

Abstract
Understanding the mechanisms underlying the acquisition and maintenance of effector function during T cell differentiation is important to unraveling how these processes can be dysregulated in the context of disease and manipulated for therapeutic intervention. Herein, we report the identification of a novel regulator of murine T cell differentiation through the evaluation of a previously unreported activity of the kinase inhibitor, BioE-1197. Specifically, we demonstrate that liver kinase B1 (LKB1)-mediated activation of salt-inducible kinases (SIKs) epigenetically regulates cytokine recall potential in effector CD8+ and Th1 cells. Evaluation of this phenotype revealed that SIK-mediated phosphorylation-dependent stabilization of histone deacetylase 7 (HDAC7) occurred during late-stage effector differentiation. HDAC7 stabilization increased nuclear HDAC7 levels which correlated with total and cytokine loci-specific reductions in the activating transcription mark, histone 3 lysine 27 acetylation (H3K27Ac). Accordingly, HDAC7 stabilization diminished transcriptional induction of cytokine genes upon re-stimulation. Inhibition of this pathway during differentiation produced effector T cells epigenetically poised for enhanced cytokine recall. This work identifies a novel target for enhancing effector T cell functionality.
INTRODUCTION
Elucidation of pathways regulating T cell differentiation is important to the advancement of our mechanistic understanding of these processes and their function in various immunological settings including cancer, autoimmunity, infectious disease, and vaccine development. T cell activation by antigen recognition initiates epigenetic reprogramming to promote transcriptional expression of genes associated with T cell differentiation and the acquisition of effector function (1–4). At the core of these induced genes, are hallmark lineage specifying transcription factors and cytokines that define each effector subset (5–7). Upon differentiation, effector T cells assist in the clearance of the immunological challenge as antigen receptor activation induces T cell degranulation resulting in the directed release of effector molecules, including lineage specific cytokines. Following resolution of the immunological insult, effector T cells contract, and long-lived memory is established (8, 9). The dynamic nature of the initial establishment and subsequent restriction of effector functionality highlights temporal differences in regulating T cell differentiation. Accordingly, the need to understand both early and late-stage T cell differentiation processes is of essential importance.
The primary pathways associated with these varied processes have been defined over many years. However, significant interest remains in identifying additional pathways that regulate T cell differentiation to further refine our understanding and ability to modulate T cell functionality. Herein, we report the identification of salt-inducible kinases (SIKs) as novel regulators of effector cytokine production in effector CD8+ and Th1 cells. We demonstrate that effector T cell SIK activity leads to phosphorylation-dependent stabilization of the class IIa histone deacetylase (HDAC), HDAC7, during the latter stages of differentiation. This enhanced stability of HDAC7 correlates with increased nuclear HDAC7 levels, reduced total and cytokine loci specific histone 3 lysine 27 acetylation (H3K27Ac) levels and restricted cytokine production. Moreover, inhibition of this pathway, using the kinase inhibitor, BioE-1197, durably programs enhanced cytokine functionality by effector T cells.
MATERIALS AND METHODS
Mice
All procedures were approved by the Johns Hopkins University Institutional Animal Care and Use Committee under protocol numbers M019M71 and M022M271. OT-I (RRID:IMSR_JAX:003831), OT-II (RRID:IMSR_JAX:004194), CD90.1 (RRID:IMSR_JAX:000406), CD45.1 (RRID:IMSR_JAX:002014), and C57Bl/6J (RRID:IMSR_JAX:000664) mice were originally acquired from The Jackson Laboratory. P14 TCR transgenic mice were kindly given by Dr. David A. Hildeman (Cincinnati Children’s Hospital Medical Center). PASK knockout (KO) mice were a generous gift from Dr. Jared Rutter (University of Utah School of Medicine) (10, 11). Mice were housed in a specific pathogen-free facility. Six- to 20- week-old male and female WT C57BL/6J, OT-I, OT-II, P14, and PASK KO mice were used as sources for primary murine CD8+ and CD4+ T cells for in vitro culture. Six- to eight-week-old male C57BL/6J mice were used as hosts for adoptive transfer experiments. Males were specifically used based on sex matching to the co-adoptively transferred congenic T cells.
Drugs
BioE-1197 was synthesized using the previously reported method (12). TMP269 (Cat# HY-18360) and YKL-05-099 (Cat# HY-101147) were purchased from MedChemExpress. HG-9-91-01 was purchased from Selleckchem (Cat# S8393). Drugs were solubilized in DMSO (ThermoFisher Scientific Cat# D128-500).
In vitro T cell culture
Primary murine T cells were isolated from the spleen and inguinal, axillary, and brachial lymph nodes of mice. Following the generation of a single-cell suspension by dissociation through a 70 μM filter (Fisherbrand, 22-363-548), T cells were enriched using negative isolation. CD8+ T cells were isolated using MojoSort Mouse CD8 T Cell Isolation Kits (BioLegend, 480008) using eight microliters of biotin-antibody cocktail and streptavidin beads per mouse. Untouched CD8+ T cells were then purified by magnetic separation using LS columns (Miltenyi Biotec Cat# 130-042-401). Naïve CD4+ murine T cells were isolated using Naïve CD4+ T Cell Isolation Kits and LS columns (Miltenyi Biotec Cat# 130-104-453) according to manufacturer’s protocol. T cells were activated and expanded in complete RPMI. Complete RPMI was prepared by supplementing RPMI with 10% FBS (Gemini BioProducts Cat# 100-106), 2mM L-Glutamine (ThermoFisher Scientific Cat# 25030-081), 10 mM HEPES (Corning Cat# 25-060-Cl), 100 U/mL penicillin/streptomycin (ThermoFisher Scientific Cat# 15140-122), 1:100 MEM NEAA (ThermoFisher Scientific Cat# 11140050), 55 μM 2-mercaptoethanol (ThermoFisher Cat# 21985023), and 50 μg/mL gentamycin (Quality Biological Cat# 120-098-661).
CD8+ and CD4+ T cells were activated with plate bound anti-CD3 (5μg/mL, BioXCell Cat# BP0001-1) and soluble anti-CD28 (2μg/mL, BioXCell Cat# BE0015-1). CD4+ T cells were additionally cultured with 10ng/mL IL-12p70 (Peprotech Cat# 210-12), 10ng/mL IFNγ (Peprotech Cat# 315-05) and 5ug/mL anti-IL-4 (BioXCell Cat# BE0045) at the time of activation for proper Th1 skewing. T cells were seeded for activation at a density of 1 × 106 cells/mL. After 48 hours, T cells were removed from the activating plate and expanded by two-fold expansion with IL-2 (10 ng/mL, Peprotech Cat# 212-12). These cells were maintained by two-fold IL-2 expansion every 48 hours thereafter, until the experimental endpoint was met. BioE-1197 was administered at the time of activation, 24 hours after activation and at each addition of IL-2 for expansion to maintain pathway inhibition during T cell activation and differentiation. The final concentration of BioE-1197 for each dosing was 50 μM based on previous studies (13, 14). The pan-SIK inhibitors, HG-9-91-01 and YKL-05-099, were administered at the same intervals as BioE-1197 at a final concentration of 25 nM and 270 nM, respectively. The class IIa HDAC inhibitor, TMP269, was administered at the time of expansion in IL-2, so as to only inhibit HDAC activity during the time frame in which BioE-1197 diminishes HDAC7 stability, at a concentration of 12.5 μM. Control differentiation conditions for all experiments received an equivalent volume of the vehicle, DMSO.
Cytokine re-stimulation
On day six after initial activation, the remaining live T cells were purified by Ficoll density centrifugation (Ficoll-Paque Plus, Cytiva Cat# 17144002). Cells were then reactivated by plate bound anti-CD3 (1μg/mL) and anti-CD28 (1μg/mL) at a density of 1 × 106 cells/mL. For intracellular cytokine staining, one hour after reactivation, Monensin and Brefeldin A protein transport inhibitors were added at a dilution factor of 1:1000 (BD, Golgi-Stop Cat# 554724, Golgi-Plug Cat# 555029). After three and half hours of total re-stimulation, cells were harvested for intracellular cytokine staining and fixed using one percent methanol free paraformaldehyde (Thermofisher Scientific Cat# 28906) for ten minutes at 37⁰ C. For ELISAs, supernatants were harvested three and a half hours after reactivation and flash frozen in liquid nitrogen. IFNγ and TNFα ELISAs (eBioscience Cat# 88-7314 and 88-7324) were performed per manufacture’s protocol.
P14 LCMV park and recall
Single cell suspensions of splenocytes from congenically-marked, littermate P14 TCR transgenic mice were generated by dissociation through a 70 μM filter. CD8+ T cells were then activated by culturing whole splenocytes in complete RPMI at a density of 4 × 106 cells/mL with LCMV gp33-41 peptide (1 μg/mL, KAVYNFATM, JHMI Synthesis & Sequencing Facility). Cells were expanded and dosed according to the procedure described in In vitro T cell culture. On day six after activation, live cells were isolated by ficoll density centrifugation (Ficoll-Paque Plus, Cytiva Cat# 17144002). Control differentiated (Thy1.1+/Thy1.2+) and BioE-1197 differentiated (Thy1.1+) CD8+ T cells were mixed 1:1 and 1 × 106 cells of each population were co-adoptively transferred into WT C57Bl/6J (Thy1.2+) mice by retro-orbital injection. 32 days after co-adoptive transfer, the mice were challenged with LCMV Armstrong (2 × 105 PFU) by intra-peritoneal injection. LCMV-Armstrong virus was a gift originally obtained from Susan Kaech (Salk Institute for Biological Studies). Five to eight days after infection, splenocytes were harvested and reactivated with gp33-41 peptide for three and a half hours with administration of Monensin and Brefeldin A (1:1000) protein transport inhibitors for ex vivo re-stimulation analysis and intracellular cytokine staining.
Flow cytometry
Viability was assessed by staining with eBioscience Fixable Viability Dye eFluor 780 (ThermoFisher Scientific Cat#, 65-0865-14). Antibodies against the following proteins were purchased: IFNγ BV786 (BD Biosciences Cat# 563773, RRID:AB_2738419), TNFα BV421 (BD Biosciences Cat# 563387, RRID:AB_2738173), CD8 BV605 (BioLegend Cat# 100744, RRID:AB_2562609), CD4 BV605 (BioLegend Cat# 100548, RRID:AB_2563054), Thy1.1 AF700 (BioLegend Cat# 202528, RRID:AB_1626241), Thy1.2 BV510 (BioLegend Cat# 140319, RRID:AB_2561395), Tbet eF660 (Thermo Fisher Scientific Cat# 50-5825-82, RRID:AB_10596655), Eomes PerCP-eFluor710 (Thermo Fisher Scientific Cat# 46-4877-42, RRID:AB_2573759), Granzyme B PE/Dazzle 594 (BioLegend Cat# 372216, RRID:AB_2728383), Perforin PE (BioLegend Cat# 154306, RRID:AB_2721639), and H3K27Ac AF488 (Cell Signaling Technology Cat# 15485, RRID:AB_2798743). Permeabilization of cells for intracellular staining after fixation was achieved using eBioscience Permeabilization Buffer (ThermoFisher Scientific Cat# 00-8333-56) per manufacturer’s instructions. To ensure proper identification of positive signal and quantification, a fluorescence minus one (FMO) was prepared and ran for every experimental protein of interest across all experimental conditions. All samples were run on a FACSCelesta (BD Biosciences) and analyzed using FlowJo_v10.6.2.
LKB1 deletion by CRISPR Cas9/sgRNA complex electroporation
Electroporation of Cas9/sgRNA complexes for target gene deletion in naïve T cells was completed as previously described (15). Briefly, naïve T cells were isolated as described above and 10 × 106 cells were used per sgRNA target. Cas9/sgRNA complexes were prepared in a total volume of five microliters containing Alt-R S.p. Cas9 Nuclease V3 (6ug, IDT Cat# 1081059) and sgRNA (0.3 nmol, Synthego) diluted in RNase free water. Complexes were allowed to form for 10 minutes at room temperature. PBS washed, naïve T cells were then pelleted and re-suspended in 20 μL of P3 solution (P3 primary cell 4D Nucleofector electroporation kit, Lonza Cat# V4XP-3032), transferred to tubes containing complexed Cas9/sgRNA, and mixed. This solution was then transferred to the electroporation cassette and electroporated with a Lonza 4D-Nucleofector System using the pulse code DS-137. Immediately after electroporation, the cells were supplemented with complete RPMI and allowed to rest at 37°C for ten minutes. Subsequently, the P3 solution was washed out with complete RPMI, the cells were pelleted, and re-suspended in fresh complete RPMI supplemented with IL-7 (10ng/mL, Peprotech Cat# 217-17) and seeded at a density of 2 × 106 cells/mL. Cells were rested in IL-7 for at least two days prior to activation to allow for genomic editing and turnover of the target protein pool. The sgRNAs utilized in this study were synthesized by Synthego. The sgRNA sequence utilized for the control sgRNA was 5’-CUCCUUUAGAUCAUUCCUUG-3’ and targets the ROSA locus. The targeted sequence for the LKB1 specific sgRNA was 5’-CCAGGCCGUCAAUCAGCUGG-3’. The LKB1 specific sgRNA was identified using Synthego’s CRISPR design tool.
Western blot
Samples were flash-frozen at the time of harvest and then lysed in radioimmunoprecipitation (RIPA) buffer supplemented with 25x protease inhibitor cocktail (Sigma-Aldrich Cat# 11836145001), 1μM Sodium Orthovanadate (Millipore Sigma Cat# 450243), 10μM Sodium Pyrophosphate (Millipore Sigma, S6422), 50μM Sodium Fluoride (Millipore Sigma Cat# 7920), 1mM Phenylmethylsulfonyl Fluoride (Millipore Sigma Cat# 1083709100), and 10μM β-glycerophosphate (MilliporeSigma Cat# G9422). RIPA consists of 100mM Tris-HCl pH 8.0 (Quality Biological Cat# 351-007-101), 300mM NaCl (MilliporeSigma Cat# S5886), 2% NP-40 (MilliporeSigma Cat# 13021), 1% Sodium Deoxycholate (MilliporeSigma Cat# 30970), and 0.2% SDS (MilliporeSigma Cat# L6026). Lysate was clarified and quantified using Pierce Coomassie Plus (Bradford) Assay (ThermoFisher Cat# 1856209) with a SpectraMax M3 plate reader (Molecular Devices). Equivalent amounts of protein were loaded and separated on 4-12% Bis-Tris Mini Protein Gels (Invitrogen) and then transferred to PVDF membrane (BioRad Cat# 1620177). Blots were blocked with five percent milk at room temperature and primary antibodies incubated in four percent BSA overnight at four degrees. Primary antibodies used in these studies include anti-phosphorylated-HDAC4/5/7 (Cell Signaling Technology Cat# 3443, RRID:AB_2118723), anti-HDAC7 (Cell Signaling Technology Cat# 33418, RRID:AB_2716756), anti-Actin (Sigma-Aldrich Cat# A2066, RRID:AB_476693), anti-LKB1 (Cell Signaling Technology Cat# 3047, RRID:AB_2198327), anti-PASK (Cell Signaling Technology Cat# 3086, RRID:AB_2159082), anti-Lamin B (Santa Cruz Biotechnology Cat# sc-6216, RRID:AB_648156), and anti-IκBα (Cell Signaling Technology Cat# 4814, RRID:AB_390781). Secondary antibodies for detection include anti-Rabbit-HRP (Cell Signaling Technology Cat# 7074, RRID:AB_2099233), anti-mouse-HRP (Cell Signaling Technology Cat# 7076, RRID:AB_330924) and anti-Goat-HRP (Santa Cruz Biotechnology Cat# sc-2354, RRID:AB_628490). Signal was detected using SuperSignal West Pico PLUS Chemiluminescent Substrate or SuperSignal West Femto Maximum Sensitivity Substrate (ThermoFisher Scientific Cat# 34580 and 34095). Images were captured with the UVP Biospectrum500 imaging system or the BioRad ChemiDoc Imaging system.
