

Abstract
Malignant migrating partial seizure of infancy (MMPSI) is a devastating and pharmacoresistant form of infantile epilepsy. MMPSI has been linked to multiple gain-of-function (GOF) mutations in the KCNT1 gene, which encodes for a potassium channel often referred to as SLACK. SLACK channels are sodium-activated potassium channels distributed throughout the central nervous system (CNS) and the periphery. The investigation described here aims to discover SLACK channel inhibitor tool compounds and profile their pharmacokinetic and pharmacodynamic properties. A SLACK channel inhibitor VU0531245 (VU245) was identified via a high-throughput screen (HTS) campaign. Structure-activity relationship (SAR) studies were conducted in five distinct regions of the hit VU245. VU245 analogs were evaluated for their ability to affect SLACK channel activity using a thallium flux assay in HEK-293 cells stably expressing wild-type (WT) human SLACK. Selected analogs were tested for metabolic stability in mouse liver microsomes and plasma-protein binding in mouse plasma. The same set of analogs was tested via thallium flux for activity versus human A934T SLACK and other structurally related potassium channels, including SLICK and Maxi-K. In addition, potencies for selected VU245 analogs were obtained using whole-cell electrophysiology (EP) assays in CHO cells stably expressing WT human SLACK through an automated patch clamp system. Results revealed that this scaffold tolerates structural changes in some regions, with some analogs demonstrating improved SLACK inhibitory activity, good selectivity against the other channels tested, and modest improvements in metabolic clearance. Analog VU0935685 represents a new, structurally distinct small-molecule inhibitor of SLACK channels that can serve as an in vitro tool for studying this target.
Keywords: KCNT1; Slack; KNa1.1; Slo2.2; MMPSI; EIMFS; 1,2,4-oxadiazoles
1. Introduction
Malignant migrating partial seizure of infancy (MMPSI) or epilepsy of infancy with migrating focal seizures (EIMFS) is a devastating and rare form of infantile epilepsy.1 MMPSI patients usually present with pharmacoresistant seizures within the first six months of life, characterized by continuous multifocal partial seizures migrating to different brain regions. MMPSI is accompanied by deterioration of psychomotor and cognitive development, visual impairment, hypotonia, and nearly 25% of the patients die within the first year of life. The etiology of MMPSI is linked to multiple gain-of-function (GOF) de novo missense heterozygous mutations in the KCNT1 gene in approximately 50% of the patients.2–4 KCNT1 encodes for the sodium (Na+)-activated potassium (K+) SLACK (Sequence Like A Calcium-Activated K+) channel. SLACK (KNa1.1, Slo2.2) is a member of the Slo family of K+ channels, which also includes Slo1 (Maxi-K, BK, or KCa1.1), Slo2.1. (SLICK), and Slo3.5–7 SLACK channels are critical regulators of electrical activity in the central nervous system (CNS) where they play an important role in regulating excitability, including afterhyperpolarization (AHP) after repetitive firing, which helps regulate the rate of action potentials.3–5, 8, 9
SLACK channels are found throughout the CNS, including the brainstem, olfactory bulb, cortical embryonic neurons, and hippocampus, as well as in the gonads, muscle tissues, cardiomyocytes, and pituitary glands.3, 5, 6, 10 SLACK channels are a tetramer of subunits comprised of six hydrophobic transmembrane domains (S1 – S6) and a pore-forming region between S5 – S6, in addition to an extended C-terminal cytoplasmic domain, which contains the regulator of potassium conductance (RCK) domain, a region that confers Na+ sensitivity to SLACK currents, as well as a nicotinamide adenine dinucleotide-binding (NAD+) domain.2, 7, 9, 11–14 Intracellular Na+ level is a key regulator of SLACK channel activity;13, 15 however, it is also regulated by the transmembrane voltage and various other factors, including direct phosphorylation of the channel by protein kinase C (PKC), or indirect modulation by protein kinase A (PKA), cytoplasmic NAD+ and ATP levels, estradiol, phosphatidyinostitol 4,5-bisphosphate, Cl− and others.13, 16, 17
Over 30 KCNT1 GOF mutations associated with MMPSI have been reported.3, 18–21 Previous studies have shown that these mutations are mainly present in the C-terminal region, which includes both the NAD+ and RCK binding domains, as well as the transmembrane pore-forming region.3, 7, 8, 14, 22, 23 Furthermore, KCNT1 GOF mutations are linked to epilepsy phenotypes other than MMPSI, such as early-onset epileptic encephalopathy (EOEE, i.e., West syndrome, and Ohtahara syndrome),23 autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE),24, 25 and others.26 27 GOF mutations in KCNT1 lead to SLACK channels with increased neuronal excitability by several potential mechanisms. Present data are most consistent with SLACK GOF mutations decreasing the activity of inhibitory interneurons resulting in an excitatory/inhibitory imbalance.14, 19 Activation of SLACK channels also results in an increased AHP amplitude, which correspondingly leads to an increased neuronal firing frequency.18, 28 It has also been noted that SLACK mutations may disrupt interactions with cell signaling pathways such as FMRP and Phactr-1.28 In fact, the cytoplasmic domain of SLACK channels interacts with developmentally relevant proteins such as the fragile X mental retardation protein (FMRP),29 cytoplasmic FMR1-interacting protein 1 (Cyfip1), and actin regulator 1 (Phactr1).30
To date, no broadly effective and safe drug therapy has been approved for the treatment of MMPSI. However, quinidine, clofilium, and bepridil inhibit SLACK currents in vitro, and were used as investigational treatments for MMPSI. Unfortunately, they have limited efficacy, and use of quinidine led to toxicity due to lack of selectivity and unfavorable pharmacokinetic properties.4, 8, 14, 20, 25, 31–37 Recently, researchers have shown increased interest in the discovery of small molecule selective SLACK channel inhibitors. To identify pore blockers, a virtual screen of 100,000 compounds was performed by researchers utilizing cryo-electron microscopy-derived chicken KNa1.1 structure. Six compounds that inhibited KNa1.1 channels at micromolar potencies were identified (compounds 1 and 2 in Figure 1 are representative examples); however, four compounds inhibited hERG (KV11.1) channels at 10 μM and showed a reduction in cell viability.38
Figure 1.

