

Abstract
Background:
The drive to expedite patient access for diseases with high unmet treatment needs has come with an increasing use of single-arm trials (SATs), especially in oncology. However, the lack of control arms in such trials creates challenges to assess and demonstrate comparative efficacy. External control (EC) arms can be used to bridge this gap, with various types of sources available to obtain relevant data.
Objective:
To examine the source of ECs in single-arm oncology health technology assessment (HTA) submissions to the National Institute for Health and Care Excellence (NICE) and the Pharmaceutical Benefits Advisory Committee (PBAC) and how this selection was justified by manufacturers and assessed by the respective HTA body.
Methods:
Single-arm oncology HTA submission reports published by NICE (England) and PBAC (Australia) from January 2011 to August 2021 were reviewed, with data qualitatively synthesized to identify themes.
Results:
Forty-eight oncology submissions using EC arms between 2011 and 2021 were identified, with most submissions encompassing blood and bone marrow cancers (52%). In HTA submissions to NICE and PBAC, the EC arm was typically constructed from a combination of data sources, with the company's justification in data source selection infrequently provided (PBAC [2 out of 19]; NICE [6 out of 29]), although this lack of justification was not heavily criticized by either HTA body.
Conclusion:
Although HTA bodies such as NICE and PBAC encourage that EC source justification should be provided in submissions, this review found that this is not typically implemented in practice. Guidance is needed to establish best practices as to how EC selection should be documented in HTA submissions.
Keywords: external controls, health technology assessment, oncology, real-world data, real-world evidence, single-arm trial
Plain language summary
What is this article about?
Diseases with high unmet needs are often assessed in trials with no comparator arm. As new treatments need to be compared with what is used in current clinical practice, data from other sources, called an external control arm, can be used to bridge the gap. The type of external control arm used and why it has been chosen should be explained when making submissions to a health technology agency.
What were the results?
Submissions made to two health technology agencies were reviewed to identify whether justification was provided for the type of external control arm used. This review demonstrated that this justification was rarely provided.
What do the results of the study mean?
These findings highlight the need for guidance to be established regarding the documentation of external control arms in submissions made to health technology agencies.
Randomized controlled trials (RCTs) are the cornerstone in determining the efficacy and safety of a new health technology against standard of care (SoC) or placebo through comparative efficacy analyses in a controlled environment [1,2]. Control arms in RCTs aim to isolate the effects of the investigated technology from those caused by other treatment-effect modifiers [1,2].
RCTs for innovative therapies can be challenging due to ethical considerations in diseases with high unmet needs, difficulty in patient recruitment (particularly in rare diseases), or early-phase trial data that reveal large efficacy benefits [3]. Regulatory bodies have recognized these challenges and, accordingly, have approved therapies with single-arm trial (SAT) data as the primary evidence base [3,4]. However, the use of SAT data may be more problematic in reimbursement assessments, given the need to reliably quantify the comparative treatment effects of a new technology over current SoC [4].
External control (EC) arms use external data that can be used to benchmark the potential relative efficacy and safety of a technology by serving as a control group for the SAT population [1]. ECs provide data from patient cohorts created outside the SAT and treated based on clinical practice and, thus, should closely emulate the experimental trial arm to minimize bias [5,6]. Careful selection of EC arm data is critical to ensure the comparability of potential prognostic factors, treatment-effect modifiers and outcomes measurements with the SAT and timely representation of current clinical practice in the location and setting of interest [7,8].
EC arms can be built from several sources: aggregate data from published studies of clinical trials or observational studies; individual patient data (IPD) from intervention or control arms in other clinical trials; and real-world evidence (RWE) generated from real-world data (RWD) (Figure 1) [9]. The use of RWE to inform an EC arm requires stringent planning and analysis to ensure comparability between the target and SAT populations [10]. This includes ensuring that the underlying therapy considered SoC has not changed drastically over time and does not differ distinctively between the EC source and the SAT [10].
Figure 1. . Sources used to establish external control arms.

Adapted with permission from [7].
Evaluating fit for purpose of different RWD sources or databases that can inform an EC arm is recognized as an important first step to address comparative effectiveness research questions to avoid potential biased selections of RWD to favor a particular outcome for a comparator of interest [11–13]. The challenges associated with this task have also been the subject of debate in healthcare decision-making [9,14]. The selection of EC arms, particularly using RWD, has been widely described to support regulatory authorization [7,15–17]; considerations have emphasized study design (e.g., differences in patient characteristics or the use of concomitant treatments) and approaches to mitigate certain types of biases in these analyses [7,15–17]. Previous publications in oncology and non-oncology indications also reviewed SAT-based health technology assessment (HTA) submissions and reported the distribution of EC arm data used, including the impact on HTA decisions and the main methodological issues across several HTA bodies [8,9,15]. However, these publications did not explicitly focus on the specifics around the selection of EC data and how it was justified by manufacturers and/or assessed and critiqued by the HTA bodies. In addition, limited guidance is available on how to best select RWD to create an EC arm for HTA submissions based on SAT data especially when multiple data sources are available to construct EC arms. Recent RWE frameworks have been developed by HTA agencies [1,12,13], and provide much needed clarity on RWE use in comparative assessments but do not explicitly reference the topics of EC arm selection and justification.
Based on the core principles of the HTA process – unbiased selection and robust quality assessment of evidence to inform decision-making [18] – this work aimed to examine what data sources manufacturers used to construct EC arms to support SAT-based oncology submissions to the National Institute for Health and Care Excellence (NICE, England) and Pharmaceutical Benefits Advisory Committee (PBAC, Australia); if any justification based on specific quality criteria was provided by the manufacturers; and whether their rationales were accepted or criticized by the HTA bodies. An additional objective was to determine, if, for those reports which provided some EC arm selection justification, manufacturers and HTA bodies placed greater importance on any specific quality criteria regarding the EC source selection to help inform manufacturers' EC planning and decisions for future SAT HTA submissions.
Methodology
A comprehensive review was conducted in August 2021 to identify oncology HTA submission documents (both primary and re-submissions) to NICE and PBAC since January 2011 in which SATs were used as the pivotal evidence (i.e., the main evidence for the intervention being assessed/has been used in support of marketing authorization). The definition of the EC arm from the US FDA E10 Choice of Control Group and Related Issues in Clinical Trials Guidance (May 2001) (namely the control group is not derived from exactly the same population as the treated population) was used to guide the inclusion of HTA submissions [1]. NICE and PBAC were selected given the availability of information these bodies provide in the manufacturers' submissions and assessment reports, to allow elaboration of data to answer our research question. Oncology was chosen as the therapeutic area of interest due to the advancement in highly effective therapies, especially in molecularly defined subsets of common cancers, and the increase in regulatory approvals of therapies based on non-randomized clinical data [3,19]. The timeframe (i.e., January 2011) coincides with the introduction of the Cancer Drugs Fund (CDF) by NICE, which provided greater flexibility for the use of RWE for decision-making [20]. A pre-defined protocol guided the search process and documentation of information (Supplementary Table 3). The NICE website was searched first to identify relevant technologies. These were identified by applying filters (technology appraisal guidance under guidance program) and applying a date restriction. These technologies were then systematically searched on the PBAC website. Only publicly available documents were reviewed; requests for missing or incomplete documents were not made. Re-submissions of technology assessments were also included as part of this review.
