

Abstract
The ability of regulatory factors to access their nucleosomal targets is modulated by nuclear proteins such as histone H1 and HMGN (previously named HMG-14/-17 family) that bind to nucleosomes and either stabilize or destabilize the higher-order chromatin structure. We tested whether HMGN proteins affect the interaction of histone H1 with chromatin. Using microinjection into living cells expressing H1–GFP and photobleaching techniques, we found that wild-type HMGN, but not HMGN point mutants that do not bind to nucleosomes, inhibits the binding of H1 to nucleosomes. HMGN proteins compete with H1 for nucleosome sites but do not displace statically bound H1 from chromatin. Our results provide evidence for in vivo competition among chromosomal proteins for binding sites on chromatin and suggest that the local structure of the chromatin fiber is modulated by a dynamic interplay between nucleosomal binding proteins.
INTRODUCTION
The structure and activity of the chromatin fiber are modulated by numerous nuclear proteins, including the linker histone H1 and the HMGN non-histones (the nomenclature of which has been modified recently and reviewed in Bustin, 2001a,b), proteins known to affect its compactness. Several types of studies suggest that histone H1 and HMGN proteins have opposite effects on the structure and activity of the chromatin fiber. Histone H1 stabilizes the higher-order chromatin structure (Thomas, 1999), hinders the access of transcriptional coactivators to DNA (Herrera et al., 2000; Cheung et al., 2002; Horn et al., 2002) and acts as a general repressor of transcription (Laybourn and Kadonaga, 1991; Wolffe et al., 1997). In contrast, HMGN proteins are nucleosome binding proteins that reduce the compaction of the chromatin fiber and enhance transcription from chromatin templates (Bustin, 2001b). In the chicken globin gene cluster, transcriptional activation is associated with depletion of H1 and enrichment in HMGN proteins (Postnikov et al., 1991). In SV40 minichromosomes, HMGN1 (HMG-14) alleviates both the chromatin compaction and the transcriptional inhibition induced by histone H1 (Ding et al., 1997). The chromatin binding sites of H1 partially overlap with those of HMGN (Alfonso et al., 1994), and contact between these proteins has been detected (Ring and Cole, 1979; Boulikas et al., 1980). The intranuclear organization of both H1 (Lever et al., 2000; Misteli et al., 2000) and HMGN (Phair and Misteli, 2000) proteins is dynamic, and their binding to any specific site on chromatin is transient. Taken together, these observations raise the possibility that these proteins compete for chromatin binding sites and that a dynamic interplay between them may play a role in modulating the structure of specific regions in the chromatin fiber.
To test whether in vivo HMGN proteins affect the interaction of H1 with chromatin, we microinjected HMGN1 and HMGN2 (HMG-17) into living cells and used fluorescence recovery after photobleaching (FRAP) to monitor the binding of H1 to chromatin (Misteli et al., 2000). The rate of FRAP is inversely related to the time that a molecule resides at its binding site. We reasoned that, if the injected HMGN and the fluorescently labeled H1 compete for the same binding sites, a fraction of the fluorescent H1 protein would have a shorter chromatin residence time, a change that will lead to an increase in the rate of recovery of fluorescence and could be detected in a FRAP experiment.
RESULTS AND DISCUSSION
To test whether competition between nuclear proteins for chromatin binding sites in vivo can be monitored by FRAP, we probed whether competition for identical binding sites between a fluorescently labeled protein and its non-labeled counterpart can be detected (Figure 1A). We injected purified, unlabeled histone H1 (Figure 2B) into the cytoplasm of cells stably expressing H10–GFP and compared the intranuclear mobility of the H10–GFP in the injected cells with that in control, non-injected cells using FRAP (Figure 1B). We have already demonstrated that H1–GFP is functionally indistinguishable from the endogenous H1 by several criteria (Misteli et al., 2000). In the conditions used, H10–GFP represents <5% of the total H1, and therefore the cellular amount of H1 is not significantly changed. In the control, non-injected cells, the time required to reach 40% (t40) of the pre-bleach fluorescence intensity was 14 s. In the H1-injected cells, the t40 was significantly reduced and was 6 s. Within 23 s after photobleaching, 47% of the signal was recovered in the control cells, whereas 66% of the signal was recovered in the injected cells (Table I). Thus, as a consequence of competition for the same binding sites, the exogenously introduced H1 increased the intranuclear mobility of the stably expressed H10-GFP.

