

Abstract
Background
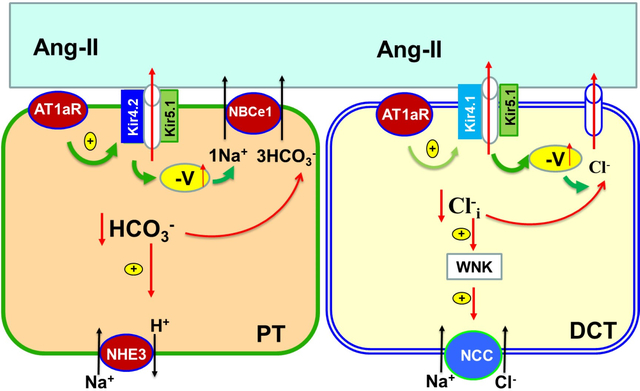
Kir4.2 and Kir4.1 play a role in regulating membrane transport in the proximal-tubule (PT) and in the distal-convoluted-tubule (DCT), respectively.
Methods
We generated kidney-tubule-specific-AT1aR-knockout (Ks-AT1aR-KO) mice to examine whether renal AT1aR regulates Kir4.2 and Kir4.1.
Results
Ks-AT1aR-KO mice had a lower systolic blood pressure (SBP) than Agtr1aflox/flox (control) mice. Ks-AT1aR-KO mice had a lower expression of Na+/H+-exchanger-3 (NHE3) and Kir4.2, a major Kir-channel in PT, than Agtr1aflox/flox mice. Whole-cell-recording also demonstrated that the membrane potential in PT of Ks-AT1aR-KO mice was lesser negative than Agtr1aflox/flox mice. The expression of Kir4.1 and Kir5.1, Kir4.1/Kir5.1-mediated K+ currents of DCT and DCT membrane potential in Ks-AT1aR-KO mice were similar to Agtr1aflox/flox mice. However, angiotensin-II perfusion for 7 days hyperpolarized the membrane potential in PT and DCT of the control mice but not in Ks-AT1aR-KO mice, while angiotensin-II perfusion did not change the expression of Kir4.1, Kir4.2 and Kir5.1. Deletion of AT1aR did not significantly affect the expression of αENaC and βENaC but increased cleaved γENaC expression. Patch-clamp-experiments demonstrated that deletion of AT1aR increased amiloride-sensitive-Na+-currents in the cortical-collecting-duct (CCD) but not in late-DCT. However, TPNQ-sensitive-ROMK-currents were similar in both genotypes.
Conclusion
AT1aR determines the baseline membrane potential of PT by controlling Kir4.2 expression/activity but AT1aR is not required for determining the baseline membrane potential of the DCT and Kir4.1/Kir5.1 activity/expression. However, AT1aR is required for angiotensin-II-induced hyperpolarization of basolateral membrane of PT and DCT. Deletion of AT1aR had no effect on baseline ROMK activity but increased ENaC activity in the CCD.
Keywords: Angiotensin II, inwardly-rectifying K+ channels, proximal tubule, DCT, CCD
Graphical Abstract
Introduction
Renal inwardly-rectifying K+ (Kir) channels play several important roles in regulating membrane transport of the renal tubules 1, 2. Kir1.1 (ROMK) is expressed in the apical membrane of TAL, late DCT, CNT) and CCD 1. ROMK plays a key role in maintaining the function of NKCC2 activity in the TAL whereas ROMK in the DCT2, CNT and CCD is responsible for mediating ENaC-dependent renal K+ excretion 3. Kir4.2 and Kir5.1 are expressed in the basolateral membrane of PT and participate in generating the negative membrane potential of the PT 4, 5. Depolarization of PT membrane potential is associated with the inhibition of NHE3 function without altering NHE3 protein expression in Kir4.2 knockout mice 4. In the distal nephron, Kir4.1 and Kir5.1 are expressed in the basolateral membrane of the DCT, CNT and CCD and play a dominant role in determining the negativity of the basolateral membrane potential of the distal nephrons 2, 6. Moreover, we and others have previously demonstrated that Kir4.1/Kir5.1 activity plays a key role in regulating the membrane transport in the DCT and CCD 7–9. For instance, the deletion of Kir4.1 in the kidney was associated with the inhibition of thiazide-sensitive NCC activity 7, 10.
A large body of evidence has demonstrated that AT1R or mouse AT1aR plays an important role in the regulation of membrane transport in the PT, DCT and CCD 11–19. However, these studies are exclusively focused on the apical membrane transport proteins such as NHE3 in the PT, thiazide-sensitive Na-Cl cotransporter (NCC) in the DCT and aquaporin-2 in the collecting duct. The role of AT1aR in the regulation of Kir-channels in the PT and distal nephrons is not understood, although Kir-channels play an important role in the regulation of the membrane transport of these nephrons. We now hypothesize that AT1aR may also regulate Kir4.2 in the PT and Kir4.1 in the DCT thereby modulating NHE3 and NCC activity. Thus, the first aim is to examine whether AT1aR regulates Kir4.2 and Kir4.1, and determines the baseline membrane potential of PT and DCT. Also, previous studies have demonstrated that angiotensin-II stimulates ENaC activity in aldosterone-sensitive distal nephron (ASDN) and regulates renal K+ excretion in the distal nephron 14, 20, 21. However, it is not clear whether AT1aR activity regulates baseline activity of ENaC and ROMK. Thus, the second aim of the present study is to explore the role of AT1aR in the ASDN in the regulation of baseline activity of ENaC and ROMK.
Material and Methods
All supporting data and detailed methods including animal preparation, electrophysiology, western blot, fluorescence immunostaining, RNAscope and qRT-PCR are available within the article and its online supplementary file.
Generating Ks-AT1aR KO mice
Mice expressing Pax8-rtTA and tet-on LC-1 transgene were crossed with Agtr1a-floxed mice (originally created in Coffman’s lab in Duke Iniversity) to generate inducible Ks-AT1aR KO mice11. Agtr1a gene deletion was conducted in 8-week-old male/female (m/f) mice homozygous for floxed Agtr1a gene and heterozygous for Pax8-rtTA/LC-1 transgene by providing doxycycline (5mg/ml, 5% sucrose) in the drinking water for 2 weeks. This was followed by at least 2 additional weeks without doxycycline treatment before performing experiments. Littermate mice of the same age and genetic background drinking 5% sucrose were used as controls (Agtr1aflox/flox). Tail DNA was PCR amplified and the primers for genotyping are shown in table s1. The method for dissecting tubules, metabolic cage and measurement of AT1aR mRNA levels by qRT-PCR are described in supplementary material.
Patch-clamp experiments
A Narishige electrode puller (Narishige, Japan) was used to make the patch-clamp pipettes from Borosilicate glass (1.7-mm OD). The resistance of the pipette was 2 MΩ when it was filled with solution containing (in mM) 140 KCl, 1.8 MgCl2 and 10 HEPES (titrated with KOH to pH 7.4).
Measurement of Kir4.1/Kir5.1-mediated K+ currents
We have used the whole-cell recording to measure Ba2+-sensitive Kir4.1/Kir5.1-mediated K+ currents in early DCT (DCT1) and late DCT (DCT2). An Axon 200A patch-clamp amplifier was used to record the whole-cell K+ currents, which were low-pass filtered at 1 KHz, digitized by an Axon interface (Digidata 1440A). The pipette solution contains (in mM) 140 KCl, 2 MgCl2, 1 EGTA and 5 HEPES (pH 7.4) with 0.5 mM MgATP, whereas the bath solution contains (in mM) 140 KCl, 1.8 CaCl2, 1.8 MgCl2, and 10 HEPES (pH 7.4). Because ROMK (Kir1.1) is expressed in the apical membrane of DCT2 but not DCT1 22, we used 400 nM Tertiapin-Q (TPNQ) (Sigma-Aldrich) in split-open DCT2 to block ROMK and then added 1 mM Ba2+ to measure whole-cell Kir4.1/Kir5.1 mediated K+ currents of DCT2. Fig. s1A is a gap-free recording showing TPNQ-sensitive currents (ROMK) only in DCT2 but not in DCT1 while Ba2+ -sensitive and TPNQ-insensitive whole-cell K+ currents (Kir4.1/Kir5.1) in both DCT1 and DCT2 (clamped at −60 mV). Fig.s1B is a whole-cell voltage-clamp recording showing Ba2+-sensitive Kir4.1/Kir5.1 currents of DCT2 measured with step-protocol from −100 to 60 mV in the presence of 400 nM TPNQ by subtracting the total currents from Ba2+-insensitive currents. Data were analyzed using the pClamp Software System 9 (Axon).
Measurement of inward-to-outward current reversal potential (I-reversal potential)
For measuring I-reversal potential, an index of cell membrane potential, using perforated whole-cell voltage-clamp in PT or DCT, the isolated PT or DCT was super-fused with a bath solution containing (in mM) 140 NaCl, 5 KCl, 1.8 CaCl2, 1.8 MgCl2, and 10 HEPES (pH 7.4). The pipette was filled with 140 mM KCl pipette solution and was then backfilled with amphotericin B (20 mg/0.1ml). After forming a high-resistance seal (>2 GΩ), the membrane capacitance was monitored until the whole-cell patch configuration was formed. The voltage at which the inward currents were zero was the I -reversal potential determined by a ramp protocol from −100 to 100 mV.
