

Abstract
Artificial chromosomes have been claimed to be the ideal vector for gene therapy, but their use has been hampered by an inability to produce stable and well designed molecules. We have used a structurally defined minichromosome to clone the human cystic fybrosis transmembrane conductance regulator (CFTR) locus. To guarantee the presence of the proper regulatory elements, we used the 320 kb yeast artificial chromosome (YAC) 37AB12 with the intact CFTR gene and upstream sequences. The resulting minichromosome was analyzed for the presence of the entire CFTR gene and for its functional activity by molecular and functional methods. We have identified clones showing the presence of both the transcript and the CFTR protein. Moreover, in the same clones, a chloride secretory response to cAMP was detected. Mitotic and molecular stability after prolonged growth without selection demonstrated that the constructs were stable. This is the first example of a structurally known minichromosome made to contain an active therapeutic gene.
INTRODUCTION
Since the construction of the first artificial chromosome (yeast artificial chromosome, YAC) and the claim that genes can be used to correct genetic defects (gene therapy), it has been hypothesized that artificial chromosomes could be the ideal vectors for gene therapy. Mammalian artificial chromosomes (MACs) can be produced by transfection of large blocks of alphoid DNA into human cells (Ebersole et al., 2000). Alternatively, they can be obtained by reducing the size of a natural chromosome with multiple rounds of fragmentation induced by the insertion of a telomeric fragment in an interstitial position (Mills et al., 1999) or by irradiation (Carine et al., 1986). The first class of molecules is referred to as de novo MACs and the latter as minichromosomes. Both types of molecules are mitotically stable, are in the 1–10 Mb size range and are mainly composed of alphoid DNA, but they are quite different from a structural point of view: (i) de novo MACs, with the exception of few linear examples (Harrington et al., 1997), are suspected to be circular, larger than the input DNAs and most probably composed of multimers of alphoid arrays (Ebersole et al., 2000); and (ii) minichromosomes maintain the structure of the parental chromosomes, are linear and smaller than the parental chromosome, are capped with functional telomeres and contain a functional centromere of reduced size (Mills et al., 1999; Yang et al., 2000; Auriche et al., 2001).
Artificial chromosomes offer some important advantages for gene transfer: they are episomal and replicate autonomously, they are stably retained in the host cells at 1–2 copies per cell, they are not immunogenic and they are suited to hosting large fragments of exogenous DNA. Moreover, they avoid detrimental mutagenic effects that could be caused by the insertion of exogenous genes in the genome and positional effects that could silence the gene itself.
Minichromosomes and de novo MACs have already been considered as vectors for delivering therapeutic genes into cultured cells. We have used a minichromosome, MC1, as a cloning and expression vector of the human IL-2 genes (Guiducci et al., 1999). Other groups have assembled de novo MACs with the human HPTR gene (Grimes et al., 2001; Mejia et al., 2001). Moreover, a small accessory chromosome has been used to clone a human HPTR minigene, and the resulting construct was demonstrated to be germ-line transmitted in mice (Voet et al., 2001).
Production of de novo MACs is relatively simple with respect to the procedures used to obtain minichromosomes, but, since the resulting molecules remain uncharacterized with respect to their structure and no information is available concerning their structural stability, we focused our attention on a minichromosome, MC1 (Auriche et al., 2001), to evaluate its therapeutic potential for the treatment of one of the most common genetic diseases, cystic fibrosis (CF). Several clinical trials have demonstrated that CF defects can be corrected by gene therapy but only for a limited time period (Alton and Geddes, 1998), partially because of the host immune response, which destroys the corrected cells. In this paper, we report the cloning and functional analysis of the human CF transmembrane conductance regulator (CFTR) gene into the minichromosome MC1.