Fig. 1. FRAP analysis of competition between proteins for binding sites in living cells. (A) Experimental approach. (1) A nuclear protein is injected into the cytoplasm of cells expressing a GFP-fusion protein. (2) Either fluorescent protein alone or a mixture of fluorescent and non-fluorescent proteins is microinjected into the cytoplasm. After the microinjected proteins enters the nucleus, the mobility of the fluorescent proteins (either as GFP-fusion products or chemically labeled) is analyzed by FRAP. (B–D) Quantitative analysis of FRAP experiments after bleaching cells containing either fluorescent protein alone (blue) or a mixture of fluorescent and non-fluorescent protein (red). The proteins analyzed are indicated in each panel. The values are the mean ± SD from at least eight cells of a typical experiment. Arrows on the abscissa point to the time required to reach either 40% (B) or 80% (C and D) of the pre-bleach fluorescence intensity. The percentages on the right of the curves indicate the difference in the percentage recovery between the curves within 23 s after photobleaching.

Fig. 2. Competition between H10–GFP and HMGN proteins for chromatin binding sites. (A) Colocalization of HMGN1 and HMGN2 with H10–GFP. Confocal images of living cells expressing H10–GFP and microinjected with fluorescently labeled HMGN proteins. Scale bar = 5 µm. (B) SDS–PAGE analysis of competitors microinjected into the H10–GFP expressing cells. (C–K) Quantitative analysis of FRAP experiments of cells expressing H10–GFP that either were not (blue) or were (red) injected with the proteins indicated in each panel. The values are the mean ± SD from at least eight cells of a typical experiment. Arrows on the abscissa point to the time required to reach 40% of the pre-bleach fluorescence intensity. The percentages on the right of the curves indicate the difference in the percentage recovery between the curves within 23 s after photobleaching. Note the differences between euchromatin and heterochromatin and that the controls in (E) and (H) did not affect the mobility of H10–GFP.
Table I. HMGN proteins increase the intranuclear mobility of H10–GFP.
| Experiment | t40 |
Recovery after 23 s |
|||
|---|---|---|---|---|---|
| Time (s) ± SD | t-test | % ± SD | t-test | ||
| H10–GFP | Mixed chromatin domains | 14.7 ± 4.1 | P < 0.005 | 46.7 ± 5.6 | P < 0.005 |
| H10–GFP + H1 | 5.7 ± 2.1 | 66.3 ± 8.5 | |||
| H10–GFP | Mixed chromatin domains | 13.7 ± 4.1 | P < 0.06 | 46.7 ± 5.6 | P < 0.02 |
| H10–GFP + HMGN1 | 9.3 ± 5.1 | 58.9 ± 10.1 | |||
| H10–GFP | Mixed chromatin domains | 12.8 ± 3.1 | P < 0.005 | 50.7 ± 6.1 | P < 0.005 |
| H10–GFP + HMGN2 | 6.0 ± 1.5 | 61.0 ± 3.6 | |||
| H10–GFP | Mixed chromatin domains | 12.5 ± 1.8 | P > 0.6 | 46.9 ± 5.0 | P > 0.6 |
| H10–GFP + H2B | 13.2 ± 3.8 | 45.8 ± 3.8 | |||
| H10–GFP | Mixed chromatin domains | 15.4 ± 3.0 | P > 0.2 | 46.0 ± 3.9 | P > 0.6 |
| H10–GFP + HMGN1-S20,24E | 13.6 ± 3.3 | 47.1 ± 6.5 | |||
| H10–GFP | Mixed chromatin domains | 12.3 ± 3.8 | P > 0.6 | 46.9 ± 6.5 | P > 0.8 |
| H10–GFP + HMGN1 (1–73) | 13.2 ± 3.4 | 46.4 ± 4.7 | |||
| H10–GFP | Euchromatin | 10.2 ± 3.5 | P < 0.005 | 55.9 ± 7.3 | P < 0.005 |
| H10–GFP + HMGN1 | 4.2 ± 1.4 | 69.0 ± 9.9 | |||
| H10–GFP | Euchromatin | 10.6 ± 2.7 | P < 0.005 | 52.2 ± 3.4 | P < 0.005 |
| H10–GFP + HMGN2 | 2.6 ± 0.6 | 79.0 ± 4.1 | |||
| H10–GFP | Heterochromatin | > 23 s | n.a. | 35.1 ± 5.8 | P < 0.005 |
| H10–GFP + HMGN1 | 10.5 ± 3.8 | 53.7 ± 8.2 | |||
| H10–GFP | Heterochromatin | > 23 s | n.a. | 36.3 ± 3.2 | P < 0.005 |
| H10–GFP + HMGN2 | 8.2 ± 1.8 | 55.9 ± 6.3 | |||
The time required to reach 40% (t40) of the pre-bleach fluorescence intensity was determined from each curve. The recoveries at 23 s are means of the last five images of each recovery curve (last 0.6 s). The results are from recovery curves of at least eight cells. The statistical significance of the differences of t40 and total recovery relative to controls was determined by Student’s t-test.