Measurement of ROMK and ENaC
We used an Axon 200A amplifier to measure whole-cell TPNQ-sensitive K+ currents (ROMK) and amiloride-sensitive Na+ currents (ENaC) using gap-free protocol in the split-open DCT2 and CCD of m/f control and Ks-AT1aR KO mice. Detailed method for measuring TPNQ-sensitive (ROMK) and amiloride-sensitive Na+ (ENaC) currents is described in supplemental material.
Statistical analysis
We used software (Sigma plot 14) for the statistical analysis. For analyzing the values between two groups we used t-test, and for comparisons of the values within the same group, we used paired t-test. We used one-way or two-way ANOVA for analyzing results of more than two groups, and Holm-Sidak test was used as post-hoc analysis. P-values <0.05 were considered statistically significant. Data are presented as the mean ± SEM.
Results
We have generated an inducible kidney-tubule-specific (Ks)-AT1aR deficient mice by crossing mice expressing Pax8-rtTA and tet-on LC-1 with Agtr1aflox/flox mice. Since no reliable AT1aR antibody is available 23, we measured Agtr1a mRNA levels in the renal cortex by qRT-PCR with the primers and method described previously by Coffman’s group 24. Fig.1A is an agarose gel showing Agtr1a mRNA expression in renal cortex of Ks-AT1aR KO mice and Agtr1aflox/flox mice (control). Fig.1B is a bar graph with scatter plots summarizing quantitative analysis of Agtr1a mRNA expression levels showing that Agtr1a mRNA to GAPDH mRNA expression in Ks-AT1aR KO mice decreased by 80±5% in comparison to Agtr1aflox/flox mice (n=5), an indication of AT1aR deletion. The deletion of AT1aR in renal tubules was also confirmed by examining Agtr1a mRNA-expression (red dots) with RNAscope-technique (Fig.1C and Fig.s2a) and it is apparent that Agtr1a mRNA was almost absent in the renal tubule of Ks-AT1aR-KO mice (Table s3 shows quantification analysis). We have also measured BP and heart rate with tail-cuff method in m/f control and Ks-AT1aR KO mice (n=8 for each group). Fig.1D is a scatter plot showing that the SBP of Ks-AT1aR KO mice (male, 102±3 mmHg; female, 101±3 mmHg) was lower than the control mice (male, 119±3 mmHg; female 115±2 mmHg). Also, the diastolic BP was lower in the Ks-AT1aR-KO than the control mice (Table s4). However, we did not find significant difference for heart rate in two genotypes. We next performed the metabolic cage study for analyzing body weight (BW), food/water intakes, urinary Na+ (UNa) /K+ (UK) excretion rate, and plasma Na+ and K+ concentrations in both male(m)/female(f) genotypes (Fig.1E). It is apparent that food/water intakes, urinary Na+/K+ excretion and plasma Na+/K+ concentrations were similar between the two genotypes. These results are consistent with previous study showing that plasma Na+ and K+ levels were similar between the control and global-AT1aR KO mice 25. However, the plasma aldosterone levels in both genders were higher in Ks-AT1aR KO than Agtr1aflox/flox mice. Also, plasma Ang-II levels were slightly higher in male Ks-AT1aR-KO mice than the control, although the difference was not significant (Fig.s2b). But, it should be noted that the plasma Ang-II level may not reflect the renal Ang-II level. 26
Fig. 1. Ks-AT1aR KO mice are hypotensive.
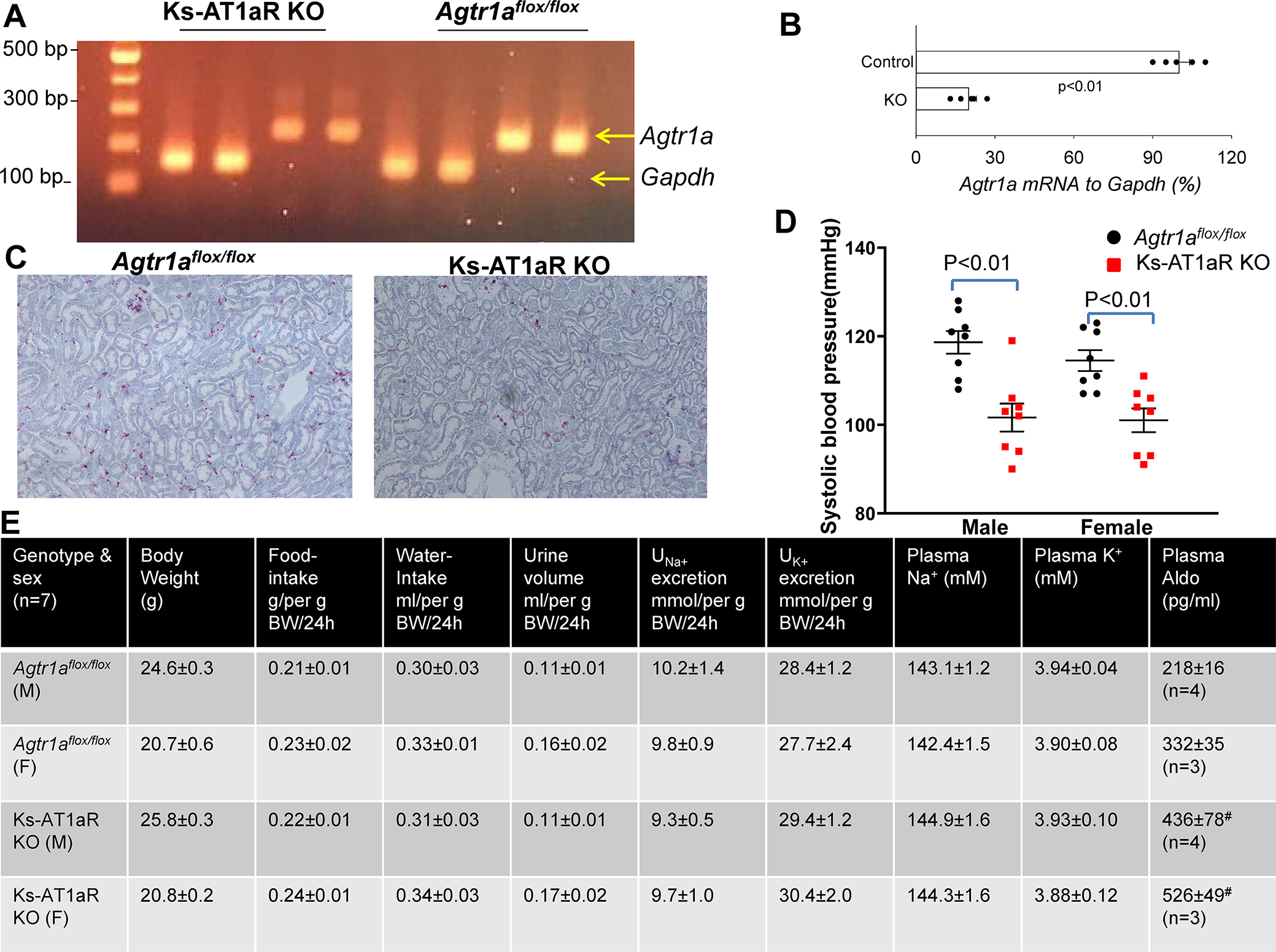
(A) An agarose gel shows the expression of Agtr1a mRNA expression level in Agtr1aflox/flox mice and in Ks-AT1aR KO mice. The mRNA was isolated from renal cortex tissue and was subjected to reverse transcription. (B) A scatter plot showing the expression of Agtr1a mRNA to Gapdh mRNA from Ks-AT1aR KO mice in comparison to Agtr1aflox/flox mice. Significance is determined by an unpaired t test. (C) Images show Agtr1a mRNA expression (red dots) in Agtr1aflox/flox and Ks-AT1aR-KO mice. Hybridization signaling are detected using fast-red staining. (D) A scatter plot shows each measurement and mean value ± SEM of systolic blood pressure measured with tail-cuff method in male/female Ks-AT1aR KO mice and Agtr1aflox/flox mice. Significance is determined by two-way ANOVA. (E) A table shows the baseline characterization of mouse phenotypes in Ks-AT1aR KO mice and Agtr1aflox/flox mice. # indicates a significant difference in comparison to the corresponding control.
Since AT1aR expression in renal tubules is predominantly located in the PT under control conditions 27, we first examined whether deletion of renal AT1aR altered the expression of Kir4.2, a major renal Kir-channel expressed in basolateral membrane of the PT 4. Fig. 2A is a western blot (Fig.s3 shows an uncut gel) showing the expression of Kir4.2, p(S552)NHE3 and total NHE3 in m/f control, 4, 28 and Ks-AT1aR KO mice and Fig.2B is a scatter blot summarizing the normalized band density of Kir4.2, pNHE3 and NHE3 expression (4 m/4f mice). Kir4.2 expression (40 kDa monomer) was lower in Ks-AT1aR KO mice (69±3% control vale for male and 63±5% control value for female) than the corresponding control mice. We have also examined the expression of Kir4.2 with fluorescence microscope (Fig. S4). We confirmed the previous report that Kir4.2 immunostaining is exclusively located in the PT4. However, the difference of fluorescence staining intensity of Kir4.2 was not obvious between the control and Ks-AT1aR KO mice, although we observed a sharp basolateral staining of Kir4.2 only in the control mice. Like PT-specific AT1aR KO mice 11, 12, the expression of pNHE3 (71±4% of the control value for male and 70±5% of the control value for female) and total NHE3 (66±4% of the control value for male and 65±5% of the control value for female) was lower in Ks-AT1aR KO mice than Agtr1aflox/flox mice. The notion that deletion of AT1aR decreased NHE3 activity was also confirmed by the observation that NHE3-inhibitor (S3226)-induced natriuresis was significantly smaller in Ks-AT1aR-KO mice than the control (Fig.s5). Since Kir4.2 has been shown to interact with Kir5.1 to form the basolateral K+ channel in the PT 4, 29, decreased Kir4.2 expression is expected to depolarize PT membrane potential. This notion is supported by measuring I- reversal potential of the PT (an index of PT membrane potential). Fig.2C is a typical trace of the whole-cell voltage clamp showing I-reversal potentials of the PT in Ks-AT1aR KO mice (red) and in Agtr1aflox/flox mice (black). Fig.2D is a scatter plot summarizing each data point of total 8 measurements (4 tubules from 3 male mice and 4 tubules from 3 female mice) and the mean value (including m/f mice). The mean value of I-reversal potentials of the PT (−51±1 mV) in the control mice is significantly more negative than Ks-AT1aR KO mice (−37±2 mV). Thus, deletion of AT1aR decreased the expression of Kir4.2 and depolarized the PT membrane potential.