RESULTS
Introduction of the intact CFTR gene into MC1
To introduce CFTR into the minichromosome, we used the yCFIRES dicistronic YAC, which contains the human CFTR locus and a picornaviral IRES-β-geo cassette fused to the 3′ end of the CFTR gene (Vassaux and Huxley, 1997). yCFIRES allows expression of both CFTR and geo (lacZ and neo activity) genes from the same mRNA molecules, thus providing an easy, although indirect, assay for CFTR expression. The YAC was introduced into CHO cells containing MC1 (CHO-MC1) by protoplast fusion (see Supplementary data available at EMBO reports Online). After ∼10 days of growth with selection, 30 G418-resistant clones appeared (P clones). All of them were initially screened by staining for the presence of the lacZ gene. Six clones showed significant X-gal staining (Figure 1A): the P34 and P36 clones showed the lowest level of activity; in P16, we observed an increase in the number of stained cells; and in P37, P38 and P39, most cells were highly stained. The presence of CFTR in these clones was shown by CFTR-PCR for exon 10 (data not shown). The remaining clones were both lacZ and CFTR-PCR negative. The CFTR gene in the selected clones was mapped by FISH analysis with a human CFTR probe that does not hybridize to both CHO and MC1 chromosomes (data not shown). Three of the six clones (P37, P38 and P39) showed unique hybridization signals unequivocally localized in MC1 (Figure 1B). The remaining clones showed multiple hybridization signals preferentially localized to the centromeric region of some hamster chromosomes, probably due to the presence of pericentromeric DNA in the transfection procedure. P16 showed hybridization also in MC1 (data not shown). Since P37, P38 and P39 showed only one hybridization signal mapped to MC1, it is reasonable to surmise that the human CFTR was integrated in the minichromosome, although we cannot exclude the integration of small portions of the gene undetectable by FISH.

Fig.1. Analysis of MC1-CFTR clones. (A) β-galactosidase staining with X-gal for the indicated clones. MC1 (CHO-MC1 cells) represents the negative control. (B) FISH analysis of the clones indicated in each panel with a CFTR probe. Chromosomes were counterstained with propidium iodide and the probe was revealed with avidin-FITC. The arrow points to the minichromosome.
MC1-CFTR contains one copy of the CFTR gene and is stable in the absence of selection
To confirm the presence of the CFTR gene in the P clones, we performed exon PCR for several selected exons (1, 2, 5, 7, 10, 13, 16, 20 and 23) spanning the entire length of the gene. Figure 2A shows analysis of P39, which revealed the expected band for each exon. Similar results were obtained with P16, P37 and P38 (data not shown). This result, together with the presence of an active β-geo cassette (β-galactosidase positivity), strongly suggest that the entire yCFIRES construct was integrated in these clones. To confirm this hypothesis, we searched for additional sequences of the original construct yCFIRES. By PCR, we demonstrated the presence of Ura3 sequences (right arm) and the Amp-ori fragment (left arm) in P16, P37, P38 and P39 (see Supplementary figure 1). P37, P38 and P39 were chosen to determine CFTR copy number and the mitotic stability of the minichromosome.

Fig. 2. Structural analysis of MC1-CFTR. (A) PCR products from the indicated CFTR exons (1, 2, 5, 7, 10, 13, 16, 20 and 23; see Methods for primers). For each exon MC1 (negative control), T84 (positive control) and P39 DNA samples are reported from left to right. (B) Logarithmic plot of the ratio T/S (CFTR band intensity/competitor band intensity) versus competitor DNA. The data refer to 15 µl aliquots of each PCR. Dashed lines represent P39 (squares) and T84 (triangles), respectively; the corresponding continuous lines represent the regression. (C) Mitotic stability of MC1-CFTR after growth with (+) and without (–) selection for the indicated number of generations. The majority of the cells contained one minichromosome per cell. (D) Alu-PCR products from MC1, P37, P38 and P39 after 60 generations of growth in the presence (+G418) and absence (–G418) of selection. MC1 represents CHO-MC1 cells.
CFTR copy number was calculated by competitive PCR. One example, representative of three independent experiments, is reported in Figure 2B, in which the intersection of the P39 and human lines with a y-value of 1 demonstrates the presence of half CFTR target in P39 with respect to human T84 cells. Similar results were obtained with P37 and P38. P39 was further analyzed by quantitative PCR using the limiting dilution approach and taking into consideration the CFTR target with respect to the actin internal standard (see Supplementary data). This analysis confirmed the presence of half CFTR target with respect to the actin standard. Taking into consideration that 90–95% of the cells contain one minichromosome per cell, the CFTR amount is consistent with the presence of one copy of the gene per cell. The mitotic stability of MC1-CFTR in clones P37, P38 and P39 was determined after 30 and 60 generations of growth with and without selection. Following in situ hybridization with Sat2 and CFTR probes, no significant difference in the number of metaphases containing the minichromosome was observed at the two time points and at the two growth conditions analyzed (Figure 2C). To monitor potential structural changes of the minichromosome after prolonged growth, we analyzed the Alu-PCR profile, which is characteristic of the minichromosome and derives from the amplification of sequences flanked by inverted Alu repeats <3 kb apart (Ledbetter et al., 1990). At the two time points, DNA samples prepared from cells with and without selection were amplified with an Alu primer. No variation of the Alu-PCR pattern was visible in any of the samples analyzed (Figure 2D). Moreover, it can be seen that the P clones showed the same profile of MC1, suggesting that CFTR integrated into the pericentromeric region of MC1, which is poor in Alu repeats. The presence of CFTR in the same DNA samples was determined by PCRs for exon 10 (data not shown).
