

SUMMARY
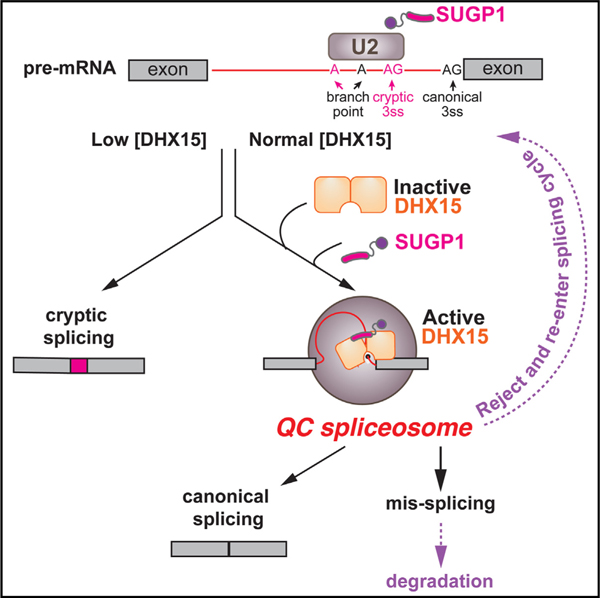
Pre-mRNA splicing is surveilled at different stages by quality control (QC) mechanisms. The leukemia-associated DExH-box family helicase hDHX15/scPrp43 is known to disassemble spliceosomes after splicing. Here, using rapid protein depletion and analysis of nascent and mature RNA to enrich for direct effects, we identify a widespread splicing QC function for DHX15 in human cells, consistent with recent in vitro studies. We find that suboptimal introns with weak splice sites, multiple branch points, and cryptic introns are repressed by DHX15, suggesting a general role in promoting splicing fidelity. We identify SUGP1 as a G-patch factor that activates DHX15’s splicing QC function. This interaction is dependent on both DHX15’s ATPase activity and on SUGP1’s U2AF ligand motif (ULM) domain. Together, our results support a model in which DHX15 plays a major role in splicing QC when recruited and activated by SUGP1.
Graphical Abstract
In brief
Pre-mRNA splicing is surveilled to ensure the appropriate splice sites are chosen. Feng et al. reveal a splicing quality control mechanism mediated by the RNA helicase DHX15 and its activator, SUGP1. This work has implications for understanding splicing fidelity and leukemia-associated mutations in DHX15 and related factors.
INTRODUCTION
Pre-mRNA splicing is a multistage process catalyzed by the spliceosome, which undergoes conformational and compositional rearrangement driven by RNA helicases.1,2 The transitions between catalytic and post-catalytic stages of splicing are mediated by four DEAH-box RNA helicases (DHXs), hsDHX16/scPrp2, hsDHX38/scPrp16, hsDHX8/scPrp22, and hsDHX15/scPrp43, which participate in the branch-formation, exon-exon-joining, mRNA-releasing, and lariat-releasing steps of the splicing cycle, respectively.3,4 Despite their structural similarity, these four helicases specifically remodel spliceosomal B*, C*, P, and intronlariat spliceosome (ILS) complexes, respectively, and act from the exterior of the spliceosome to pull on either the small nuclear RNA (snRNA) or pre-mRNA to unwind snRNA-mRNP duplex within the spliceosome core.5,6 This stage specificity of each DHX is likely achieved through recruitment or activation mechanisms by different G-patch proteins (GPs), but these interactions are not well understood.7,8
GPs contain a short (~45 amino acids) flexible glycine-rich motif, termed the G-patch domain, which can bind and activate DHX helicases.9 Structurally, two recent studies have revealed that the G-patch domain binds to the side opposite the helicase’s RNA tunnel, which likely confines the helicase in a semi-open state with improved RNA affinity and hence increased RNA-dependent ATPase and helicase activity.10,11 Because of the low intrinsic RNA affinity and hence RNA-stimulated ATP hydrolysis and RNA unwinding activities of purified DHX helicases alone,12 recombinant DHX-G-patch chimeric proteins have often been used in in vitro splicing and reconstituted spliceosome disassembly experiments.13–15
SUGP1, previously known as SF4, is an early spliceosomal component, likely involved in bridging the SF3b complex in U2 snRNP with the 3′ss recognition U2AF heterodimer.16,17 Recently, SUGP1 mutations in cancer have been linked to cryptic splicing.18,19 It is suspected that SUGP1 represses cryptic 3′ss usage by acting as a DHX activator through its G-patch domain. However, the identity of this speculated DHX helicase has not been confirmed.
DHX15, which is commonly mutated in acute myeloid leukemia (AML),20,21 is canonically known for its spliceosome disassembly role at the end of the splicing cycle to facilitate the recycling of spliceosomal components and extraction of excised intron lariats.13,22 Additionally, recent studies in cell-free systems have identified a quality control (QC) function for DHX15 (Figure 1A). One model for splicing QC is that splicing intermediates that are processed more slowly will be rejected by upstream DHXs from processive splicing and subsequently subjected to DHX15-mediated disassembly.23,24 Besides this rejection route, recent work with DHX15-immunodepleted HeLa nuclear extract has identified that DHX15 also surveils early spliceosome assembly.25 However, whether and how widely DHX15 functions in splicing QC in human cells remains largely unclear.
Figure 1. Genome-wide identification of introns regulated by DHX15-mediated splicing QC.
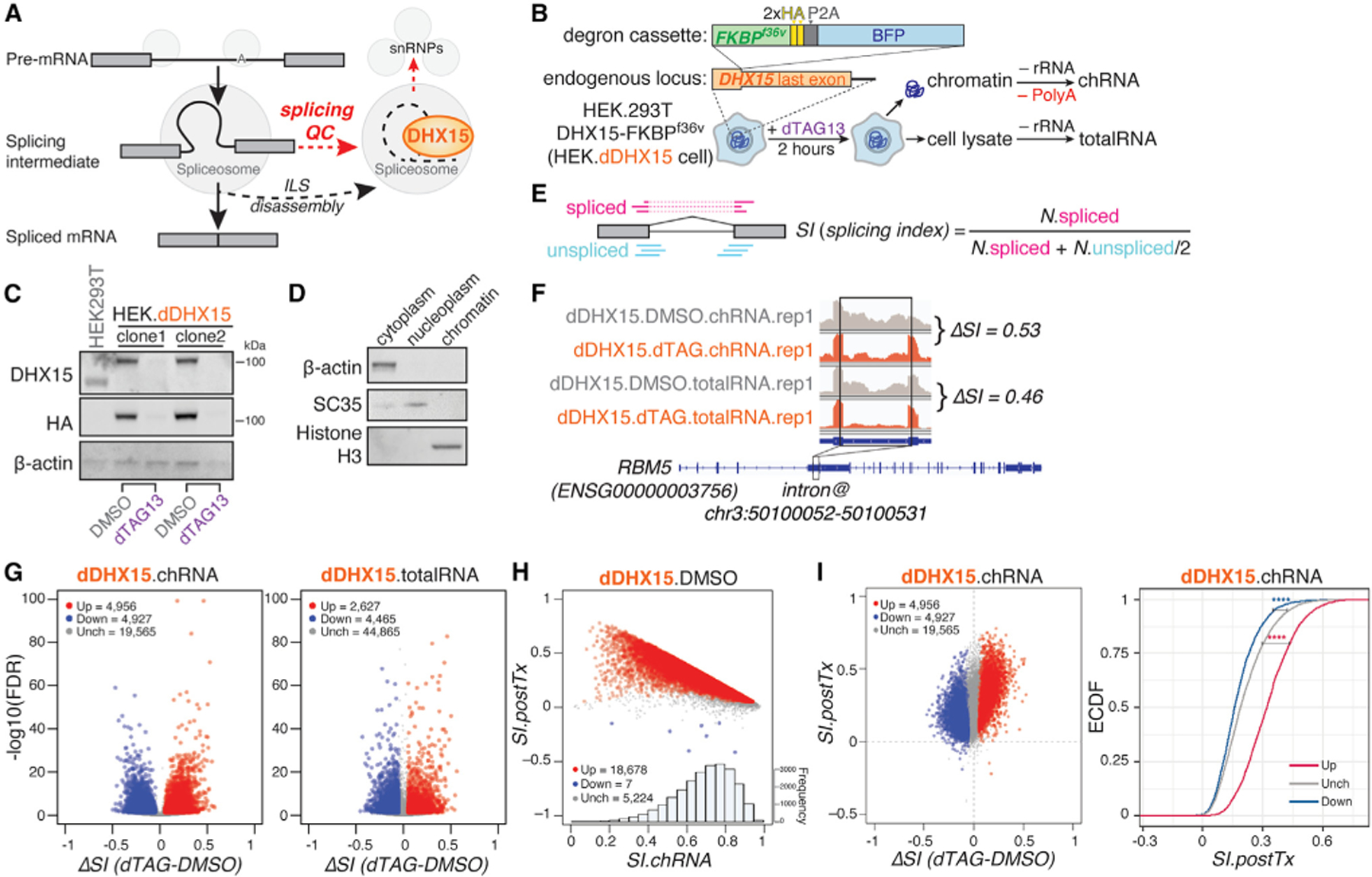
(A) Diagram of DHX15’s canonical role in disassembling of intron-lariat spliceosome (ILS) at the end of the splicing cycle and its proposed role in disassembling aberrant splicing intermediates during splicing QC.
(B) Schematic of HEK.dDHX15 cell line construction and total and nascent chromatin-associated RNA-seq experiments following rapid dTAG13-induced proteolysis of endogenously FKBPF36V degron-tagged DHX15.
(C) Immunoblots of total cell lysates from parental HEK293T.A2 cells and two monoclonal HEK.dDHX15 cell lines were treated with DMSO versus dTAG13 at 100 nM for 2 h.
(D) Immunoblots of cytoplasm, nucleoplasm, and chromatin fractions, collected upon the preparation of chromatin-associated nascent RNA. β-actin, cytoplasmic marker; SC35, nucleoplasm and nuclear speckle marker; Histon H3, chromatin-associated protein marker.
(E) Illustration of how SI is computed by taking the ratio between spliced exon-exon junction reads and normalized total counts of spliced plus unspliced junction-spanning (exon-intron and intron-exon junction) reads.
(F) Example chRNA and totalRNA-seq read coverage of the indicated RBM5 intron in control (DMSO) versus dTAG13-treated HEK.dDHX15 cells. ΔSI is shown.
(G) Volcano plots of introns with altered splicing efficiency across six biological replicates upon dTAG13-induced DHX15 depletion. Red/blue, introns exhibiting significant (false discovery rate [FDR] ≤ 0.05) increases/decreases of SI ≥ 0.05; gray, introns exhibiting insignificant or unaltered changes in SI.
(H) Scatterplot between chromatin-associated splicing efficiency index (SI.ch) and post-transcriptional splicing efficiency index (SI.postTx) across six biological replicates.
(I) Scatterplot (left) and empirical cumulative distribution function (eCDF) plot (right) of DHX15-altered introns and their splicing efficiency changes between steady-state and chromatin-associated nascent state (SI.postTx), across six biological replicates. Magenta/navy (up/down), introns exhibiting significant (FDR ≤ 0.05) increases/decreases of SI ≥ 0.05 upon dTAG13-induced DHX15 depletion in HEK.dDHX15 cells; gray (Unch), introns exhibiting insignificant or unaltered changes in SI. Statistical significance is calculated by Welch’s t test, indicated by asterisks ****p < 0.0001), unless otherwise indicated. See also Figure S1.
In this study, we induced rapid proteolysis of endogenous DHX15, which allowed us to assess immediate impacts on transcripts by high-throughput sequencing of total and chromatin-associated nascent RNAs. This analysis identified two classes of intron substrates: one, whose splicing is promoted by DHX15, is jointly regulated by upstream exon-joining helicase DHX38; the other, whose splicing is repressed by DHX15, includes many suboptimal and cryptic introns, overlapping with introns regulated by the GP, SUGP1. Using a variety of biochemical and molecular genetic approaches, we were able to build a model of how SUGP1 may recruit DHX15 to disassemble spliceosomes assembled at weak and cryptic splice sites.
RESULTS
Genome-wide identification of DHX15-regulated splicing QC introns
To achieve rapid and efficient depletion of endogenous DHX15, and to assess its primary effect on splicing QC, we engineered HEK.dDHX15 cells (Figures 1B, S1A, and S1B) in which all three copies of DHX15 gene in HEK293T.A2 parental background are endogenously tagged with FKBPF36V degron tags,26 using CRISPR-Cas9 gene editing.27 Treating the HEK.dDHX15 cells with the inducer dTAG13 for 2 h resulted in almost complete depletion of DHX15 by western analysis (Figure 1C). We then performed high-throughput sequencing of rRNA-depleted RNAs extracted from total cell lysates (totalRNA), as well as poly(A)-depleted chromatin-associated RNAs (chRNAs), from HEK.dDHX15 cells treated with DMSO versus dTAG13 for 2 h, to assess impacts on splicing and expression (Figures 1B–1D).
To measure intron excision efficiency changes upon DHX15 depletion, for each intron with sufficient read coverage, we calculated its splicing index (SI).28,29 SI measures splicing completeness by taking the ratio between read counts across exon-exon junctions (spliced reads) and normalized total counts of junction-spanning reads (including exon-exon, exon-intron, and intron-exon junctions, representing spliced + unspliced reads) (Figure 1E). For each intron, the difference of SI values between depletion and control conditions, ΔSI, assesses the direction and magnitude of changes in intron excision upon DHX15 depletion. For example, RBM5 intron 5 has positive ΔSI, supporting increased splicing efficiency when DHX15 is depleted (Figure 1F). For brevity, we refer to introns with significant positive and negative ΔSI as ‘‘DHX15-suppressed’’ and ‘‘DHX15-enhanced’’ introns, respectively.
Widespread changes in splicing were observed upon DHX15 depletion. From totalRNA sequencing (totalRNA-seq), we identified 4,465 introns with decreased splicing and 2,627 introns with increased splicing, while, from chRNA sequencing (chRNA-seq), representing nascent RNA, 4,927 introns had decreased splicing and 4,956 introns had increased splicing (Figure 1G). We also calculated the efficiency of post-transcriptional splicing for each intron by taking the difference of SI values between total-RNA, representing predominantly mature RNA, and chRNA, representing nascent RNA as SI.postTx = SI.totalRNA – SI.chRNA. In DMSO-treated HEK.dDHX15 cells, 5,224 (22%) introns detected in both total and chromatin RNA sets were spliced at similar efficiency, whereas 18,678 (78%) introns showed higher SI in total RNA, suggesting some degree of post-transcriptional splicing (Figure 1H). Considering this measure of post-transcriptional splicing, the two groups of DHX15-sensitive introns we identified are likely affected at different stages of the mRNA life cycle (Figure 1I). DHX15-enhanced introns (negative ΔSI) are more co-transcriptionally processed, while DHX15-suppressed introns (positive ΔSI) are more post-transcriptionally processed.