Nuclear and cytosolic fractionation
At time of fractionation, the remaining live T cells were purified by Ficoll density centrifugation (Ficoll-Paque Plus, Cytiva Cat# 17144002), counted and equal cell number was used between treatment groups. Buffer A consists of 10mM HEPES (Corning Cat# 25-060-Cl), 10mM KCl (Quality Biological 351-044-101), 0.1 mM EGTA (Amresco Cat# 0372), and 0.1 mM EDTA (Corning Cat# 46-034-Cl), and was prepared and supplemented with 25x protease inhibitor cocktail (Sigma-Aldrich Cat# 11836145001), 1mM DTT (Millipore Sigma Cat# D9779), 1μM Sodium Orthovanadate (Millipore Sigma Cat# 450243), and 1mM Phenylmethylsulfonyl Fluoride (Millipore Sigma Cat# 1083709100). Cells were allowed to swell on ice with buffer A for fifteen minutes. An equivalent volume of buffer A, supplemented with 2% NP-40 (MilliporeSigma Cat# 13021), was then added and cells were vortexed, vigorously for 30 seconds. Nuclei were then pelleted at 15,000 rpm for 30 seconds. The supernatant containing the cytosolic fraction was then transferred to a new tube and the nuclear pellet was lysed in RIPA and subsequently clarified. Equal volumes were loaded between samples, separated on 4–12% Bis-Tris Mini Protein Gels (Invitrogen) and subjected to western blot. Fractionation was confirmed using the nuclear and cytosolic loading controls Lamin B and IκBα, respectively.
RNA isolation and qRT-PCR for mRNA transcript abundance
Cellular pellets were flash frozen upon harvest. Pellets were solubilized in Trizol (ThermoFisher Scientific Cat# 15596018) and RNA was extracted by addition of chloroform (FisherScientific Cat# C298-500) based on manufacture’s protocol. In brief, after mixing by inversion, layers were separated by centrifugation at four degrees Celsius. The upper, aqueous layer was collected and RNA was precipitated using isopropanol (FisherScientific Cat# A416-500) at −20° C overnight. The next day, RNA was pelleted and washed with 70% ethanol two times (FisherScientific Cat# 2016012). RNA pellets were allowed to air dry for 10 minutes at room temperature and then were re-suspended in water. RNA was quantitated using the Spectra Max M3 (Molecular Devices) plate reader and equivalent amounts of cDNA were generated per condition using ProtoScript II Reverse Transcriptase (NEB Cat# M0368L) and random hexamers (Qiagen Cat# 79236) according to manufacturer’s protocol. qRT-PCR for Ifng (ThermoFisher Scientific Cat# 4331182, Taqman probe: Mm01168134_m1), Tnf (ThermoFisher Scientific Cat# 4331182, Taqman probe: Mm00443258_m1), Gzmb (ThermoFisher Scientific Cat# 4331182, Taqman probe: Mm00442834_m1), Prf1 (ThermoFisher Scientific Cat# 4331182, Taqman probe: Mm00812512_m1), and Hdac7 (ThermoFisher Scientific Cat# 4331182, Taqman probe: Mm00469527_m1) were normalized to Rn18s (Thermofisher Scientific Cat# 4310893E) and run on a Step One Plus real time PCR system (Applied Biosystems) using EagleTaq universal master mix (SigmaAldrich Cat# 7260288190). Fold change differences in target gene expression were calculated by 2−ΔΔCt.
H3K27Ac chromatin immunoprecipitation followed by sequencing (ChIP-Seq)
Day six CD8+ T cells differentiated in a vehicle control or BioE-1197 were prepared as described above. Following Ficoll density centrifugation, the remaining live cells were fixed with one percent methanol free paraformaldehyde (Thermofisher Scientific Cat# 28906) at a density of 1 × 106 cells/mL in complete RPMI for 10 minutes at room temperature, while shaking. Fixation was terminated by addition of glycine (Millipore Sigma Cat# G7126) to a final concentration of 125 μM. Samples were incubated on ice for five minutes and then pelleted at 2000 rpm and washed two times with cold PBS. Pellets were flash frozen with liquid N2 and stored at −80⁰ C.
ChIP-Seq was performed as has been previously detailed (16). In brief, nuclei were harvested by sequential lysis in lysis buffer one (LB1), two (LB2), and three (LB3) which were supplemented with protease inhibitor (ThermoFisher Scientfic Cat# 78429; 1X final concentration added right before use). LB1 consists of 50mM HEPES-KOH (pH 7.5, MilliporeSigma Cat# H4034 & P9333), 140mM NaCl (MilliporeSigma Cat# 7653), 1mM EDTA (pH 8.0, MilliporeSigma Cat# E6758), 10% glycerol (MilliporeSigma Cat# G5516), 0.5% Igepal CA-630 (MilliporeSigma Cat# I8896), 0.25% Triton X-100 (MilliporeSigma Cat# T8787) and KOH to adjust pH to 7.5. LB2 consists of 10mM Tri-HCl pH 8.0 (Quality Biological Cat# 351-007-101), 200 mM NaCl, 1mM EDTA pH 8.0, 0.5 mM EGTA and HCl to adjust pH to 8.0. LB3 consists of 10mM Tris-HCl (pH 8.0), 100 mM NaCl, 1mM EDTA, 0.5 mM EGTA, 0.1% Sodium deoxycholate (MilliporeSigma Cat# 30970), 0.5% N-lauroylsarcosine (MilliporeSigma Cat# L7414) and HCl to adjust pH to 8.0. Cells were re-suspended in LB1, rotated for 10 minutes at four degrees Celsius, pelleted at 2000 rpm, re-suspended in LB2 and rotated for 5 minutes at four degrees Celsius. Samples were then pelleted at 2000 rpm and re-suspended in LB3. Nuclei were then sonicated at four degrees Celsius using a Fisher 150E Sonic Dismembrator with settings: 50% amplitude, 30 s ON, 30 s OFF for 12 min total time. Following sonication, samples were pelleted to remove cellular debris at 20,000g for 10 minutes at four degrees. The supernatant was transferred and the remainder was utilized for ChIP with anti-H3K27Ac (Abcam Cat# ab4729, RRID:AB_2118291).
50 μL Protein A beads (Thermo Fisher Scientific Cat # 10002D) were used per IP and transferred to a 2 mL tube on a magnetic stand. Beads were washed twice with blocking buffer (BB: 0.5% BSA in PBS), then resuspended in 100 μL BB per IP. For each sample, 3μg of anti-H3K27Ac antibody (Abcam Cat# ab4729, RRID:AB_2118291) was added to the beads, and the mixture was incubated with rotation at room temperature for one to three hours. The 2 mL tube was then placed on a magnetic rack and washed three times with BB, before resuspending in 50 μL BB per IP. 50 μL of beads in BB were transferred to each IP sample. Samples were immunoprecipitated overnight at four degrees Celsius. The next day, samples were transferred to a 1.5 mL LoBind tube on a magnetic stand, washed six times with RIPA buffer (50 mM HEPES, 500 mM LiCl, 1 mM EDTA, 1% Igepal CA-630, 0.7% Na-Deoxycholate, pH to 7.5 using KOH) and one time with TBS buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl). Chromatin IP samples were reverse crosslinked overnight in elution buffer (50mM Tris-HCl pH 8.0, 10 mM EDTA pH 8.0, and 1% SDS). DNA was extracted from reverse crosslinked chromatin samples by incubation with RNaseA (Thermo Fisher Scientific Cat# EN0531) for 15 min at 37C; and proteinase K (Thermo Fisher Scientific Cat# AM2546) for 1h at 65C. DNA was subsequently purified using MinElute (Qiagen Cat# 28004) purification.
DNA libraries were prepared and amplified using NEBNext Ultra II DNA Library Prep Kit for Illumina (New England BioLabs Cat# E7103L) and dual-indexed eith the NEBNext Multiplex Oligos for Illumina (New England BioLabs Cat# E6440S). Final libraries were pooled, quantified with QuBit (Thermo), Bioanalyzer (Agilent) and qPCR (BioRad), then sequenced on a Novoseq SP 100 flow cell (Illumina) using paired-end 2×50bp reads at the Johns Hopkins Single Cell and Transcriptomics Core.
ChIP-Seq data analysis
Reads were demultiplexed using bcl2fastq, and were aligned to the mm9 genome using bowtie2 (17). Samtools (18) was used to filter for mapping quality >= 25, remove singleton reads, convert to BAM format, remove potential PCR duplicates, and index reads. Deeptools (19) was then used to perform a running window average (binsize of 10 bp) of BAM files, normalized by RPKM and converted into the final bigwig files. These bigwig files were plotted using PyGenomeTracks (20) with standard input parameters for Control and BioE tracks, and the subtraction operation used for Enrichment tracks. Data files associated with this analysis have been deposited at GEO (GSE242482 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE242482).
Kinase domain alignment
The protein sequences for the kinase domain of PASK and the LKB1 activated members of the ARK family were acquired from Uniprot and aligned using T-Coffee (21). The alignment was visualized and colored for percent identity using Jalview (22).
Data presentation and analysis
For flow cytometric analysis of in vitro cultured CD8+ or CD4+ T cells prior to or during re-stimulation, representative dot plots and quantification of cytokine, transcription factor, and histone marks were prepared by gating for lymphocytes, single cells, and then either CD8+ or CD4+. For flow cytometric analysis of in vitro cultured CD8+ or CD4+ T cells during differentiation, quantification of H3K27Ac histone marks was achieved by gating for lymphocytes, single cells, live cells, and then either CD8+ or CD4+. In vivo co-adoptively transferred CD8+ T cells were identified by gating for lymphocytes, single cells, live cells, CD8+, and Thy1.1+. Then, vehicle control cells (Thy1.1+/1.2+) were distinguished from BioE-1197 (Thy1.1+) treated cells by gating for Thy1.2 expression. For all flow cytometry data, gates were set for proteins of interest based on an FMO. For quantification of gMFI values, the FMO for each experimental condition was subtracted from the full stain gMFI values to account for any baseline differences in fluorescence between treatment groups. Flow data for gMFI are presented as a fold change to the average of the control condition for each individual experiment.
Western blot signal intensity was quantified by densitometry with Visionworks software (Analytik Jena). For each western blot sample quantified, a background densitometric reading was measured within the lane of that sample and subtracted from the total signal. Phosphorylated HDAC7 and total HDAC7 signal cannot be stripped from the membrane so they are probed on separate, duplicate western blots from the same experimental lysate. Western blots are presented as a fold change to the control of each independent experiment. Fold change quantification of phosphorylated and total HDAC7 over time during differentiation in control and BioE-1197 differentiated T cells were calculated to the day one control data. This strategy was employed to quantitate change over time as the signal intensity in some experiments of the naïve (day 0) control was too low to be able to accurately calculate a fold change (an extremely small value, or zero).
For all statistical analyses, normality of the collected data set was evaluated. If, normality was confirmed or could not be evaluated due to too small of sample size, then the appropriate parametric statistical test was utilized to compare differences between treatment groups. If the normality assumption of a data set was not met, then the non-parametric statistical test was employed. Experiment specific statistical tests are described in their respective figure legends. All statistical analyses were conducted using Graphpad Prism version 9.5.0.
RESULTS
BioE-1197 stably enhances effector cytokine production independent of PASK
We sought to evaluate the role of the PAS domain-containing serine/threonine kinase (PASK)/WD repeat domain 5 (WDR5) pathway in regulating T cell differentiation. Mammalian target of rapamycin complex 1 (mTORC1) dependent activation of PASK has been shown to initiate epigenetic induction of WDR5 mediated effector differentiation programs across multiple cell types (13, 14). The conserved nature of this pathway prompted us to query whether T cells also utilize PASK to regulate their effector differentiation programs. To this end, CD8+ T cells were activated and expanded with IL-2 in either the presence of a vehicle control or the previously described PASK inhibitor, BioE-1197 (23). Effector differentiation was assessed by evaluating cytokine recall potential through the measurement of effector cytokine production upon re-stimulation, six days after initial activation. CD8+ T cells differentiated in the presence of BioE-1197 had significantly enhanced IFNγ and TNFα effector cytokine production when re-stimulated compared to control differentiated T cells based on increases in both the percent of cells positive for intracellular cytokine staining and geometric mean fluorescence intensity (gMFI) (Fig. 1 A–C). This enhanced cytokine production was further confirmed by assessing cytokine release in response to re-stimulation by ELISA (Fig. 1D).
Figure 1. Administration of BioE-1197 during T cell differentiation durably enhances effector cytokine production upon re-stimulation in CD8+ T cells, independent of PASK.