Having shown that P37, P38 and P39 contain only one copy of the CFTR and that the CFTR signal localized to the minichromosome, it is reasonable to surmise that not more than one copy of the CFTR gene was integrated into MC1.
CFTR activity in MC1-CFTR clones
RT–PCR for two different regions of CFTR mRNA revealed no amplification products for CHO-MC1 cells (see Supplementary figure 3) and the expected products for P16, P34, P36, P37, P38 and P39 and the positive control (T84 cells), indicating that a substantial amount of CFTR transcript was produced in the P clones. Northern blot analysis revealed the full-length transcript in the positive control (T84) and in P16, P37, P38 and P39, although at different levels. Additional bands did not appear even after prolonged exposure, suggesting that only the full-length transcript was present. No signal was detected in P34 and P36, probably due to a much lower level of transcription (Figure 3A), nor in the negative control CHO-MC1 or CHO cells. Human CFTRp was detected by immunoprecipitation of cell lysates with an antibody, specific for the human protein and directed to the R-domain of CFTRp (MATG 1105; Dechecchi et al., 1993), phosphorylation with [γ-32P]ATP and analysis by SDS–PAGE (Figure 3B). P37, P38 and P39 produced mature, glycosylated CFTRp that could be phosphorylated in vitro. A similar CFTR profile was seen in the positive control HT29 cells, whereas no corresponding band was observed in the negative controls (CHO and CHO-MC1 lysates). P16 was negative to this assay (data not shown), possibly due to an insufficient amount of CFTR mRNA, as expected from the low level of mRNA it showed in the northern blot (Figure 3A).
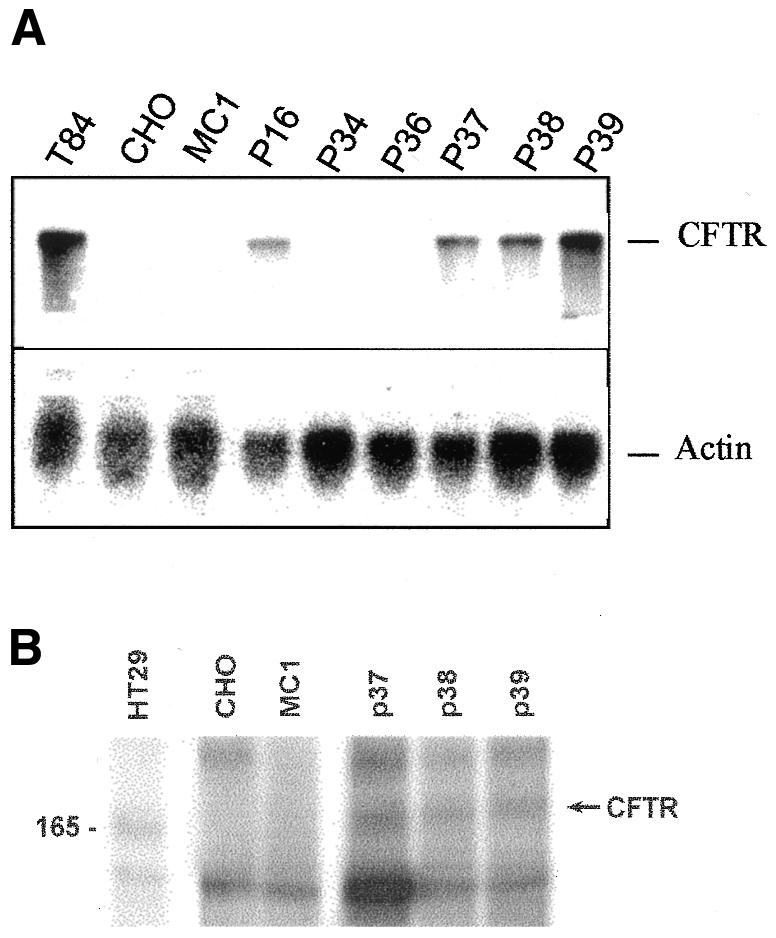
Fig. 3. CFTR activity in MC1-CFTR clones. (A) Northern blot analysis of the indicated RNA samples using a CFTR probe (upper panel). RNA loading control using an actin probe (lower panel). (B) SDS–PAGE of cell lysates immunoprecipitated with MATG1105 and phosphorylated with [γ-32P]ATP. The arrow points to mature CFTR, and the molecular marker on the left side of the figure is indicated in kilodaltons. Samples: T84 (A) and HT29 (B) cells, positive controls; CHO and MC1 (CHO-MC1 cells), negative controls.