Representative SLACK channel inhibitors
A lead optimization campaign that culminated in characterization and in vivo studies of 1,2,4-oxadiazole tool compound 3 was recently reported.39 Additional analogs of 3 and related scaffolds have been disclosed in the patent literature as well.40 The activity of 3 was assessed in cell lines stably expressing human KCNT1 GOF variants and revealed IC50 values ranging from 221 nM to 1768 nM versus numerous human and mouse SLACK variants. Compound 3 was evaluated in brain slices from wild-type (WT) mice and mice homozygous for SLACK variant P905L (Kcnt1L/L), and a significant reduction in the firing was observed in neurons from Kcnt1L/L mice at 10 μM. However, 3 was an inhibitor of the hERG channel (IC50 = 11.9 μM). Likewise, it displayed 74% displacement at GABAA Cl– channels in a binding displacement assay, raising the possibility that GABAergic activity contributed to the observed anti-seizure effect.17, 39, 41
Our early research on SLACK inhibitors started with a high-throughput screen (HTS) using a thallium (TI+) flux assay in HEK-293 cells stably expressing WT human SLACK and a 110,000 member compound library. These efforts resulted in the discovery of the 2-amino-N-phenylacetamide VU0606170 (VU170) (4), which was found to be a selective SLACK channel inhibitor with low micromolar potency versus WT and select mutant SLACK channels (Figure 1). VU170 also effectively decreased the frequency of spontaneous, synchronized calcium (Ca2+) oscillations in a cortical neuronal/glial co-culture. Furthermore, the discovery of VU170 provided further evidence that SLACK channels can be effectively inhibited by small molecules.7 Finally, we recently disclosed the structure-activity relationship (SAR) studies that were executed in five distinct regions around VU170, which unfortunately revealed relatively flat SAR. Selected VU170 analogs were tested versus the A934T SLACK mutant revealing a potency on par with WT. In addition, results in whole-cell electrophysiology studies with the same compounds mirrored those obtained with the TI+ flux assay.17
Higher quality tool compounds that selectively target SLACK channels are needed to further study the role of SLACK modulation on KCNT1-associated epilepsies. As a result, we pursued a new, distinct hit series for optimization towards an improved SLACK channel tool compound. The same cell-based HTS campaign also led to the identification of 1,2,4-oxadiazole hit compound VU0531245 (VU245, 5), which inhibited WT SLACK channels with an IC50 value of 2.1 μM (Figure 1). In addition, compound 5 demonstrated high permeability in a MDCK-MDR1 bidirectional permeability assay (31.1 × 10−6 cm/sec) with no evidence of P-glycoprotein (Pgp)-mediated efflux (efflux ratio = 0.65). It likewise possessed calculated properties consistent with CNS penetrant molecules (MW = 391 g/mol, cLog P = 2.3, tPSA = 81, H-bond donors = 0, H-bond acceptors = 7).42–44 Nonetheless, the hit compound 5 suffered from high metabolic clearance in mouse liver microsomes (<10% remaining after 10 minutes), which hinders its usefulness for in vivo studies in mouse models of MMPSI, which are a relevant species given the prevalence of genetic models of epilepsy in that species.45 For example, see the successful generation of a mouse line bearing SLACK mutations by Quraishi et al.18 Still, the scaffold was viewed as highly synthetically tractable, and it was thus selected as a starting point for the discovery of SLACK tool compounds. Herein, we report the discovery, synthesis, SAR studies, in vitro pharmacokinetic, selectivity assessment, and electrophysiology studies within this series of 1,2,4-oxadiazoles as novel SLACK inhibitors.
