

SUMMARY
Dysfunctional mitochondria are removed via multiple pathways, such as mitophagy, a selective autophagy process. Here, we identify an intracellular hybrid mitochondria-lysosome organelle (termed the mitochondria-lysosome-related organelle [MLRO]), which regulates mitochondrial homeostasis independent of canonical mitophagy during hepatocyte dedifferentiation. The MLRO is an electron-dense organelle that has either a single or double membrane with both mitochondria and lysosome markers. Mechanistically, the MLRO is likely formed from the fusion of mitochondria-derived vesicles (MDVs) with lysosomes through a PARKIN-, ATG5-, and DRP1-independent process, which is negatively regulated by transcription factor EB (TFEB) and associated with mitochondrial protein degradation and hepatocyte dedifferentiation. The MLRO, which is galectin-3 positive, is reminiscent of damaged lysosome and could be cleared by overexpression of TFEB, resulting in attenuation of hepatocyte dedifferentiation. Together, results from this study suggest that the MLRO may act as an alternative mechanism for mitochondrial quality control independent of canonical autophagy/mitophagy involved in cell dedifferentiation.
In brief
Hepatocyte dedifferentiation is a common feature for chronic liver diseases such as alcohol-associated hepatitis and metabolic-dysfunction-associated steatohepatitis. Ma et al. report that hepatocyte dedifferentiation is associated with MLRO induction that can be reversed by TFEB-mediated removal of leaky lysosomes.
Graphical Abstract
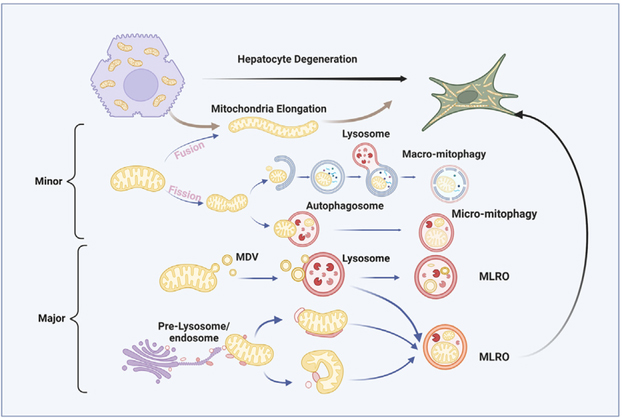
INTRODUCTION
Mitochondria play critical roles in regulating cellular redox homeostasis, lipid metabolism, energy production, and cell death. Mitochondrial dysfunction is associated with various acute and chronic diseases, including tissue injury, metabolic syndrome, cardiovascular and neurogenerative diseases, and cancer.1–4 To maintain cellular function, tight regulation of mitochondrial homeostasis and quality is essential. Mitochondria are highly dynamic organelles that constantly undergo fission and fusion, processes orchestrated by a number of GTPases, including dynamin-related protein 1 (DRP1) in mitochondrial fission and mitofusin 1 (MFN1) and mitofusin 2 (MFN2) and OPA1 in mitochondrial outer and inner membrane fusion, respectively.4,5 The fission process may assist in repair of damaged mitochondria by segregating damaged mitochondrial components, including proteins, mtDNA, and lipids. Conversely, increased mitochondrial fusion may help save and exchange materials between healthy mitochondria.4,5
In addition to mitochondrial fission and fusion, emerging evidence indicates that cells can use multiple distinct yet redundant mechanisms to regulate mitochondrial quality control and turnover.6–9 First, outer mitochondrial membrane proteins can be ubiquitinated and degraded by the ubiquitin-proteasome system via mitochondrial-associated degradation (MAD), similar to ER-associated degradation (ERAD).9,10 Second, mitochondria have unique AAA ATPase family proteases that localize to both sides of the inner membrane, which can degrade unfolded or oxidized proteins within the matrix and intermembrane space, areas not accessible to cytosolic proteases.11,12 Third, damaged mitochondria can be enveloped by autophagosomes to trigger autolysosome-based degradation via mitophagy, a widely studied process that removes entire mitochondria through both PINK1-PARKIN-dependent and independent pathways.8,9,13 Fourth, damaged mitochondria can undergo morphological remodeling to form mitochondrial spheroids and acquire lysosomal markers for possible degradation, which is negatively regulated by PARKIN and requires MFN1 and MFN2.14,15 Fifth, mitochondria can be enclosed within micro vesicles or migrasomes and extruded outside of cells as an extracellular vesicle in processes such as mitocytosis and autophagic secretion of mitochondria (ASM), which serves as a mitochondrial quality control mechanism and is important for inter-organ cross-talk.16–19 Sixth, a portion of mitochondria can bud off and form mitochondria-derived vesicles (MDVs) under certain conditions such as oxidative stress, which can further fuse with either peroxisomes or lysosomes to degrade sequestered oxidized mitochondrial proteins.20–22
In the present study, using prolonged cultured primary mouse hepatocytes undergoing dedifferentiation, we identified an organelle likely derived from MDVs fusing with a lysosome that form a hybrid “mitochondria-lysosome-like structure.” This organelle, which we term the mitochondria-lysosome-related organelle (MLRO), is morphologically distinct from the mitophagosome. Overexpression of transcription factor EB (TFEB) promoted MLRO clearance. The MLRO may aid in mitochondria degradation during cell dedifferentiation independent of autophagy and mitophagy.
RESULTS
Prolonged cultured primary mouse hepatocytes undergo dynamic remodeling associated with increased mitochondrial elongation
To study mitochondrial morphological changes in cultured primary hepatocytes, we cultured isolated mouse hepatocytes for different lengths of time up to 7 days. Well-attached hepatocytes on the first day of culture exhibited characteristic polygonal shapes with one or two spherical nuclei. These cells flattened and lost their typical polygonal features in culture. By day 5, hepatocytes adopted a flattened stellate shape, possibly indicating elevated mobility in this dedifferentiation remodeling process. Eventually, these dedifferentiated hepatocytes developed spindle-shaped fibroblastic on day 7 of culture (Figure 1A). In addition to morphological transformation, α-smooth muscle actin (α-SMA), a fibroblast marker, increased dramatically in both protein and mRNA levels from day 1 to day 7 (Figure 1B). In contrast, hepatic cytochrome P450 (CYP) enzymes, such as CYP2E1 and CYP2B10, markedly decreased from day 1, becoming undetectable by day 3 of culture, suggesting that hepatocyte drug metabolizing enzymes depletion in culture. Similarly, hepatocyte nuclear factor 4 alpha (HNF-4a), a transcription factor that regulates gene expression for hepatocyte identity, declined at day 1 and became almost undetectable in culture compared with freshly isolated hepatocytes (Figure 1B). Unsurprisingly, we found one of the cytoskeleton components, beta-actin, increased in remodeled hepatocytes, which may be associated with the fibroblast-like features of these dedifferentiated hepatocytes (Figure 1B). Consistent with the protein changes, mRNA levels of Cyp2e1, Cyp2b10, and Hnf4a-P1 (adult form) decreased but Hnf4a-P2 (fetal form) and Acta2 increased in cultured hepatocytes of days 1 and 2 (Figure 1C). Together, prolonged mouse hepatocyte culture leads to progressive fibroblastic transformation, which is associated with reduced hepatic metabolic functions and elevated fibroblastic features. Cultured hepatocytes at day 1 were enriched with mitochondria, which filled the bulk of the cytoplasm and were mostly visible as “dot-shaped” structures (Figure 1D). However, tubular mitochondria became evident on day 3 and persisted through day 7 of culture, particularly in the cytoplasm periphery (Figure 1D). Transmission electron microscopy (TEM) analysis further confirmed the morphological transformation of mitochondria from ovoid to elongated architecture in hepatocytes during remodeling (Figure 1E). Protein levels of DRP1 increased, whereas levels of MFN1 and MFN2 decreased in culture (Figure 1F), likely an adaptive compensatory response to restore mitochondria size homeostasis by increasing mitochondria fission and decreasing fusion. Using a 14C-labeled fatty acid oxidation (FAO) assay, we found that mitochondrial FAO increased by approximately 30% in day 3 hepatocytes but decreased by 50% in day 5 hepatocytes compared with day 1 hepatocytes (Figure 1G), suggesting that elongated mitochondria may initially act as an adaptive response but may become maladaptive for FAO during the prolonged culture course. Taken together, these data indicate that prolonged cultured hepatocytes undergo dynamic morphological remodeling associated with increased elongated mitochondria.
Figure 1. Fibroblastic transformation of prolonged cultured primary mouse hepatocytes associated with increased elongated mitochondria.
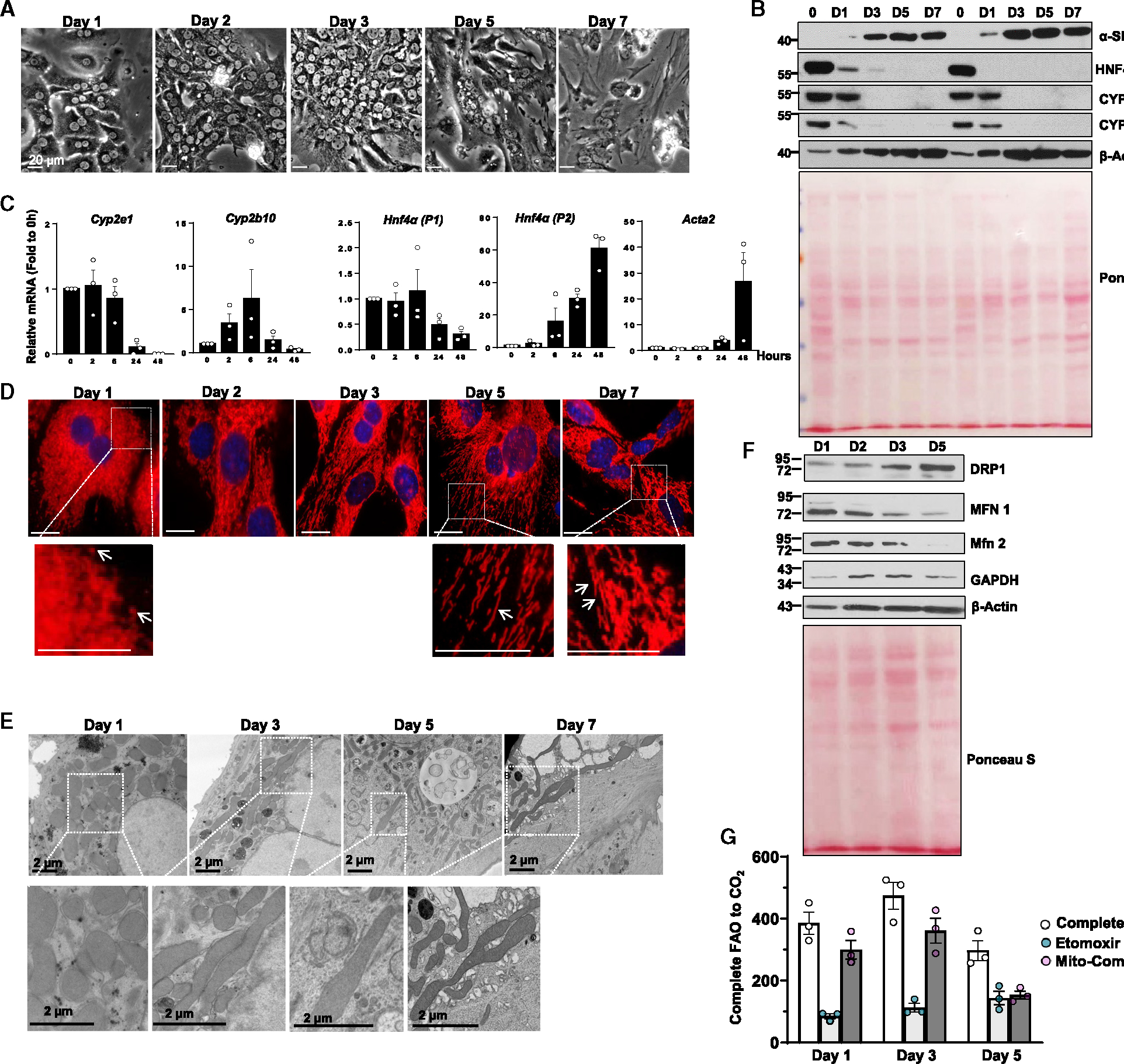
(A) Representative bright-field images of primary mouse hepatocytes cultured for indicated times.