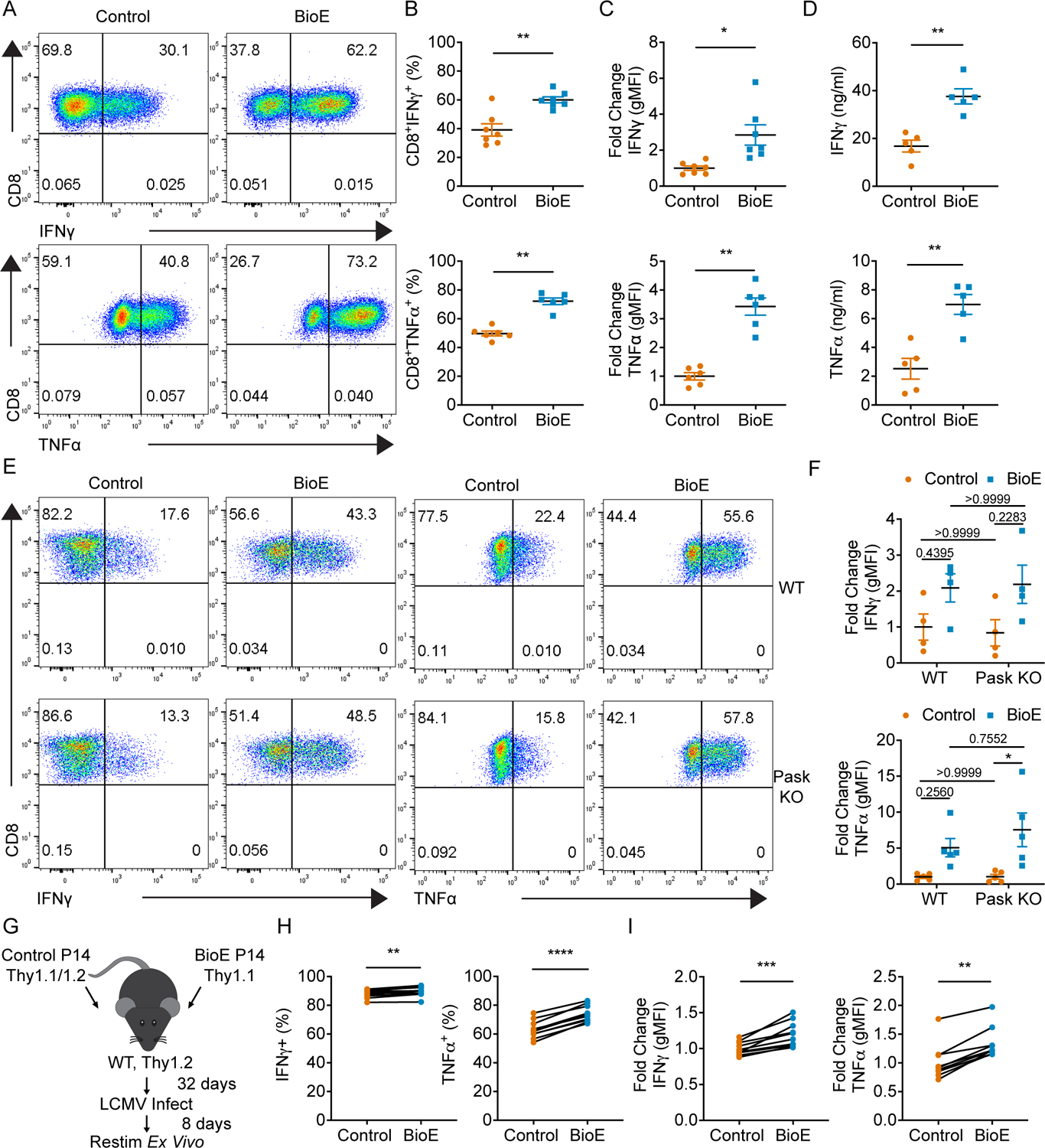
(A) Representative intracellular cytokine staining dot plots of IFNγ (top) and TNFα (bottom) production upon re-stimulation of CD8+ T cells on day six after activation and differentiation in control (DMSO) or BioE-1197 (50μM) conditions. (B) Quantification of the percent of IFNγ (top) and TNFα (bottom) cytokine positive cells represented in A across independent experiments. (C) Quantification of the fold change in geometric mean fluorescence intensity (gMFI) of IFNγ (top) and TNFα (bottom) production by CD8+ T cells represented in A across independent experiments. (D) Quantification of IFNγ (top) and TNFα (bottom) production in the supernatants of re-stimulated CD8+ T cells across independent experiments by ELISA. (E) Representative dot plots of IFNγ (left) and TNFα (right) production upon re-stimulation of WT (top) or PASK knockout (KO) (bottom) CD8+ T cells differentiated in control (DMSO) or BioE-1197 (50μM) conditions. (F) Quantification of the fold change in gMFI of IFNγ (top) and TNFα (bottom) production by WT and PASK KO CD8+ T cells represented in E across independent experiments. IFNγ: Fgenotype p = 0.3640, FBioE-1197 p = 0.0012. TNFα: Fgenotype p = 0.9442, FBioE-1197 p = 0.0133. (G) Schematic representing the experimental design for park and recall co-adoptive transfer of control (DMSO) and BioE-1197 (50μM) differentiated P14 CD8+ T cells. (H) Quantification of the percent of cytokine positive cells for IFNγ and TNFα upon ex vivo peptide re-stimulation and intracellular cytokine staining across ten mice. (I) Quantification of the fold change in gMFI for IFNγ (left) and TNFα (right) upon ex vivo peptide re-stimulation and intracellular cytokine staining across ten mice. Each dot represents values from an independent experiment, summary data are presented as the mean (black line) with SEM error bars (B-D, F). Each dot represents an individual mouse within one independent experiment and presented results are representative of two independent experiments (H, I). Paired t test (B, C, D). Two-way ANOVA, Sidak’s multiple comparisons (F). Paired t test (H, I – IFNγ). Wilcoxon test (I-TNFα). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
While BioE-1197 enhanced cytokine production in CD8+ T cells, there was no observed increase in expression of the lineage specifying transcription factor, T-bet, by either percent or gMFI intensity (Fig. S1 A–C). Additionally, other effector molecules associated with the CD8+ T cell effector differentiation program, Granzyme B and Perforin, did not exhibit BioE-1197-dependent regulation like the cytokine effector molecules. There was no observed difference in Granzyme B expression between control and BioE-1197 differentiated CD8+ T cells either by percent or gMFI (Fig. S1 D–F), and while there was a statistically significant increase in Perforin expression by percent between control and BioE-1197 treated cells (16.5% versus 21.8%), there was no difference by gMFI and these changes were minimal compared to those observed for cytokine production (Fig. S1 G–I). Together these data suggest BioE-1197 was acting to selectively regulate the CD8+ T cell effector cytokine program.
Despite the pronounced ability of the PASK inhibitor, BioE-1197, to enhance effector cytokine production, differentiated PASK knockout CD8+ T cells did not have elevated IFNγ and TNFα cytokine production upon re-stimulation. Moreover, differentiation of PASK knockout CD8+ T cells in the presence of BioE-1197 still enhanced cytokine production to levels similar to that of wild type (WT) cells (Fig. 1 E–F). These data indicated that BioE-1197 enhances CD8+ T cell effector cytokine production in a PASK-independent manner, thus illuminating a novel mechanism of BioE-1197 involved in regulation of cytokine recall potential in CD8+ T cells.
In addition to revealing a novel target of BioE-1197, these findings suggested BioE-1197 produced a potentially durable phenotype as the cells were washed and no BioE-1197 was added during the re-stimulation assay to actively increase cytokine production. While the exact half-life of BioE-1197 is unknown, these data could suggest that the PASK-independent pathway inhibited by BioE-1197 is active during T cell differentiation, rather than during re-stimulation. Inhibition of this pathway during the course of differentiation could, therefore, result in the stable programming of enhanced cytokine recall, instead of directly enhancing cytokine production during recall. To further assess the sustainability of BioE-1197 enhanced cytokine programming, we generated CD8+ lymphocytic choriomeningitis virus (LCMV) specific P14 T cells treated with either a vehicle or BioE-1197 in vitro and co-adoptively transferred these cells, at a 1:1 ratio, into naïve WT C57BL/6J hosts. We let these T cells rest in the naive hosts for 32 days and then recalled the T cells with LCMV infection. After challenge, we evaluated cytokine production by ex vivo peptide re-stimulation assays (Fig. 1 G). After this prolonged rest period, BioE-1197 differentiated CD8+ T cells still demonstrated enhanced cytokine production compared to their control differentiated counterparts (Fig. 1 H–I) without any observed differential expression of T-bet or eomes, transcription factors known to regulate effector cytokine production (Fig S1 J–K). The stable elevation of enhanced cytokine production by BioE-1197 differentiated cells in this park and recall model further supports the hypothesis that the novel target of BioE-1197, capable of regulating effector cytokine production, is active during CD8+ T cell differentiation. Thus, we sought to identify the unknown pathway inhibited by BioE-1197 during effector differentiation that regulates cytokine recall potential in CD8+ T cells.
BioE-1197 activity is dependent on AMP activated protein kinase (AMPK) related kinases (ARKs)
To elucidate how BioE-1197 stably enhances effector CD8+ T cell cytokine production upon re-stimulation, we sought to identify candidate kinases that could be inhibited by BioE-1197. To this end, we utilized the protein basic local alignment search tool (BLAST) to query the amino acid sequence corresponding to the kinase domain of PASK to identify similar kinases. This analysis revealed 13 members of the ARK family as potential targets of BioE-1197 (Fig. S2). Based on this result, if BioE-1197 enhanced cytokine recall was dependent on ARK family inhibition, it could be due to the selective inhibition of a specific member of this family or the collective inhibition of all/many members of this family. As activation of each of these ARK family members is mediated by liver kinase B1 (LKB1) T-loop phosphorylation (24), we first sought to evaluate whether BioE-1197 enhanced cytokine recall was dependent on ARKs by knocking out the shared upstream activating kinase, LKB1, in naïve CD8+ T cells. While prior studies have demonstrated important roles for LKB1 in T cell development, survival and activation, these studies have not evaluated cytokine recall potential of LKB1 deficient T cells (25–28). Therefore, to knockout LKB1, naïve T cells were electroporated with either control or LKB1 specific Cas9/single guide RNA (sgRNA) complexes (15). Cells were then rested in IL-7 to maintain viability of these primary cells while allowing for genomic editing and LKB1 protein turnover prior to activation. Upon activation, control or LKB1 sgRNA edited T cells were differentiated in a vehicle control or BioE-1197 conditions and cytokine recall was evaluated. Particularly, by culturing LKB1 deficient cells in BioE-1197 differentiation conditions we would be able to assess whether LKB1 was necessary for enhanced effector cytokine production.
In these experiments, LKB1 deficient CD8+ T cells exhibited diminished survival compared to control edited CD8+ T cells (Fig. 2 A–B). While this is a phenotype previously described for LKB1 deficient CD8+ T cells (25–27), it is not a phenotype observed with BioE-1197 treatment (Fig. 2 A–B), suggesting BioE-1197 is not likely functioning to inhibit all LKB1 activated ARKs. Upon re-stimulation, vehicle control treated LKB1 deficient CD8+ T cells show no increase in IFNγ or TNFα cytokine production (Fig. 2 C–D), suggesting LKB1 deletion is not sufficient to elevate cytokine. However, BioE-1197 treated LKB1 deficient CD8+ T cells cannot elevate cytokine recall to the same extent as control edited CD8+ T cells differentiated in BioE-1197 (Fig. 2 C–D), suggesting LKB1 is necessary for BioE-1197 enhanced cytokine production. While not to the same extent as control edited cells, there is a minor increase in cytokine production in LKB1 deficient CD8+ T cells differentiated in BioE-1197 compared to vehicle control conditions. This slight increase could be due to incomplete knockout of LKB1 using CRISPR/Cas9 in a polyclonal setting, leaving some residual pathway activity to be regulated by BioE-1197 (Fig. 2 E). Nonetheless, the diminished ability of BioE-1197 to elevate cytokine production upon re-stimulation in LKB1 deficient cells compared to control edited cells suggests BioE-1197 enhanced cytokine recall is highly dependent on LKB1 activated ARK family member(s).
Figure 2. BioE-1197 activity is dependent on ARKs.
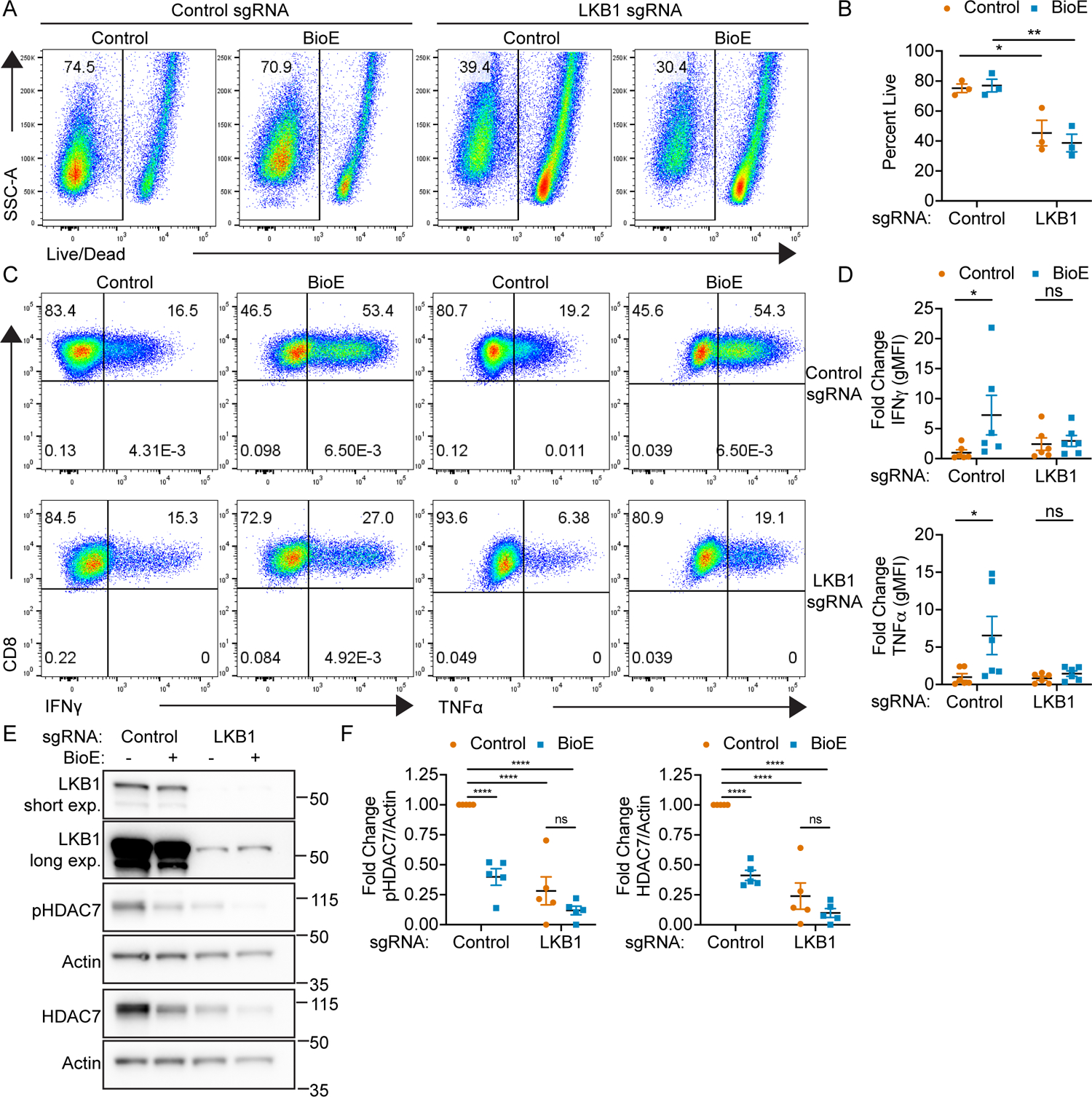
(A) Representative dot plots of Live/Dead staining of control or LKB1 sgRNA edited CD8+ T cells on day six after initial activation and differentiation in control (DMSO) or BioE-1197 (50 μM) conditions. (B) Quantification of percent live cells on day six after activation represented in A across independent experiments. (C) Representative dot plots of IFNγ (left) or TNFα (right) production in control (top) or LKB1 sgRNA (bottom) edited CD8+ T cells on day six after activation and differentiation in control (DMSO) or BioE-1197 (50μM) conditions. (D) Quantification of fold change in IFNγ (top) and TNFα (bottom) gMFI represented in C across independent experiments. (E) Representative western blot of control or LKB1 sgRNA edited CD8+ T cells on day six after activation and differentiation in control (DMSO) or BioE-1197 (50μM) conditions. (F) Quantification of the fold change in pHDAC7/Actin (left) and HDAC7/Actin (right) represented in E across independent experiments. Each dot represents values from an independent experiment, summary data are presented as the mean (black line) with SEM error bars (B, D, F). Two-way ANOVA, Sidak’s multiple comparison test (B, D). Two-way ANOVA, Tukey’s multiple comparisons test (F). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, ns = not significant.