Fig. 2. Deletion of AT1aR decreases Kir4.2 expression.
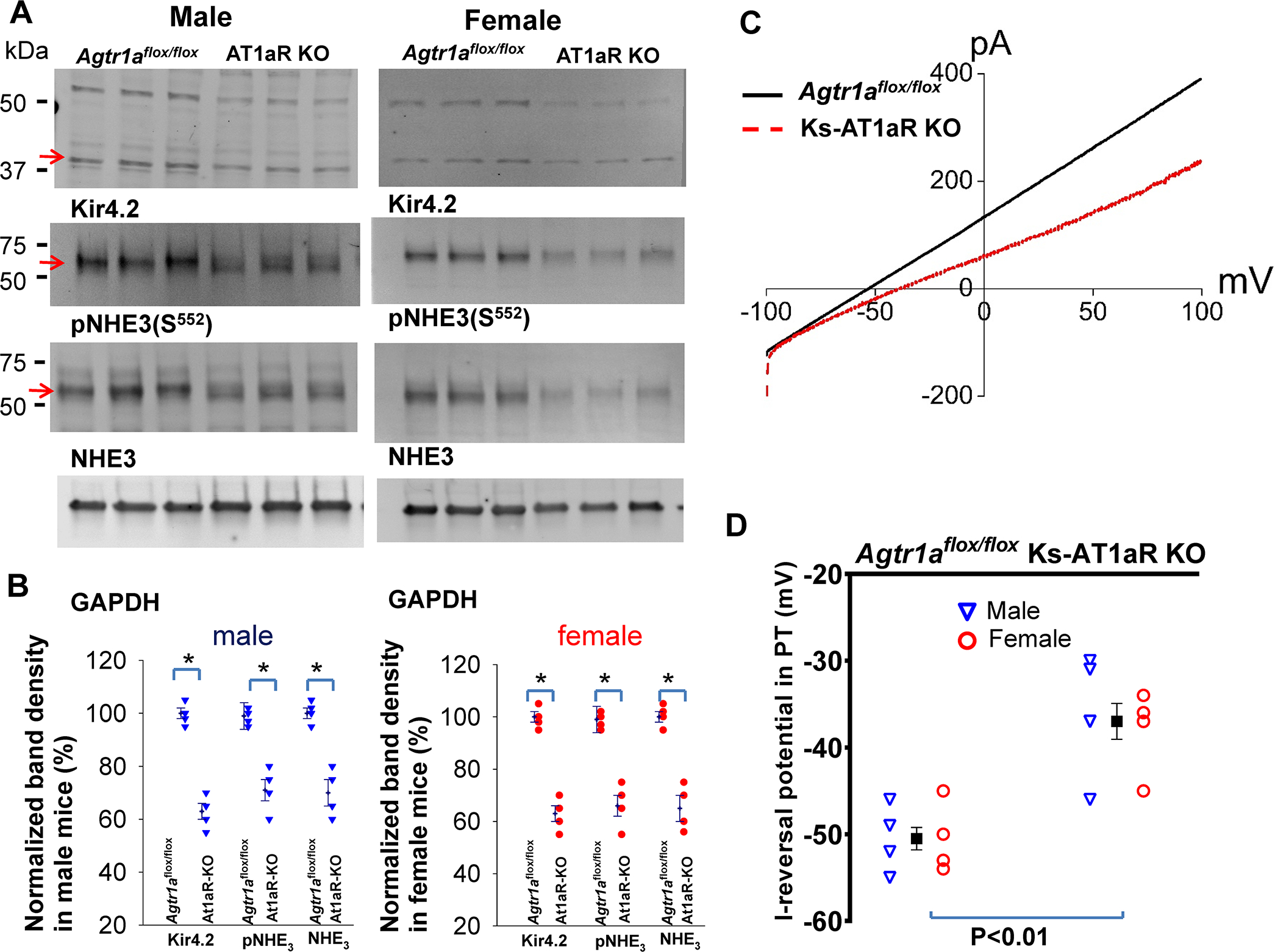
(A) A set of western blot shows the expression of Kir4.2, phosphor (P)-NHE3 (S552), total NHE3 and GAPDH in male/female Ks-AT1aR KO mice and Agtr1aflox/flox mice. An arrow indicates the band used for calculating normalized-band-density. (B) A scatter plot summarizes mean value (at the left column) and each data point of the normalized band density for male mice (blue triangle) and female mice (red circle) in AT1aR KO mice and Agtr1aflox/flox mice. Kir4.2 monomer at around 40 kDa was used to calculate band density. Significance is determined by an unpair test. (C) A set of recordings showing I-reversal potential of the PT measured with whole-cell voltage clamp in male AT1aR KO mice (red) and Agtr1aflox/flox mice (black). (D) A scatter plot summarizing each data point of I-reversal potential of the PT in male (blue triangle)/female (red circle) of AT1aR KO mice and Agtr1aflox/flox mice. The mean value includes data from both genders. The significance is determined by an unpaired t test.
We next examined whether deletion of AT1aR affects the expression of Kir4.1 and Kir5.1, two major renal Kir-channels in the basolateral membrane from the DCT, CNT to CCD 2, 6. Fig.3A is a western blot (Fig. s6 shows an uncut gel) showing the expression of Kir4.1 and Kir5.1 in m/f Ks-AT1aR KO mice and Agtr1aflox/flox mice. It is apparent that the deletion of AT1aR either did not decrease (male) or even slightly increased (female) the expression of Kir4.1 and Kir5.1 comparing to Agtr1aflox/flox mice. However, the modest increase in Kir4.1 of female AT1aR-KO mice may have no physiological significance because Kir4.1/Kir5.1-mediated-K+ currents were similar between female control and AT1aR-KO-mice (Fig. 3B and 3C). Fig3B is a set of whole-cell recordings showing Kir4.1/Kir5.1-mediated K+ currents measured with step protocol from −100 to 60 mV in DCT1 of male control and Ks-AT1aR KO mice. Fig.3C is a scatter plot showing each data point measured at −60 mV in DCT1 and DCT2 of m/f control and Ks-AT1aR KO mice (4 tubules from 3 male mice and 4–5 tubules from 3 female mice) and the mean value (including m/f mice). Because there is no significant difference between the two genders, we pooled the results. The Kir4.1/Kir5.1-mediated K+ currents of the DCT1 (control, 1269±31 pA; AT1aR KO, 1298±59 pA) and DCT2 (control, 1192±45 pA; AT1aR KO, 1214±37 PA) were similar between two genotypes. We next measured the I-reversal potentials of the DCT in two genders. Fig. 3D is a set of recordings showing I-reversal potential of DCT1 and DCT2 measured with whole-cell voltage-clamp. Fig.3E is a scatter plot summarizing each data point measured in the DCT1 and DCT2 from m/f Ks-AT1aR KO and Agtr1aflox/flox mice (4–5 tubules from 3 male mice and 4–5 tubules from 3 female mice) and the mean value (including m/f mice). We observed similar membrane potentials in DCT1 (control, 63±1 mV; AT1aR KO, 62±1 mV) or DCT2 (control, 61±1 mV; AT1aR KO, 62±1 mV) between two genotypes. Finally, immunostaining images have also demonstrated a similar Kir4.1 expression in the control and Ks-AT1aR KO mice (Fig. s7). Thus, AT1aR is not required for determining the baseline activity of Kir4.1/Kir5.1 in the DCT.
Fig. 3. Deletion of AT1aR did not affect Kir4.1/Kir5.1-heterotetramer activity.