2. Results and Discussion
2.1. Chemistry
Our medicinal chemistry strategy targeted 5 different regions of VU245 as illustrated in Figure 2. Initially, we explored SAR within ring A utilizing the synthetic route shown in Scheme 1. First, commercially available nitrile 6 was treated with hydroxylamine hydrochloride in basic medium to afford amidoxime 7, which was subsequently reacted with 1-(tert-butoxycarbonyl)azetidine-3-carboxylic acid 8 in the presence of the coupling agent HATU, followed by cyclization at high temperature to afford 1,2,4-oxadiazole 9. Next, cleavage of the BOC-protecting group via 4M HCl in 1,4-dioxane yielded the secondary amine 10 in the form of its hydrochloride salt. Penultimate intermediate 10 was reacted with different sulfonyl chlorides under basic conditions to generate 5 and the library compounds 11 – 25. The synthesis of related ring A analogs 26 - 30 is described in the Supplementary Material.
Figure 2.

Regions for SAR development in the VU0531245 scaffold.
Scheme 1.

Reagents and conditions: (a) NH2OH·HCl, NaHCO3, MeOH, 60 °C, 100%; (b) 1-(tert-butoxycarbonyl)azetidine-3-carboxylic acid (8), HATU, DIEA, DCM, NMP, 140 °C, 55%; (c) 4M HCl in 1,4-dioxane, 99%; (d) ArSO2Cl, NEt3, DCM, 17 – 79%.
SAR around the linker region was evaluated by replacing the sulfonamide with alkyl, urea, and carbamate groups as illustrated in Scheme 2. The methylene (31) and ethylene (32) linkers were prepared by reductive amination of amine 10 with 2-ethoxybenzaldehyde and 2-ethoxyacetophenone, respectively. Amide linkers 33 and 34 were synthesized by coupling amine 10 with 2-ethoxybenzoic acid and 2-ethoxyphenyl acetic acid, respectively. The urea linker in 35 was generated by reacting the amine intermediate 10 with 2-ethoxyisocyanate. Finally, the carbamate linker 36 was synthesized by reacting 4-nitrophenyl chloroformate with amine 10, followed reaction with 2-ethoxyphenol in the presence of potassium hydroxide under reflux conditions (Scheme 2).
Scheme 2.

Reagents and conditions: (a) DIEA, AcOH, NaBH(AcO)3, DCM, 2-ethoxybenzaldehyde (31, X = CH2, 44%), 2-ethoxyacetophenone, (32, X = CHCH3, 52%); (b) DIEA, HATU, DCM, 2-ethoxybenzoic acid (33, n = 0, 16%), 2-ethoxyphenyl acetic acid (34, n = 1, 6%); (c) 2-ethoxyphenyl isocyanate, DIEA, DCM, 13%; (d) ClCOOC6H4-4-(NO2), 2-ethoxyphenol, KOH, PhMe, reflux, 20%.
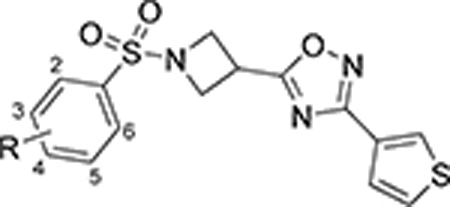
Next, ring D was systematically examined by replacing the 3-thiophene ring with unsubstituted and substituted phenyl rings, pyridines, and other 5-membered ring heterocycles. This library was prepared as outlined in Scheme 3. The amine 37 was reacted with 2-ethoxybenzene sulfonyl chloride to afford the ester 38, which was subsequently hydrolyzed in the presence of aqueous lithium hydroxide to yield the carboxylic acid 39. Acid 39 was reacted with assorted amidoximes to afford oxadiazoles 40 – 68 utilizing a variety of different synthetic conditions (Scheme 3).
Scheme 3.

Reagents and conditions: (a) 2-ethoxybenzene sulfonyl chloride, NEt3, DCM, 88%; (b) LiOH, H2O, THF, 95%; (c) 40, 59 ArC(NH)NHOH, HATU, DIEA, DMF, 140 °C, 6 - 40%; (d) 41 – 49, 53 – 58, 63 – 65 ArC(NH)NHOH, EDC, HOBt, DIEA, 1,4-dioxane, reflux, 5 –24%, (e) 50 – 52 ArC(NH)NHOH, CDI (2X), DMF, 80 °C, 39 – 43% or (f) 60 – 62, 66 – 68 ArC(NH)NHOH, CDI, NaOH, DMSO, 27 – 65%.
