomere protection by TPP1/POT1 requires tethering to TIN2, Mol. Cell 44 (2011) 647–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li B, de Lange T, Rap1 affects the length and heterogeneity of human telomeres, Mol. Biol. Cell 14 (2003) 5060–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Amiard S, et al. , A topological mechanism for TRF2-enhanced strand invasion, Nat. Struct. Mol. Biol 14 (2007) 147–154. [DOI] [PubMed] [Google Scholar]
- [30].Benarroch-Popivker D, et al. , TRF2-mediated control of telomere DNA topology as a mechanism for chromosome-end protection, Mol. Cell 61 (2016) 274–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fouché N, et al. , The basic domain of TRF2 directs binding to DNA junctions irrespective of the presence of TTAGGG repeats, J. Biol. Chem 281 (2006) 37486–37495. [DOI] [PubMed] [Google Scholar]
- [32].Nora GJ, Buncher NA, Opresko PL, Telomeric protein TRF2 protects Holliday junctions with telomeric arms from displacement by the Werner syndrome helicase, Nucleic Acids Res 38 (2010) 3984–3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Poulet A, et al. , TRF2 promotes, remodels and protects telomeric Holliday junctions, EMBO J 28 (2009) 641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tomáška Ľ, Cesare AJ, AlTurki TM, Griffith JD, Twenty years of t-loops: a case study for the importance of collaboration in molecular biology, DNA Repair 94 (2020), 102901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Griffith JD, et al. , Mammalian telomeres end in a large duplex loop, Cell 97 (1999) 503–514. [DOI] [PubMed] [Google Scholar]
- [36].Doksani Y, Wu JY, de Lange T, Zhuang X, Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation, Cell 155 (2013) 345–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gong Y, de Lange T, A Shld1-controlled POT1a provides support for repression of ATR signaling at telomeres through RPA exclusion, Mol. Cell 40 (2010) 377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Denchi EL, de Lange T, Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1, Nature 448 (2007) 1068–1071. [DOI] [PubMed] [Google Scholar]
- [39].Churikov D, Price CM, Pot1 and cell cycle progression cooperate in telomere length regulation, Nat. Struct. Mol. Biol 15 (2008) 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sfeir A, et al. , Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication, Cell 138 (2009) 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pannunzio NR, Watanabe G, Lieber MR, Nonhomologous DNA end-joining for repair of DNA double-strand breaks, J. Biol. Chem 293 (2018) 10512–10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Truong LN, et al. , Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells, Proc. Natl. Acad. Sci. USA 110 (2013) 7720–7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ly D Van, et al. , Telomere loop dynamics in chromosome end protection, Mol. Cell 71 (510–525) (2018), e6. [DOI] [PubMed] [Google Scholar]
- [44].Takai H, Smogorzewska A, de Lange T, DNA damage foci at dysfunctional telomeres, Curr. Biol 13 (2003) 1549–1556. [DOI] [PubMed] [Google Scholar]
- [45].Okamoto K, et al. , A two-step mechanism for TRF2-mediated chromosome end protection, Nature 494 (2013) 502–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ribes-Zamora A, Indiviglio SM, Mihalek I, Williams CL, Bertuch AA, TRF2 interaction with Ku heterotetramerization interface gives insight into c-NHEJ prevention at human telomeres, Cell Rep 5 (2013) 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sfeir A, de Lange T, Removal of shelterin reveals the telomere end-protection problem, Science 336 (2012) 593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Mateos-Gomez PA, et al. , Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination, Nature 518 (2015) 254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ceccaldi R, et al. , Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair, Nature 518 (2015) 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Rai R, et al. , The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres, EMBO J 29 (2010) 2598–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Liddiard K, Grimstead JW, Cleal K, Evans A, Baird DM, Tracking telomere fusions through crisis reveals conflict between DNA transcription and the DNA damage response, NAR Cancer (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Takata M, et al. , Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells, EMBO J 17 (1998) 5497–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Scully R, Panday A, Elango R, Willis NA, DNA double-strand break repair-pathway choice in somatic mammalian cells, Nat. Rev. Mol. Cell Biol 20 (2019) 698–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sobinoff AP, et al. , BLM and SLX4 play opposing roles in recombination-dependent replication at human telomeres, EMBO J 36 (2017) 2907–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Celli GB, Denchi EL, de Lange T, Ku70 stimulates fusion of dysfunctional telomeres yet protects chromosome ends from homologous recombination, Nat. Cell Biol 8 (2006) 885–890. [DOI] [PubMed] [Google Scholar]
- [56].Denchi EL, de Lange T, Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1, Nature 448 (2007) 1068–1071. [DOI] [PubMed] [Google Scholar]
- [57].Wu L, et al. , Pot1 deficiency initiates DNA damage checkpoint activation and aberrant homologous recombination at telomeres, Cell 126 (2006) 49–62. [DOI] [PubMed] [Google Scholar]
- [58].Wang RC, Smogorzewska A, de Lange T, Homologous recombination generates T-loop-sized deletions at human telomeres, Cell 119 (2004) 355–368. [DOI] [PubMed] [Google Scholar]
- [59].Zhu X-D, et al. , ERCC1/XPF removes the 3’ overhang from uncapped telomeres and represses formation of telomeric DNA-containing double minute chromosomes, Mol. Cell 12 (2003) 1489–1498. [DOI] [PubMed] [Google Scholar]
- [60].Sarek G, Vannier J-B, Panier S, Petrini JHJ, Boulton SJ, TRF2 recruits RTEL1 to telomeres in S phase to promote t-loop unwinding, Mol. Cell 57 (2015) 622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Londoño-Vallejo JA, Der-Sarkissian H, Cazes L, Bacchetti S, Reddel RR, Alternative lengthening of telomeres is characterized by high rates of telomeric exchange, Cancer Res 64 (2004) 2324–2327. [DOI] [PubMed] [Google Scholar]
