

SUMMARY
Ubiquitination of proliferating cell nuclear antigen (PCNA) at lysine 164 (K164) activates DNA damage tolerance pathways. Currently, we lack a comprehensive understanding of how PCNA K164 ubiquitination promotes genome stability. To evaluate this, we generated stable cell lines expressing PCNAK164R from the endogenous PCNA locus. Our data reveal that the inability to ubiquitinate K164 causes perturbations in global DNA replication. Persistent replication stress generates under-replicated regions and is exacerbated by the DNA polymerase inhibitor aphidicolin. We show that these phenotypes are due, in part, to impaired Fanconi anemia group D2 protein (FANCD2)-dependent mitotic DNA synthesis (MiDAS) in PCNAK164R cells. FANCD2 mono-ubiquitination is significantly reduced in PCNAK164R mutants, leading to reduced chromatin association and foci formation, both prerequisites for FANCD2-dependent MiDAS. Furthermore, K164 ubiquitination coordinates direct PCNA/FANCD2 colocalization in mitotic nuclei. Here, we show that PCNA K164 ubiquitination maintains human genome stability by promoting FANCD2-dependent MiDAS to prevent the accumulation of under-replicated DNA.
Graphical Abstract
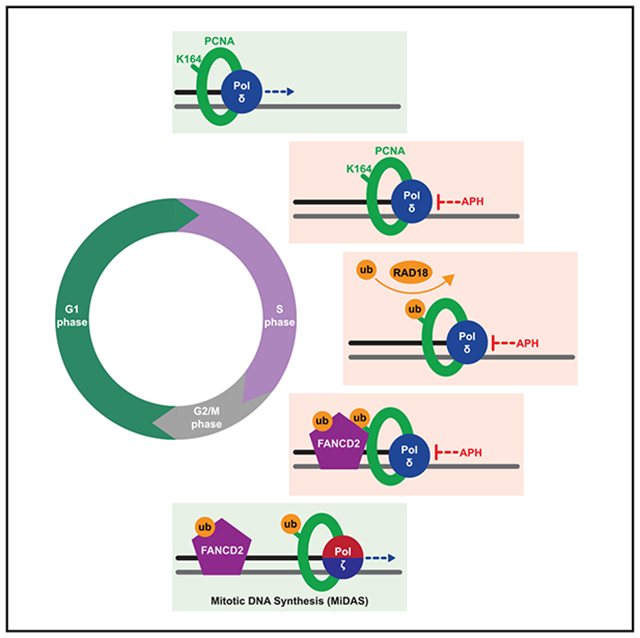
In brief
Leung et al. show that FANCD2-dependent mitotic DNA synthesis (MiDAS) in non-transformed human cells relies on PCNA lysine 164 (K164) ubiquitination. Specifically, PCNA K164 ubiquitination promotes FANCD2 ubiquitination and recruitment to under-replicated regions and coordinates interactions in mitotic nuclei to promote MiDAS.
INTRODUCTION
Maintenance of genome integrity is an intricate process that requires an extended protein network and coordination of multiple cellular pathways.1 Proper DNA replication is essential for genome maintenance and ensures precise duplication without leaving sequences under-replicated or replicated more than once. Endogenous and exogenous sources of DNA damage can impede DNA synthesis, stall replication forks, and generate aberrant fork structures that can give rise to chromosomal alterations.2,3 In response to DNA lesions, proliferating cell nuclear antigen (PCNA) is ubiquitinated at the conserved lysine 164 (K164) residue, activating DNA damage tolerance (DDT) pathways.4 Mono-ubiquitination at K164 by the E3 ubiquitin ligase radiation sensitive 18 (RAD18) activates the translesion synthesis (TLS) DDT pathway.5,6 TLS is catalyzed by specialized low-fidelity DNA polymerases to bypass DNA lesions and fill single-stranded DNA (ssDNA) gaps.7 Mono-ubiquitinated PCNA can be further modified by K63-linked poly-ubiquitin chains to activate the template-switching (TS) DDT pathway. TS utilizes a recombination-like mechanism by which the nascent DNA of the sister chromatid serves as a template for replication.8 Given the importance of PCNA K164 ubiquitination, it is not surprising that mutating this residue renders cells hypersensitive to DNA damage.9-11 However, how this modification functions in maintaining human genome stability under unperturbed conditions is not understood.
Regions of the genome that are particularly reliant on DDT pathways include late-replicating or so-called “hard-to-replicate” loci, including common fragile sites (CFSs). Failure to complete DNA replication leads to the accumulation of late replication intermediates and under-replicated regions. As the cell cycle proceeds, these regions are marked by the formation of Fanconi anemia (FA) group D2 protein (FANCD2) foci.12,13 If left unresolved, these aberrant structures can cause DNA bridges in anaphase and/or are sequestered into p53-binding protein 1 (53BP1) nuclear bodies (NBs) during the subsequent G1 phase. One mechanism to resolve these issues and to ensure proper chromosome segregation is mitotic DNA synthesis (MiDAS).14,15 MiDAS utilizes a recombination-like mechanism similar to break-induced replication (BIR) to complete DNA synthesis at under-replicated loci in G2/M phase. Human cancer cells can utilize RAD52- or FANCD2-dependent MiDAS15; however, we demonstrated that in non-transformed (i.e., non-cancerous) human cells, the RAD52-dependent mechanism is absent, and instead, MiDAS relies on FANCD2.16,17 Interestingly, a recent study found that PCNA K164 ubiquitination by RAD18 is required for MiDAS in cancer cell lines but did not investigate its necessity in non-transformed human cells.18
In this study, we demonstrate that PCNA K164 ubiquitination is critical for accurate DNA replication and genome stability in non-transformed human cells. Loss of PCNA K164 ubiquitination in human telomerase reverse transcriptase (hTERT) immortalized retinal pigment epithelium-1 (RPE-1) (referred to subsequently as RPE-1) cells disrupts global DNA replication, causing chronic replication stress. This leads to under-replicated regions that are transmitted to daughter cells and marked by 53BP1 NBs. The repair of these regions by MiDAS is compromised and is directly linked to decreased FANCD2 mono-ubiquitination and chromatin association. Finally, we show that K164 ubiquitination is required for PCNA/FANCD2 colocalization in mitotic nuclei. Taken together, our data show that PCNA ubiquitination at K164 is not only important for progressive S-phase DNA synthesis but promotes FANCD2-dependent MiDAS to ensure complete genome duplication prior to cell division.
RESULTS
Stable PCNAK164R mutants are severely sensitive to lesion-induced DNA damage
Previous studies investigating the role of PCNA ubiquitination in human cells relied on ectopic expression of PCNAK164R or PCNA-ubiquitin fusions in cells expressing wild-type PCNA, with or without knockdown of endogenous PCNA.10,11,18 To understand the importance of PCNA K164 modification when stably expressed from the endogenous locus, we utilized CRISPR-Cas9-mediated gene targeting to knock in an A>G mutation in the codon for K164 of PCNA in RPE-1 cells (Figure S1A). PCR analyses identified mono- and biallelic targeting of PCNA (Figures S1B and S1C), and sequencing confirmed one homozygous PCNAK164R (2B10) and two hemizygous PCNAK164R/− (A1 and B1) clonal cell lines. Regardless of whether mutant cell lines carried one or two functional PCNAK164R alleles, both total PCNA expression (Figure S1D) and the amount of chromatin-bound PCNA was comparable to wild type (Figure 1A). These observations demonstrate that a single PCNAK164R allele is sufficient to maintain wild-type expression levels in RPE-1 cells. Finally, western blot analyses of PCNA ubiquitination after UV irradiation showed a dose-dependent increase in wild-type cells but not in the PCNAK164R cell lines (Figure 1A), functionally validating these mutant cell lines.
Figure 1. PCNAK164R mutant cell lines have increased sensitivity to DNA damage.

(A) Western blot analyses of chromatin-associated PCNA, ubiquityl-PCNA (K164), phospho-RPA32 (S33), and γH2AX, with or without 20 and 40 J/m2 UV treatment, with histone H2AX as the loading control from wild-type RPE-1 and PCNAK164R cells. Quantified band intensities were normalized to loading controls.
(B) Western blot analyses of whole-cell extracts from wild-type RPE-1 and PCNAK164R cells for phospho-p53 (S15), p53, and p21, with or without 20 and 40 J/m2 UV treatment, with GAPDH or tubulin as the loading control. Quantified intensities of phospho-p53 (S15), p53, and p21 levels were normalized to loading controls.
(C) Comparison of drug sensitivity comparing average percentage of viability in RPE-1 wild-type and PCNAK164R cell lines. Each drug and concentration tested is indicated. Error bars indicate standard deviation, and statistical significance was calculated using Student’s t test with *p < 0.05, **p < 0.01, and ***p < 0.001; n = 9–12 replicate wells across three biological replicates for all data points.
(D) Representative cell-cycle distributions based on DNA content (DAPI) of RPE-1 wild-type and PCNAK164R cell lines treated with MMC (20 nM) or MMS (20 μM) for 48 h.
(E) Cell-cycle distributions of RPE-1 wild-type and PCNAK164R cell lines treated with MMC or MMS from three biological replicates. Percentage of each population in G1 (green), S (purple), or G2/M phase (gray) is shown. Error bars indicate standard deviation, and statistical significance was calculated using Student’s t test with *p < 0.05, **p <0.01, and ***p < 0.001; n = 6 replicate wells across three biological replicates. For wild type, statistics represent the comparison of each cell-cycle phase in drug-treated (MMC, MMS) versus untreated (UT) cell lines. For PCNAK164R cell lines, statistics represent the comparison of each cell-cycle phase versus wild type within each treatment group (UT, MMC, MMS).
(F) Histogram (left, middle) and quantification of mean fluorescent intensity (right) of EdU staining of S-phase cells from RPE-1 wild-type (blue) and PCNAK164R cells (maroon) treated with MMC and MMS; n = 6 across three biological replicates. Error bars indicate standard deviation, and significance was calculated using two-way ANOVA with *p < 0.05, **p < 0.01, and ***p < 0.001.
