


Abstract
The Ume6p–Sin3p–Rpd3p complex negatively regulates expression of genes containing a Ume6p binding site. However, these regulatory proteins also function independently to regulate gene expression both negatively and positively. The model system for this combinatorial regulation is the yeast phospholipid biosynthetic pathway. Sin3p negatively regulates the INO1, CHO1, CHO2 and OPI3 genes while Ume6p negatively regulates the INO1 gene and positively regulates the other genes. We have suggested that the positive regulation results from indirect effects on expression of the INO2 transcriptional activator gene. Here, we demonstrate that the effect of Ume6p on INO2 gene expression is also indirect. We also show that Rpd3p is a negative regulator of phospholipid biosynthetic gene expression. The ability of Ume6p, Sin3p and Rpd3p to differentially regulate expression of the phospholipid biosynthetic genes affects phospholipid composition. A sin3 mutant strain lacks detectable levels of phosphatidylethanolamine and elevated levels of phosphatidylcholine (PC) and a rpd3 mutant strain has reduced levels of PC. These alterations in membrane composition suggest that there may exist additional differences in regulation of phospholipid biosynthetic gene expression and that membrane compositions may be coordinated with other biological processes regulated by Ume6p, Sin3p and Rpd3p.
INTRODUCTION
The Ume6p–Sin3p–Rpd3p complex has received considerable attention due to its role in repression of gene expression (1–4). Ume6p interacts with specific DNA sequences found in the promoters of a diverse set of yeast genes (5,6). Sin3p functions as a co-repressor by recruiting Rpd3p to the DNA-bound Ume6p. Rpd3p deacetylates histones H3 and H4, resulting in repression of gene expression (2,3). This model is now well established and the roles of Sin3p and Rpd3p in repression are of particular interest because they are conserved in other eukaryotes (4). However, it is also well established that these proteins function independently of each other to effect both positive and negative regulation. One of the best examples of this combinatorial control is the phospholipid biosynthetic pathway. The UME6 and SIN3 genes are required for both positive and negative regulation of phospholipid biosynthetic gene expression. Here we have examined the roles of the UME6, SIN3 and RPD3 genes in regulating transcription of the phospholipid biosynthetic genes and the consequent effect on membrane composition.
Transcriptional regulation of phospholipid biosynthesis has been extensively studied in yeast (reviewed in 7,8). Transcription of the INO1, CHO1, CHO2 and OPI3 structural genes is repressed by growth in the presence of inositol and choline and derepressed by growth in their absence. This regulation is dictated by a conserved element present in the respective promoters called the UASINO element. This element is a binding site for the products of the INO2 and INO4 regulatory genes (9,10). Ino2p and Ino4p belong to the basic helix–loop–helix (bHLH) family of proteins, which dimerize through the HLH domains and interact with DNA via the basic regions (11). The precise mechanism for derepression of gene expression has not been elucidated, although it is known that derepression of the INO2 gene is required (12). The INO2 gene is autoregulated in response to inositol and choline in a pattern identical to that of its target genes (13). Conversely, repression of the phospholipid biosynthetic genes does not require repression of INO2 transcription (12). However, it does require the OPI1 gene, which encodes a leucine zipper protein that is glutamine-rich. Strains deleted for the OPI1 gene express the phospholipid biosynthetic genes at constitutive derepressed levels (7,8).
Previous studies have suggested that Ume6p and Sin3p can function as both repressors and activators of transcription, in concert or independently. Ume6p binds to a DNA sequence referred to as URS1, which is present in a large number of yeast genes, including the INO1 phospholipid biosynthetic gene (5,6,14). The UME6 gene is required for the negative regulation of meiotic genes (15–17), heat shock genes (18), arginine catabolic genes (19) and the INO1 gene (14). While its ability to repress expression of the meiotic genes and the INO1 gene is SIN3-dependent, the effect on the arginine catabolic genes is SIN3-independent. Ume6p can also positively regulate some early meiotic genes during sporulation (20,21), a DNA repair gene (22), several phospholipid biosynthetic genes (CHO1, CHO2 and OPI3) (14) and the INO2 regulatory gene (14). The ability to function as an activator of early meiotic gene expression is dependent on the IME1 gene product, which has been shown to interact with Ume6p (23). We have previously suggested that the role of Ume6p on CHO1, CHO2 and OPI3 gene expression is indirect via regulation of INO2 gene expression (14). Unlike the situation with the early meiotic genes, the CHO1 gene is not regulated by the IME1 gene (14). Consequently, activation of INO2 expression (and CHO1 expression) must involve a novel IME1-independent mechanism.
Sin3p is a negative regulator of the early meiotic genes (24), the HO gene (25), the TRK2 gene (26) and several phospholipid biosynthetic genes (27). In the case of the HO gene, repression does not require the UME6 gene. In the case of the CHO1, CHO2 and OPI3 phospholipid biosynthetic genes, repression does not require a URS1 element or the UME6 gene but does require the UASINO element (28). While most of the evidence suggests that the SIN3 gene encodes a negative regulator, SIN3 is also a positive regulator of the STE6 gene (29).