For NICE, terminated, superseded submissions, or multiple technology submissions were excluded. No PBAC submissions were excluded as only completed submissions were identified. Non-oncology submissions or oncology submissions assessing the complications of cancer or radiotherapies and surgeries were excluded, as they were beyond the scope of this review. Two independent reviewers assessed the eligibility of submissions for inclusion; any disagreements were resolved by a third reviewer.
A data extraction template was developed in Microsoft Excel® and, subsequently, piloted. The template was designed to capture: a summary of each HTA submission including year, target population in the decision problem, and reimbursement decision; EC data source using pre-established categories (Table 1 for definitions) [15]; any identification of/justification for the EC data source selection provided by the manufacturers; and any critique by the HTA bodies of the manufacturer's choice of EC data source. We mapped the manufacturers' justifications and HTA critiques regarding the EC arm source selection based on the relevant quality criteria developed by Thorlund et al., 2020 (Table 2) [10]. These quality criteria provide guidance on what questions researchers should ask when critically evaluating the EC evidence to be used for comparative assessments of SATs. Results were synthesized qualitatively to identify themes in individual HTA submissions and provide insights to answer the review's research question. Full data extractions are available upon request. Information on statistical methodologies and EC considerations were not extracted as part of this research as this was beyond the scope of this review.
Table 1. . Categories of external controls.
| External control | Definition | Examples from NICE TAs |
|---|---|---|
| Concurrent | Group based on patient-level data collected at the same time as the treatment arm but in another setting (e.g., group at another institution observed at the same time, or a group at the same institution but outside the study) | “The company… provide a comparison of vismodegib with BSC in the CS through the use of a landmark analysis approach where the relative treatment effect of responders compared with non-responders is used as a surrogate…” (TA489) |
| Retrospectively collected, real-world data (natural history studies) | Patient-level data collected retrospectively as part of clinical practice (e.g., from existing medical records, or from a previously conducted registry) | “…based on patient level data (PLD) from… a European Chart Review… a large observational retrospective study commissioned…” (TA491) |
| Published data | Aggregate data derived from the published literature, but without access to subject-level data | “…it was found that only one study (a retrospective cohort study by Ou et al.) provides evidence on BSC…” (TA395) |
| Previous clinical study | Patient-level data from an arm of a previously completed clinical study in the same indication and/or patient population | “As the IMPRESS trial was an AstraZeneca trial, access to the individual patient level data (IPD)… enabled a comparison in the population referred to in the decision problem.” (TA653) |
| Baseline-controlled study | Historical data derived from a patient's baseline (e.g., collected over time prior to initiation of treatment, and patients' status on therapy is compared with status before therapy [observed changes from baseline or between study periods]) | – |
Adapted with permission from [15].
BSC: Best supportive care; CS: Company submission; NICE: National Institute for Health and Care Excellence; TA: Technology appraisal.
Table 2. . Synthetic control quality checklist – external control data source questions.
| Item number | Key question | Criteria for judgement |
|---|---|---|
| 1 | Was the original data collection process similar to that of the clinical trial? | State whether patients are from large, well-conducted randomized controlled trials or high-quality prospective cohort studies, and whether patient characteristics are similar to the target population |
| 2 | Was the external control population sufficiently similar to the clinical trial population? | State how the external population is similar with regard to key characteristics, such as (but not limited to): age, geographic distribution, performance status, treatment history, and sex |
| 3 | Did the outcome definitions of the external control match those of the clinical trial? | State whether the outcomes are measured similarly or not |
| 4 | Was the synthetic control dataset sufficiently reliable and comprehensive? | State whether there are sufficient sample sizes and covariates that can create comparable control groups |
| 5 | Were there any other major limitations to the dataset? | State any other aspects of the dataset that would limit the reliability and validity of comparisons |
Adapted with permission from [10].
Results
Review of appraisals & summary of trends
Twenty-nine oncology submissions to NICE and 19 to PBAC (Supplementary Table 1) using SATs as the pivotal evidence base were identified. Fifteen submissions to NICE [21–35] and 10 to PBAC [36–45] were for treatments for blood and bone marrow cancers. The other 14 NICE submissions were on non-small cell lung cancer (NSCLC; n = 5) [46–50], skin cancers (carcinomas; n = 3) [51–53], bladder cancer (n = 2) [54,55], tumor agnostic (n = 2) [56,57], liver cancer (n = 1) [58] and breast cancer (n = 1) [59]. The other nine PBAC submissions were on NSCLC (n = 4) [60–63], carcinomas (n = 3) [64–66], colorectal cancer (n = 1) [67] and tumor agnostic (n = 1) [68].
The target population in the HTA assessment was identified using a biomarker in 15 NICE submissions [21,22,25,28,33,46–50,54,56–59] and nine PBAC submissions [36,38,39,60–63,67,68]. Eleven NICE submissions [21,25,27,28,30–33,35,52,58] and zero PBAC submissions were in orphan diseases (three NICE submissions had orphan drug status at the time of submission but were subsequently withdrawn). A summary of single-arm trial oncology submissions by external control data source is provided in Table 3, for NICE submissions and Table 4, for PBAC submissions.
Table 3. . NICE single-arm trial oncology submissions by external control data source and external control manufacturer's justification, patient and disease characteristics and final decision-making.
| External comparator source | Total NICE SAT submissions | SAT submissions with a biomarker-defined patient population | SAT submissions in an orphan disease indication | Final HTA recommendations | Any EC justification described in the SAT submission | ||
|---|---|---|---|---|---|---|---|
| Positive (routine use) | Use only in CDF | Negative | |||||
| Published, aggregate data derived from an RCT or a SAT only | 4 (13.8%) | 3 (10.3%) | 2 (6.9%) | 2 (6.9%) | 2 (6.9%) | – | 4 |
| IPD data derived from an RCT or a SAT only | 2 (6.9%) | 1 (3.4%) | – | 1 (3.4%) | – | 1 (3.4%) | 1 (3.4%) |
| Published, aggregate data derived from observational studies only | 3 (10.3%) | 1 (3.4%) | 2† (6.9%) | 2 (6.9%) | – | 1‡ (3.4%) | 3 (10.3%) |
| IPD data derived from RWD only | – | – | – | – | – | – | – |
| Concurrent internal controls only | 1 (3.4%) | – | – | – | – | 1 (3.4%) | 1 (3.4%) |
| Multiple sources (combination of the above) | 19 (65.5%) | 9 (31.0%) | 7† (17.2%) | 9 (31.0%) | 10 (34.5%) | – | 19 (65.5%) |
Three submissions had orphan drug status at the time of submission which has since been withdrawn.
One submission assessed two populations, one of which received a negative recommendation and one which was recommended in the CDF.
CDF: Cancer Drugs Fund; EC: External control; HTA: Health technology assessment; IPD: Individual patient data; NICE: National Institute for Health and Care Excellence; RCT: Randomized controlled trial; RWD: Real-world data; SAT: Single-arm trial.