To understand the effects of stable expression of PCNAK164R on the response to UV exposure, we examined phosphorylated replication protein A (pRPA32) at S33 and phosphorylated histone H2AX (γH2AX) at S139 as readouts for an ataxia telangiectasia and Rad3-related (ATR)-mediated replication stress response and DNA double-stranded breaks (DSBs), respectively. We found a dose-dependent increase in pRPA32 and γH2AX in all cell lines, with significantly elevated levels in the PCNAK164R mutants (Figure 1A). Due to the observed increases in replication stress and DSB markers, we investigated the expression of p53, as well as its phosphorylated form, p-p53 (S15). Although both markers showed a modest increase in the untreated mutant cell lines, the p53 target p21 was not significantly activated (Figure 1B). These observations suggest that the inability to ubiquitinate K164 confers increased susceptibility to UV-induced replication stress but that endogenous DNA damage does not trigger a robust checkpoint response. Next, we wanted to characterize DNA damage resistance in stable PCNAK164R cell lines in response to other DNA lesions. Utilizing luminescent cell viability assays, we observed that PCNAK164R mutants were highly sensitive to methyl methanesulfonate (MMS), mitomycin C (MMC), and 4-nitroquinoline 1-oxide (4NQO) (Figure 1C). To assess the effects of MMC and MMS on the cell cycle, we utilized quantitative flow cytometry. We determined the cell-cycle distribution using DAPI staining for total DNA content in combination with a 30 min 5-ethynyl-2′-deoxyuridine (EdU) pulse to label S-phase cells. These analyses revealed a G2/M-phase arrest in PCNAK164R mutants when challenged with MMC or MMS (Figures 1D and 1E) and a significant decrease in S-phase DNA synthesis (Figure 1F). Taken together, these data suggest that PCNA K164 post-translational modification, presumably ubiquitination, is required in RPE-1 cells for tolerance of lesion-induced replication stress, consistent with previous literature.4
Unperturbed DNA replication and cell-cycle progression are altered in PCNAK164R mutants
Although markers of replication stress and DNA damage were not significantly increased without exogenous genotoxic stress, we noted a growth defect in PCNAK164R mutants under unperturbed conditions (Figure 2A). To understand this defect, we utilized complementary techniques to evaluate DNA replication and cell-cycle progression. First, using single-cell quantitative chromatin flow cytometry, we observed small changes in the cell-cycle distribution of PCNAK164R cells compared to wild type (Figures 2B and S2A), with an increase in the G2/M-phase population and a proportional decrease in the G1-phase population. PCNAK164R mutants also displayed a mild decrease in S-phase EdU incorporation (Figures 2C and S2B) and MCM2 loading during late G1 phase (Figures 2D and S2C), and although small, these changes were statistically significant. However, western blot analyses of these mutants did not detect a significant change in chromatin-bound MCM2 or ORC2 (Figure S2D), suggesting that the differences observed by flow cytometry may be a result of a particularly stringent gating strategy (Figure S2C). Similar to our RPE-1 cells, when we compared transformed wild-type 293T cells with PCNAK164R mutants, shown previously to express equivalent amounts of wild-type or mutant PCNA,19 we found small but detectable perturbations in cell-cycle distribution, S-phase EdU incorporation, and MCM2 loading in late G1 phase (Figure S3). Second, we utilized single-molecule DNA combing analyses, outlined schematically in Figure S2E, to assess DNA replication in RPE-1 cells more closely. Although replication fork speed was unaltered (Figure 2E), we observed increased interorigin distances (IODs) in PCNAK164R mutants (Figure 2F). The average IOD in wild-type cells was ~102 kb, whereas the average in PCNAK164R mutants was increased to ~140 kb (Figure 2F), which equates to at least 30% fewer origins. Taken together, these analyses suggest that DNA replication and cell-cycle progression in PCNAK164R mutants are modestly disrupted under unperturbed conditions. Consistent with this, using a combination of Annexin V and propidium iodide staining, we observed a small increase in apoptosis in two out of three PCNAK164R mutants in comparison to wild type, although only one cell line showed a statistically significant increase (Figures 2G and S2F). Based on these data, we conclude that the inability to ubiquitinate PCNA at K164 causes mild, chronic replication stress that reduces cell proliferation and may result in cell death.
Figure 2. The PCNA-K164R mutation disrupts DNA replication without exogenous genotoxic stress.

(A) Average cell proliferation rate in PCNAK164R cell lines normalized to wild type. For each cell line, n = 9 wells across three biological replicates. Error bars indicate standard deviation, and significance was calculated using one-way ANOVA with *p < 0.05, **p < 0.01, and ***p < 0.001.
(B) Cell-cycle distribution of RPE-1 wild-type and PCNAK164R cell lines from four biological replicates. Percentage of each population in G1 (green), S (purple), or G2/M phase (gray) is shown. Error bars indicate standard deviation, and significance was calculated using Student’s t test with *p < 0.05, **p < 0.01, and ***p < 0.001.
(C) Quantification of mean fluorescent intensity of EdU staining in S-phase cells from RPE-1 wild-type (blue) and PCNAK164R cells (maroon); n = 12 across four biological replicates. Error bars indicate standard deviation, and significance was calculated using Student’s t test with *p < 0.05, **p < 0.01, and ***p < 0.001.
(D) Quantification of the percentage of MCM2-stained cells in late G1 from RPE-1 wild-type (blue) and PCNAK164R cells (maroon); n = 12 across four biological replicates. Error bars indicate standard deviation, and significance was calculated using Student’s t test with ***p < 0.001.
(E) Fork speed quantification from two biological replicates in RPE-1 wild-type (blue) and PCNAK164R cells (maroon). Average IOD and number (n) quantified are listed. Significance was calculated by Kruskal-Wallis with Dunn’s multiple comparison test with ***p < 0.001.
(F) IOD quantification from two biological replicates in RPE-1 wild-type (blue) and PCNAK164R cells (maroon). Average IOD and number (n) quantified are listed. Significance was calculated by Kruskal-Wallis with Dunn’s multiple comparison test with ***p < 0.001.
(G) Percentage of apoptotic cells in RPE-1 wild-type (blue) and PCNAK164R populations. Error bars indicate standard deviation, and significance was calculated by Kruskal-Wallis with Dunn’s multiple comparison test with *p < 0.05; n = 12 replicates across four biological replicates.
PCNAK164R mutants display DNA under-replication and suppressed MiDAS activation
Chronic replication stress primarily affects late-replicating and hard-to-replicate regions.20 The inability to complete replication of these regions can generate late-replicating intermediates (LRIs) and under-replicated DNA. The persistence of LRIs in mitosis may result in the formation of chromatin bridges during anaphase.21 If these bridges persist, they can disrupt the spindle apparatus and cause aberrant chromosome segregation, including lagging chromosomes.21 When we analyzed the frequency of anaphase abnormalities including DAPI-positive bridges and lagging chromosomes, these events were extremely rare in wild-type and PCNAK164R mutant RPE-1 cells (Figure 3A). However, exacerbating replication stress with a low dose (300 nM) of the DNA polymerase inhibitor aphidicolin (APH) caused a significant increase in anaphase abnormalities, especially in PCNAK164R mutants (Figure 3A). One fate of under-replicated regions that are inherited by daughter cells is their sequestration into G1-phase 53BP1 NBs.22 Quantification of 53BP1 NBs in G1 nuclei revealed that PCNAK164R mutants had a 3-fold increase in 53BP1 NBs under unperturbed conditions (Figure 3B). Interestingly, low-dose APH treatment (300 nM) led to an increase in 53BP1 NBs in wild-type cells but not PCNAK164R mutants (Figure 3B). A similar phenotype was observed when we performed the same experiment in wild-type and PCNAK164R 293T cell lines (Figure S4A). These data suggest that PCNAK164R mutants have reached the maximal number of 53BP1 NBs under unperturbed conditions and/or that these mutants upregulate alternative mechanisms to resolve under-replication in response to APH. Unexpectedly, RPE-1 PCNAK164R mutants were not more sensitive to APH than wild-type cells (Figure 3C). A similar response was observed following exposure to hydroxyurea (HU) (Figure S4B). These data show that although K164 ubiquitination is broadly important for the cellular response to replication stress, it is only acutely essential for viability in response to certain, chemically modified, lesions. Furthermore, we speculate that the ability of PCNAK164R mutants to tolerate aberrant chromosome structures, at least over relatively few population doublings, likely comes at the expense of reduced genome stability.
Figure 3. Phenotypes associated with DNA under-replication are increased in PCNAK164R mutants.

(A) (Top) Representative images of DAPI-stained chromosomes during anaphase: (left) normal, (middle) chromatid bridge, and (right) lagging chromosome. Scale bar, 5 μm. (Bottom) Percentage of abnormal anaphases in UT and APH-treated cells in wild-type (blue) and PCNAK164R lines (maroon). Number (n) of anaphases quantified is listed. Error bars indicate standard deviation, and significance was calculated using Student’s t test with *p < 0.05.
(B) (Top) Image of 53BP1 NB and cyclin A staining in RPE-1 cell lines. DAPI (blue), 53BP1 NB (green), and cyclin A (pink) are indicated. Scale bar, 20 μm. (Bottom) 53BP1 NB quantification of UT and APH-treated cells in wild type (blue) and PCNAK164R lines (maroon). Number (n) of nuclei quantified is listed. Error bars indicate standard deviation, and significance was calculated using Kruskal-Wallis with Dunn’s multiple comparison test.
(C) Comparison of APH sensitivity based on the average percentage viability at each APH concentration in RPE-1 wild-type and PCNAK164R cell lines. Concentrations tested are indicated. Error bars indicate standard deviation, and statistical significance was calculated using Student’s t test when comparing wild-type cells with each respective PCNAK164R cell line with *p < 0.05, **p < 0.01, and ***p < 0.001; n = 9–12 replicate wells across three biological replicates for all data points.
An increase in under-replicated genomic regions can lead to the formation of FANCD2 foci during, and activation of MiDAS as the cells’ final attempt to complete DNA replication prior to, cell division.16,18 Whereas transformed cells utilize RAD52 and FANCD2 for MiDAS, non-transformed cell lines do not require RAD52 and are dependent on FANCD2.16 To visualize APH-induced MiDAS, we pulse labeled cells with EdU and quantified the number of EdU and FANCD2 foci in nuclei that stained positive for histone H3 phosphorylation at serine 10 (pH3-S10), a hallmark of mitotic chromosomes (Figure S5A).15,16 Given the altered replication dynamics in PCNAK164R cells, we suspected that these mutants would upregulate MiDAS to minimize under-replicated DNA passing through mitosis. Surprisingly, RPE-1 PCNAK164R cells had significantly fewer FANCD2 foci (~2- to 3-fold) and a corresponding decrease in EdU foci (~1.5- to 3.5-fold) compared to wild-type cells (Figures 4A-4C). Similar results were seen in 293T PCNAK164R mutant cells (Figures 4D and 4E), suggesting a shared aspect of FANCD2-dependent MiDAS regulation in transformed and non-transformed cell types.