Many introns and genes are sensitive to both DHX38 and DHX15
In the yeast Saccharomyces cerevisiae, there is evidence for a ‘‘rejection’’ pathway in which DHX38 homolog Prp16 can proofread 5′ splice-site cleavage by rejecting substrates that proceed slowly through this step, followed by discarding of associated splicing machinery by DHX15 homolog Prp43 and potential reentry of the transcript to splicing.30–32 To ask whether a similar pathway may exist in human cells, we generated an HEK.dDHX38 cell line, tagging endogenous DHX38 gene copies with degron tags, and performed total- and chromatin-associated RNA-seq following 2 h of DMSO or dTAG13 treatment (Figures 2B and S1C). Upon dTAG13-induced DHX38 depletion, we observed a global decrease in the efficiency of intron excision (Figure 2C), consistent with DHX38’s canonical role in promoting spliceosomal C to C* complex transition. From dDHX38.chRNA-seq, most DHX38-enhanced introns (negative ΔSI) were sensitive to DHX38 depletion alone. These introns had reduced ratios of exon-intron to intron-exon junctions relative to unchanged introns (Figure S1F), suggesting that they may proceed more slowly through the second step of splicing following DHX38 depletion. Furthermore, more than 2,000 introns were enhanced by both DHX38 and DHX15, an almost 2-fold enrichment over background (Figures 2D and 2F), and these introns also showed evidence of slow second-step progression. This observation supports the existence of a similar rejection pathway in human cells in which DHX38 rejects slowly splicing intermediates, triggering DHX15-mediated disassembly. At the gene level, we observed that the majority of genes containing DHX15-enhanced introns (82%) also contained DHX38-enhanced introns (Figures 2E and 2F) and that these genes are enriched for RNA metabolic and splicing functions (Figure 2G). This enrichment could indicate that the activity of the splicing machinery is altered in response to perturbation of splicing QC pathways, perhaps in ways that compensate for decreased proofreading activity.
Figure 2. Shared introns and genes sensitive to DHX38 and DHX15 depletion.
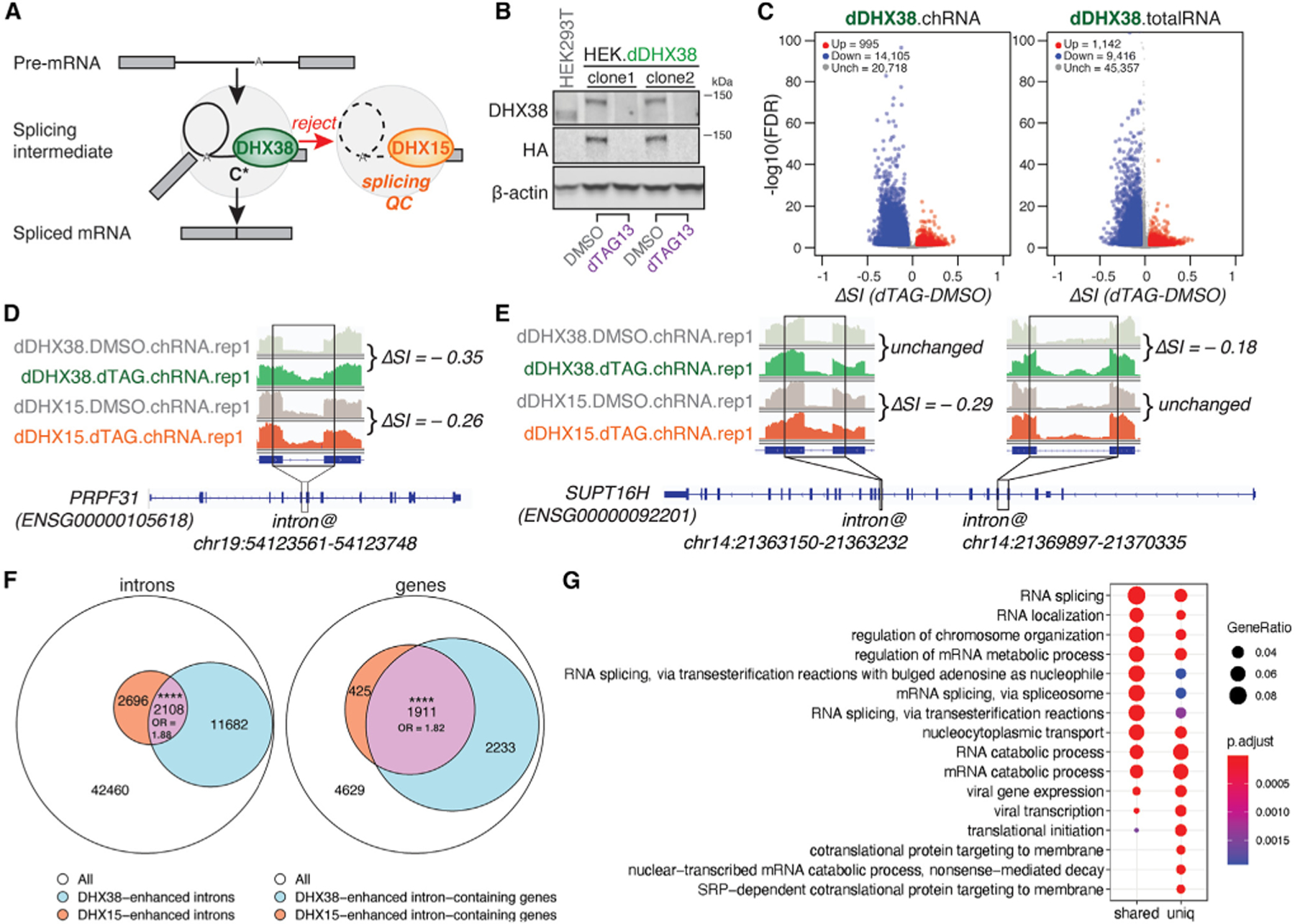
(A) Diagram of splicing QC via the rejection route: DHX15 dissembles splicing intermediates rejected by exon-joining helicase DHX38 to facilitate spliceosomal components recycling and intron-lariat degradation.
(B) Immunoblots of total cell lysates from parental HEK293T.A2 cells and two monoclonal HEK.dDHX38 cell lines were treated with DMSO versus dTAG13 at 100 nM for 2 h.
(C) Volcano plots of introns with altered splicing efficiency averaged across six biological replicates upon dTAG13-induced DHX38 depletion. Red/blue, introns exhibiting significant (FDR ≤ 0.05) increases/decreases of SI ≥ 0.05; gray, introns exhibiting insignificant or unaltered changes in SI. Left, nascent RNA-seq; right, total RNA-seq.
(D) Nascent RNA-seq read coverage of the indicated PRPF31 intron in control (DMSO) versus dTAG13-treated HEK.dDHX38 and HEK.dDHX15 cells. ΔSI is shown by taking the SI difference between dTAG13-treated and control cells.
(E) Nascent RNA-seq read coverage of SUPT16H’s two introns (as indicated) with different sensitivity to dTAG13-mediated DHX38 versus DHX15 depletions. (F and G) (F) Venn diagram of shared introns (left) with decreased splicing efficiency, and shared genes (right) with introns that exhibit decreased splicing efficiency upon DHX15 and DHX38 depletion, across six biological replicates each. The size of the intersection and odds ratios (ORs) are shown. Statistical significance of the intersection is calculated by Hypergeometric test in R (****p < 0.0001) (G) Gene Ontology (GO) enrichment of the shared and unique substrate genes between DHX38 and DHX15. Enriched Biological Process (BP) GO terms are shown. Adjusted p value was calculAdjusted p value was calculated by Benjamini-Hochberg method. Seeated by Benjamini-Hochberg method. See also Figure S1.
DHX15 represses the splicing of suboptimal and cryptic introns
It was notable that DHX15 depletion promotes the splicing of thousands of introns (Figure 1G). We hypothesized that these introns may be quality controlled at a pre-catalytic splicing step, such that, after DHX15-mediated disassembly, these transcripts have another chance to re-enter the splicing cycle, possibly using different splice sites, perhaps post-transcriptionally (Figure 1I). Indeed, DHX15-suppressed introns (positive ΔSI) have weaker 5′ and 3′ splice sites (Figure 3A) and are more likely to have multiple and distal branchpoint sequences (BPSs) (Figures 3B and 3C). In yeast, suboptimal introns were less favored for A-complex assembly driven by the spliceosome DEAD-box helicase scPrp5/hDDX46.33–35 Thus, this class of introns may undergo less-efficient early spliceosome assembly, which could be a trigger for rejection, as recently proposed.25 By contrast, DHX15-enhanced introns (negative ΔSI) had similar splice-site strength and BPS features relative to unchanged introns (Figures 3A–3C).
Figure 3. DHX15 represses the splicing of suboptimal and cryptic introns.
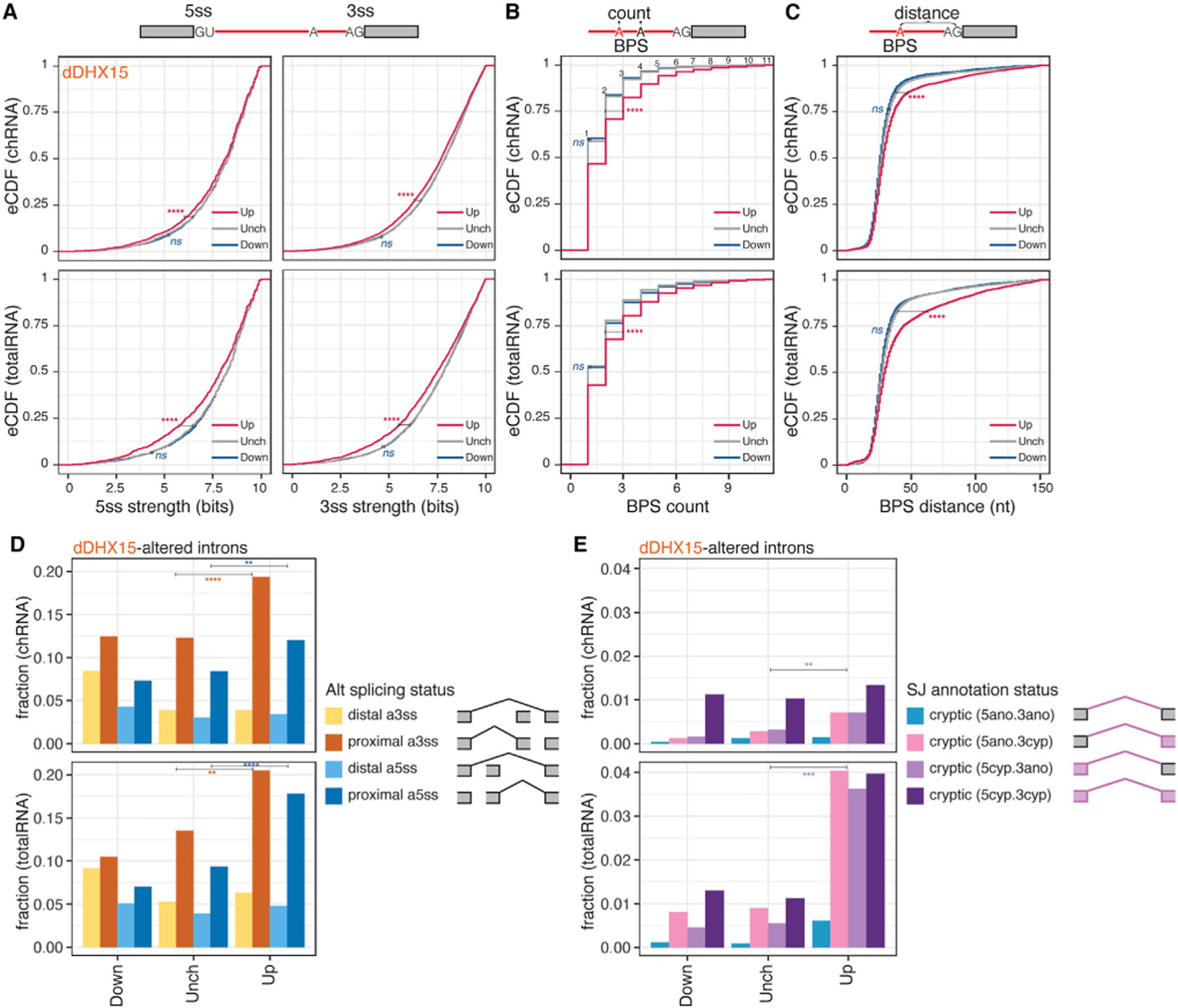
(A) eCDF of splice sites strength MaxEntScan scores. Magenta/navy (up/down), introns exhibiting significant (FDR ≤ 0.05) increases/decreases of SI ≥ 0.05 upon dTAG13-induced DHX15 depletion in HEK.dDHX15 cells; gray (Unch), introns exhibiting insignificant or unaltered changes in SI. 5ss, 5′ splice site; 3ss, 3′ splice site.
(B) Similar to (A), the distribution of counts of branchpoint site (BPS) per intron.
(C) Similar to (B), the distribution of distances between BPS and 3ss for each BPS-3ss pair. Statistical significance in (A)–(C) is calculated by Welch’s t test, indicated by asterisks ****p < 0.0001; ns, not significant) unless otherwise indicated.
(D and E) (D) Fraction of proximal versus distal alternative 3ss and 5ss usage and (E) fraction of cryptic splicing status in the down, up, or unchanged intron groups upon dTAG13-induced DHX15 depletion. SJ, splicing junction. Statistical significance in (D)–(E) is calculated by chi-squared test, indicated by asterisks (****p < 0.0001, ***p< 0.001, **p < 0.01) unless otherwise indicated. See also Figure S2.
We next sought to explore how the repression of suboptimal introns by DHX15 is related to alternative splice-site choice. Introns were classified into constitutive introns whose 5′ and 3′ splice-site pairing is consistently observed, versus introns with alternative 5′ or 3′ splice sites. We noticed that DHX15 depletion promotes the splicing of suboptimal alternative 5′ and alternative 3′ splice-site introns (Figure S2). Especially for alternative splice sites, we observed increases in proximal alternative 5′ and 3′ splice sites, including those flanking cassette exons, following DHX15 depletion (Figure 3D). These observations suggest that DHX15 affects both constitutive and alternative introns, repressing the usage of proximal suboptimal and alternative splice sites.
If suboptimal introns are repressed by DHX15-mediated splicing QC, we hypothesized that DHX15 depletion might trigger cryptic splicing (i.e., splicing of unannotated splice sites or splice site pairs). We thus categorized all detected introns based on whether their splice sites and site pairing were annotated in the GENCODE database (hg38, v28) and found that cryptic splicing increased dramatically following DHX15 depletion (Figure 3E). The relative impact on cryptic splicing was more profound in totalRNA than chRNA, suggesting that splicing at the affected cryptic sites often occurs post-transcriptionally (Figure 1I). These observations are consistent with the idea that, under normal conditions, many cryptic splice sites are initially recognized by splicing machinery but proceed slowly through spliceosome assembly and are discarded by QC pathways involving DHX15, but they are able to eventually complete splicing when this factor is depleted.