(B) Total cell lysates from indicated mouse hepatocytes were subjected to western blot analysis. Reversible Ponceau S staining was applied as loading controls.
(C) Total mRNA was extracted from cultured mouse hepatocytes followed by real-time qPCR analysis. Data are presented as mean ± SE (n = 3 independent experiments).
(D) Immunofluorescence staining of TOMM20 in cultured mouse hepatocytes at indicated time points. Arrows denote elongated mitochondria.
(E) Representative images of transmission electron microscopy analysis of hepatocytes ad indicated time points.
(F) Total cell lysates from primary mouse hepatocytes cultured for indicated times were subjected to western blot analysis. Reversible Ponceau S staining was applied to indicate equal loadings.
(G) Mouse hepatocytes were cultured at indicated time points, and mitochondria FAO was measured as described in STAR Methods. Data are presented as mean ± SE (n = 3 independent experiments).
Increased fragmented mitochondria that are colocalized with lysosomal markers in prolonged cultured hepatocytes
We next examined the time course of autophagy and mitophagy flux in primary cultured hepatocytes using GFP-RFP-LC3 and COX8-GFP-mCherry analysis, respectively. The principle of tandem fluorescence assays is based on the property of green fluorescence to be quenched within acidic cellular compartments (such as low pH autolysosomes), while mCherry/RFP fluorescence is relatively stable.23,24 We found increased numbers of red-only puncta from GFP-RFP-LC3-infected cells, increased endogenous LC3-II levels and decreased SQSTM1/p62 levels as well as increased GFP-LC3 puncta in the presence of chloroquine in a time-dependent manner (Figures S1A–S1D), suggesting increased autophagic flux during prolonged hepatocyte culture. TEM analysis for double-membraned autophagosomes and single-membraned late autolysosomes enveloping mitochondria (so-called mitophagosomes) is the gold standard for visualizing mitophagy.13,23 Although we did observe the typical “mitophagosome” structures, the number of “mitophagosomes” was surprisingly very low in cultured hepatocytes (Figures S1E and S1F). In COX8-GFP-mCherry-expressing cells, while most mitochondria were yellow (indicating presence of green and red fluorescence), the number of red-only mitochondria was significantly increased by day 3 and sustained to day 7 (Figure 2A). None of the red-only mitochondria were elongated, and almost all of the red-only fragmented mitochondria colocalized with LAMP1-positive compartments (Figure 2A), suggesting that the red-only fragmented mitochondria were acidic and spatially associated with autolysosomal or lysosomal-like compartments. Interestingly, all these red-only mitochondria were TOMM20 negative (Figure S2A), indicating that some of the outer mitochondrial membrane proteins might have already been degraded before reaching the lysosome. No significant differences were found in proteasome activity between day 1 and day 3 hepatocytes (Figure S2B). It is likely some of the outer mitochondrial membrane proteins such as TOMM20 is ubiquitinated and degraded via proteasome without increasing overall proteasome activity. A careful morphological analysis of the COX8-GFP-mCherry images revealed at least two distinct forms of LAMP1-associated mitochondrial structures, which we have defined them as type I and type II structures. Type I structures are fragmented yellow mitochondria colocalized with LAMP1-positive compartments (Figures 2C, S2C, and S2D, white circles with white arrowheads), which may reflect an early fragmented mitochondria (likely MDVs)-lysosome contact event in which LAMP1-positive compartments are not acidic enough to completely quench the mitochondrial green fluorescence. On the other hand, type II structures, which appear purple on immunofluorescence (IF), are red-only fragmented mitochondria colocalized with LAMP1-positive compartments that are acidic and completely quench the mitochondrial green fluorescence (Figures 2C, S2C, and S2D, red circles with white arrows), which may reflect a late event of either direct mitochondria-lysosome fusion or fusion of an autophagosome-enveloped fragmented mitochondria with a lysosome. Indeed, most red-only COX8- and LAMP1-positive vesicles were also LysoTracker positive, indicating their acidic nature (Figure S2E). Super-resolution microscopy analysis revealed some LAMP1-positive only structures (presumably lysosomes) in close proximity to or in direct contact with yellow mitochondria (Figure S3A, white arrows), suggesting a possible mitochondria and lysosome “hit and run” event. Time-lapse live imaging analysis of COX8-GFP-mCherry-expressing hepatocytes clearly shows that a small portion of mitochondria bud off from yellow mitochondria (likely MDVs) and turn into red-only mitochondria (Video S1, circled events). To further determine whether type I and type II structures would be generated by transient contact between mitochondria and lysosome/endosome in a “hit and run” process, COX8-GFP-mCherry-expressing hepatocytes were further labeled with deep red dextran, a fluorescent marker of fluidphase endocytosis that can enter endosome/lysosome compartments, and subjected to time-lapse confocal microscopy. Recordings of frequent, temporary interactions between blue lysosomes/endosomes (represented by blue pseudo-color) and yellow mitochondria were observed. Ultimately, the yellow mitochondria transformed into red-only mitochondria (Videos S2–S4, circled events; Figure S3B, arrows and arrowheads). Moreover, almost all the red-only mitochondria were also dextran positive, implying direct interaction or possible fusion of endo-lysosomal compartments with these fragmented mitochondria, resulting in the quenching of green GFP signals. Nanoparticles are endocytosed by cells and enter endo-lysosomal compartments, a process that can be visualized on TEM. Using a BSA-conjugated gold particle labeling and TEM approach, we observed that gold-labeled endo-lysosomal compartments were closely associated with MDVs (Figure S3C), further supporting a possible lysosome-MDV “hit and run” process.
Figure 2. Increased acidic fragmented mitochondrial compartments that are colocalized with a lysosomal marker in prolonged cultured hepatocytes.
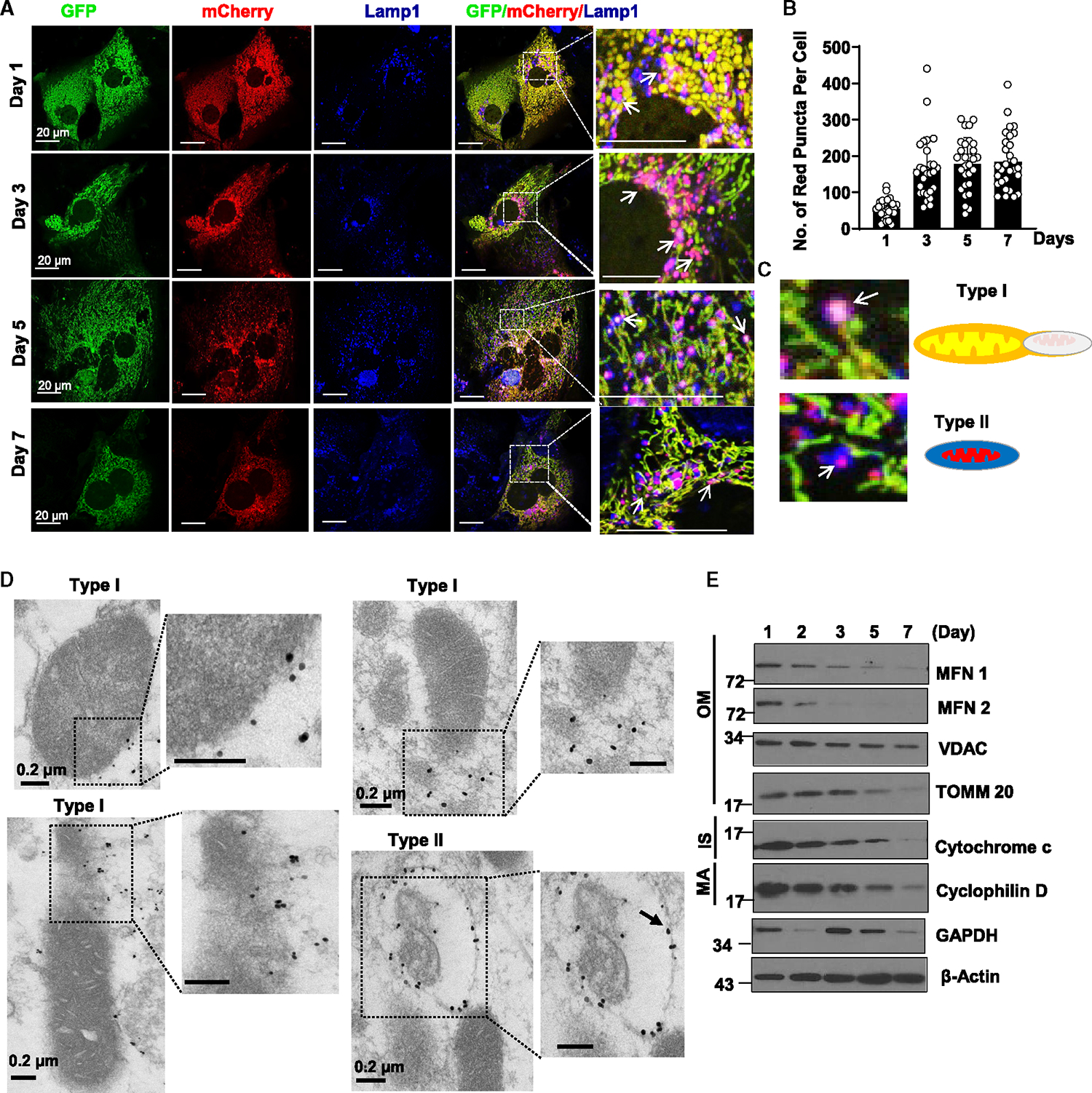
(A) Primary mouse hepatocytes were infected with adenovirus-COX8-GFP-mCherry (multiplicity of infection [MOI] = 10) from seeding for overnight. Cells grown on coverslips were fixed in 4% paraformaldehyde (PFA) at indicated time points followed by LAMP1 immunofluorescence staining. Representative images from confocal microscopy analysis are shown. Arrows denote the colocalization of red-only mitochondria from COX8-GFP-mCherry assay with the lysosomal marker LAMP1.
(B) Quantification data for the number of red-only mitochondria in each cell from COX8-GFP-mCherry assay. Data are presented as mean ± SD. At least 15 fields from 2 or 3 independent mouse hepatocytes isolation were quantified.
(C) Schemes for type I and type II LAMP1-associated mitochondrial structures.
(D) Representative images of immunogold electron microscopy using an anti-LAMP1 antibody from day 3 cultured mouse hepatocytes.
(E) Total cell lysates from primary mouse hepatocytes cultured for indicated times were subjected to western blot analysis. *p < 0.05, two-tailed Student’s t test.