The RPD3 gene is required for repression of IME2, SPO13 and INO1 transcription (1–3). Moreover, studies have shown that Ume6p specifically recruits the Sin3p–Rpd3p dimer to the URS1 element of the INO1 promoter, which controls deacetylation of histones H3 and H4. Here we report the role of the RPD3 gene in regulating phospholipid biosynthesis, which is mediated through the UASINO element. We also establish a novel pathway for UME6 positive regulation that involves control of INO2 transcription. Lastly, we have evaluated the effects of mutations in each of the three regulatory proteins on membrane composition. The mutant phenotypes suggest additional differential control.
MATERIALS AND METHODS
Yeast strains, plasmids and growth conditions
The yeast strains used in this study were: FT5 (MATα, ura3-52, trp1-Δ63, his3-Δ200, leu2::PET56) (30), ume6Δ (ume6::LEU2), sin3Δ (sin3::HIS3) and rpd3Δ (rpd3::HIS3), which are isogenic to FT5 (1,30); AMP1008 (MATα, GAL1-IME1::TRP1, gal80::LEU2, arg6, ura3, leu2, trp1, lys2, ho::LYS2) (20) and the isogenic AMP1170 (rim16-12) (15); BRS1001 (MATa, ade2-1, his3-11,15, leu2-3-112, can1-100, trp1-1, ura3-1) (this laboratory); BRS2009 (MATa, ade2-1, his3-11,15, leu2-3-112, can1-100, trp1-1, ura3-1, ume6::LEU2) (14); BRS2013 (MATa, ade2-1, his3-11,15, leu2-3-112, can1-100, trp1-1, ura3-1, ume6::LEU2, ura3::pGAL1-INO2::URA3, ino2::TRP1) (this study); BRS1005 (MATa/MATα, ade1/ade1, ino1-13/ino1-13) (this laboratory). Yeast cultures were grown at 30°C in complete synthetic medium (31) containing 2% glucose (w/v) either supplemented with 75 µM inositol and 1 mM choline (I+C+) or lacking inositol and choline (I–C–). Where indicated, galactose (varying concentrations) and 2% raffinose were used as carbon sources. The X-gal solid medium has been described (27).
Plasmids pTL101, pGAL1-INO2 and pLG669-Z have been described (12,32,33). Plasmid pJH330 contains 543 bp of the sequences upstream of the INO1 gene and 132 codons of the INO1 gene fused in-frame to the lacZ reporter gene in YEp357R (34). This plasmid was constructed by inserting a 1.0 kb PstI–BglII fragment containing the INO1 sequences into the PstI–BamHI sites of YEp357R. Yeast transformations were performed using a routine procedure (35).
Opi plate assay
The Opi test is a cross-feeding assay that assesses the ability of yeast strains to excrete inositol into the growth medium. A modification of the original plate assay was used (36,37).
RNA analyses
Total yeast RNA was isolated using a glass bead disruption and hot phenol extraction procedure (38). Northern blot hybridizations were performed as described previously (39). RNA probes (cRNA) were synthesized using the Riboprobe Combination System SP6/T7 (Promega, Madison, WI) from plasmids (27) linearized with a restriction enzyme as follows (shown as plasmid/restriction enzyme/RNA polymerase) for the indicated (parenthesized) probe: pAB309Δ/EcoRI/SP6 (TCM1); pAS103/HindIII/T7 (CHO1); pJH310/HindIII/T7 (INO1); pPLg/BamHI/SP6 (ACT1). Results were visualized using a phosphorimager and quantitated using ImageQuant from Molecular Dynamics Inc. (Sunnyvale, CA).
β-Galactosidase assays
Yeast transformants were assayed for β-galactosidase activity as previously described (40). Units of β-galactosidase activity are defined as (OD420/min/mg total protein) × 1000. Total protein concentration was determined using a Bio-Rad Protein Assay Kit (Bio-Rad, Rockville Center, NY).
Determination of steady-state phospholipid composition
Steady-state labeling of phospholipids with [32P]orthophosphate (NEN DuPont, Boston, MA) was performed essentially as described previously (41). Cells were grown in the presence of [32P]orthophosphate (50 µCi, 9000 Ci/mmol) for at least five generations and harvested at a density of 5 × 107 cells/ml. Labeled cells were suspended in 5 ml of cold 5% trichloroacetic acid and incubated on ice for 20 min. Cells were pelleted and washed twice with distilled H2O and 1 ml of polar solvent (40% ethanol, 13.9% diethyl ether, 2.8% pyridine, 0.027% ammonium hydroxide and 0.01% butylated hydroxytoluene in 2:1 chloroform:methanol) was added. Lipids were extracted by incubation at 60°C for 20 min (42). After cooling to room temperature, a 0.5 vol of distilled H2O and 5 vol of 2:1 chloroform:methanol containing 0.005% butylated hydroxytoluene were added and the mixture was vortexed (43). Solvents were then fractionated by centrifugation (1500 g, 10 min) and the lower layer was transferred to a dram vial and dried under a stream of nitrogen gas. The lipid pellet was dissolved in 20 µl of 2:1 chloroform:methanol, spotted onto chromatography paper (Whatman SG81 treated with 2% EDTA, pH 7.4) and separated by 2-dimensional chromatography (44). The solvent for the first dimension was chloroform:methanol:2.8% ammonium hydroxide:dH2O (66:27:3:0.8) and the solvent for the second dimension was chloroform:methanol:acetic acid:dH2O (32:4:5:1). Phospholipids were visualized by autoradiography and quantitated by liquid scintillation.