Given the observed dependence of BioE-1197 enhanced cytokine recall on LKB1 activated ARK family member(s), we hypothesized that if BioE-1197 is functioning as an ARK family kinase inhibitor, then BioE-1197 differentiated T cells should have diminished phosphorylation of known ARK family substrates. Notably, many members of the LKB1 activated ARK family have been described to phosphorylate class IIa HDACs across numerous cell types (29–35). Given the broad ability of LKB1 activated ARKs to phosphorylate class IIa HDACs, we sought to further confirm BioE-1197 was functioning to inhibit ARK family member(s) during CD8+ T cell differentiation by evaluating class IIa HDAC phosphorylation. To this end, control and LKB1 sgRNA edited CD8+ T cells differentiated in control or BioE-1197 conditions were assessed for phosphorylation of class IIa HDACs on day six after activation. Control sgRNA edited CD8+ T cells exhibited distinct phosphorylation of the class IIa HDAC, HDAC7, that was expectedly diminished in LKB1 deficient CD8+ T cells. In addition to a reduction in phosphorylated HDAC7, LKB1 deficient CD8+ T cells also exhibited a decrease in total HDAC7 (Fig. 2 E–F). The concurrent reduction in phosphorylated and total HDAC7 protein in LKB1 deficient T cells was not unexpected as the evaluated HDAC7 phosphorylation site, S178, has been described to be critical for the stability of HDAC7 protein (36–38). Therefore, these results indicate LKB1 deficient CD8+ T cells have diminished phosphorylation dependent stabilization of HDAC7. These experiments additionally demonstrated that BioE-1197 differentiated CD8+ T cells exhibit diminished phosphorylation dependent stabilization of HDAC7 as these cells also have reduced phosphorylated and total HDAC7 (Fig. 2 E–F). The observed reduction in phosphorylated and total HDAC7 in either LKB1 deficient or BioE-1197 differentiated CD8+ T cells could be slightly enhanced by BioE-1197 treatment of LKB1 deficient T cells, although the effect did not reach statistical significance. This could also be explained by incomplete LKB1 deletion using electroporation of CRISPR/Cas9 sgRNA on a polyclonal population allowing for BioE-1197 mediated inhibition of residual LKB1 dependent pathway activity in the combination treatment (Fig. 2 E–F). Together, the reduced ability of BioE-1197 to elevate cytokine recall in LKB1 deficient T cells and the diminished phosphorylation of the known ARK substrate, HDAC7, further support BioE-1197 is acting as an inhibitor of ARK family member(s) during T cell differentiation. Additionally, these data reveal the inactivation of ARKs by either LKB1 deletion or BioE-1197 inhibition, during differentiation, leads to inhibition of phosphorylation dependent stabilization of HDAC7.
ARK family SIKs regulate HDAC7 phosphorylation dependent stabilization and effector cytokine production
While numerous ARK family members have been described to phosphorylate class IIa HDACs, a previous report has specifically identified the ARK family member, SIK1, as a regulator of HDAC7 phosphorylation dependent stabilization in cardiomyocytes (36). Given the necessity of LKB1 activated ARKs in BioE-1197 cytokine regulation and the observed inhibition of HDAC7 stability in LKB1 deficient and BioE-1197 treated CD8+ T cells, we sought to evaluate whether SIKs were responsible for HDAC7 stability and cytokine recall capacity in differentiating CD8+ T cells. To this end we employed two pan-SIK inhibitors, HG-9-91-01 and YKL-05-099, capable of inhibiting SIK1, 2 and 3. CD8+ T cells differentiated in the presence of either pan-SIK inhibitor reduced phosphorylation dependent stabilization of HDAC7, as both phosphorylated and total HDAC7 levels were decreased in HG-9-91-01 and YKL-05-099 differentiated effectors compared to control differentiation conditions (Fig. 3 A–B). Moreover, CD8+ T cells differentiated in the presence of pan-SIK kinase inhibition consistently exhibited elevated lineage specific cytokine production of IFNγ and TNFα upon re-stimulation (Fig. 3 C–D). These findings indicate inhibition of ARK family SIKs, during T cell differentiation, is sufficient to reproduce the effects observed with BioE-1197 with respect to HDAC7 stabilization and cytokine recall potential and further support BioE-1197 is functioning as a selective ARK family inhibitor.
Figure 3. SIKs regulate phosphorylation dependent stabilization of HDAC7 and lineage specific cytokine production in CD8+ T cells.
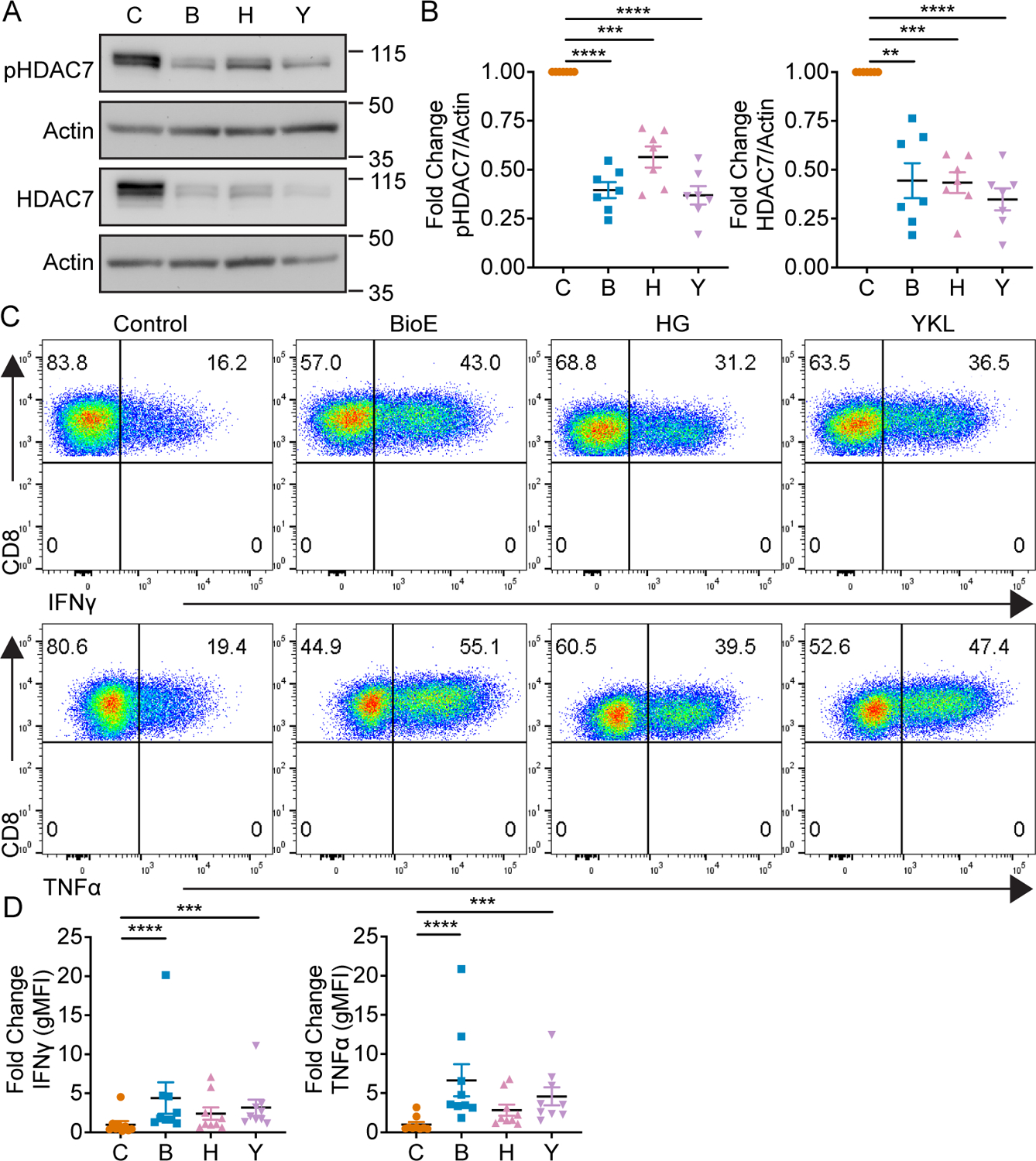
(A) Representative western blot of phosphorylated and total HDAC7 by CD8+ T cells on day four after activation and differentiation in either control (DMSO), BioE-1197 (50 μM), HG-9-91-01 (HG, 25 nM) or YKL-05–099 (YKL, 270 nM) differentiation conditions. (B) Quantification of the fold change in phosphorylated (left) and total HDAC7 (right) production in CD8+ T cells represented in A across independent experiments. (C) Representative intracellular cytokine staining dot plots of IFNγ (top) and TNFα (bottom) production by CD8+ T cells on day six after activation and differentiation in either control (DMSO), BioE-1197 (50 μM), HG-9-91-01 (HG, 25 nM) or YKL-05–099 (YKL, 270 nM) conditions. (D) Quantification of the fold change in gMFI of IFNγ (left) and TNFα (right) production in CD8+ T cells represented in C across independent experiments. Each dot represents values from an independent experiment, summary data are presented as the mean (black line) with SEM error bars (B, D). Repeated measures one-way ANOVA, Dunnet’s multiple comparisons test (B). Friedman test, Dunn’s multiple comparisons test (D). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, ns = not significant.
Phosphorylation dependent stabilization enhances nuclear availability of HDAC7
These collective data revealed LKB1 dependent activation of SIKs regulates HDAC7 phosphorylation dependent stabilization during T cell differentiation. To further confirm this is truly differential HDAC7 protein stability, we assessed Hdac7 mRNA transcript levels in CD8+ T cells activated and differentiated in either control or BioE-1197 conditions. On day six after differentiation, there was no difference in the mRNA expression of Hdac7 reflective of the differences we observed at the protein level (Fig. 4 A), further supporting the differential protein levels of HDAC7 observed between control and BioE-1197 differentiated CD8+ T cells is due to differential protein stability.
Figure 4. Phosphorylation dependent stabilization enhances nuclear availability of HDAC7 and epigenetically restricts effector cytokine loci.
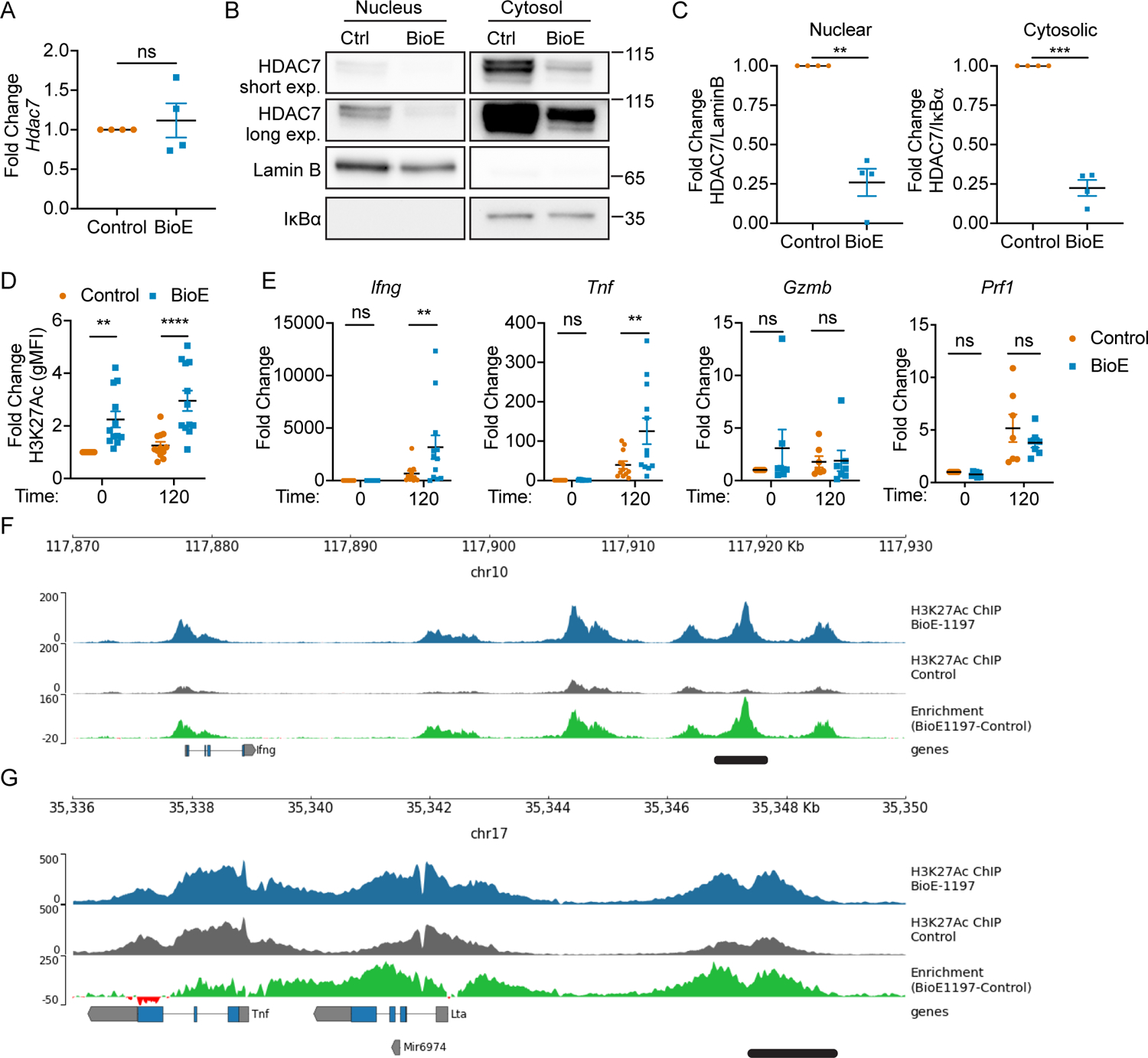
(A) Fold change in HDAC7 mRNA levels of CD8+ T cells on day six after activation and differentiation in control (DMSO) or BioE-1197 (50μM) conditions. (B) Representative western blot of HDAC7 from subcellular fractionation of CD8+ T cells on day three after activation and differentiation in control (DMSO) or BioE-1197 (50μM) conditions. Lamin B and IκBα serve as nuclear and cytosolic compartment loading controls, respectively. (C) Quantification of the fold change in HDAC7 localization represented in B across independent experiments in CD8+ T cells on day three after activation and differentiation in control (DMSO) or BioE-1197 (50μM) conditions. (D) Quantification of the fold change in global H3K27Ac levels for control (DMSO) and BioE-1197 (50μM) differentiated CD8+ T cells prior to re-stimulation and two hours after re-stimulation on day six after activation and differentiation across independent experiments. (E) Fold change of Ifng, Tnf, Gzmb, and Prf1 mRNA transcript abundance prior to re-stimulation and two hours after re-stimulation in CD8+ T cells on day six after activation and differentiation in control (DMSO) or BioE-1197 (50μM) across independent experiments. (F) H3K27Ac mark deposition within the Ifng loci in control and BioE-1197 (50μM) differentiated CD8+ T cells on day six after activation and differentiation. (G) H3K27Ac mark deposition within the Tnf loci in control and BioE-1197 (50μM) differentiated CD8+ T cells on day six after activation and differentiation. Each dot represents values from an independent experiment, summary data are presented as the mean (black line) with SEM error bars (A, C, D, E). Results are representative of three, independent biological replicates, additional replicates presented in Supplemental Figure 3 (F,G). Paired t-test (A,C). Two-way ANOVA, Sidak’s multiple comparisons test (D, E). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, ns = not significant.
Given HDAC7 functions as a transcriptional repressor (39) and BioE-1197 inhibition of phosphorylation dependent stabilization of HDAC7 results in reduced protein levels of this transcriptional repressor, we hypothesized the differential stability of HDAC7 in control and BioE-1197 differentiated cells may be responsible for differential cytokine production. Specifically, we anticipated diminished HDAC7 protein levels in BioE-1197 differentiated effector T cells should limit the amount of HDAC7 available in the nucleus of these cells to mediate transcriptional repression thus enhancing cytokine production. However, while phosphorylation of HDAC7 at S178 is stabilizing for HDAC7 protein, it is also known to disrupt the regular cycling of HDAC7 into and out of the nucleus, as the stabilizing interaction with 14-3-3 occurs within the cytosol (40–42). Therefore, to determine whether HDAC7 stability underlies differential cytokine production, we first assessed whether diminished stability of HDAC7 would result in diminished nuclear availability of HDAC7. Control and BioE-1197 differentiated CD8+ T cells were generated and nuclear and cytosolic localization of HDAC7 was assessed after activation by subcellular fractionation and western blot. This analysis revealed inhibition of HDAC7 stabilization with BioE-1197 impacts HDAC7 levels both within the cytosolic and nuclear compartments, with BioE-1197 treated effector T cells having lower nuclear levels of HDAC7 compared to control differentiated effector T cells (Fig. 4 B). Normalization of HDAC7 levels to subcellular compartment loading controls and calculation of a fold change relative to the control condition, for each compartment, further reveals BioE-1197 inhibition equivalently reduces both nuclear and cytosolic HDAC7 levels (Fig. 4 C). These observations suggest, during T cell differentiation phosphorylation dependent stabilization of HDAC7 functions to increase the total amount of HDAC7 in the cell and, therefore, the amount of HDAC7 that can be imported into the nucleus to mediate transcriptional repression.