Figure 4. FANCD2-dependent MiDAS is impaired when PCNA K164 cannot be ubiquitinated.

(A) Representative images of phospho-H3 S10-stained nuclei (blue), EdU foci (green), and FANCD2 (red) foci from APH-treated RPE-1 wild-type and PCNAK164R cell lines. EdU foci almost exclusively (>95%) colocalized with FANCD2 foci. Scale bar, 5 μm.
(B) FANCD2 foci quantification from two biological replicates in RPE-1 wild-type (blue), PCNAK164R (maroon), RAD18−/− (salmon), and RAD18−/−;PCNAK164R (pink) cells treated with 300 nM APH. Number (n) of nuclei quantified is listed. Significance was calculated by Kruskal-Wallis with Dunn’s multiple comparison test with ***p < 0.001. FANCD2 foci overlapped with EdU foci in 98% of nuclei.
(C) EdU foci quantification from two biological replicates in RPE-1 wild-type (blue), PCNAK164R (maroon), RAD18−/− (salmon), and RAD18−/−;PCNAK164R (pink) cells treated with 300 nM APH. Number (n) of nuclei quantified is listed. Significance was calculated by Kruskal-Wallis with Dunn’s multiple comparison test with ***p < 0.001.
(D) FANCD2 foci quantification from two biological replicates in 293T wild-type (blue) and PCNAK164R cells (maroon) treated with 450 nM APH. Number (n) of nuclei quantified is listed. Significance was calculated by one-way ANOVA with Tukey’s multiple comparison test with ***p < 0.001.
(E) EdU foci quantification from two biological replicates in 293T wild-type (blue) and PCNAK164R cells (maroon) treated with 450 nM APH. Number (n) of nuclei quantified is listed. Significance was calculated by one-way ANOVA with Tukey’s multiple comparison test with ***p < 0.001.
(F) Western blot analyses of chromatin associated PCNA, with or without 40 J/m2 UV treatment, with histone H2AX as the loading control from RPE-1 wildtype, PCNAK164R, and reverted PCNAK164 cells.
(G) FANCD2 foci quantification from two biological replicates in RPE-1 wild-type (blue), PCNAK164R (maroon), and reverted PCNAK164 (purple) cells treated with 300 nM APH. Number (n) of nuclei quantified is listed. Significance was calculated by Kruskal-Wallis with Dunn’s multiple comparison test with ****p < 0.001.
(H) EdU foci quantification from two biological replicates in RPE-1 wild-type (blue), PCNAK164R (maroon), and reverted PCNAK164 (purple) cells treated with 300 nM APH. Number (n) of nuclei quantified is listed. Significance was calculated by Kruskal-Wallis with Dunn’s multiple comparison test with ****p < 0.001.
To further evaluate the contribution of PCNA K164 ubiquitination in non-transformed cells, we generated RPE-1 RAD18−/− single and RAD18−/−:PCNAK164R double mutants (Figure S5B) and examined them for mitotic EdU and FANCD2 foci. RAD18−/− cells displayed approximately 40% fewer foci compared to wild type. However, RAD18−/−:PCNAK164R double mutants had similar levels of FANCD2 and EdU foci as the PCNAK164R single mutants, consistent with RAD18 acting upstream of PCNA ubiquitination (Figures 4A-4C and S5B). The number of FANCD2 foci in RAD18−/− cells was elevated in comparison with PCNAK164R and RAD18−/−:PCNAK164R double mutants (Figure 4B). These results agree with reports that three additional E3 ubiquitin ligases, ring finger protein 8 (RNF8), Cullin-4-RING-ligase (CRL4)-Ddb1-Cdt2 (CRL4Cdt2), and ring finger and WD repeat domain 3 (RFWD3), are capable of mono-ubiquitinating PCNA.23-25 Therefore, in the absence of RAD18, PCNA ubiquitination by alternative E3 ligases may partially activate MiDAS. In contrast, when PCNA is unable to be ubiquitinated at K164, MiDAS is severely compromised.
To demonstrate that the MiDAS defects in RPE-1 cell lines were due specifically to the inability to ubiquitinate PCNA K164, we utilized CRISPR-Cas9-mediated gene targeting to revert the mutation in the codon for K164 at the endogenous PCNA locus in RPE-1 cells (Figure S5C). Western blot analyses of these reverted cell lines following UV exposure demonstrated a complete restoration in the ability to ubiquitinate PCNA K164 (Figure 4F). Furthermore, we found that both FANCD2 foci formation and EdU incorporation in pSer10-histone H3-positive nuclei were rescued in reverted cell lines (Figures 4G and 4H). Taken together, these data indicate that PCNA K164 plays a critical role in activating FANCD2-dependent MiDAS in response to under-replicated DNA.
PCNA K164 is an important regulator of FANCD2 mono-ubiquitination to promote MiDAS
FANCD2 has emerged as a central player in MiDAS, particularly in non-transformed human cell lines.16 The mono-ubiquitination of FANCD2, catalyzed by the E3 ligase FANCL, is needed for robust DNA association26,27 and is a prerequisite for focus formation. To confirm this, we performed MiDAS experiments in FANCD2 and FANCL null RPE-1 cell lines (Figures S6A and S6B). As expected, FANCD2 foci were undetectable in nuclei of FANCD2−/− or FANCL−/− mutants (Figure S6C). However, MiDAS was not completely abolished, implying that an alternative FANCD2-independent pathway exists in RPE-1 cells. Next, we examined whether K164 ubiquitination correlated with FANCD2 mono-ubiquitination. Under unperturbed conditions, FANCD2 mono-ubiquitination was drastically reduced in comparison to wild type in asynchronous RPE-1 PCNAK164R cells (Figure 5A) but not in 293T PCNAK164R cells (Figure S6E). Upon low-level APH-induced replication stress identical to that used to evaluate MiDAS, FANCD2 mono-ubiquitination was elevated in RPE-1 and 293T wild-type cells but not in the PCNAK164R mutants (Figures 5A and S6E). This phenotype was completely rescued in reverted RPE-1 clones (Figure 5B), confirming the link between PCNA K164 ubiquitination and FANCD2 ubiquitination in response to replication stress.
Figure 5. PCNA K164 is required for robust FANCD2 ubiquitination and chromatin association.

(A) Western blot analyses of FANCD2 with Ku86 or Tubulin as the loading control in nuclear fractions (top) or whole-cell extracts (bottom) from RPE-1 wild-type and PCNAK164R cells with or without 300 nM APH treatment. Quantified band intensities were normalized to loading controls.
(B) Western blot analyses of FANCD2 with Ku86 as the loading control in whole-cell extracts (bottom) from RPE-1 wild-type, PCNAK164R, and reverted PCNAK164 cells with or without 300 nM APH treatment. Quantified band intensities were normalized to loading controls.
(C) Representative cell-cycle distributions based on propidium iodide staining from RPE-1 wild-type, PCNAK164R, and reverted PCNAK164 cells with or without 300 nM APH treatment.
(D) Cell-cycle distributions of RPE-1 wild-type, PCNAK164R and reverted PCNAK164 cell lines from two biological replicates. Percentage of each population in G1 (green), S (purple), or G2/M phase (gray) is shown. Error bars indicate standard deviation, and significance was calculated using Student’s t test with *p < 0.05, **p < 0.01, and ***p < 0.001. Statistics in white text represent comparisons between each cell-cycle phase versus wild type in each treatment condition. Statistics in yellow text represent comparisons between each cell-cycle phase in reverted PCNAK164 cell lines versus their parental PCNAK164R mutant with 300 nM APH treatment.
The observed decrease in FANCD2 mono-ubiquitination was not due to an overall reduction in FANCD2 expression (Figures 5A and S6E). Since FANCD2 is mono-ubiquitinated in S phase,28 and since the S-phase distribution of wild-type and PCNAK164R mutants was similar (Figure 2B), these differences were likely not due to cell-cycle variations. To confirm this, we analyzed the cell-cycle distribution of wild-type, PCNAK164R, and reverted cell lines following low-dose APH exposure (300 nM). As expected, the percentage of each population in S phase was nearly identical across all cell lines (Figures 5C and 5D). Importantly, the accumulation in G1 phase observed in PCNAK164R cells following APH treatment was rescued in the reverted cell lines (Figure 5D). These data are consistent with the notion that PCNA K164 ubiquitination promotes FANCD2 ubiquitination and chromatin loading in S phase to facilitate DNA synthesis in mitosis. Finally, to understand the relationship between PCNA and FANCD2 specifically during MiDAS, we utilized proximity ligation assays (PLAs) to quantify close-proximity protein colocalization in pH3-S10-positive nuclei (Figure 6A). Interestingly, the number of PLA foci in wild-type nuclei was significantly higher than in PCNAK164R mutants, which was identical to the FANCD2 null control, with foci formation returning to wild-type levels in the reverted cell lines (Figure 6B). Furthermore, overlapping PLA and EdU foci were extremely rare (Figure 6C), suggesting that PCNA/ FANCD2 colocalization occurs prior to, but not during, the DNA synthesis step of MiDAS. Taken together, we propose a model in which PCNA K164 ubiquitination activates MiDAS first by promoting FANCD2 ubiquitination and chromatin association in S phase, followed by direct close-proximity colocalization to mark under-replicated genomic loci that engage MiDAS prior to cell division (Figure 6D).
Figure 6. PCNA K164 ubiquitination promotes its colocalization with FANCD2 in mitotic nuclei.

(A) Representative images of phospho-H3 S10-stained nuclei (blue), EdU foci (green), and PLA foci (red) from APH-treated RPE-1 cells. Scale bar, 5 μm.
(B) PLA foci quantification from at least three biological replicates in RPE-1 wild-type (blue), FANCD2−/− (green), PCNAK164R (maroon), and reverted PCNAK164 (purple) cells treated with 300 nM APH. Number (n) of nuclei quantified is listed. Significance was calculated by Kruskal-Wallis with Dunn’s multiple comparison test with ****p > 0.001.