Table 4. . Pharmaceutical Benefits Advisory Committee single-arm trial oncology submissions by external control data source and external control manufacturer's justification, patient and disease characteristics and final decision-making.
| EC source | Total PBAC SAT submissions, n (%) | SAT submissions with a biomarker-defined patient population | SAT submissions in an orphan disease indication | Final HTA recommendations | Any EC justification described in the SAT submission | |
|---|---|---|---|---|---|---|
| Positive | Negative | |||||
| Published, aggregate data derived from an RCT or a SAT only | 7 (36.8%) | 5 (26.3%) | – | 5 (26.3%) | 2 (10.5%) | 2 (10.5%) |
| IPD data derived from an RCT or a SAT only | – | – | – | – | – | – |
| Published, aggregate data derived from observational studies only | 5 (26.3%) | – | – | 2 (10.5%) | 1 (5.3%) | 3 (15.8%) |
| IPD data derived from RWD only | – | – | – | 2 (10.5%) | 1 (5.3%) | |
| Concurrent internal controls only | – | – | – | – | – | – |
| Multiple sources (combination of the above) | 7 (36.8%) | 4 (15.8%) | – | 7 (36.8%) | – | 2 (10.5%) |
EC: External control; HTA: Health technology assessment; IPD: Individual patient data; PBAC: Pharmaceutical Benefits Advisory Committee; RCT: Randomized controlled trial; RWD: Real-world data; SAT: Single-arm trial.
The number of oncology HTA submissions using SATs as the pivotal evidence base increased from 2017 to 2019 (Figure 2). For submissions to both agencies for the same technology and indication, three submissions went to PBAC 3 years earlier than NICE (2014 vs 2017) [36,37,69], and four submissions went to PBAC 4 years earlier (2015 vs 2019) [38–40,70].
Figure 2. . Number of oncology submissions from 2014 to 2021 per health technology assessment body with a single-arm trial as the pivotal evidence.
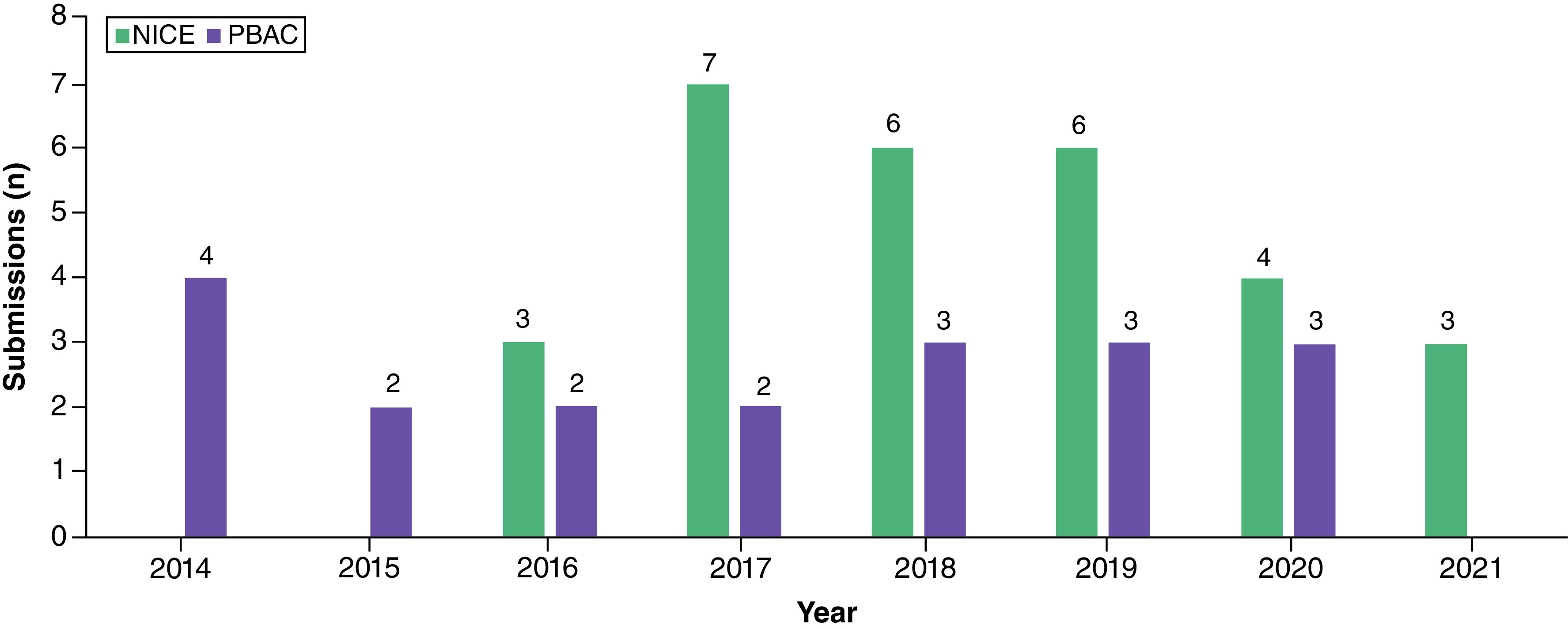
NICE: National Institute for Health and Care Excellence; PBAC: Pharmaceutical Benefits Advisory Committee.
In terms of reimbursement decisions, 14 submissions to NICE resulted in positive recommendations for routine use [21–24,28,33,34,46–50,52,58], 12 for use within the CDF [26,27,30,31,33,35,51,53,54,56,57,59], and three were not recommended [29,51,55]. One NICE submission which was not recommended for routine use had a second population in which the technology was recommended as part of the CDF [29]. Sixteen PBAC submissions [36–45,60–63,65,66] were recommended for routine use, and three were not recommended [64,68,71].
Justification by manufacturers & critiques by HTA bodies of EC data source in NICE submissions
Study design
Among the 29 retrieved NICE submissions, four submissions (13.8%) constructed an EC arm using aggregated interventional data only [30,48,57,58], 2 (6.9%) used IPD from an RCT or a SAT only [47,54], three (10.3%) solely relied on aggregated RWD only (observational studies) [29,33,52], 1 (3.4%) was from a concurrent internal control only [51] and 19 used a combination of sources (Table 3) [21–24,26–28,31,33–35,46,49–51,53,54,56,59]. Justifications based on study design were not frequently reported; however, the most common source to build a control arm was aggregate data, which were derived from published literature based on previous interventional studies (n = 18) [21,22,25,27,30–32,35,46,48–50,54–59] or retrospective observational data (n = 17) [21–24,26,28,29,31–33,35,46,49,52,53,56,59].
In the submissions that used concurrent internal controls, manufacturers justified this EC selection based on the lack of any relevant RCT and SAT comparator data through a systematic literature review [24,28,34,51]. However, the use of concurrently conducted internal controls (by means of non-responders/intra-patient comparison) was considered unreliable by the decision-making committees in all submissions, mainly due to the challenges of conducting parallel and controlled assignment of patients to different treatment arms.