- [62].Lex K, et al. , Telomere shortening produces an inflammatory environment that increases tumor incidence in zebrafish, Proc. Natl. Acad. Sci. USA 117 (2020) 15066–15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Artandi SE, et al. , Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice, Nature 406 (2000) 641–645. [DOI] [PubMed] [Google Scholar]
- [64].Chin L, et al. , p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis, Cell 97 (1999) 527–538. [DOI] [PubMed] [Google Scholar]
- [65].Blasco MA, et al. , Telomere shortening and tumor formation by mouse cells lacking telomerase RNA, Cell 91 (1997) 25–34. [DOI] [PubMed] [Google Scholar]
- [66].Revy P, Kannengiesser C, Bertuch AA, Genetics of human telomere biology disorders, Nat. Rev. Genet 24 (2023) 86–108. [DOI] [PubMed] [Google Scholar]
- [67].Armanios M, Blackburn EH, The telomere syndromes, Nat. Rev. Genet 13 (2012) 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Armanios M, The role of telomeres in human disease, Annu. Rev. Genom. Hum. Genet 23 (2022) 363–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Hartmann D, et al. , Telomerase gene mutations are associated with cirrhosis formation, Hepatology 53 (2011) 1608–1617. [DOI] [PubMed] [Google Scholar]
- [70].O’Sullivan JN, et al. , Chromosomal instability in ulcerative colitis is related to telomere shortening, Nat. Genet 32 (2002) 280–284. [DOI] [PubMed] [Google Scholar]
- [71].Jonassaint NL, Guo N, Califano JA, Montgomery EA, Armanios M, The gastrointestinal manifestations of telomere-mediated disease, Aging Cell 12 (2013) 319–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Kinouchi Y, et al. , Telomere shortening in the colonic mucosa of patients with ulcerative colitis, J. Gastroenterol 33 (1998) 343–348. [DOI] [PubMed] [Google Scholar]
- [73].Harapas CR, et al. , Organellar homeostasis and innate immune sensing, Nat. Rev. Immunol 22 (2022) 535–549. [DOI] [PubMed] [Google Scholar]
- [74].Tomalka JA, Suthar MS, Diamond MS, Sekaly RP, Innate antiviral immunity: how prior exposures can guide future responses, Trends Immunol 43 (2022) 696–705. [DOI] [PubMed] [Google Scholar]
- [75].Coquel F, et al. , SAMHD1 acts at stalled replication forks to prevent interferon induction, Nature 557 (2018) 57–61. [DOI] [PubMed] [Google Scholar]
- [76].Rongvaux A, et al. , Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA, Cell 159 (2014) 1563–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].West AP, et al. , Mitochondrial DNA stress primes the antiviral innate immune response, Nature 520 (2015) 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Brégnard C, et al. , Upregulated LINE-1 activity in the fanconi anemia cancer susceptibility syndrome leads to spontaneous pro-inflammatory cytokine production, EBioMedicine 8 (2016) 184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Miller KN, et al. , Cytoplasmic DNA: sources, sensing, and role in aging and disease, Cell 184 (2021) 5506–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Taffoni C, et al. , Nucleic acid immunity and DNA damage response: new friends and old foes, Front. Immunol 12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Capper R, et al. , The nature of telomere fusion and a definition of the critical telomere length in human cells, Genes Dev 21 (2007) 2495–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Liddiard K, et al. , Sister chromatid telomere fusions, but not NHEJ-mediated inter-chromosomal telomere fusions, occur independently of DNA ligases 3 and 4, Genome Res 26 (2016) 588–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Liddiard K, et al. , DNA Ligase 1 is an essential mediator of sister chromatid telomere fusions in G2 cell cycle phase, Nucleic Acids Res 47 (2019) 2402–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Celli GB, de Lange T, DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion, Nat. Cell Biol 7 (2005) 712–718. [DOI] [PubMed] [Google Scholar]
- [85].Cleal K, Baird DM, Catastrophic endgames: emerging mechanisms of telomere-driven genomic instability, Trends Genet. TIG 36 (2020) 347–359. [DOI] [PubMed] [Google Scholar]
- [86].McClintock B, The behavior in successive nuclear divisions of a chromosome broken at meiosis, Proc. Natl. Acad. Sci. USA 25 (1939) 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Hoffelder DR, et al. , Resolution of anaphase bridges in cancer cells, Chromosoma 112 (2004) 389–397. [DOI] [PubMed] [Google Scholar]
- [88].Terradas M, Martín M, Tusell L, Genescà A, Genetic activities in micronuclei: Is the DNA entrapped in micronuclei lost for the cell? Mutat. Res 705 (2010) 60–67. [DOI] [PubMed] [Google Scholar]
- [89].Pampalona J, Soler D, Genescà A, Tusell L, Telomere dysfunction and chromosome structure modulate the contribution of individual chromosomes in abnormal nuclear morphologies, Mutat. Res. Mol. Mech. Mutagen 683 (2010) 16–22. [DOI] [PubMed] [Google Scholar]
- [90].Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T, Chromothripsis and kataegis induced by telomere crisis, Cell 163 (2015) 1641–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Umbreit NT, et al. , Mechanisms generating cancer genome complexity from a single cell division error, eaba0712, Science 368 (2020). eaba0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Sun L, Wu J, Du F, Chen X, Chen ZJ, Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway, Science 339 (2013) 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW, Catastrophic nuclear envelope collapse in cancer cell micronuclei, Cell 154 (2013) 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Mackenzie KJ, et al. , cGAS surveillance of micronuclei links genome instability to innate immunity, Nature 548 (2017) 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Liu S, et al. , Nuclear envelope assembly defects link mitotic errors to chromothripsis, Nature 561 (2018) 551–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Lahaye X, et al. , NONO detects the nuclear HIV capsid to promote cGAS-mediated innate immune activation, e22, Cell 175 (2018) 488–501. e22. [DOI] [PubMed] [Google Scholar]