SUGP1 and DHX15 repress overlapping sets of cryptic introns
DHX15 and other DExH-box RNA helicases have very low intrinsic RNA substrate specificity in vitro.15 During ILS disassembly, hDHX15/scPrp43 is recruited as a component of the NineTeen complex-related (NTR) complex and activated by G-patch factor hTFIP11/scNtr1.8,36 Fusing the G-patch domain from scNtr1 to the C terminus of DHX15 promotes the disassembly of biochemically stalled splicing intermediates.13 However, the endogenous GP partner(s) of DHX15 during splicing QC remain unclear.
Twenty-two genes encode GPs in the human genome (excluding the 13 G-patch domain-containing human retrovirus K proteins), with different subcellular localizations, lengths, and G-patch domain locations (Figure 4A). Many are potential interaction partners with DHX15 according to BioGRID annotation, based on yeast two-hybrid screening.37 We collected published RNA-seq data from ENCODE and GEO from knockdown experiments for nine GPs. Each GP knockdown results in significant splicing changes, measured by ΔSI. The splicing of more than 1,000 introns was affected in five such knockdowns (Figure 4B) (SUGP1, GPKOW, TFIP11, RBM17, and RBM10), all with previously described roles in splicing.9
Figure 4. SUGP1 and DHX15 repress splicing of overlapping sets of cryptic introns.
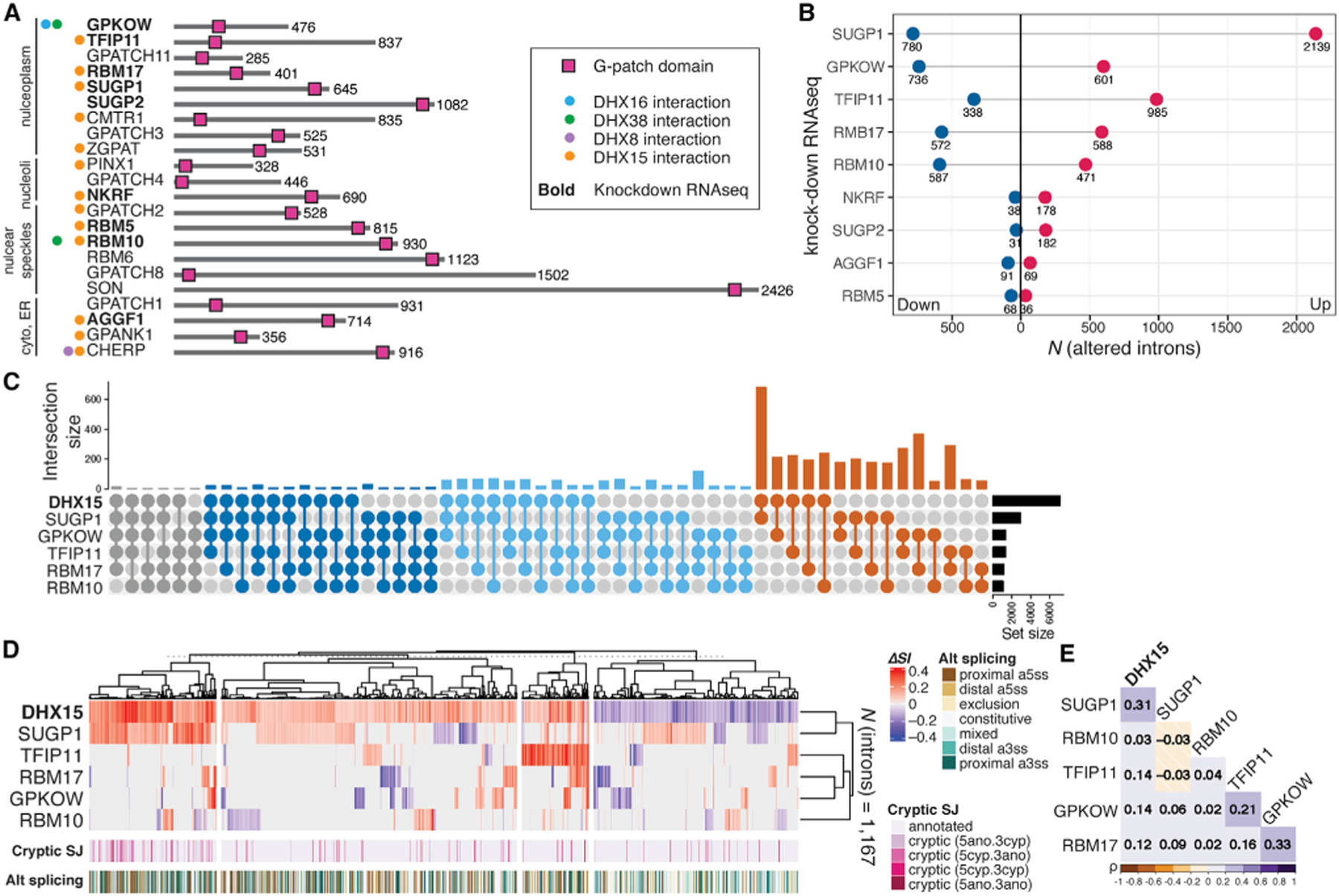
(A) Diagram of the 22 human G-patch domain-containing proteins annotated in ProRule (PRU00092), excluding the 13 retroviral genes. G-patch factors are grouped together based on their subcellular localizations annotated in the Human Protein Atlas.38 Spliceosomal DHX interaction partners (colored dots) are based on BioGRID annotations.37
(B) The number of introns with altered splicing efficiency upon knockdown of corresponding GPs. Magenta/navy (up/down), counts of introns exhibiting significant changes in splicing (|ΔSI| ≥ 0.05, FDR ≤ 0.05) upon corresponding GP factor knockdown.
(C) Intersection UpSet plot of altered introns (|ΔSI| ≥ 0.05, FDR ≤ 0.05) upon depletion of DHX15 and KD of GPs SUGP1, GPKOW, TFIP11, RBM17, and RBM10.
(D) Heatmap and hierarchical clustering of 1,167 introns with altered splicing efficiency (|ΔSI| ≥ 0.05, FDR ≤ 0.05) upon depletion of DHX15 that are also altered in any one of the five GP knockdown experiments. Clustering distance = 1 – Pearson’s correlation. Cryptic_SJ, cryptic intron status annotated based on their splicing junctions.
(E) Heatmap of Pearson correlation matrix constructed on the introns as in (D). See also Figure S3.
We then asked whether introns affected by these GPs are also affected by DHX15. We assayed their ΔSI profiles in comparison to our DHX15 depletion totalRNA-seq data by intersection size and Pearson correlation clustering (Figures 4C–4E). Intron clustering revealed a highly significant overlap in the cryptic introns activated by DHX15 and SUGP1 (Figure 4D). This observation led us to hypothesize that SUGP1 promotes the ability of DHX15 to repress cryptic splice sites.
A role for SUGP1 in repressing cryptic 3′ss choice was previously studied in the context of the SF3B1 cancer mutation K700E.18,19 During revision of this work, a study appeared39 concluding that DHX15 is functionally related to SF3B1.K700E. However, the overlap between DHX15- and SF3B1.K700E-sensitive splicing changes in that study was not claimed to be statistically significant, and we observed no significant overlap of altered splicing between DHX15 depletion and SF3B1.K700E expression, nor any clustering of altered cryptic neojunctions (Figures S3A–S3B). These observations contrast with the significant functional coupling we observed between DHX15 and SUGP1. Therefore, we do not find evidence of a relationship to SF3B1 and argue that DHX15-SUGP1’s splicing QC role is more general than that proposed by Zhang et al.39 This recent study also provided structural data confirming the direct interaction of DHX15 with SUGP1, as well as evidence of activation of DHX15 by SUGP1, consistent with our findings.
We further expanded our search for RBPs involved in suppressing splicing of cryptic neojunctions by extending our analysis to the entire shRNA-seq set from the ENCODE consortium, which includes over 200 RBP knockdown (KD) experiments in two cell lines, HepG2 and K562.40 To assay usage of cryptic neojunctions, we computed the ratio between neojunction read counts and the normalized total spliced read counts across the neojunction 5′ or 3′ss (Figure S3C). These analyses identified 2,230 and 14,553 altered neojunctions in HepG2 and K562 sets, respectively. The difference in counts between the cell lines resulted primarily from the KD of the spliceosomal RNA helicase AQR, performed only in K562 cells, which elicited usage of an enormous number of neojunctions (12,247), few of which were shared with other RBPs (Figures S3E and S3G). As expected, KD of the pair of EJC components MAGOH and EIF4A3, and of the U2AF complex components U2AF1 and U2AF2, yielded highly overlapping subsets of neojunctions in both cell lines (Figures S3F and S3G). However, none of the neojunctions observed in any of the other KDs overlapped with those observed following DHX15 depletion (Figures S3D and S3E). This lack of shared neojunction targets between other assayed RBPs and DHX15 most likely reflects functional divergence, with these cryptic neojunctions not being surveilled by DHX15, although cell type differences may also contribute.
DHX15’s NTPase activity is required for its interaction with SUGP1
We next sought to test whether DHX15-SUGP1’s functional interaction can be explained by their physical interaction inside living cells. To capture protein-protein interactions (PPIs) in intact cells that are likely transient and unstable, we used the PPI-dependent split-APEX (sAPEX) proximity labeling methodology,41 in which APEX activity is only reconstituted upon direct interaction of the two client proteins. Used in this manner, the assay therefore provides a test of interaction rather than cellular proximity. This approach allowed us to test for interaction between SUGP1 and DHX15 in situ and to enrich for biotinylated proteins located within the APEX radius of 10–20 nm42 (Figures 5A and 5B). We observed strong biotinylation signal for wild-type (WT) DHX15 with AP tag, co-transfected with EX-tagged SUGP1 (Figure 5B), confirming the interaction of these proteins.
Figure 5. Interaction between DHX15 and SUGP1 requires DHX15’s NTPase activity.
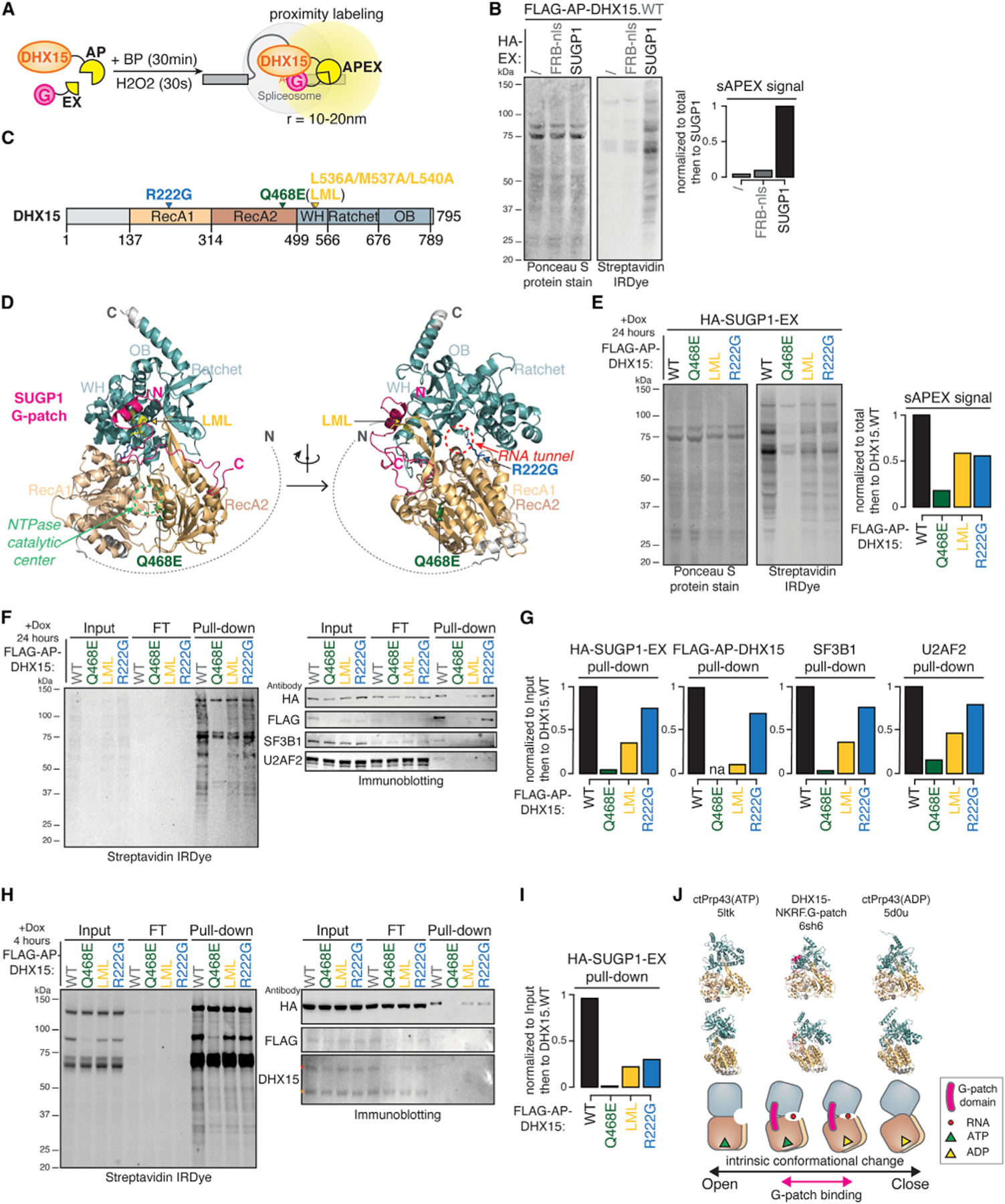
(A) Diagram of split-APEX (sAPEX) proximity labeling experiments with AP-tagged DHX15 and EX-tagged SUGP1. BP, biotin-phenol; APEX, ascorbate peroxidase. Upon short incubation of H2O2, DHX15-SUGP1 interaction-dependent reconstitution of APEX activity oxidizes BP into biotin-phenoxyl radicals, which then biotinylated proteins within a radius of several nanometers.
(B) Protein blots (left) and quantification (right) of biotinylated proteins labeled by DHX15-SUGP1 interaction-reconstituted sAPEX activity. Ponceau S protein stain, loading control for total protein; streptavidin IRDye, detection of biotinylated proteins; FRB-nls, FRB control protein fused with an SV40 NLS.
(C) Diagram of DHX15’s primary domain structure and sites of mutations tested in (E), (F), and (H).
(D) Predicted protein complex structure of DHX15 interaction with SUGP1 G-patch domain by ColabFold.43 Arrows, sites of three mutations, and the corresponding functional centers. Colors of domains and mutations match (C).