To further examine the nature of these LAMP1-associated mitochondrial structures, we next performed immunogold TEM analysis using an anti-LAMP1 antibody. As shown in Figure 2D, we found three distinct LAMP1-labeled structures that were closely associated with mitochondria, which we termed types I and II. Type I structures were LAMP1-positive particles that were adjacent or localized to the outer mitochondrial membrane and appeared to have normal mitochondrial structure. Some type I structures were LAMP1-positive fragmented mitochondria that were in the process of budding off or leaving the parental mitochondria, which are likely MDVs. Type II structures were LAMP1-positive vesicles enclosing mitochondria-like structures, which appeared to be undergoing some sort of degradation (Figure 2D). It is difficult to correlate type I structures with the confocal microscopy data (Figure 2C) because of the resolution limitations of confocal microscopy. However, type I and II structures from the immuno-TEM analysis likely correlate with type I and II structures identified from the COX8-GFP-mCherry confocal and super-resolution microscopy analysis (Figures S2 and S3). Type I and type II structures were reminiscent of the structures that were positive for the BSA-gold particles (Figures S3C–S3E). Western blot analysis showed decreased levels of mitochondrial outer membrane (MFN1, MFN2, TOMM20, and VDAC), intermembrane space (cytochrome c), and matrix proteins (cyclophilin D) in hepatocytes after prolonged culture (Figure 2E), suggesting possible mitochondrial degradation during prolonged culture of hepatocytes. Together, these data indicate that prolonged cultured hepatocytes develop increased numbers of fragmented, acidic mitochondria associated with lysosomal markers likely derived from “hit and run” interactions between mitochondria and lysosomes, which are destined for degradation. Mitochondrial fragmentation is a prerequisite for the generation of red-only COX8 puncta.
Formation of the MLRO independent of ATG5 in cultured hepatocytes
ATG5 conjugates with ATG12 and forms a complex with ATG16, functioning as an E3-like ubiquitin ligase to regulate LC3-PE conjugation and autophagosome formation.25,26 We determined whether ATG5-mediated macroautophagy is involved in the formation of LAMP1-positive red-only mitochondria in COX8-GFP-mCherry-expressing hepatocytes. Like in wild-type (WT) hepatocytes, all the red-only mitochondria also colocalized with LAMP1 in ATG5 knockout (KO) hepatocytes (Figure 3A). There were very few red-only mitochondria on day 1, though the number was markedly increased by days 3, 5, and 7 in cultured ATG5 KO hepatocytes, the number of red-only mitochondria in day 5 and 7 cultured ATG5 KO hepatocytes being comparable with that of WT (Figure 3B), suggesting a chronologic transition of macroautophagy-dependent to macroautophagy-independent mechanisms for the formation of COX8-red-only mitochondria. Western blot analysis showed a lack of ATG12-ATG5 conjugates and LC3-II but increased LC3-I and SQSTM1/p62 in ATG5 KO hepatocytes (Figure S3F), confirming successful deletion of ATG5 and resultant macroautophagy deficiency in ATG5 KO hepatocytes. Moreover, levels of mitochondrial outer membrane (MFN2 and VDAC), intermembrane space (cytochrome c), and matrix (cyclophilin D) proteins were markedly decreased as early as day 2 in cultured WT hepatocytes but were barely changed until days 5 and 7 in cultured ATG5 KO hepatocytes (Figure 3C). These data indicate that ATG5-mediated macroautophagy is likely required for early phase mitophagy/mitochondrial turnover in cultured hepatocytes but is dispensable for late-phase mitochondrial turnover in prolonged cultured hepatocytes, suggesting an adaptive alternative pathway of mitochondrial turnover in the absence of ATG5. To further characterize the nature of the red-only mitochondria, we performed time course electron microscopy (EM) studies on prolonged cultured WT and ATG5 KO hepatocytes. We found that approximately 8% and 20% of mitochondria had electron-dense small membrane structures in close contact with the outer mitochondrial membrane on days 1 and 3, respectively (Figure 4A). Sometimes multiple such membrane structures were found attached to the same mitochondria in both WT and ATG5 KO hepatocytes (Figure 4, white arrows). In most cases, these electron-dense membrane structures adopted vesicle-like shapes (Figures S4A and S4C, white arrows), though some appeared as elongated membranes closely surrounding the outer mitochondrial membrane (Figures S4A and S4C, black arrows), and such structures are believed to be type I mitochondrial-related structures as defined earlier. High magnification analysis of these structures revealed that the electron-dense vesicles had continuous membrane connections with mitochondrial membranes (Figure S4A, boxed area, white arrowhead), suggesting that they are most likely MDVs. Interestingly, electron-dense “lysosome-like” structures approximately 0.5–1 μm in diameter markedly accumulated within day 5 and 7 cultured WT and ATG5 KO hepatocytes. Unlike lysosomes, which are single-membrane-bound organelles, many of these structures appeared to be double-membrane bound and contained undegraded electron-dense onion-like membranes with other heterogeneous content (Figures 4, S4B, and S4D, red arrows). However, some of these electron-dense structures also seemed to be single-membrane-bound enveloping undegraded membranes or membrane whorls (Figures 4, S4B, and S4D, black arrows), likely representing end-stage structures originated from double-membrane electron-dense structures undergoing more extensive degradation of the inner membranes. Immunogold TEM analysis showed that these electron-dense structures are LAMP1 positive, confirming that they are likely “lysosome-like” structures (Figure 4C), which we termed type II structures. The number of type I structures increased at day 3 and declined at days 5 and 7, whereas the number of type II structures increased in a time-dependent manner in both WT and ATG5 KO hepatocytes (Figure S4E). Correlative light and EM analysis showed that several red-only COX8 positive puncta were overlapped with the type II structures (Figures S5A and S5B), indicating type II lysosome-like structures are indeed derived from mitochondria. Through analysis of serial cellular sections, we found that formation of electrondense structures associated with mitochondria (type I mitochondria-related structure) occurred on almost all mitochondria (Figure S5C). Thus, the number of type I mitochondria-related structures that we quantified is most likely an underestimate, as only a single cellular section was used for quantification. High-magnification analysis again showed the continuity of the electron-dense vesicle membrane with the mitochondrial membrane, suggesting these type I structures are indeed MDVs. Nevertheless, we also found similar electron-dense membrane vesicle structures enriched in the Golgi complex (Figure S5D, white arrows), as well as in association with the cellular cytoskeleton (Figure S5D, red arrows). However, the nature of these vesicles associated with Golgi and cytoskeleton remain unclear. Increased number of red-only COX 8 puncta as well as similar type I and II mitochondria-related structures were also found in prolonged cultured human hepatocytes (Figures S5E and S5F). Taken together, these data suggest that large numbers of type I structures (likely MDVs) are formed and may travel and fuse with lysosomes to induce the formation of a hybrid organelle in prolonged cultured hepatocytes independent of canonical autophagy, which we term the MLRO). The formation of the MLRO may represent an alternative mitochondrial turnover mechanism independent of macroautophagy.
Figure 3. The mitochondria-lysosome-related organelle is formed independent of Atg5.
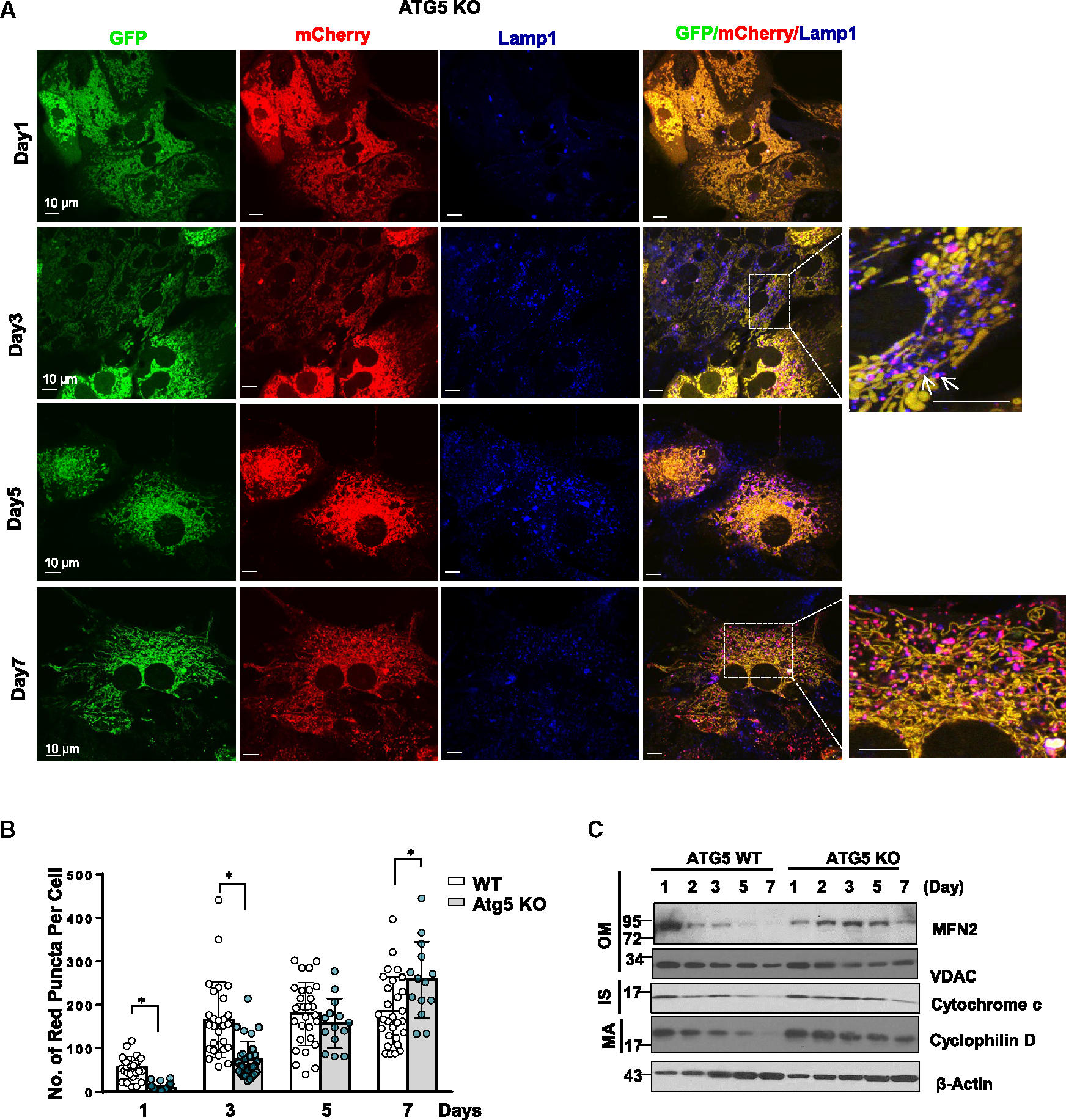
(A) Atg5 KO primary mouse hepatocytes were infected with adenovirus-Cox8-GFP-mCherry (MOI = 10) from seeding for overnight. Cells grown on coverslips were fixed in 4% PFA at indicated time points followed by Lamp1 immunofluorescence staining. Images were captured using confocal microscopy. Arrows denote the colocalization of red-only mitochondria in Cox8-GFP-mCherry assay with lysosomal marker LAMP1.
(B) Quantification data for the number of red-only mitochondria in each cell from Cox8-GFP-mCherry assay. Data are presented as mean ± SD. At least 15 fields from two independent mouse hepatocytes perfusion were quantified.
(C) WT and Atg5 KO mouse hepatocytes were cultured for indicated times. Total cell lysates were subjected to western blot analysis. *p < 0.05, two-tailed Student’s t test.
Figure 4. Ultrastructure of the mitochondria-lysosome-related organelle (MLRO) by electron microscopy.