Population
The EC arm closely matched the target population in the decision problem in 19 of the 29 SAT oncology submissions, according to the manufacturer [21–23,25–27,29–33,35,46–49,52–55]. In the other nine submissions, three used a proxy population [50,58,59], two used a pooled dataset across a mixed population to maximize the sample size [56,57], and four used non-concurrent internal control groups from non-responders [24,28,34,51]. In all cases, the manufacturers stated the lack of evidence in the population of interest to justify the EC selection. For proxy populations, manufacturers relied on clinical feedback to support this selection. No justification was provided for the other types of EC sources. In all submissions, NICE cited concerns in the source selection of EC populations due to potential heterogeneity with the target populations. For the two submissions in which manufacturers used a pooled dataset across a mixed population, the committee noted that there was no further evidence supporting the comparability of these populations, and no formal assessment that could be conducted to determine the direction of bias in the results [56,57].
Outcome
One submission explicitly discussed differences regarding outcome definitions between SAT and EC source [50]. No specific critiques were recorded by NICE on this matter. However, in one submission, where data informing the EC were pooled across studies, different studies were selected by the manufacturers to inform each end point (overall survival, progression-free survival) in the clinical assessments. NICE noted that this approach was unacceptable as data should only be pooled from studies in which all time-to-event endpoints of interest were reported, to preserve within-study correlation between outcomes [35].
Reliability & comprehensiveness
In 10 submissions, manufacturers noted issues with the sample size in the selected EC data (given the nature of rare diseases) and a lack of reporting of prognostic variables for these groups [24,30,33–35,48,49,51,53,59]. The NICE committee felt that small sample sizes were subject to increased uncertainty when considering the subsequent estimated treatment effect. When there was an explicit exclusion of relevant prognostic variables, reweighting analysis was criticized to directly affect the precision and confidence of results in the indirect treatment comparisons (ITC).
Other limitations
In 11 submissions, manufacturers justified the use of a proxy comparator (e.g., pooling data to form a blended comparator) due to either a lack of SoC or the need to increase the sample size of the EC arm [32–35,47–49,53,56,57,59]. The NICE committee felt this approach lacked clinical validation, given this was not reflective of the comparator in the decision problem. A summary of manufacturer's justification for single-arm trial oncology submissions to NICE, by external control data source is provided in Table 5.
Table 5. . NICE single-arm trial oncology submissions by external control data source and manufacturer's justification according to external control data sources questions.
| EC source | Total NICE SAT submissions, n (%) | EC data source questions | ||||
|---|---|---|---|---|---|---|
| Study design: Was the original data collection process similar to that of the clinical trial? | Population: Was the EC population sufficiently similar to the clinical trial population? | Outcomes: Did the outcome definitions of the EC match those of that clinical trial? | Reliability and comprehensiveness: Was the synthetic control dataset sufficiently reliable and comprehensive? | Other limitations: Were there any other major limitations to the dataset? | ||
| Published, aggregate data derived from an RCT or a SAT only | 4 (13.8%) | 4 (13.8%) | 3 (10.3%) | 1 (3.4%) | 3 (10.3%) | 1 (3.4%) |
| IPD data derived from an RCT or a SAT only | 2 (6.9%) | 1 (3.4%) | 1 (3.4%) | – | 1 (3.4%) | – |
| Published, aggregate data derived from observational studies only | 3 (10.3%) | – | 2 (6.9%) | – | 1 (3.4%) | 1 (3.4%) |
| IPD data derived from RWD only | – | – | – | – | – | – |
| Concurrent internal controls only | 1 (3.4%) | – | 1 (3.4%) | – | 1 (3.4%) | – |
| Multiple sources (combination of the above) | 19 (65.5%) | 6 (20.7%) | 12 (41.4%) | – | 10 (34.5%) | 8 (27.6%) |
EC: External control; IPD: Individual patient data; NICE: National Institute for Health and Care Excellence; RCT: Randomized controlled trial; RWD: Real-world data; SAT: Single-arm trial.
Justification by manufacturers & critiques by HTA bodies of EC data source in PBAC submissions
Study design
Of 19 SAT-based submissions to PBAC, seven (36.8%) constructed an EC using aggregated interventional data only [40,60–62,66–68], five (26.3%) relied on RWD only (observational studies using aggregate data n = 3 [45,64,65]; RWE IPD n = 2 [37,41]) and seven (36.8%) used a combination of sources (Table 5) [36,38,39,42–44,63]. Aggregate data were used in most submissions to build a control arm and were commonly derived from published literature from interventional studies (n = 11) [36,40,42,43,60–63,66,68,71] or retrospective observational studies (n = 9) [36,38,39,42–44,63–65]. Retrospective IPD RWE (i.e., from registries) formed the EC arm in four submissions [37,39,41,45]. The least common EC source was concurrently conducted internal controls (by means of non-responders/intra-patient comparison; n = 1) [40].
Although no submissions provided specific justification for the selection of EC based on study design, PBAC generally noted that the nature of study designs providing data to build the EC arm, with respect to observational and non-randomized trials, introduced a high risk of bias.
Population
Four submissions used an EC arm in a proxy population [40,43,44,63]; PBAC criticized this choice because the EC population was considered heterogenous compared with the target population in the SAT, and imbalances in prognostic factors in the two populations which may not have been accounted for. No justification was provided in the remaining 15 submissions, although heterogeneity between the target population and EC population was either acknowledged by the manufacturer [67], or criticized by PBAC [36,38,39,71].
Outcome
No manufacturer justifications were provided for this topic. However, PBAC noted that heterogeneity surrounding end point definition and assessment criteria, as well as the duration and follow-up of comparator studies, may lead to biased comparative treatment-effect estimates for SATs. Pooled treatment-effect estimates for EC arm (informed by multiple studies) were considered unreliable due to the high risk of bias.
Reliability & comprehensiveness
The manufacturers offered no justifications for EC selection based on these criteria. One submission justified the small sample size of its comparator arm (n = 7 from a pool of 2312 from registry data) due to the rarity of the condition, which manufacturers felt reflected the proposed PBAC population (i.e., population under evaluation) [67]. PBAC disagreed, noting the sample size was likely to be much larger than what was presented in the manufacturer's justification.
Overall, PBAC raised concerns about the EC sources of limited sample size and when clinically relevant prognostic factors and confounders were not accounted for in ITCs, thus limiting the confidence in the comparative estimates.
Other limitations
In three submissions, historical data were used to construct the EC arm, as it was justified to be the best available EC evidence by the manufacturers [44,63,66]. PBAC noted that the EC arm using historical data was unlikely to reflect current clinical practice or the population in Australia. These issues were not addressed by the manufacturer, as in some instances, PBAC acknowledged that due to the rarity of the population, no better data would be expected [66]. A summary of manufacturer's justification for single-arm trial oncology submissions to PBAC, by external control data source is provided in Table 6.