Indirect immunofluorescence was performed with an antibody directed against the first extracellular loop of human CFTRp and specific for the human protein (MATG1031; Demolombe et al., 1996). Figure 4 shows that P37, P38 and P39 presented CFTRp at intracellular compartments reminiscent of either Golgi or endoplasmic reticulum, as suggested by the diffused staining forming a halo around the nuclei (Figure 4). P16 did not reveal any signals, confirming the absence of sufficient amounts of transcript and protein in this clone (data not shown). P39 also exhibited a plasma membrane staining (arrows in Figure 4D). This result was confirmed by confocal microscopy, which revealed most of the CFTRp around the nuclei and a subfraction on the plasma membrane (data not shown). To determine exactly how many cells of different clones had CFTRp on the plasma membrane, we have developed a cytofluorimetric assay on viable cells. P37, P38 and P39 presented plasma membrane staining in ∼3–4% of cells. To determine whether part of the CFTRp was localized on vesicles below the plasma membrane, we treated cells with 5 µM forskolin, which has been shown to induce CFTRp transport from intracellular vesicles to the cell surface (Prince et al., 1994). Whereas P37 and P38 did not show any significant change in CFTRp production, P39 exhibited a 60% increase in the CFTRp surface signal (see Supplementary figure 4). This result strongly suggests that the CFTRp produced in P39 may traffic to plasma membrane and possibly this process will be more efficient in the appropriate cellular background, i.e. human differentiated epithelial cells.
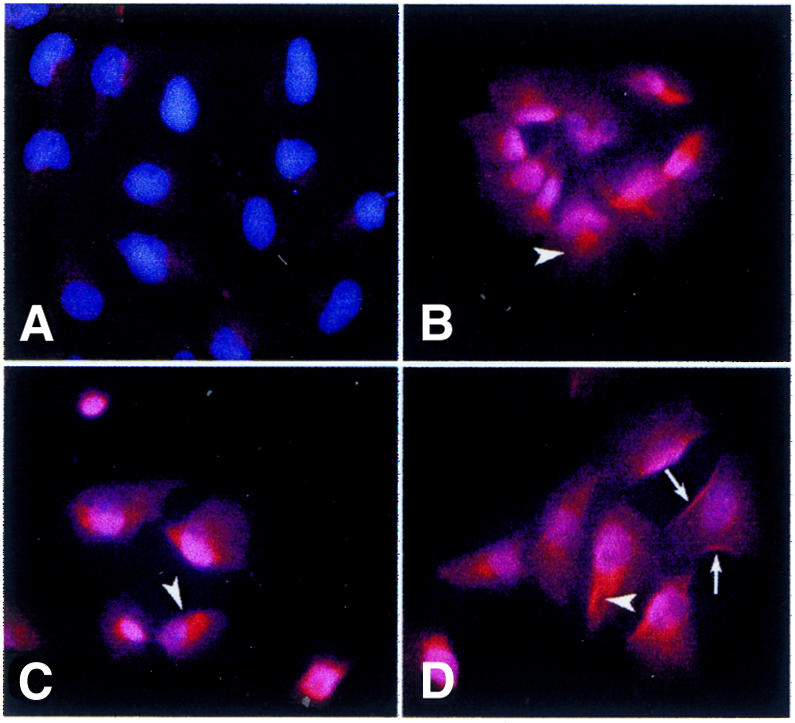
Fig. 4. CFTR subcellular localization in MC1-CFTR clones. (A) CHO-MC1; (B–D) P37–P39. Cells were immunostained with MATG1031, revealed with a second antibody (Texas Red) and counterstained with Hoechst 33258. Arrowheads point to specific staining around nuclei in a position reminiscent of Golgi; arrows point to plasma membrane staining.
To determine possible clonal variation of P39 with respect to CFTRp production and processing, we carried out indirect immunofluorescence as previously described. Comparative analysis of six subclones, derived from single cells, with the original P39 did not show any substantial difference (data not shown).