(E) Protein blots (left) and quantification (right) of biotinylated proteins labeled by WT versus mutant DHX15-SUGP1 interaction-reconstituted split-APEX activity.
(F) Protein blots of biotinylated proteins labeled in (E), enriched by streptavidin-coated bead pull-down experiments. Cell lysates were collected after 24 h of AP-DHX15 and EX-SUGP1 co-transfection. WT and mutant AP-DHX15 expression induced by doxycycline addition for 24 h.
(G) Quantification of pull-downs in (F).
(H) Similar to (F), except that WT and mutant AP-DHX15 expressions were induced for 4 h. FLAG-AP-DHX15 detectable in the inputs on both anti-FLAG and anti-DHX15 blots. Red asterisk, FLAG-AP-DHX15 band; orange asterisk, endogenous DHX15 band.
(I) Quantification of pull-downs in (H).
(J) Model. Top row, hDHX15/ctPrp43 at ATP-bound open (PDB: 5ltk), G-patch domain-bound semi-open (PDB: 6sh6), and ADP-bound closed (PDB: 5dou) states. Middle row, side view of the open, semi-open, and closed states. Bottom row, cartoon representations of the structures, the G-patch domain (pink) binds to DHX15 at a semi-open state.
To explore aspects of DHX15’s interactions and function, we designed three mutants based on DHX15’s domain structure and predicted DHX15-SUGP1.G-patch 3D structures generated by ColabFold43 (Figures 5C and 5D). We constructed doxycycline-inducible plasmids expressing AP-tagged DHX15, with WT or one of three DHX15 mutants—Q468E, the “LML” triple mutant L536A/M537A/L540A, or R222G—designed to perturb three different aspects of DHX15’s normal function. Q468E is an ATPase-dead dominant-negative mutant.15 The LML compound mutation substitutes with alanines the conserved L536/M537/L540 residues flanking the glycine-rich region at DHX15’s G-patch interaction surface.11 Finally, the R222G mutant disrupts the protein’s ability to translocate RNA substrates15 and was recently identified as a leukemogenic mutant in AML.20,21
Following transfection and induction of mutant AP-DHX15 together with EX-tagged full-length SUGP1, we noticed that both the G-patch interaction-mutant (LML) and the RNA translocation mutant (R222G) have reduced levels of biotinylated proteins (Figure 5E), suggesting that they may interact more weakly with or form a less stable complex with SUGP1. Because the equivalent scPrp43_Q423E dominant-negative mutant was previously used in vitro to enrich for stalled intron-lariat intermediates,30 we expected that Q468E mutant might enrich for stalled early QC spliceosomes in cells and would enhance sAPEX labeling with SUGP1. However, AP-tagged DHX15_Q468E unexpectedly failed to induce biotinylation when expressed with EX-tagged SUGP1 (Figure 5E).
We considered three potential explanations for this observation. First, the dominant-negative Q468E mutant may cause cell death after 24-h induced expression. Second, the Q468E mutation may destabilize DHX15 protein. Finally, the Q468E mutant may be locked in the ATP-bound open state,15 which may not bind SUGP1 effectively. Alternatively, SUGP1 may bind open-state DHX15, but this interaction is unstable due to absence of a required ATP-hydrolysis-driven conformational change of DHX15.
To explore these alternatives, we performed streptavidin pull-down experiments after sAPEX labeling, with cells induced to express either WT or the three mutant DHX15s for 4 h (Figures 5H and 5I) or 24 h (Figures 5F and 5G). After 4-h induction, we were able to detect the Q468E mutant at a similar level as the WT, LML, and R222G mutants from the input (Figure 5H), implying that the Q468E mutant is not destabilized or toxic after 4 h. However, Q468E still failed to interact with SUGP1 (Figure 5I), favoring the explanation that SUGP1 cannot bind DHX15’s ATP-bound open state. However, after 24-h induction, we failed to detect FLAG-AP-DHX15.Q468E from the input cell lysates, whereas WT DHX15 and the LML and R222G mutants were expressed at similar levels, suggesting that prolonged expression of dominant-negative Q468E likely causes cell death or protein destabilization.
We also found that the G-patch interaction surface mutant LML led to a substantial decrease in SUGP1 interaction to 22% after 4 h and 36% after 24 h of expression and that the leukemogenic mutant R222G reduced SUGP1 interaction more moderately (to 31% after 4 h, 75% after 24 h). Known SUGP1 interactors SF3B1 and U2AF2 in the early spliceosomal A complex were also enriched in the streptavidin pull-downs when expressing WT DHX15 but at substantially decreased levels when expressing LML (36% for SF3B1, 47% for U2AF2) and moderately decreased levels when expressing R222G (76% for SF3B1, 80% for U2AF2).
Together, our results are consistent with the working model that SUGP1 may activate DHX15 by promoting or stabilizing a semi-open state, rather than an open or closed state, to drive ATPase and helicase activity (Figure 5J).
SUGP1’s ULM domain is required for DHX15 recruitment
It was important to understand the determinants of the SUGP1-DHX15 interaction. SUGP1 contains two tandem SURP RNA-binding domains, a short and flexible U2AF ligand motif (ULM), and a C-terminal G-patch domain, flanked and connected by unstructured regions (Figure 6A). This flexible yet functionally varied domain structure underlies its function in cryptic 3′ splice-site selection,17 in which it was proposed to bridge branch-point-binding U2 snRNP with the 3′ splice-site-binding U2AF2/U2AF1 heterodimer and to recruit a DExH RNA helicase to disassemble early spliceosomal A complexes assembled on cryptic introns.
Figure 6. SUGP1 recruits DHX15 via its ULM domain.
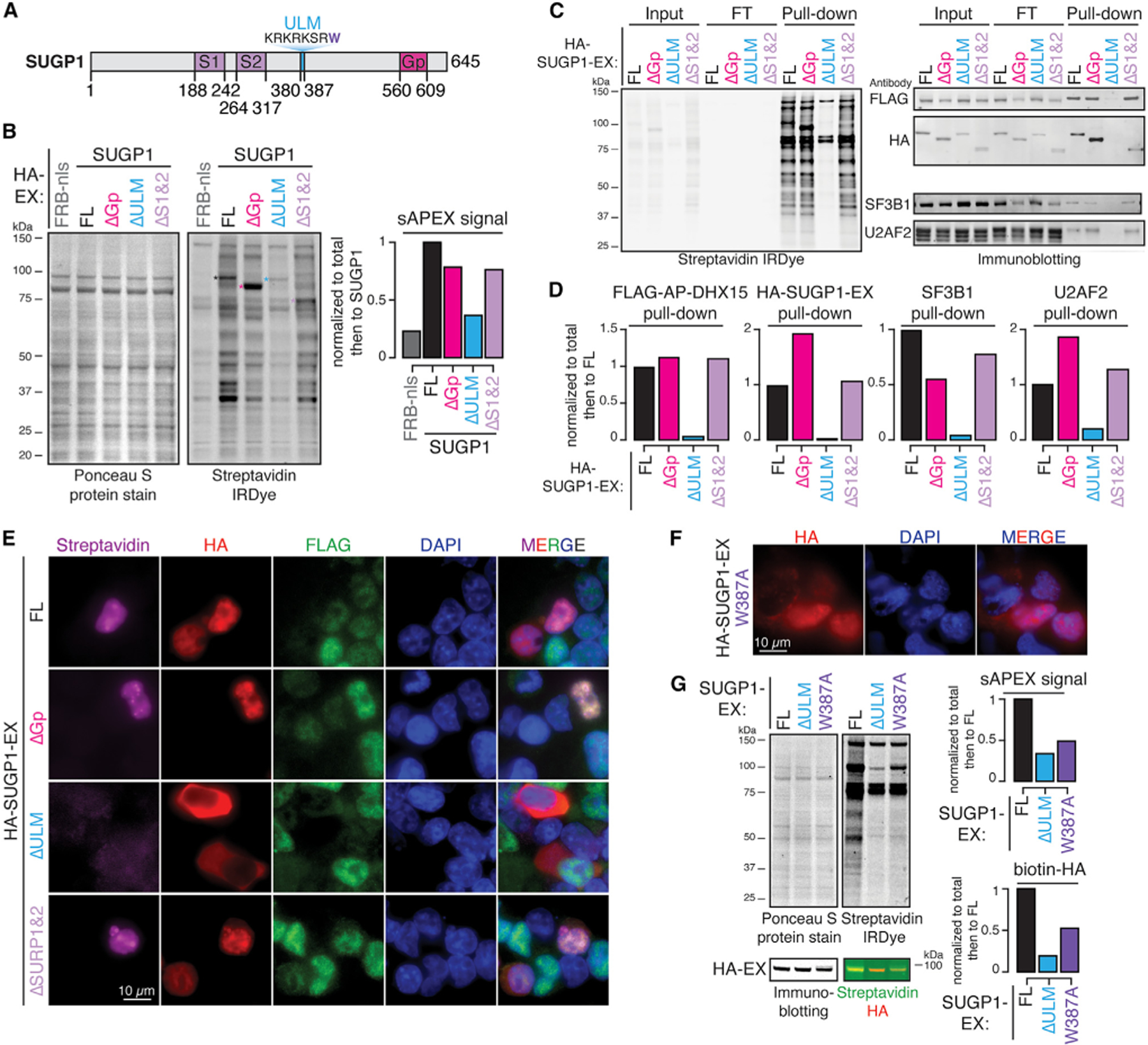
(A) Diagram of SUGP1’s primary domain structure.
(B) Protein blots and quantification of biotinylated proteins labeled by FL versus truncated SUGP1-DHX15 split-APEX activity. Ponceau S protein stain, loading control for total protein; streptavidin IRDye, detection of biotinylated proteins; FRB-nls, FRB control protein fused with an SV40 NLS; ΔGp, G-patch domain truncation; ΔULM, ULM truncation; ΔS1&2, SURP1 and SURP2 truncation; colored asterisks, corresponding bands to FL and truncated HA-SUGP1-EX.
(C) Protein blots of biotinylated proteins labeled in (B) enriched by streptavidin pull-down experiments.
(D) Quantification of pull-downs in (C).
(E) Fluorescent microscopy images of biotinylation signals (streptavidin, magenta), antibodies detecting HA-tagged SUGP1-EX, FL versus truncations (HA, red), FLAG-tagged AP-DHX15 (FLAG, green), nuclear DNA dye (DAPI, blue), and merged channels.
(F) Fluorescent microscopy images of antibody detecting HA-tagged SUGP1.W387A-EX (HA, red), nuclear DNA dye (DAPI, blue), and merged channels.
(G) Protein blots and quantification of biotinylated proteins labeled by full-length (FL), ΔULM, versus W387A mutant SUGP1-DHX15 interaction-reconstituted split-APEX activity. Ponceau S protein stain, loading control for total protein; streptavidin IRDye, detection of biotinylated proteins. Biotinylated HA was detected by merging the streptavidin channel (green) with the HA channel (red). See also Figure S4.
To test which domain of SUGP1 is required for DHX15 recruitment, we constructed an inducible FLAG-AP-DHX15.WT polyclonal HEK293T.A2 cell line and performed sAPEX-based proximity labeling experiments with plasmids containing full-length (FL) HA-SUGP1-EX or versions with specific deletions in SUGP1. Surprisingly, deleting the G-patch domain (ΔGp) or the two RNA-binding SURP domains (ΔS1, ΔS2) only modestly reduced sAPEX-mediated biotinylation signal. However, the ULM deletion (ΔULM) dramatically reduced biotinylated protein (Figure 6B). Streptavidin pull-down confirmed undetectable binding between DHX15 and SUGP1ΔULM (Figures 6C and 6D).
The ULM is composed of a stretch of positively charged lysine (K) and arginine (R) residues followed by a highly conserved tryptophan (W387 in SUGP1) (Figure 6A). The KR repeat may function as a monopartite nuclear localization signal (NLS), while the W residue is critical for mediating binding to U2AF homology motifs (UHMs).44,45 To separate these two potential functions, we first assayed the localization of FL versus truncated SUGP1-EX by immunofluorescence microscopy and then tested the localization and sAPEX-labeling efficiency of a W387A mutant of SUGP1-EX. HA-SUGP1-EX expressing FL, ΔGP, and ΔSURP1,2 are nuclear localized (Figure 6E, HA), and all three produce bright foci with FLAG-AP-DHX15 in cell nuclei (Figure 6E, streptavidin). Upon deletion of the ULM, SUGP1 fails to localize to the nucleus and thus fails to reconstitute sAPEX signal (Figure 6E). However, the SUGP1 mutation W387A localizes to the nucleus (Figure 6F) yet still fails to reconstitute sAPEX-mediated biotinylation (Figure 6G). The ~2-fold reduction of biotinylated total protein and biotinylated HA-SUPG1 comparing W387A mutant to the FL SUGP1 protein suggests that the UHM-binding ability of SUGP1’s ULM is important for its interaction with DHX15.
Deleting the G-patch domain of SUGP1 alone (ΔGp) resulted in a modest reduction of SUGP1-DHX15 interaction-mediated biotinylation (Figure 6B), suggesting that other regions outside of the G-patch domain are able to mediate DHX15 interaction. Regions flanking SUGP1’s G-patch domain contain multiple cancer hotspot mutations, including L515P, G515V, K542Rfs*3, R625T, P636L, and R642W. These cancer mutants promote cryptic splicing to various degrees,18,19 suggesting a possible regulatory role of these G-patch flanking regions in mediating DHX15 interaction or activation. We therefore deleted the flanking regions around the G-patch domain. Deleting the C-terminal 515–645 region of SUGP1 (ΔC) still only modestly decreased SUGP1-DHX15 interaction-mediated total biotinylation but reduced HA-SUGP1-EX pull-down to 31% (Figures S4A and S4B). This result, together with the 1.9-fold increase of HA-SUGP1-EX pull-down when deleting the G-patch domain alone, suggests that the flanking regions also contribute to DHX15 binding. Activation of DHX15 upon G-patch binding is likely to result in rapid spliceosome disassembly, while SUGP1ΔGp may interact with DHX15 without triggering spliceosome disassembly, resulting in stronger protein labeling.
SUGP1’s G-patch domain alone can bind DHX15 but not recruit it to nuclear foci
We next sought to test whether SUGP1’s G-patch domain is sufficient to reconstitute APEX activity with DHX15 by performing sAPEX-labeling experiments with polyclonal HEK.FLAG-AP-DHX15.WT cells transfected with HA-EX constructs expressing FL SUGP1 or SUGP1’s G-patch domain alone fused to an NLS (Gp-nls). The G-patch domain alone increased total biotinylated protein levels by 3.2-fold compared to FL SUGP1 (Figure 7A). However, Gp-reconstituted biotinylation signals were uniformly distributed in the nucleus instead of forming the bright foci observed with FL SUGP1 (Figure 7B). These observations suggest that SUGP1’s G-patch domain is sufficient to bind DHX15 in an unrestricted manner but not to recruit it to nuclear foci.