Table 6. . Pharmaceutical Benefits Advisory Committee single-arm trial oncology submissions by external control data source and manufacturer's justification according to external control data sources questions.
| EC source | Total PBAC SAT submissions, n (%) | EC data source questions | ||||
|---|---|---|---|---|---|---|
| Study design: Was the original data collection process similar to that of the clinical trial? | Population: Was the EC population sufficiently similar to the clinical trial population? | Outcomes: Did the outcome definitions of the EC match those of that clinical trial? | Reliability and comprehensiveness: Was the synthetic control dataset sufficiently reliable and comprehensive? | Other limitations: Were there any other major limitations to the dataset? | ||
| Published, aggregate data derived from an RCT or a SAT only | 7 (36.8%) | – | 1 (5.3%) | – | – | 1 (5.3%) |
| IPD data derived from an RCT or a SAT only | – | – | – | – | – | – |
| Published, aggregate data derived from observational studies only | 5 (26.3%) | – | – | – | – | – |
| IPD data derived from RWD only | – | – | – | – | 1 (5.3%) | – |
| Concurrent internal controls only | – | – | – | – | – | – |
| Multiple sources (combination of the above) | 7 (36.8%) | – | 3 (15.8%) | – | – | 2 (10.5%) |
EC: External control; IPD: Individual patient data; NICE: National Institute for Health and Care Excellence; RCT: Randomized controlled trial; RWD: Real-world data; SAT: Single-arm trial.
Discussion
The methods to select and use EC arms to support SAT regulatory and reimbursement submissions have been rapidly evolving [9]. Previous research has focused on EC methodological issues in regulatory and HTAs such as: trial design that may incorporate EC data in oncology [8], descriptive statistics of EC source use in SAT-based HTA submissions [9], or general methodological considerations to improve rigor in the generation of EC arms using RWD [7]. The current study, however, aimed to investigate if, and how the selected EC source was justified in manufacturers' SAT-based oncology HTA submissions, and whether and how HTA bodies criticized the manufacturers' choices. More transparency is needed on this topic as it is unclear if the lack of information regarding the justification of EC source was not considered by the manufacturers in the first place or was not mentioned by the HTA bodies (who may have considered this topic during the committee's discussion) but was not included in the documentation. Overall, our research found that manufacturers' rationales were limited in NICE submissions and rarely reported in those for PBAC, and the lack thereof was not routinely challenged by either HTA body. When EC selection critiques were mapped for the same health technologies assessed by both NICE and PBAC, it was found that patient heterogeneity, lack of local population representation, and limited sample size were the three top concerns (Supplementary Table 2).
Across the reviewed submissions, there was a trend for manufacturers to select an EC arm that closely matched the SAT population in terms of baseline characteristics and the use of comparator regimens. The choices of study design (RCT, RWE), data source and outcome definition were not usually justified for included EC arms, and it seems that the assessment committee tended to provide feedback on these SAT elements only in tandem with other evidence limitations. When RWE was used to build EC arms, HTA bodies looked more extensively at the quality, locality, and representativeness of clinical practices of these data compared with other sources (e.g., RCTs).
In terms of the other assessment domains (reliability and comprehensiveness), EC data were scrutinized mainly when they could have impacted the precision of ITC results (e.g., sample size and consideration of all prognostic variables and effect modifiers). Our review indicated that aspects of EC data sources closely linked to the analytical methods of constructing comparative clinical assessments for SAT submissions were more highlighted in HTA critiques than the rationale for the EC data source.
Similar to our findings, a previous review of NICE single technology appraisal oncology submissions noted that a key criticism was the lack of clear justification of the similarities between trial population and patients considered in RWE study [72]. It is unclear though how these concerns about patient similarity can be justified when multiple EC sources are available, and how the lack of hierarchical criteria (e.g., what factors determine an acceptable level of ‘patient similarity’ and how this trades off with other considerations such as data quality) can compromise transparency and consistency in HTA decision-making.
Importantly, the quality of EC data is a key driver for the credibility of SAT comparative analyses, which can pose threats to the validity of estimated relative treatment effects, independent of the statistical methodologies used. It is well recognized that there are challenging trade-offs when selecting the most appropriate EC data source to build comparator arms in SATs. For example, it is considered nearly impossible to balance all unknown or unmeasured confounders when historical RWE data are used to build the EC arm. In these cases, trade-offs should be justified by manufacturers and considered by decision-makers by making trade-offs between accepting lower precision of treatment-effect estimates vs widening patient inclusion criteria to a more heterogeneous population to increase precision of treatment-effect estimates. Another example is related to differences in outcomes/end point collection methods (e.g., assessor [independent review committee, investigator] and assessment criteria of response and/or progression-free survival endpoints) in an SAT vs in EC sources and how this may impact the decision around the EC data source selection. Issues around data locality and transferability of evidence from other jurisdictions were rarely discussed in the included HTA submissions, except for the reference to current practice for a given healthcare system. Overall, the lack of manufacturer's justification in the selection of a specific data source to construct an EC arm can directly lead to increased uncertainty and lack of trust for the results of the comparative efficacy analyses.
Formal approaches for estimating the potential direction, magnitude, and uncertainly of bias associated with measures of treatment effects in the HTA decision-making process have been a long-standing practice (e.g., probabilistic sensitivity analyses in cost-effectiveness analyses and quantitative bias analyses in comparative efficacy/effectiveness research) and formally recommended in the recent SAT guidance from various organizations (e.g., FDA, NICE) [12]. In addition, RWE-specific methods such as using a target trial emulation framework [73] may also help categorize and mimic the characteristics of SATs to identify the most comparable EC source by clearly describing the trade-offs; however, it is recognized that a target trial emulation framework may be more challenging to be implemented in SATs [12]. This may influence manufacturers' practices in future SATs submissions as well as HTA critiques.
The findings from our review can also inform early generation plans to support SAT clinical programs; early insight on the potential challenges in the available EC arm can help manufacturers identify alternative evidence generation or analytical strategies to mitigate some of these future problematic situations. For example, building up a strong justification regarding the EC data identification and selection in the base-case analysis can involve back-up validation exercises using data from separate sources (e.g., natural history study, case report forms, registries) as scenario analyses.
The recent explosion of RWE frameworks as published by HTA bodies (e.g., NICE [12], Canadian Agency for Drugs and Technologies in Health [13]) and working groups (e.g., RWD in Asia for HTA and Reimbursement [74]), as well as reporting guidance from international societies (e.g., International Society for Pharmacoepidemiology/The Professional Society for Health Economics and Outcomes Research) [13,75] offers some guidance around RWE use to build EC arms for SAT-based HTA submissions. However, this still leaves unanswered questions regarding the selection and justification of other data sources, or blended comparators across different study designs, on top of problems of adherence to multiple guidance documents for manufacturers preparing for multiple countries submissions. Interestingly, the US FDA draft guidance [11] on considerations for the design and conduct of externally controlled trials provides some clarity for the EC arm selection process during SAT planning, recommendations that can be also applicable to drive EC arm selection decisions and criteria in the HTA setting even though different decision criteria are applied for the technology assessments between regulatory and HTA agencies.