- [97].Liu H, et al. , Nuclear cGAS suppresses DNA repair and promotes tumorigenesis, Nature 563 (2018) 131–136. [DOI] [PubMed] [Google Scholar]
- [98].Gentili M, et al. , The N-terminal domain of cGAS determines preferential association with centromeric DNA and innate immune activation in the nucleus, e13, Cell Rep 26 (2019) 2377–2393. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Jiang H, et al. , Chromatin-bound cGAS is an inhibitor of DNA repair and hence accelerates genome destabilization and cell death, EMBO J 38 (2019), e102718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Chen H, et al. , cGAS suppresses genomic instability as a decelerator of replication forks, Sci. Adv (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Cui S, et al. , Nuclear cGAS functions non-canonically to enhance antiviral immunity via recruiting methyltransferase Prmt5, Cell Rep 33 (2020), 108490. [DOI] [PubMed] [Google Scholar]
- [102].Pathare GR, et al. , Structural mechanism of cGAS inhibition by the nucleosome, Nature 587 (2020) 668–672. [DOI] [PubMed] [Google Scholar]
- [103].Chen Y-A, et al. , Extrachromosomal telomere repeat DNA is linked to ALT development via cGAS-STING DNA sensing pathway, Nat. Struct. Mol. Biol 24 (2017) 1124–1131. [DOI] [PubMed] [Google Scholar]
- [104].Groff-Vindman C, Cesare AJ, Natarajan S, Griffith JD, McEachern MJ, Recombination at long mutant telomeres produces tiny single- and double-stranded telomeric circles, Mol. Cell. Biol 25 (2005) 4406–4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Cesare AJ, Griffith JD, Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops, Mol. Cell. Biol 24 (2004) 9948–9957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Compton SA, Choi J-H, Cesare AJ, Ozgür S, Griffith JD, Xrcc3 and Nbs1 are required for the production of extrachromosomal telomeric circles in human alternative lengthening of telomere cells, Cancer Res 67 (2007) 1513–1519. [DOI] [PubMed] [Google Scholar]
- [107].Pickett HA, Cesare AJ, Johnston RL, Neumann AA, Reddel RR, Control of telomere length by a trimming mechanism that involves generation of t-circles, EMBO J 28 (2009) 799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Pickett HA, Henson JD, Au AYM, Neumann AA, Reddel RR, Normal mammalian cells negatively regulate telomere length by telomere trimming, Hum. Mol. Genet 20 (2011) 4684–4692. [DOI] [PubMed] [Google Scholar]
- [109].Deng Z, Dheekollu J, Broccoli D, Dutta A, Lieberman PM, The origin recognition complex localizes to telomere repeats and prevents telomere-circle formation, Curr. Biol. CB 17 (2007) 1989–1995. [DOI] [PubMed] [Google Scholar]
- [110].Li JSZ, et al. , TZAP: a telomere-associated protein involved in telomere length control, Science 355 (2017) 638–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Moreno SP, et al. , TZAP overexpression induces telomere dysfunction and ALT-like activity in ATRX/DAXX-deficient cells, iScience 26 (2023), 106405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Rivera T, Haggblom C, Cosconati S, Karlseder J, A balance between elongation and trimming regulates telomere stability in stem cells, Nat. Struct. Mol. Biol 24 (2017) 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Henson JD, et al. , DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity, Nat. Biotechnol 27 (2009) 1181–1185. [DOI] [PubMed] [Google Scholar]
- [114].Silva B, et al. , FANCM limits ALT activity by restricting telomeric replication stress induced by deregulated BLM and R-loops, Nat. Commun 10 (2019) 2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Lu R, et al. , The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT), Nat. Commun 10 (2019) 2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Loe TK, et al. , Telomere length heterogeneity in ALT cells is maintained by PML-dependent localization of the BTR complex to telomeres, Genes Dev 34 (2020) 650–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Zhang J-M, Yadav T, Ouyang J, Lan L, Zou L, Alternative lengthening of telomeres through two distinct break-induced replication pathways, Cell Rep 26 (955–968) (2019), e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Andreeva L, et al. , cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders, Nature 549 (2017) 394–398. [DOI] [PubMed] [Google Scholar]
- [119].Li X, et al. , Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization, Immunity 39 (2013) 1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Du M, Chen ZJ, DNA-induced liquid phase condensation of cGAS activates innate immune signaling, Science 361 (2018) 704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Ishikawa H, Barber GN, STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling, Nature 455 (2008) 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Ishikawa H, Ma Z, Barber GN, STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity, Nature 461 (2009) 788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Kang J, et al. , Post-translational modifications of STING: a potential therapeutic target, Front. Immunol 13 (2022), 888147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Schneider WM, Chevillotte MD, Rice CM, Interferon-stimulated genes: a complex web of host defenses, Annu. Rev. Immunol 32 (2014) 513–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Abe T, Barber GN, Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1, J. Virol 88 (2014) 5328–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Fitzgerald KA, et al. , IKKε and TBK1 are essential components of the IRF3 signaling pathway, Nat. Immunol 4 (2003) 491–496. [DOI] [PubMed] [Google Scholar]
- [127].Fang R, et al. , NEMO-IKKβ are essential for IRF3 and NF-κB activation in the cGAS-STING pathway, J. Immunol 199 (2017) 3222–3233. [DOI] [PubMed] [Google Scholar]
- [128].Crow YJ, Manel N, Aicardi-Goutières syndrome and the type I interferonopathies, Nat. Rev. Immunol 15 (2015) 429–440. [DOI] [PubMed] [Google Scholar]
- [129].Yasutomo K, et al. , Mutation of DNASE1 in people with systemic lupus erythematosus, Nat. Genet 28 (2001) 313–314. [DOI] [PubMed] [Google Scholar]