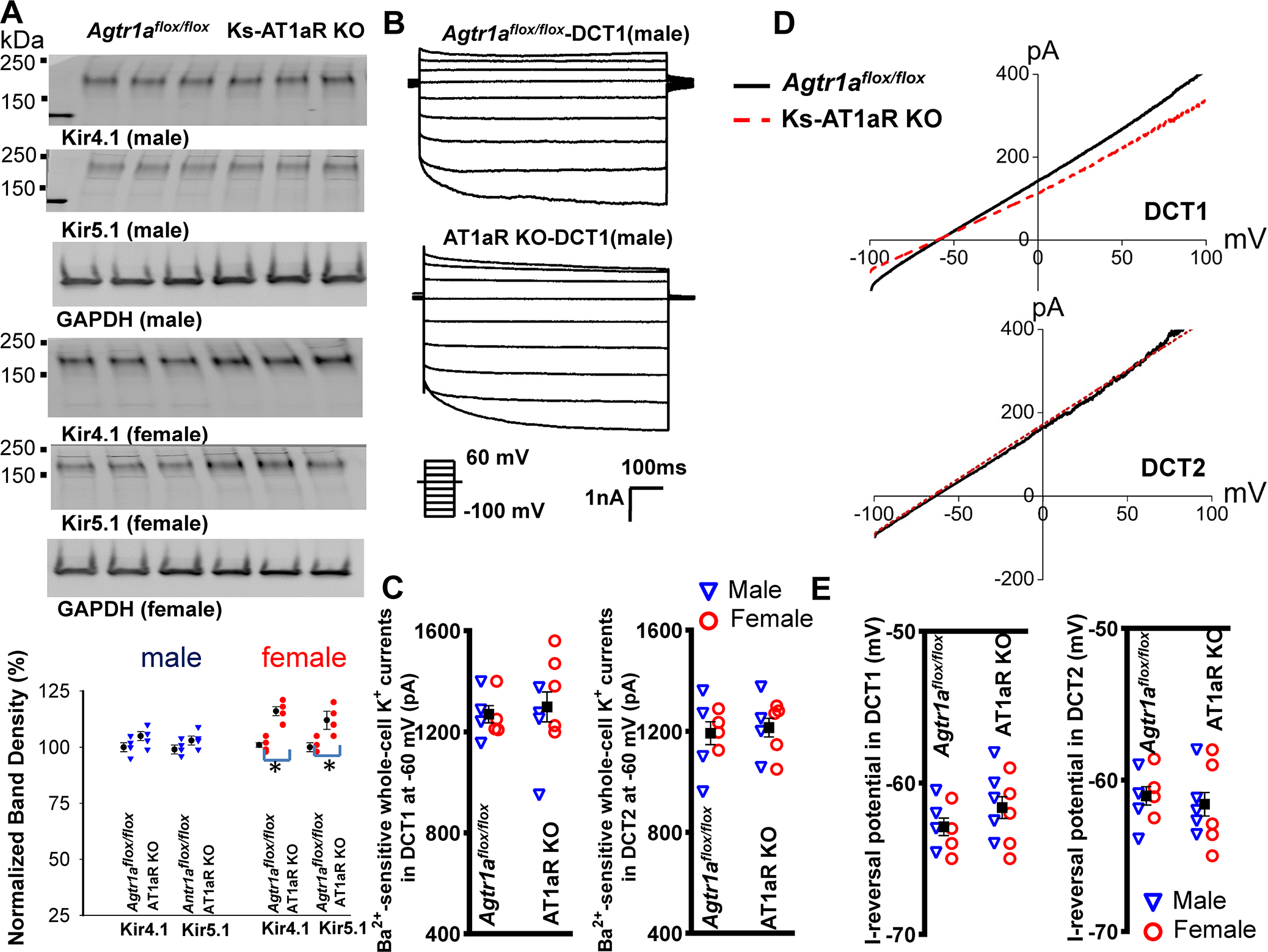
(A) A set of western blot shows the expression of Kir4.1, Kir5.1 and GAPDH in male/female Ks-AT1aR KO mice and Agtr1aflox/flox mice. A scatter plot in the bottom panel summarizes mean value (at the left column) and each data point of the normalized band density for male (blue triangle)/ female mice (red circle) in AT1aR KO mice and Agtr1aflox/flox mice. (B) Two traces show Ba2+-sensitive Kir4.1/Kir5.1-mediated K+ currents measured with whole-cell-recording with step-protocol from −100 to 60 mV in DCT1 of male AT1aR KO mice and Agtr1aflox/flox mice. (C) Two scatter plots summarize the experiments in which the whole-cell Ba2+-sensitive Kir4.1/Kir5.1-mediated-K+ currents were measured at −60 mV in the DCT1 and DCT2, respectively. Blue triangle and red circle represent each data point measured at male and female mice, respectively. The mean value of each group is shown in the middle (including both genders). (D) A set of traces show I-reversal potential measured with whole-cell voltage-clamp from −100 to 100 mV in DCT1 and DCT2 of male AT1aR KO (red) and Agtr1aflox/flox mice (black). (E) Two scatter plots summarize the experiments in which I-reversal potential was measured in DCT1 and DCT2 of male (blue triangle) and female (red circle) AT1aR KO mice and Agtr1aflox/flox mice. The mean value of each group is shown in the middle (including both genders).
To examine whether stimulation of AT1aR was able to increase the expression of Kir4.1, Kir4.2 and Kir5.1, and to hyperpolarize the membrane in both PT and DCT, we treated the control mice with angiotensin-II through subcutaneously installed osmotic pump for 7 days at 200 ng/Kg/min by following the method published previously19. It was reported that neither renal cortical angiotensinogen abundance nor urinary angiotensinogen excretion was altered in the mice received Ang-II perfusion at 200 ng/Kg/min for 3 days. Moreover, we observed that Ang-II perfusion for 7-days did not raise the BP (data not shown), although plasma Ang-II levels in WT mice with Ang-II perfusion were higher than untreated mice (Fig.s2b). After the treatment of angiotensin-II, the renal tissues were harvested for the immunoblot and for the electrophysiological experiments. Fig. 4A is a western blot (Fig.s8 shows uncut gel) showing the effect of angiotensin-II perfusion on Kir4.2, Kir5.1, Kir4.1 and NHE3. Fig. 4B is a bar graph with scatter plots summarizing the experiments. We observed that angiotensin-II perfusion had no significant effect on the expression of Kir4.2, Kir5.1 and Kir4.1. In contrast, angiotensin-II perfusion significantly increased the expression of pNHE3, NHE3, tNCC and pNCC (Fig.4A and Fig. s8). We next measured the I-reversal potentials in the PT and DCT of m/f control mice. Fig.4C is a set of recordings showing I-reversal potentials measured in the PT and DCT1 of male control mice (Fig. s9 shows the recording in female mice). Fig. 4D is a bar graph with scatter plots summarizing the results of the above experiments in m/f Agtr1aflox/flox mice. As expected, angiotensin-II perfusion hyperpolarizes the membrane potential of PT from −49±1 mV to −63±2 mV (5–6 tubules of 3 male mice) and from −50±2 mV to −62±3 mV (5–6 tubules of 3 female mice). Angiotensin-II perfusion also hyperpolarizes DCT membrane from −61±1 mV to −74±1 mV (n=5 tubules of three male mice) and from −61±2 mV to −73±2 mV (5 tubules of three female mice). Moreover, angiotensin-II-induced hyperpolarization of PT and DCT depends on AT1aR because angiotensin-II has no effect on the membrane potential of PT and DCT in Ks-AT1aR KO mice (Fig.s9). Thus, although AT1aR determines the baseline basolateral membrane potential only in the PT, AT1aR is required for angiotensin-II-induced hyperpolarization of the basolateral membrane in both PT and DCT.
Fig.4. Chronic Angiotensin-II perfusion hyperpolarizes PT and DCT membrane.