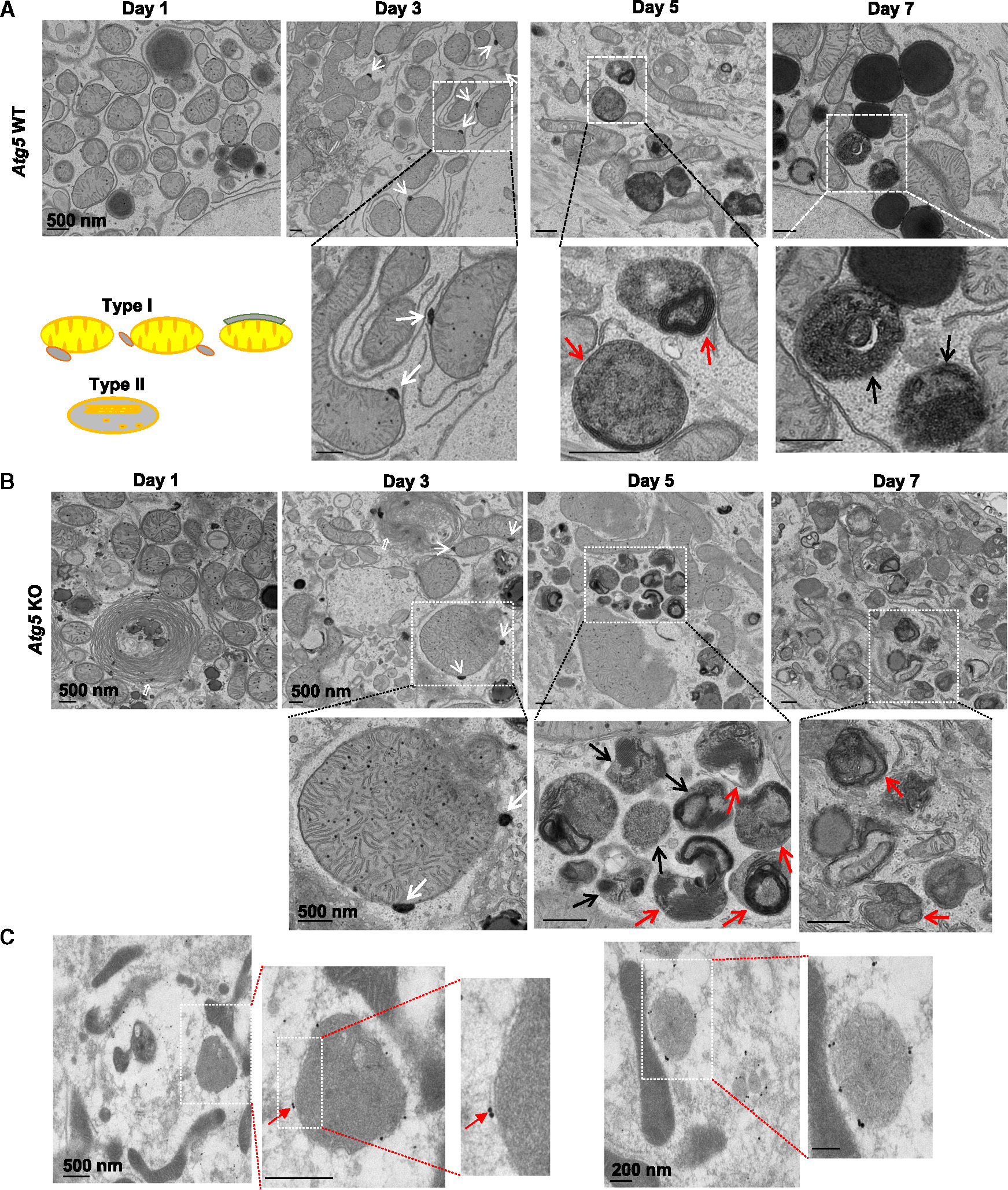
(A and B) Representative images of transmission electron microscopic examination of day 1 to day 7 cultured WT (A) and Atg5 KO (B) mouse hepatocytes. White arrows denote the contacts of electron-dense membrane structure with mitochondria. Red arrows denote the double-membrane electron-dense structure that contains heterogeneous undegraded contents. Black arrows denote the end-stage single-membrane MLRO structures.
(C) Representative images of immunogold electron microscopy (EM) using an anti-LAMP1 antibody in day 3 cultured mouse hepatocytes. Red arrows denote the LAMP1-positive MLRO structures.
PARKIN is quickly lost in cultured hepatocytes and is not required for MLRO formation in prolonged cultured hepatocytes
To investigate whether PARKIN is required for the formation of the MLRO in prolonged cultured hepatocytes, we first determined the time-dependent changes of PARKIN in primary cultured mouse hepatocytes as it is well known that levels of PARKIN protein are undetectable in most immortalized and cancer cells.27 The protein levels of PARKIN were higher in freshly isolated mouse hepatocytes but were undetectable after one day of culture and remained so on days 2, 3, and 5 (Figure S6A). A more detailed time course study showed that the levels of PARKIN started to decline at 6 h and were almost undetectable at 20 h of culture. No PARKIN signal was detected in hepatocytes isolated from PARKIN KO mice, indicating specificity of the PARKIN antibody (Figure S6B). mRNA levels of Prkn (the PARKIN gene name) did not change during prolonged culture, suggesting that PARKIN is regulated mainly at the protein level (Figure S6C). As levels of PARKIN are undetectable in hepatocytes after 24 h of culture, while MLRO increased in prolonged cultured hepatocytes, PARKIN is likely dispensable for the induction of the MLRO. To confirm that the induction of the MLRO is indeed PARKIN independent, primary cultured hepatocytes isolated from PARKIN KO mice were subjected to COX8-GFP-mCherry assay, western blot, and TEM analysis. We found significantly reduced numbers of COX8-red-only mitochondria in day 1 cultured PARKIN KO hepatocytes, though this number significantly increased to be comparable with WT hepatocytes in prolonged culture (Figures 5A and 5B). Both type I and type II MLRO structures were detected in PRKIN KO hepatocytes, the number of type II MLRO being lower on day 1 but markedly increased by days 3 and 5 (Figures 5C and 5D). Western blot analysis showed decreased levels of outer mitochondrial membrane (MFN1, MFN2, and TOMM20), intermembrane space (cytochrome c), and matrix proteins (cyclophilin D) by days 5 and 7 in cultured WT and PARKIN KO hepatocytes (Figure 5E). Together, these data indicate that formation of the MLRO is independent of PARKIN.
Figure 5. PARKIN is dispensable for MLRO formation in prolonged cultured hepatocytes.
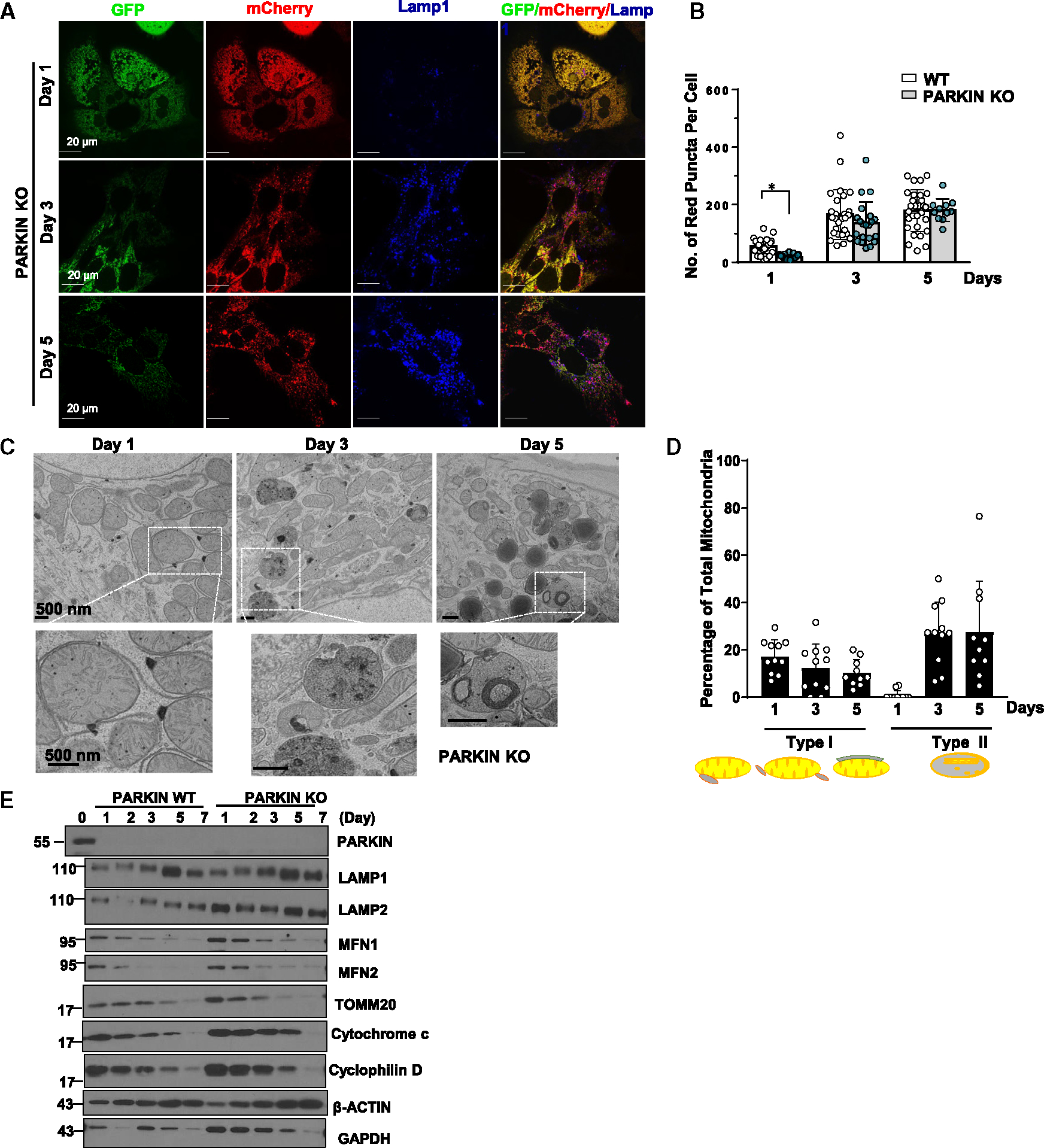
(A) PARKIN KO primary mouse hepatocytes were infected with adenovirus-Cox8-GFP-mCherry (MOI = 10) from seeding for overnight. Cells grown on coverslips were fixed in 4% PFA at indicated time points followed by LAMP1 immunofluorescence staining. Images were captured using confocal microscopy.
(B) Quantification data for the number of red-only mitochondria in each cell from Cox8-GFP-mCherry assay. Data are presented as mean ± SD. At least 15 fields from two independent mouse hepatocytes perfusion were quantified.
(C) Day 1 to day 5 cultured PARKIN KO mouse hepatocytes were examined using transmission electron microscopy (TEM).
(D) Type I and type II MLRO structure were quantified from (C) and normalized by the total mitochondria number in each field.
(E) WT and PARKIN KO primary mouse hepatocytes were cultured for indicated times. Total cell lysates were subjected to western blot analysis. *p < 0.05, two-tailed Student’s t test.