The diminished nuclear HDAC7 protein levels in BioE-1197 differentiated cells further support the hypothesis that differential HDAC7 stability between control and BioE-1197 differentiated cells could lead to altered HDAC7 transcriptional repression and therefore cytokine regulation. HDAC7 transcriptional repression can be mediated epigenetically by the removal of the activating chromatin transcription mark, H3K27Ac. Therefore, we sought to assess whether differential nuclear HDAC7 levels in control and BioE-1197 differentiated T cells would specifically result in differential total H3K27Ac marks and epigenetic alterations in these conditions. To this end, control and BioE-1197 differentiated effector T cells were assessed for total H3K27Ac levels by flow cytometry on day six prior to and during re-stimulation. Notably, BioE-1197 treated CD8+ T cells have elevated H3K27Ac levels at baseline on day six compared to control differentiated CD8+ T cells, and during re-stimulation, H3K27Ac levels remain higher in BioE-1197 treated CD8+ T cells than control differentiated cells (Fig. 4 D). These data indicate control differentiated CD8+ T cells exhibit stabilized HDAC7 which correlates with low H3K27Ac levels. Alternatively, inhibition of HDAC7 stabilization by BioE-1197 during differentiation reduces nuclear HDAC7 levels correlating with increased H3K27Ac activating transcription marks, globally.
BioE-1197 administration transcriptionally enhances cytokine recall
Collectively these data demonstrate that enhanced HDAC7 stability during T cell differentiation correlates with an epigenetic restriction of H3K27Ac marks. These data, thus, support a model whereby, T cells utilize LKB1 activated SIKs to mediate phosphorylation dependent stabilization of HDAC7 to increase HDAC7 activity by increasing its nuclear availability. BioE-1197 differentiated cells, however, cannot stabilize HDAC7 through LKB1 mediated SIK activity. Therefore, these cells have diminished nuclear HDAC7 levels and elevated H3K27Ac marks. If this model were true and capable of regulating cytokine recall due to an epigenetically altered genomic landscape, one would expect BioE-1197 differentiated effector CD8+ T cells to have elevated cytokine transcript abundance upon re-stimulation in addition to the observed increases in cytokine at the protein level. To confirm this, we activated and differentiated CD8+ T cells in control or BioE-1197 differentiation conditions. On day six after activation, these cells were re-stimulated and effector molecule mRNA transcript abundance was assessed. Indeed, BioE-1197 differentiated CD8+ T cells have elevated cytokine transcript abundance two hours after re-stimulation compared to control differentiated effectors (Fig. 4 E), further supporting the proposed model. Importantly, granzyme B and perforin mRNA, which were not impacted by BioE-1197 treatment at the protein level, did not exhibit differences at the transcript level (Fig. 4 E). These data indicate LKB1 dependent SIK activity regulates HDAC7 stability to selectively regulate transcriptional recall of effector cytokine production in differentiating CD8+ T cells.
BioE-1197 epigenetically poises cytokine loci for enhanced recall potential
To assess whether the observed transcriptional regulation of effector cytokine production was through direct regulation of H3K27Ac levels within the cytokine gene loci, we performed H3K27Ac ChIP-Seq on day six after activation for control and BioE-1197 differentiated CD8+ T cells. Analysis of the Ifng loci revealed enhanced H3K27Ac marks in BioE-1197 differentiated CD8+ T cells compared to control treated cells. Of note, BioE-1197 treated cells particularly exhibited enhanced H3K27Ac marks across biological replicates within a previously reported, T cell receptor signaling responsive, conserved non-coding sequence, +40 kb, from the transcription start site denoted by the black bar (Figure 4F, S3A) (43, 44). Additionally, the Tnf loci exhibited enhanced H3K27Ac marks in BioE-1197 differentiated CD8+ T cells, also, in a previously reported T cell receptor signaling responsive conserved non-coding sequence, −9 kb, from the transcription start site denoted by the black bar (Figure 4G, S3B) (45, 46). Importantly, the Tbx21 loci, which showed no differences in expression upon treatment with BioE-1197, exhibited no enhancement of H3K27Ac marks within BioE-1197 differentiated cells (Figure S3C). Moreover, other cytokine loci not associated with the CD8+ T cell effector program, including the IL-17A and IL-4 loci, showed no H3K27Ac deposition in both control and BioE-1197 differentiated CD8+ T cells (Figure S4). Together, these data indicate stabilization of HDAC7, during T cell differentiation, results in selective reductions of H3K27Ac levels in known, distant regulatory elements of the lineage specific effector cytokine loci. These reduced H3K27Ac marks, in turn, result in transcriptionally blunted cytokine recall potential.
BioE-1197 regulates effector cytokine programs in CD4+ Th1 cells
These data identified LKB1 dependent SIK activity induces HDAC7 stability to epigenetically regulate effector cytokine programs in CD8+ T cells. Given this effector cytokine program is shared by CD4+ Th1 cells, we sought to determine whether this pathway was also active in Th1 cells. CD4+ naïve T cells were activated and skewed to a Th1 lineage, and treated with a vehicle control or BioE-1197. Th1 cells differentiated in the presence of BioE-1197 also exhibited elevated IFNγ and TNFα production in response to re-stimulation (Fig. 5 A–C). Th1 skewed CD4+ T cells differentiated in the presence of BioE-1197 exhibited statistically significant increases in the expression of the lineage specifying transcription factor, T-bet, by percent (mean: 97.6 vs 99.1) and gMFI (mean: 1 vs 1.256) but the magnitude of these differences was quite small and unlikely to explain the differences in observed cytokine production (Fig. 5 D–F). Moreover, BioE-1197 differentiated Th1 cells exhibit similarly diminished HDAC7 phosphorylation dependent stabilization and nuclear availability compared to control differentiated cells (Fig. 5 G–H), and this Th1 phosphorylation dependent stabilization of HDAC7 and effector cytokine production is also dependent on ARK family SIKs (Fig. 6 A–D).
Figure 5. BioE-1197 enhances effector cytokine production and regulates HDAC7 stability and localization in CD4+ Th1 cells.
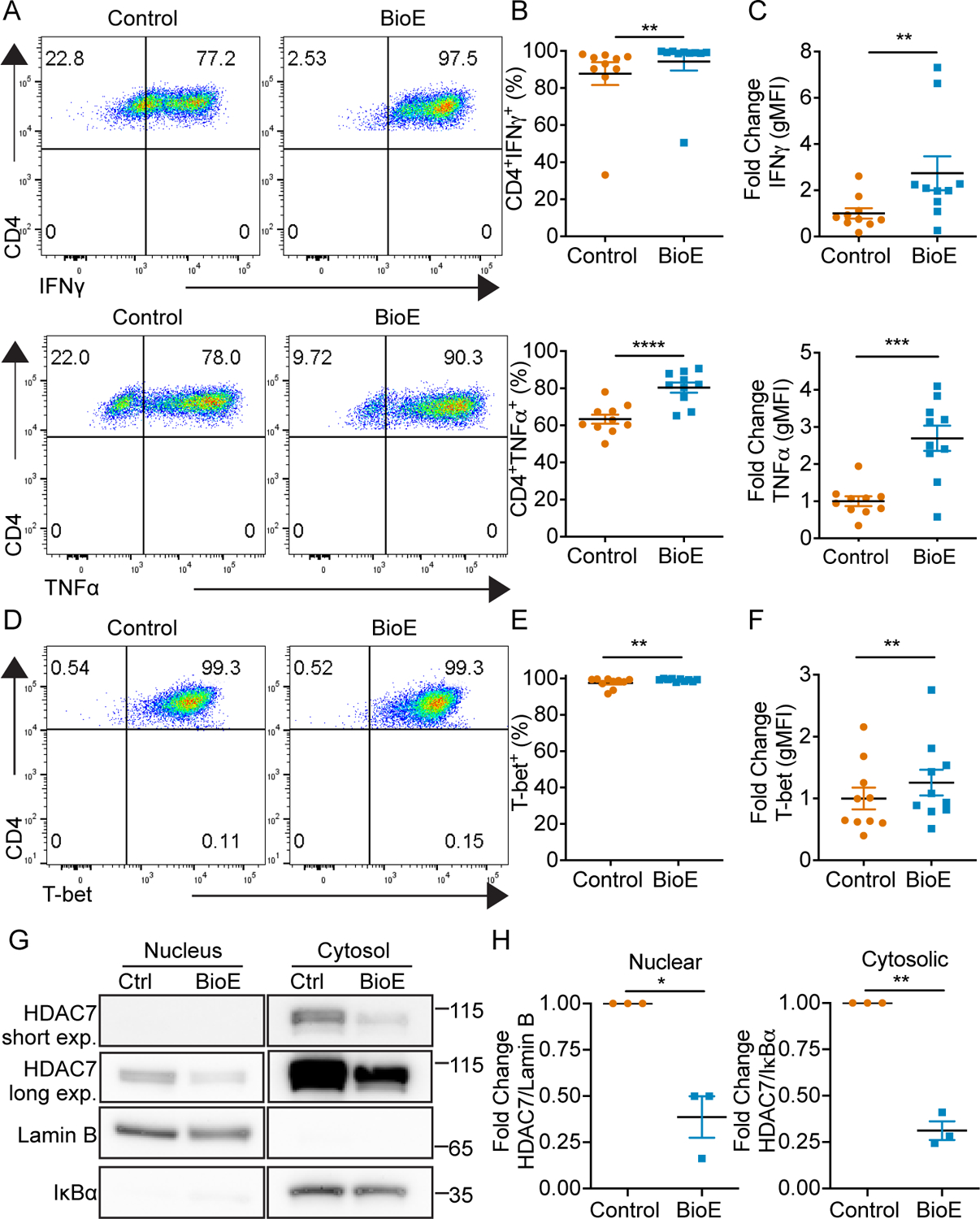
(A) Representative intracellular cytokine staining dot plots of IFNγ (top) and TNFα (bottom) production upon re-stimulation of Th1 cells on day six after activation and differentiation in control (DMSO) or BioE-1197 (50μM) conditions. (B) Quantification of the percent of IFNγ (top) and TNFα (bottom) cytokine positive Th1 cells represented in A across independent experiments. (C) Quantification of the fold change in gMFI of IFNγ (top) and TNFα (bottom) production by Th1 cells represented in A across independent experiments. (D) Representative intracellular staining dot plots of T-bet in Th1 cells on day six after activation and differentiation in control (DMSO) or BioE-1197 (50μM) conditions. (E) Quantification of the percent of T-bet positive cells represented in D across independent experiments. (F) Quantification of the fold change in gMFI of T-bet by CD8+ T cells represented in D across independent experiments. (G) Representative western blot of HDAC7 from subcellular fractionation of Th1 cells on day three after activation and differentiation in control (DMSO) or BioE-1197 (50μM) conditions. Lamin B and IκBα serve as nuclear and cytosolic compartment loading controls, respectively. (H) Quantification of the fold change in HDAC7 localization represented in G across independent experiments in Th1 cells on day three after activation and differentiation in control (DMSO) or BioE-1197 (50μM) condition. Each dot represents values from an independent experiment, summary data are presented as the mean (black line) with SEM error bars (B-C, E-F, H). Wilcoxon test (B-IFNγ, C-IFNγ, E). Paired t-test (B-TNFα, C-TNFα, F, H). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, ns = not significant.
Figure 6. SIKs regulate phosphorylation dependent stabilization of HDAC7 and lineage specific cytokine production in Th1 cells.
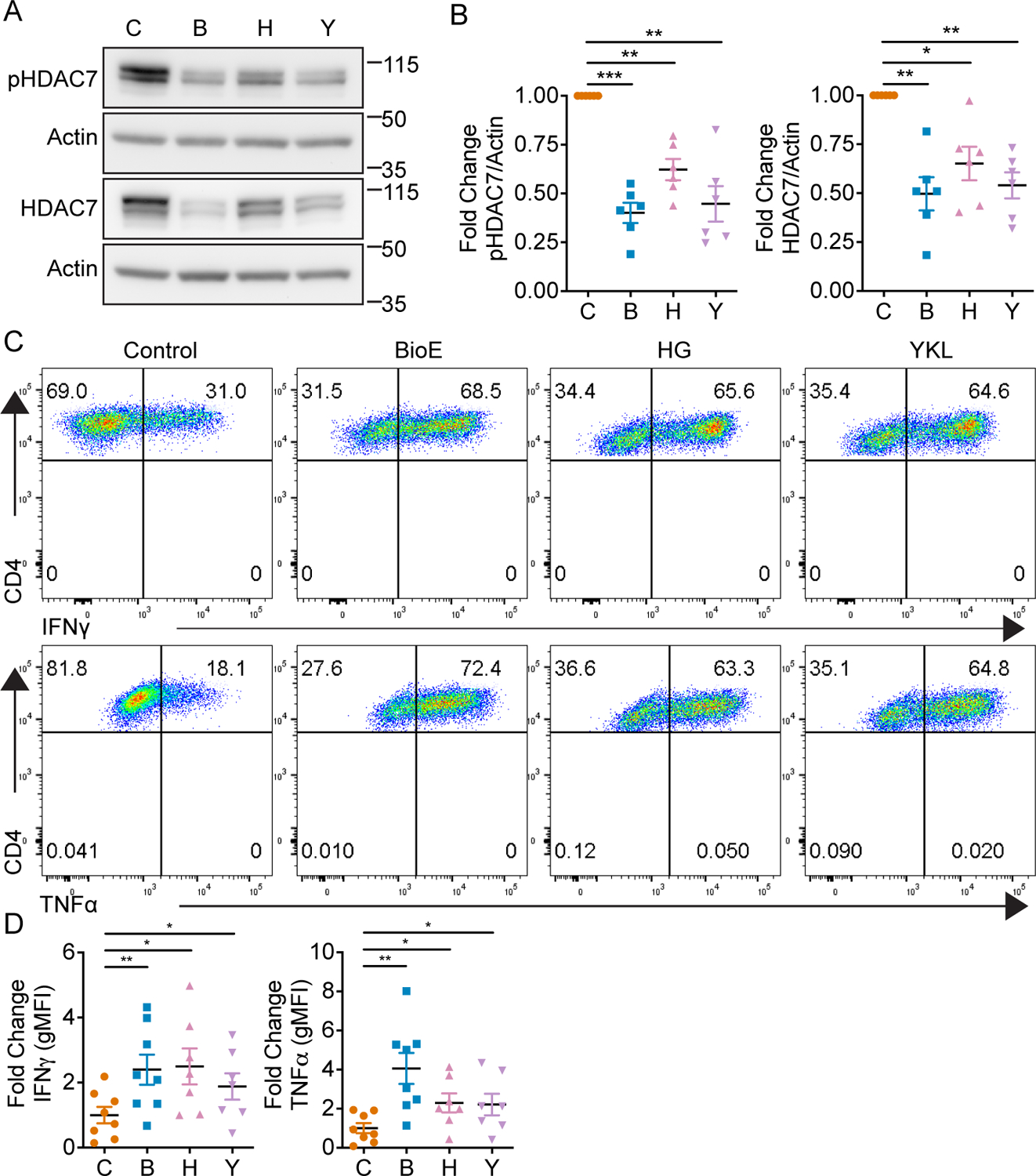
(A) Representative western blot of phosphorylated and total HDAC7 by Th1 cells on day four after activation and differentiation in either control (DMSO), BioE-1197 (50 μM), HG-9-91-01 (HG, 25 nM) or YKL-05-099 (YKL, 270 nM) differentiation conditions. (B) Quantification of the fold change in phosphorylated (left) and total HDAC7 (right) production in Th1 cells represented in A across independent experiments. (C) Representative intracellular cytokine staining dot plots of IFNγ (top) and TNFα (bottom) production by Th1 cells on day six after activation and differentiation in either control (DMSO), BioE-1197 (50 μM), HG-9-91-01 (HG, 25 nM) or YKL-05–099 (YKL, 270 nM) conditions. (D) Quantification of the fold change in gMFI of IFNγ (top) and TNFα (bottom) production in Th1 cells represented in C across independent experiments. Each dot represents values from an independent experiment, summary data are presented as the mean (black line) with SEM error bars (B, D). Repeated measures one-way ANOVA, Dunnet’s multiple comparisons test (B). Mixed effects analysis, Dunnet’s multiple comparisons test (D). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, ns = not significant.