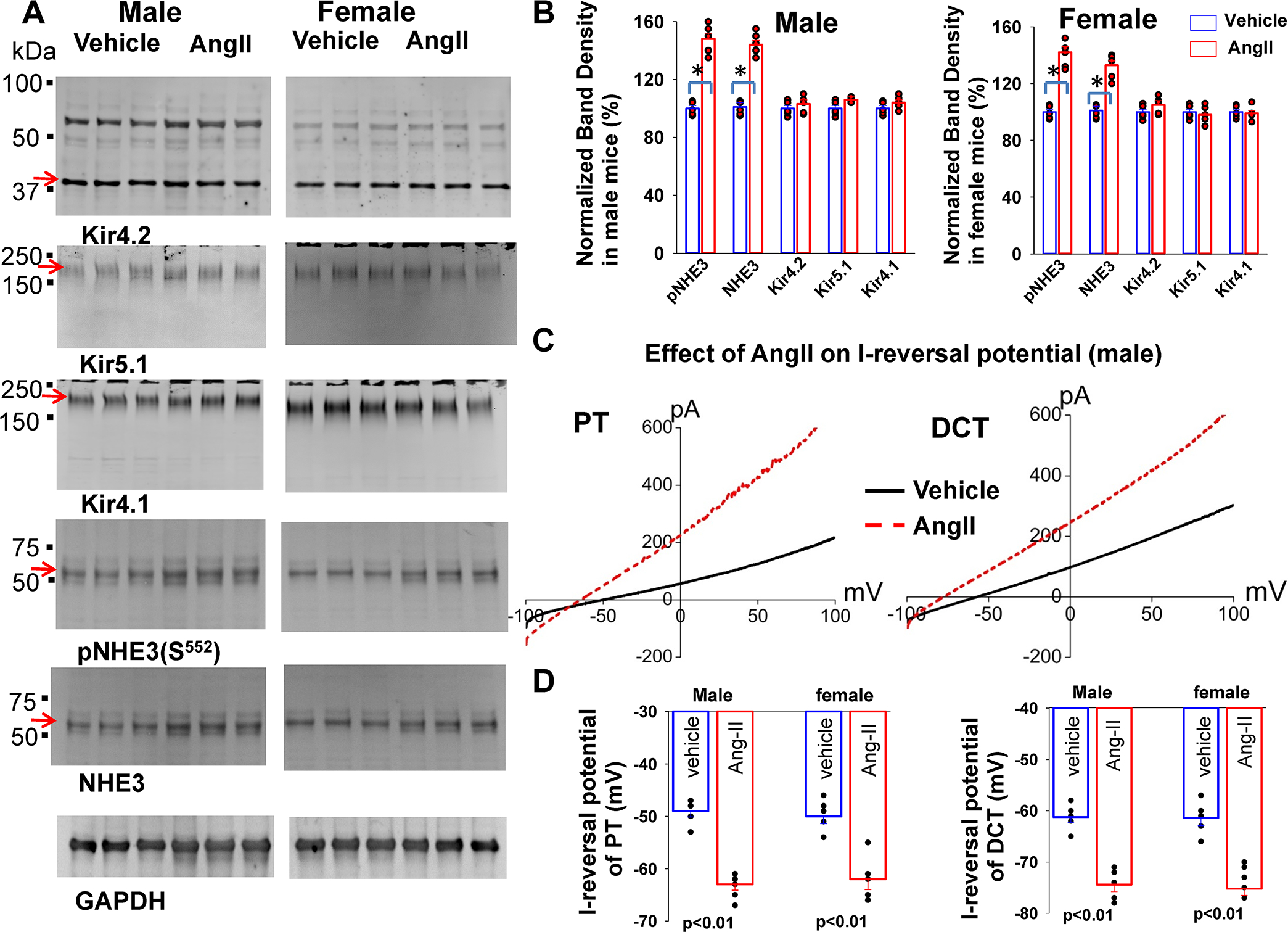
(A) A set of western blot shows the expression of Kir4.2, Kir4.1, Kir5.1, pNHE3 (S552), total NHE3 and GAPDH in male and female control mice treated with angiotensin-II for 7 days (at 200 ng/min/ Kg) through an osmotic pump. An arrow indicates the band used for calculating normalized-band-density. (B) A bar graph with a scatter plot summarizes mean value and each data point of the normalized band density for male/female control mice treated with vehicle (blue bar) and angiotensin-II (red bar). The significance is determined by an unpair t test. (C) ) A set of I-reversal potential traces measured with whole-cell voltage clamp from −100 to 100 mV in the PT and DCT1 of male control mice treated with angiotensin-II for 7 days (red) and vehicle (black), respectively. (D) A bar graph with a scatter plot summarizes the results of experiments in which I-reversal potentials were measured in the PT and DCT1 of m/f control mice treated with vehicle (blue) and angiotensin-II (red). Significance is determined by two-way ANOVA.
We next examined whether deletion of AT1aR affected the baseline ROMK activity by measuring TPNQ-sensitive-K+ currents (ROMK) in the DCT2 and initial CNT (DCT2/CNT) and in the CCD with the whole-cell recording. Fig.5A is a set of traces showing TPNQ-sensitive ROMK-mediated-K+ currents measured with gap-free protocol at −40 mV in the DCT2/CNT (top panel) and in the CCD (bottom panel) of male control and Ks-AT1aR KO mice. Fig.5B is a scatter blot summarizing the results of the above experiments measured at −40 mV in the DCT2/CNT and CCD of m/f control and Ks-AT1aR KO mice. The mean TPNQ-sensitive ROMK currents were 1008± 65 pA in DCT2/CNT (6 tubules of four m/f Agtr1aflox/flox mice) and were 940±67 pA (5–6 tubules of three-four m/f AT1aR KO mice). Thus, deletion of AT1aR had no effect on ROMK channel activity in DCT2/CNT. The similar results were obtained in the CCD because ROMK currents in the CCD were similar in both genotypes (465±36 pA, 5–6 tubules of four m/f control mice; 444±27 pA, 4–5 tubules of three m/f AT1aR KO mice).
Fig. 5. Deletion of AT1aR did not affect ROMK.
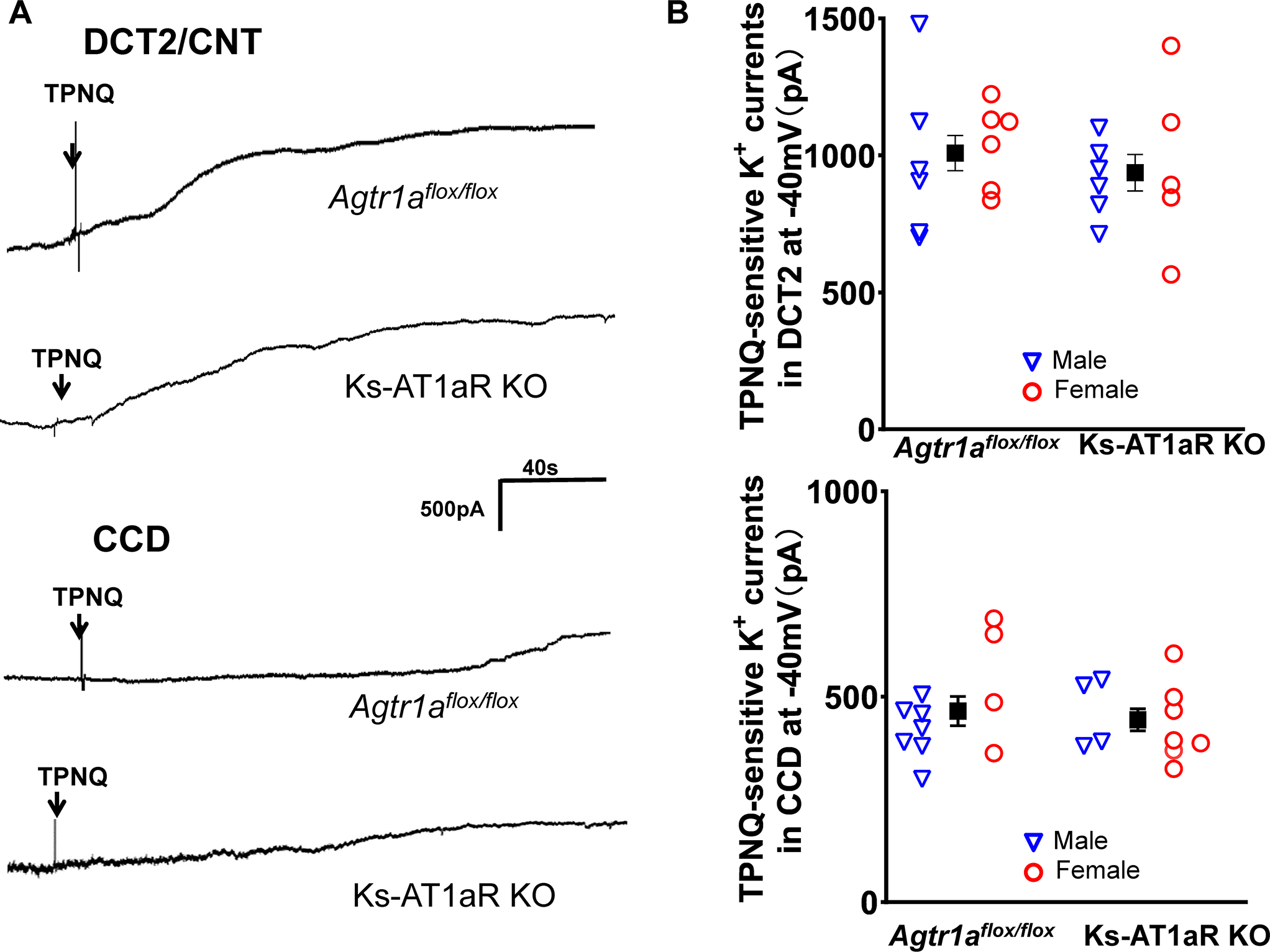
(A) A set of whole-cell recordings shows TPNQ (400 nM)-sensitive K+ currents (ROMK) measured with gap-free-protocol at −40 mV in the DCT2/CNT and in the CCD of the control and Ks-AT1aR KO mice. (B) A set of scatter plots summarizes the results of TPNQ-sensitive ROMK currents measured at −40 mV in the DCT2/CNT and in the CCD of male (blue triangle) and female (red circle) control and Ks-AT1aR KO mice, respectively. The mean value of each group is shown in the middle (including both genders). The significance is determined by an unpair t test.
We next examined ENaC expression/activity in the control and Ks-AT1aR KO mice. Fig. 6A is a western blot showing the expression of αENaC, βENaC and γENaC of m/f control and Ks-AT1aR KO mice (Fig.s10 shows an uncut gel). Deletion of AT1aR did not significantly affect the expression of αENaC and βENaC but slightly increased expression of cleaved γENaC. We have also used the whole-cell-recording to measure the amiloride-sensitive Na+ currents in the DCT2/CNT and in the CCD. Fig. 6B shows two sets of the traces measured with the gap-free protocol at −60 mV in the DCT2/CNT and in the CCD of male control and Ks-AT1aR KO mice, respectively. The mean amiloride-sensitive Na+ currents in DCT2/CNT in Agtr1aflox/flox mice were 210±9 pA (5 tubules from three male mice and 7 tubules from five female mice) were similar to Ks-AT1aR KO mice ( 229±16 pA, 4 tubules from three m/f mice) (Fig.6C). However, the mean amiloride-sensitive Na+ currents in the CCD of Ks-AT1aR KO mice (90±7 pA, 4–5 tubules from three m/f mice) were larger than Agtr1aflox/flox mice ( 50±4 pA, 5 tubules from three male mice and 6 tubules from four female mice). Thus, deletion of AT1aR increased ENaC activity in the CCD but not in the DCT2/CNT, presumably induced by high aldosterone levels (Fig. 1D). This view is also supported by the observation that spironolactone-treatment abolished the difference of amiloride-sensitive Na+-currents of the CCD between two genders (Fig.s11).