In a modification of this approach (Figure 1A, path 2), we microinjected into the cytoplasm of mouse embryonic fibroblasts either fluorescently labeled HMGN protein alone or the same amount of labeled HMGN mixed with an 8-fold excess of unlabeled HMGN. Fluorescence microscopy on living cells revealed that the protein entered the nucleus within 15 min (data not shown). Because the chromatin residence time of HMGN is significantly shorter than that of H10 (Lever et al., 2000; Misteli et al., 2000; Phair and Misteli, 2000), we measured the differences at t80, i.e. the time necessary to recover 80% of the pre-bleach fluorescence intensity. FRAP measurements indicate that the presence of unlabeled HMGN1 decreased the mean t80 of the labeled protein from 6.3 to 3.8 s and that the presence of unlabeled HMGN2 decreased the t80 of HMGN2 from 6.2 to 4.0 s (Figure 1C and D). For both HMGN proteins, an 8-fold increase in the amount of injected nuclear protein reproducibly increased the amount of fluorescence recovered after 23 s from ∼90 to 95% (Figure 1C and D), indicating that the excess HMGN decreased the relative amount of the HMGN that is immobile. These experiments demonstrate that FRAP can be used to assess competition between chromatin binding proteins for common binding sites in living cells.
To test whether HMGN proteins compete with H10 for shared binding sites, we injected either HMGN1 or HMGN2 into the cytoplasm of cells stably expressing H10–GFP. Confocal fluorescence microscopy of living cells revealed that the injected HMGN proteins colocalize with the stably expressed H10–GFP (Figure 2A). Because the recovery kinetics of H1 relate to the degree of chromatin compaction (Lever et al., 2000; Misteli et al., 2000), we photobleached H10–GFP in either euchromatin, heterochromatin or in regions containing both euchromatin and heterochromatin (Figure 2; Table I). We defined heterochromatin as regions that were strongly labeled by H10–GFP. These regions were also strongly stained by antibody against methylated lysine 9 of H3 (data not shown, but see Jenuwein and Allis, 2001), Hoechst and HP1 antibody (Misteli et al., 2000). Euchromatin regions were defined as weakly stained by H10–GFP and Hoechst (Misteli et al., 2000). These regions were strongly stained by antibody against acetylated lysine 9 of H3 (data not shown, but see Jenuwein and Allis, 2001). HMGN proteins affected the mobility of H10 in all chromatin regions to a significant degree (Table I), suggesting that the two proteins compete for binding sites in chromatin. In the regions containing both condensed and de-condensed chromatin, microinjection of HMGN proteins reduced the t40 of H10–GFP from 13–14 to 6–9 s and the signal recovered after 23 s increased from 46–50 to >60% (Figure 2C and D). In euchromatin, microinjection of HMGN reduced the t40 of H10 from 11–13 to 3–4 s (Figure 2F and G). In heterochromatin, where the t40 of H10 is ∼28 s (data not shown; Misteli et al., 2000), microinjection of HMGN1 or HMGN2 reduced the t40 to <10 s (Figure 2I and J). Control microinjections indicate that histone H2B, which binds to chromatin non-specifically, did not affect the mobility of H10 (Figure 2E). More significantly, an HMGN1 double point mutant in which two serines in the nucleosomal binding domain were mutated to glutamic acid (HMGN1-S20,24E), which enters the nucleus but does not bind to nucleosomes (Prymakowska-Bosak et al., 2001), did not affect the mobility of H10 (Figure 2H). Likewise, an HMGN1 deletion mutant that lacks the C-terminal region that interacts with the N-terminus of histone H3 (Ding et al., 1997; Trieschmann et al., 1998) also failed to affect the mobility of H10 (Figure 2K). Thus, the increase in H10 mobility is specific to HMGN and is contingent on the ability of HMGN to bind to nucleosomes and interact with the tail of histone H3.