Electrophoretic mobility shift assay (EMSA)
Recombinant MBP and MBP–Ume6p were generated as described (5). Binding reactions were carried out using three fragments of the INO2 promoter or a 26 bp oligonucleotide containing a URS1 element from the SPO13 gene using conditions described previously (5).
Mutagenesis
The ume6Δ strain transformed with pTL101 was mutagenized with EMS to 30% survival as described previously (27).
RESULTS
UME6, SIN3 and RPD3 repress INO1 transcription
It is now well established that the URS1 element binds Ume6p, which interacts with Sin3p and Rpd3p to repress target gene expression (1,3). Consistent with this model, we showed previously that the URS1 element in the INO1 promoter is required for repression of INO1–lacZ and INO1–cat reporter genes (14,45) and that UME6 is a negative regulator of INO1 expression (45). Moreover, a mutant (cpe1) defective in repression of an INO1–lacZ reporter gene (27) was found to be allelic to sin3 mutations. These observations suggested that RPD3 could be a negative regulator of INO1.
To test this possibility, we examined INO1 expression in a rpd3 mutant strain by northern blot hybridization, by quantitating INO1–lacZ expression and by assaying for an Opi– phenotype (46). We had previously observed that a ume6Δ mutant strain displays an Opi– phenotype (14). Here, we found that the sin3Δ and rpd3Δ mutant strains also display an Opi– phenotype (Fig. 1A). Consistent with the Opi– phenotype, repression of both INO1 transcription (Fig. 1B) and INO1–lacZ expression (data not shown) is defective in all three mutant strains. The three assays also show that the defect in INO1 expression is most pronounced in the ume6Δ strain and least pronounced in the rpd3Δ strain. This graded defect in repression has been observed by others using a heterologous reporter system that contained either the INO1 or the IME2 URS1 elements (1).
Figure 1.
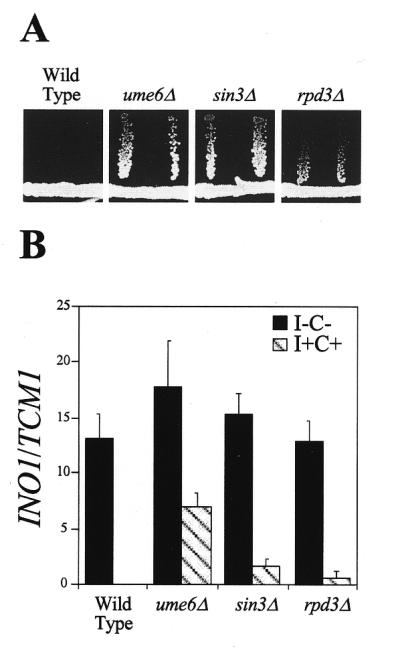
Repression of INO1 transcription is defective in ume6Δ, sin3Δ and rpd3Δ mutant strains. (A) Opi test. Mutants (horizontal) are streaked on 1.2% agar medium lacking inositol and allowed to grow for 72 h. A tester strain (inositol auxotroph, ino1) is streaked away (vertical) from the original streak and allowed to grow for 72 h (37). (B) INO1 transcription quantitated by northern blot hybridization and normalized using TCM1. I–C–, derepressing medium lacking inositol and choline (solid); I+C+, repressing medium containing 75 µM inositol and 1 mM choline (hatched).
UME6, SIN3 and RPD3 regulation through the CHO1 UASINO element
We had previously shown that SIN3 is a negative regulator of the INO1, CHO1, CHO2 and OPI3 phospholipid biosynthetic genes in spite of the fact that only the INO1 promoter contains the URS1 element (27). Later, it was shown that SIN3 functioned through two UASINO elements present in the INO1 promoter (28). The interaction between Sin3p and Ume6p prompted an examination of phospholipid gene expression in a ume6Δ strain. Unexpectedly, the UME6 gene was found to be a positive regulator of CHO1, CHO2 and OPI3 and a negative regulator of INO1 (14).
Since the three genes that were positively regulated by the UME6 gene all contain the UASINO element, we assayed the effect of a ume6Δ mutation on expression of a lacZ reporter gene driven by the UASINO element from the CHO1 promoter (pTL101) (32). The pTL101 plasmid contains the lacZ gene driven by a CYC1 promoter lacking the CYC1 UAS element but containing the CHO1 UASINO element (inserted using a 28 bp oligonucleotide).