Human CFTR is functional in MC1-CFTR clones
The presence of functional CFTR protein at the plasma membrane of those clones showing intracellular and plasma membrane CFTRp was determined by means of the cAMP-stimulated 36Cl– efflux assay (Figure 5). We looked for a difference between the response of P clones and CHO-MC1 cells. After an interval at rest, cAMP stimulation was obtained by the addition of the membrane-permeable cAMP analog 8-(4-chlorophenylthio)adenosine-3′-5′-cyclic monophosphate (CPT-cAMP) The 36Cl– efflux exhibited by P37 was at the limit of detection (data not shown), whereas P38 and P39 showed a significant efflux, statistically different from control cells, but their behavior was substantially different. P38 showed a faster efflux from the beginning of the experiment and no response after the addition of CPT-cAMP; the efflux was significantly reduced by 100 µM glibenclamide, a well-known inhibitor of CFTRp (Sheppard and Welsh, 1992), suggesting that this clone was constitutively active (Figure 5A and C). Conversely, the rate of 36Cl– efflux of P39 at rest was lower and similar to that of CHO-MC1 cells (Figure 5B). Application of CPT-cAMP caused an efflux increase significantly higher than that found in control cells, and this stimulated efflux was completely abolished when glibenclamide was present in the bathing solution (Figure 5D). 36Cl– efflux experiments performed on three P39 subclones showed an activity that was not different from the original clone P39 (data not shown). This assay showed CFTR activity in the three clones analyzed, although P39, which revealed a cAMP-regulated Cl– efflux, was the best functional clone, in agreement with its higher CFTR mRNA and protein levels.

Fig. 5 36Cl– efflux from cells stimulated with 500 µM CPT-cAMP. (A) P38 (n = 8) and MC1 (n = 20); (B) P39 (n = 10) and CHO-MC1 (n = 20); (C) P38 in the absence (n = 8) and presence (n = 4) of glibenclamide; (D) P39 in the absence (n = 10) and presence (n = 6) of glibenclamide. P38 in the absence (filled circles) and presence (open circles) of glibenclamide; P39 in the absence (filled squares) and presence (open squares) of glibenclamide; CHO-MC1 (triangles). Vertical bars are SEM. Asterisks indicate statistically significant differences (*P < 0.05, **P < 0.01, ***P < 0.005 and ****P < 0.001).
DISCUSSION
We report the assembly and functional analysis of a minichromosome containing the entire human CFTR locus (MC1-CFTR). Using a simple protocol, based on Sat2 cotransfection and subsequent yeast protoplast fusion, we obtained 13% integration of CFTR in the minichromosome. We used cotransfection, since it is a simple and reproducible procedure to insert genes into chromosomes (Raimondi et al., 1996; Guiducci et al., 1999), but, due to the extension of the region in which we directed the CFTR gene (Sat2 is ∼3 Mb long in MC1), the isolated clones may differ from each other, at least in the position of the CFTR gene with respect to the MC1 centromere and telomeres. Nonetheless, the finding of the CFTR full-length transcript and the protein of the expected sizes demonstrates that the gene was integrated in an intact form in a region permissive for expression, most probably the Sat2 pericentromere. Moreover, since the MC1-CFTR minichromosomes analyzed were mitotically highly stable even after prolonged growth in the absence of selection and did not show structural changes (although micro-lesions cannot be excluded), the insertion of the therapeutic gene did not modify the functional properties of the minichromosome.
It was previously demonstrated that CHO cells do not express endogenous CFTR but, nonetheless, can support the expression of either an integrated human CFTR locus with its own regulatory sequences (Mogayzel et al., 1997) or a cDNA construct with an heterologous promoter (Barriere et al., 2001), suggesting that the human gene is insensitive to CHO control. In the present work, the functional activity of CFTR cloned in MC1 and propagated as a free minichromosome in CHO cells was demonstrated at the various functional levels: mRNA transcription, protein translation and processing to plasma membrane, and functional activity of the chloride channel. In all these experiments, the CFTR detected in P37, P38 and P39 cells correlates to that of the human cells (T84 and HT29) used as positive controls. Strikingly, P16, which revealed a lower amount of CFTR mRNA with respect to P37, P38 and P39, did not show any protein and was negative to functional analysis. No activity was found in the negative control, which consisted of CHO cells containing the parental MC1. Although we cannot exclude a possible effect of the endogenous gene, the fact that functional CFTR activity was detected only in those clones that contained the human CFTR transcript and protein does not support this hypothesis but strongly indicates the presence of a functional human CFTR.