Previous FRAP analysis indicated that, while most of the nuclear H1 is mobile and continuously exchanging, a small fraction is more tightly bound and is significantly less mobile (Lever et al., 2000; Misteli et al., 2000). When we performed FRAP analysis over a period of 4 min, we noticed that HMGN1 has a relatively small effect on the mobility of H10–GFP in heterochromatin (Figure 3A) and no effect on the mobility of H10–GFP in euchromatin (Figure 3B). The HMGN1 effect reflects its stronger binding to heterochromatin (Figure 3C). The similarity in the recovery plateau reached in both control and injected cells (Figure 3A and B) indicates that HMGN did not affect significantly the mobility of the H10 fraction that is more statically bound to chromatin (cf. Figure 2C). Our finding that HMGN proteins enhance the mobility of the rapidly exchanging H10 to a significantly larger extent than that of the less mobile H10 fraction suggests that HMGN proteins affect the mobility of H10 by dynamically competing for binding sites on nucleosomes rather than by displacing tightly bound H10 from its chromatin binding sites.

Fig. 3. Loss of competition between HMGN1 and H10–GFP in cells treated with compounds that affect the interaction of these proteins with chromatin. (A) FRAP analysis of the effect of HMGN1 on the mobility of H10–GFP in heterochromatin. (B) FRAP analysis of the effect of HMGN1 on the mobility of H10–GFP in euchromatin. Note that HMGN does not affect the immobile fraction of H10–GFP. (C) FRAP analysis of HMGN1 in euchromatin and heterochromatin. (D) FRAP analysis indicating that Act-D treatment decreases the residence time of HMGN proteins on chromatin. (E) FRAP analysis indicating that Act-D treatment abolishes the ability of HMGN proteins to reduce the residence time of H10–GFP on chromatin. (F) FRAP analysis indicating that DRB abolishes the ability of HMGN proteins to reduce the residence time of H10–GFP on chromatin.
To further confirm that HMGN proteins compete with H1 for binding sites on chromatin, we performed FRAP analysis on cells treated with either 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB), a kinase inhibitor that decreases the mobility of H1 (Lever et al., 2000), or actinomycin D (Act-D), a DNA intercalator that displaces HMGN proteins from chromatin (Schroter et al., 1985). Act-D treatment significantly increased the mobility of HMGN1, and the t80 decreased from 5.3 to 3 s (Figure 3D). It also abolished the effect of HMGN on the mobility of H10–GFP (Figure 3E; cf. Figure 2C), an expected result since this DNA intercalator releases the H1 and HMGN proteins from chromatin. Likewise, consistent with the expected effects of the interaction of H10 with nucleosomes, treatment of cells with DRB also abolished the effect of the injected HMGN on the intranuclear mobility of H10 (Figure 3F; cf. Figure 2C). Thus, treatments that interfere with the ability of HMGN proteins to bind to nucleosomes or enhance the interaction of H10 with chromatin diminish the ability of HMGN proteins to compete with H10 for nucleosome binding sites.
In this study, we combined microinjection and FRAP techniques to study competition between proteins for common chromatin binding sites in living cells. This approach is suitable for studying competition between any proteins that share binding sites. Our findings provide evidence that in living cells HMGN proteins compete with histone H1 for binding sites on chromatin. HMGN mutants that do not bind to nucleosomes do not compete with H1, and drugs that prevent the binding of HMGN proteins to chromatin abolish the ability of HMGN proteins to displace H1. These findings argue strongly that the effects of HMGN proteins on the chromatin residence time of H1 is due to competition for binding sites on nucleosomes. The possible consequence of this competition is schematically illustrated in Figure 4. The scheme takes into account the well-established finding that histone H1 stabilizes the higher-order chromatin structure (Thoma and Koller, 1977; van Holde, 1988), whereas HMGN proteins destabilize this structure and reduce the compactness of the chromatin fiber (reviewed in Bustin, 2001b). Recent FRAP experiments also demonstrated that the residence time of H1 is limited (Lever et al., 2000; Misteli et al., 2000). As illustrated in the scheme, our results raise the possibility that a dynamic interplay between HMGN proteins and histone H1 on the surface of the nucleosome affects the chromatin structure at selected sites. The residence time of H1 on chromatin is significantly longer than that of HMGN proteins, and thus the binding of H1 to chromatin is stronger than that of HMGN proteins. A temporary increase in the local concentration of HMGN proteins could weaken the interaction of H1 at selected chromatin loci, thereby ‘unfolding’ chromatin and providing a window of opportunity for the binding of regulatory factors to their chromatin targets. Indeed, in the nucleus HMGN proteins are organized into distinct foci, and clusters of contiguous HMGN-containing nucleosomes have been detected in the chromatin fiber (Postnikov et al., 1997).