Fig.6. Deletion of AT1aR slightly stimulates ENaC in the CCD.
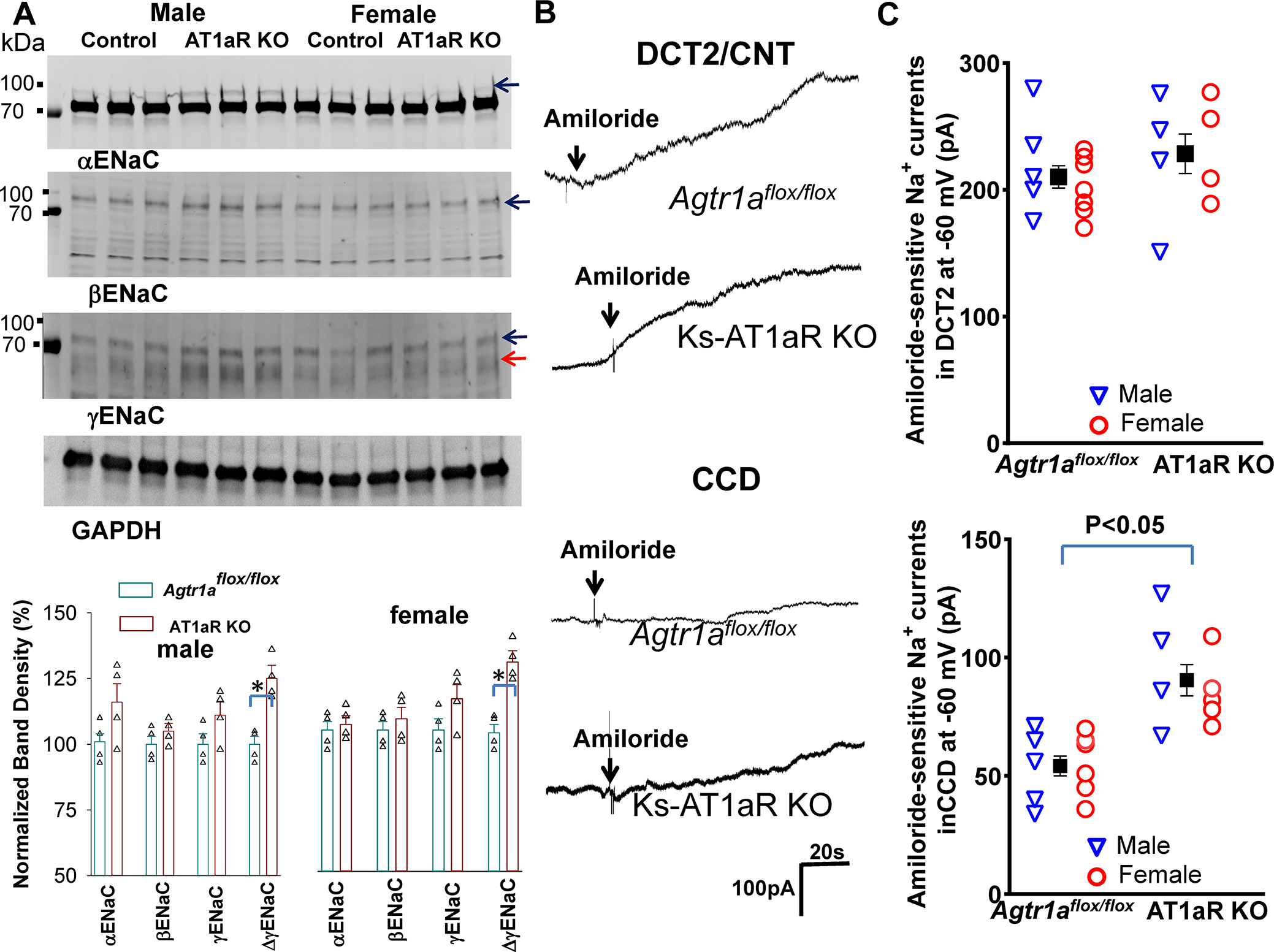
(A) A set of western blot shows the expression of αENaC, βENaC, γENaC, cleaved γENaC (indicated by a red arrow) and GAPDH in m/f Ks-AT1aR KO mice and Agtr1aflox/flox mice. The normalized band density of ENaC expression (The band used to calculate the normalized band density is indicated by arrows) is summarized in a bar graph with a scatter plot (bottom panel). Since the band which has higher molecular weight than the main band has been shown to be aldosterone-sensitive 50, this band was used to calculate αENaC density. (B) A set of whole-cell recordings shows amiloride-sensitive Na+ currents (ENaC) measured with gap-free-protocol at −60 mV in the DCT2/CNT and CCD of the control and Ks-AT1aR KO mice. (C) A set of scatter plots summarizes the results of amiloride-sensitive ENaC currents measured at −60 mV in the DCT2/CNT and in the CCD of male (Blue triangle)/female (red circle) control and Ks-AT1aR KO mice, respectively. The mean value of each group is shown in the middle (including male and female). Significance is determined by unpaired t test.
Discussion
Mouse AT1aR is considered to be the closest homologue to the human AT1R and plays an important role in mediating the effect of angiotensin-II on renal function 30. Although AT1aR mRNA in mouse renal tubules is predominantly expressed in the PT under the control conditions 27, 31, a large body of functional evidence has suggested the presence of AT1aR at protein level in the DCT and CCD 14, 15, 20, 32–35. For instance, angiotensin-II has been shown to stimulate NCC activity in the DCT and ENaC in the CCD 33–37. Global knockout of AT1aR has been shown to decrease blood pressure and to inhibit NHE3 expression/activity 13, 38. Similar phenotypes have been observed in PT-specific AT1aR KO mice which have low systolic blood pressure and decreased NHE3 expression/activity 11, 12. Now we have also shown that Ks-AT1aR KO mice had lower systolic blood pressure and NHE3 expression than the control mice. However, the main novel finding of the present study is to demonstrate that deletion of AT1aR in renal tubules decreased the expression of renal Kir4.2 and depolarized the membrane potential of the PT, suggesting that AT1aR plays a key role in determining the baseline membrane basolateral membrane potential through controlling the expression of Kir4.2. Kir4.2 interacts with Kir5.1 to form basolateral Kir4.2/Kir5.1-heterotetramer which determines the negativity of PT membrane potential 4, 5. The role of Kir4.2 in determining the PT membrane potential has been previously confirmed by Bignon et al showing that Kir4.2-KO mice had a lower PT membrane potential than the corresponding control mice 4.
PT basolateral membrane potential provides the driving force for HCO3− exit through electrogenic 1Na+/3HCO3− cotransporter (NBCe1). Thus, depolarization is expected to inhibit HCO3− exit thereby causing HCO3− retention inside PT cells. High intracellular HCO3− concentrations are expected to alkalinize the intracellular pH thereby inhibiting NHE3, which plays an important role in the maintain Na+ and HCO3− absorption in the PT 39, 40. The notion that the basolateral K+ channel activity of the PT is able to regulate NHE3 function is also suggested by the finding that deletion of Kir4.2 in the PT inhibited HCO3− absorption in the PT, presumably due to depolarization of PT basolateral membrane-induced inhibition of NHE34. Because NHE3 expression in Kir4.2 KO mice was similar to the control mice, the finding suggests that AT1aR plays a role in modulating baseline Na+ and HCO3− absorption in the PT not only through modulating NHE3 protein expression but also by PT membrane potential. Not only determining PT baseline membrane potential, AT1aR is also responsible for angiotensin-II perfusion-induced hyperpolarization of the membrane potential of the PT since the deletion of AT1aR abolishes the effect of angiotensin-II. Also, we confirmed the previous finding that stimulation of renal AT1R by angiotensin-II perfusion stimulates the NHE3 41, 42. We speculate that angiotensin-II induced hyperpolarization of PT should increase Na+-HCO3− transport by NHE3.
AT1aR plays an important role in mediating Na+ and HCO3− absorption in the PT through NHE3 43, 44. This is supported by previous finding using micro-perfusion technique showing that the baseline HCO3− absorption was decreased by about one-third in global AT1aR KO mice comparing to the control 44. However, we have observed that urinary Na+ excretion (ENa) in Ks-AT1aR KO mice was similar to the control mice. We speculate that Na+ transport in the nephron segments other than PT may compensate decreased Na+ transport due to NHE3 inhibition thereby maintaining a normal baseline renal Na+ reabsorption. This view is also supported by the finding that amiloride-sensitive Na+ currents in the CCD is increased in the Ks-AT1aR KO mice. Moreover, the effect of AT1aR-deletion on baseline ENa may be different between mouse models in which whether Agtr1a gene is deleted by conditional knockout or constitutive deletion. Zhou et al have reported that the mice (B6.129P2 background) with constitutive deletion of AT1aR in the PC had increased baseline-ENa 17.