(C) Quantification of overlapping PLA and EdU foci from at least three biological replicates in RPE-1 wild-type (blue), FANCD2−/− (green), PCNAK164R (maroon), and reverted PCNAK164 (purple) cells treated with 300 nM APH. Number (n) of nuclei quantified is listed.
(D) In wild-type (PCNAK164) cells (left), PCNA is ubiquitinated at K164 in response to replication stress by RAD18. During S phase, this facilitates the switch from processive DNA polymerase to TLS polymerases (for example, Pol η) to complete DNA synthesis. At alternative loci, ubiquitinated PCNA promotes the ubiquitination and chromatin association of FANCD2. During G2/M phase, PCNA recruits subunits of TLS Pol ζ to FANCD2-marked foci to initiate MiDAS and then facilitates processive replication through association with components of Pol δ. In mutant (PCNAK164R) cells (right), PCNA ubiquitination is impaired. Subsequently, TLS polymerase and FANCD2 recruitment to stressed replication forks is reduced in S and G2/M phases, causing the persistence of under-replicated genomic loci.
DISCUSSION
In this study, we utilized non-transformed human cell lines stably expressing mutant PCNAK164R from the endogenous locus to fill gaps in our understanding of PCNA’s role(s) in maintaining genome stability. We present evidence that PCNAK164R mutants experience chronic replication stress, producing under-replicated regions that are not rescued by MiDAS. We propose that these defects are due, at least in part, to the inability of unmodified PCNA to appropriately recruit and/or activate TLS DNA polymerases and facilitate stable chromatin binding of FANCD2 in S and G2/M phases of the cell cycle.
Replication stress, whether acute or chronic, can be caused by many exogenous and endogenous sources that disrupt the normal DNA replication program.20 Consistent with previous studies in mammalian cells, RPE-1 PCNAK164R mutants were acutely sensitive to DNA lesions induced by UV, MMS, and MMC (Figure 1),10,11,29,30 implying that TLS was significantly impaired. In this context, PCNA K164 ubiquitination is critical for the recruitment of specialized DNA polymerases that bypass lesions and fill ssDNA gaps. Interestingly, K164-dependent gap filling is also critical under normal growth conditions, helping to prevent nascent DNA degradation at stalled forks and promoting Okazaki fragment maturation.19 Here, we find that PCNAK164R mutants experience chronic replication stress that causes perturbations in the global replication program, including a significant reduction in origin activation as indicated by increased IODs (Figure 2F). This chronic replication stress significantly slowed proliferation (Figures 2, S2, and S3) without eliciting a DNA damage response (DDR; Figure 1B). These data are consistent with previous observations where mild replication stress caused ssDNA accumulation and under-replication of CFSs, but not a DDR, in checkpoint-proficient cells.31 We observed that PCNAK164R mutants accumulated under-replicated DNA that was sequestered into G1-phase 53BP1 NBs (Figures 3B and S4A) and an increase in anaphase abnormalities in the presence of low-dose APH (Figure 3A). As APH has classically been used to stimulate under-replication and breakage at CFSs, these genomic loci may be even more sensitive to replication stress in PCNAK164R mutants.
Human cells employ MiDAS to promote genome stability by completing DNA synthesis at under-replicated loci prior to cell division. However, MiDAS in cancerous and non-transformed cell models is not identical.16,17 A recent study found that RAD18-mediated PCNA K164 ubiquitination, and not sumoylation, was critical for MiDAS in human cancer cell lines.18 We extend these findings to 293T cancer cells and, importantly, non-transformed RPE-1 cells, as PCNAK164R mutants in both backgrounds were defective in MiDAS (Figure 4). We observed a similar defect in RPE-1 RAD18−/− cells (Figures 4B and 4C), whereas RAD18−/−:PCNAK164R double mutants had no additional defect, confirming that RAD18 and PCNA function in the same MiDAS pathway. In our study, RAD18-dependent K164 ubiquitination only partially accounted for the MiDAS defect, suggesting that other E3 ligases may modify K164 to activate MiDAS. Using cancer cell lines, Wu et al. proposed that K164 ubiquitination was required to first recruit components of TLS polymerase zeta (Pol ζ) to initiate MiDAS, which were then exchanged for subunits of the processive replicative Pol delta (Pol δ).18 Interestingly, Bergoglio et al. found that the absence of TLS Pol eta (Pol η) increased MiDAS as the result of impaired gap filling and CFS under-replication in S phase.32 Taken together, these data suggest that in the absence of PCNA K164 ubiquitination, appropriate spatial and temporal recruitment of TLS polymerases is lost, generating ssDNA gaps and under-replicated regions in S phase that cannot be rescued by DNA synthesis in G2/M phase.
FANCD2 is a canonical marker of under-replicated loci, including CFSs, and is the main regulator of MiDAS in non-transformed cells.16,17 FANCD2 exists as a homodimer and in response to replication stress one subunit is exchanged for FANCI, forming a FANCD2-FANCI heterodimer. Ubiquitination of this DNA-bound complex by the FA core complex locks it onto DNA.26,27 Specifically, FANCD2 is mono-ubiquitinated during S phase by FANCL.33 We demonstrate here that mono-ubiquitination of FANCD2 is significantly reduced in PCNAK164R cells, despite wild-type FANCD2 expression levels (Figures 5A and S6E). This phenotype was present during unperturbed and APH treatment conditions and cannot be explained by a reduction in the S-phase population (Figure 5D). Furthermore, in mitotic pH3-S10-positive nuclei, the number of FANCD2, EdU, and PCNA/ D2 close-proximity colocalization (PLA) foci were each significantly reduced in PCNAK164R mutants and rescued with restoration of wild-type PCNA expression (Figures 4 and 6). Based on these observations, we propose that FANCD2 is recruited to or stabilized at under-replicated regions by PCNA. In particular, we propose that ubiquitinated PCNA is required for robust FANCD2 recruitment/stabilization, although in the present study, we cannot delineate between the role(s) of mono- or poly-ubiquitinated PCNA. Reciprocally, we speculate that FANCD2 might prevent unloading of ubiquitinated PCNA from stalled forks in S phase to promote MiDAS at under-replicated loci. One possibility is that PCNA directly recruits FANCD2 through its PCNA-interacting peptide (PIP) box22 and/or through its coupling of ubiquitin conjugation to endoplasmic reticulum degradation (CUE) domain.34 In addition, evidence suggests that FANCL recruitment also requires RAD18-mediated PCNA K164 ubiquitination, which stimulates FANCD2 mono-ubiquitination,35 and that loss or depletion of RAD18 impairs FANCD2 foci formation in response to replication stress.36 Consistent with the notion that PCNA recruits FANCD2 and activates the FA pathway, PCNA and FANCD2 colocalize following HU- or APH-induced replication stress.37 Finally, FA proteins participate in a BIR-like mechanism, similar to MiDAS, to alleviate replication stress during S phase.38 Based on these data, we propose that, as for TLS polymerases, spatial and temporal recruitment and activation of FANCD2 are lost in PCNAK164R mutants.
Our findings support a model that applies to both untreated and APH-treated cells in which PCNA K164 ubiquitination prevents the accumulation of under-replicated DNA in multiple ways (Figure 6D). First, during S phase, replication forks stall under stress, and PCNA recruits TLS polymerases, including but perhaps not limited to Pol η, to proceed with DNA synthesis and prevent under-replication. PCNA also promotes robust origin activation, through an undefined mechanism, to avoid perpetuating ssDNA gaps and under-replicated regions to subsequent cell cycles. At loci that remain under-replicated, PCNA promotes the ubiquitination and chromatin association of FANCD2. Next, as cells progress to G2/M phase, PCNA recruits subunits of TLS Pol ζ to FANCD2-marked loci to initiate MiDAS and then facilitate processive replication through association with components of Pol δ. When the ability to ubiquitinate PCNA K164 is reduced or abolished as it is in RAD18 and PCNA mutants, respectively, TLS polymerase and FANCD2 recruitment are impaired. Notably, PCNA directly interacts with the E3 ubiquitin ligase TRAF-interacting protein (TRAIP)39,40 and the DNA helicase regulator of telomere length 1 (RTEL1),41 two proteins with demonstrated roles in MiDAS.42,43 Future studies may be warranted to understand whether the contributions of PCNA K164 ubiquitination in suppressing under-replication and promoting MiDAS also include the regulation of these two factors. Ultimately, our data place PCNA K164 ubiquitination at a key position in pathways that not only resolve replication stress in S phase but rescue DNA synthesis at particularly sensitive loci prior to cell division.
Limitations of the study
Our study provides strong evidence for the contribution of PCNA K164 ubiquitination in preventing the generation of under-replicated DNA and in promoting FANCD2-dependent MiDAS in human cells. However, gaps in our knowledge remain. First, our data cannot distinguish whether PCNA K164 promotes MiDAS through mono- or poly-ubiquitination. Given the mechanistic similarities between TS, BIR, and MiDAS, evaluating the requirements of PCNA mono- and poly-ubiquitination would be informative. Second, we do not understand the mechanism underlying reduced replication initiation in PCNAK164R mutants. Using a particularly stringent gating strategy for late-G1-phase cells, we found a statistically significant reduction in the amount of chromatin-bound MCM2. However, western blot analyses of chromatin-bound MCM2 and ORC2 did not detect any differences (Figure S2D), suggesting that any biological effect on origin licensing is relatively small. Interestingly, Madireddy et al. reported that FANCD2 was required for efficient replication initiation at the FRA16D locus in lymphoblast and fibroblast cell lines, an effect that may occur genome wide.44 Establishing that PCNA ubiquitination promotes FANCD2 recruitment for robust origin firing either genome wide or at specific loci is an enticing model but, at the moment, remains speculative.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Please direct requests for resources and reagents to Anja-Katrin Bielinsky, Ph.D. (azu3jn@virginia.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Data and code availability
The original data within the paper will be available from the lead contact upon request.