DRP1 is dispensable for the induction of the MLRO in prolonged cultured hepatocytes
Because we found that red-only mitochondria were all fragmented in the COX8-GFP-mCherry assay, we next determined whether DRP1 is required for the formation of the MLRO. We found marked increase in the numbers of red-only mitochondria in day 3 cultured DRP1 KO hepatocytes (Figure 6A). The number of red-only mitochondria in DRP1 KO hepatocytes was slightly lower than WT on day 1, but much higher than in WT hepatocytes after 3–7 days of culture (Figures 6A and 6B). Notably, these red-only mitochondria were also fragmented and colocalized with lysosomal marker LAMP1 in DRP1 KO hepatocytes (Figure 6A). The levels of DRP1 increased in day 5 and 7 cultured WT hepatocytes but were undetectable in DRP1 KO hepatocytes. In comparison, the levels of MFN1 and MFN2 were lower in DRP1 KO hepatocytes than in WT hepatocytes and decreased in both types of hepatocytes during culture (Figure 6C). It is possible that decreased levels of MFN1 and MFN2 were an adaptive response to the increased mitochondrial elongation during culture. TEM analysis revealed an accumulation of type I and type II MLRO structures in DRP1 KO hepatocytes (Figures 6D and 6E). Together, these data indicate that mitochondria can be fragmented independent of DRP1, and that DRP1 is dispensable for the formation of the MLRO in prolonged cultured hepatocytes.
Figure 6. DRP1 is not required for the induction of MLRO in prolonged cultured mouse hepatocytes.
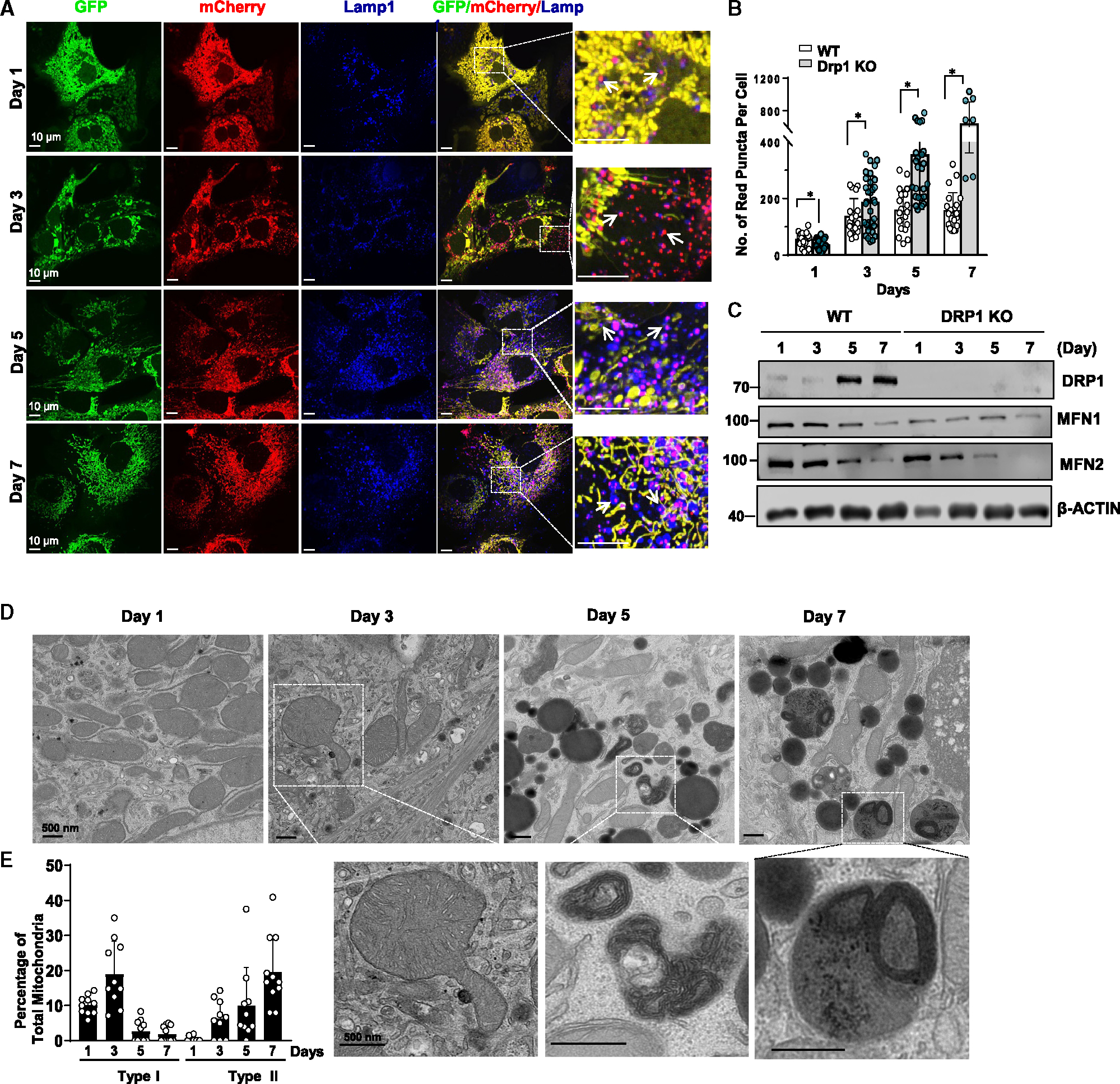
(A) DRP1 KO primary mouse hepatocytes were infected with adenovirus-COX8-GFP-mCherry (MOI = 10) from seeding for overnight. Cells grown on coverslips were fixed in 4% PFA at indicated time points followed by LAMP1 immunofluorescence staining. Representative images of confocal microscopy are shown. Arrows denote the colocalization of red-only mitochondria in COX8-GFP-mCherry assay with a lysosomal marker LAMP1.
(B) Quantification data for the number of red-only mitochondria in each cell from COX8-GFP-mCherry assay. Data are presented as mean ± SD. At least 15 fields from two independent mouse hepatocyte isolations were quantified.
(C) Total cell lysates were extracted from primary hepatocytes isolated from WT and liver-specific DRP1 KO mice that were cultured for indicated time points followed by western blot analysis.
(D) Representative TEM images from day 1 to day 7 cultured DRP1 KO mouse hepatocytes are shown.
(E) Type I and type II MLRO structures were quantified from (D) and normalized by the total mitochondria number in each field. *p < 0.05, two-tailed Student’s t test.
Inhibition of lysosomal degradation by leupeptin increases the accumulation of the MLRO
To determine whether blocking lysosomal degradation would extend MLRO accumulation, hepatocytes were treated with the lysosome protease inhibitor leupeptin. We found that the number of COX8-red-only mitochondria increased significantly with leupeptin treatment compared with untreated control hepatocytes (Figures S6D and S6E). TEM analysis revealed markedly increased numbers of type II MLRO structures in leupeptin-treated cells (Figure S6F). Some of the MLRO adopted elongated mitochondria-like shapes or resembled partially degraded mitochondria (Figure S6G, arrow), further supporting the concept that the MLRO may originate from mitochondria. Together, these data indicate that inhibition of lysosomal function promotes MLRO accumulation in hepatocytes.
Sorting nexin 9 is not required for MLRO formation, but overexpression of TFEB inhibits the accumulation of the MLRO
It has been reported that the presence of sorting nexin 9 (SNX9) was necessary for the formation of MDV and its transportation to late endosomes/lysosomes.28 However, upon infecting hepatocytes with adenovirus-shRNA (short hairpin RNA) Snx9 and adenovirus-null, the number of red-only COX8 was found to be similar, indicating that SNX9 may not be required for MLRO formation (Figures S7A and S7B). If MLRO is a hybrid organelle of mitochondria and lysosomes, we next asked whether MLRO would have normal or impaired lysosomal function. Galectin-3 (Gal3), a β-galactoside-binding cytosolic lectin that recognizes and binds galactose-containing glycans on the luminal membrane of lysosomes, is one of the best markers for measuring endo-lysosomal damage, as it forms discernible puncta on damaged lysosomes.29 We found increased number of Gal3 puncta in day 3 cultured hepatocytes compared with day 2 cultured hepatocytes (Figures S8A and S8B). More important, RFP-positive Gal3 puncta were all LAMP1-positive, and approximately 30% Gal3-positive puncta are also MitoTracker deep red positive (Figure S8C), supporting that the MLRO may be reminiscent of damaged lysosomes. Western blot analysis revealed increased levels of TFEB, the master regulator of lysosomal biogenesis, as well as increased levels of lysosomal membrane proteins LAMP1 and LAMP2 in primary cultured hepatocytes in a time-dependent manner (Figure S8D), suggesting a possible increased lysosomal biogenesis as an adaptive compensatory response. Overexpression of TFEB in hepatocytes increased the expression of a group of lysosomal genes, including Vatp6v1d, Vatp60e1, Vatp6v1h, Lamp1, Lamp2, and mitochondria biogenesis gene Ppargc1a (Figure S9A). Overexpression of TFEB also increased cathepsin B activities (Figure S9B) and decreased the number of RFP-positive Gal3 puncta in hepatocytes (Figures S9C and S9D).
We next determined whether overexpression of TFEB would affect the formation of the MLRO. The number of COX8-red-only mitochondria dramatically decreased in TFEB-overexpressed hepatocytes compared with the control group (Figures 7A and 7B). Moreover, TEM analysis revealed increased accumulation of type I but markedly decreased type II MLRO structures in TFEB-overexpressed hepatocytes compared with control hepatocytes (Figures 7C and 7D). Results from western blot analysis showed successful overexpression of TFEB in hepatocytes (Figure 7E). The levels of several mitochondria outer membrane (MFN1, MFN2, TOMM20, and VDAC), intermembrane space (cytochrome c), and matrix (cyclophilin D) proteins all increased in TFEB overexpressed hepatocytes (Figure 7E), suggesting a possible increase in mitochondria mass in hepatocytes with TFEB overexpression. Moreover, overexpression of TFEB increased protein levels of HNF4α but decreased α-SMA (Figure 7E). Together, these results indicate that overexpression of TFEB decreases the number of MLROs likely via increased MLRO clearance.
Figure 7. Overexpression of TFEB inhibits the formation of MLRO in prolonged cultured mouse hepatocytes.