- [130].Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E, Lingner J, Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends, Science 318 (2007) 798–801. [DOI] [PubMed] [Google Scholar]
- [131].Bah A, Wischnewski H, Shchepachev V, Azzalin CM, The telomeric transcriptome of Schizosaccharomyces pombe, Nucleic Acids Res 40 (2012) 2995–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Greenwood J, Cooper JP, Non-coding telomeric and subtelomeric transcripts are differentially regulated by telomeric and heterochromatin assembly factors in fission yeast, Nucleic Acids Res 40 (2012) 2956–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Luke B, et al. , The Rat1p 5′ to 3′ exonuclease degrades telomeric repeat-containing RNA and promotes telomere elongation in saccharomyces cerevisiae, Mol. Cell 32 (2008) 465–477. [DOI] [PubMed] [Google Scholar]
- [134].Schoeftner S, Blasco MA, Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II, Nat. Cell Biol 10 (2008) 228–236. [DOI] [PubMed] [Google Scholar]
- [135].Diman A, Decottignies A, Genomic origin and nuclear localization of TERRA telomeric repeat-containing RNA: from darkness to dawn, FEBS J 285 (2018) 1389–1398. [DOI] [PubMed] [Google Scholar]
- [136].Porro A, Feuerhahn S, Reichenbach P, Lingner J, Molecular dissection of telomeric repeat-containing RNA biogenesis unveils the presence of distinct and multiple regulatory pathways, Mol. Cell. Biol 30 (2010) 4808–4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Nergadze SG, et al. , CpG-island promoters drive transcription of human telomeres, RNA N. Y. N 15 (2009) 2186–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Porro A, et al. , Functional characterization of the TERRA transcriptome at damaged telomeres, Nat. Commun 5 (2014) 5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Wang Z, Lieberman PM, The crosstalk of telomere dysfunction and inflammation through cell-free TERRA containing exosomes, RNA Biol 13 (2016) 690–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Nassour J, et al. , Telomere-to-mitochondria signalling by ZBP1 mediates replicative crisis, Nature 614 (2023) 767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Feuerhahn S, Iglesias N, Panza A, Porro A, Lingner J, TERRA biogenesis, turnover and implications for function, FEBS Lett 584 (2010) 3812–3818. [DOI] [PubMed] [Google Scholar]
- [142].Lee YW, Arora R, Wischnewski H, Azzalin CM, TRF1 participates in chromosome end protection by averting TRF2-dependent telomeric R loops, Nat. Struct. Mol. Biol 25 (2018) 147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Deng Z, Norseen J, Wiedmer A, Riethman H, Lieberman PM, TERRA RNA binding to TRF2 facilitates heterochromatin formation and ORC recruitment at telomeres, Mol. Cell 35 (2009) 403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Balk B, et al. , Telomeric RNA-DNA hybrids affect telomere-length dynamics and senescence, Nat. Struct. Mol. Biol 20 (2013) 1199–1205. [DOI] [PubMed] [Google Scholar]
- [145].Pfeiffer V, Crittin J, Grolimund L, Lingner J, The THO complex component Thp2 counteracts telomeric R-loops and telomere shortening, EMBO J 32 (2013) 2861–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Wang Z, et al. , Telomeric repeat-containing RNA (TERRA) constitutes a nucleoprotein component of extracellular inflammatory exosomes, Proc. Natl. Acad. Sci. USA 112 (2015) E6293–E6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Beishline K, et al. , CTCF driven TERRA transcription facilitates completion of telomere DNA replication, Nat. Commun 8 (2017) 2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Arnoult N, Van Beneden A, Decottignies A, Telomere length regulates TERRA levels through increased trimethylation of telomeric H3K9 and HP1α, Nat. Struct. Mol. Biol 19 (2012) 948–956. [DOI] [PubMed] [Google Scholar]
- [149].Cusanelli E, Romero CAP, Chartrand P, Telomeric noncoding RNA TERRA is induced by telomere shortening to nucleate telomerase molecules at short telomeres, Mol. Cell 51 (2013) 780–791. [DOI] [PubMed] [Google Scholar]
- [150].Farnung BO, Brun CM, Arora R, Lorenzi LE, Azzalin CM, Telomerase efficiently elongates highly transcribing telomeres in human cancer cells, PLoS One 7 (2012), e35714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Lalonde M, Chartrand P, TERRA, a multifaceted regulator of telomerase activity at telomeres, J. Mol. Biol 432 (2020) 4232–4243. [DOI] [PubMed] [Google Scholar]
- [152].Moravec M, et al. , TERRA promotes telomerase-mediated telomere elongation in Schizosaccharomyces pombe, EMBO Rep 17 (2016) 999–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Petti E, et al. , SFPQ and NONO suppress RNA:DNA-hybrid-related telomere instability, Nat. Commun 10 (2019) 1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Arora R, et al. , RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells, Nat. Commun 5 (2014) 5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Silva B, Arora R, Bione S, Azzalin CM, TERRA transcription destabilizes telomere integrity to initiate break-induced replication in human ALT cells, Nat. Commun 12 (2021) 3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Atianand MK, Caffrey DR, Fitzgerald KA, Immunobiology of Long Noncoding RNAs, Annu. Rev. Immunol 35 (2017) 177–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Flores-Concha M, Oñate ÁA, Long non-coding RNAs in the regulation of the immune response and trained immunity, Front. Genet 11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Takaoka A, et al. , DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response, Nature 448 (2007) 501–505. [DOI] [PubMed] [Google Scholar]