HDAC7 stabilization and H3K27Ac restriction occurs in late-stage T effector differentiation
These experiments reveal LKB1 activated ARK family SIKs regulate CD8+ and Th1 phosphorylation dependent stabilization of HDAC7 and effector cytokine production. Given the unexpected observations that these differentiating CD8+ and Th1 cells engage this pathway to epigenetically constrict cytokine production, we sought to evaluate the kinetics of HDAC7 phosphorylation dependent stabilization during differentiation to understand why T cells engage this negative regulatory pathway. CD8+ and Th1 cells were activated and expanded in either control or BioE-1197 differentiation conditions and phosphorylation dependent stabilization of HDAC7 was assessed by western blot. Phosphorylation dependent stabilization of HDAC7 in control differentiated CD8+ and Th1 cells increased after activation as these cells underwent differentiation, while CD8+ and Th1 cells differentiated in the presence of BioE-1197 could not undergo phosphorylation dependent stabilization of HDAC7 (Fig. 7 A–B). Notably, in both CD8+ and Th1 cells, dramatic differences in HDAC7 stability between control and BioE-1197 differentiated cells begin to be observed on day three after activation, following the expansion of these cells in IL-2. Interestingly, in CD4+ Th1 cells there are also subtle differences between control and BioE-1197 conditions for HDAC7 early during differentiation that are not observed in CD8+ T cells. Nonetheless, these findings indicate inhibition of phosphorylation dependent stabilization during the differentiation process leads to the largest differences in total HDAC7 levels in the later stages of differentiation, during expansion in IL-2, for both CD8+ and Th1 cells.
Figure 7. Phosphorylation dependent stabilization of HDAC7 occurs during late-stage differentiation of CD8+ and Th1 effector.
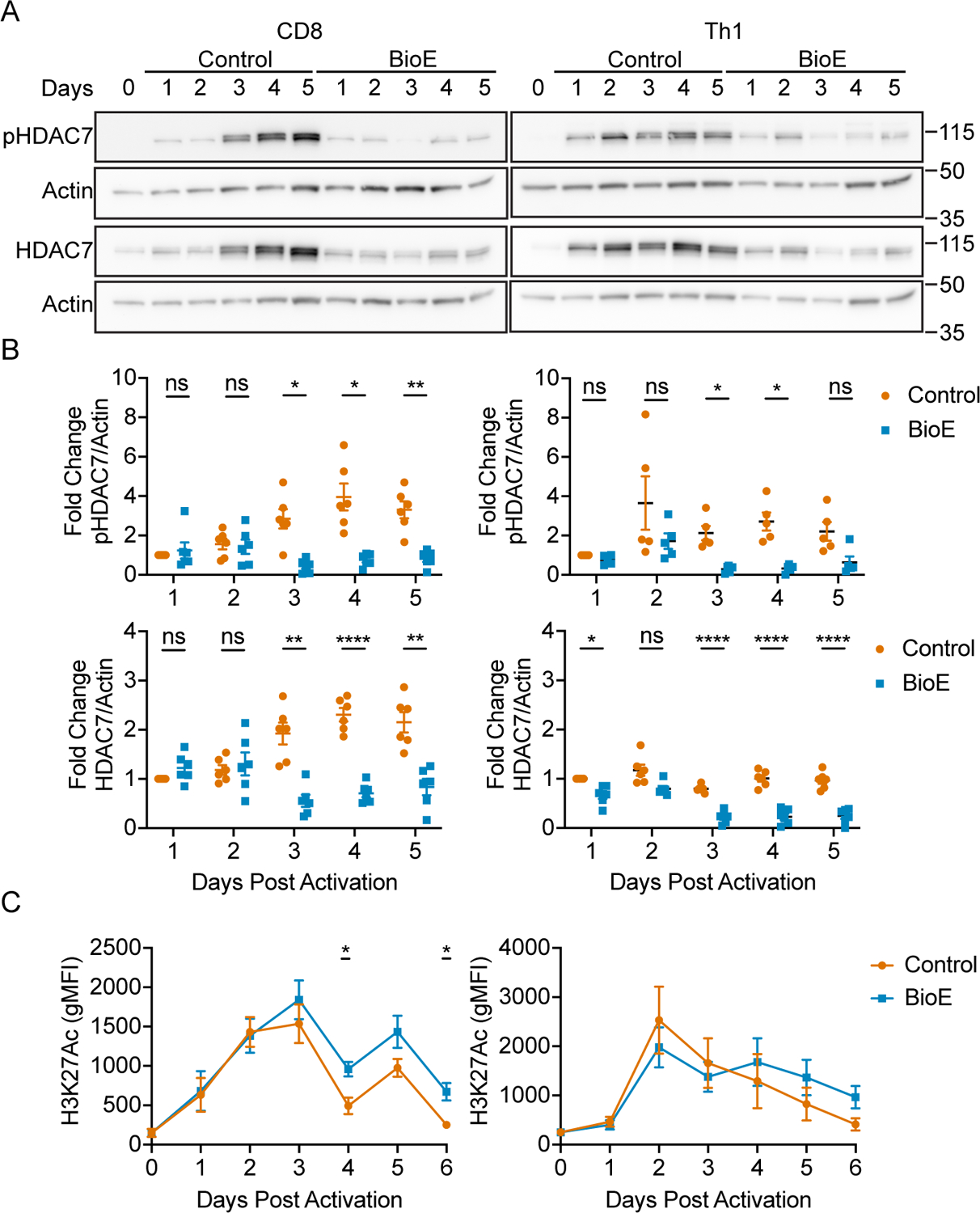
(A) Representative western blots for control (DMSO) and BioE-1197 (50μM) differentiated CD8+ (left) and Th1 (right) cells for phosphorylated and total HDAC7. (B) Quantification of the fold change in phosphorylated (top) and total (bottom) HDAC7 in CD8+ (left) and Th1 (right) cells represented in A across independent experiments. (C) Quantification of global H3K27Ac marks over time in CD8+ (left) and Th1 (right) cells differentiated in control (DMSO) and BioE-1197 (50μM) conditions. Each dot represents values form an independent experiment, summary data are presented as the mean (black line) with SEM error bars (B). The mean and standard error of measurement (SEM) are presented for 12 independent experiments (C). Two-way ANOVA, Sidak’s multiple comparisons test (B). Mixed effects analysis, Sidak’s multiple comparisons test (C). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, ns = not significant.
Given these results revealed HDAC7 stabilization occurs during the late stages of T effector differentiation, we additionally sought to evaluate the kinetics of the global activating transcription mark, H3K27Ac, over time during differentiation. In general, H3K27Ac levels increase over the naïve condition in CD8+ and Th1 cells during their initial activation but begin to decline as differentiation progresses (Fig 7 C); however, the exact regulation of these marks, on a total scale, exhibits cell type specific patterns. In CD8+ T cells, the initial accumulation of H3K27Ac correlates with low levels of phosphorylated and total HDAC7. Upon day three, when these CD8+ T cells begin to exhibit differential HDAC7 stability, global H3K27Ac levels begin to decline in both control and BioE-1197 differentiated cells. However, in BioE-1197 differentiated CD8+ T cells, which have low levels of HDAC7, you observe elevated H3K27Ac marks compared to the control condition during the late-stages of differentiation. This general decline over the late-stages of differentiation is additionally marked by fluctuations in H3K27Ac levels with the high points and low points occurring 24 and 48 hours after IL-2 exposure, respectively. Alternatively, in CD4+ Th1 cells, the initiation of H3K27Ac decline begins earlier. However, during late-stage differentiation, BioE-1197 differentiated cells trend toward higher H3K27Ac marks, though they are not statistically significant. This could be due to fewer genes being regulated by this pathway in CD4+ T cells making resolution of these differences on a total scale more difficult. This is supported by the observation that there does seem to be selectivity in the effector genes regulated by this pathway. Moreover, CD4+ Th1 cells do not exhibit the late-stage H3K27Ac fluctuations associated with the CD8+ T cells. Nonetheless, in both populations epigenetic restriction, marked by H3K27Ac decline, is associated with late-stage differentiation during IL-2 expansion.
Late-stage class IIa HDAC inhibition is sufficient to elevate cytokine recall
Next, we sought to assess whether this late-stage epigenetic restriction associated with HDAC7 stabilization is responsible for alterations in effector cytokine production in CD8+ and Th1 cells. To this end, we hypothesized if late-stage HDAC7 stabilization occurs to epigenetically regulate cytokine production then, inhibition of HDAC7 deacetylase activity during the late stages of differentiation should be sufficient to elevate cytokine production. To test this hypothesis, we activated and differentiated CD8+ and CD4+ T cells in the presence of TMP269, a class IIa HDAC inhibitor, given the lack of a HDAC7 specific inhibitor. TMP269 was added at the time of expansion in IL-2, starting at day 2 after activation, so as to specifically inhibit deacetylase activity during the time frame in which control and BioE-1197 differentiated cells have differential HDAC7 stability. Upon re-stimulation, TMP269 differentiated CD8+ and Th1 cells have elevated lineage specific cytokine production with cytokine levels similar to that of BioE-1197 differentiated effectors (Fig. 8 A–D). These observations indicate inhibition of class IIa HDACs during the late stages of differentiation is sufficient to elevate cytokine recall upon re-stimulation, suggesting epigenetic constriction of cytokine genes during late-stage differentiation is active.
Figure 8. Late-stage class IIa HDAC inhibition elevates effector cytokine production.
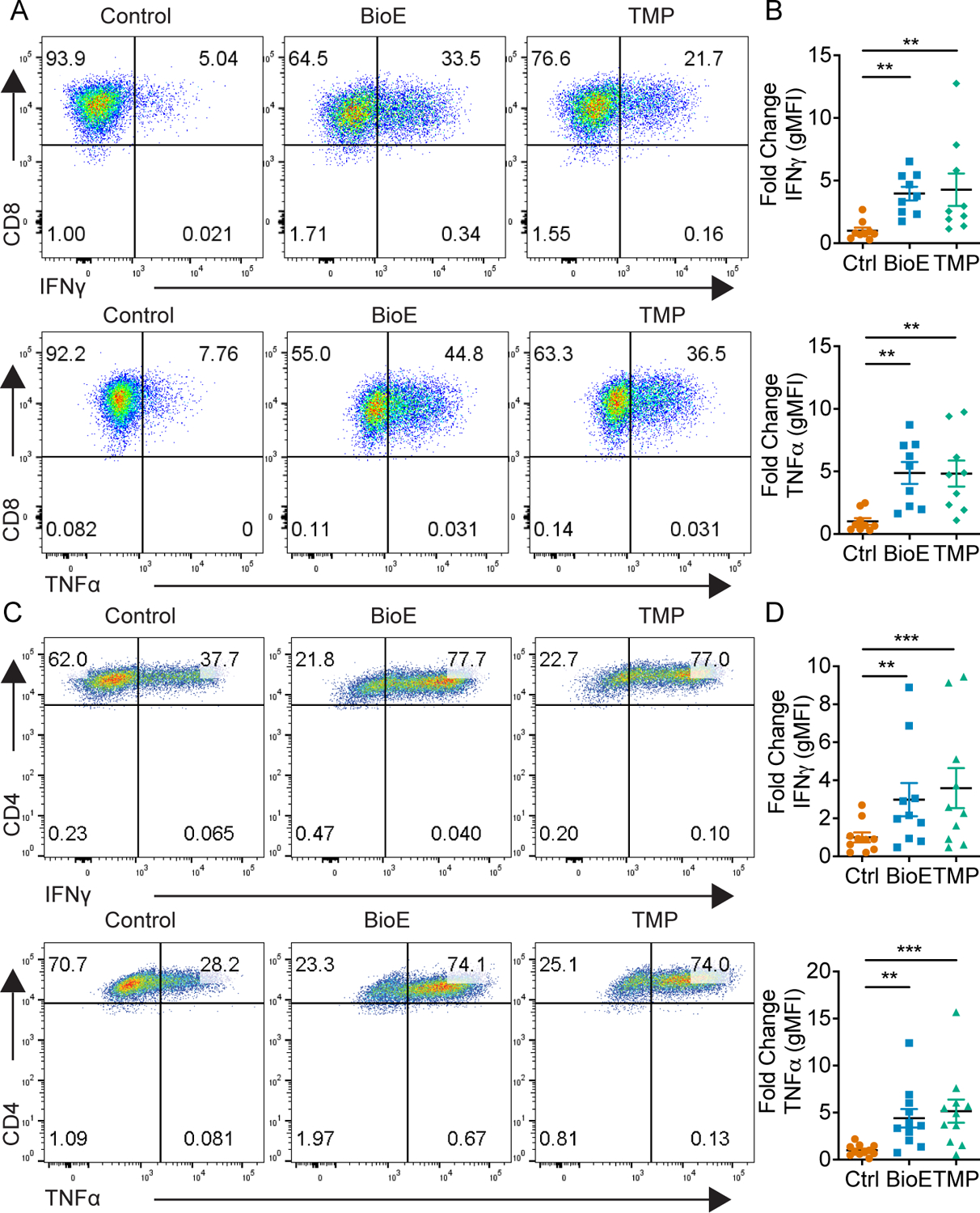
(A) Representative dot plots of IFNγ (top) and TNFα (bottom) production by CD8+ T cells on day six after activation and differentiation in either control (DMSO), BioE-1197 (50μM), or TMP269 (TMP, 12.5 μM) conditions. (B) Quantification of the fold change in gMFI of IFNγ (top) and TNFα (bottom) production in CD8+ T cells represented in A across independent experiments. (C) Representative dot plots of IFNγ (top) and TNFα (bottom) production by Th1 cells on day six after activation and differentiation in either control (DMSO), BioE-1197 (50μM), or TMP269 (TMP, 12.5 μM)) conditions. (D) Quantification of the fold change in gMFI of IFNγ (top) and TNFα (bottom) production in Th1 cells represented in C across independent experiments. Each dot represents values from an independent experiment, summary data are presented as the mean (black line) with SEM error bars (B,D). Friedman test, Dunn’s multiple comparisons test (B,D). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Collectively, these observations identify LKB1 dependent SIK activation as a regulator of phosphorylation dependent stabilization of HDAC7 in late-stage T cell differentiation. This enhanced HDAC7 stability increases nuclear availability of HDAC7 by increasing the amount of HDAC7 available for nuclear import. This enhanced nuclear HDAC7 protein availability leads to a reduction in H3K27Ac marks within effector cytokine loci and, therefore, restricts cytokine recall at the transcriptional level (Figure 9A). BioE-1197 mediated inhibition of this pathway results in reduced HDAC7 stability and, therefore, diminished nuclear HDAC7 levels, enabling the persistence of H3K27Ac marks within effector cytokine loci. Thus, BioE-1197 mediated inhibition epigenetically equips effector T cells to have enhanced effector cytokine production upon recall (Figure 9B).