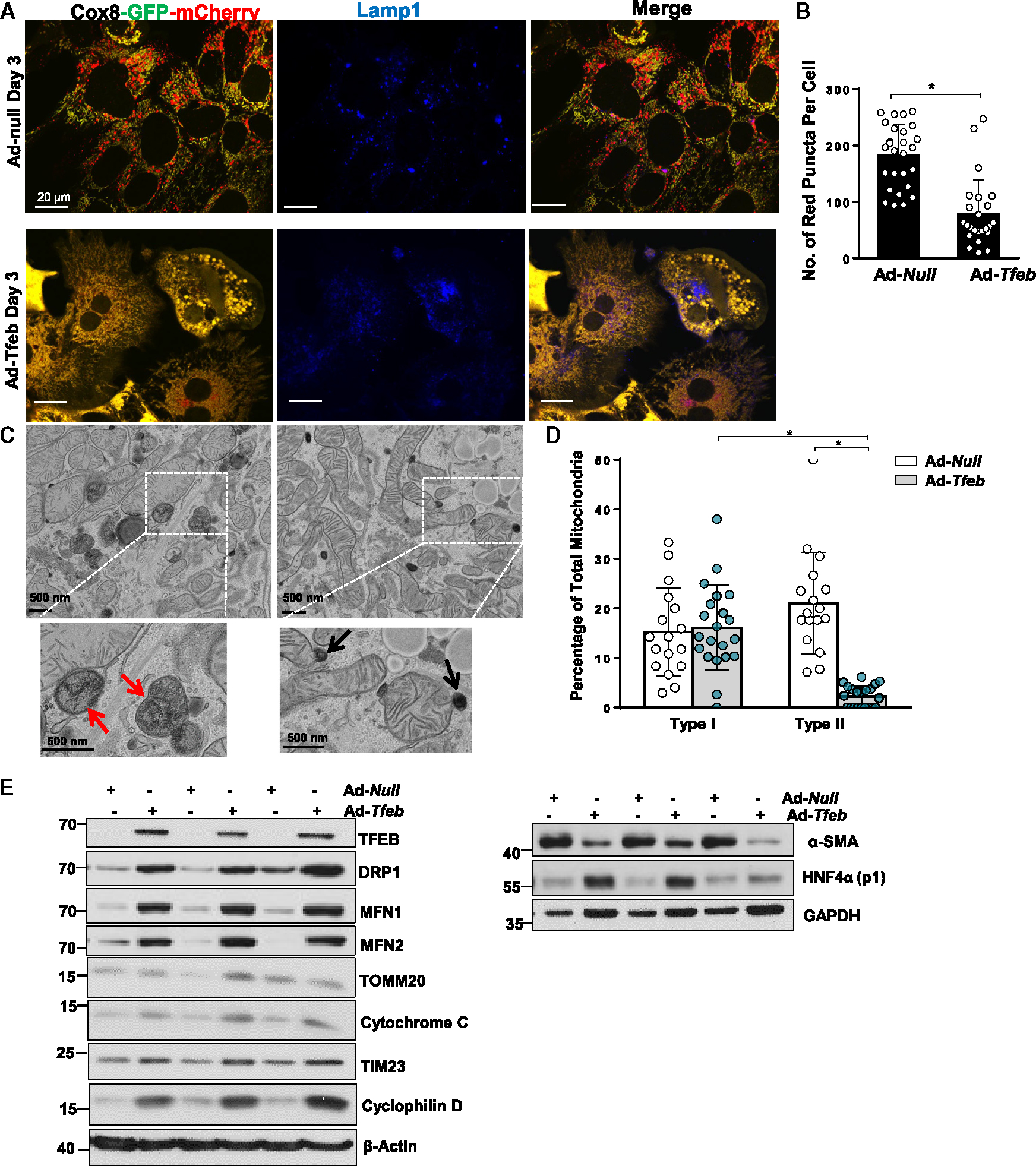
(A) Primary mouse hepatocytes were infected with adenovirus (Ad)-COX8-GFP-mCherry (MOI = 10) and adenovirus-TFEB (MOI = 1) or adenovirus-null from seeding for overnight. Cells grown on coverslips were fixed in 4% PFA on Day3 followed by LAMP1 immunofluorescence staining. Representative images of confocal microscopy are shown.
(B) Quantification data for the number of red-only mitochondria in each cell from COX8-GFP-mCherry assay. Data are presented as mean ± SD. At least 15 fields of cells from three independent hepatocytes perfusion were quantified.
(C) Represented EM image of day 3 cultured mouse hepatocytes with TFEB overexpression are shown.
(D) Type I and type II MLRO structure were quantified from Ad-null- and Ad-TFEB-infected mouse hepatocytes at day 3 and normalized by the total mitochondria number in each field.
(E) Total cell lysate from day 3 cultured mouse hepatocytes with or without TFEB overexpression were subjected to western blot analysis. *p < 0.05, one-way ANOVA with Bonferroni’s post hot test for (D), two-tailed Student’s t test for (B).
Induction of the MLRO during the adipocyte differentiation and diet-induced metabolic-dysfunction-associated steatohepatitis
Mouse 3T3-L1 fibroblasts can be differentiated into adipocytes in vitro. During the differentiation process, 3T3-L1 cells undergoes dramatic cellular and organelle remodeling, including the formation of lipid droplets and removal of excess mitochondria. As the adipocyte differentiation process is somewhat reminiscent of (although not completely identical to) our cultured hepatocyte remodeling, we next determined whether MLRO would also occur during the 3T3-L1 cell differentiation. Following the culture of 3T3-L1 cells in the adipocyte induction medium (IM) for 9 days, the accumulation of lipid droplets was very evident either by phase-contrast microscopy or oil red O staining (Figures S10A and S10B), suggesting successful transforming 3T3-L1 fibroblasts into adipocyte-like cells. EM analysis showed a decreased number of mitochondria with increased electrondense vesicles with enwrapped membrane residue structures inside, which were morphologically similar to MLRO in hepatocytes (Figure S10C). As hepatocytes dedifferentiation is a common feature in alcohol-associated hepatitis and non-alcoholic steatohepatitis (NASH, also called metabolic-dysfunction-associated steatohepatitis), we wondered whether MLRO would occur in NASH conditions. Mice fed with a choline-deficient, L-amino acid-defined, high-fat diet (CDAHFD) developed typical NASH with increased accumulation of lipid droplets, inflammation, and decreased HNF4α-positive hepatocytes (Figures S11A and S11B). TEM analysis showed increased the number of type I and type II MLRO in CDAHFD-fed mice compared with chow-diet-fed mice (Figures S11C and S11D). Western blot analysis showed that isolated mitochondria from chow-diet-fed mouse livers contained LAMP1, which were further increased in CDAHFD-fed mice (Figure S11E).Taken together, induction of the MLRO may occur during the adipocyte differentiation process and in hepatocytes of NASH undergo dedifferentiation, similar to primary cultured hepatocytes.
DISCUSSION
In this study, we have identified a hybrid intracellular organelle that we have termed the MLRO, which is likely a hybrid mitochondria-lysosome structure and serves as an alternative pathway for mitochondrial degradation during hepatocyte dedifferentiation. The MLRO is distinct from currently known mitochondrial quality control mechanisms such as mitophagy.
Mitophagy, whether PARKIN dependent or independent, requires the formation of double-membrane autophagosomes that carry the enveloped mitochondria to lysosomes, where mitochondria are degraded by lysosomal enzymes. Unlike canonical mitophagy, the formation of the MLRO bypasses the autophagosome step and likely involves “hit and run” contact between MDVs and a lysosome. We propose that the type I electron-dense membrane structures may originate from mitochondria and are most likely MDVs in nature as some of these type I membrane structures clearly show the membrane continuity with mitochondrial membranes. Although earlier studies showed that MDV formation requires PINK1-PARKIN and the trafficking of MDVs to lysosomes,30,31 more recent evidence suggest that MDVs were negatively regulated by PINK1-PARKIN for mitochondrial antigene presentation.32 Similarly, the formation of the MLRO is PARKIN independent although it is unclear whether the MLRO would also function for antigen presentation in addition to mitochondria degradation.
Earlier data showed that TOMM20-negative MDV formation is DRP1 independent, yet more recent data suggest that DRP1 is required for TOMM20-positive MDV formation.20,33 The MLRO is TOMM20 negative and DRP1 independent, suggesting some mechanistic similarities with the TOMM20-negative MDV. Cancer cells lacking ATG7 or FIP200 also have increased MDV formation, which requires SNX9 to act as an adaptive response for cancer cell survival.28 However, we found that knocking down Snx9 did not affect the formation of MLRO in primary cultured mouse hepatocytes (data not shown). Together, our data support the notion that the MLRO derives from MDVs that fuse with a lysosome to form a mitochondria-lysosome hybrid organelle, which is mechanistically and morphologically distinct from canonical mitophagy. Although MLROs look morphologically like lysosomes, the diameter of MLROs generally ranges from 1 to 1.5 μm, which is larger than lysosomes (which generally range from 0.5 to 1 μm). The formation of the MLRO is most likely involving lysosomal structural remolding due to the receiving of one or multiple MDVs. Functionally, MLRO formation is associated with mitochondrial protein degradation and inhibition of lysosomal degradation, resulting in MLRO accumulation, suggesting that the MLRO likely acts as a mitochondrial quality control mechanism to regulate mitochondrial homeostasis and maintain cellular function. A large amount of cellular energy and resources, such as lipid and proteins, are required for autophagosome formation, trafficking, and fusion with lysosomes. From an evolutionary viewpoint, the formation of MLRO is more energy and resource efficient even in the presence of autophagy proteins. Although we have provided evidence that MLRO formation is independent of PARKIN, DRP1, ATG5, and SNX9, the positive regulators of MLRO formation remains elusive and in need of future investigation.
Although we have provided compelling evidence to support the MLRO as a hybrid cellular organelle that may regulate mitochondrial homeostasis, we must ask, what are the potential physiological function and pathological relevance of MLRO? Hepatocyte dedifferentiation is very common in the late stage of chronic liver diseases such as alcohol-associated hepatitis and NASH, which leads to liver failure.34,35 Primary cultured hepatocytes undergo dedifferentiation during culture though the mechanisms are still poorly understood. Primary cultured hepatocytes lose hepatocyte features, including decreases in hepatic transporters, albumin production, metabolic enzymes such as CYP, and hepatocyte identity transcription factor HNF4α. These biochemical and functional changes are also associated with hepatocyte morphological remodeling, including changes in cell shape, polarity, cytoskeletal organization, mitochondrial fusion, mitophagy, and cell-cell contacts.36–39 A previous study showed that hepatocytes in culture generate increased numbers of LysoTracker positive digestive organelles associated with progressive mitochondria loss.37 However, detailed characterization of these acidic organelles was lacking, particularly at the ultrastructure level. These increased LysoTracker-positive acidic compartments observed in prolonged cultured hepatocytes likely contained mitochondrial markers similar to those of the MLRO that we observed in the present study. Our study has linked the MLRO to hepatocyte dedifferentiation although how exactly the MLRO contributes to the dedifferentiation remains unclear.
Perhaps one of the most intriguing findings in the present study was that overexpression of TFEB eliminated the number of COX8-red-only mitochondria and type II MLROs in primary cultured hepatocytes. TFEB is a master regulator of cellular organelle homeostasis via control of autophagy-related gene expression, as well as lysosome and mitochondria biogenesis.40,41 Recent evidence elucidates the critical role of TFEB in regulating liver development, live cell fate, and differentiation,42 suggesting that TFEB could play an important role in cytosolic organelle remodeling and dedifferentiation in hepatocytes. Indeed, we found that overexpression of TFEB increases levels of both mitochondrial fission (DRP1) and fusion (MFN1 and MFN2) proteins, as well as both outer and inner mitochondria proteins. Increased mitochondrial protein is likely because of increased mitochondrial biogenesis in TFEB-overexpressed hepatocytes. Damaged or dysfunctional lysosomes can be removed by lysophagy and replaced via lysosome biogenesis. Indeed, a decrease of MLROs in TFEB-overexpressed hepatocyte is likely due to enhanced clearance of MLROs rather than lysosomal biogenesis, though the exact mechanisms remain to be determined. Notably, overexpression of TFEB also increases HNF-4a but decreases α-SMA, suggesting that TFEB overexpression may help retain some hepatocyte features and inhibit dedifferentiation in culture. An increase in MLROs is also associated with adipocyte differentiation, suggesting that formation of MLROs may play a general role in cell differentiation and dedifferentiation.
In conclusion, we identify MLRO as a hybrid cellular organelle that may derive from MDVs and act as an alternative adaptive mechanism for mitochondrial quality control independent of canonical autophagy/mitophagy. Our results provide insights into cytosolic organelle remodeling and quality control in cellular dedifferentiation, which may be helpful for diseases with increased hepatocyte dedifferentiation such as alcohol-associated hepatitis and NASH.
Limitations of the study
Our time-lapse confocal imaging data showed a portion of mitochondrion budding off yellow mitochondria (labeled with COX8-GFP-mCherry) turning in to red. However, it should be noted that the current confocal microscopy technique is unable to precisely distinguish between the type I and type II structures at high resolution. We acknowledge this limitation, as we were unable to provide direct evidence to clearly demonstrate the conversion of type I to type II structures. Additionally, there is a possibility that a red-only mitochondrion may move from the down to the up plane, as we could focus on only one confocal Z section plane for the time-lapse imaging. Multiple images from multi-plane Z sections followed by three-dimensional (3D) reconstruction may be able to provide more insights on the nature of the biogenesis of red-only COX8 puncta.