- [159].Zhang T, et al. , Influenza Virus Z-RNAs induce ZBP1-mediated necroptosis, Cell 180 (1115–1129) (2020), e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Thapa RJ, et al. , DAI senses influenza A virus genomic RNA and activates RIPK3-dependent cell death, Cell Host Microbe 20 (2016) 674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Sridharan H, et al. , Murine cytomegalovirus IE3–dependent transcription is required for DAI/ZBP1–mediated necroptosis, EMBO Rep 18 (2017) 1429–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Maelfait J, et al. , Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis, EMBO J 36 (2017) 2529–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Jiao H, et al. , Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation, Nature 580 (2020) 391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Wang R, et al. , Gut stem cell necroptosis by genome instability triggers bowel inflammation, Nature 580 (2020) 386–390. [DOI] [PubMed] [Google Scholar]
- [165].Kesavardhana S, et al. , The Zα2 domain of ZBP1 is a molecular switch regulating influenza-induced PANoptosis and perinatal lethality during development, J. Biol. Chem 295 (2020) 8325–8330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Karki R, et al. , ADAR1 restricts ZBP1-mediated immune response and PANoptosis to promote tumorigenesis, Cell Rep 37 (2021), 109858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Ha SC, et al. , The crystal structure of the second Z-DNA binding domain of human DAI (ZBP1) in complex with Z-DNA reveals an unusual binding mode to Z-DNA, Proc. Natl. Acad. Sci. U.S.A 105 (2008) 20671–20676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Deigendesch N, Koch-Nolte F, Rothenburg S, ZBP1 subcellular localization and association with stress granules is controlled by its Z-DNA binding domains, Nucleic Acids Res 34 (2006) 5007–5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Schwartz T, Behlke J, Lowenhaupt K, Heinemann U, Rich A, Structure of the DLM-1-Z-DNA complex reveals a conserved family of Z-DNA-binding proteins, Nat. Struct. Biol 8 (2001) 761–765. [DOI] [PubMed] [Google Scholar]
- [170].Kaiser WJ, Upton JW, Mocarski ES, Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors, J. Immunol 181 (2008) 6427–6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Rebsamen M, et al. , DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB, EMBO Rep 10 (2009) 916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Balachandran S, Mocarski ES, Viral Z-RNA triggers ZBP1-dependent cell death, Curr. Opin. Virol 51 (2021) 134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Nogusa S, et al. , RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus, Cell Host Microbe 20 (2016) 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Guo H, et al. , Species-independent contribution of ZBP1/DAI/DLM-1-triggered necroptosis in host defense against HSV1, Cell Death Dis 9 (2018) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Li S, et al. , SARS-CoV-2 Z-RNA activates the ZBP1-RIPK3 pathway to promote virus-induced inflammatory responses, Cell Res 33 (2023) 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Wittig B, Dorbic T, Rich A, Transcription is associated with Z-DNA formation in metabolically active permeabilized mammalian cell nuclei, Proc. Natl. Acad. Sci. USA 88 (1991) 2259–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Rich A, Zhang S, Timeline: Z-DNA: the long road to biological function, Nat. Rev. Genet 4 (2003) 566–572. [DOI] [PubMed] [Google Scholar]
- [178].Zheng M, Kanneganti T-D, The regulation of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis), Immunol. Rev 297 (2020) 26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Hubbard NW, et al. , ADAR1 mutation causes ZBP1-dependent immunopathology, Nature 607 (2022) 769–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Zhang T, et al. , ADAR1 masks the cancer immunotherapeutic promise of ZBP1-driven necroptosis, Nature 606 (2022) 594–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Jiao H, et al. , ADAR1 averts fatal type I interferon induction by ZBP1, Nature 607 (2022) 776–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].de Reuver R, et al. , ADAR1 prevents autoinflammation by suppressing spontaneous ZBP1 activation, Nature 607 (2022) 784–789. [DOI] [PubMed] [Google Scholar]
- [183].Herbert A, Mendelian disease caused by variants affecting recognition of Z-DNA and Z-RNA by the Zα domain of the double-stranded RNA editing enzyme ADAR, Eur. J. Hum. Genet 28 (2020) 114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Herbert A, et al. , A Z-DNA binding domain present in the human editing enzyme, double-stranded RNA adenosine deaminase, Proc. Natl. Acad. Sci. USA 94 (1997) 8421–8426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Samuel CE, Adenosine deaminase acting on RNA (ADAR1), a suppressor of double-stranded RNA-triggered innate immune responses, J. Biol. Chem 294 (2019) 1710–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Kuriakose T, Kanneganti T-D, ZBP1: Innate sensor regulating cell death and inflammation, Trends Immunol 39 (2018) 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Rothenburg S, Schwartz T, Koch-Nolte F, Haag F, Complex regulation of the human gene for the Z-DNA binding protein DLM-1, Nucleic Acids Res 30 (2002) 993–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Ponnusamy K, et al. , The innate sensor ZBP1-IRF3 axis regulates cell proliferation in multiple myeloma, Haematologica 107 (2021) 721–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Xu L-G, et al. , VISA is an adapter protein required for virus-triggered IFN-beta signaling, Mol. Cell 19 (2005) 727–740. [DOI] [PubMed] [Google Scholar]
- [190].Meylan E, et al. , Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus, Nature 437 (2005) 1167–1172. [DOI] [PubMed] [Google Scholar]
- [191].Kawai T, et al. , IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction, Nat. Immunol 6 (2005) 981–988. [DOI] [PubMed] [Google Scholar]
- [192].Seth RB, Sun L, Ea C-K, Chen ZJ, Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3, Cell 122 (2005) 669–682. [DOI] [PubMed] [Google Scholar]
- [193].Peisley A, Wu B, Yao H, Walz T, Hur S, RIG-I forms signaling-competent filaments in an ATP-dependent, ubiquitin-independent manner, Mol. Cell 51 (2013) 573–583. [DOI] [PubMed] [Google Scholar]