Figure 9. LKB1 activated SIK function mediates phosphorylation dependent stabilization of HDAC7 and lineage specific cytokine recall in CD8+ and Th1 cells.
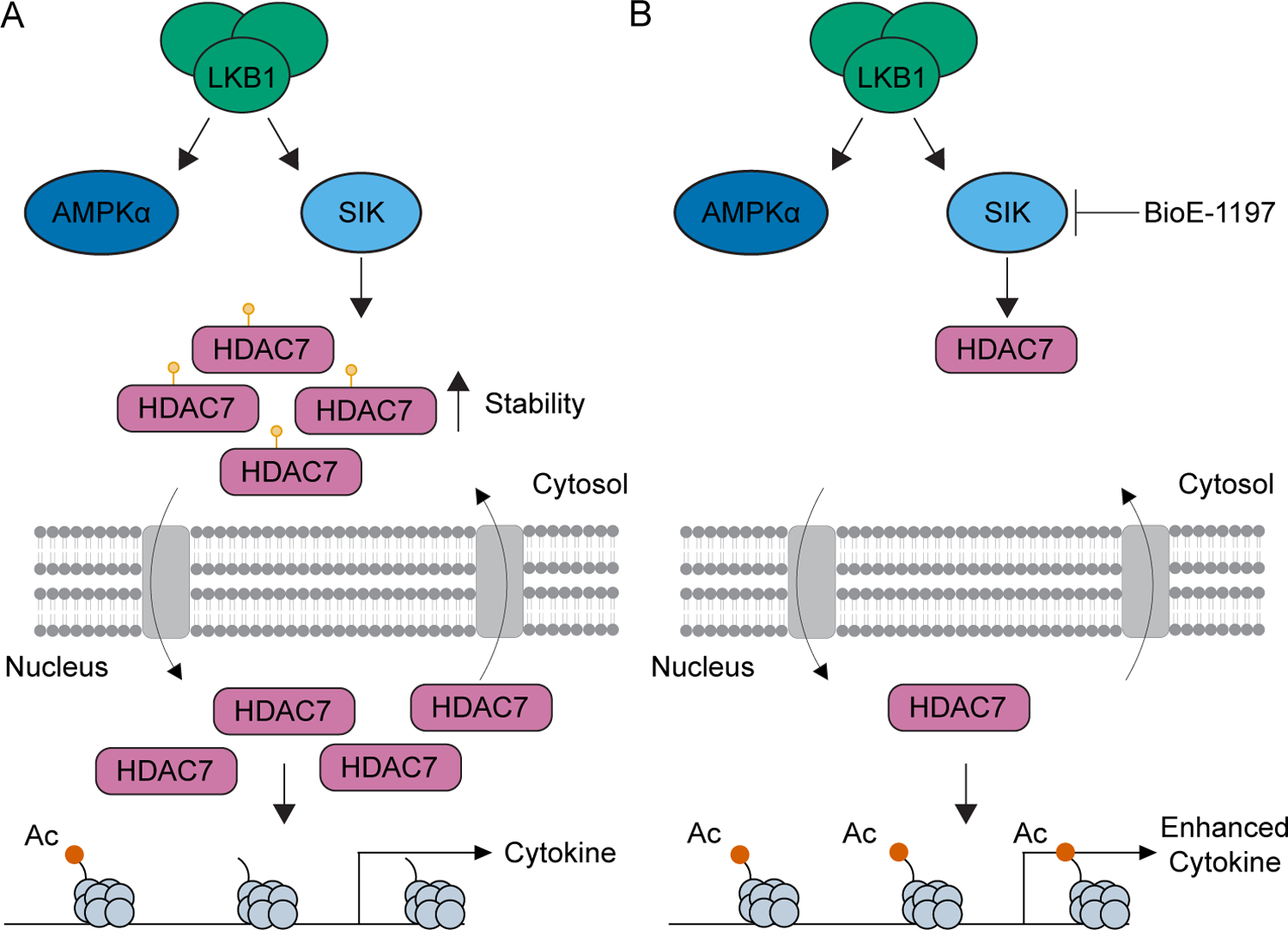
(A) Model depicting LKB1-SIK mediated phosphorylation dependent stabilization of HDAC7 leading to enhanced HDAC7 nuclear import due to increased HDAC7 protein levels, removal of H3K27Ac marks and restriction of effector cytokine production, epigenetically. (B) Model depicting BioE-1197 mediated inhibition of SIK dependent HDAC7 phosphorylation dependent stabilization which maintains low nuclear HDAC7 levels compared to the control condition, persistence of H3K27Ac levels, and the generation of cells epigenetically poised for enhanced effector cytokine production.
DISCUSSION
Herein, we report the identification of a novel activity of the kinase inhibitor, BioE-1197, in CD8+ and Th1 cells. While previously developed as a PASK inhibitor, we demonstrate that exposure to BioE-1197 in T cells leads to the durable programming of effector CD8+ and Th1 cells with enhanced cytokine recall in a PASK-independent manner. The durable nature of this enhanced cytokine production was illustrated by co-adoptive transfer park and recall experiments in which even after a 32-day rest in naïve hosts, BioE-1197 differentiated CD8+ T cells maintain elevated cytokine production upon recall. While cytokine production is a key feature of T cell effector function, the importance of effector cytokine production, specifically to anti-tumor immunity, has been recently highlighted. Some preclinical models of immunotherapy have been shown to be reliant on IFNγ effector cytokine production as opposed to other effector molecules such as perforin or granzymes (47–49). Likewise, IFN-γ signaling signatures have been shown to track with clinical response in patients treated with immunotherapy checkpoint blockade (50). Given these findings, the observed ability of BioE-1197 to enhance effector T cell functionality, specifically by modulating cytokine production, has encouraging immunotherapeutic potential. Particularly, the ability to produce long-term, enhanced cytokine recall following transient differentiation in the presence of BioE-1197 could be of great utility for the generation and expansion of autologous or engineered adoptive cellular immunotherapies.
Given the therapeutic potential of this observed phenotype, we sought to identify the pathway targeted by BioE-1197 in T cells. Using CRISPR/Cas9 mediated LKB1 deletion, we demonstrated LKB1 activated ARK family members were necessary for the observed BioE-1197 cytokine recall phenotype. Whether all 13 LKB1 activated ARK family kinases are active and have roles in T cell differentiation is unknown. To date, AMPKα1 is the only ARK, demonstrated by T cell specific deletion, to have important functions in effector T cell activation and metabolism (26, 51, 52). As AMPKα1 deletion can recapitulate some, but not all, phenotypes associated with T cell specific LKB1 deletion, it is likely other ARKs are active during T cell differentiation (26). Our studies demonstrated BioE-1197 was unlikely to be inhibiting all ARKs active during T cell differentiation, as BioE-1197 differentiated CD8+ T cells do not exhibit the diminished survival associated with LKB1 deletion. Therapeutically, these data demonstrate a selective advantage of BioE-1197 over targeting upstream molecules, as BioE-1197 can preserve cell viability while enhancing cytokine production. While these analyses revealed dependence of BioE-1197 on some LKB1 activated ARKs, LKB1 deletion was not sufficient to elevate cytokine recall. The observed insufficiency of LKB1 deletion to enhance cytokine recall potential could be overshadowed by the inactivation of other ARK family members during T cell differentiation. This possibility is supported by the observation that pan SIK inhibition, downstream of LKB1 activation, is sufficient to elevate cytokine production. These collective observations suggest individual members of the ARK family modulate distinct aspects of T cell differentiation following LKB1 mediated activation as opposed to these kinases functioning redundantly downstream of LKB1, and identify a need to further evaluate the roles of other ARK family members in effector T cell activation and differentiation. Our data specifically indicate that SIKs are capable of regulating lineage specific cytokine production, but more work is needed to determine whether this phenotype is dependent on a specific SIK or the collective function of SIKs during differentiation. Previous work has identified SIK2 and SIK3 to be critical regulators of thymocyte development, and SIK dependent functions in T regulatory cells have been described (53–56). However, to our knowledge, this is the first illustration of SIK dependent function in effector T cell differentiation.
Deletion of LKB1, pan SIK inhibition, and BioE-1197 treatment all lead to reductions in phosphorylated and total HDAC7, a known ARK family substrate. SIKs are known to mediate HDAC7 phosphorylation, which regulates HDAC7 stability, and SIK1 mediated phosphorylation dependent stabilization of HDAC7 has been previously reported (29, 30, 34, 36, 37). These observations suggest LKB1 mediated SIK activation during T cell differentiation leads to phosphorylation dependent stabilization of HDAC7. Kinetic analysis of this phosphorylation event throughout differentiation reveals phosphorylation increases after T cell activation. Moreover, inhibition of this phosphorylation event leads to a reduction in HDAC7 stability specifically during the latter part of T cell differentiation. A previous report has documented HDAC7 phosphorylation in CD8+ T cells. However, this prior study suggested HDAC7 phosphorylation to be constitutive and independent of T cell receptor activation in CD8+ T cells as there was no further increase in HDAC7 phosphorylation between naïve and activated T cells over 30 minutes (57). The expanded timing of our kinetic analysis, however, reveals phosphorylation of HDAC7 to be observed long after T cell activation, with changes observed over longer time frames, on the order of days, during differentiation and IL-2 expansion. Moreover, BioE-1197 exposure reveals that diminished HDAC7 phosphorylation particularly impacts HDAC7 stability during the later stages of differentiation, with robust differences becoming evident on day three after activation. These observations reveal a timed specificity in regulating HDAC7 stability during late-stage T effector differentiation and identify new potential areas of research to understand what intrinsic or extrinsic signals initiate SIK mediated stabilization of HDAC7 specifically within this window.
The observed differential stability of HDAC7 in control and BioE-1197 differentiated T cells suggested potential differences in nuclear accumulation of HDAC7, given increasing the total HDAC7 pool, through phosphorylation dependent stabilization, would increase the total amount of HDAC7 available for nuclear import. However, phosphorylated HDAC7 is reported to be primarily sequestered in the cytosol as the stabilizing interaction with 14-3-3 simultaneously blocks its nuclear import signal (36, 37, 40–42). Therefore, while phosphorylation stabilizes the total HDAC7 protein pool, phosphorylated HDAC7 would also be heavily restricted to the cytosol. Experimentally, we were able to show, however, phosphorylation dependent stabilization of HDAC7 simultaneously regulates HDAC7 protein levels in both the cytosol and the nucleus. Therefore, while phosphorylation is known to restrict HDAC7 to the cytosol, the simultaneous stabilization of HDAC7 increases the total amount of HDAC7 in the cell and therefore the amount of HDAC7 available for nuclear import, given phosphorylation of HDAC7 is a reversible phenomenon (58). These data highlight an additional layer of complexity to consider in interpreting the functionality of HDAC7 protein based on phosphorylation status.
Our studies additionally revealed the stabilization mediated increase in nuclear HDAC7 correlates with a decline in total H3K27Ac activating transcription marks within the late-stages of T effector differentiation. This observation suggests, as T cells continue to differentiate, their activating epigenetic landscape becomes constrained to tone down effector cytokine production. Administration of BioE-1197 during differentiation was able to increase H3K27Ac levels at a global level in CD8+ T cells, and, in turn, elevate cytokine production at both the protein and mRNA transcript level. Evaluation of these H3K27Ac marks at the end of differentiation by ChIP-Seq revealed BioE-1197 differentiated CD8+ T cells have enhanced H3K27Ac marks specifically within the cytokine loci compared to control differentiated T cells. In both the IFNγ and TNFα loci, enrichment of H3K27Ac levels, across biological replicates, in previously described T cell receptor dependent conserved non-coding sequences was evident. The +40 kb site for IFNγ has been specifically described to mediate RelA binding in response to re-stimulation to regulate IFNγ transcript production (43, 44), while the −9kb site for TNFα is known to mediate T cell receptor dependent NFAT binding (45, 46). The observed enrichment of these H3K27Ac marks specifically within these distant regulatory elements, which have known binding sites for T cell receptor activated transcription factors, further explains how this BioE-1197 mediated phenotype could function independent of lineage specifying transcription factors known to regulate effector cytokine function. These observations, therefore, additionally raise the question of whether other T effector programs additionally use this pathway to regulate their cytokine programs.
The evident H3K27Ac enrichment within these sites in BioE-1197 differentiated CD8+ T cells supports epigenetic restriction of these distant regulatory elements is ongoing during late-stage T effector differentiation. These data additionally suggest BioE-1197 can block, to some extent, the observed restriction in effector functionality potentially through epigenetics. Further confirmation of this epigenetic mechanism is rooted in the observed sufficiency of class IIa HDAC inhibition, with TMP269, to elevate lineage specific cytokine production to the same extent as BioE-1197 administration when only administered during late-stage differentiation. An underlying epigenetic mechanism also explains the durability of BioE-1197 enhanced cytokine recall potential observed in the co-adoptive transfer park and recall experiments. Together these data suggest phosphorylation dependent stabilization of HDAC7 during late-stage T effector differentiation functions to restrict cytokine recall potential and inhibition of this stabilization, with BioE-1197, epigenetically programs cells for lasting, enhanced cytokine recall.
Previous work in CD8+ T cells has demonstrated overexpression of a triple non-phosphorylatable HDAC7 mutant, compared to un-transduced cells, results in a reduction in IFNγ production (57). However, in our study, drug mediated inhibition of HDAC7 phosphorylation enhanced IFNγ production. These results are not incongruent in that retroviral mediated overexpression can overcome the stability phenotype associated with HDAC7 phosphorylation status by continuously replacing the turned over, non-phosphorylatable HDAC7 population. The turnover of the non-phosphorylatable HDAC7 is supported by a previous report that the non-phosphorylatable mutant is expressed at lower levels than its WT counterpart (59). Therefore, forced overexpression, by replacing the destabilized protein, is able to enhance nuclear accumulation of HDAC7 over the control despite diminished protein stability leading to a reduction in cytokine production. Likewise, in our study, control differentiated cells, in which stabilization leads to increased nuclear HDAC7 total protein, have reduced cytokine production compared to BioE-1197 differentiated cells. Therefore, in both data sets, when HDAC7 nuclear availability is increased there is a correlative decrease in IFNγ cytokine production.
This study, through evaluation of a previously unreported function of BioE-1197, identifies a novel pathway capable of regulating effector cytokine functionality in CD8+ and Th1 cells. The data presented within reveal LKB1 activated SIKs mediate HDAC7 phosphorylation dependent stabilization and T cell cytokine recall potential. Moreover, inhibition of this pathway leads to the production of effector T cells with stably enhanced cytokine recall potential. The targeted interruption of SIK activity represents a particularly advantageous strategy for epigenetically modulating T cell functionality as this offers enhanced selectivity over other epigenetic therapies, such as class specific HDAC inhibitors, which would be active broadly, across many cell types. The work presented within defines a novel therapeutic target for enhancing T cell functionality, and illuminates new avenues of research in understanding the processes that regulate late-stage effector T cell differentiation.
Supplementary Material
KEY POINTS.
LKB1-mediated SIK activation regulates HDAC7 stability.
HDAC7 stabilization correlates with epigenetic restriction of cytokine loci.
Inhibition of this pathway epigenetically poises cells for enhanced cytokine recall.
ACKNOWLEDGEMENTS
The authors would like to thank Dr. Elizabeth A. Thompson for her careful revision and feedback during the preparation of this manuscript, and Drs. Anjana Rao, Patrick G. Hogan, and Srikanth Battu for their advice on H3K27Ac ChIP-Seq.