This paper does not report custom code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL DETAILS AND STUDY PARTICIPANT DETAILS
Cell lines
Non-transformed p53-proficient hTERT RPE-1 cells (ATCC, CRL-4000, RRID:CVCL_4388) were grown in Dulbecco’s Modified Eagle Medium: Nutrient Mixture F12 (DMEM/F12, Gibco 11320) supplemented with 10% fetal bovine serum (FBS, Sigma F4135) and 1% Penicillin-Streptomycin (Gibco 15140). Transformed p53-deficient 293T cells (ATCC, CRL-3216, RRID:CVCL_0063) were grown in Dulbecco’s Modified Eagle Medium (DMEM, Gibco 11995) supplemented with 10% FBS and 1% Pen Strep. Cells were cultured at 37°C and 5% CO2.
METHOD DETAILS
Cell line generation using CRISPR/Cas9 gene editing
To generate PCNAK164R cells, a guide RNA (gRNA) targeting PCNA exon 5 was designed (gRNA: 5′- ATACGTGCAAATTCACCAGA -3′) and cloned into a CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR associated 9) plasmid (hSpCas9(BB)-2A-GFP/PX458; Addgene plasmid #48138). Utilizing golden gate cloning, a double stranded donor plasmid containing the desired K164R mutation was constructed. Silent mutations were introduced into the donor plasmid to generate a novel restriction enzyme recognition site. Parental RPE-1 wildtype cells were transfected with the CRISPR/Cas9 plasmid containing the PCNA gRNA and the donor plasmid containing the K164R mutation via electroporation (Neon Transfection System, Invitrogen MPK5000; 1530 V, 10 ms, 3 pulses). Two days post-transfection GFP-expressing cells were collected by flow cytometry and subcloned. Subclones were screen for correct targeting by PCR amplification and restriction enzyme digestion (Forward: 5′- TGGCGCTAGTATTTGAAGCA -3′, Reverse: 5′- ACTTGGGATCCAATTCTGTCTACT -3′, Restriction Enzyme: EcoRI, NEB R3101). Specific mutations were identified by Sanger sequencing (Sequencing: 5′- AGGTGTTGCCTTTTAAGAAAGTGAGG -3′). 293T PCNAK164R lines were generated as described previously.28
To generate RAD18−/− and RAD18−/−:PCNAK164R mutant cell lines, a gRNA targeting RAD18 exon 2 (gRNA: 5′- AGACAATAGATGATTTGCTG -3′) was designed such that DNA cleavage would disrupt an endogenous restriction enzyme recognition site and was subsequently cloned into a CRISPR/Cas9 plasmid as described above. RPE-1 wildtype cells and PCNAK164R 2B10 cells were transfected with the CRISPR/Cas9 plasmid containing the RAD18 gRNA using the Neon Transfection System. Two days post-transfection GFP-expressing cells were collected by flow cytometry and subcloned. Subclones were screen for correct targeting by PCR amplification and restriction enzyme digestion (Forward: 5′- GTAGTACCATGCCGAAAGCAC -3′, Reverse: 5′- GGAACCACCTATCTGTTATCC -3′, Restriction Enzyme: TseI, NEB R0591). Knockout lines were identified by Sanger sequencing (Sequencing: 5′- CTACCTCATGTAAAAATCGC -3′) and Tracking of Indels by Decomposition (TIDE) analyses67.
To generate PCNAK164/− reversion cell lines, a synthetic sgRNA (Synthego, 5′-GUAAUUUCUUGUGCAAGAGA-3′) specifically targeting the R164 allele on exon 5 of PCNA and an 89 nt single-stranded oligodeoxynucleotide (ssODN) donor with a point mutation to revert the arginine (AGA) back to a lysine (AAA) (5′-GCCGAGATCTCAGCCATATTGGAGATGCTGTTGTAATTTCCTGCGCAAAAGACGGAGTGAAATTTTCTGCTAGTGGAGAACTTGGAAAT-3′) were designed. A silent mutation upstream of the R164K site was introduced into the ssODN to generate a novel FspI restriction site for screening purposes. PCNAK164R/− A1 and B1 cells were transfected with the synthetic sgRNA, CleanCap Cas9 mRNA (TriLink), and the ssODN using the Neon Transfection System (Invitrogen). Knock-ins were identified in monoclonal populations by PCR amplification using the primer pair (PCNA_Exon5_TIDE_ScrF: 5′-TCTTGTTCCCTGGATGGTGC-3’; PCNA_Exon5_TIDE_ScrR: 5′-ACACGTGCTGAATTTGTATTCCC-3′) followed by FspI (NEB R0135L) restriction enzyme digestion and further confirmed by Sanger sequencing.
Cell proliferation
Cells were plated at 100,000 cells per well (RPE-1) or 125,000 cells per well (293T) in 6-well plates. Cell counts were performed 3-day after seeding using Trypan Blue (Invitrogen T10282) on Countess slides (Invitrogen C10283) using a Countess automated cell counter (Invitrogen C20181).
Cell viability assay
RPE-1 cells were plated at 500 cells (wildtype) or 800 cells (PCNAK164R) per well in 96-well plates and allowed to recover for 24 h. Stock solutions of each drug were prepared in sterile 1X phosphate-buffered saline (PBS), water or dimethyl sulfoxide (DMSO) as appropriate and further diluted in growth medium. Cells were allowed to grow for 96 h in drug containing medium (methyl methanesulfonate, Acros Organics 156890250, mitomycin C, Sigma M4287, 4-nitroquinoline 1-oxide, Sigma N8141, hydroxyurea, Acros Organics 151680250; aphidicolin, Sigma A0781) and cell viability was measured with the CellTiter-Glo Luminescent Cell Viability Assay (Promega G7572) following manufacturer’s instructions. The viability of drug treated cells was normalized to the average viability of the untreated control cells for each cell line. Plates were imaged using a GloMax Discover Microplate Reader (Promega). Analysis and statistical tests were performed using Microsoft Excel.
Protein extraction, nuclear fractionation and western blotting
For preparation of whole cell extracts, cells were lysed in NETN (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% NP-40 and protease inhibitors) buffer for 10 min at 4°C and then centrifuged at 12,000 rpm for 10 min at 4°C. Cleared lysates were collected and protein concentrations were determined using Bradford protein assay (Bio-Rad 5000006). Lysates were then mixed with SDS loading buffer and denatured at 95°C before fractionation by SDS-PAGE and analyses by Western blot. Nuclear fractions were isolated as previously described68. Briefly, extracts were prepared by lysis in Buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 0.1% Triton X-100 and protease inhibitors) for 5 min at 4°C. Insoluble nuclear proteins were isolated by centrifugation at 1300 g at 4°C and chromatin bound proteins were subsequently released by sonication after being resuspended in TSE buffer (20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 2 mM EDTA, 0.1% SDS, 0.1% Triton X-100 and protease inhibitors). Remaining insoluble factors were cleared by centrifugation at 17000 g at 4°C. Protein concentrations of nuclear fractions were determined using Bradford protein assay prior to fractionation by SDS-PAGE and Western blot analyses. Primary antibodies were incubated in 5% BLOT-QuickBlocker (G-Biosciences 786-011) as follows: mouse anti-PCNA (Abcam, ab29; 1:3000), rabbit anti-Ubiquityl-PCNA (Lys164) (Cell Signaling, D5C7P, 13439; 1:1000), rabbit anti-RPA32 (S33) (Bethyl, A300-246A; 1:2000), rabbit anti-γH2AX (Bethyl, A300-081A; 1:2000), rabbit anti-H2AX (Bethyl, A300-082A; 1:5000), rabbit anti-p-p53 (S15) (Cell Signaling, 9284S; 1:500), mouse anti-p53 (Santa Cruz, sc-126; 1:2000), rabbit anti-p21 (Santa Cruz, sc-397 clone C19; 1:1000), rabbit anti-FANCD2 (Abcam, ab108928; 1:2000), mouse anti-FANCL (Proteintech, 66639-1-Ig; 1:1,000), rabbit anti-RAD18 (Bethyl, A300-340A; 1:1000), rabbit anti-MCM2 (Cell Signaling, 4007S; 1:1000; BD Biosciences, 610701; 1:1000; Proteintech, 10513-1-AP; 1:1000), mouse anti-GAPDH (GeneTex, GTX627408; 1:10000), mouse anti-Ku86 (Santa Cruz, B-1, sc-5280; 1:500), mouse anti-α-tubulin (Millipore, T9026, clone DM1A; 1:10000), rabbit anti-ORC2 (Thermo Fisher, PA5-110063, 1:1000), rabbit anti-Histone H4 (Abcam, ab10158, 1:200,000). Secondary antibodies were incubated in 5% BLOT-QuickBlocker as follows: goat anti-mouse HRP conjugate (BioRad, 1706516; 1:5000), donkey anti-rabbit HRP conjugate (Amersham, NA9340; 1:5000). Detection was performed using WesternBright Quantum detection kit (K-12042-D20). Quantification was performed using FIJI and Microsoft Excel. Image preparation was performed using Adobe Photoshop.
FACS analysis
For flow cytometry analyses of cell cycle distribution, DNA synthesis and origin licensing, RPE-1 and 293T cells were treated as described previously.36 Briefly, cells were treated with methyl methanesulfonate (20 μM) or mitomycin C (20 nM) for 48 h when applicable and incubated with 10 μM EdU (Lumiprobe, 20540) for 30 min before harvesting with trypsin. Cells were then washed with cold 1X PBS and lysed in CSK (10 mM PIPES pH 7.0, 300 mM sucrose, 100 mM NaCl, 3 mM MgCl2 hexahydrate) with 0.5% Triton X-100, then fixed in PBS with 4% paraformaldehyde (Electron Microscopy Sciences) for 15 min. Cells were labeled with AF647-azide (Life Technologies, A10277) in 100 mM ascorbic acid, 1 mM CuSO4, and PBS to detect EdU for 30 min at room temperature (RT). Cells were then washed and incubated with anti-MCM2 antibody (BD Biosciences, #610700; 1:200) in 1% bovine albumin serum (BSA) in PBS with 0.1% NP-40 for 1 h at 37°C. Next, cells were washed and labeled with donkey anti-mouse AF488 secondary antibody (Invitrogen, A11029; 1:1000) in 1% BSA in PBS with 0.1% NP-40 for 1 h at 37°C. Lastly, cells were washed and incubated in DAPI (Life Technologies, D1306; 1 μg/mL) and RNAse A (Sigma, R6513; 100 ng/mL) for 1 h at 37°C. Samples were processed on an LSR II (BD Biosciences) flow cytometer and analyzed with FlowJo v10.6.1 and Microsoft Excel.