- [194].Peisley A, et al. , Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition, Proc. Natl. Acad. Sci. USA 108 (2011) 21010–21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Cadena C, Hur S, Filament-like assemblies of intracellular nucleic acid sensors: commonalities and differences, Mol. Cell 76 (2019) 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Horner SM, Liu HM, Park HS, Briley J, Gale M, Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus, Proc. Natl. Acad. Sci 108 (2011) 14590–14595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Dixit E, et al. , Peroxisomes are signaling platforms for antiviral innate immunity, Cell 141 (2010) 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Seth RB, Sun L, Ea C-K, Chen ZJ, Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3, Cell 122 (2005) 669–682. [DOI] [PubMed] [Google Scholar]
- [199].Gack MU, et al. , TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity, Nature 446 (2007) 916–920. [DOI] [PubMed] [Google Scholar]
- [200].Oshiumi H, et al. , The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection, Cell Host Microbe 8 (2010) 496–509. [DOI] [PubMed] [Google Scholar]
- [201].Oshiumi H, Recent advances and contradictions in the study of the individual roles of ubiquitin ligases that regulate RIG-I-Like receptor-mediated antiviral innate immune responses, Front. Immunol 11 (2020) 1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Jiang X, et al. , Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response, Immunity 36 (2012) 959–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Rehwinkel J, Gack MU, RIG-I-like receptors: their regulation and roles in RNA sensing, Nat. Rev. Immunol 20 (2020) 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Hou F, et al. , MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response, Cell 146 (2011) 448–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Peisley A, Wu B, Xu H, Chen ZJ, Hur S, Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I, Nature 509 (2014) 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Refolo G, Vescovo T, Piacentini M, Fimia GM, Ciccosanti F, Mitochondrial interactome: a focus on antiviral signaling pathways, Front. Cell Dev. Biol 8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Seth RB, Sun L, Ea C-K, Chen ZJ, Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3, Cell 122 (2005) 669–682. [DOI] [PubMed] [Google Scholar]
- [208].Martadinata H, Phan AT, Structure of human telomeric RNA (TERRA): stacking of two G-quadruplex blocks in K(+) solution, Biochemistry 52 (2013) 2176–2183. [DOI] [PubMed] [Google Scholar]
- [209].Collie GW, et al. , Electrospray mass spectrometry of telomeric RNA (TERRA) reveals the formation of stable multimeric G-quadruplex structures, J. Am. Chem. Soc 132 (2010) 9328–9334. [DOI] [PubMed] [Google Scholar]
- [210].Agarwala P, Pandey S, Maiti S, The tale of RNA G-quadruplex, Org. Biomol. Chem 13 (2015) 5570–5585. [DOI] [PubMed] [Google Scholar]
- [211].Mei Y, et al. , TERRA G-quadruplex RNA interaction with TRF2 GAR domain is required for telomere integrity, Sci. Rep 11 (2021) 3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Missiroli S, et al. , Mitochondria-associated membranes (MAMs) and inflammation, Cell Death Dis 9 (2018) 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Franceschi C, et al. , Inflamm-aging. An evolutionary perspective on immunosenescence, Ann. N. Y. Acad. Sci 908 (2000) 244–254. [DOI] [PubMed] [Google Scholar]
- [214].Jurk D, et al. , Chronic inflammation induces telomere dysfunction and accelerates ageing in mice, Nat. Commun 2 (2014) 4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Zhang J, et al. , Ageing and the telomere connection: An intimate relationship with inflammation, Ageing Res. Rev 25 (2016) 55–69. [DOI] [PubMed] [Google Scholar]
- [216].Kordinas V, Ioannidis A, Chatzipanagiotou S, The telomere/telomerase system in chronic inflammatory diseases. Cause or Effect? Genes 7 (2016) 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Nassour J, Schmidt TT, Karlseder J, Telomeres and cancer: resolving the paradox, Annu. Rev. Cancer Biol 5 (2021) 59–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Wright WE, Shay JW, The two-stage mechanism controlling cellular senescence and immortalization, Exp. Gerontol 27 (1992) 383–389. [DOI] [PubMed] [Google Scholar]
- [219].Hayflick L, The limited in vitro lifetime of human diploid cell strains, Exp. Cell Res 37 (1965) 614–636. [DOI] [PubMed] [Google Scholar]
- [220].Harley CB, Vaziri H, Counter CM, Allsopp RC, The telomere hypothesis of cellular aging, Exp. Gerontol 27 (1992) 375–382. [DOI] [PubMed] [Google Scholar]
- [221].Counter CM, et al. , Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity, EMBO J 11 (1992) 1921–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].d’Adda di Fagagna F, et al. , A DNA damage checkpoint response in telomere-initiated senescence, Nature 426 (2003) 194–198. [DOI] [PubMed] [Google Scholar]
- [223].Herbig U, Jobling WA, Chen BPC, Chen DJ, Sedivy JM, Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21CIP1, but Not p16INK4a, Mol. Cell 14 (2004) 501–513. [DOI] [PubMed] [Google Scholar]
- [224].Kaul Z, Cesare AJ, Huschtscha LI, Neumann AA, Reddel RR, Five dysfunctional telomeres predict onset of senescence in human cells, EMBO Rep 13 (2011) 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Hemann MT, Strong MA, Hao LY, Greider CW, The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability, Cell 107 (2001) 67–77. [DOI] [PubMed] [Google Scholar]
- [226].Karlseder J, Smogorzewska A, de Lange T, Senescence induced by altered telomere state, not telomere loss, Science 295 (2002) 2446–2449. [DOI] [PubMed] [Google Scholar]