This work is supported by grants from NIBIB (J.D.P.: 5P41EB018239-03), the Johns Hopkins Institute for Clinical and Translational Research (T.T. & B.S.S.: UL1 TR001079), and the Bloomberg-Kimmel Institute for Cancer Immunotherapy (J.D.P.). T.H. acknowledges support from the National Institutes of Health (grant U01 DK 127432). A.M.-G. is a Howard Hughes Medical Institute (HHMI) Awardee of the Life Sciences Research Foundation. T.H. is an investigator of the HHMI.
Abbreviations
- AMPK
AMP activated protein kinase
- ARKs
AMPK related kinases
- FMO
fluorescence minus one
- gMFI
geometric mean fluorescence intensity
- HDAC7
histone deacetylase 7
- H3K27Ac
Histone 3 lysine 27 acetylation
- KO
knockout
- LCMV
Lymphocytic choriomeningitis virus
- LKB1
liver kinase B1
- mTORC1
Mammalian target of rapamycin complex 1
- PASK
PAS domain-containing serine/threonine kinase
- RIPA
radioimmunoprecipitation
- sgRNA
single guide RNA
- SIKs
salt-inducible kinases
- WDR5
WD repeat domain 5
- WT
wild type
Footnotes
Conflicts of Interest
C.H.P. and J.D.P. are current employees of Calico Life Sciences LLC. The authors have no financial conflicts of interest to report with reference to the work presented within this manuscript.
REFERENCES
- 1.Crompton JG, Narayanan M, Cuddapah S, Roychoudhuri R, Ji Y, Yang W, Patel SJ, Sukumar M, Palmer DC, Peng W, Wang E, Marincola FM, Klebanoff CA, Zhao K, Tsang JS, Gattinoni L, and Restifo NP. 2016. Lineage relationship of CD8(+) T cell subsets is revealed by progressive changes in the epigenetic landscape. Cell Mol Immunol 13: 502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rohaan MW, Wilgenhof S, and Haanen J. 2019. Adoptive cellular therapies: the current landscape. Virchows Arch 474: 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scharer CD, Barwick BG, Youngblood BA, Ahmed R, and Boss JM. 2013. Global DNA methylation remodeling accompanies CD8 T cell effector function. J Immunol 191: 3419–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT, Schones DE, Peng W, Sun HW, Paul WE, O’Shea JJ, and Zhao K. 2009. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30: 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruterbusch M, Pruner KB, Shehata L, and Pepper M. 2020. In Vivo CD4(+) T Cell Differentiation and Function: Revisiting the Th1/Th2 Paradigm. Annu Rev Immunol 38: 705–725. [DOI] [PubMed] [Google Scholar]
- 6.Saravia J, Chapman NM, and Chi H. 2019. Helper T cell differentiation. Cell Mol Immunol 16: 634–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang N, and Bevan MJ. 2011. CD8(+) T cells: foot soldiers of the immune system. Immunity 35: 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang JT, Wherry EJ, and Goldrath AW. 2014. Molecular regulation of effector and memory T cell differentiation. Nat Immunol 15: 1104–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farber DL, Yudanin NA, and Restifo NP. 2014. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol 14: 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hao HX, Cardon CM, Swiatek W, Cooksey RC, Smith TL, Wilde J, Boudina S, Abel ED, McClain DA, and Rutter J. 2007. PAS kinase is required for normal cellular energy balance. Proc Natl Acad Sci U S A 104: 15466–15471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katschinski DM, Marti HH, Wagner KF, Shibata J, Eckhardt K, Martin F, Depping R, Paasch U, Gassmann M, Ledermann B, Desbaillets I, and Wenger RH. 2003. Targeted disruption of the mouse PAS domain serine/threonine kinase PASKIN. Mol Cell Biol 23: 6780–6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCall JM, McKearn J, Romero DL, and Clair M. 2011. Preparation of heterocyclic compounds for inhibiting PAS Kinase. PCT Int. Pat. Appl. WO 2011/028947, 2011. [Google Scholar]
- 13.Kikani CK, Wu X, Paul L, Sabic H, Shen Z, Shakya A, Keefe A, Villanueva C, Kardon G, Graves B, Tantin D, and Rutter J. 2016. Pask integrates hormonal signaling with histone modification via Wdr5 phosphorylation to drive myogenesis. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kikani CK, Wu X, Fogarty S, Kang SAW, Dephoure N, Gygi SP, Sabatini DM, and Rutter J. 2019. Activation of PASK by mTORC1 is required for the onset of the terminal differentiation program. Proc Natl Acad Sci U S A 116: 10382–10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nüssing S, House IG, Kearney CJ, Chen AXY, Vervoort SJ, Beavis PA, Oliaro J, Johnstone RW, Trapani JA, and Parish IA. 2020. Efficient CRISPR/Cas9 Gene Editing in Uncultured Naive Mouse T Cells for In Vivo Studies. J Immunol 204: 2308–2315. [DOI] [PubMed] [Google Scholar]
- 16.Wienert B, Wyman SK, Yeh CD, Conklin BR, and Corn JE. 2020. CRISPR off-target detection with DISCOVER-seq. Nat Protoc 15: 1775–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langmead B, and Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramírez F, Dündar F, Diehl S, Grüning BA, and Manke T. 2014. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42: W187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopez-Delisle L, Rabbani L, Wolff J, Bhardwaj V, Backofen R, Grüning B, Ramírez F, and Manke T. 2021. pyGenomeTracks: reproducible plots for multivariate genomic datasets. Bioinformatics 37: 422–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Notredame C, Higgins DG, and Heringa J. 2000. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol 302: 205–217. [DOI] [PubMed] [Google Scholar]
- 22.Waterhouse AM, Procter JB, Martin DM, Clamp M, and Barton GJ. 2009. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 25: 1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu X, Romero D, Swiatek WI, Dorweiler I, Kikani CK, Sabic H, Zweifel BS, McKearn J, Blitzer JT, Nickols GA, and Rutter J. 2014. PAS kinase drives lipogenesis through SREBP-1 maturation. Cell Rep 8: 242–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lizcano JM, Göransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Mäkelä TP, Hardie DG, and Alessi DR. 2004. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. Embo j 23: 833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao Y, Li H, Liu H, Zheng C, Ji H, and Liu X. 2010. The serine/threonine kinase LKB1 controls thymocyte survival through regulation of AMPK activation and Bcl-XL expression. Cell Res 20: 99–108. [DOI] [PubMed] [Google Scholar]
- 26.MacIver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, Rathmell JC, and Jones RG. 2011. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J Immunol 187: 4187–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tamás P, Macintyre A, Finlay D, Clarke R, Feijoo-Carnero C, Ashworth A, and Cantrell D. 2010. LKB1 is essential for the proliferation of T-cell progenitors and mature peripheral T cells. Eur J Immunol 40: 242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zarrouk M, Rolf J, and Cantrell DA. 2013. LKB1 mediates the development of conventional and innate T cells via AMP-dependent kinase autonomous pathways. PLoS One 8: e60217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abend A, Shkedi O, Fertouk M, Caspi LH, and Kehat I. 2017. Salt-inducible kinase induces cytoplasmic histone deacetylase 4 to promote vascular calcification. EMBO Rep 18: 1166–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berdeaux R, Goebel N, Banaszynski L, Takemori H, Wandless T, Shelton GD, and Montminy M. 2007. SIK1 is a class II HDAC kinase that promotes survival of skeletal myocytes. Nat Med 13: 597–603. [DOI] [PubMed] [Google Scholar]
- 31.Dequiedt F, Martin M, Von Blume J, Vertommen D, Lecomte E, Mari N, Heinen MF, Bachmann M, Twizere JC, Huang MC, Rider MH, Piwnica-Worms H, Seufferlein T, and Kettmann R. 2006. New role for hPar-1 kinases EMK and C-TAK1 in regulating localization and activity of class IIa histone deacetylases. Mol Cell Biol 26: 7086–7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGee SL, van Denderen BJ, Howlett KF, Mollica J, Schertzer JD, Kemp BE, and Hargreaves M. 2008. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes 57: 860–867. [DOI] [PubMed] [Google Scholar]
- 33.Mihaylova MM, Vasquez DS, Ravnskjaer K, Denechaud PD, Yu RT, Alvarez JG, Downes M, Evans RM, Montminy M, and Shaw RJ. 2011. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell 145: 607–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Linden AM, Nolan KM, and Sengupta P. 2007. KIN-29 SIK regulates chemoreceptor gene expression via an MEF2 transcription factor and a class II HDAC. Embo j 26: 358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walkinshaw DR, Weist R, Kim GW, You L, Xiao L, Nie J, Li CS, Zhao S, Xu M, and Yang XJ. 2013. The tumor suppressor kinase LKB1 activates the downstream kinases SIK2 and SIK3 to stimulate nuclear export of class IIa histone deacetylases. J Biol Chem 288: 9345–9362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hsu A, Duan Q, McMahon S, Huang Y, Wood SA, Gray NS, Wang B, Bruneau BG, and Haldar SM. 2020. Salt-inducible kinase 1 maintains HDAC7 stability to promote pathologic cardiac remodeling. J Clin Invest 130: 2966–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li X, Song S, Liu Y, Ko SH, and Kao HY. 2004. Phosphorylation of the histone deacetylase 7 modulates its stability and association with 14-3-3 proteins. J Biol Chem 279: 34201–34208. [DOI] [PubMed] [Google Scholar]
- 38.Jing X, Sui WH, Wang S, Xu XF, Yuan RR, Chen XR, Ma HX, Zhu YX, Sun JK, Yi F, Chen ZY, and Wang Y. 2017. HDAC7 Ubiquitination by the E3 Ligase CBX4 Is Involved in Contextual Fear Conditioning Memory Formation. J Neurosci 37: 3848–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin M, Kettmann R, and Dequiedt F. 2007. Class IIa histone deacetylases: regulating the regulators. Oncogene 26: 5450–5467. [DOI] [PubMed] [Google Scholar]
- 40.Gao C, Li X, Lam M, Liu Y, Chakraborty S, and Kao HY. 2006. CRM1 mediates nuclear export of HDAC7 independently of HDAC7 phosphorylation and association with 14-3-3s. FEBS Lett 580: 5096–5104. [DOI] [PubMed] [Google Scholar]
- 41.Kao HY, Verdel A, Tsai CC, Simon C, Juguilon H, and Khochbin S. 2001. Mechanism for nucleocytoplasmic shuttling of histone deacetylase 7. J Biol Chem 276: 47496–47507. [DOI] [PubMed] [Google Scholar]
- 42.Nishino TG, Miyazaki M, Hoshino H, Miwa Y, Horinouchi S, and Yoshida M. 2008. 14-3-3 regulates the nuclear import of class IIa histone deacetylases. Biochem Biophys Res Commun 377: 852–856. [DOI] [PubMed] [Google Scholar]
- 43.Balasubramani A, Mukasa R, Hatton RD, and Weaver CT. 2010. Regulation of the Ifng locus in the context of T-lineage specification and plasticity. Immunol Rev 238: 216–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Balasubramani A, Shibata Y, Crawford GE, Baldwin AS, Hatton RD, and Weaver CT. 2010. Modular utilization of distal cis-regulatory elements controls Ifng gene expression in T cells activated by distinct stimuli. Immunity 33: 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Falvo JV, Tsytsykova AV, and Goldfeld AE. 2010. Transcriptional control of the TNF gene. Curr Dir Autoimmun 11: 27–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsytsykova AV, Rajsbaum R, Falvo JV, Ligeiro F, Neely SR, and Goldfeld AE. 2007. Activation-dependent intrachromosomal interactions formed by the TNF gene promoter and two distal enhancers. Proc Natl Acad Sci U S A 104: 16850–16855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Allard B, Pommey S, Smyth MJ, and Stagg J. 2013. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res 19: 5626–5635. [DOI] [PubMed] [Google Scholar]
- 48.Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, and Smyth MJ. 2011. Anti-TIM3 antibody promotes T cell IFN-γ-mediated antitumor immunity and suppresses established tumors. Cancer Res 71: 3540–3551. [DOI] [PubMed] [Google Scholar]
- 49.Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, Huang Q, Liu J, Takeda K, Teng MWL, Sachsenmeier K, and Smyth MJ. 2016. Co-inhibition of CD73 and A2AR Adenosine Signaling Improves Anti-tumor Immune Responses. Cancer Cell 30: 391–403. [DOI] [PubMed] [Google Scholar]
- 50.Grasso CS, Tsoi J, Onyshchenko M, Abril-Rodriguez G, Ross-Macdonald P, Wind-Rotolo M, Champhekar A, Medina E, Torrejon DY, Shin DS, Tran P, Kim YJ, Puig-Saus C, Campbell K, Vega-Crespo A, Quist M, Martignier C, Luke JJ, Wolchok JD, Johnson DB, Chmielowski B, Hodi FS, Bhatia S, Sharfman W, Urba WJ, Slingluff CL Jr., Diab A, Haanen J, Algarra SM, Pardoll DM, Anagnostou V, Topalian SL, Velculescu VE, Speiser DE, Kalbasi A, and Ribas A. 2020. Conserved Interferon-γ Signaling Drives Clinical Response to Immune Checkpoint Blockade Therapy in Melanoma. Cancer Cell 38: 500–515.e503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, Raissi TC, van der Windt GJ, Viollet B, Pearce EL, Pelletier J, Piccirillo CA, Krawczyk CM, Divangahi M, and Jones RG. 2015. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 42: 41–54. [DOI] [PubMed] [Google Scholar]
- 52.Mayer A, Denanglaire S, Viollet B, Leo O, and Andris F. 2008. AMP-activated protein kinase regulates lymphocyte responses to metabolic stress but is largely dispensable for immune cell development and function. Eur J Immunol 38: 948–956. [DOI] [PubMed] [Google Scholar]
- 53.He N, Fan W, Henriquez B, Yu RT, Atkins AR, Liddle C, Zheng Y, Downes M, and Evans RM. 2017. Metabolic control of regulatory T cell (Treg) survival and function by Lkb1. Proc Natl Acad Sci U S A 114: 12542–12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Medina CB, Chiu YH, Stremska ME, Lucas CD, Poon I, Tung KS, Elliott MR, Desai B, Lorenz UM, Bayliss DA, and Ravichandran KS. 2021. Pannexin 1 channels facilitate communication between T cells to restrict the severity of airway inflammation. Immunity 54: 1715–1727.e1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mock JR, Dial CF, Tune MK, Norton DL, Martin JR, Gomez JC, Hagan RS, Dang H, and Doerschuk CM. 2019. Transcriptional analysis of Foxp3+ Tregs and functions of two identified molecules during resolution of ALI. JCI Insight 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nefla M, Darling NJ, van Gijsel Bonnello M, Cohen P, and Arthur JSC. 2021. Salt inducible kinases 2 and 3 are required for thymic T cell development. Sci Rep 11: 21550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Navarro MN, Goebel J, Feijoo-Carnero C, Morrice N, and Cantrell DA. 2011. Phosphoproteomic analysis reveals an intrinsic pathway for the regulation of histone deacetylase 7 that controls the function of cytotoxic T lymphocytes. Nat Immunol 12: 352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parra M, Mahmoudi T, and Verdin E. 2007. Myosin phosphatase dephosphorylates HDAC7, controls its nucleocytoplasmic shuttling, and inhibits apoptosis in thymocytes. Genes Dev 21: 638–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dequiedt F, Kasler H, Fischle W, Kiermer V, Weinstein M, Herndier BG, and Verdin E. 2003. HDAC7, a thymus-specific class II histone deacetylase, regulates Nur77 transcription and TCR-mediated apoptosis. Immunity 18: 687–698. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.