Figure 7. SUGP1’s G-patch domain is sufficient to bind DHX15 but not to recruit it to nuclear foci.
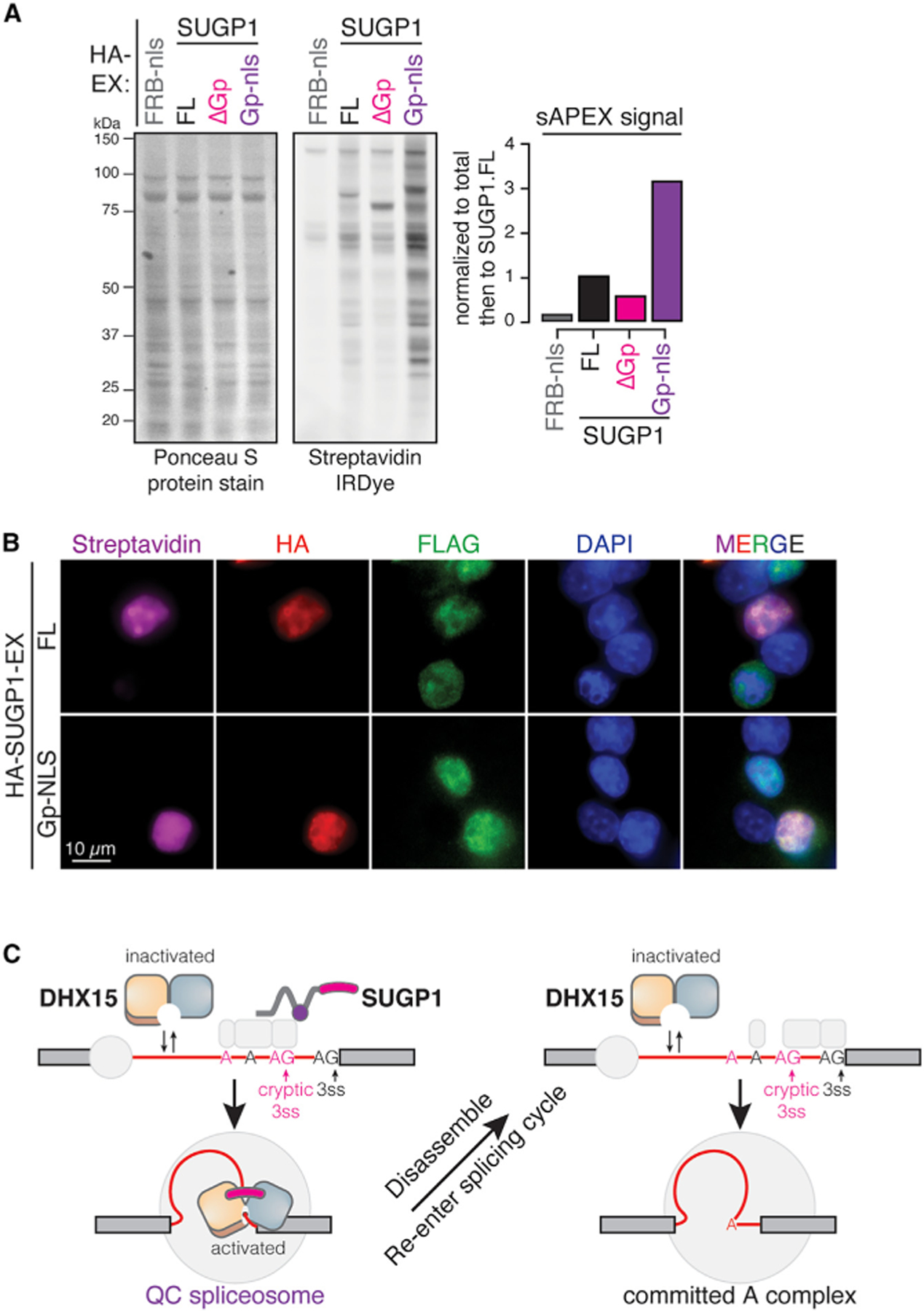
(A) Protein blots and quantification of biotinylated proteins labeled by FL versus truncated SUGP1-DHX15 interaction-reconstituted split-APEX activity. Ponceau S protein stain, loading control for total protein; streptavidin IRDye, detection of biotinylated proteins; FRB-nls, FRB control protein fused with an SV40 NLS; ΔGp, G-patch domain truncation; Gp-nls, G-patch domain alone fused with an SV40 NLS.
(B) Fluorescent microscopy images of biotinylation signals (streptavidin, magenta), antibodies detecting HA-tagged SUGP1-EX, FL versus Gp-nls (HA, red), FLAG-tagged AP-DHX15 (FLAG, green), nuclear DNA dye (DAPI, blue), and merged channels.
(C) A sampling-and-recruitment model of DHX15-SUGP1 interaction during early-splicing QC.
DISCUSSION
Collectively, our observations concerning mutants of DHX15 and SUGP1 and their interactions suggest a model of early splicing QC (Figure 7C). In this model, DHX15 by itself spontaneously samples potential RNA clients and rapidly dissociates.15 When SUGP1 is recruited to a 3′ splice site via ULM-UHM-mediated interactions with U2AF (or another UHM protein) in an early spliceosome complex, its C-terminal region binds DHX15 in a semi-open state, and its G-patch domain activates DHX15’s helicase activity, which may then commit the associated early spliceosomal complex to disassembly. After disassembly, the intron-containing RNA could potentially re-engage in splice-site selection and spliceosome assembly or be degraded. We propose that DHX15 and SUGP1 promote splicing fidelity by preferentially disassembling complexes on suboptimal substrates.
Specificity for suboptimal substrates could derive from a simple timing mechanism and/or from more complex substrate-dependent activity.23 If DHX15 disassembles A complexes after a certain period of time, then canonical substrates that proceed quickly through subsequent steps could evade disassembly so that DHX15 would preferentially act on slower-progressing sub-optimal substrates. The time required for the steps required to activate DHX15, including interaction with RNA and activation by a G-patch factor, may enable such a kinetic proofreading mechanism. Alternatively (or in addition), disassembly may preferentially occur on suboptimal substrates. For example, DHX15 may preferentially reject suboptimal substrates through regulation of its ATPase activity or as a result of stability differences between spliceosomes associated with optimal and suboptimal substrates. Our data raise the possibility, discussed below, that differential recruitment and activation may contribute to substrate specificity.
Previous studies have found that G-patch factors can influence DHX15’s localization and local abundance, likely contributing to DHX15’s multifunctionality in regulating mRNA processing, rRNA processing, and even viral RNA.46,47 In the absence of a GP, DHX15 is expected to sample and dissociate from RNAs rapidly due to its wide range of conformations when not constrained by a GP factor. In addition to SUGP1, four other G-patch factors also occur in A-complex spliceosomes: RBM5, RBM10, RBM17, and CHERP.16,48–50 RBM17 also interacts with DHX15 and contributes to alternative splicing.51 We observed minimal overlap between introns affected by RBM17 and DHX15, but substantial overlap and correlation between introns repressed by DHX15 and SUGP1 (Figure 4D), suggesting that SUGP1 is the primary G-patch partner of DHX15 for splicing QC. Productive targeting is likely to rely on the flexible yet multifunctional domain structure of SUGP1 and its previously proposed function in repressing cryptic 3′ splice sites.17–19 SUGP1 may perform this function by preventing the U2 snRNP scaffold protein SF3B1 from accessing upstream cryptic branch sites, thus preventing the use of associated cryptic 3′ splice sites.17 An orthogonal study52 using mutagenesis of spliceosome components in haploid cells and BioID labeling also supports interaction between SUGP1 and DHX15 and implicates it in splicing fidelity.
Here, we found using mutagenesis and sAPEX that DHX15 recruitment/binding is blocked by a point mutation that inhibits the UHM-binding ability of SUGP1’s ULM domain. UHM-ULM interaction is a widely used mechanism for early step spliceosome assembly, for example between U2AF1’s UHM and U2AF2’s ULM during 3′ splice site recognition.53 Which UHM partner(s) may be responsible for the SUGP1-DHX15 interaction remains unclear. The five known human UHM-containing genes—U2AF1, U2AF2, PUF60, RBM17/SPF45, and RBM39/CAPERα—all function during early spliceosome assembly near the BPS-polypyrimidine-3′ splice-site region. Optimal and suboptimal 3′ splice sites are expected to interact differently with these factors.54,55 This leads to the intriguing possibility that differential activity of DHX15 on suboptimal introns results at least in part from differential recruitment of SUGP1 to these introns by specific UHM-containing factors. Further studies are needed to test this conjecture and to better understand the cis-acting elements and protein factors involved in suboptimal and cryptic intron QC by DHX15.
Splicing-derived cryptic neojunctions have recently emerged as a potential source for the generation of neoantigens,56–58 with potential for therapeutic interventions, including T cell therapy and cancer vaccines. Here, in addition to DHX15-SUGP1 co-regulated cryptic neojunctions, we also identified thousands of neojunctions regulated by other RBPs, including the BAQR spliceosomal helicase AQR,59 as well as EJC and U2AF components. The potential linkage between cancer-associated mutations in these splicing regulators and the expression of potential neojunction-derived cancer neoantigens warrants further investigation.
We observed that the R222G mutation of DHX15, associated with AML, appears to weaken but not abolish its interaction with SUGP1. Based on our findings, this could result in less-efficient splicing QC by DHX15, likely resulting in less accurate but perhaps faster splicing, as commonly observed when restricting kinetic proofreading schemes60; increased speed might benefit tumors that are often sensitive to splicing throughput.61 On the other hand, less-efficient recruitment of DHX15 by SUGP1 might shift the nuclear distribution of DHX15 toward other locations and activities, such as ribosome biogenesis, perhaps enabling more rapid tumor growth.
Limitations of the study
As our study was performed with cultured human cells, the physiological role of DHX15-mediated splicing QC may differ in different cell or tissue types, healthy or diseased. Our proposed sampling-and-recruitment model of DHX15-SUGP1-mediated early splicing QC needs further mechanistic investigation and testing in biochemical systems. Whether and how leukemia-associated DHX15 may contribute to the development of cancer via dysregulation of splicing QC, especially in the context of the R222G mutant, remains to be explored.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents and resources should be directed to and will be fulfilled by the lead contact, Christopher Burge (cburge@mit.edu)
Materials availability
All materials generated in this study are available from the lead contact upon request and completion of an MTA.
Data and code availability
All RNA-seq data sets generated in this manuscript have been deposited in the public functional genomics data repository Gene Expression Omnibus (GEO) and are publicly available as of the date of the publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Anti-HA | MilliporeSigma | Cat#: 3F10; RRID:AB_2314622 |
| Anti-FLAG (M2) | MilliporeSigma | Cat#: F3165; RRID:AB_259529 |
| Anti-DHX15 | Abcam | Cat#: Ab254591; RRID:AB_2892059 |
| Anti-β-actin (AC-15) | MilliporeSigma | Cat#: A5441; RRID:AB_476744 |
| Anti-H3 | Abcam | Cat#: Ab1791; RRID:AB_302613 |
| Anti-SC35 | Abcam | Cat#: Ab204916; RRID:AB_2909393 |
| Anti-SF3B1 | Abcam | Cat#: Ab170854 |
| Anti-U2AF2 | Invitrogen | Cat#: PA530442; RRID:AB_2547916 |
| Anti-U2AF1 | Proteintech | Cat#: 10334-1 AP; RRID:AB_2211314 |
| IRdye 800CW Sterptavidin | LI-COR | Cat#: 926-32230 |
| Anti-mouse Alexa Fluor 488 | ThermoFisher | Cat#: A28175; RRID:AB_2536161 |
| Anti-rat Alexa Fluor 647 | ThermoFisher | Cat#: A21247; RRID:AB_141778 |
| NeutrAvidin- DyLight 594 | ThermoFisher | Cat#: 22842 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| dTAG13 | Tocris Bioscience | Fisher Scientific Cat#: 66-055 |
| α-amanitin | Santa Cruz Biotechnology | Cat#: Sc-202440A |
| cOmplete™ ULTRA protease inhibitor | Roche | MilliporeSigma Cat#: 589297001 |
| PhosStop | Roche | MilliporeSigma Cat#: 4906837001 |
| Hygromycin | Millipore | Cat#: 400052 |
| Lipofectamine 3000 | Invitrogen | Cat#: L3000008 |
| Proteinase K | Viagen | Cat#: 501-PK |
| Direct PCR reagent | Viagen | Cat#: 301-C |
| Direct-zol | Zymo Research | Cat#: R2052 |
| SUPERaseIn | Life Technologies | Cat#: AM2694 |
| Tri reagent | Zymo Research | Cat#: R2050-1-200 |
| Doxycyclin | MilliporeSigma | Cat#: D9891 |
| Biotin Phenol | MilliporeSigma | Cat#: SML2135-250MG |
| H2O2 | MilliporeSigma | Cat#: H1009-100ML |
| Streptavidin-coated magnetic beads | Pierce | Thermo Scientific Cat#: 88817 |
| DAPI | ThermoFisher | Cat#: 62248 |
|
| ||
| Critical commercial assays | ||
|
| ||
| NEBuilder HiFi DNA assembly cloning kit | NEB | Cat#: E5520S |
| NEB OneTaq Quick-Load 2X Mastermix | NEB | Cat#: M0486L |
| NEBNext Poly(A) mRNA magnetic isolation module | NEB | Cat#: E7490L |
| NEBNext Ultra II Directional RNA kit | NEB | Cat#: E7760L |
| NEBNext rRNA Depletion Kit | NEB | Cat#: E6310X |
|
| ||
| Deposited data | ||
|
| ||
| Human reference genome NCBI release 28, GRCh38 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| SUGP1 KD | Liu et al.19 | GEO: GSE159304 |
| RBM10 KD | Wang et al.62 | GEO: GSE44976 |
| SUGP2 KD | ENCODE Consortium | ENCLB206KMT, ENCLB331AGB |
| NKRF KD | ENCODE Consortium | ENCLB552FZS, ENCLB950ZAG |
| TFIP11 KD | ENCODE Consortium | ENCLB144PBT, ENCLB483ITG |
| RBM17 KD | ENCODE Consortium | ENCLB012PNW, ENCLB719FFS |
| GPKOW KD | ENCODE Consortium | ENCLB168TMK, ENCLB908ZJN |
| RBM5 (CRISPR KO) | ENCODE Consortium | ENCLB036OZM, ENCLB293XAV |
| AGGF (CRISPR KO) | ENCODE Consortium | ENCLB710LCN, ENCLB644OBV |
| Raw and analyzed data | This Paper | GEO: GSE241712 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK293T.A2 | Khandelia et al.63 | N/A |
| HEK.FLAG-AP-DHX15.WT | This paper | N/A |
| HEK.dDHX15 | This paper | N/A |
| HEK.dDHX38 | This paper | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S1 | N/A | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pSpCas9-gRNA-GFP | Ran et al.27 | Cat# PX458; no. 48138 |
| pUC19-DHX15- FKBPF36V-2xHA_P2A_BFP | This paper | N/A |
| pUC19-DHX15- FKBPF36V-2xHA_P2A_HygR | This paper | N/A |
| pUC19-DHX38- FKBPF36V-2xHA_P2A_BFP | This paper | N/A |
| pRD-FLAG-AP-DHX15 (wt and mutants) | This paper | N/A |
| pX304-HA-SUGP1-EX (full-length and truncations) | This paper | N/A |
| pCAGGS-nlCre | Khadelia et al.63 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| STAR version 2.7.3.a | Dobin et al.64 | https://github.com/alexdobin/STAR |
| RSEM v1.3.3 | Li et al.65 | https://github.com/deweylab/RSEM |
| Samtools V5.2.5 | Li et al.66 | http://www.htslib.org/ |
| clusterProfiler v4.0 | Yu et al.67 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| MaxEntScan | Yeo et al.68 | http://hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html |
| ComplexHeatmap v.2.2.0 | Gu et al.69 | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| Corrplot v0.92 | https://github.com/taiyun/corrplot | |
| ColabFold | Mirdita et al.43 | https://github.com/sokrypton/ColabFold |
| Pymol v2.5.2 | https://pymol.org | |
| Nikon NIS-Elements Imaging Software | Nikon | N/A |
| FIJI | Schindelin et al.70 | https://imagej.net/software/fiji/ |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT
Cell lines
Cell lines used in the study: HEK293T.A2 parental cells; HEK.dDHX15 degron cells; HEK.dDHX38 degron cells; HEK.FLAG-AP-DHX15.WT cells.