The data demonstrate that the positive role of UME6 on CHO1 expression is exerted through the UASINO element, since lacZ expression was decreased in the ume6Δ strain (Fig. 2). This pattern of expression mirrors that of the native CHO1 gene in that CHO1 transcription was also decreased in the ume6Δ strain (Fig. 3; 14). However, CHO1 expression in the ume6Δ strain was previously shown to be unresponsive to the presence of inositol and choline while lacZ expression from pTL101 was still sensitive to inositol and choline (Fig. 2). This could be due to differences in the sensitivities of northern blot hybridizations and β-galactosidase assays. Alternatively, the decreased activity of the UASINO element in the ume6Δ mutant strain may abate the response to inositol and choline when the UASINO element is in its native context. This possibility is supported by the observation that lacZ expression is regulated 18-fold by the CHO1 UASINO element in pTL101 while CHO1 and CHO1–lacZ expression is regulated 3–4-fold (14,31,47). The data further show that SIN3 is a negative regulator through the CHO1 UASINO, as it is through the two UASINO elements in the INO1 promoter (28). In addition, RPD3 was found to function as a negative regulator through the CHO1 UASINO element. Therefore, this data identifies two new mechanisms for the action of Ume6p, Sin3p and Rpd3p in addition to the URS1-mediated repression mechanism: Sin3p–Rpd3p-mediated negative regulation through the UASINO element; and Ume6p-mediated positive regulation through the UASINO element.
Figure 2.

Ume6p functions as a positive regulator and Sin3p and Rpd3p function as negative regulators through the CHO1 UASINO element. β-Galactosidase activity from transformants containing pTL101 (32). I–C–, solid; I+C+, hatched.
Figure 3.

Overexpression of INO2 rescues a ume6Δ phenotype (absence of CHO1 derepression). Northern blot showing CHO1 transcript levels in a wild-type strain (BRS1001) and an isogenic ume6Δ, ino2Δ strain containing a GAL1 promoter–INO2 fusion. Northerns were probed with CHO1- and ACT1-specific cRNA probes. The lower panel shows a control experiment demonstrating the ume6Δ phenotype (14). Lanes marked + indicate RNA purified from cells grown in medium containing 75 µM inositol and 1 mM choline while the lanes marked – contain RNA from cells grown in the absence of these precursors. The RNA samples used in the upper panel northerns were grown in medium containing various concentrations of galactose plus 2% raffinose. The RNA samples used in the lower panel northern were grown in medium containing glucose.
INO2 overexpression restores regulation of CHO1 expression in a ume6Δ strain
Because UME6 functions as a positive regulator of UASINO-containing genes, we previously suggested that Ume6p either directly interacted with the UASINO element or that it regulated expression of the genes encoding the UASINO-binding proteins Ino2p and Ino4p. We have shown that INO2–cat expression is decreased in the ume6Δ strain but INO4–cat expression is unaffected (14).
Therefore, the decreased inositol/choline response of the CHO1 gene in a ume6Δ strain could be due to decreased INO2 expression. To test this, we examined expression of the CHO1 gene in a ume6Δ, ino2Δ strain containing the INO2 gene driven by the GAL1 promoter (12). We have previously used this GAL1–INO2 construct, which expresses INO2 in a galactose-dependent but inositol/choline-independent manner (12). Here we used it to determine if overexpression of the INO2 gene restores regulation of CHO1 gene expression in a ume6Δ strain. The data show that CHO1 expression increased as a function of galactose concentration (Fig. 3; BRS2013). Moreover, regulation of CHO1 expression in response to inositol and choline was more evident at higher galactose concentrations when INO2 expression was increased. Galactose did not affect CHO1 expression in a strain containing a wild-type INO2 gene (Fig. 3; BRS1001)
To further test if INO2 overexpression rescues the CHO1 phenotype, we devised a screen for mutants that express wild-type (or greater) levels of lacZ from the pTL101 plasmid in a ume6Δ mutant strain. The goal of this screen was to identify mutants that overexpress the lacZ reporter, possibly as a result of increased INO2 transcription. The biggest problem that we anticipated was the isolation of bypass suppressors that would increase lacZ expression from the reporter without increasing INO2 expression. Optimally, we would want to use a reporter system driven by the INO2 promoter. Unfortunately, the INO2 promoter is too weak to drive expression of the lacZ reporter gene (unpublished observation).
As described above, in a ume6Δ strain lacZ expression from pTL101 is 70% lower than in the isogenic wild-type strain (I–C– medium) (Fig. 2). Fortunately, this 3-fold difference in β-galactosidase activity is easily observed by plate assay. A wild-type strain turns blue on X-gal I–C– medium after a 24 h incubation whereas the ume6Δ strain requires 4 days to turn light blue. This enormous difference in plate phenotype is presumably due to the fact that the β-galactosidase activity generated by the ume6Δ strain is at the lower limit of sensitivity of the plate assay.
We treated the ume6Δ strain containing pTL101 with EMS and screened for colonies that turned blue after 24 h on X-gal I–C– medium. As a control, mock-mutagenized cells were plated and found to turn light blue after 4 days. Candidate mutant colonies were assayed for β-galactosidase activity by a liquid assay which revealed that those that had turned blue after 24 h yielded wild-type (or greater) levels of activity (Fig. 4A). Northern blot hybridization was performed to identify mutants that overexpressed the INO2 transcript. Among the original 11 mutant strains, five were found to express wild-type (or greater) levels of the INO2 transcript (data not shown). All five mutants are recessive and have been designated rescuers of ume6Δ (rum). All five mutant strains were grown on 5"-FOA plates to select for colonies lacking the pTL101 reporter plasmid and re-transformed with pTL101 to ensure that the phenotype was not plasmid borne. We also examined expression of a CYC1 promoter-driven lacZ reporter (pLG669-Z) in the rum mutant strains to determine if any of the rum mutants identify general transcription factors. The data show that some of the rum mutants (rum6, rum11 and rum18) display an increase in expression of the lacZ gene relative to the ume6Δ parental strain (Fig. 4B). However, the increase is modest relative to the pTL101 phenotype (Fig. 4A) (1.5-fold for the pLG669-Z reporter versus 3.5–6.5-fold for the pTL101 reporter). Moreover, the rum4 and rum14 mutants were indistinguishable from the ume6Δ parental strain when assayed for the pLG669-Z reporter. We have also determined that the rum mutants are not complemented by the OPI1, SIN3 and RPD3 genes, when they are present on a centromeric plasmid (data not shown).