Establishing trade-off decision criteria (e.g., between data locality vs quality) for robust EC source selection and reliable SAT analysis, although multifactorial and possibly dependent on the decision problem, will enable manufacturers to guide the EC selection based on clear, consistent, and acceptable practices. Until then, HTA bodies may need to follow FDA approaches in transparently documenting decisions regarding key methodological topics (such as the FDA commitment to record types of surrogate outcomes used in submissions) for topics such as the EC data source selection that can guide future submissions, transparently demonstrate consistency in decision-making, and remove delays in the submission process.
Limitations
The current findings were informed by a 2021 review; therefore, recent submissions to NICE and PBAC (particularly those based after the publication of 2022 NICE RWE framework) have not been captured. Research-related information in PBAC's public summary documents was sparse compared with information available from NICE; therefore, interpretation of findings from the current review does not fully reflect what may have changed as a result of recently published HTA RWE guidance documents. For example, explicit reference to bias quantification methods (as referenced by the NICE RWE framework) was not systematically extracted through the manufacturers' submissions but was only captured if mentioned by the committee's critique. Furthermore, the justification of EC selection to inform ITCs may be reported as part of supplementary HTA submission materials, which are often not publicly available; no effort was made to obtain additional information from any stakeholders though individual contacts. In addition, manufacturers may have previously consulted respective HTA bodies as part of early scientific advice regarding the EC study design and therefore did not clearly document their decisions in their submissions, if aligned with that advice. Of note, the interpretation of findings across these HTA submissions was qualitative, speculative in some instances when the information regarding the research question was indicative but not clear, and some individual interpretation of data collection may be unavoidable. Finally, the findings were based on SAT-based oncology submissions to NICE and PBAC which may not be generalizable to other indications and HTA agencies.
Conclusion
EC data source selection and justification seems to have been an underassessed and underreported area in SAT oncology submissions to both NICE and PBAC. An unbiased selection of the most representative and fit-for-purpose EC data source will allow transparency and trust in the results of the comparative treatment effects of SATs beyond exclusive focus on quality concerns of EC data source. Further research should explore whether the use of step-wise selection criteria (e.g., quality, locality), including those provided by recent RWE frameworks developed by HTA agencies, can guide the choice and justification of EC sources to be used in SAT-based HTA submissions including also informing decisions around the early evidence generation planning activities for the clinical development of new products.
Summary points.
In diseases with high unmet need, single-arm trials (SATs) are often used to support submissions of new health technologies. In the absence of comparative data for SATs, data from externals controls (ECs), outside the SAT setting, are needed to build the comparative effectiveness estimates for the new technologies vs comparators established in clinical practice.
The present review aimed to identify how the EC data source used in SAT oncology health technology assessment (HTA) submissions was identified and justified by the manufacturers, and how this was critiqued by the decision-makers.
Overall findings showed that EC data selection was rarely justified by manufacturers on SAT submissions to the National Institute for Health and Care Excellence (NICE) and was seldom reported in Pharmaceutical Benefits Advisory Committee (PBAC) oncology submissions.
This lack of rationale for the selection of EC data sources was not routinely challenged by either HTA agency which can lead to increased uncertainty on whether alternative data sources may have provided less biased evidence for the relative treatment analyses compared with what was selected by the manufacturers.
Aggregate data were the most common data source used to build EC arms in both NICE and PBAC submissions. When real-world evidence (RWE) was used to construct EC arms, HTA bodies looked more extensively at the quality, locality, and representativeness of clinical practices of these data compared with other sources (e.g., randomized controlled trials).
HTA bodies more widely critiqued the aspects of EC data sources closely linked to the analytical methods of constructing comparative clinical assessments for SAT submissions than the rationale for the selection of EC data source.
These findings can also inform early generation plans to support SAT clinical programs and provide manufacturers with insights on the potential challenges in the selection of ECs, thus avoiding delays in patient access to new technologies.
Guidance needs to be more explicit regarding the selection and justification of data sources to build EC arms for SAT-based HTA submissions, or use of blended comparators across different study designs, as well as how to handle submissions across multiple localities.
There is a need to establish clear and consistent trade-off decision criteria (e.g., data locality vs quality) to guide manufacturers in the selection of the most robust and reliable EC source for SAT submissions.
Supplementary Material
Acknowledgments
The authors thank Andrea Garcia, who made a significant contribution toward data collection required for the manuscript, and Colleen Dumont for writing and editorial support of this manuscript.
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: https://bpl-prod.literatumonline.com/doi/10.57264/cer-2023-0140
Author contributions
K Appiah, M Rizzo, G Sarri and L Hernandez were responsible for the conceptualization and interpretation of data and findings of the study. KA collected the data presented and drafted the initial manuscript. All authors reviewed, edited and approved each draft of the manuscript.
Disclaimer
This article reflects the views and opinions of the authors and not necessarily those of the organizations to which individuals are affiliated.
Financial disclosure
Writing and editorial support was funded by Takeda Pharmaceuticals America, Inc. The authors have received no other financial and/or material support for this research or the creation of this work apart from that disclosed.
Competing interests disclosure
K Appiah, M Rizzo, G Sarri are full-time employees of Cytel. L Hernandez is a full-time employee of Takeda Pharmaceuticals America, Inc. The authors have no other competing interests or relevant affiliations with any organization/entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Writing disclosure
Writing and editorial support was funded by Takeda Pharmaceuticals America, Inc.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit https://creativecommons.org/licenses/by-nc-nd/4.0/
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.ICH. E10: choice of control group and related issues in clinical trials (2000). https://database.ich.org/sites/default/files/E10_Guideline.pdf ; • A guideline outlining control groups in clinical trials.
- 2.US FDA. Draft Guidance: Demonstrating Substantial Evidence of Effectiveness for Human Drug and Biological Products Guidance for Industry (2019). https://www.fda.gov/media/133660/download
- 3.Tenhunen O, Lasch F, Schiel A, Turpeinen M. Single-arm clinical trials as pivotal evidence for cancer drug approval: a retrospective cohort study of centralized european marketing authorizations between 2010 and 2019. Clin. Pharmacol. Ther. 108(3), 653–660 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Goring S, Taylor A, Müller K et al. Characteristics of non-randomised studies using comparisons with external controls submitted for regulatory approval in the USA and Europe: a systematic review. BMJ Open 9(2), e024895 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hernán MA, Robins JM. Using big data to emulate a target trial when a randomized trial is not available. Am. J. Epidemiol. 183(8), 758–764 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Franklin JM, Pawar A, Martin D et al. Nonrandomized real-world evidence to support regulatory decision making: process for a randomized trial replication project. Clin. Pharmacol. Ther. 107(4), 817–826 (2020). [DOI] [PubMed] [Google Scholar]; • A review in which real-world evidence was used to emulate data from randomized trials.
- 7.Burcu M, Dreyer NA, Franklin JM et al. Real-world evidence to support regulatory decision-making for medicines: considerations for external control arms. Pharmacoepidemiol. Drug Saf. 29(10), 1228–1235 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Provides definitions of different types of external control arms.
- 8.Mishra-Kalyani PS, Amiri Kordestani L, Rivera DR et al. External control arms in oncology: current use and future directions. Ann. Oncol. 33(4), 376–383 (2022). [DOI] [PubMed] [Google Scholar]; • Provides an introduction to the concepts of external control arm use in oncology.