One area of uncertainty is how LAMP1-positive vesicles make their way to MDV/mitochondria. TEM data reveal electron-dense vesicles near the Golgi complex and microtubules, suggesting that LAMP1-positive vesicles may travel along the microtubules to reach their destination. Another question that remains unanswered is how these vesicles fuse with MDV/mitochondria to form the MLRO. Although SNX9, RAB7, and syntaxin-17 have been implicated in MDV delivery to lysosomes, our findings suggest that SNX9 may not be necessary for MLRO formation. Further research is needed to fully understand the roles of RAB7 and syntaxin-17 in MLRO formation.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Wen-Xing Ding (wxding@kumc.edu).
Materials availability
This paper does not have newly generated materials.
Data and code availability
Data reported in this paper is willing to share after published, either through an online repository or upon request from the lead contact.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Primary hepatocytes culture
C57BL/6J wildtype mice and PARKIN knockout (KO) mice were obtained from The Jackson Laboratories (Bar Harbor, ME). Dnm1l Flox/Flox (F/F) mice (C57BL/6/129) and Atg5 Flox/Flox were crossed with albumin-Cre mice (Alb-Cre, C57BL/6J) (Jackson Laboratory) for six to nine generations to generate liver-specific DRP1 KO and ATG5 KO mice as we described previously.43,44 All animals were specific pathogen-free (SPF) and maintained in a barrier rodent facility under standard experimental conditions. All procedures were approved by the Institutional Animal Care and Use Committee of the University of Kansas Medical Center. Murine hepatocytes were isolated by retrograde, nonrecirculating perfusion of livers with 0.05% Collagenase Type IV (Sigma, C5138) from 2 to 3 months old mice as described previously 45. Cells were cultured in William’s medium E with 10% fetal bovine serum, 2 mM glutamine as well as routine antibiotics supplements for 2 h for attachment, which was designated as Day 0. Afterward, the culture medium was removed and cells were washed with PBS and cultured in the same medium without serum for designated times up to 7 days. All cells were maintained in a 37°C incubator with 5% CO2. Adenovirus was added to the cell suspension before seeding as needed.
3T3-L1 Cell culture and adipogenesis induction
3T3-L1 cells were maintained at 37°C in a humidified 5% CO2 atmosphere in DMEM (Hyclone) supplemented with 10% (v/v) FBS, penicillin, and streptomycin (100 U/ml), and glutamine (100 μg/mL). To induce adipogenesis, 3T3-L1 cells were seeded and propagated to confluence. Forty-eight hours after 100% confluence, which was designated as Day 0, cells were incubated in a differentiation medium [10 μg/mL insulin, 10 μM troglitazone, 1 μM dexamethasone, 0.5 mM 3-isobutyl-1-methylxanthine (IBMX)] for 3 days. On Day 3, cells were switched to a maintenance medium (5 μg/mL insulin, 10 μM troglitazone) and the maintenance medium was replaced every three days.
METHOD DETAILS
Oil red O staining
Oil Red O stock solution and Millipore water (v/v = 6:4) were mixed well and filtered through 125 mm filter paper to make a working solution before use. Cells were washed once with PBS, fixed with 4% PFA at RT for 1 h, and washed with 60% 2-propanol twice for 5 min. Then the cells were counterstained with hematoxylin for 1 min and washed with PBS.
Confocal and super-resolution microscopy
To examine autophagic flux or mitophagy, primary hepatocytes were seeded on a coverslip in a 12 well-plate (2 × 105 per well) and infected with adenovirus-RFP-GFP-LC3 (10 multiplicity of infection (MOI)) or adenovirus-COX8-GFP-mCherry (10 MOI) overnight. Some cells were treated with chloroquine (40 μM), Leupeptin (40 μM), or Bafilomycin A1 (200 nM) for 6 or 24 h as needed. After treatment, cells were fixed on designated time points with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 2 h at room temperature or kept at 4°C for microscopy. Fluorescence images were acquired under Nikon A1R confocal or Leica SP8 STED super-resolution microscope. For the Dextran staining, 10 μg/mL Dextran-Alexa Fluor 647 (10,000 MW, Invitrogen, D22914) was added to the cell culture medium on Day 1 for 16 h and washed out. Time-Lapse live cell confocal imaging was acquired using Leica SP8 STED super-resolution microscope.
Immunofluorescence staining
Cells grown on coverslips were fixed with 4% PFA in PBS for 2 h at room temperature and permeabilized in 0.2% Triton X-100/PBS for 15 min, followed by blocking in 2% BSA/PBS for 1 h. Cells were incubated with primary antibodies overnight in a humidified chamber at 4°C followed by extensive washing before the addition of secondary antibodies for 1 h at room temperature. Nuclear was stained by Hoechst 33342 as needed. Cells were mounted in PermaFluor Aqueous Mounting Medium (Thermo Scientific, TA-030-FM) followed by confocal microscopy. For double-color immunofluorescence, two first antibodies were co-incubated followed by Alexa Fluor 488/549 secondary antibodies. All the primary and secondary antibodies used in this study were listed in Table S1.
Electron microscopy
Cells grown on plastic coverslips were fixed with 2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) followed by 1% OsO4. After dehydration, thin sections were stained with uranyl acetate and lead citrate for observation under a JEM 1016CX electron microscope. For the BSA-Gold labeling, 5–10 μL of BSA-gold tracer (Aurion, 25487) was added to the cell culture medium for 16 h before the cells were fixed. For immunoelectron microscopy, 4% PFA fixed cells on coverslips were permeabilized in 0.2% Triton X-100/PBS for 15 min followed by blocking in 2% BSA/PBS for 1 h. Lamp1 primary antibody was applied for 2 h at RT followed by extensive washing before the addition of ImmunoGold Goat-anti-Rat IgG (Aurion, 25181) for 2 h at RT. A sliver enhancement step was conducted following the manufacturer’s instruction (Aurion, 500.033) before OsO4 was applied.
Correlative light and EM analysis
Primary mouse hepatocytes were cultured in 35 mm gridded MatTek dishes and infected with adenovirus-COX8-GFP-mCherry (10 MOI) from seeding for indicated time. At Day 3, cells were fixed with 4% PFA and 20 X phase contrast and fluorescence images of the cells were taken followed by 60 X oil lens fluorescence images of fluorescence microscopy. Cells were then fixed with 2% glurataldehyde (EM grade) in 0.2M HEPES followed EM analysis. Phase contrast, fluorescence and EM images were overlayed using the Adobe Photoshop Elements software.
Fatty acid oxidation assay
Fatty acid oxidation by primary hepatocytes was determined as previously described with minor modifications 46. Hepatocytes were starved of serum and then 12-well plates were washed with warm PBS. Next, the cells were incubated in triplicate with a 14C-labeled FAO reaction medium consisting of DMEM-low glucose (lnvitrogen), 0.5 μCi/mL [1–14C] palmitate, 50 μM palmitate, 0.5% BSA, 1 mM carnitine, and 12.5 mM HEPES (pH −7.4) at 37°C for 3 h. To inhibit hepatocyte CPT-1a, parallel wells were treated with Etomoxir (100 μM). CPT-1-mediated FAO was calculated by subtracting FAO in the presence of Etomoxir from absolute FAO. After 3 h, the medium from each well was collected and a sample of the medium was dispensed into a sealed trapping device. The 14CO2 was removed from the media sample by the addition of perchloric acid and trapped in NaOH. This was then collected and analyzed by liquid scintillation counting to determine complete FAO to CO2. The acidified medium was collected, refrigerated, and centrifuged (16,000 g, 4°C). An aliquot was analyzed by liquid scintillation counting to determine the acid-soluble metabolites (ASMs) of FAO. Finally, the cells were rinsed three times with ice-cold Krebs-Henseleit buffer and lysed with SDS lysis buffer. The protein concentration of the lysate was determined by BCA assay.
Immunoblot analysis
Cells were washed in PBS and lysed in radioimmunoprecipitation assay (RIPA) buffer [1% NP40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl (lauryl) sulfate] 20 μg of protein from each sample were separated by SDS-PAGE gel and transferred to PVDF membranes. Membranes were probed using indicated primary and secondary antibodies and developed with SuperSignal West Pico Plus Chemiluminescent Substrate (Thermo Scientific, 34579) and Immobilon Western Chemiluminescent HRP Substrate (Millipore, WBKLS0500). All the primary and secondary antibodies used in this study were listed in Table S1.
RNA isolation and real-time qPCR
Total cell RNA was isolated using GeneJET RNA Purification Kit (Thermo Scientific, K0732) and reverse transcribed into cDNA by RevertAid reverse transcriptase (Fermentas, EP0442). Quantitative PCR was performed using SYBR Green chemistry (Bio-Rad,1725124). Primer sequences (5ʹ–3ʹ) used in the present study were listed in Table S2. All the results were normalized to 18s and expressed as fold change over the Control group.
Proteasome and cathepsin B activity assay
Cells were washed in PBS and lysed in M2 lysis buffer [50 mM Tris-base (pH 7.4), 130 mM NaCl, 10% glycerol, 0.5% NP-40, 0.5 mM EDTA, 0.5 mM EGTA, 1 mM phenylmethylsulfonyl fluoride (PMSF)] for 10 min on ice. Proteasome activity from total cell lysate was measured using Suc-LLVY-AMC substrate, cathepsin B activity was measured using Z-Phe-Arg-AMC (50 μM) substrate. AMC release was measured in fluorometer using 380/460 excitation/emission test filter.
Mouse NASH model and liver mitochondria purification
Male 2–3 months-old C57BL/6J mice were fed with a chow or a choline-deficient, L-amino acid-defined, high-fat diet (CDAHFD) for 6 weeks to induce typical NASH. Paraffin-embedded liver sections were stained with hematoxylin and eosin (H&E) and immunostaining for HNF4α was performed as we described previously 47. Fine-cut liver tissues were fixed with 2% glutaraldehyde in phosphate buffer 0.1 mol/L (pH 7.4) followed by 1% OsO4. After dehydration, thin sections were stained with uranyl acetate and lead citrate for observation under a JEM 1016CX electron microscope (JEOL, Tokyo, Japan). Images were acquired digitally. Highly purified liver mitochondria were isolated from the chow and CDAHFD-fed mouse livers according to the protocol with some modifications 48. Briefly mouse livers were homogenized in isolation buffer 1 (IB1, 225 mM mannitol, 75mM sucrose, 30 mM Tris-HCl pH 7.4) and centrifuged at 740g for 5 min at 4°C. The supernatant was collected and centrifuged at 9,000g for 10 min at 4°C. The pellet was collected that contained the crude mitochondria and further gently resuspended in the ice-cold IB2 (225 mM mannitol, 75mM sucrose, 0.5% BSA and 30 mM Tris-HCl pH 7.4) followed by centrifuging at 10,000g for 10 min at 4°C. The supernatant was discarded and the crude mitochondria pellet was gently resuspended in mitochondria resuspending buffer (MRB, 250 mM mannitol, 5 mM HEPES (pH 7.4), and 0.5 mM EGTA), and centrifuged at 10,000g for 10 min at 4°C. Eight mL Percoll medium was then added to the ultracentrifuge tubes and layered the collected crude mitochondria on top of the Percoll medium and centrifuged at 95,000g for 30 min at 4°C. A dense band containing purified mitochondria at the bottom of the tubes were collected for Western blot analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Experimental data were subjected to a Student t-test or one-way ANOVA analysis with Bonferroni post hoc test where appropriate. Mean ± SD was used for the quantitative data. A p value less than 0.05 was considered significant.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| See Table S1 for a list of antibodies | ||
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Collagenase Type IV | Sigma | C5138 |
| Dextran-Alexa Fluor 647 (10,000 MW) | Invitrogen | D22914 |
| PermaFluor Aqueous Mounting Medium | Thermo Scientific | TA-030-FM |
| BSA-gold tracer | Aurion | 25487 |
| SuperSignal West Pico Plus Chemiluminescent Substrate | Thermo Scientific | 34579 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| 3T3-L1 Cell line | ATCC | CL-173 |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S2 for a list of oligonucleotides | ||
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6J mouse | Jackson Laboratories | 000664 |
| Atg5 flox | Riken | RBRC02975 |
| Drp1 flox | Johns Hopkins University | Dr. Hiromi Sesaki Lab |
|
| ||
| Software and algorithms | ||
|
| ||
| SPSS | KU Information Technology | N/A |
Highlights.