Figure 4.

The rum mutations rescue the defect in lacZ expression from pTL101 caused by the ume6Δ mutant. (A) β-Galactosidase activity from transformants containing pTL101 (32). I–C–, solid; I+C+, open. (B) β-Galactosidase activity from transformants containing pLG669-Z (33)
UME6 regulates INO2 gene expression indirectly
The data above shows that UME6 is a positive regulator of INO2. This is not unreasonable given that Ume6p is known to function as both a repressor and activator of transcription (20). However, several conditions are required for Ume6p activator function (20), namely: (i) it functions as an activator in meiotic cells; (ii) it requires the URS1 element; (iii) it requires the IME1 gene. None of these conditions are met in our experiments with the INO2 gene (14).
Consequently, Ume6p might regulate INO2 expression via an indirect mechanism that does not require its activator function. To test this we examined CHO1 transcription in a rim16-12 mutant strain (AMP1170). This allele has a mutation in the UME6 gene that eliminates activator function but does not affect repressor function (20). The data show that CHO1 expression in the rim16-12 strain was indistinguishable from that of the isogenic wild-type strain (Fig. 5). The response to inositol and choline was also observed in the rim16-12 strain. These data suggest that either UME6 is regulating INO2 expression indirectly or directly by a novel IME1-independent activation mechanism.
Figure 5.

The Ume6p transcriptional activation activity is not required for CHO1 expression. Quantitative northern blot hybridization using a CHO1-specific cRNA probe normalized using a TCM1-specific cRNA probe. I–C–, solid; I+C+, hatched.
To test if Ume6p directly interacts with the promoter of the INO2 gene, we performed EMSA using recombinant MBP–Ume6p. We were unable to detect binding of the recombinant MBP–Ume6p to three different fragments of the INO2 promoter (Fig. 6) even upon prolonged exposure (five times longer than the exposure shown in Fig. 6). The three overlapping fragments used in this study encompass 500 bp of the INO2 promoter that are responsive to mutations in the UME6 gene (14). However, the recombinant protein was fully competent for complex formation with a binding site from the SPO13 gene (Fig. 6).
Figure 6.

Recombinant MBP–Ume6p does not form complexes with restriction fragments from the INO2 promoter. Three different restriction fragments from the INO2 promoter were combined with recombinant MBP–Ume6p. The regions covered by each of the restriction fragments are shown relative to the 500 bp immediately upstream of the INO2 start codon and UES and UASINO elements (B.P.Ashburner, D.A.Eiznhamer and J.M.Lopes, unpublished data). As a control, an oligonucleotide containing the URS1 element from the SPO13 gene was used in reactions with either MBP or MBP–Ume6p.
Mutations in the UME6, SIN3 and RPD3 genes yield altered membrane phospholipid compositions
Membrane phospholipid composition can be altered by different growth conditions and by mutations in genes that regulate phospholipid biosynthetic gene expression (reviewed in 7,8). One of the more dramatic effects is seen in strains harboring mutations in the OPI1 negative regulatory gene. The membranes of wild-type strains typically contain 28–30% phosphatidylinositol (PI) when grown in medium containing inositol and 10–12% PI when grown in medium lacking inositol. The is due to the fact that the enzyme which synthesizes PI (PI synthase) is limited for inositol when cells are grown in medium lacking inositol (48). Strains harboring mutant opi1 alleles contain elevated levels of PI when cells are grown in medium lacking inositol. This effect is due to overexpression of the INO1 gene that is required for de novo synthesis of inositol (7,8).
Because the ume6, sin3 and rpd3 mutant strains display the Opi– phenotype and because they differentially affect expression of the phosphatidylcholine (PC) biosynthetic genes, we examined the effects of these mutant alleles on phospholipid composition. There are three salient changes in the phospholipid compositions of these strains. The first is an increase in the amount of PI in cells grown in the absence of inositol (Table 1). This phenotype is not unexpected since the opi1 mutant strains have a similar phenotype (7,8). Interestingly, the effect of the mutant strains on PI composition (Table 1) is graded, as is the effect on INO1 expression (Fig. 1). Another phenotype is the reduced level of PC in the rpd3 mutant strain. This reduction in PC was observed in cells grown in the presence and absence of inositol (Table 1). A third phenotype is the complete absence of phosphatidylethanolamine (PE) in the sin3 mutant strain. Again, this phenotype was observed regardless of the presence or absence of inositol (Table 1). The absence of PE may account for the increase in PC or it may be due to decreased synthesis of PE or a combination of the two events. Regardless of the explanation for this phenotype, this is the first observation of a mutant that specifically affects PE levels.