- 9.Patel D, Grimson F, Mihaylova E et al. Use of external comparators for health technology assessment submissions based on single-arm trials. Value Health 24(8), 1118–1125 (2021). [DOI] [PubMed] [Google Scholar]; •• A previously published health technology assessment (HTA) review on the use of external control arms for single-arm trial based submissions.
- 10.Thorlund K, Dron L, Park JJH, Mills EJ. Synthetic and external controls in clinical trials – a primer for researchers. Clin. Epidemiol. 12, 457–467 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.US FDA. Considerations for the Design and Conduct of Externally Controlled Trials for Drug and Biological Products: Guidance for Industry (2023). https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-design-and-conduct-externally-controlled-trials-drug-and-biological-products
- 12.National Institute for Health and Care Excellence (NICE). ECD9: NICE real-world evidence framework (2022). https://www.nice.org.uk/corporate/ecd9/chapter/overview ; •• A framework which outlines key considerations and the implementation of real-world evidence within NICE guidance.
- 13.Canada's Drug and Health Technology Agency (CADTH). Guidance for Reporting Real-World Evidence (2023). https://www.cadth.ca/sites/default/files/RWE/MG0020/MG0020-RWE-Guidance-Report-Secured.pdf
- 14.Jaksa A, Louder A, Maksymiuk C et al. A comparison of seven oncology external control arm case studies: critiques from regulatory and health technology assessment agencies. Value Health 25(12), 1967–1976 (2022). [DOI] [PubMed] [Google Scholar]; •• A previously published review of the use of external control arms in FDA submissions.
- 15.Jahanshahi M, Gregg K, Davis G et al. The use of external controls in FDA regulatory decision making. Ther. Innov. Reg. Sci. 55(5), 1019–1035 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hashmi M, Rassen J, Schneeweiss S. Single-arm oncology trials and the nature of external controls arms. J. Comp. Eff. Res. 10(12), 1053–1066 (2021). [DOI] [PubMed] [Google Scholar]
- 17.Seeger JD, Davis KJ, Iannacone MR et al. Methods for external control groups for single arm trials or long-term uncontrolled extensions to randomized clinical trials. Pharmacoepidemiol. Drug Saf. 29(11), 1382–1392 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.HTAi. Health Technology Assessment International (HTAi) (2021). https://htai.org/
- 19.Simon R, Blumenthal GM, Rothenberg ML et al. The role of nonrandomized trials in the evaluation of oncology drugs. Clin. Pharmacol. Ther. 97(5), 502–507 (2015). [DOI] [PubMed] [Google Scholar]
- 20.NHS England Cancer Drugs Fund. Cancer Drugs Fund (2019). https://www.england.nhs.uk/cancer/cdf/
- 21.National Institute for Health and Care Excellence (NICE). TA401: bosutinib for previously treated chronic myeloid leukaemia (2016). https://www.nice.org.uk/guidance/ta401
- 22.National Institute for Health and Care Excellence (NICE). TA451: ponatinib for treating chronic myeloid leukaemia and acute lymphoblastic leukaemia (2017). https://www.nice.org.uk/guidance/ta451
- 23.National Institute for Health and Care Excellence (NICE). TA462: nivolumab for treating relapsed or refractory classical Hodgkin lymphoma (2017). https://www.nice.org.uk/guidance/ta462
- 24.National Institute for Health and Care Excellence (NICE). TA478: brentuximab vedotin for treating relapsed or refractory systemic anaplastic large cell lymphoma (2017). https://www.nice.org.uk/guidance/ta478
- 25.National Institute for Health and Care Excellence (NICE). TA487: venetoclax for treating chronic lymphocytic leukaemia (2017). https://www.nice.org.uk/guidance/ta487
- 26.National Institute for Health and Care Excellence (NICE). TA491: ibrutinib for treating Waldenstrom's macroglobulinaemia (2017). https://www.nice.org.uk/guidance/ta491
- 27.National Institute for Health and Care Excellence (NICE). TA510: daratumumab monotherapy for treating relapsed and refractory multiple myeloma (2018). https://www.nice.org.uk/guidance/ta510
- 28.National Institute for Health and Care Excellence (NICE). TA524: brentuximab vedotin for treating CD30-positive Hodgkin lymphoma (2018). https://www.nice.org.uk/guidance/ta524
- 29.National Institute for Health and Care Excellence (NICE). TA540: pembrolizumab for treating relapsed or refractory classical Hodgkin lymphoma (2018). https://www.nice.org.uk/guidance/ta540
- 30.National Institute for Health and Care Excellence (NICE). TA554: tisagenlecleucel for treating relapsed or refractory B-cell acute lymphoblastic leukaemia in people aged up to 25 years (2018). https://www.nice.org.uk/guidance/ta554
- 31.National Institute for Health and Care Excellence (NICE). TA559: axicabtagene ciloleucel for treating diffuse large B-cell lymphoma and primary mediastinal large B-cell lymphoma after 2 or more systemic therapies (2019). https://www.nice.org.uk/guidance/ta559
- 32.National Institute for Health and Care Excellence (NICE). TA567: tisagenlecleucel for treating relapsed or refractory diffuse large B-cell lymphoma after 2 or more systemic therapies (2019). https://www.nice.org.uk/guidance/ta567
- 33.National Institute for Health and Care Excellence (NICE). TA589: blinatumomab for treating acute lymphoblastic leukaemia in remission with minimal residual disease activity (2019). https://www.nice.org.uk/guidance/ta589
- 34.National Institute for Health and Care Excellence (NICE). TA604: idelalisib for treating refractory follicular lymphoma (2019). https://www.nice.org.uk/guidance/ta604
- 35.National Institute for Health and Care Excellence (NICE). TA677: autologous anti-CD19-transduced CD3+ cells for treating relapsed or refractory mantle cell lymphoma (2021). https://www.nice.org.uk/guidance/ta677
- 36.PBAC. Public Summary Document (PSD) November 2014 PBAC Meeting: ponatinib (2014). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2014-11/ponatinib-psd-11-2014
- 37.PBAC. Public Summary Document (PSD) March 2014 PBAC Meeting: Brentuximab Vedotin (2014). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2014-03/brentuximab-vedotin-psd-03-2014
- 38.PBAC. Public Summary Document (PSD) November 2015 PBAC Meeting: blinatumomab (2015). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2015-11/blinatumomab-blincyto-psd-november-2015
- 39.PBAC. Public Summary Document (PSD) March 2015 PBAC Meeting: Brentuximab Vedotin (2015). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2015-03/brentuximab-vedotin-adcetris-psd-03-2015
- 40.PBAC. Public Summary Document (PSD) March 2015 PBAC Meeting: idelalisib (2015). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2015-03/idelalisib-zydelig-2-psd-03-2015
- 41.PBAC. Public Summary Document (PSD) November 2015 PBAC Meeting: pralatrexate (2015). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2015-11/pralatrexate-folotyn-psd-11-2015