HEK293T.A2 cells were a gift from Eugene V. Makeyev.63 HEK.dDHX15 and HEK.dDHX38 cells were genetically engineered using the CRISPR/Cas9 system. HEK.FLAG-AP-DHX15.WT cells were engineered using the Cre-LoxP system as previously described.63
Growth conditions: All cells were cultured at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) with high glucose (4.5 g/L) (Gibco 11965118) supplemented with 10% fetal bovine serum (FBS) (Gibco A3160402).
METHOD DETAILS
HEK.dDHXs cell line generation via CRISPR/Cas9 genome editing
Gene-specific gRNA-encoding oligonucleotides were cloned into the pSpCas9-gRNA-GFP plasmid (Addgene PX458; no. 48138) targeting the C terminus coding region of the endogenous DHX15 and DHX38 genes, using BbsI restriction digestion and ligation. The oligo sequences used for cloning are provided (Table S1). DNA repair template plasmids containing DHX15-FKBPF36V-2xHA_P2A_BFP, DHX15-FKBPF36V-2xHA_P2A_HygR, and DHX38-FKBPF36V-2xHA_P2A_BFP were synthesized with 800bp dsDNA homology arms by Genewiz and assembled using the NEBuilder HiFi DNA assembly cloning kit (NEB E5520S).
To generate the endogenously tagged lines, one million HEK293T.A2 cells were co-transfected with the pSpCas9-gRNA-GFP plasmids and the pUC19-FKBPF36V-P2A-selection repair template plasmids at 1:1 ratio (Figure S1A). Transfection of HEK293T.A2 cells was performed using Lipofectamine3000 (Invitrogen L3000008) according to the manufacturer’s instructions.
Two days post-transfection, cells were sorted for the expression of Cas9-GFP. Five days post GFP sorting, cells were sorted again for BFP, and serially diluted in 100 µg/ml Hygromycin-containing (Millipore 400052) growth media to allow single-cell clone formation. HEK.dDHX38 line was generated without a second HygR repair template, and the BFP-positive cells were serially diluted to grow in regular growth media. One week post sorting, viable single clone colonies were picked using a stereoscope into a 96-well plate. Two days after clone-picking, 80% of the cells were collected into a 96-well PCR plate for genotyping. For genotyping, cells were lysed in direct PCR reagent (Viagen 301-C) supplemented with 1 μL proteinase K (Viagen 501-PK), and further used as templates in genotyping PCR reactions (NEB OneTaq Quick-Load 2xMasterMix, M0486L) (Figures S1B and S1C). Clones with homozygous degron tags were expanded and used for RNA-seq experiments.
Total and chromatin-associated RNA isolation
Total and chromatin-associated RNA samples (totalRNA and chRNA) were collected in a paired manner. For each replicate, 5 million cells were seeded on a 15cm plate containing one 25 mm plastic coverslip (Thermanox, VWR 100500-878). At 24 h after seeding, 6 replicates of each condition were treated with DMSO or 100 nM dTAG13 (Tocris Bioscience, Fisher Scientific 66-055) for 2 h. Post-treatment, cells growing on coverslips were collected by transferring into PBS-containing 6-well plates for on-plate TRI reagent lysis, and Directi-zol (Zymo R2052) column purification of totalRNA. The remaining ~10 million cells on each 15cm plate were used for cell fractionation as described previously,71 with minor modifications.
Every procedure was performed in RNase-free environment, buffers were pre-chilled to 4°C, and samples were kept on ice. For each replicate, cells were washed on plate with 10mL ice-cold PBS and scraped and collected in 1mL ice-cold PBS supplemented with 10U/ml SUPERase.In (Life Technologies AM2694). Cells were then resuspended gently in 200 μL cytoplasmic lysis buffer (10 mM Tris, pH:7.0, 150 mM NaCl, 0.15% v/v NP-40) supplemented with 25 μM α-amanitin (Santa Cruz Biotechnology sc-202440A), 10 U/mL SUPERase.In, 1X cOmplete ULTRA protease inhibitor (Roche, MilliporeSigma 5892970001), and 1X PhosSTOP (Roche, MilliporeSigma 4906837001) and incubated on ice for 5 min. To separate the cytosolic fraction from the nuclei, the lysed cells were layered over 500 µL sucrose buffer (10 mM Tris, pH:7.0, 150 mM NaCl, 25% w/v Sucrose) supplemented with 25 μM α-amanitin, 20 U/mL SUPERase.In, 1X protease inhibitor, and 1X PhosSTOP, and centrifuged at 16,000 x g for 10 min at 4°C. After complete removal of the supernatant, the nuclei pellet was resuspended in 800 μL of nuclei wash buffer (0.1% v/v Triton X-100, 1mM EDTA, in 1xPBS) supplemented with 25 μM α-amanitin, 40 U/mL SUPERase.In, 1X protease inhibitor, and 1X PhosSTOP, and centrifuged at 7,000 x g for 1 min at 4°C. Washed nuclei were then resuspended in 200 μL glycerol buffer (20 mM Tris, pH:8.0, 75mM NaCl, 0.5mM EDTA, 50% v/v glycerol) supplemented with 0.85 mM DTT, 25 μM α-amanitin, 10 U/mL SUPERase.In, 1X protease inhibitor, and 1X PhosSTOP. Next, 200 μL of nuclei lysis buffer (1% v/v NP-40, 20mM HEPES, pH:7.5, 300mM NaCl, 0.2mM EDTA, 1M Urea) supplemented with 1 mM DTT, 25 μM α-amanitin, 10 U/mL SUPERase.In, 1X protease inhibitor, and 1X PhosSTOP, were added, mixed by pulsed vortex, and incubated on ice for 2 min. To separate the nucleoplasmic fraction from the chromatin, the lysed nuclei were centrifuged at 18,500 x g for 2 min at 4°C. After complete removal of the supernatant, the chromatin pellet was resuspended in 50 μL chromatin resuspension solution (1x PBS supplemented with 1mM DTT, 25 μM α-amanitin, 20 U/mL SUPERase. In, 1X protease inhibitor and 1X PhosSTOP) before RNA extraction. To facilitate homogeneous RNA lysis, the chromatin pellet was lysed with 1mL TRI reagent and immediately mixed thoroughly by passing through a 1 mL syringe with a 23G needle, followed by Direct-zol column purification of chRNA.
Protein assays by Western blots
For dTAG13-mediated DHX15 and DHX38 proteolysis confirmation, total cell lysates collected in 1x RIPA buffer (Cell Signaling Technology ab156034) supplemented with 1X protease inhibitor and 1X PhosSTOP were collected according to manufacturer’s instructions. Briefly, cells were lysed on ice for 5 min and sonicated by Bioruptor (Diagenode, B01020001) at the high setting for 5 min with 30 s intervals at 4°C. The sonicated samples were then cleared by centrifuge at 15,000 x g for 10 min at 4°C. Protein concentrations in the cleared supernatants were measured using Pierce 660nm protein assay reagent (Thermo Scientific, 22660). For fractionation confirmation, cytoplasmic, nucleoplasmic, and chromatin fractions collected from the cell fractionation procedures above were assayed by using the volume ratio between the buffers to match the same number of input cells. Chromatin protein lysates were extracted by incubating chromatin pellets with RIPA lysis buffer followed by sonication as above. For APEX labeling, total cell lysates were collected in 1x RIPA buffer supplemented with 1X protease inhibitor, 1X PhosSTOP, and 1X quencher mixture (10 mM Sodium L-ascorbate, 5 mM Trolox, and 10 mM Sodium Azide).
Primary antibodies used in this study include anti-HA (3F10, MilliporeSigma 11867423001), anti-FLAG (M2, MilliporeSigma F3165), anti-DHX15 (Abcam ab254591), anti-β-actin (AC-15, MilliporeSigma A5441), anti-H3 (Abcam ab1791), anti-SC35 (Abcam ab204916), anti-SF3B1 (Abcam ab170854), anti-U2AF2 (Invitrogen PA5-30442), anti-U2AF1 (Proteintech 10334-1-AP), and IRDye 800CW Streptavidin (LI-COR 926-32230).
RNA sequencing library preparation
Six replicates of totalRNA and chRNA were collected as above. RNA-seq library preparation and sequencing were performed by MIT BioMicro Center. Briefly, for chRNA set, poly(A) depletion was achieved by taking the unbound supernatant after chRNA was incubated with the oligo-d(T) beads from the NEBNext Poly(A) mRNA magnetic isolation module (NEB, E7490L). Then, paired-end, rRNA-depleted, dual-indexed libraries of totalRNA and poly(A)-depleted chRNA were prepared using the NEBNext Ultra II Directional RNA kit (E7760L) with ribosomal RNA (rRNA) depletion module (E6310X). Indexed libraries were sequenced on the Illumina NovaSeq S2 200 platform, resulting in ~20 million paired-end 2 x 100 bp reads per replicate.
Protein structure prediction and visualization
Protein sequences of DHX15 (UniProt O43143) and SUGP1’s (UniProt Q8IWZ8) G-patch domain (VENIGYQMLMKMGW KEGEGLGSEGQGIKNPVNKGTTTVDGAGFGIDRP) were used as input sequences for protein complex structure prediction using the ColabFold AlphaFold2_mmseqs2 notebook.43 The predicted complex structure was visualized using Pymol v2.5.2.
sAPEX proximity labeling
For Western blot analysis, HEK293T.A2 cells were co-transfected with AP-tagged DHX15 and EX-tagged SUGP1 at 1:1 ratio for 24 h. AP-DHX15 expression is induced by doxycycline (MilliporeSigma D9891), for 24 or 4 h as specified. For fluorescent imaging, inducible HEK.AP-DHX15 cells cultured on coverslips were used to allow uniform expression of AP-DHX15. This polyclonal cell line was generated by co-transfecting HEK293T.A2 cells with Cre-expression construct (pCAGGS-nlCre) and inducible AP-DHX15 construct (pRD-AP-DHX15) at 1:200 ratio, followed by a 7-day puromycin selection, based on the recombination-mediated cassette exchange (RMCE) protocol.63
APEX labeling was performed following published protocol,72 with minor modifications. Briefly, following transfection and induction, to allow biotin substrate intake, cells were incubated at tissue culture condition with growth medium containing 500 μM biotin-phenol (MilliporeSigma SML2135-250MG) for 30 min. APEX labeling was initiated by adding fresh H2O2 (Sigma, H1009-100ML) to the cultured cells at a final concentration of 1mM with gentle agitation for 30 s at room temperature. The H2O2-containing medium was quickly poured into a waste collection beaker, and the APEX labeling reaction was quickly quenched by the addition of pre-chilled quenching solution (10 mM Sodium L-ascorbate, 5 mM Trolox, and 10 mM Sodium Azide in 1X PBS). After removing the quenching solution by aspiration, cells were washed twice more with the quenching solution. After washes, cells were lysed with RIPA buffer for western blots or fixed on coverslips for microscopy.
Streptavidin pulldown
To enrich biotinylated protein from the total protein lysates, 60 μL streptavidin-coated magnetic beads (Pierce, Thermo Scientific 88817) were used for each 1 mg of total protein lysates collected after APEX labeling. Prior to the incubation, the streptavidin beads were prepared as follows. Streptavidin beads were fully resuspended in stock solution by pulsed vortex and then washed twice with pre-chilled 1X RIPA buffer. The washed beads were then aliquoted to protein lysates samples to a total volume of 500 μL and the mixtures were incubated with rotation at 4°C overnight. Post incubation, flow-through samples for western blotting analysis were saved in 4X LDS SampleBuffer (Invitrogen, ThermoFisher Scientific NP0007) after DynaMag-2 (Invitrogen, ThermoFisher Scientific 12321D) magnetic rack-based pull-down. The beads were subsequently washed twice with 500 μL of pre-chilled 1X RIPA buffer, once with 500 μL of 1 M KCl, once with 500 μL of 0.1 M Na2CO3, once with 500 μL of 2 M urea in 10 mM Tris-HCl (pH 8.0), and twice with 500 μL 1X RIPA buffer. For western blotting analysis, the enriched proteins were eluted by boiling the beads in 50 μL of 2X LDS SampleBuffer supplemented with 20 mM DTT and 2 mM biotin.
Immunofluorescence microscopy
Inducible HEK.AP-DHX15 cells cultured on coverslips in 24-well plates were transfected, induced, and labeled as described above. Cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences, Fisher Scientific 50-980-487) in 1X PBS at room temperature for 15 min. Cells were then washed with 1X PBS for three times and permeabilized with 0.3% Triton-100-containing 1X PBS at room temperature for 5 min. Cells were then washed three times with PBS and blocked at room temperature for 1 h with 10% goat serum (ThermoFisher Scientific, 50062Z). Primary antibodies used in this study were diluted in blocking buffer (10% goat serum) at 1:100 dilution. Primary incubation was done by flipping the corresponding coverslips on top of each 20μL of diluted primary anti-bodies on a parafilm-covered humidity chamber at 4°C overnight. After washing three times with 1X PBS, cells were incubated with secondary antibodies (anti-mouse-Alexa Fluor 488 and anti-rat-Alexa Fluor 647) and NeutrAvidin-DyLight 594 (ThermoFisher Scientific 22842) in blocking buffer (10% goat serum) at 1:500 dilution at room temperature for 1 h. Cells were then incubated with 1 μg/mL DAPI (ThermoFisher Scientific 62248) -containing 1X PBS at room temperature for 5min, washed three times with 1X PBS, and mounted on slides with Vectashield (Vector Laboratories, H-1200-10) before imaging. Fluorescence microscopy was performed with a Nikon spinning-disk confocal microscope with X60 oil-immersion objectives. All images were collected with Nikon NIS-Elements imaging software and processed using ImageJ (Fiji).70
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-seq processing and quantification
For the HEK.dDHX15 and HEK.dDHX38 totalRNA and chRNA sequencing experiments, reads were mapped to the GRCh38 using the GENCODE v28 annotations with STAR version 2.7.3a,64 with ENCODE standard options invoked the argument ‘–twopassMode Basic’ to allow a second pass of junction identification. Gene expression levels were quantified using RSEM v1.3.3.65 DESeq2 v1.26.073 was then used to quantify differential expression and compute distance clustering between replicates (Figures S1D and S1E).