- [227].Cesare AJ, Hayashi MT, Crabbe L, Karlseder J, The telomere deprotection response is functionally distinct from the genomic DNA damage response, Mol. Cell 51 (2013) 141–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Ivanov A, et al. , Lysosome-mediated processing of chromatin in senescence, J. Cell Biol 202 (2013) 129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Dou Z, et al. , Cytoplasmic chromatin triggers inflammation in senescence and cancer, Nature 550 (2017) 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Takahashi A, et al. , Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells, Nat. Commun 9 (2018) 1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Glück S, et al. , Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence, Nat. Cell Biol 19 (2017) 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Yang H, Wang H, Ren J, Chen Q, Chen ZJ, cGAS is essential for cellular senescence, Proc. Natl. Acad. Sci 114 (2017) E4612–E4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Fumagalli M, et al. , Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation, Nat. Cell Biol 14 (2012) 355–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM, Cellular senescence in aging primates, Science 311 (2006) 1257. [DOI] [PubMed] [Google Scholar]
- [235].Victorelli S, et al. , Senescent human melanocytes drive skin ageing via paracrine telomere dysfunction, EMBO J 38 (2019), e101982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Martínez-Zamudio RI, et al. , Senescence-associated β-galactosidase reveals the abundance of senescent CD8+ T cells in aging humans, Aging Cell 20 (2021), e13344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Farr JN, et al. , Identification of Senescent Cells in the Bone Microenvironment. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res 31 (2016) 1920–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Birch J, et al. , DNA damage response at telomeres contributes to lung aging and chronic obstructive pulmonary disease, Am. J. Physiol. Lung Cell. Mol. Physiol 309 (2015) L1124–L1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Anderson R, et al. , Length-independent telomere damage drives post-mitotic cardiomyocyte senescence, EMBO J 38 (2019), e100492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Ogrodnik M, et al. , Obesity-induced cellular senescence drives anxiety and impairs neurogenesis, Cell Metab 29 (2019) 1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Jones DL, Rando TA, Emerging models and paradigms for stem cell ageing, Nat. Cell Biol 13 (2011) 506–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Rossi DJ, Jamieson CHM, Weissman IL, Stems cells and the pathways to aging and cancer, Cell 132 (2008) 681–696. [DOI] [PubMed] [Google Scholar]
- [243].Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL, Cellular senescence and the senescent secretory phenotype: therapeutic opportunities, J. Clin. Investig 123 (2013) 966–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Xu M, et al. , Senolytics improve physical function and increase lifespan in old age, Nat. Med 24 (2018) 1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Baker DJ, et al. , Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders, Nature 479 (2011) 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Baker DJ, et al. , Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan, Nature 530 (2016) 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Cesare AJ, Karlseder J, A three-state model of telomere control over human proliferative boundaries, Curr. Opin. Cell Biol 24 (2012) 731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Nassour J, et al. , Autophagic cell death restricts chromosomal instability during replicative crisis, Nature 565 (2019) 659–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM, The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of, Cell (1990) p53. [DOI] [PubMed] [Google Scholar]
- [250].Davoli T, de Lange T, Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells, Cancer Cell 21 (2012) 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Denton D, et al. , Autophagy, not apoptosis, is essential for midgut cell death in drosophila, Curr. Biol 19 (2009) 1741–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Yin VP, Thummel CS, A balance between the diap1 death inhibitor and reaper and hid death inducers controls steroid-triggered cell death in Drosophila, Proc. Natl. Acad. Sci. USA 101 (2004) 8022–8027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Arakawa S, et al. , Role of Atg5-dependent cell death in the embryonic development of Bax/Bak double-knockout mice, Cell Death Differ 24 (2017) 1598–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Mizushima N, Levine B, Autophagy in mammalian development and differentiation, Nat. Cell Biol 12 (2010) 823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Xie C, et al. , Neuroprotection by selective neuronal deletion of Atg7 in neonatal brain injury, Autophagy 12 (2016) 410–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Matsui Y, et al. , Distinct roles of autophagy in the heart during ischemia and reperfusion, Circ. Res 100 (2007) 914–922. [DOI] [PubMed] [Google Scholar]
- [257].Zheng Y, Liu J, Li X, Xu L, Xu Y, RNA interference-mediated downregulation of Beclin1 attenuates cerebral ischemic injury in rats, Acta Pharmacol. Sin 30 (2009) 919–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Koike M, et al. , Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury, Am. J. Pathol 172 (2008) 454–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Fulda S, Kögel D, Cell death by autophagy: emerging molecular mechanisms and implications for cancer therapy, Oncogene 34 (2015) 5105–5113. [DOI] [PubMed] [Google Scholar]
- [260].Bialik S, Dasari SK, Kimchi A, Autophagy-dependent cell death – where, how and why a cell eats itself to death, jcs215152, J. Cell Sci 131 (2018). jcs215152. [DOI] [PubMed] [Google Scholar]
- [261].Jones RE, et al. , Escape from telomere-driven crisis is DNA ligase III dependent, Cell Rep 8 (2014) 1063–1076. [DOI] [PubMed] [Google Scholar]
- [262].Letsolo BT, Rowson J, Baird DM, Fusion of short telomeres in human cells is characterized by extensive deletion and microhomology, and can result in complex rearrangements, Nucleic Acids Res 38 (2010) 1841–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Cleal K, Jones RE, Grimstead JW, Hendrickson EA, Baird DM, Chromothripsis during telomere crisis is independent of NHEJ, and consistent with a replicative origin, Genome Res 29 (2019) 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Li Y, et al. , Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia, Nature 508 (2014) 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Hadi K, et al. , Distinct classes of complex structural variation uncovered across thousands of cancer genome graphs, Cell 183 (197–210) (2020), e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Chin K, et al. , In situ analyses of genome instability in breast cancer, Nat. Genet 36 (2004) 984–988. [DOI] [PubMed] [Google Scholar]