For flow cytometry analyses of apoptosis, RPE-1 wildtype and PCNAK164R cells were seeded in 6-well plates at 75,000 cells per well and allowed to proliferate for approximately 72 h. Cells were collected, washed twice with 1X PBS and stained using the APC Annexin V apoptosis detection kit (Biolegend 640932) according to the manufacturer’s instructions. Samples were processed on an LSRII (BD Biosciences) flow cytometer. Apoptotic cells were identified by annexin V staining while cell viability was determined by PI staining. Data was analyzed using FlowJo v10.6.1 and GraphPad Prism 8.
DNA combing
For genome-wide analyses of DNA replication, wildtype and PCNAK164R RPE-1 and 293T cells were plated at 40% confluency in 10 cm plates 24 h prior to labeling. Cells were incubated with 25 or 100 μM IdU (Sigma C6891) for 30 min, rinsed twice with pre-warmed medium and then incubated with 100 μM or 200 μM CldU (Sigma I7125) for 30 min. Approximately 250,000 cells were embedded in 0.5% agarose plugs (NuSieve GTG Agarose, Lonza, 50080) and digested for 48 h in plug digestion solution (10 mM Tris-HCl, pH 7.5, 1% Sarkosyl, 50 mM EDTA and 2 mg/mL Proteinase K). Plugs were then melted in 50 mM MES pH 5.7 (Calbiochem #475893) and digested overnight with β-agarase (NEB M0392). DNA was then subsequently combed onto commercially available vinyl silane-coated coverslips (Genomic Vision COV-001). Integrity of combed DNA for all samples was quality checked via staining with YOYO-1 (Invitrogen Y3601). Combed coverslips were baked at 60°C for 2–4 h, cooled to RT and stored at −20°C. DNA was denatured in 0.5 M NaOH and 1 M NaCl for 8 min at RT. All antibody staining was performed in 2% BSA in PBS-Triton (0.1%). Primary antibodies included rabbit anti-ssDNA (IBL 18731), mouse anti-BrdU/IdU (BD Biosciences 347580; clone B44) and rat anti-BrdU/CldU (Abcam, ab6326; BU1/75 (ICR1)). Secondary antibodies included goat anti-mouse Cy3.5 (Abcam ab6946), goat anti-rat Cy5 (Abcam ab6565) and goat anti-rabbit BV480 (BD Horizon #564879). Imaging was performed using Genomic Vision EasyScan service. Images were blinded and analyzed using the Genomic Vision FiberStudio software. Data/statistical analyses were performed in Microsoft Excel and GraphPad Prism 8.
Immunostaining
For immunofluorescent staining of 53BP1 nuclear bodies and Cyclin A, wildtype and PCNAK164R RPE-1 and 293T cells were seeded onto fibronectin (Sigma F4759) coated coverslips at 100,000 cells per coverslip and allowed to recover for 24 h. Cells were then treated with 300 nM APH (Sigma A0781) for 24 h. After, cells were washed with PBS containing 0.9 mM CaCl2 and 0.49 mM MgCl2 (PBS-Ca2+/Mg2+) and fixed in PBS-Ca2+/Mg2+ with 3.7% formaldehyde (Fisher Scientific F79-500) for 10 min at RT. Next, cells were washed twice with PBS-Ca2+/Mg2+, permeabilized with 0.1% Triton X-100 for 5 min at RT, and subsequently blocked in ABDIL (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 2% BSA, 0.2% Fish Gelatin, 0.1% NaN3) for 1 h at RT. Coverslips were incubated with rabbit anti-53BP1 (Abcam, ab36823; 1:500) and mouse anti-Cyclin-A (Santa Cruz; sc-271682 clone B8; 1:200) primary antibodies overnight at 4°C. Next day, coverslips were washed with PBS-Ca2+/Mg2+ containing 0.1% Tween 20 and incubated with Alexa Fluor 488 donkey anti-rabbit (Invitrogen, A21206; 1:1000) and Alexa Fluor 594 goat anti-mouse (Invitrogen, A11032; 1:000) secondary antibodies for 1 h at RT. Lastly, coverslips were washed, stained with DAPI (Life Technologies, D1306; 1 μg/mL), and mounted on microscope slides with Vectashield anti-fade reagent (Vector Laboratories, H1000). Between 30 and 40 fields per coverslip were imaged on a Zeiss Spinning Disc confocal. Images were scored using FIJI and statistical analyses were performed in GraphPad Prism 8.
For immunofluorescent staining of FANCD2 and EdU foci, wildtype and PCNAK164R RPE-1 and 293T lines were seeded onto fibronectin coated coverslips and treated with 300 or 450 nM APH when applicable. Cells were then washed with PBS and incubated with 20 μM EdU for 30 min before fixation with 10% formalin. Fixed cells on coverslips were then subjected to a Click-Chemistry Reaction (20 μM Biotin-Azide, 10 mM sodium ascorbate, and 2 mM CuSO4 in PBS) at RT for 1 h. After, coverslips were washed with PBS and incubated with rabbit anti-FANCD2 (Abcam, ab108928; 1:250) and mouse anti-phospho-Histone H3 (S10) (Cell Signaling; 9706S; 1:200) primary antibodies in PBS containing 0.3% Triton X-100 and 1% BSA at 4°C overnight. The next day, coverslips were washed with PBS and incubated with Alexa Fluor 488 Streptavidin (Thermo Fisher, S32354; 1:100), Alexa Fluor 350 anti-mouse (Thermo Fisher, A11045; 1:100) and Alexa Fluor anti-rabbit (Thermo Fisher, A31632; 1:1000) secondary antibodies at RT for 1 h. Lastly, coverslips were washed with PBS and mounted on microscope slides with Prolong Gold anti-fade reagent (Thermo Fisher, P36931). EdU and FANCD2 foci were scored using a Zeiss Axio Imager A1 fluorescent microscope and EVOS FL imaging system (ThermoFisher AMF43000). Between 300 and 400 cells per cell line were scored in a blinded manner per experiment. Statistical analyses were performed in GraphPad Prism 8.
For immunofluorescent staining of anaphases, wildtype and PCNAK164R RPE-1 cells were seeded onto coverslips at 200,000 cells per coverslip and allowed to recover for 24 h. Cells were then treated with 300 nM APH for 24 h. After, cells were washed with PBS and fixed with 10% formalin for 15 min. Fixed cells on coverslips were then washed with PBS, stained with DAPI and mounted on microscope slides with Vectashield anti-fade reagent (Vector Laboratories H1000). Between 200 and 300 anaphases per cell line were scored per experiment in a blinded manner using the EVOS FL imaging system (ThermoFisher AMF43000). Statistical analyses were performed in GraphPad Prism 8.
Proximity ligation assay
For immunofluorescent staining of PCNA/FANCD2 PLA and EdU foci, wildtype, PCNAK164R and PCNAK164 RPE-1 cells were seeded onto coverslips and treated with 300 nM APH. Cells were then incubated with 20 μM EdU for 30 min. Cells were then pre-extracted with ice-cold 0.5% Triton X-100 in 1x PBS for 15 min on ice, fixed with ice-cold methanol with 5% glacial acetic acid for 15 min on ice, and permeabilized with 0.1% Triton X-100 in 1x PBS for 10 min at room temp. PLA staining was peformed using a Duolink In Situ Red Starter Kit (Millipore Sigma, DUO92101) Coverslips were washed twice in 1x PBS at room temp and blocked with Duoilink Blocking Solution (DUO82007) at 37°C for 1 h. Coverslips were then incubated with mouse anti-PCNA (Abcam, ab29; 1:250) and rabbit anti-FANCD2 (Abcam, ab108928; 1:250) diluted in Duolink Antibody Diluent (DUO82008) at 4°C overnight. After two 5 min washes in 1x Duolink Wash Buffer A at room temp, coverslips were incubated with anti-mouse minus (DUO92004) and anti-rabbit plus (DUO92002) PLA probes at 37°C for 1 h. After two 5 min washes in 1x Duolink Wash Buffer A at room temp, coverslips were incubated with ligation buffer containing ligase (DUO92008) for 30 min at 37°C. After two 5 min washes in 1x Duolink Wash Buffer A at room temp, coverslips were incubated with amplification buffer with polymerase (DUO92008) for 100 min at 37°C. Coverslips were next washed twice for 10 min in 1x Duolink Wash Buffer B, once for 1 min in 0.01x Duolink Wash Buffer B, and once for 1 min in 1x PBS. Coverslips were then subjected to a Click-Chemistry Reaction (20 μM Biotin-Azide, 10 mM sodium ascorbate, and 2 mM CuSO4 in PBS) at room temp for 1 h. After one 5 min wash in 1x PBS at room temp, coverslips were incubated with rat anti-phospho-Histone H3 (S10) (Active Motif, 61623; 1:200) diluted in 0.3% Triton X-100 and 1% BSA in 1x PBS at 4°C overnight. Next, coverslips were washed once for 5 min in 1x PBS and then incubated with Alexa Fluor 488 Streptavidin (Thermo Fisher, S32354; 1:100) and Alexa Fluor 350 anti-rat (Invitrogen, A21093; 1:250) diluted in 0.3% Triton X-100 and 1% BSA in 1x PBS at room temp for 1 h. Lastly, coverslips were washed twice for 5 min with 1x PBS and mounted on microscope slides with Prolong Gold anti-fade reagent (Thermo Fisher, P36931) or Vectashield anti-fade reagent (Vector Laboratories, H1000). Foci were scored in a blinded manner using a Zeiss Axio Imager A1 fluorescent microscope or EVOS M5000 imaging system (ThermoFisher, A40486). Statistical analyses were performed in GraphPad Prism 9.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses
Statistical details of experiments including the statistical test used, value of n, what n represents, dispersion and precision measures (e.g., average, standard deviation), as well as how significance was defined is indicated in each figure and corresponding figure legend. The software used for statistical analysis of each type of experiment is indicated in the corresponding Method Details sections.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| α-tubulin (Clone DM1A) | Sigma | T9026 RRID:AB_477593 |
| γH2AX | Bethyl | A300-081A RRID:AB_203288 |
| 53BP1 | Abcam | ab36823 RRID:AB_722497 |
| CldU (BU1/75) (ICR1) | Abcam | ab6326 RRID:AB_305426 |
| Cyclin-A (Clone B8) | Santa Cruz | sc-271682 RRID:AB_10709300 |
| Donkey anti-mouse AF488 | Invitrogen | A11029 RRID:AB_2534088 |
| Donkey anti-rabbit AF488 | Invtrogen | A21206 RRID:AB_2535792 |
| FANCD2 | Abcam | ab108928 RRID:AB_10862535 |
| FANCL | Proteintech | 66639-1-Ig RRID:AB_2881998 |
| GAPDH | GeneTex | GTX627408 RRID:AB_11174761 |
| Goat anti-mouse HRP conjugate | BioRad | 1706516 RRID:AB_11125547 |
| Goat anti-mouse IgG H&L (Cy3.5) | Abcam | ab6946 RRID:AB_955045 |
| Goat anti-mouse AF350 | Invitrogen | A11045 RRID:AB_14275 |
| Goat anti-mouse AF594 | Invitrogen | A11032 RRID:AB_2534091 |
| Goat anti-rabbit IgG (BV480) | BD Horizon | 564879 RRID:AB_2738997 |
| Goat anti-rabbit AF594 | Thermo Fisher | A31632 RRID:AB_2813889 |
| Goat anti-rat IgG H&L (Cy5) | Abcam | ab6565 RRID:AB_955063 |
| Goat anti-rat IgG H + L AF350 | Invitrogen | A21093 RRID:AB_10563602 |
| Histone H2AX | Bethyl | A300-082A RRID:AB_203287 |
| Histone H4 | Abcam | ab10158 RRID:AB_296888 |
| Ku86 (Clone B-1) | Santa Cruz | sc-5280 RRID:AB_672929 |
| IdU (Clone B44) | BD Bioscience |
BD347580 RRID:AB_10015219 |
| MCM2 (BM28) | BD Biosciences | 610701 RRID:AB_398024 |
| MCM2 | Cell Signaling | 4007 RRID:AB_2142134 |
| MCM2 | Proteintech | 10513-1-AP RRID:AB_2142131 |
| ORC2 | Thermo Fisher | PA5-110063 RRID:AB_2855474 |
| p21 (clone 19) | Santa Cruz | sc-397 RRID:AB_632126 |
| p53 | Santa Cruz | sc-126 RRID:AB_628082 |
| p-p53 (S15) | Cell Signaling | 9284 RRID:AB_331464 |
| p-Histone H3 (S10) | Cell Signaling | 9706 RRID:AB_331748 |
| p-Histon H3 (S10) | Active Motif | 61623 RRID:AB_2793707 |
| PCNA | Abcam | ab29 RRID:AB_303394 |
| RAD18 | Bethyl | A301-340A RRID:AB_937974 |
| RPA32 (S33) | Bethyl | A300-246A RRID:AB_2180847 |
| ssDNA | Genomic Vision | IBL18731 RRID:AB_2341405 |
| Streptavidin AF488 | Thermo Fisher | S32354 RRID:AB_2315383 |
| ubiquityl-PCNA (Lys164) | Cell Signaling | 13439 RRID:AB_2798219 |
| Vinculin | Abcam | ab18058 RRID:AB_444215 |
| Chemicals, peptides, and recombinant proteins | ||
| 4-nitroquinoline 1-oxide (4NQO) | Sigma | N8141 |
| 16% paraformaldehyde (MeOH free) | Electron Microscopy Sciences | 15710 |
| AF647-azide | Life Technologies | A10277 |
| Aphidicolin | Sigma | A0781 |
| Biotin-azide | Cayman Chemical | 13040 |
| BLOT-QuickBlocker | G-Biosciences | 786–001 |
| Bovine serum albumin | Sigma | A3059 |
| CldU | Sigma | I7125 |
| Copper sulfate | Thermo Fisher | 197722500 |
| DAPI | Life Technologies | D1306 |
| DMEM | Gibco | 11995 |
| DMEM/F12 | Gibco | 11320 |
| EcoRI | New England Biolabs | R3101 |
| EdU | Lumiprobe | 20540 |
| Fetal Bovine Serum | Sigma | F4135 |
| Fibronectin | Sigma | F4759 |
| Formaldehyde, 37% | Fisher Scientific | F79-500 |
| Formalin | RPI | F10800–500.0 |
| FspI | New England Biolabs | R0135 |
| Hydroxyurea | Acros Organics | 151680250 |
| IdU | Sigma | C6891 |
| Methyl methanesulfonate | Acros Organics | 156890250 |
| Mitomycin C | Sigma | M4287 |
| MES, Free Acid, ULTROL Grade | Millipore | 475893 |
| NuSieve GTG Agarose | Lonza | 50080 |
| Penicillin-Streptomycin | Gibco | 15140 |
| Prolong Gold | Thermo Fisher | P36931 |
| RNase A | Sigma | R6513 |
| Sodium ascorbate | Sigma | A4034 |
| TseI | New England Biolabs | R0591 |
| Vectashield | Vector Laboratories | H-1000 |
| YOYO-1 | Invitrogen | &3601 |
| Critical commercial assays | ||
| APC Annexin V Apoptosis Detection Kit | Biolegend | 640932 |
| CellTiter-Glo | Promega | G7572 |
| Duolink In Situ Red Starter Kit | Millipore Sigma | DUO92101 |
| EasyScan Service | Genomic Vision | https://store.genomicvision.com/services/77-easyscan.html |
| WesternBright Quantum Detection Kit | Advansta | K-12042-D20 |
| Experimental models: Cell lines | ||
| hTERT RPE-1 | ATCC | CRL-4000 RRID:CVCL_4388 |
| 293T | ATCC | CRL-3216 RRID:CVCL_0063 |
| Oligonucleotides | ||
| PCNA exon 5 gRNA: 5′-ATACGTGCAAATTCACCAGA-3′ | Integrated DNA Technologies | N/A |
| PCNA screen FW: 5′-TGGCGCTAGTATTTGAAGCA-3′ | Integrated DNA Technologies | N/A |
| PCNA screen RV: 5′-ACTTGGGATCCAATTCTGTCTACT-3′ | Integrated DNA Technologies | N/A |
| PCNA sequencing: 5′-AGGTGTTGCCTTTTAAGAAAGTGAGG-3′ | Integrated DNA Technologies | N/A |
| PCNA reversion sgRNA: 5′-GUAAUUUCUUGUGCAAGAGA-3′ | Synthego Corporation | N/A |
| PCNA ssODN: 5′-GCCGAGATCTCAGCCATATTGGAGATGCTGTTGTAATTTCCTGCGCAAAAGACGGAGTGAAATTTTCTGCTAGTGGAGAACTTGGAAAT-3′ | Integrated DNA Technologies | N/A |
| PCNA_Exon5_TIDE_ScrF: 5′-TCTTGTTCCCTGGATGGTGC-3′ | Integrated DNA Technologies | N/A |
| PCNA_Exon5_TIDE_ScrR: 5′-ACACGTGCTGAATTTGTATTCCC-3′ | Integrated DNA Technologies | N/A |
| RAD18 exon 2 gRNA: 5′-AGACAATAGATGATTTGCTG-3′ | Integrated DNA Technologies | N/A |
| RAD18 screen FW: 5′-GTAGTACCATGCCGAAAGCAC-3′ | Integrated DNA Technologies | N/A |
| Additional oligonucleotide information is in Table S1 | N/A | N/A |
| Recombinant DNA | ||
| hSpCas9(BB)-2A-GFP (PX458) | Addgene | 48138 RRID:Addgene481318 |
| Software and algorithms | ||
| Adobe Photoshop | Adobe | https://www.adobe.com/products/photoshop.html; RRID:SCR_014199 |
| FIJI | ImageJ | https://imagej.net/software/fiji/; RRID:SCR_00228 5 |
| FlowJo | BD Biosciences | https://www.flowjo.com/solutions/flowjo; RRID:SCR_008520 |
| FiberStudio | Genomic Vision | http://www.genomicvision.com/products/molecular-combing-platform/software/ |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com; RRID: SCR_002798 |
| Microsoft Excel | Microsoft | https://www.microsoft.com/en-gb/; RRID: SCR_016137 |
| TIDE | Brinkman et al.45 | https://tide.nki.nl/ |
| Other | ||
| Countess Automated Cell Counter | Invitrogen | C20181 |
| Countess Slides | Invitrogen | C10283 |
| DNA combing coverslips | Genomic Vision | COV-001 |
| EVOS FL Imaging System | Thermo Fisher | AMF43000 |
| EVOS M5000 Imaging System | Thermo Fisher | A40486 |
| GloMax Discover Microplate Reader | Promega | GM3000 |
| LSR II Flow Cytometer | BD Biosciences | N/A |
| Molecular Combing System | Genomic Vision | MCS-001 |
| Neon Transfection System | Invitrogen | MPK5000 |
| Zeiss Azio Imager A1 Microscope | Zeiss | N/A |
| Zeiss Spinning Disk Confocal Microscope | Zeiss | N/A |
Highlights.