To further explore SAR around ring C, 5-membered heterocycles other than 1,2,4-oxadiazole were prepared. 1,3,4-Oxadiazole 81 was synthesized according to Scheme 4. First, carboxylic acid 8 was converted into hydrazide 78, followed by coupling with 4-fluorobenzoic acid and cyclization with Burgess reagent to afford the intermediate 79. BOC-deprotection using 4M HCl in 1,4-dioxane generated amine 80 in the form of its HCl salt, which was further reacted with 2-ethoxybenzene sulfonyl chloride to afford final compound 81. Next, 1,3,4-triazole 86 was prepared by reacting the hydrazide intermediate 78 with 4-fluorobenzonitrile at 160 °C in the presence of potassium carbonate to yield intermediate 82. The triazole nitrogen atom was then protected with a benzyl group to afford 83, followed by BOC-deprotection to generate amine 84 in the form of its HCl salt. Reaction of 84 with 2-ethoxybenzene sulfonyl chloride generated intermediate 85, which was subsequently subjected to catalytic hydrogenolysis to yield final compound 86 (Scheme 4).
Scheme 4.

Reagents and conditions: (a) CDI, THF, N2H4·H2O, 87%; (b) 4-fluorobenzoic acid, HATU, DIEA, Burgess reagent, THF, 82%; (c) 4M HCl in 1,4-dioxane, 93%; (d) 2-ethoxybenzene sulfonyl chloride, NEt3, DCM, 14%, (e) 4-fluorobenzonitrile, K2CO3, n-BuOH, 160 °C, 31%; (f) BnBr, K2CO3, DMF, 64%; (g) 4M HCl in 1,4-dioxane, 100%; (h) 2-ethoxybenzene sulfonyl chloride, DIEA, DCM, 54%; (i) 20% Pd(OH)2/C, MeOH, H-cube®, 25%.
Isoxazole analog 92 was prepared from the alcohol 87 (Scheme 5). Alcohol 87 was oxidized utilizing pyridine-sulfur trioxide complex into aldehyde 88, which was converted via Bestmann-Ohira reagent into alkyne 89. Cyclization of 89 with 4-fluorobenzaldehyde oxime via bis(trifluoroacetoxy)iodo)benzene (PIFA) generated isoxazole intermediate 90, which was BOC-deprotected using 4M HCl in 1,4-dioxane to yield amine 91 in the form of its HCl salt. Reaction of 91 with 2-ethoxybenzene sulfonyl chloride provided isoxazole analog 92. For the synthesis of 1,2,3-triazole analog 95, click chemistry between alkyne 89 and 1-azido-4-fluorobenzene yielded 1,2,3-triazole 93 as outlined in Scheme 5. BOC deprotection in the presence of 4M HCl in 1,4-dioxane to generate the amine 94 in the form of its HCl salt, which was further reacted with 2-ethoxybenzene sulfonyl chloride to yield analog 95.
Scheme 5.

Reagents and conditions: (a) Pyridine-sulfur trioxide complex, NEt3, DCM, DMSO, 60%; (b) Bestmann-Ohira reagent, K2CO3, MeOH, 72%; (c) 4-fluorobenzaldehyde oxime, PIFA, MeOH, H2O, 52%; (d) 4M HCl in 1,4-dioxane, 100%; (e) 2-ethoxybenzene sulfonyl chloride, DIEA, DCM, 55%; (f) 1-azido-4-fluorobenzene, sodium ascorbate, CuSO4, tert-BuOH, H2O, 56%; (g) 4M HCl in 1,4-dioxane, 100%; (h) 2-ethoxybenzene sulfonyl chloride, NEt3, DCM, 31%.
For the synthesis of oxazole analog 97, carboxylic acid intermediate 39 was reacted with 2-amino-1-(4-fluorophenyl)ethenone hydrochloride to yield intermediate 96 as illustrated in Scheme 6. Dehydration of 96 was accomplished under reflux conditions in phosphorus oxychloride (POCl3) to generate oxazole analog 97. Imidazole analogs 99 and 100 were generated by reacting carboxylic acid 39 with 2-bromo-4’-fluoroacetophenone (98) under basic conditions in the presence of ammonium acetate at reflux to yield imidazole 99. N-methylation of 99 was accomplished with methyl iodide to afford analog 100.
Scheme 6.

Reagents and conditions: (a) 2-amino-1-(4-fluorophenyl)ethenone hydrochloride, EDC, HOBt, DIEA, DCM, DMF, 6%; (b) POCl3, reflux, 25%; (c) Cs2CO3, EtOH, 2-bromo-4’-fluoroacetophenone (98), DMF, NH4OAc, PhMe, reflux, 63%, (d) MeI, K2CO3, DMF, 78%.
Certain analogs with different alkoxy or alkyl amine groups at position 2 of the phenyl ring were prepared according to Scheme 7. First, intermediate 104 (synthesis described in Supplementary Material) was reacted with the corresponding alcohols or amines via nucleophilic aromatic substitution to yield analogs 112 – 118. On the other hand, analogs 109 and 110 were prepared via nucleophilic aromatic substitution of intermediate 104 with allyl alcohol to yield intermediate 105. Removal of the allyl group by palladium catalysis generated phenol intermediate 106, which was further reacted with the tosylated intermediates 107 and 108 to afford analogs 109 and 110, respectively.