The second finding of the present study is that angiotensin-II perfusion hyperpolarized the DCT membrane potential, an effect was completely absent in Ks-AT1aR KO mice. This suggests that AT1aR in the DCT mediates angiotensin-II-induced stimulation of Kir4.1/Kir5.1. Since Kir4.1/Kir5.1 activity of the DCT determines the NCC activity/expression 2, we speculate that angiotensin-II perfusion-induced stimulation of Kir4.1/Kir5.1 should contribute the stimulatory effect of angiotensin-II on NCC 14. We need additional experiments to prove this hypothesis. Although the stimulation of AT1aR activates the Kir4.1/Kir5.1 of the DCT, AT1aR may not be essential for determining the baseline activity of Kir4.1 and Kir5.1 in the DCT because deletion of AT1aR had no effect on Kir4.1/Kir5.1 activity/expression and DCT membrane potential. This is in sharp contrast to AT1aR of the PT which determines the baseline activity of Kir4.2/Kir5.1. We speculate that dietary K+-intake or plasma K+-concentration rather than AT1aR is the primary factor determining the baseline Kir4.1/Kir5.1 expression/activity in the DCT. It is well documented that Kir4.1/Kir5.1 of the DCT is a key member of “K+-sensor-system’ in the kidney 6, 45. A large body of evidence have indicated that DCT Kir4.1/Kir5.1 and NCC work in concert with ENaC and ROMK in the ASDN to regulate renal K+ excretion and maintaining K+ homeostasis 6, 7, 45, 46. Because plasma K+ concentrations were similar between the control and AT1aR KO mice, deletion of AT1aR had no effect on baseline Kir4.1/Kir5.1 activity/expression in DCT. The mechanism by which Ang-II stimulates Kir4.1 and Kir4.2 is not explored. AT1aR activation is known to increase phospholipase-C-dependent PtdIns(4,5)P2 generation which is a potent stimulator of inwardly-rectifying-K+ channel 47–49, Further experiments are required to explore whether PtdIns(4,5)P2 or other factors mediates the effect of angiotensin-II on Kir4.1 or Kir4.2.
Our previous experiments have demonstrated that the inhibition of AT1aR with losartan decreased baseline-activity of ENaC in the DCT2/CNT but to a lesser degree on ENaC in the CCD 20, suggesting the role of AT1aR in regulating baseline activity of ENaC in the DCT2/CNT. However, we did not observe decreased ENaC activity in DCT2/CNT of Ks-AT1aR KO mice. It is possible that discrepancy between two studies is due to different time intervals of AT1aR inhibition. In previous experiments, amiloride-sensitive Na+ currents in the DCT2/CNT were measured in the mice treated with losartan for 3 days. In contrast, we have now measured ENaC activity in the mice at least two weeks after AT1aR deletion. Consequently, an increase in aldosterone may compensate for ENaC inhibition induced by AT1aR deletion. The argument is also supported by the finding that ENaC activity in the CCD was slightly increased in Ks-AT1aR KO mice, presumably due to increased aldosterone levels.
Supplementary Material
Pathophysiological Novelty and Relevance.
1). What is new?
AT1aR determines the baseline Kir4.2 expression in the PT and the deletion of AT1aR depolarizes the PT membrane potential.
AT1aR mediates angiotensin-II-induced hyperpolarization of PT membrane.
AT1aR mediates angiotensin-II-induced hyperpolarization of DCT membrane.
2). What is relevant?
We identify a novel mechanism by which AT1aR regulates baseline NHE3 function by targeting proximal-tubule Kir4.2/Kir5.1 activity.
Our study indicates that angiotensin-II-induced-stimulation of Kir4.2/Kir5.1 in PT and Kir4.1/Kir5.1 in DCT may be involved in angiotensin-II-induced-stimulation of NHE3 and thiazide-sensitive-Na-Cl-cotransporter.
Clinical/Pathophysiological Implications?
AT1aR determines the baseline membrane potential of PT by controlling Kir4.2 expression/activity but AT1aR does not determine the baseline Kir4.1/Kir5.1 activity/expression. However, AT1aR is required for angiotensin-II-induced hyperpolarization of basolateral membrane of PT and DCT.
Perspective.
AT1aR plays an important role in determining the baseline expression of Kir4.2 and basolateral membrane potential of the PT. Since the basolateral membrane potential of the PT regulates electrogenic NBCe1 activity, inhibition of Kir4.2 expression/activity should contribute to the compromised PT membrane transport in Ks-AT1aR mice. Also, although AT1aR has no effect on the baseline expression of Kir4.1 and DCT membrane potential, angiotensin-II-induced hyperpolarization of DCT membrane should contribute to the stimulation of NCC. Thus, targeting Kir4.2 or Kir4.1 could offset the effect of angiotensin-II on NHE3 and NCC, respectively.
Sources of Funding:
The work is supported by NIH grant RO1DK133220 (WHW/DHE) and RO1DK136491 (DHL). Yu-Xiao is supported by NSF Heilongjiang Province#LH2022C105.
List of Nonstandard Abbreviations
- PT
Proximal-tubule
- TAL
Thick ascending limb
- DCT
Distal convoluted tubule
- CNT
connecting tubule
- CCD
Cortical collecting duct
- ROMK
Renal outer medullary potassium channel
- NKCC2
Type 2 Na+-K+-2Cl− transporter
- Ks-AT1aR KO mice
Kidney-tubule-specific-AT1aR-knockout mice
Footnotes
Disclosure No
References
- 1.Hebert SC, Desir G, Giebisch G, Wang W. Molecular diversity and regulation of renal potassium channels. Physiol. Rev. 2005;85:319–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang W-H, Lin D-H. Inwardly rectifying k+ channels 4.1 and 5.1 (kir4.1/kir5.1) in the renal distal nephron. American Journal of Physiology-Cell Physiology. 2022;323:C277–C288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang L, Xu Y, Gravotta D, Frindt G, Weinstein AM, Palmer LG. Enac and romk channels in the connecting tubule regulate renal k+ secretion. Journal of General Physiology. 2021;153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bignon Y, Pinelli L, Frachon N, Lahuna O, Figueres L, Houillier P, Lourdel Sp, Teulon J, Paulais M. Defective bicarbonate reabsorption in kir4.2 potassium channel deficient mice impairs acid-base balance and ammonia excretion. Kidney International. 2020;97:304–315 [DOI] [PubMed] [Google Scholar]
- 5.Tucker SJ, Imbrici P, Salvatore L, D’Adamo MC, Pessia M. Ph dependence of the inwardly rectifying potassium channel, kir5.1, and localization in renal tubular epithelia. J. Biol. Chem. 2000;275:16404–16407 [DOI] [PubMed] [Google Scholar]
- 6.Su XT, Ellison DH, Wang WH. Kir4.1/kir5.1 in the dct plays a role in the regulation of renal k+ excretion. American Journal of Physiology-Renal Physiology. 2019;316:F582–F586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuevas CA, Su XT, Wang MX, Terker AS, Lin DH, McCormick JA, Yang C-L, Ellison DH, Wang WH. Potassium sensing by renal distal tubules requires kir4.1. J. Am. Soc. Nephrol. 2017;28:1814–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palygin O, Levchenko V, Ilatovskaya DV, Pavlov TS, Pochynyuk OM, Jacob HJ, Geurts AM, Hodges MR, Staruschenko A. Essential role of kir5.1 channels in renal salt handling and blood pressure control. JCI Insight. 2017;2:pil 92331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomilin VN, Zaika O, Subramanya AR, Pochynyuk O. Dietary k+ and clgêæ independently regulate basolateral conductance in principal and intercalated cells of the collecting duct. Pfl++gers Archiv - European Journal of Physiology. 2018;470:339–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang C, Wang L, Zhang J, Su X-T, Lin DH, Scholl UI, Giebisch G, Lifton RP, Wang WH. Kcnj10 determines the expression of the apical na-cl cotransporter (ncc) in the early distal convoluted tubule (dct1). Proc Natl Acad Sci USA. 2014;111:11864–11869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gurley SB, Riquier-Brison ADM, Schnermann J, Sparks MA, Allen AM, Haase VH, Snouwaert JN, Le TH, McDonough AA, Koller BH, Coffman TM. At1a angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metabolism. 2011;13:469–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nelson JW, McDonough AA, Xiang Z, Ralph DL, Robertson JA, Giani JF, Bernstein KE, Gurley SB. Local and downstream actions of proximal tubule angiotensin ii signaling on na+ transporters in the mouse nephron. American Journal of Physiology-Renal Physiology. 2021;321:F69–F81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Hatano R, Xu S, Wan L, Yang L, Weinstein AM, Palmer L, Wang T. Gender difference in kidney electrolyte transport. I. Role of at1a receptor in thiazide-sensitive na+-cl− cotransporter activity and expression in male and female mice. American Journal of Physiology-Renal Physiology. 2017;313:F505–F513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Lubbe N, Lim CH, Fenton RA, Meima ME, Jan Danser AH, Zietse R, Hoorn EJ. Angiotensin ii induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter independent of aldosterone. Kidney International. 2011;79:66–76 [DOI] [PubMed] [Google Scholar]
- 15.Stegbauer J, Gurley SB, Sparks MA, Woznowski M, Kohan DE, Yan M, Lehrich RW, Coffman TM. At1 receptors in the collecting duct directly modulate the concentration of urine. Journal of the American Society of Nephrology. 2011;22:2237–2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sandberg MB, Riquier ADM, Pihakaski-Maunsbach K, McDonough AA, Maunsbach AB. Ang ii provokes acute trafficking of distal tubule na+-cl− cotransporter to apical membrane. American Journal of Physiology-Renal Physiology. 2007;293:F662–F669 [DOI] [PubMed] [Google Scholar]