Table 1. Phospholipid composition of ume6Δ, sin3Δ and rpd3Δ mutant strains.
| Strains | Medium | PIa | PSa | PEa | PCa | PAa |
|---|---|---|---|---|---|---|
| Wild-type | I–C– | 12.5 1.3 | 9.5 1.2 | 6.0 2.3 | 51.1 7.5 | 2.3 0.6 |
| ume6Δ | I–C– | 17.8 1.5 | 10.6 2.1 | 7.0 2.0 | 50.7 4.0 | 1.6 0.1 |
| sin3Δ | I–C– | 15.7 3.2 | 12.5 2.5 | NDb | 56.3 2.5 | 1.1 0.0 |
| rpd3Δ | I–C– | 13.3 2.1 | 15.0 2.6 | 10.0 1.0 | 43.7 1.2 | 2.0 0.0 |
| Wild-type | I+C+ | 32.3 5.1 | 7.9 1.7 | 6.4 1.0 | 41.8 2.6 | 1.0 0.2 |
| ume6Δ | I+C+ | 28.3 4.1 | 7.8 0.6 | 8.6 1.0 | 46.1 1.4 | 0.8 0.2 |
| sin3Δ | I+C+ | 27.7 1.5 | 8.0 1.0 | ND | 55.3 2.9 | 0.9 0.1 |
| rpd3Δ | I+C+ | 36.8 9.4 | 7.0 0.0 | 4.0 2.6 | 31.7 9.0 | 0.9 0.2 |
aPI, phosphatidylinositol; PS, phosphatidylserine; PE, phosphatidylethanolamine; PC, phosphatidylcholine; PA, phosphatidic acid.
bND, not detectable.
DISCUSSION
The role of Ume6p, Sin3p and Rpd3p in regulating phospholipid biosynthesis was examined. It had been previously demonstrated that UME6 and SIN3 are required for repression of the INO1 gene and that this repression requires the URS1 element (1,3,14,27,45). Data presented here demonstrate that RPD3 is also required for repression of the INO1 gene (Fig. 1). Curiously, the most striking phenotype of a rpd3 mutant strain is the Opi– phenotype, which usually correlates with INO1 overexpression (46). Although both the INO1 transcript levels and INO1–lacZ activity are increased in the rpd3 mutant strain, the degree of overexpression is modest relative to the Opi– phenotype. Consequently, the discrepancy between the Opi– phenotype and the defect in INO1 transcription suggests that the rpd3 mutant may affect some other genes involved in inositol production or excretion.
The data also reveal that Rpd3p represses transcription through the UASINO element (Fig. 2), which is present in most of the phospholipid biosynthetic genes (7,8). This observation is consistent with the previous finding that Sin3p represses transcription through the UASINO element (28). At present it is not possible to determine if the effect of Sin3p and Rpd3p through the UASINO element is direct or indirect, although there is no evidence to support the model of direct binding to the UASINO element. One possibility is that Sin3p may tether Rpd3 to proteins already bound at the UASINO element, such as Ino2p and Ino4p. Curiously, the mammalian Mad and Mxi1 proteins interact with the mammalian Sin3p homolog (49,50). Mad, Mxi1, Ino2p and Ino4p are all members of the basic helix–loop–helix family of proteins (7,8,11). Another possibility is that the effect may be indirect by regulating INO2 transcription. However, if this were the case the effect might be redundant since the INO2 promoter also contains a UASINO element (D.A.Eiznhamer and J.M.Lopes, unpublished data). Nevertheless, the ability of Sin3p to repress transcription in the absence of Ume6p has been previously documented for the HO gene (25). The UASINO-lacZ reporter (Fig. 2) should be a useful tool in genetic screens to define the precise mechanism for Sin3p–Rpd3p repression of phospholipid biosynthetic gene expression.
We previously reported that UME6 is a positive regulator of phospholipid biosynthetic gene expression (14). Here we show that the positive regulation is exerted through the UASINO element (Fig. 2). We have also shown that this regulation is indirectly mediated by controlling the amount of INO2 expression. Overexpression of the INO2 gene using the GAL1 promoter (Fig. 3) or in the rum mutant strains (Fig. 4) rescues the CHO1 mutant phenotype. While Ume6p can function as a positive regulator, it usually requires an interaction with Ime1p, which provides the transcriptional activation domain (23). However, the IME1 gene is not required for proper regulation of CHO1 transcription (14). This has led us to consider two models for Ume6p positive regulation. One model predicts that Ume6p directly interacts with the INO2 promoter and associates with a novel protein to activate INO2 transcription. However, we have not been able to detect binding of Ume6p to the INO2 promoter (Fig. 6). Moreover, a comprehensive deletion analysis of the INO2 promoter has identified only two positive cis-acting sequences; a TATA-like element and a UASINO element (D.A.Eiznhamer and J.M.Lopes, unpublished data). Deletion of these elements from the INO2 promoter completely eliminates INO2 transcription, which is a very different phenotype from that observed in ume6 mutant strains. Furthermore, neither of these two positive cis-acting sites contain sequences which conform to the Ume6p binding site (51). Thus, we favor a second model that predicts an indirect effect of Ume6p on INO2 transcription through an intermediate positive regulatory gene. This intermediate gene may be present among the rum mutants described in this manuscript. Cloning of the RUM gene(s) coupled with mobility shift assays using the INO2 promoter and extracts from rum mutant strains will help distinguish between the models. Furthermore, we have isolated a larger collection of rum mutants using an INO2 promoter–HIS3 reporter which will also help define the UME6-mediated regulation of INO2 gene expression.