Scheme 7.

Reagents and conditions: (a) allyl alcohol, Cs2CO3, 70 °C, 47%; (b) Pd(PPh3)4, K2CO3, MeOH, 77%; (c) Cs2CO3, DMF, 120 °C, 20 min, μW, 107 (109, 70%), 108 (110, 56%); (d) TsCl, DMAP, NEt3, DCM, (107, 70%), (108, 75%); (e) alkyl alcohol or alkyl amine, Cs2CO3, DMF, 16 – 52%.
A library of bicyclic ring analogs in the ring A region (119 – 122) was prepared by following a synthetic route similar to that shown in Scheme 1 (detailed synthesis described in the Supplementary Material). However, analogs 125, 126, 129, 130, 132, and 133 were prepared according to the synthetic route outlined in Scheme 8.46 First, intermediate 123 (synthesis described in the Supplementary Material) was refluxed in ammonium hydroxide to yield amine 124. Cyclization of 124 with either 2-bromo-1,1-diethoxyethane or 1-bromo-2,2-dimethoxypropane under basic conditions afforded the imidazo[1,2-a]pyridine analogs 125 and 126, respectively. Synthesis of [1,2,4]triazolo[1,5-a]pyridine analogs 129 and 130 began with intermediate 124, which was treated with either dimethylformamide dimethyl acetal or dimethylacetamide dimethyl acetal to afford the amidoxime intermediates 127 and 128, respectively. Next, cyclization of the amidoxime intermediates was achieved through treatment with trifluoroacetic anhydride (TFAA) to yield the [1,2,4]triazolo[1,5-a]pyridine analogs 129 and 130. Finally, synthesis of [1,2,4]triazolo[4,3-a]pyridine analogs was initiated from intermediate 123. Nucleophilic aromatic substitution of 123 with hydrazine under microwave irradiation afforded intermediate 131. Cyclization of 131 via microwave heating in either trimethyl orthoformate or trimethyl orthoacetate provided [1,2,4]triazolo[4,3-a]pyridines 132 and 133, respectively.
Scheme 8.

Reagents and conditions: (a) NH4OH, 1,4-dioxane, 100 °C, 42%; (b) R = H, BrCH2CH(OC2H5)2, 21% or R = CH3, CH3C(OCH3)2CH2Br, 12%; (c) 127: R = H, DMF.DMA, isopropanol, 80 °C, NH2OH·HCl, 50 °C; 128: R = CH3, DMA.DMA, isopropanol, 80 °C, NH2OH·HCl; (d) TFAA, THF; 129 (R = H), 22%; 130 (R = CH3), 11%; (e) N2H4.H2O, ethanol, μW, 120 °C, 15 min, 90%; (f) 132: R = H, HC(OCH3)3, μW, 180 °C, 20 min, 47%; or 133: R = CH3, CH3C(OCH3)3, μW, 180 °C, 59%.
2.2. Pharmacology
2.2.1. Structure Activity Relationship Studies
Optimization of the hit compound and subsequent SAR elucidation was achieved via our Tl+ flux assay in HEK-293 cells stably expressing WT human SLACK to determine the potency (IC50) and efficacy of all new analogs.7 This assay uses Tl+ as a surrogate for potassium ions along with an intracellular dye that exhibits fluorescent activity upon binding Tl+ ions. Thus, fluorescence activity is used as the read out for this assay. Our investigation began with a SAR study of ring A by systematically scanning all positions of the phenyl ring employing common functional groups (11 – 25). Different electron withdrawing and electron donating groups led to mode switching from an inhibitor into an activator, or loss of activity, as most of the analogs were either weak activators of the channel or inactive (Table 1). The same observation was noted in our earlier hit optimization effort,17 and is consistent with the possibility that the tested analogs modulate SLACK function by a mechanism other than pore blockade. Interestingly, while 2-methoxy analog 23 was a weak activator, extending the alkoxy chain at position 2 of the phenyl ring into n-propyl (26) or i-propyl (27) maintained the SLACK inhibitory activity seen with 5, while the n-butyl (28) led to a loss of potency. On the other hand, replacing the oxygen with nitrogen (29) or carbon (30) led to a loss of activity, suggesting that the oxygen atom, in addition to a medium length alkyl chain (2–3 carbons) are necessary for optimal SLACK inhibitory activity. Next, replacing the sulfonamide linker with other linkers such as methylene (31), ethylene (32), amide (33, 34), urea (35), and carbamate (36) were examined (Table 2). It was noted that alternative linkers led to mode switching, resulting in weak activators, and that the sulfonamide was preferred for SLACK inhibitory activity. We postulate that the loss of activity upon changing the linker region is due to its unique properties, including the pyramidal structure adopted by the sulfonamide nitrogen atom, which presumably affects the orientation of the different functional groups that participate in the binding of the molecules to their binding sites.47
Table 1.