- [267].Roger L, et al. , Extensive telomere erosion in the initiation of colorectal adenomas and its association with chromosomal instability, JNCI J. Natl. Cancer Inst 105 (2013) 1202–1211. [DOI] [PubMed] [Google Scholar]
- [268].Lin TT, et al. , Telomere dysfunction and fusion during the progression of chronic lymphocytic leukemia: evidence for a telomere crisis, Blood 116 (2010) 1899–1907. [DOI] [PubMed] [Google Scholar]
- [269].Djojosubroto MW, Choi YS, Lee H-W, Rudolph L, Telomeres and telomerase in aging, regeneration and cancer, Mol. Cells 15 (2003) 164–175. [PubMed] [Google Scholar]
- [270].Demanelis K, et al. , Determinants of telomere length across human tissues, eaaz6876, Science 369 (2020). eaaz6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Daniali L, et al. , Telomeres shorten at equivalent rates in somatic tissues of adults, Nat. Commun 4 (2013) 1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].Sabharwal S, et al. , Telomere length dynamics in early life: the blood-and-muscle model, FASEB J 32 (2018) 529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Hewitt G, et al. , Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence, Nat. Commun 3 (2012) 708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [274].Lee HW, et al. , Essential role of mouse telomerase in highly proliferative organs, Nature 392 (1998) 569–574. [DOI] [PubMed] [Google Scholar]
- [275].Anchelin M, et al. , Premature aging in telomerase-deficient zebrafish, Dis. Model. Mech 6 (2013) 1101–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Henriques CM, Carneiro MC, Tenente IM, Jacinto A, Ferreira MG, Telomerase is required for zebrafish lifespan, PLoS Genet 9 (2013), e1003214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Carneiro MC, et al. , Short telomeres in key tissues initiate local and systemic aging in zebrafish, PLoS Genet 12 (2016), e1005798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [278].Rudolph KL, et al. , Longevity, stress response, and cancer in aging telomerase-deficient mice, Cell 96 (1999) 701–712. [DOI] [PubMed] [Google Scholar]
- [279].Chakravarti D, et al. , Telomere dysfunction activates YAP1 to drive tissue inflammation, Nat. Commun 11 (2020) 4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Rudolph KL, Chang S, Millard M, Schreiber-Agus N, DePinho RA, Inhibition of experimental liver cirrhosis in mice by telomerase gene delivery, Science 287 (2000) 1253–1258. [DOI] [PubMed] [Google Scholar]
- [281].Franceschi C, Garagnani P, Vitale G, Capri M, Salvioli S, Inflammaging and ‘Garb-aging’, Trends Endocrinol. Metab. TEM 28 (2017) 199–212. [DOI] [PubMed] [Google Scholar]
- [282].Risques RA, et al. , Ulcerative colitis is a disease of accelerated colon aging: evidence from telomere attrition and DNA damage, Gastroenterology 135 (2008) 410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].van Heek NT, et al. , Telomere shortening is nearly universal in pancreatic intraepithelial neoplasia, Am. J. Pathol 161 (2002) 1541–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Laish I, et al. , Telomere dysfunction in nonalcoholic fatty liver disease and cryptogenic cirrhosis, Cytogenet. Genome Res 150 (2016) 93–99. [DOI] [PubMed] [Google Scholar]
- [285].Birch J, et al. , Telomere dysfunction and senescence-associated pathways in bronchiectasis, Am. J. Respir. Crit. Care Med 193 (2016) 929–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Coppé J-P, Desprez P-Y, Krtolica A, Campisi J, The senescence-associated secretory phenotype: the dark side of tumor suppression, Annu. Rev. Pathol 5 (2010) 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [287].Chien Y, et al. , Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity, Genes Dev 25 (2011) 2125–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Rodier F, et al. , Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion, Nat. Cell Biol 11 (2009) 973–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Birch J, Gil J, Senescence and the SASP: many therapeutic avenues, Genes Dev 34 (2020) 1565–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [290].Hayashi MT, Cesare AJ, Rivera T, Karlseder J, Cell death during crisis is mediated by mitotic telomere deprotection, Nature 522 (2015) 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [291].Dou Z, et al. , Cytoplasmic chromatin triggers inflammation in senescence and cancer, Nature 550 (2017) 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [292].Furman D, et al. , Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states, Nat. Med 23 (2017) 174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].Furman D, et al. , Chronic inflammation in the etiology of disease across the life span, Nat. Med 25 (2019) 1822–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [294].Pereira CA, et al. , Mitochondrial DNA promotes NLRP3 Inflammasome activation and contributes to endothelial dysfunction and inflammation in type 1 diabetes, Front. Physiol 10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [295].West AP, et al. , Mitochondrial DNA stress primes the antiviral innate immune response, Nature 520 (2015) 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [296].Lei Y, et al. , Cooperative sensing of mitochondrial DNA by ZBP1 and cGAS promotes cardiotoxicity, e22, Cell 186 (2023) 3013–3032. e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [297].Pinti M, et al. , Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”, Eur. J. Immunol 44 (2014) 1552–1562. [DOI] [PubMed] [Google Scholar]
- [298].Zhang Q, et al. , Circulating mitochondrial DAMPs cause inflammatory responses to injury, Nature 464 (2010) 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [299].Caielli S, et al. , Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus, J. Exp. Med 213 (2016) 697–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.