For the published G-patch factor knock-down (KD) RNAseq experiments, raw sequencing data from SUGP1 KD (GSE159304)18 and RBM10 KD (GSE44976)62 experiments were downloaded using the NCBI SRA toolkit. ENCODE shRNA-mediated KD RNAseq in HepG2 cells for SUGP2 (ENCLB206KMT, ENCLB331AGB), NKRF (ENCLB552FZS, ENCLB95-ZAG), TFIP11 (ENCLB144PBT, ENCLB483ITG), RMB17 (ENCLB012PNW, ENCLB719FFS), GPKOW (ENCLB168TMK, ENCLB908ZJN), CRISPR/Cas9-mediated KD RNAseq in HepG2 cells for RBM5 (ENCLB036OZM, ENCLB293XAV) and AGGF (ENCLB710LCN, ENCLB644OBV), and their corresponding non-specific shRNA or gRNA controls were downloaded from ENCODE.62,74 These raw fastq data were then aligned and quantified using STAR and RSEM as above.
Splicing index (SI), post-Tx SI, and EIE analysis
Splicing junctions detected by STAR (SJ.out.tab) were filtered using samtools v5.2.566 with the following criteria: (1) remove reads with undefined strands; (2) restrict to uniquely mapped reads; (3) for each replicate, use a cut-off of ≥10 reads per junction. These splicing junction-defined intron boundaries were then used to construct a BED file, by converting the 1-based intron start and end to the 0-based, half-open BED format, to allow counting of paired exon-intron and intron-exon junction reads using bedtools.75 In each replicate, for each filtered splicing junction-defined intron, the counts of exon-exon, intron-exon, and exon-intron junction reads were then compiled into a table to compute Splicing Index (SI), which is the ratio between spliced exon-exon junction read counts and the total number of spliced and normalized unspliced junction reads (Figure 1E); as well as the EIE index, which is the log2 ratio between exon-intron and intron-exon junction reads. For the HEK.dDHX15 and HEK.dDHX38 totalRNA and chRNA sequencing experiments, splicing junctions that are supported by < 10 unique exon-exon junction reads per replicate, introns <50 nt, and junctions present in <3 replicates were removed from the downstream analysis. For each intron, to calculate ΔSI and post-Tx SI, a 2x2 Fisher exact test was performed using R v3.6.3, between the two comparing conditions (DMSO versus dTAG, or chRNA versus totalRNA) and the spliced versus unspliced junction counts, and the false discovery rate (FDR) was estimated from the resulting p values.
GO enrichment analysis
For GO analysis, a background set of genes expressed in our HEK.dDHX15 and HEK.dDHX38 experiments were selected by using the cutoff of TPM or FPKM ≥10 from the RSEM outputs. GO enrichment analysis was performed by running the enrichGO function with clusterProfiler v4.0,75,67 with a Benjamini-Hochberg adjusted p value cutoff at 0.05.
Splice sites and BPS analysis
Splicing junction-defined intron boundaries were used to extra the 9mer sequences around 5’ss and 23mer sequences around 3’ss to compute the MaxEntScan scores.68 BPS annotation supported by RNAseq reads spanning lariat BPS-5’ss junctions were previously determined from 17,164 RNA sequencing data sets.76
Splicing junction-defined intron classification
For a given 5’ss-3’ss pair identified in our RNAseq results (supported by ≥ 10 uniquely mapped reads), alternative splicing status is classified as following: (1) if no alternative pairing donor or acceptor was identified, it is classified as constitutive; (2) if one or multiple alternative 3’ss (a3ss) was identified to pair with same 5’ss, based on their distance, the closest pair is classified as proximal a3ss, the rest are classified as distal a3ss; (3) conversely, if one or multiple alternative 5’ss (a5ss) was identified to pair with same 3’ss, based on their distance, the closest pair is classified as proximal a5ss, the rest are classified as distal a5ss; (4) if the 5’ss is a distal a5ss, and at the same time the 3’ss is a distal a3ss, this pairing is classified as exclusion; (5) the remaining cases are classified as mixed.
For a given 5’ss-3’ss pair identified in our RNAseq results (supported by ≥ 10 uniquely mapped reads), cryptic splicing status is classified as following: (1) intron 5’ss-3’ss pairings not annotated in GENCODE v28 annotation (basic) are all classified cryptic junctions; (2) cryptic junctions were then further classified based on whether the 5’ss or 3’ss is annotated or cryptic.
Heatmap and hierarchical clustering
An unsupervised hierarchical clustering of the 1,167 introns with altered splicing efficiency (ΔSI ≥ 0.05, FDR adjust p.value <0.05) in our HEK.dDHX15 experiments and the G-patch factor KD experiments was performed using ComplexHeatmap v2.2.0,69 with (1 – Pearson’s correlation) as the clustering distance. Upset plot of the intersection size of introns with altered splicing were also performed using make_comb_mat function with ComplexHeatmap. Correlation heatmap generated using corrplot v0.92.
Neo-splicing index (neoSI) analysis across ENCODE shRNA KD experiments
Raw RNAseq fastq files from the ENCODE shRNA KD experiments were downloaded and mapped as previously described.40 Splicing junctions detected by STAR (SJ.out.tab) were filtered with the following criteria to extract unannotated neojunctions: (1) remove reads with undefined strand; (2) restrict to uniquely mapped reads; (3) filter column 6 value to be 0 to extract unannotated splice junctions; (4) for each cell type (HepG2 or K562), use a cutoff of ≥3 reads per junction in any given shRNA experiment. These neojunction-defined intron boundaries were then used to extract the 5’ss and 3’ss coordinates. To extract all spliced reads (annotated and unannotated) spanning the neojunction 5’ss and 3’ss, each SJ.out.tab file was filtered to allow counting of donor and acceptor-spanning spliced reads using custom R scripts. Two replicates were combined, for each filtered neojunction, the counts of neojunction, donor spliced, and acceptor-spliced reads were then compiled into a table to compute neo-Splicing Index (SI), which is the ratio between spliced neojunction read counts and the normalized total number of spliced donor and acceptor reads (Figure S4C). For each neojunction, to calculate ΔneoSI, a 2x2 Fisher exact test was performed in R v3.6.3, between the RBP shRNA KD experiment and the matched non-targeting shRNA control experiment and the false discovery rate (FDR) was estimated from the resulting p values.
Supplementary Material
Highlights.
The DHX15 RNA helicase mediates widespread splicing quality control in human cells
Many RNA transcripts are sensitive toward both DHX38 and DHX15
Repression of suboptimal and cryptic introns by DHX15 is promoted by SUGP1
DHX15’s ATPase activity and SUGP1’s ULM domain are required for their interaction
ACKNOWLEDGMENTS
We thank the MIT BioMicro Center for Illumina NovaSeq library preparation and sequencing, the MIT Koch Institute (KI) Microscopy Core Facility for microscope support, and the KI Flow Cytometry Core Facility for cell sorting services. We thank members of the C.B. laboratory for their helpful discussions, especially Michael McGurk. Q.F. was supported by a postdoctoral fellowship from the Jane Coffin Childs Memorial Fund. This work was funded by grants from the NIH (GM085319 and HG002439 to C.B.B.). The sequencing work done at MIT BioMicro was supported in part by the Koch Institute Support (Core) Grant P30-CA14051 from the NCI.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113223.
DECLARATION OF INTERESTS
The authors declare no competing interests. Received: November 14, 2022
REFERENCES
- 1.Staley JP, and Guthrie C (1998). Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell 92, 315–326. [DOI] [PubMed] [Google Scholar]
- 2.Liu Y-C, and Cheng S-C (2015). Functional roles of DExD/H-box RNA helicases in Pre-mRNA splicing. J. Biomed. Sci 22, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Bortoli F, Espinosa S, and Zhao R (2021). DEAH-Box RNA Helicases in Pre-mRNA Splicing. Trends Biochem. Sci 46, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cordin O, Hahn D, and Beggs JD (2012). Structure, function and regulation of spliceosomal RNA helicases. Curr. Opin. Cell Biol 24, 431–438. [DOI] [PubMed] [Google Scholar]
- 5.Semlow DR, Blanco MR, Walter NG, and Staley JP (2016). Spliceosomal DEAH-Box ATPases Remodel Pre-mRNA to Activate Alternative Splice Sites. Cell 164, 985–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strittmatter LM, Capitanchik C, Newman AJ, Hallegger M, Norman CM, Fica SM, Oubridge C, Luscombe NM, Ule J, and Nagai K (2021). psiCLIP reveals dynamic RNA binding by DEAH-box helicases before and after exon ligation. Nat. Commun 12, 1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roy J, Kim K, Maddock JR, Anthony JG, and Woolford JL Jr. (1995). The final stages of spliceosome maturation require Spp2p that can interact with the DEAH box protein Prp2p and promote step 1 of splicing. RNA 1, 375–390. [PMC free article] [PubMed] [Google Scholar]
- 8.Tanaka N, Aronova A, and Schwer B (2007). Ntr1 activates the Prp43 helicase to trigger release of lariat-intron from the spliceosome. Genes Dev 21, 2312–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bohnsack KE, Ficner R, Bohnsack MT, and Jonas S (2021). Regulation of DEAH-box RNA helicases by G-patch proteins. Biol. Chem 402, 561–579. [DOI] [PubMed] [Google Scholar]
- 10.Hamann F, Schmitt A, Favretto F, Hofele R, Neumann P, Xiang S, Urlaub H, Zweckstetter M, and Ficner R (2020). Structural analysis of the intrinsically disordered splicing factor Spp2 and its binding to the DEAH-box ATPase Prp2. Proc. Natl. Acad. Sci. USA 117, 2948–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Studer MK, Ivanović L, Weber ME, Marti S, and Jonas S (2020). Structural basis for DEAH-helicase activation by G-patch proteins. Proc. Natl. Acad. Sci. USA 117, 7159–7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanner NK, and Linder P (2001). DExD/H box RNA helicases: from generic motors to specific dissociation functions. Mol. Cell 8, 251–262. [DOI] [PubMed] [Google Scholar]
- 13.Fourmann J-B, Dybkov O, Agafonov DE, Tauchert MJ, Urlaub H, Ficner R, Fabrizio P, and Lührmann R (2016). The target of the DEAH-box NTP triphosphatase Prp43 in Saccharomyces cerevisiae spliceosomes is the U2 snRNP-intron interaction. Elife 5, e15564. 10.7554/eLife.15564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fourmann J-B, Tauchert MJ, Ficner R, Fabrizio P, and Lührmann R (2017). Regulation of Prp43-mediated disassembly of spliceosomes by its cofactors Ntr1 and Ntr2. Nucleic Acids Res 45, 4068–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tauchert MJ, Fourmann J-B, Lührmann R, and Ficner R (2017). Structural insights into the mechanism of the DEAH-box RNA helicase Prp43. Elife 6, e21510. 10.7554/eLife.21510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Behzadnia N, Golas MM, Hartmuth K, Sander B, Kastner B, Deckert J, Dube P, Will CL, Urlaub H, Stark H, and Lührmann R (2007). Composition and three-dimensional EM structure of double affinity-purified, human prespliceosomal A complexes. EMBO J 26, 1737–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J, Ali AM, Lieu YK, Liu Z, Gao J, Rabadan R, Raza A, Mukherjee S, and Manley JL (2019). Disease-Causing Mutations in SF3B1 Alter Splicing by Disrupting Interaction with SUGP1. Mol. Cell 76, 82–95.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alsafadi S, Dayot S, Tarin M, Houy A, Bellanger D, Cornella M, Wassef M, Waterfall JJ, Lehnert E, Roman-Roman S, et al. (2021). Genetic alterations of SUGP1 mimic mutant-SF3B1 splice pattern in lung adenocarcinoma and other cancers. Oncogene 40, 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Z, Zhang J, Sun Y, Perea-Chamblee TE, Manley JL, and Rabadan R (2020). Pan-cancer analysis identifies mutations in that recapitulate mutant SF3B1 splicing dysregulation. Proc. Natl. Acad. Sci. USA 117, 10305–10312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faber ZJ, Chen X, Gedman AL, Boggs K, Cheng J, Ma J, Radtke I, Chao J-R, Walsh MP, Song G, et al. (2016). The genomic landscape of core-binding factor acute myeloid leukemias. Nat. Genet 48, 1551–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pan L, Li Y, Zhang H-Y, Zheng Y, Liu X-L, Hu Z, Wang Y, Wang J, Cai Y-H, Liu Q, et al. (2017). DHX15 is associated with poor prognosis in acute myeloid leukemia (AML) and regulates cell apoptosis via the NF-kB signaling pathway. Oncotarget 8, 89643–89654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toroney R, Nielsen KH, and Staley JP (2019). Termination of pre-mRNA splicing requires that the ATPase and RNA unwindase Prp43p acts on the catalytic snRNA U6. Genes Dev 33, 1555–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Semlow DR, and Staley JP (2012). Staying on message: ensuring fidelity in pre-mRNA splicing. Trends Biochem. Sci 37, 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koodathingal P, and Staley JP (2013). Splicing fidelity: DEAD/H-box ATPases as molecular clocks. RNA Biol 10, 1073–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maul-Newby HM, Amorello AN, Sharma T, Kim JH, Modena MS, Prichard BE, and Jurica MS (2022). A model for DHX15 mediated disassembly of A-complex spliceosomes. RNA 28, 583–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nabet B, Roberts JM, Buckley DL, Paulk J, Dastjerdi S, Yang A, Leggett AL, Erb MA, Lawlor MA, Souza A, et al. (2018). The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol 14, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herzel L, Straube K, and Neugebauer KM (2018). Long-read sequencing of nascent RNA reveals coupling among RNA processing events. Genome Res 28, 1008–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drexler HL, Choquet K, and Churchman LS (2020). Splicing Kinetics and Coordination Revealed by Direct Nascent RNA Sequencing through Nanopores. Mol. Cell 77, 985–998.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mayas RM, Maita H, Semlow DR, and Staley JP (2010). Spliceosome discards intermediates via the DEAH box ATPase Prp43p. Proc. Natl. Acad. Sci. USA 107, 10020–10025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koodathingal P, Novak T, Piccirilli JA, and Staley JP (2010). The DEAH box ATPases Prp16 and Prp43 cooperate to proofread 5’ splice site cleavage during pre-mRNA splicing. Mol. Cell 39, 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tseng C-K, Liu H-L, and Cheng S-C (2011). DEAH-box ATPase Prp16 has dual roles in remodeling of the spliceosome in catalytic steps. RNA 17, 145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu Y-Z, and Query CC (2007). Competition between the ATPase Prp5 and branch region-U2 snRNA pairing modulates the fidelity of spliceosome assembly. Mol. Cell 28, 838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang W-W, and Cheng S-C (2015). A novel mechanism for Prp5 function in prespliceosome formation and proofreading the branch site sequence. Genes Dev 29, 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Z, Rigo N, Dybkov O, Fourmann J-B, Will CL, Kumar V, Urlaub H, Stark H, and Lührmann R (2021). Structural insights into how Prp5 proofreads the pre-mRNA branch site. Nature 596, 296–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsai R-T, Fu R-H, Yeh F-L, Tseng C-K, Lin Y-C, Huang Y-H, and Cheng S-C (2005). Spliceosome disassembly catalyzed by Prp43 and its associated components Ntr1 and Ntr2. Genes Dev 19, 2991–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hegele A, Kamburov A, Grossmann A, Sourlis C, Wowro S, Weimann M, Will CL, Pena V, Lührmann R, and Stelzl U (2012). Dynamic protein-protein interaction wiring of the human spliceosome. Mol. Cell 45, 567–580. [DOI] [PubMed] [Google Scholar]