Ring A analogs
| ||||
|---|---|---|---|---|
|
| ||||
| No. | R | Modea | IC50 / EC50 (μM)b | Efficacy (%)b,c |
|
| ||||
| 5 | 2-OEt | Inh | 2.1 | 102 |
| 11 | 2-F | Act | 4.8 | 30 |
| 12 | 3-F | Act | >10 | 24 |
| 13 | 4-F | Inactive | ||
| 14 | 2-Cl | Act | 7.5 | 47 |
| 15 | 3-Cl | Inactive | ||
| 16 | 4-Cl | Inactive | ||
| 17 | 2-CF3 | Act | 6.0 | 36 |
| 18 | 3-CF3 | Act | 6.2 | 61 |
| 19 | 4-CF3 | Inactive | ||
| 20 | 2- Me | Act | 6.1 | 44 |
| 21 | 3- Me | Act | >10 | <10 |
| 22 | 4- Me | Inactive | ||
| 23 | 2-OMe | Act | 8.1 | 29 |
| 24 | 3-OMe | Inactive | ||
| 25 | 4-OMe | Act | 3.2 | 51 |
| 26 | 2-O(n-Pr) | Inh | 2.6 | 87 |
| 27 | 2-O(i-Pr) | Inh | 3.7 | 103 |
| 28 | 2-O(n-Bu) | Inactive | ||
| 29 | 2-NHEt d | Inactive | ||
| 30 | 2-n-Pr d | Inh | >10 | 79 |
Inh = inhibitor; Act = activator
Concentration-response curve (CRC) from Tl+ flux assay in HEK-293 cells expressing WT SLACK
Amplitude of response in the presence of 30 μM test compound as a percentage of the maximum response for VU0606170 (inhibitors) or loxapine (activators)
Synthesis described in Supplementary Material
Table 2.
Linker Library Analogs
| No. | Modea | IC50 / EC50 (μM)b | Efficacy (%)b,c |
|---|---|---|---|
|
| |||
| 31 | Act | >10 | 31 |
| 32 | Act | >10 | <10 |
| 33 | Act | >10 | 18 |
| 34 | Act | >10 | 37 |
| 35 | Act | >10 | <10 |
| 36 | Act | >10 | 50 |
Inh = inhibitor; Act = activator; NA = not active
Concentration-response curve (CRC) from Tl+ flux assay in HEK-293 cells expressing WT SLACK
Amplitude of response in the presence of 30 μM test compound as a percentage of the maximum response for VU0606170 (inhibitors) or loxapine (activators)
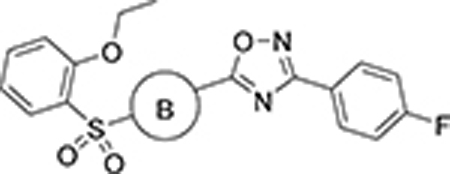
Next, we moved our attention to replacement of the 3-thiophenyl group in the ring D region of VU245, which is considered a structural alert due to its potential metabolism into reactive metabolites such as thiophene S-oxide and thiophene epoxide,48 and potentially contributes to the high clearance observed in VU245. In order to avoid these metabolic liabilities, the bioisosteric replacement of the 3-thiophene with a phenyl ring was explored, in addition to installing different functional groups (analogs 40 – 58, Table 3).49 The unsubstituted phenyl ring (40) led to a loss of potency; however, other modifications were generally tolerated, such as the ortho- and para-substituted halogens (41, 43, 44, and 46), while others led to mode switching or loss of potency. Next, we decided to further explore the effect of 2,4-di-substituted halogens on the phenyl ring (56 – 58), which retained good to moderate potency. Additionally, several heterocycles at the same position were prepared and tested (59 – 68). Interestingly, the SLACK channel activity showed a preference for the 5-membered (59 – 64) over the 6-membered heterocycles (65 – 68). Notably, 5-fluorothiophen-2-yl and 4-isothiazolyl rings demonstrated comparable SLACK inhibitory activity to the hit compound. The 4-fluorophenyl was selected as the basis for subsequent SAR elucidation, since the fluorine introduced minimal changes to the physiochemical properties while blocking a potential metabolic site. Likewise, this functional group offered a better balance of potency and lipophilicity than other similarly potent analogs. For instance, the cLogP value for 43 was 2.6 versus 3.3 and 3.0 for 49 and 58, respectively.44 Additionally, the 4-fluorophenyl group was expected to mitigate the potential toxicity concerns and offer improved metabolic stability compared to its 3-thiophene counterpart.
Table 3.