The clonal variation we found by the functional method and the low percentage of CHO cells with plasma membrane CFTR could be due to limiting factors that promote efficient protein traffic. In fact, CHO cells may not recapitulate the normal events occurring in differentiated human epithelial cells. But clonal variations could also be due to differences in the relative position of the gene with respect to the functional elements (centromere and telomeres) of the minichromosome and/or to small sequence rearrangements. Of the three clones made to contain CFTR-MC1, two showed limited CFTR activity, and the remaining one, P39, showed the presence of a cAMP-regulated channel specifically inactivated by a CFTRp inhibitor, demonstrating that it contains a functionally active CFTR gene. Of particular interest is the fact that P39 clones derived from single cell were almost identical in terms of protein immunolocalization and Cl– efflux.
The minichromosome in P39 represents a unique tool for in vitro and in vivo CF gene therapy and for studying the regulation of CFTR expression and function. Utilization of this molecule in gene therapy approaches is hindered at the moment by the absence of efficient and feasible transfer methods and the limited experimentation made to date with such types of molecules. Nonetheless, recent advances in chromosome transfer (de Jong et al., 2001) and stem cells production and manipulation open new possibilities for MC1-CFTR application.
METHODS
Cell lines.
The hamster cell line CHO-MC1 containing the minichromosome MC1 and the parental CHO are described in Guiducci et al. (1999). T84 and HT29 colon carcinoma human cells expressing high levels of CFTR were used as positive controls (Yoshimura et al., 1991).
β-galactosidase staining and PCR.
β-galactosidase staining for the presence of the lacZ gene was carried out according to standard procedures. PCRs were carried out with 100 ng DNA samples and primers specific for the CFTR exons. The primers for exons 1, 2, 5, 7, 10, 13, 16 and 23 were those reported in Zielenski et al. (1991); exon 20 was amplified with 20DJ5 (GTCACAGAAGTGATCCCATC) and 20DJB3 (CTGGCTAAGCCCTTTTGCTC). Competitive PCR was carried out according to Hirano et al. (2002), with minor modifications. Each 25 µl reaction contained 100 ng of DNA sample, 4 pmol of each primer (20DJ5 and 20DJB3), 5 µl of each competitor dilution, 1× Taq buffer and 0.25 IU of Taq (Ampli-Taq from Perkin Elmer, Applied Biosystems, Foster City, CA). The heterologous competitor DNA (see Supplementary data) was prepared by 2-fold serial dilutions of the 2 ng/µl stock solution. Three aliquots of each PCR (2.5, 5 and 15 µl) were analyzed by gel electrophoresis, stained with ethidium bromide and quantitated by an image analyzer (Kodak 1D; Eastman Kodak, New Haven, CT). Three independent experiments were performed with P39 and T84 DNAs. The ratio of the target (CFTR) to the standard (competitor) band intensity (T/S) was plotted against the amount of competitor, in a double logarithmic graphical representation. The amount of the target can be read at the intersection of the line with a y-value of 1. The regression line was calculated to take into consideration the experimental errors.
FISH and immunofluorescence.
In situ hybridization and immunofluorescence detection of CFTRp cDNA probe, purified from pBQ6.2 (Rommens et al., 1991), on metaphase chromosomes was performed according to Auriche et al. (2001).
CFTR mRNA and protein analysis.
RNA extracts were prepared by the guanidinium-isothiocyanate procedure, and northern blot analysis was carried out according to standard procedures. RNA filters were hybridized to the CFTR exon 11–13 probe; RNA loading was controlled using an actin probe.
Immunoprecipitation with MATG 1105 (Transgene, Strasbourg, France) and phosphorylation of CFTRp in vitro were performed as described by Galietta et al. (2000). For indirect immunofluorescence, after plating onto chamber slides, cells were fixed in 3% PAF, 2% sucrose, and permeabilized in HEPES buffer (20 mM HEPES pH 7.4, 0.3 M sucrose, 0.05 M NaCl, 3 mM MgCl2) containing 0.5% Triton X-100. Cells were incubated with anti-CFTR antibody MATG 1031 (1:200; Transgene) in PBS, 2% BSA, for 30 min at 37°C, washed, and decorated with Texas Red- conjugated secondary anti-mouse antibody (1:1000; Molecular Probes, Eugene, OR). Nuclei were counterstained with Hoechst 33258 (1:3000). Slides were mounted with 20% Mowiol (Calbiochem, La Jolla, CA). The omission of primary antibody resulted in the absence of staining.