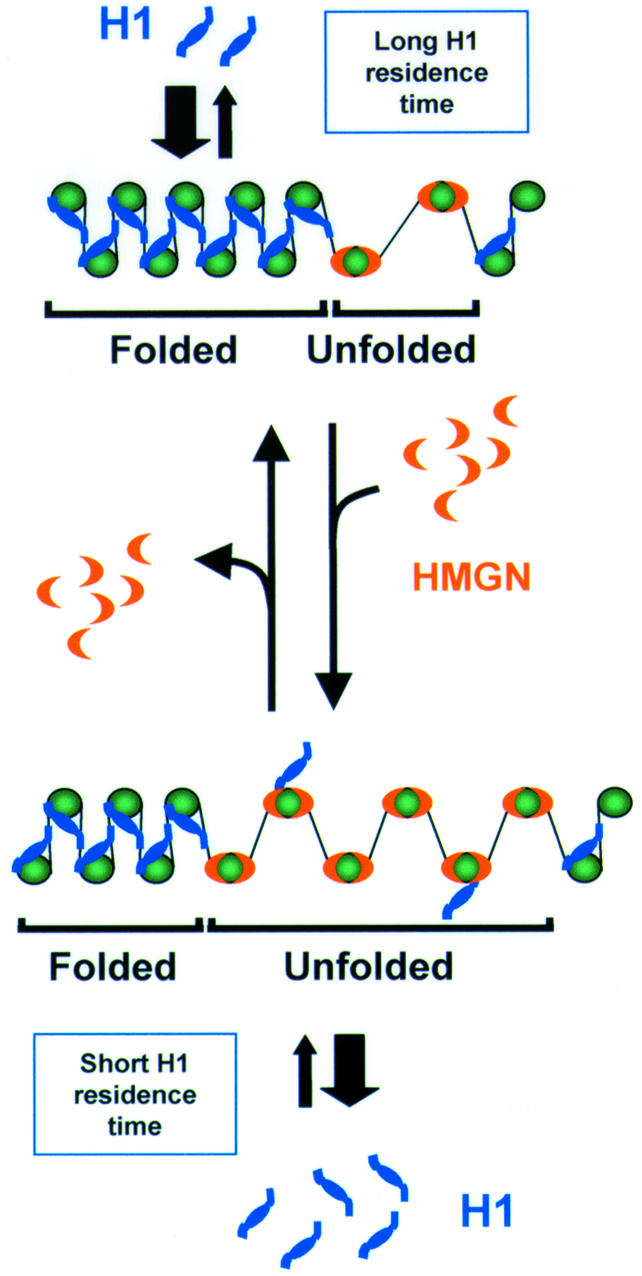
Fig. 4. Dynamic interplay between HMGN proteins and histone H1 on the surface of the nucleosome. The interaction of H1 with nucleosomes stabilizes the higher-order chromatin structure and promotes the formation of a compacted, folded chromatin fiber. An increase in the local concentration of HMGN proteins, which compete with H1 for shared binding sites on the nucleosome (Alfonso et al., 1994), decreases the H1 residence time at selected chromatin loci and promotes the unfolding of the chromatin fiber.
H1 inhibits the accessibility of regulatory factors to their nucleosomal targets (Herrera et al., 2000; Cheung et al., 2002; Horn et al., 2002) and therefore plays an important role in regulating gene expression in the context of chromatin. By decreasing the residence time of H1 on chromatin, HMGN proteins may reduce the inhibitory effects of the higher-order chromatin structure. Thus, the dynamic behavior of chromatin binding proteins in the cell nucleus plays a role in regulating the structure and activity of the chromatin fiber.