Ring D library compounds
| ||||
|---|---|---|---|---|
|
| ||||
| No. | R | Modea | IC50 / EC50 (μM)b | Efficacy (%)b,c |
|
| ||||
| 40 | H | Inh | >10 | 51 |
| 41 | 2-F | Inh | 7.9 | 88 |
| 42 | 3-F | Inactive | ||
| 43 | 4-F | Inh | 3.2 | 105 |
| 44 | 2-Cl | Inh | 8.0 | 81 |
| 45 | 3-Cl | Inactive | ||
| 46 | 4-Cl | Inh | 11 | 94 |
| 47 | 2-CF3 | Act | 1.9 | 66 |
| 48 | 3-CF3 | Inh | 11 | 48 |
| 49 | 4-CF3 | Inh | 3.8 | 95 |
| 50 | 2-Me | Act | >10 | <10 |
| 51 | 3- Me | Inh | >10 | <10 |
| 52 | 4- Me | Inactive | ||
| 53 | 2-OMe | Act | >10 | 18 |
| 54 | 3-OMe | Act | >10 | <10 |
| 55 | 4-OMe | Act | 7.1 | 21 |
| 56 | 2,4-difluoro | Inh | 6.5 | 93 |
| 57 | 4-Cl,2-F | Inh | 7.2 | 99 |
| 58 | 2-Cl,4-F | Inh | 2.1 | 95 |
| 59 | thiophen-2-yl | Inh | 6.9 | 99 |
| 60 | 5-fluorothiophen-2-yl | Inh | 4.8 | 105 |
| 61 | 2-thiazolyl | Inactive | ||
| 62 | 4-isothiazolyl | Inh | 6.6 | 106 |
| 63 | 2-furanyl | Inh | >10 | 44 |
| 64 | 2-pyridyl | Inactive | ||
| 65 | 3-pyridyl | Inactive | ||
| 66 | 4-pyridyl | Inactive | ||
| 67 | 4-fluoro-2-pyridyl | Inactive | ||
| 68 | 2-pyrimidinyl | Inactive | ||
Inh = inhibitor; Act = activator; NA = not active
Concentration-response curve (CRC) from Tl+ flux assay in HEK-293 cells expressing WT SLACK
Amplitude of response in the presence of 30 μM test compound as a percentage of the maximum response for VU0606170 (inhibitors) or loxapine (activators)
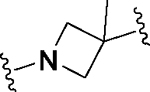
Having identified a potential replacement for the 3-thiophene ring, we turned our attention to the ring B region (Table 4). Installing a methyl on the azetidine ring (69) led to mode switching, while fluorine substitution (70) reduced inhibitor potency. Subsequently, we attempted to explore the effect of changing ring B and the sulfonamide bond together in order to eliminate any potential glutathione S-transferase-mediated cleavage of the sulfonamide.50 We flipped the azetidine ring orientation (71), installed a cyclobutane ring both in the cis (73a) and trans (73b) configurations, and replaced the azetidine with a benzene ring (77). However, none of these attempts proved successful, which accords with our earlier observations about the critical configuration adopted by the sulfonamide for optimal SLACK inhibitory activity. Expanding the azetidine ring into pyrrolidine in both R (74) and S (75) configurations was detrimental for SLACK activity. Finally, installing a piperidine ring (76) in place of the azetidine largely retained SLACK inhibitory activity (Table 4).
Table 4.
Ring B library compoundsd
| ||||
|---|---|---|---|---|
|
| ||||
| No. | B | Modea | IC50 / EC50 (μM)b | Efficacy (%)b,c |
|
| ||||
| 69 |
|
Act | 4.0 | 14 |
| 70 |
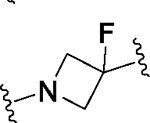
|
Inh | 9.2 | 98 |
| 71 |
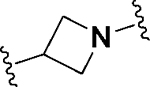
|
Inh | >10 | 24 |
| 73a |
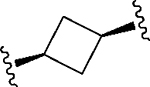
|
Inactive | ||
| 73b |
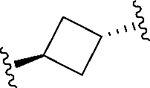
|
Inh | >10 | 52 |
| 74 |
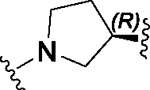
|
Inh | >10 | 31 |
| 75 |
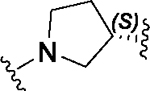
|
Inh | >10 | 63 |
| 76 |
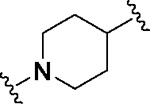
|
Inh | 4.9 | 105 |
| 77 |
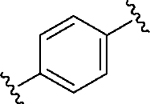
|
Inactive | ||
Inh = inhibitor; Act = activator; NA = not active
Concentration-response curve (CRC) from Tl+ flux assay in HEK-293 cells expressing WT SLACK
Amplitude of response in the presence of 30 μM test compound as a percentage of the maximum response for VU0606170 (inhibitors) or loxapine (activators)
Synthesis described in Supplementary Material
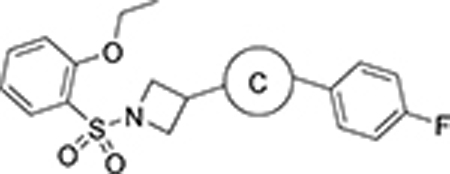
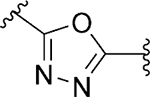
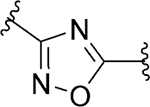
Subsequently, with the aim to explore modifications of the 1,2,4-oxadiazole core (ring C), we tested several five-membered heterocycles at that position (Table 5). It was noted that even the most closely related 1,2,4-oxadiazole regioisomer 101 led to a loss of SLACK activity. Not surprisingly, other 5-membered heterocycles, including isoxazole isomers (92 and 102) and N-methyl imidazole 100 also led to a loss of activity. On the other hand, heterocycles such as 1,3,4-oxadiazole 81, oxazole 97, triazole 86, imidazole 99, pyrazole 103, and triazole 95 exhibited mode switching. Overall, the results demonstrated that the SAR around ring C region was narrow and that the 1,2,4-oxadiazole found in the VU245 hit was optimal for SLACK inhibitory activity. Thus, we chose to maintain that core for further SAR exploration.