Measurements of 36Cl– efflux.
Confluent CHO cells were incubated for 40 min at 37°C with a saline solution containing 1 µCi of 36Cl–. The loading medium was then discarded, and cells were washed with ice-cold 36Cl–-free medium. Experiments started with the addition of 1 ml of saline solution (see Supplementary data). Every minute, the efflux medium was replaced by another milliliter of solution. CPT-cAMP was applied to the solution 3 min after the beginning and maintained up to the end of the experiment. Cells were then lysed with 0.25 M NaOH. The radioactivity present in efflux samples and lysates was determined by liquid scintillation. The time course of 36Cl– efflux was expressed by plotting fractional efflux (FE) versus time. FE is the 36Cl– released in a single efflux interval normalized by the amount of residual 36Cl– at the beginning of that interval. For more details, see Supplementary data.
Supplementary data.
Supplementary data are available at EMBO reports Online.
Supplementary Material
Acknowledgments
ACKNOWLEDGEMENTS
Thanks are due to C. Huxley for providing yCFIRES and to Lorella Ranucci for technical support. D.C. received a fellowship from Associazione Lombarda Fibrosi Cistica. This work was supported by Telethon-Onlus grant N A.164 and partially by MURST (Modalità alternative di transgenesi animale: ricerche scientifiche sulla costruzione di mini- e micro-cromosomi artificiali) and “fondazione per la ricerca sulla fibrosi cistica”.
REFERENCES
- Alton E. and Geddes, D. (1998) Cystic fibrosis clinical trials. Adv. Drug Deliv. Rev., 30, 205–217. [DOI] [PubMed] [Google Scholar]
- Auriche C., Donini, P. and Ascenzioni, F. (2001) Molecular and cytological analysis of a 5.5 Mb minichromosome. EMBO rep., 2, 102–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barriere H., Poujeol, C., Tauc, M., Blasi, J.M., Counillon, L. and Poujeol, P. (2001) CFTR modulates programmed cell death by decreasing intracellular pH in Chinese hamster lung fibroblasts. Am. J. Physiol. Cell Physiol., 281, C810–C824. [DOI] [PubMed] [Google Scholar]
- Carine K., Solus, J., Waltzer, E., Manch-Citron, J., Hamkalo, B.A. and Scheffler, I.E. (1986) Chinese hamster cells with a minichromosome containing the centromeric region of human chromosome 1. Somat. Cell Mol. Genet., 12, 479–491. [DOI] [PubMed] [Google Scholar]
- Dechecchi M.C., Tamanini, A., Berton, G. and Cabrini, G. (1993) Protein kinase C activates chloride conductance in c127 cells stably expressing the cystic fibrosis gene. J. Biol. Chem., 268, 11321–11325. [PubMed] [Google Scholar]
- de Jong G., Telenius, A., Vanderbyl, S., Meitz, A. and Drayer, J. (2001) Efficient in-vitro transfer of a 60-Mb mammalian artificial chromosome into murine and hamster cells using cationic lipids and dendrimers. Chromosome Res., 9, 475–485. [DOI] [PubMed] [Google Scholar]
- Demolombe S., Baro, I., Bebok, Z., Clancy, J.-P., Sorscher, E.J., Thomas-Soumarmon, A., Pavirani, A. and Escande D. (1996) A method for the rapid detection of recombinant CFTR during gene therapy in cystic fibrosis. Gene Ther., 3, 685–694. [PubMed] [Google Scholar]
- Ebersole T.A., Ross, A., Clark, E., McGill, N., Schindelhauer, D., Cooke, H. and Grimes, B. (2000) Mammalian artificial chromosome formation from circular alphoid input DNA does not require telomere repeats. Hum. Mol. Genet., 9, 1623–1631. [DOI] [PubMed] [Google Scholar]
- Galietta L.J., Folli, C., Marchetti, C., Romano, L., Carpani, D., Conese, M. and Zegarra-Moran, O. (2000) Modification of transepithelial ion transport in human cultured bronchial epithelial cells by interferon-γ. Am. J. Physiol. Lung Cell. Mol. Physiol., 278, L1186–L1194. [DOI] [PubMed] [Google Scholar]