It is a little more difficult to reconcile the effect of the ume6 mutant on INO1 expression. This promoter contains both the URS1 element and two UASINO elements (45,52). Consequently, the ume6 mutant should result in increased expression through the URS1 element and decreased expression through the UASINO element. The fact that only the former phenotype is observed suggests that the URS1-mediated regulation is epistatic to UASINO-mediated regulation. Alternatively, the lowered amount of Ino2p in the ume6Δ mutant may be sequestered to the UASINO elements of the INO1 promoter. In support of this, it is known that the UASINO elements of the INO1 promoter are able to drive significantly greater levels of lacZ expression relative to the UASINO element from the CHO1 promoter (40; Fig. 2).
The consequence of mutations in the three regulatory genes on phospholipid composition had not been previously examined. The rpd3 and sin3 mutants displayed novel profiles. The rpd3 mutant yielded a substantial decrease in the level of PC (Table 1). This suggests that rpd3 may affect expression of the CHO2 or OPI3 genes in a manner different from its effect on the CHO1 gene. Alternatively, the rpd3 mutant may affect expression of genes in the salvage pathway or the PC turnover pathway (7,8). One of the more striking phenotypes is the complete absence of PE in the sin3 mutant strain (Table 1). It has been demonstrated that sin3 mutant strains overexpress the CHO1, CHO2 and OPI3 genes (27). One possible explanation for the phenotype is that overexpression of the CHO2 and OPI3 genes depletes the cell of PE at the expense of PC. Another possibility is that the PSD1 and PSD2 genes, which convert PS to PE, are down-regulated. These two genes had not been cloned when the sin3 effect on CHO1, CHO2 and OPI3 transcription was tested (7,27). Regardless of the mechanism, the finding of a condition where PE is completely absent from the membrane is interesting and raises the possibility that PE may be required for certain biological processes. Interestingly, Mcm1p and Sin3p function as positive regulators in the mating response pathway (29) and the PSD1 and PSD2 promoters contain Mcm1p binding sites (J.M.Lopes, unpublished observation). It remains to be determined if PE levels play a role in biological processes such as mating.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Kyle Gardenour and Jay Koepke for helpful discussions and critical reading of the manuscript. We thank Kevin Struhl (Harvard Medical School) and Aaron Mitchell (Columbia University) for providing strains and Randy Strich (Fox Chase Cancer Center) for providing the MBP–Ume6p-expressing strain. This work was supported by a grant (RPG-97-002-01) from the American Cancer Society to J.M.L.
REFERENCES
- 1.Kadosh D. and Struhl,K. (1997) Cell, 89, 365–371. [DOI] [PubMed] [Google Scholar]
- 2.Kadosh D. and Struhl,K. (1998) Mol. Cell. Biol., 18, 5121–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rundlett S.E., Carmen,A.A., Suka,N., Turner,B.M. and Grunstein,M. (1998) Nature, 392, 831–835. [DOI] [PubMed] [Google Scholar]
- 4.Grunstein M. (1997) Nature, 389, 349–352. [DOI] [PubMed] [Google Scholar]
- 5.Strich R., Surosky,R.T., Steber,C., Dubois,E., Messenguy,F. and Esposito,R.E. (1994). Genes Dev., 8, 796–810. [DOI] [PubMed] [Google Scholar]
- 6.Luche R., Smart,W.C., Sumrada,R.A. and Cooper,T.G. (1993) Mol. Cell. Biol., 13, 5749–5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenberg M.L and Lopes,J.M. (1996) Microbiol. Rev., 60, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Henry S.A. and Patton-Vogt,J.L. (1998) Prog. Nucleic Acids Res., 61, 133–179. [DOI] [PubMed] [Google Scholar]
- 9.Ambroziak J. and Henry,S.A. (1994) J. Biol. Chem., 269, 15344–15349. [PubMed] [Google Scholar]
- 10.Nikoloff D.M. and Henry,S.A. (1994) J. Biol. Chem., 269, 7402–7411. [PubMed] [Google Scholar]
- 11.Amati B. and Land,H. (1994) Curr. Opin. Genes Dev., 4, 102–108. [DOI] [PubMed] [Google Scholar]