PCNA K164 ubiquitination promotes mitotic DNA synthesis in non-transformed human cells
Robust FANCD2 ubiquitination, chromatin association, and foci formation rely on PCNA K164
PCNA and FANCD2 association in G2/M-phase nuclei requires PCNA ubiquitination at K164
ACKNOWLEDGMENTS
We would like to thank the Bielinsky lab for helpful discussions. We would like to thank the University of Minnesota Flow Cytometry Core Facility, the University of Minnesota Imaging Center (RRID SCR_020997), and the Masonic Cancer Center Cytogenomics Shared Resource. We would like to thank Sanjiv Ramana and Madison Hawrysz for their experimental contributions and David Largaespada for sharing research equipment and facilities. This work was supported by the National Institutes of General Medicine Sciences (NIGMS R35-GM141805 to A.-K.B., R01-GM134681 to A.K.B. and G.-L.M., and R01-GM138833 to N.S.), the National Cancer Institute (T32-CA009138 to W.L., R.M.B., and C.B.R.), and the ARCS Foundation (to C.B.R.). The Masonic Cancer Center Shared Resources are supported by NIH P30 CA077598.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113523.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Barnes R, and Eckert K (2017). Maintenance of Genome Integrity: How Mammalian Cells Orchestrate Genome Duplication by Coordinating Replicative and Specialized DNA Polymerases. Genes 8, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Técher H, Koundrioukoff S, Nicolas A, and Debatisse M (2017). The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat. Rev. Genet 18, 535–550. [DOI] [PubMed] [Google Scholar]
- 3.Tubbs A, and Nussenzweig A (2017). Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 168, 644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leung W, Baxley RM, Moldovan GL, and Bielinsky AK (2018). Mechanisms of DNA Damage Tolerance: Post-Translational Regulation of PCNA. Genes 10, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, and Jentsch S (2002). RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419, 135–141. [DOI] [PubMed] [Google Scholar]
- 6.Davies AA, Huttner D, Daigaku Y, Chen S, and Ulrich HD (2008). Activation of ubiquitin-dependent DNA damage bypass is mediated by replication protein a. Mol. Cell 29, 625–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sale JE, Lehmann AR, and Woodgate R (2012). Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol 13, 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Becker JR, Gallo D, Leung W, Croissant T, Thu YM, Nguyen HD, Starr TK, Brown GW, and Bielinsky AK (2018). Flap endonuclease overexpression drives genome instability and DNA damage hypersensitivity in a PCNA-dependent manner. Nucleic Acids Res. 46, 5634–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arakawa H, Moldovan GL, Saribasak H, Saribasak NN, Jentsch S, and Buerstedde JM (2006). A role for PCNA ubiquitination in immunoglobulin hypermutation. PLoS Biol. 4, e366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Niimi A, Brown S, Sabbioneda S, Kannouche PL, Scott A, Yasui A, Green CM, and Lehmann AR (2008). Regulation of proliferating cell nuclear antigen ubiquitination in mammalian cells. Proc. Natl. Acad. Sci. USA 105, 16125–16130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qin Z, Lu M, Xu X, Hanna M, Shiomi N, and Xiao W (2013). DNA-damage tolerance mediated by PCNA*Ub fusions in human cells is dependent on Rev1 but not Polη. Nucleic Acids Res. 41, 7356–7369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan KL, Palmai-Pallag T, Ying S, and Hickson ID (2009). Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol 11, 753–760. [DOI] [PubMed] [Google Scholar]
- 13.Kawabata T, Luebben SW, Yamaguchi S, Ilves I, Matise I, Buske T, Botchan MR, and Shima N (2011). Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Mol. Cell 41, 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minocherhomji S, Ying S, Bjerregaard VA, Bursomanno S, Aleliunaite A, Wu W, Mankouri HW, Shen H, Liu Y, and Hickson ID (2015). Replication stress activates DNA repair synthesis in mitosis. Nature 528, 286–290. [DOI] [PubMed] [Google Scholar]
- 15.Bhowmick R, Minocherhomji S, and Hickson ID (2016). RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 64, 1117–1126. [DOI] [PubMed] [Google Scholar]
- 16.Graber-Feesl CL, Pederson KD, Aney KJ, and Shima N (2019). Mitotic DNA synthesis is differentially regulated between cancer and noncancerous cells. Mol. Cancer Res 17, 1687–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Traband EL, Hammerlund SR, Shameem M, Narayan A, Ramana S, Tella A, Sobeck A, and Shima N (2023). Mitotic DNA synthesis in untransformed human cells preserves common fragile site stability via a FANCD2-driven mechanism that requires HELQ. J. Mol. Biol 435, 168294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu W, Barwacz SA, Bhowmick R, Lundgaard K, Gonçalves Dinis MM, Clausen M, Kanemaki MT, and Liu Y (2023). Mitotic DNA synthesis in response to replication stress requires the sequential action of DNA polymerases zeta and delta in human cells. Nat. Commun 14, 706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thakar T, Leung W, Nicolae CM, Clements KE, Shen B, Bielinsky AK, and Moldovan GL (2020). Ubiquitinated-PCNA protects replication forks from DNA2-mediated degradation by regulating Okazaki fragment maturation and chromatin assembly. Nat. Commun 11, 2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Magdalou I, Lopez BS, Pasero P, and Lambert SAE (2014). The causes of replication stress and their consequences on genome stability and cell fate. Semin. Cell Dev. Biol 30, 154–164. [DOI] [PubMed] [Google Scholar]
- 21.Chan YW, Fugger K, and West SC (2018). Unresolved recombination intermediates lead to ultra-fine anaphase bridges, chromosome breaks and aberrations. Nat. Cell Biol 20, 92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Howlett NG, Harney JA, Rego MA, Kolling FW, and Glover TW (2009). Functional interaction between the Fanconi Anemia D2 protein and proliferating cell nuclear antigen (PCNA) via a conserved putative PCNA interaction motif. J. Biol. Chem 284, 28935–28942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang S, Chea J, Meng X, Zhou Y, Lee EYC, and Lee MYWT (2008). PCNA is ubiquitinated by RNF8. Cell Cycle 7, 3399–3404. [DOI] [PubMed] [Google Scholar]
- 24.Terai K, Abbas T, Jazaeri AA, and Dutta A (2010). CRL4(Cdt2) E3 ubiquitin ligase monoubiquitinates PCNA to promote translesion DNA synthesis. Mol. Cell 37, 143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gallina I, Hendriks IA, Hoffmann S, Larsen NB, Johansen J, Colding-Christensen CS, Schubert L, Sellés-Baiget S, Fábián Z, Kühbacher U, et al. (2021). The ubiquitin ligase RFWD3 is required for translesion DNA synthesis. Mol. Cell 81, 442–458.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alcón P, Shakeel S, Chen ZA, Rappsilber J, Patel KJ, and Passmore LA (2020). FANCD2-FANCI is a clamp stabilized on DNA by mono-ubiquitination of FANCD2 during DNA repair. Nat. Struct. Mol. Biol 27, 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang R, Wang S, Dhar A, Peralta C, and Pavletich NP (2020). DNA clamp function of the monoubiquitinated Fanconi anaemia ID complex. Nature 580, 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, and D’Andrea AD (2002). S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood 100, 2414–2420. [DOI] [PubMed] [Google Scholar]
- 29.Vujanovic M, Krietsch J, Raso MC, Terraneo N, Zellweger R, Schmid JA, Taglialatela A, Huang JW, Holland CL, Zwicky K, et al. (2017). Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol. Cell 67, 882–890.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hendel A, Krijger PHL, Diamant N, Goren Z, Langerak P, Kim J, Reissner T, Lee KY, Geacintov NE, Carell T, et al. (2011). PCNA ubiquitination is important, but not essential for translesion DNA synthesis in mammalian cells. PLoS Genet. 7, e1002262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koundrioukoff S, Carignon S, Técher H, Letessier A, Brison O, and Debatisse M (2013). Stepwise activation of the ATR signaling pathway upon increasing replication stress impacts fragile site integrity. PLoS Genet. 9, e1003643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leung W, Baxley R, Thakar T, Rogers C, Buytendorp J, Wang L, Tella A, Moldovan G, Shima N, and Bielinsky A (2020). PCNA-K164 ubiquitination facilitates origin licensing and mitotic DNA synthesis. Preprint at bioRxiv. 2020.2006.2025.172361. [Google Scholar]
- 33.Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, Oostra AB, Yan Z, Ling C, Bishop CE, et al. (2003). A novel ubiquitin ligase is deficient in Fanconi anemia. Nat. Genet 35, 165–170. [DOI] [PubMed] [Google Scholar]
- 34.Rego MA, Kolling FW, Vuono EA, Mauro M, and Howlett NG (2012). Regulation of the Fanconi anemia pathway by a CUE ubiquitin-binding domain in the FANCD2 protein. Blood 120, 2109–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geng L, Huntoon CJ, and Karnitz LM (2010). RAD18-mediated ubiquitination of PCNA activates the Fanconi anemia DNA repair network. J. Cell Biol 191, 249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wells JP, Chang EYC, Dinatto L, White J, Ryall S, and Stirling PC (2022). RAD18 opposes transcription-associated genome instability through FANCD2 recruitment. PLoS Genet. 18, e1010309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Howlett NG, Taniguchi T, Durkin SG, D’Andrea AD, and Glover TW (2005). The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum. Mol. Genet 14, 693–701. [DOI] [PubMed] [Google Scholar]
- 38.Xu X, Xu Y, Guo R, Xu R, Fu C, Xing M, Sasanuma H, Li Q, Takata M, Takeda S, et al. (2021). Fanconi anemia proteins participate in a break-induced-replication-like pathway to counter replication stress. Nat. Struct. Mol. Biol 28, 487–500. [DOI] [PubMed] [Google Scholar]
- 39.Feng W, Guo Y, Huang J, Deng Y, Zang J, and Huen MSY (2016). TRAIP regulates replication fork recovery and progression via PCNA. Cell Discov. 2, 16016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoffmann S, Smedegaard S, Nakamura K, Mortuza GB, Räschle M, Ibañez de Opakua A, Oka Y, Feng Y, Blanco FJ, Mann M, et al. (2016). TRAIP is a PCNA-binding ubiquitin ligase that protects genome stability after replication stress. J. Cell Biol 212, 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vannier JB, Sandhu S, Petalcorin MIR, Wu X, Nabi Z, Ding H, and Boulton SJ (2013). RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science 342, 239–242. [DOI] [PubMed] [Google Scholar]
- 42.Sonneville R, Bhowmick R, Hoffmann S, Mailand N, Hickson ID, and Labib K (2019). TRAIP drives replisome disassembly and mitotic DNA repair synthesis at sites of incomplete DNA replication. Elife 8, e48686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu W, Bhowmick R, Vogel I, Özer Ö, Ghisays F, Thakur RS, Sanchez de Leon E, Richter PH, Ren L, Petrini JH, et al. (2020). RTEL1 suppresses G-quadruplex-associated R-loops at difficult-to-replicate loci in the human genome. Nat. Struct. Mol. Biol 27, 424–437. [DOI] [PubMed] [Google Scholar]
- 44.Madireddy A, Kosiyatrakul ST, Boisvert RA, Herrera-Moyano E, García-Rubio ML, Gerhardt J, Vuono EA, Owen N, Yan Z, Olson S, et al. (2016). FANCD2 Facilitates Replication through Common Fragile Sites. Mol. Cell 64, 388–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brinkman EK, Chen T, Amendola M, and van Steensel B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original data within the paper will be available from the lead contact upon request.
This paper does not report custom code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.