- Grimes B.R., Schindelhauer, D., McGill, N.I., Ross, A., Ebersole, T.A. and Cooke, H.J. (2001) Stable gene expression from a mammalian artificial chromosome. EMBO rep., 2, 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiducci C., Ascenzioni, F., Auriche, C., Piccolella, E., Guerrini, A.M. and Donini, P. (1999) Use of a human minichromosome as a cloning and expression vector for mammalian cells. Hum. Mol. Genet., 8, 1417–1424. [DOI] [PubMed] [Google Scholar]
- Harrington J.J., van Bokkeln, G., Mays, R.W., Gustashaw, K. and Willard, H.F. (1997) Formation of de novo centromeres and construction of first-generation human artificial chromosomes. Nat. Genet., 15, 345–355. [DOI] [PubMed] [Google Scholar]
- Hirano T., Haque, M. and Utiyama, H. (2002) Theoretical and experimental dissection of competitive PCR for accurate quantification of DNA. Anal. Biochem., 303, 57–65. [DOI] [PubMed] [Google Scholar]
- Ledbetter S.A., Garcia-Heras, J. and Ledbetter, D.H. (1990) ‘PCR-karyotype’ of human chromosomes in somatic cell hybrids. Genomics, 8, 614–622. [DOI] [PubMed] [Google Scholar]
- Mejia J.E., Willmot, A., Levy, E., Earnshaw, C. and Larin, Z. (2001) Functional complementation of a genetic deficiency with human artificial chromosomes. Am. J. Hum. Genet., 69, 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills W., Critcher, R., Lee, C. and Farr, C. (1999) Generation of a 2.4 Mb human centromere based minichromsome by targeted telomere-associated chromosome fragmentation in DT40. Hum. Mol. Genet., 8, 751–761. [DOI] [PubMed] [Google Scholar]
- Mogayzel P.J., Henning, K.A., Bittner, M.L., Novotny, E.A., Schwiebert, E.M., Guggino, W.B., Jiang, Y. and Rosenfeld, M.A. (1997) Functional human CFTR produced by stable chinese hamster ovary cell line derived using yeast artificial chromosomes. Hum. Mol. Genet., 6, 59–68. [DOI] [PubMed] [Google Scholar]
- Prince L.S., Workman, R.B. and Marchase, R.B. (1994) Rapid endocytosis of the cystic fibrosis transmembrane conductance regulator chloride channel. Proc. Natl Acad. Sci. USA, 91, 5192–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondi E., Balzaretti, M., Moralli, D., Vagnarelli, P., Tredici, F., Bensi, M. and De Carli, L. (1996) Gene targeting to the centromeric DNA of a human minichromosome. Hum. Gene Ther., 7, 1103–1109. [DOI] [PubMed] [Google Scholar]
- Rommens J.M., Dho, S., Bear, C.E., Kartner, N., Kennedy, D., Riordan, J.R., Tsui, L.C. and Foskett, J.K. (1991) cAMP-inducible chloride conductance in mouse fibroblast lines stably expressing the human cystic fibrosis transmembrane conductance regulator. Proc. Natl Acad. Sci. USA, 88, 7500–7504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard D.N. and Welsh, M.J. (1992) Effects of ATP-sensitive K+ channel regulators on cystic fibrosis transmembrane conductance regulator chloride currents. J. Gen. Physiol., 100, 573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassaux G. and Huxley, C. (1997) A dicistronic construct allows easy detection of human CFTR expression from YAC DNA in human cells. Nucleic Acids Res., 25, 4167–4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voet T., Vermeesch, J., Carens, A., Durr, J., Labaere, C., Duhamel, H., David, G. and Marynen, P. (2001) Efficient male and female germline transmission of a human chromosomal vector in mice. Genome Res., 11, 124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.W., Pendon, C., Yang, J., Haywood, N., Chand, A. and Brown, W.R.A. (2000) Human mini-chromosomes with minimal centromeres. Hum. Mol. Genet., 9, 1891–1902. [DOI] [PubMed] [Google Scholar]
- Yoshimura K., Nakamura, H., Trapnell, B.C., Dalemans, W., Pavirani, A., Lecocq, J.P. and Crystal, R.G. (1991) The cystic fibrosis gene has a ‘housekeeping’-type promoter and is expressed at low levels in cells of epithelial origin. J. Biol. Chem., 266, 9140–9144. [PubMed] [Google Scholar]
- Zielenski J., Rozmahel, R., Bozon, D., Kerem, B.S., Grzelczak, Z., Riordan, J.R., Rommens, J. and Tsui, L.C. (1991) Genomic DNA sequence of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics, 10, 214–228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.