- 12.Ashburner B.P. and Lopes,J.M. (1995) Proc. Natl Acad. Sci. USA, 92, 9722–9726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ashburner B.P. and Lopes,J.M. (1995) Mol. Cell. Biol., 15, 1709–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jackson J.C. and Lopes,J.M. (1996) Nucleic Acids Res., 24, 1322–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bowdish K.S. and Mitchell,A.P. (1993) Mol. Cell. Biol., 13, 2172–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buckingham L.E., Wang,H.-T., Elder,R.T., McCarroll,R.M., Slater,M.R. and Esposito,R.E. (1990) Proc. Natl Acad. Sci. USA, 87, 9406–9410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vershon A.K., Hollingsworth,N.M. and Johnson,A.D. (1992) Mol. Cell. Biol., 12, 3706–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park H.-O. and Craig,E.A. (1989) Mol. Cell. Biol., 9, 2025–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park H.-D., Luche,R.M. and Cooper,T.G. (1992) Nucleic Acids Res., 20, 1909–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bowdish K.S., Yuan,H.E. and Mitchell,A.P. (1995) Mol. Cell. Biol., 15, 2955–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steber C.M. and Esposito,R.E. (1995) Proc. Natl Acad. Sci. USA, 92, 12490–12494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sweet D.H., Jang,Y.K. and Sancar,G.B. (1997) Mol. Cell. Biol., 17, 6223–6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rubin-Bejerano I., Mandel,S., Robzyk,K. and Kassir,Y. (1996) Mol. Cell. Biol., 16, 2518–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strich R., Slater,M.R. and Esposito,R.E. (1989) Proc. Natl Acad. Sci. USA, 86, 10018–10022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sternberg P.W., Stern,M.J., Clark,I. and Herskowitz,I. (1987) Cell, 48, 567–577. [DOI] [PubMed] [Google Scholar]
- 26.Vidal M., Buckley,A., Yohn,C., Hoeppner,D. and Gaber,R. (1995) Proc. Natl Acad. Sci. USA, 92, 2370–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hudak K., Lopes,J.M. and Henry,S.A. (1994) Genetics, 136, 475–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slekar K.H. and Henry,S.A. (1995) Nucleic Acids Res., 23, 1964–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H., Reynolds-Hager,L. and Stillman,D.J. (1994) Mol. Gen. Genet., 245, 675–685. [DOI] [PubMed] [Google Scholar]
- 30.Tzamarias D. and Struhl,K. (1994) Nature, 369, 758–761. [DOI] [PubMed] [Google Scholar]
- 31.Kelly B.L. and Greenberg,M.L. (1990) Biochim. Biophys. Acta, 1046, 144–150. [DOI] [PubMed] [Google Scholar]
- 32.Bailis A.M., Lopes,J.M., Kohlwein,S.D. and Henry,S.A. (1992) Nucleic Acids Res., 20, 1411–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guarente L. and Ptashne,M. (1981) Proc. Natl Acad. Sci. USA, 78, 2199–2203. [Google Scholar]
- 34.Myers A.M., Tzagoloff,A., Kinney,D.M. and Lusty,C.J. (1986) Gene, 45, 299–310. [DOI] [PubMed] [Google Scholar]
- 35.Chen D.-C., Yang,B.-C. and Kuo,T.-T. (1992) Curr. Genet., 21, 83–84. [DOI] [PubMed] [Google Scholar]
- 36.Greenberg M.L., Reiner,B. and Henry,S.A. (1982) Genetics, 100, 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGee T.P., Skinner,H.B. and Bankaitis,V.A. (1994) J. Bacteriol., 176, 6861–6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elion E.A. and Warner,J.R. (1984) Cell, 39, 663–673. [DOI] [PubMed] [Google Scholar]
- 39.Hirsh J.P. and Henry,S.A. (1986) Mol. Cell. Biol., 6, 3320–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lopes J.M., Hirsch,J.P., Chorgo,P.A., Schulze,K.L. and Henry,S.A. (1991) Nucleic Acids Res., 19, 1687–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Atkinson K.D., Jensen,B., Kolat,A.I., Storm,E.M., Henry,S.A. and Fogel,S. (1980) J. Bacteriol., 141, 558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Atkinson K.D. and Ramirez,R.M. (1984) J. Bacteriol., 160, 80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanson B.A. and Lester,R.L. (1980) J. Lipid Res., 21, 309–315. [PubMed] [Google Scholar]
- 44.Steiner M.R. and Lester,R.L. (1972) Biochim. Biophys. Acta, 260, 222–243. [DOI] [PubMed] [Google Scholar]
- 45.Lopes J.M., Schulze,K.L., Yates,J.W., Hirsch,J.P. and Henry,S.A. (1993) J. Bacteriol., 175, 4235–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swede M.J., Hudak,K.A., Lopes,J.M. and Henry,S.A. (1992) Methods Enzymol., 209, 21–34. [DOI] [PubMed] [Google Scholar]
- 47.Bailis A.M., Poole,M.A., Carman,G.M. and Henry,S.A. (1987) Mol. Cell. Biol., 7, 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kelley M.J., Bailis,A.M., Henry,S.A. and Carman,G.M. (1988) J. Biol. Chem., 263, 18078–18085. [PubMed] [Google Scholar]
- 49.Ayer D.E., Lawrence,Q.A. and Eisenman,R.N. (1995) Cell, 80, 767–776. [DOI] [PubMed] [Google Scholar]
- 50.Schreiber-Agus N., Chin,L., Chen,K., Torres,R., Rao,G., Guida,P., Akoultchi,A.I. and DePinho,R.A. (1994) Cell, 80, 777–786. [DOI] [PubMed] [Google Scholar]
- 51.Anderson S.F., Steber,C.M., Esposito,R.E. and Coleman,J.E. (1995) Protein Sci., 4, 1832–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koipally J., Ashburner,B.P., Bacchawat,N., Gill,T., Hung,G., Henry,S.A. and Lopes,J.M. (1996) Yeast, 12, 653–665. [DOI] [PubMed] [Google Scholar]