- 42.PBAC. Public Summary Document (PSD) November 2016 PBAC Meeting: brentuximab vedotin (ASCT naïve) (2016). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2016-11/brentuximab-asct-naive-psd-november-2016
- 43.PBAC. Public Summary Document (PSD) July 2017 PBAC Meeting: pembrolizumab (2017). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2017-07/pembrolizumab-psd-july-2017
- 44.PBAC. Public Summary Document (PSD) November 2018 PBAC Meeting: pembrolizumab (PMBCL) (2018). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2018-11/Pembrolizumab-PMBCL
- 45.PBAC. Public Summary Document (PSD) July 2018 PBAC Meeting: blinatumomab (2018). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2018-07/Blinatumomab-psd-july-2018
- 46.National Institute for Health and Care Excellence (NICE). TA395: ceritinib for previously treated anaplastic lymphoma kinase positive non-small-cell lung cancer (2016). https://www.nice.org.uk/guidance/ta395
- 47.National Institute for Health and Care Excellence (NICE). TA416: Osimertinib for treating EGFR T790M mutation-positive advanced non-small-cell lung cancer (2016). https://www.nice.org.uk/guidance/ta653
- 48.National Institute for Health and Care Excellence (NICE). TA571: brigatinib for treating ALK-positive advanced non-small-cell lung cancer after crizotinib (2019). https://www.nice.org.uk/guidance/ta571
- 49.National Institute for Health and Care Excellence (NICE). TA628: lorlatinib for previously treated ALK-positive advanced non-small-cell lung cancer (2020). https://www.nice.org.uk/guidance/ta628
- 50.National Institute for Health and Care Excellence (NICE). TA643: entrectinib for treating ROS1-positive advanced non-small-cell lung cancer (2020). https://www.nice.org.uk/guidance/ta643
- 51.National Institute for Health and Care Excellence (NICE). TA489: vismodegib for treating basal cell carcinoma (2017). https://www.nice.org.uk/guidance/ta489
- 52.National Institute for Health and Care Excellence (NICE). TA517: avelumab for treating metastatic Merkel cell carcinoma (2018). https://www.nice.org.uk/guidance/ta517
- 53.National Institute for Health and Care Excellence (NICE). TA592: cemiplimab for treating metastatic or locally advanced cutaneous squamous cell carcinoma (2019). https://www.nice.org.uk/guidance/ta592
- 54.National Institute for Health and Care Excellence (NICE). TA492: atezolizumab for untreated PD-L1-positive locally advanced or metastatic urothelial cancer when cisplatin is unsuitable (2017). https://www.nice.org.uk/guidance/ta492
- 55.National Institute for Health and Care Excellence (NICE). TA530: nivolumab for treating locally advanced unresectable or metastatic urothelial cancer after platinum-containing chemotherapy (2018). https://www.nice.org.uk/guidance/ta530
- 56.National Institute for Health and Care Excellence (NICE). TA630: larotrectinib for treating NTRK fusion-positive solid tumours (2020). https://www.nice.org.uk/guidance/ta630
- 57.National Institute for Health and Care Excellence (NICE). TA644: entrectinib for treating NTRK fusion-positive solid tumours (2020). https://www.nice.org.uk/guidance/ta644
- 58.National Institute for Health and Care Excellence (NICE). TA722: pemigatinib for treating relapsed or refractory advanced cholangiocarcinoma with FGFR2 fusion or rearrangement (2021). https://www.nice.org.uk/guidance/ta722
- 59.National Institute for Health and Care Excellence (NICE). TA704: trastuzumab deruxtecan for treating HER2-positive unresectable or metastatic breast cancer after 2 or more anti-HER2 therapies (2021). https://www.nice.org.uk/guidance/ta704
- 60.PBAC. Public Summary Document (PSD) March 2020 PBAC Meeting: entrectinib (2020). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2020-03/entrectinib-capsule-200-mg-rozlytrek
- 61.PBAC. Public Summary Document (PSD) November 2019 PBAC Meeting: brigatinib (2019). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2019-11/brigatinib-tablet-30-mg-tablet-90-mg-tablet-180-mg-pack
- 62.PBAC. Public Summary Document (PSD) November 2019 PBAC Meeting: lorlatinib (2019). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2019-11/lorlatinib-tablet-25-mg-tablet-100-mg-lorviqua
- 63.PBAC. Public Summary Document (PSD) November 2017 PBAC Meeting: crizotinib (2017). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2017-11/crizotinib-psd-november-2017
- 64.PBAC. Public Summary Document (PSD) November 2020 PBAC Meeting: cemiplimab (2020). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2020-11/cemiplimab-solution-for-i-v-infusion-350-mg-in-7-ml-libta
- 65.PBAC. Public Summary Document (PSD) July 2018 PBAC Meeting: avelumab (2018). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2018-07/Avelumab-psd-july-2018
- 66.PBAC. Public Summary Document (PSD) March 2016 PBAC Meeting: vismodegib (2016). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2016-03/vismodegib-erivedge-psd-03-2016
- 67.PBAC. Public Summary Document (PSD) March 2019 PBAC Meeting: pembrolizumab (2019). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2019-03/pembrolizumab-solution-psd-march-2019
- 68.PBAC. Public Summary Document (PSD) November 2020 PBAC Meeting: larotrectinib (2020). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2020-11/larotrectinib-capsule-25-mg-capsule-100-mg-oral-solution
- 69.PBAC. Public Summary Document (PSD) July 2015 PBAC Meeting: ponatinib (2015). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2015-07/ponatinib-psd-july-2015
- 70.PBAC. Public Summary Document (PSD) November 2015 PBAC Meeting: idelalisib (follicular lymphoma) (2015). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2015-11/idelalisib-zydelig-follicular-lymphoma-psd-11-2015
- 71.PBAC. Public Summary Document (PSD) March 2020 PBAC Meeting: pembrolizumab (PMBCL) (2020). https://www.pbs.gov.au/pbs/industry/listing/elements/pbac-meetings/psd/2020-03/pembrolizumab-pmbcl-solution-concentrate-for-iv-infusio
- 72.Bullement A, Podkonjak T, Robinson MJ et al. Real-world evidence use in assessments of cancer drugs by NICE. Int. J. Technol. Assess. Health Care. 36(4), 388–394 (2020). [DOI] [PubMed] [Google Scholar]
- 73.Hernán MA, Sauer BC, Hernández-Díaz S, Platt R, Shrier I. Specifying a target trial prevents immortal time bias and other self-inflicted injuries in observational analyses. J. Clin. Epidemiol. 79, 70–75 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lou J, Kc S, Toh KY et al. Real-world data for health technology assessment for reimbursement decisions in Asia: current landscape and a way forward. Int. J. Tech. Assess. Health Care 36(5), 474–480 (2020). [DOI] [PubMed] [Google Scholar]
- 75.Berger ML, Sox H, Willke RJ et al. Good practices for real-world data studies of treatment and/or comparative effectiveness: recommendations from the joint ISPOR–ISPE Special Task Force on real-world evidence in health care decision making. Pharmacoepidemiol. Drug Saf. 26(9), 1033–1039 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.