METHODS
Cell lines and purified proteins. Experiments were performed with either a BALB/c 3T3 cell line expressing H10–GFP (Misteli et al., 2000) or in mouse embryonic fibroblasts. Histones, HMGN and HMGN mutants were prepared as described previously (Bustin, 1973; Bustin et al., 1991; Ding et al., 1997; Prymakowska-Bosak et al., 2001). HMGN proteins were fluorescently labeled at a –SH group introduced at position 88 in HMGN1 and 82 in HMGN2 (Hock et al., 1998). Protein concentrations were determined by the Bradford assay (Bio-Rad) and by Coomassie Blue staining of SDS–PAGE of the protein in the solutions used for microinjection.
Microinjection in cultured cells. Cells were plated 2 days prior to injection onto Labtek Chambered Coverglass II (Nalgen) in DMEM without phenol-red (Biofluids). Before injection, the medium was replaced with DMEM fortified with 5 mM HEPES pH 7.4. The final concentration of labeled proteins was 0.05 mM and of unlabeled proteins was 0.4 mM (except histone H1, at 0.2 mM) in 50 mM Tris–HCl pH 7.4 (injection buffer). Fluorescent Texas Red labeled high-molecular dextran (Molecular Probes) was added to the injection mixture to identify injected cells. Freshly prepared solutions were clarified by centrifugation at 10 000 g for 30 min at 4°C. In select experiments, the medium contained either Act-D (Sigma) at 5 µg/ml or DRB (Calbiochem) at 100 nM. Cells were microinjected at 37°C using Micromanipulator 5171 and Microinjector 5246 (Eppendorf). Microinjections were performed under air pressure using sterile glass capillaries (Femtotips, Eppendorf). After injection, cells were incubated at 37°C, 5% CO2, for at least 30 min before performing FRAP experiments.
Microscopy and FRAP. Microscopy and FRAP were performed with a Zeiss LSM 510 confocal microscope using the 488 nm line of an argon laser and the 543 nm line of an HeNe laser as described previously (Misteli et al., 2000; Phair and Misteli, 2000). Typically, five pre-bleach images were acquired, followed by a single bleach pulse of 152 ms using a spot of 2 µm in diameter. Single images were then collected with a 152 ms interval. For imaging, the laser power was set to 0.1% of a 25 mW argon laser (488 nm line); and for bleaching, the laser power was set to 100%. FRAP recovery curves were generated from background-subtracted images. The total fluorescence was determined for each image and compared to the initial total fluorescence to determine the amount of fluorescence lost during the bleach and imaging. The fluorescence intensity in the bleach area was normalized to the initial fluorescence in the bleach area. In a typical experiment, several spots, in 8–15 cells, were used for FRAP. Each experiment was repeated at least three times. Student’s t-test was used to determine the statistical significance of the results.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Susan H. Garfield, manager of the Confocal Microscopy Core Facility, Laboratory of Experimental Carcinogenesis, Center for Cancer Research, National Cancer Institute.
REFERENCES
- Alfonso P.J., Crippa, M.P., Hayes, J.J. and Bustin, M. (1994) The footprint of chromosomal proteins HMG-14 and HMG-17 on chromatin subunits. J. Mol. Biol., 236, 189–198. [DOI] [PubMed] [Google Scholar]
- Boulikas T., Wiseman, J.M. and Garrard, W.T. (1980) Points of contact between histone H1 and the histone octamer. Proc. Natl Acad. Sci. USA, 77, 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin M. (1973) Arrangement of histones in chromatin. Nat. New Biol., 245, 207–209. [DOI] [PubMed] [Google Scholar]
- Bustin M. (2001a) Revised nomenclature for high mobility group (HMG) chromosomal proteins. Trends Biochem. Sci., 26, 152–153. [DOI] [PubMed] [Google Scholar]
- Bustin M. (2001b) Chromatin unfolding and activation by HMGN(*) chromosomal proteins. Trends Biochem. Sci., 26, 431–437. [DOI] [PubMed] [Google Scholar]