- 38.Thul PJ, Åkesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Björk L, Breckels LM, et al. (2017). A subcellular map of the human proteome. Science 356, eaal3321. 10.1126/science.aal3321. [DOI] [PubMed] [Google Scholar]
- 39.Zhang J, Huang J, Xu K, Xing P, Huang Y, Liu Z, Tong L, and Manley JL (2022). DHX15 is involved in SUGP1-mediated RNA missplicing by mutant SF3B1 in cancer. Proc. Natl. Acad. Sci. USA 119, e2216712119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Nostrand EL, Freese P, Pratt GA, Wang X, Wei X, Xiao R, Blue SM, Chen J-Y, Cody NAL, Dominguez D, et al. (2020). A large-scale binding and functional map of human RNA-binding proteins. Nature 583, 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han Y, Branon TC, Martell JD, Boassa D, Shechner D, Ellisman MH, and Ting A (2019). Directed Evolution of Split APEX2 Peroxidase. ACS Chem. Biol 14, 619–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lam SS, Martell JD, Kamer KJ, Deerinck TJ, Ellisman MH, Mootha VK, and Ting AY (2015). Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 12, 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, and Steinegger M (2022). ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kielkopf CL, Rodionova NA, Green MR, and Burley SK (2001). A novel peptide recognition mode revealed by the X-ray structure of a core U2AF35/U2AF65 heterodimer. Cell 106, 595–605. [DOI] [PubMed] [Google Scholar]
- 45.Galardi JW, Bela VN, Jeffery N, He X, Glasser E, Loerch S, Jenkins JL, Pulvino MJ, Boutz PL, and Kielkopf CL (2022). A UHMULM interface with unusual structural features contributes to U2AF2 and SF3B1 association for pre-mRNA splicing. J. Biol. Chem 298, 102224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heininger AU, Hackert P, Andreou AZ, Boon K-L, Memet I, Prior M, Clancy A, Schmidt B, Urlaub H, Schleiff E, et al. (2016). Protein cofactor competition regulates the action of a multifunctional RNA helicase in different pathways. RNA Biol 13, 320–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Inesta-Vaquera F, Chaugule VK, Galloway A, Chandler L, Rojas-Fernandez A, Weidlich S, Peggie M, and Cowling VH (2018). DHX15 regulates CMTR1-dependent gene expression and cell proliferation. Life Sci. Alliance 1, e201800092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Agafonov DE, Deckert J, Wolf E, Odenwälder P, Bessonov S, Will CL, Urlaub H, and Lührmann R (2011). Semiquantitative proteomic analysis of the human spliceosome via a novel two-dimensional gel electrophoresis method. Mol. Cell Biol 31, 2667–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharma SS, Maharshi S, Damianov A, Rio DC, and Black DL (2008). Polypyrimidine tract binding protein controls the transition from exon definition to an intron defined spliceosome. Nat. Struct. Mol. Biol 17, 183–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hartmuth K, Urlaub H, Vornlocher H-P, Will CL, Gentzel M, Wilm M, and Lührmann R (2002). Protein composition of human prespliceosomes isolated by a tobramycin affinity-selection method. Proc. Natl. Acad. Sci. USA 99, 16719–16724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Maio A, Yalamanchili HK, Adamski CJ, Gennarino VA, Liu Z, Qin J, Jung SY, Richman R, Orr H, and Zoghbi HY (2018). RBM17 Interacts with U2SURP and CHERP to Regulate Expression and Splicing of RNA-Processing Proteins. Cell Rep 25, 726–736.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beusch I, Rao B, Studer MK, Luhovska T, Šukyte V, Lei S, Oses-Prieto J, SeGraves E, Burlingame A, Jonas S, and Madhani HD (2023). Targeted high-throughput mutagenesis of the human spliceosome reveals its in vivo operating principles. Mol. Cell 83, 2578–2594.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Loerch S, and Kielkopf CL (2016). Unmasking the U2AF homology motif family: a bona fide protein–protein interaction motif in disguise. RNA 22, 1795–1807. 10.1261/rna.05795.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hastings ML, Allemand E, Duelli DM, Myers MP, and Krainer AR (2007). Control of pre-mRNA splicing by the general splicing factors PUF60 and U2AF(65). PLoS One 2, e538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fukumura K, Yoshimoto R, Sperotto L, Kang H-S, Hirose T, Inoue K, Sattler M, and Mayeda A (2021). SPF45/RBM17-dependent, but not U2AF-dependent, splicing in a distinct subset of human short introns. Nat. Commun 12, 4910–4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kahles A, Lehmann K-V, Toussaint NC, Hüser M, Stark SG, Sachsenberg T, Stegle O, Kohlbacher O, Sander C, and Cancer Genome Atlas Research Network; and Rätsch G (2018). Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 34, 211–224.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu SX, De Neef E, Thomas JD, Sabio E, Rousseau B, Gigoux M, Knorr DA, Greenbaum B, Elhanati Y, Hogg SJ, et al. (2021). Pharmacologic modulation of RNA splicing enhances anti-tumor immunity. Cell 184, 4032–4047.e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Merlotti A, Sadacca B, Arribas YA, Ngoma M, Burbage M, Goudot C, Houy A, Rocañíın-Arjó A, Lalanne A, Seguin-Givelet A, et al. (2023).Noncanonical splicing junctions between exons and transposable elements represent a source of immunogenic recurrent neo-antigens in patients with lung cancer. Sci. Immunol 8, eabm6359. [DOI] [PubMed] [Google Scholar]
- 59.Schmitzová J, Cretu C, Dienemann C, Urlaub H, and Pena V (2023). Structural basis of catalytic activation in human splicing. Nature 617, 842–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murugan A, Huse DA, and Leibler S (2012). Speed, dissipation, and error in kinetic proofreading. Proc. Natl. Acad. Sci. USA 109, 12034–12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee SC-W, and Abdel-Wahab O (2016). Therapeutic targeting of splicing in cancer. Nat. Med 22, 976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Y, Gogol-Döring A, Hu H, Fröhler S, Ma Y, Jens M, Maaskola J, Murakawa Y, Quedenau C, Landthaler M, et al. (2013). Integrative analysis revealed the molecular mechanism underlying RBM10-mediated splicing regulation. EMBO Mol. Med 5, 1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Khandelia P, Yap K, and Makeyev EV (2011). Streamlined platform for short hairpin RNA interference and transgenesis in cultured mammalian cells. Proc. Natl. Acad. Sci. USA 108, 12799–12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. (2021). clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2, 100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yeo G, and Burge CB (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol 11, 377–394. [DOI] [PubMed] [Google Scholar]
- 69.Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- 70.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mayer A, and Churchman LS (2016). Genome-wide profiling of RNA polymerase transcription at nucleotide resolution in human cells with native elongating transcript sequencing. Nat. Protoc 11, 813–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hung V, Udeshi ND, Lam SS, Loh KH, Cox KJ, Pedram K, Carr SA, and Ting AY (2016). Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat. Protoc 11, 456–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Luo Y, Hitz BC, Gabdank I, Hilton JA, Kagda MS, Lam B, Myers Z, Sud P, Jou J, Lin K, et al. (2020). New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res 48, D882–D889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pineda JMB, and Bradley RK (2018). Most human introns are recognized via multiple and tissue-specific branchpoints. Genes Dev 32, 577–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All RNA-seq data sets generated in this manuscript have been deposited in the public functional genomics data repository Gene Expression Omnibus (GEO) and are publicly available as of the date of the publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Anti-HA | MilliporeSigma | Cat#: 3F10; RRID:AB_2314622 |
| Anti-FLAG (M2) | MilliporeSigma | Cat#: F3165; RRID:AB_259529 |
| Anti-DHX15 | Abcam | Cat#: Ab254591; RRID:AB_2892059 |
| Anti-β-actin (AC-15) | MilliporeSigma | Cat#: A5441; RRID:AB_476744 |
| Anti-H3 | Abcam | Cat#: Ab1791; RRID:AB_302613 |
| Anti-SC35 | Abcam | Cat#: Ab204916; RRID:AB_2909393 |
| Anti-SF3B1 | Abcam | Cat#: Ab170854 |
| Anti-U2AF2 | Invitrogen | Cat#: PA530442; RRID:AB_2547916 |
| Anti-U2AF1 | Proteintech | Cat#: 10334-1 AP; RRID:AB_2211314 |
| IRdye 800CW Sterptavidin | LI-COR | Cat#: 926-32230 |
| Anti-mouse Alexa Fluor 488 | ThermoFisher | Cat#: A28175; RRID:AB_2536161 |
| Anti-rat Alexa Fluor 647 | ThermoFisher | Cat#: A21247; RRID:AB_141778 |
| NeutrAvidin- DyLight 594 | ThermoFisher | Cat#: 22842 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| dTAG13 | Tocris Bioscience | Fisher Scientific Cat#: 66-055 |
| α-amanitin | Santa Cruz Biotechnology | Cat#: Sc-202440A |
| cOmplete™ ULTRA protease inhibitor | Roche | MilliporeSigma Cat#: 589297001 |
| PhosStop | Roche | MilliporeSigma Cat#: 4906837001 |
| Hygromycin | Millipore | Cat#: 400052 |
| Lipofectamine 3000 | Invitrogen | Cat#: L3000008 |
| Proteinase K | Viagen | Cat#: 501-PK |
| Direct PCR reagent | Viagen | Cat#: 301-C |
| Direct-zol | Zymo Research | Cat#: R2052 |
| SUPERaseIn | Life Technologies | Cat#: AM2694 |
| Tri reagent | Zymo Research | Cat#: R2050-1-200 |
| Doxycyclin | MilliporeSigma | Cat#: D9891 |
| Biotin Phenol | MilliporeSigma | Cat#: SML2135-250MG |
| H2O2 | MilliporeSigma | Cat#: H1009-100ML |
| Streptavidin-coated magnetic beads | Pierce | Thermo Scientific Cat#: 88817 |
| DAPI | ThermoFisher | Cat#: 62248 |
|
| ||
| Critical commercial assays | ||
|
| ||
| NEBuilder HiFi DNA assembly cloning kit | NEB | Cat#: E5520S |
| NEB OneTaq Quick-Load 2X Mastermix | NEB | Cat#: M0486L |
| NEBNext Poly(A) mRNA magnetic isolation module | NEB | Cat#: E7490L |
| NEBNext Ultra II Directional RNA kit | NEB | Cat#: E7760L |
| NEBNext rRNA Depletion Kit | NEB | Cat#: E6310X |
|
| ||
| Deposited data | ||
|
| ||
| Human reference genome NCBI release 28, GRCh38 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| SUGP1 KD | Liu et al.19 | GEO: GSE159304 |
| RBM10 KD | Wang et al.62 | GEO: GSE44976 |
| SUGP2 KD | ENCODE Consortium | ENCLB206KMT, ENCLB331AGB |
| NKRF KD | ENCODE Consortium | ENCLB552FZS, ENCLB950ZAG |
| TFIP11 KD | ENCODE Consortium | ENCLB144PBT, ENCLB483ITG |
| RBM17 KD | ENCODE Consortium | ENCLB012PNW, ENCLB719FFS |
| GPKOW KD | ENCODE Consortium | ENCLB168TMK, ENCLB908ZJN |
| RBM5 (CRISPR KO) | ENCODE Consortium | ENCLB036OZM, ENCLB293XAV |
| AGGF (CRISPR KO) | ENCODE Consortium | ENCLB710LCN, ENCLB644OBV |
| Raw and analyzed data | This Paper | GEO: GSE241712 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK293T.A2 | Khandelia et al.63 | N/A |
| HEK.FLAG-AP-DHX15.WT | This paper | N/A |
| HEK.dDHX15 | This paper | N/A |
| HEK.dDHX38 | This paper | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S1 | N/A | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pSpCas9-gRNA-GFP | Ran et al.27 | Cat# PX458; no. 48138 |
| pUC19-DHX15- FKBPF36V-2xHA_P2A_BFP | This paper | N/A |
| pUC19-DHX15- FKBPF36V-2xHA_P2A_HygR | This paper | N/A |
| pUC19-DHX38- FKBPF36V-2xHA_P2A_BFP | This paper | N/A |
| pRD-FLAG-AP-DHX15 (wt and mutants) | This paper | N/A |
| pX304-HA-SUGP1-EX (full-length and truncations) | This paper | N/A |
| pCAGGS-nlCre | Khadelia et al.63 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| STAR version 2.7.3.a | Dobin et al.64 | https://github.com/alexdobin/STAR |
| RSEM v1.3.3 | Li et al.65 | https://github.com/deweylab/RSEM |
| Samtools V5.2.5 | Li et al.66 | http://www.htslib.org/ |
| clusterProfiler v4.0 | Yu et al.67 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| MaxEntScan | Yeo et al.68 | http://hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html |
| ComplexHeatmap v.2.2.0 | Gu et al.69 | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| Corrplot v0.92 | https://github.com/taiyun/corrplot | |
| ColabFold | Mirdita et al.43 | https://github.com/sokrypton/ColabFold |
| Pymol v2.5.2 | https://pymol.org | |
| Nikon NIS-Elements Imaging Software | Nikon | N/A |
| FIJI | Schindelin et al.70 | https://imagej.net/software/fiji/ |