Dedifferentiated hepatocyte induces mitochondria-lysosome-related organelle (MLRO)
The MLRO is a hybrid organelle formed by lysosome fusion with mitochondria-derived vesicles
The MLRO is independent of autophagy/mitophagy machinery and negatively regulated by TFEB
ACKNOWLEDGMENTS
This study was supported in part by NIH grants R01 DK102142, R01 AG072895, and R37 AA020518 (W.-X.D.). The authors thank Larysa Stroganova at KUMC EM Research Laboratory for her excellent technical assistance on the electron microscopy studies. The authors also thank Dr. Noboru Mizushima at the University of Tokyo for providing Atg5flox/fox mice, and Dr. Han-Ming Shen from University of Macau for critical reading of the manuscript. The Leica SP8 STED was supported by NIH grant S10 OD 023625 at KUMC.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113291.
REFERENCES
- 1.Sorrentino V, Menzies KJ, and Auwerx J (2018). Repairing Mitochondrial Dysfunction in Disease. Annu. Rev. Pharmacol. Toxicol. 58, 353–389. 10.1146/annurev-pharmtox-010716-104908. [DOI] [PubMed] [Google Scholar]
- 2.Lin MT, and Beal MF (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 3.Ma X, McKeen T, Zhang J, and Ding WX (2020). Role and Mechanisms of Mitophagy in Liver Diseases. Cells 9. 10.3390/cells9040837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan DC (2020). Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 15, 235–259. 10.1146/annurev-pathmechdis-012419-032711. [DOI] [PubMed] [Google Scholar]
- 5.Giacomello M, Pyakurel A, Glytsou C, and Scorrano L (2020). The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 21, 204–224. 10.1038/s41580-020-0210-7. [DOI] [PubMed] [Google Scholar]
- 6.Ni HM, Williams JA, and Ding WX (2015). Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 4, 6–13. 10.1016/j.redox.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williams JA, and Ding WX (2018). Mechanisms, pathophysiological roles and methods for analyzing mitophagy - recent insights. Biol. Chem. 399, 147–178. 10.1515/hsz-2017-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pickles S, Vigié P, and Youle RJ (2018). Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 28, R170–R185. 10.1016/j.cub.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ng MYW, Wai T, and Simonsen A (2021). Quality control of the mitochondrion. Dev. Cell 56, 881–905. 10.1016/j.devcel.2021.02.009. [DOI] [PubMed] [Google Scholar]
- 10.Karbowski M, and Youle RJ (2011). Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr. Opin. Cell Biol. 23, 476–482. 10.1016/j.ceb.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leonhard K, Guiard B, Pellecchia G, Tzagoloff A, Neupert W, and Langer T (2000). Membrane protein degradation by AAA proteases in mitochondria: extraction of substrates from either membrane surface. Mol. Cell 5, 629–638. 10.1016/s1097-2765(00)80242-7. [DOI] [PubMed] [Google Scholar]
- 12.Arnold I, and Langer T (2002). Membrane protein degradation by AAA proteases in mitochondria. Biochim. Biophys. Acta 1592, 89–96. 10.1016/s0167-4889(02)00267-7. [DOI] [PubMed] [Google Scholar]
- 13.Ding WX, and Yin XM (2012). Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol. Chem. 393, 547–564. 10.1515/hsz-2012-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ding WX, Guo F, Ni HM, Bockus A, Manley S, Stolz DB, Eskelinen EL, Jaeschke H, and Yin XM (2012). Parkin and mitofusins reciprocally regulate mitophagy and mitochondrial spheroid formation. J. Biol. Chem. 287, 42379–42388. 10.1074/jbc.M112.413682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding WX, Li M, Biazik JM, Morgan DG, Guo F, Ni HM, Goheen M, Eskelinen EL, and Yin XM (2012). Electron microscopic analysis of a spherical mitochondrial structure. J. Biol. Chem. 287, 42373–42378. 10.1074/jbc.M112.413674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puhm F, Afonyushkin T, Resch U, Obermayer G, Rohde M, Penz T, Schuster M, Wagner G, Rendeiro AF, Melki I, et al. (2019). Mitochondria Are a Subset of Extracellular Vesicles Released by Activated Monocytes and Induce Type I IFN and TNF Responses in Endothelial Cells. Circ. Res. 125, 43–52. 10.1161/CIRCRESAHA.118.314601. [DOI] [PubMed] [Google Scholar]
- 17.Crewe C, Funcke JB, Li S, Joffin N, Gliniak CM, Ghaben AL, An YA, Sadek HA, Gordillo R, Akgul Y, et al. (2021). Extracellular vesicle-based interorgan transport of mitochondria from energetically stressed adipocytes. Cell Metabol. 33, 1853–1868.e11.e1811. 10.1016/j.cmet.2021.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiao H, Jiang D, Hu X, Du W, Ji L, Yang Y, Li X, Sho T, Wang X, Li Y, et al. (2021). Mitocytosis, a migrasome-mediated mitochondrial quality-control process. Cell 184, 2896–2910.e13. 10.1016/j.cell.2021.04.027. [DOI] [PubMed] [Google Scholar]
- 19.Tan HWS, Lu G, Dong H, Cho YL, Natalia A, Wang L, Chan C, Kappei D, Taneja R, Ling SC, et al. (2022). A degradative to secretory autophagy switch mediates mitochondria clearance in the absence of the mATG8-conjugation machinery. Nat. Commun. 13, 3720. 10.1038/s41467-022-31213-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, Andrade-Navarro MA, and McBride HM (2008). Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. 18, 102–108. 10.1016/j.cub.2007.12.038. [DOI] [PubMed] [Google Scholar]
- 21.Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, and McBride HM (2012). A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 22, 135–141. 10.1016/j.cub.2011.11.057. [DOI] [PubMed] [Google Scholar]
- 22.Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA, and McBride HM (2012). Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS One 7, e52830. 10.1371/journal.pone.0052830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, Abeliovich H, Abildgaard MH, Abudu YP, Acevedo-Arozena A, et al. (2021). Guidelines for the use and interpretation of assays for monitoring autophagy 17 (1), 1–382, 4th edition 17. 10.1080/15548627.2020.1797280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma X, and Ding WX (2021). A fluorescence imaging based-assay to monitor mitophagy in cultured hepatocytes and mouse liver. Liver Res. 5, 16–20. 10.1016/j.livres.2020.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizushima N (2020). The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol. 63, 1–10. 10.1016/j.ceb.2019.12.001. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, and Ohsumi Y (2001). The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 20, 5971–5981. 10.1093/emboj/20.21.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW, and Yin XM (2010). Nix Is Critical to Two Distinct Phases of Mitophagy, Reactive Oxygen Species-mediated Autophagy Induction and Parkin-Ubiquitin-p62-mediated Mitochondrial Priming. J. Biol. Chem. 285, 27879–27890. 10.1074/jbc.M110.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Towers CG, Wodetzki DK, Thorburn J, Smith KR, Caino MC, and Thorburn A (2021). Mitochondrial-derived vesicles compensate for loss of LC3-mediated mitophagy. Dev. Cell 56, 2029–2042.e5. 10.1016/j.devcel.2021.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y, and Yoshimori T (2013). Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 32, 2336–2347. 10.1038/emboj.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McLelland GL, Lee SA, McBride HM, and Fon EA (2016). Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J. Cell Biol. 214, 275–291. 10.1083/jcb.201603105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McLelland GL, Soubannier V, Chen CX, McBride HM, and Fon EA (2014). Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 33, 282–295. 10.1002/embj.201385902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, Fazel A, Bergeron JJ, Trudeau LE, Burelle Y, et al. (2016). Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell 166, 314–327. 10.1016/j.cell.2016.05.039. [DOI] [PubMed] [Google Scholar]
- 33.König T, Nolte H, Aaltonen MJ, Tatsuta T, Krols M, Stroh T, Langer T, and McBride HM (2021). MIROs and DRP1 drive mitochondrial-derived vesicle biogenesis and promote quality control. Nat. Cell Biol. 23, 1271–1286. 10.1038/s41556-021-00798-4. [DOI] [PubMed] [Google Scholar]
- 34.Bou Saleh M, Louvet A, Ntandja-Wandji LC, Boleslawski E, Gnemmi V, Lassailly G, Truant S, Maggiotto F, Ningarhari M, Artru F, et al. (2021). Loss of hepatocyte identity following aberrant YAP activation: A key mechanism in alcoholic hepatitis. J. Hepatol. 75, 912–923. 10.1016/j.jhep.2021.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berasain C, Arechederra M, Argemí J, Fernández-Barrena MG, and Avila MA (2023). Loss of liver function in chronic liver disease: An identity crisis. J. Hepatol. 78, 401–414. 10.1016/j.jhep.2022.09.001. [DOI] [PubMed] [Google Scholar]
- 36.Arterburn LM, Zurlo J, Yager JD, Overton RM, and Heifetz AH (1995). A morphological study of differentiated hepatocytes in vitro. Hepatology 22, 175–187. [PubMed] [Google Scholar]
- 37.Rodriguez-Enriquez S, Kai Y, Maldonado E, Currin RT, and Lemasters JJ (2009). Roles of mitophagy and the mitochondrial permeability transition in remodeling of cultured rat hepatocytes. Autophagy 5, 1099–1106. 10.4161/auto.5.8.9825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rambold AS, Cohen S, and Lippincott-Schwartz J (2015). Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 32, 678–692. 10.1016/j.devcel.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fu D, Mitra K, Sengupta P, Jarnik M, Lippincott-Schwartz J, and Arias IM (2013). Coordinated elevation of mitochondrial oxidative phosphorylation and autophagy help drive hepatocyte polarization. Proc. Natl. Acad. Sci. USA 110, 7288–7293. 10.1073/pnas.1304285110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raben N, and Puertollano R (2016). TFEB and TFE3: Linking Lysosomes to Cellular Adaptation to Stress. Annu. Rev. Cell Dev. Biol. 32, 255–278. 10.1146/annurev-cellbio-111315-125407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. (2011). TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433. 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pastore N, Huynh T, Herz NJ, Calcagni A, Klisch TJ, Brunetti L, Kim KH, De Giorgi M, Hurley A, Carissimo A, et al. (2020). TFEB regulates murine liver cell fate during development and regeneration. Nat. Commun. 11, 2461. 10.1038/s41467-020-16300-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ni HM, Boggess N, McGill MR, Lebofsky M, Borude P, Apte U, Jaeschke H, and Ding WX (2012). Liver-specific loss of Atg5 causes persistent activation of Nrf2 and protects against acetaminophen-induced liver injury. Toxicol. Sci. 127, 438–450. 10.1093/toxsci/kfs133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang X, Wang H, Ni HM, Xiong A, Wang Z, Sesaki H, Ding WX, and Yang L (2017). Inhibition of Drp1 protects against senecionine-induced mitochondria-mediated apoptosis in primary hepatocytes and in mice. Redox Biol. 12, 264–273. 10.1016/j.redox.2017.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper is willing to share after published, either through an online repository or upon request from the lead contact.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.