- Bustin M., Becerra, P.S., Crippa, M.P., Lehn, D.A., Pash, J.M. and Shiloach, J. (1991) Recombinant human chromosomal proteins HMG-14 and HMG-17. Nucleic Acids Res., 19, 3115–3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung E., Zarifyan, A.S. and Kraus, W.L. (2002) Histone H1 represses estrogen receptor α transcriptional activity by selectively inhibiting receptor-mediated transcription initiation. Mol. Cell. Biol., 22, 2463–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H.F., Bustin, M. and Hansen, U. (1997) Alleviation of histone H1-mediated transcriptional repression and chromatin compaction by the acidic activation region of chromosomal protein HMG-14. Mol. Cell. Biol., 17, 5843–5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera J.E., West, K.L., Schiltz, R.L., Nakatani, Y. and Bustin, M. (2000) Histone H1 is a specific repressor of core histone acetylation in chromatin. Mol. Cell. Biol., 20, 523–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock R., Scheer, U. and Bustin, M. (1998) Chromosomal proteins HMG-14 and HMG-17 are released from mitotic chromosomes and imported into the nucleus by active transport. J. Cell Biol., 143, 1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn P.J., Carruthers, L.M., Logie, C., Hill, D.A., Solomon, M.J., Wade, P.A., Imbalzano, A.N., Hansen, J.C. and Peterson, C.L. (2002) Phosphorylation of linker histones regulates ATP-dependent chromatin remodeling enzymes. Nat. Struct. Biol., 9, 263–267. [DOI] [PubMed] [Google Scholar]
- Jenuwein T. and Allis, D.C. (2001) Translating the histone code. Science, 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- Laybourn P.J. and Kadonaga, J.T. (1991) Role of nucleosomal cores and histone H1 in regulation of transcription by RNA polymerase II. Science, 254, 238–245. [DOI] [PubMed] [Google Scholar]
- Lever M.A., Th’ng, J.P., Sun, X. and Hendzel, M.J. (2000) Rapid exchange of histone H1.1 on chromatin in living human cells. Nature, 408, 873–876. [DOI] [PubMed] [Google Scholar]
- Misteli T., Gunjan, A., Hock, R., Bustin, M. and Brown, D. (2000) Dynamic binding of histone H1 to chromatin in living cells. Nature, 408, 877–881. [DOI] [PubMed] [Google Scholar]
- Phair R.D. and Misteli, T. (2000) High mobility of proteins in the mammalian cell nucleus. Nature, 404, 604–609. [DOI] [PubMed] [Google Scholar]
- Postnikov Y.V., Shick, V.V., Belyavsky, A.V., Khrapko, K.R., Brodolin, K.L., Nikolskaya, T.A. and Mirzabekov, A.D. (1991) Distribution of high mobility group proteins 1/2, E and 14/17 and linker histones H1 and H5 on transcribed and non-transcribed regions of chicken erythrocyte chromatin. Nucleic Acids Res., 19, 717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postnikov Y.V., Herrera, J.E., Hock, R., Scheer, U. and Bustin, M. (1997) Clusters of nucleosomes containing chromosomal protein HMG-17 in chromatin. J. Mol. Biol., 274, 454–465. [DOI] [PubMed] [Google Scholar]
- Prymakowska-Bosak M., Misteli, T., Herrera, J.E., Shirakawa, H., Birger, Y., Garfield, S. and Bustin, M. (2001) Mitotic phosphorylation prevents the binding of HMGN proteins to chromatin. Mol. Cell. Biol., 21, 5169–5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring D. and Cole, R.D. (1979) Chemical cross-linking of H1 to the nucleosomal histones. J. Biol. Chem., 254, 11688–11695. [PubMed] [Google Scholar]
- Schroter H., Maier, G., Ponstingl, H. and Nordheim, A. (1985) DNA intercalators induce specific release of HMG 14, HMG 17 and other DNA-binding proteins from chicken erythrocyte chromatin. EMBO J., 4, 3867–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoma F. and Koller, T. (1977) Influence of histone H1 on chromatin structure. Cell, 12, 101–107. [DOI] [PubMed] [Google Scholar]
- Thomas J.O. (1999) Histone H1: location and role. Curr. Opin. Cell Biol., 11, 312–317. [DOI] [PubMed] [Google Scholar]
- Trieschmann L., Martin, B. and Bustin, M. (1998) The chromatin unfolding domain of chromosomal protein HMG-14 targets the N-terminal tail of histone H3 in nucleosomes. Proc. Natl Acad. Sci. USA, 95, 5468–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Holde K.E. (1988) Chromatin. Springer, New York, NY.
- Wolffe A.P., Khochbin, S. and Dimitrov, S. (1997) What do linker histones do in chromatin? BioEssays, 19, 249–255. [DOI] [PubMed] [Google Scholar]