- 17.Li XC, Leite APO, Zheng X, Zhao C, Chen X, Zhang L, Zhou X, Rubera I, Tauc M, Zhuo JL. Proximal tubule-specific deletion of angiotensin ii type 1a receptors in the kidney attenuates circulating and intratubular angiotensin ii-induced hypertension in pt-agtr1a−/− mice. Hypertension. 2021;77:1285–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He P, Klein J, Yun CC. Activation of na+/h+ exchanger nhe3 by angiotensin ii is mediated by inositol 1,4,5-triphosphate (ip3) receptor-binding protein released with ip3 (irbit) and ca2+/calmodulin-dependent protein kinase ii*. Journal of Biological Chemistry. 2010;285:27869–27878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nguyen MTX, Han J, Ralph DL, Veiras LC, McDonough AA. Short-term nonpressor angiotensin ii infusion stimulates sodium transporters in proximal tubule and distal nephron. Physiological Reports. 2015;3:e12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu P, Gao Z, Zhang D, Duan X, Terker AS, Lin D, Ellison DH, Wang W. Effect of angiotensin ii on enac in the distal convoluted tubule and in the cortical collecting duct of mineralocorticoid receptor deficient mice. Journal of the American Heart Association. 2020;9:e014996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang T, Giebisch G. Effects of angiotensin ii on electrolyte transport in the early and late distal tubule in rat kidney. Am. J. Physiol. 1996;271:F143–F149 [DOI] [PubMed] [Google Scholar]
- 22.Zhang D-D, Zheng J-Y, Duan X-P, Lin D-H, Wang W-H. Romk channels are inhibited in the aldosterone-sensitive distal nephron of renal tubule nedd4–2-deficient mice. American Journal of Physiology-Renal Physiology. 2022;322:F55–F67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herrera M, Sparks MA, Alfonso-Pecchio AR, Harrison-Bernard LM, Coffman TM. Response to lack of specificity of commercial antibodies leads to misidentification of angiotensin type-1 receptor protein. Hypertension. 2013;61:e32–e32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sparks MA, Stegbauer J, Chen D, Gomez JA, Griffiths RC, Azad HA, Herrera M, Gurley SB, Coffman TM. Vascular type 1a angiotensin ii receptors control bp by regulating renal blood flow and urinary sodium excretion. Journal of the American Society of nephrology. 2015;26:2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J, Xu S, Yang L, Yang J, Wang CJ, Weinstein AM, Palmer LG, Wang T. Sex difference in kidney electrolyte transport ii: Impact of k+ intake on thiazide-sensitive cation excretion in male and female mice. American Journal of Physiology-Renal Physiology. 2019;317:F967–F977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Navar LG, Mitchell KD, Harrison-Bernard LM, Kobori H, Nishiyama A. Review: Intrarenal angiotensin ii levels in normal and hypertensive states. Journal of the Renin-Angiotensin-Aldosterone System. 2001;2:S176–S184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen L, Chou C-L, Knepper MA. Targeted single-cell rna-seq identifies minority cell types of kidney distal nephron. Journal of the American Society of Nephrology. 2021;32:886–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Onishi A, Fu Y, Patel R, Darshi M, Crespo-Masip M, Huang W, Song P, Freeman B, Kim YC, Soleimani M, Sharma K, Thomson SC, Vallon V. A role for tubular na+/h+ exchanger nhe3 in the natriuretic effect of the sglt2 inhibitor empagliflozin. American Journal of Physiology-Renal Physiology. 2020;319:F712–F728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pessia M, Imbrici P, D’Adamo MC, Salvatore L, Tucker SJ. Differential ph sensitivity of kir4.1 and kir4.2 potassium channels and their modulation by heteropolymerisation with kir5.1. J. Physiol. 2001;532:359–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forrester SJ, Booz GW, Sigmund CD, Coffman TM, Kawai T, Rizzo V, Scalia R, Eguchi S. Angiotensin ii signal transduction: An update on mechanisms of physiology and pathophysiology. Physiological Reviews. 2018;98:1627–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schrankl J, Fuchs M, Broeker K, Daniel C, Kurtz A, Wagner C. Localization of angiotensin ii type 1 receptor gene expression in rodent and human kidneys. American Journal of Physiology-Renal Physiology. 2021;320:F644–F653 [DOI] [PubMed] [Google Scholar]
- 32.Brooks HL, Allred AJ, Beutler KT, Coffman TM, Knepper MA. Targeted proteomic profiling of renal na(+) transporter and channel abundances in angiotensin ii type 1a receptor knockout mice. Hypertension. 2002;39:470–473 [DOI] [PubMed] [Google Scholar]
- 33.San Cristobal P, Pacheco-Alvarez D, Richardson C, Ring AM, Vazquez N, Rafiqi FH, Chari D, Kahle KT, Leng Q, Bobadilla NA, Hebert SC, Alessi DR, Lifton RP, Gamba G. Angiotensin ii signaling increases activity of the renal na-cl cotransporter through a wnk4-spak-dependent pathway. Proceedings of the National Academy of Sciences. 2009;106:4384–4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shibata S, Arroyo JP, Casta+¦eda-Bueno Ma, Puthumana J, Zhang J, Uchida S, Stone KL, Lam TT, Lifton RP. Angiotensin ii signaling via protein kinase c phosphorylates kelch-like 3, preventing wnk4 degradation. Proceedings of the National Academy of Sciences. 2014;111:15556–15561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen D, Stegbauer J, Sparks MA, Kohan D, Griffiths R, Herrera M, Gurley SB, Coffman TM. Impact of angiotensin type 1a receptors in principal cells of the collecting duct on blood pressure and hypertension. Hypertension. 2016;67:1291–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen MTX, Lee DH, Delpire E, McDonough AA. Differential regulation of na+ transporters along nephron during ang ii-dependent hypertension: Distal stimulation counteracted by proximal inhibition. American Journal of Physiology-Renal Physiology. 2013;305:F510–F519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Veiras LC, Han J, Ralph DL, McDonough AA. Potassium supplementation prevents sodium chloride cotransporter stimulation during angiotensin ii hypertension. Hypertension. 2016;68:904–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ito M, Oliverio MI, Mannon PJ, Best CF, Maeda N, Smithies O, Coffman TM. Regulation of blood pressure by the type 1a angiotensin ii receptor gene. Proceedings of the National Academy of Sciences. 1995;92:3521–3525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schultheis PJ, Clarke LL, Meneton P, Miller ML, Soleimani M, Gawenis LR, Riddle TM, Duffy JJ, Doetschman T, Wang T, Giebisch G, Aronson PS, Lorenz JN, Shull GE. Renal and intestinal absorptive defects in mice lacking the nhe3 na+/h+ exchanger. Nat. Genet. 1998;19:282–285 [DOI] [PubMed] [Google Scholar]
- 40.Fenton RA, Poulsen SB, de la Mora Chavez S, Soleimani M, Dominguez Rieg JA, Rieg T. Renal tubular nhe3 is required in the maintenance of water and sodium chloride homeostasis. Kidney International. 2017;92:397–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riquier-Brison ADM, Leong PKK, Pihakaski-Maunsbach K, McDonough AA. Angiotensin ii stimulates trafficking of nhe3, napi2, and associated proteins into the proximal tubule microvilli. American Journal of Physiology-Renal Physiology. 2010;298:F177–F186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.du Cheyron D, Chalumeau C, Defontaine N, Klein C, Kellermann O, Paillard M, Poggioli J. Angiotensin ii stimulates nhe3 activity by exocytic insertion of the transporter: Role of pi 3-kinase. Kidney International. 2003;64:939–949 [DOI] [PubMed] [Google Scholar]
- 43.Du Z, Ferguson W, Wang T. Role of pkc and calcium in modulation of effects of angiotensin ii on sodium transport in proximal tubule. American Journal of Physiology-Renal Physiology. 2003;284:F688–F692 [DOI] [PubMed] [Google Scholar]
- 44.Du Z, Wan L, Yan Q, Weinbaum S, Weinstein AM, Wang T. Regulation of glomerulotubular balance: Ii: Impact of angiotensin ii on flow-dependent transport. American journal of physiology. Renal physiology. 2012;303 11:F1507–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoorn EJ, Gritter M, Cuevas CA, Fenton RA. Regulation of the renal nacl cotransporter and its role in potassium homeostasis. Physiological Reviews. 2020;100:321–356 [DOI] [PubMed] [Google Scholar]
- 46.Terker A-S, Zhang C, McCormick J-A, Lazelle R-A, Zhang C, Meermeier N-P, Siler D-A, Park H-J, Fu Y, Cohen D-M, Weinstein A-M, Wang WH, Yang CL, Ellison D-H. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metabolism. 2015;21:39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hansen SB, Tao X, MacKinnon R. Structural basis of pip2 activation of the classical inward rectifier k+ channel kir2.2. Nature. 2011;477:495–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang C-L, Feng S, Hilgemann DW. Direct activation of inward rectifier potassium channels by pip2 and its stabilization by gbg. Nature. 1998;391:803–806 [DOI] [PubMed] [Google Scholar]
- 49.Tsilosani A, Gao C, Zhang W. Aldosterone-regulated sodium transport and blood pressure. Frontiers in Physiology. 2022;13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mutchler SM, Shi S, Whelan SCM, Kleyman TR. Validation of commercially available antibodies directed against subunits of the epithelial na+ channel. Physiological Reports. 2023;11:e15554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.