Table 5.
Ring C library compounds
| ||||
|---|---|---|---|---|
|
| ||||
| No. | C | Modea | IC50 / EC50 (μM)b | Efficacy (%)b,c |
|
| ||||
| 81 |
|
Act | >10 | <10 |
| 101 d |
|
Inh | >10 | 45 |
| 92 |
|
Inactive | ||
| 102 d |
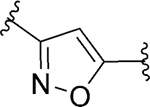
|
Inactive | ||
| 97 |
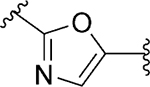
|
Act | 10 | 34 |
| 86 |
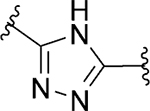
|
Act | >10 | 37 |
| 99 |
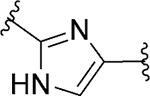
|
Act | 4.1 | 31 |
| 100 |
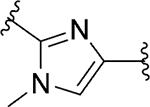
|
Inh | >10 | 47 |
| 103 d |
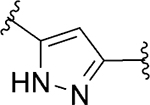
|
Act | >10 | 24 |
| 95 |
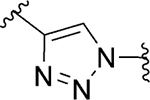
|
Act | >10 | 41 |
Inh = inhibitor; Act = activator; NA = not active
Concentration-response curve (CRC) from Tl+ flux assay in HEK-293 cells expressing WT SLACK
Amplitude of response in the presence of 30 μM test compound as a percentage of the maximum response for VU0606170 (inhibitors) or loxapine (activators)
Synthesis described in Supplementary Material
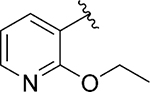
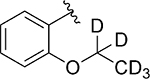
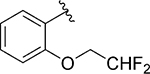
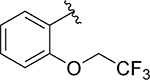
Having identified some tolerated modifications around VU245, we revisited the 2-ethoxy group at ring A, which while proving ideal for SLACK inhibitory activity, could represent a metabolic liability through P450-mediated dealkylation.51 As a result, several approaches were followed in an attempt to address this potential metabolic instability. Table 6 shows a selection of analogs (109 – 118) with alkoxy replacements that were introduced in this exercise. Replacing the benzene ring with a 3-pyridyl to lower lipophilicity as a means to improve metabolic stability52 in 111 led to a loss of activity. However, the isosteric replacement of the protium atoms of the ethoxy group with deuterium in 109 retained SLACK activity and offered an opportunity to slow down P450-mediated metabolism.53 In another attempt to increase metabolic stability, the trifluoroethyl isostere was installed at the same position;54 however, it led to a slight loss of potency (113). Surprisingly, the difluoromethyl group (112) led to mode switching and loss of activity. Pleasingly, this position was found tolerant of different aliphatic and alicyclic groups such as methyl cyclopropyl (114), methyl cyclobutyl (110), and cyclopentyl (116); however, the oxetanyl substitution at this position (115) led to mode switching, similar to the pattern observed in the first library (Table 1). This observation is potentially explained by the effect of electronic properties on mode switching, as electron density affects molecular geometry and binding characteristics.55 Replacing the oxygen with nitrogen in our first library (Table 1) introduced a new hydrogen bond donor and led to a loss of SLACK activity; interestingly, eliminating this hydrogen as in pyrrolidine 117 was tolerated. Nonetheless, expanding the ring to piperidine 118 led to a loss of activity. Taken together, SAR exploration within ring A in both libraries (Tables 1 and 6) implies that a hydrogen-bond acceptor at position 2 of the phenyl ring is critical for SLACK inhibitory activity, and this region is tolerant of only small alkyl chains.
Table 6.
Ring A Further Optimization
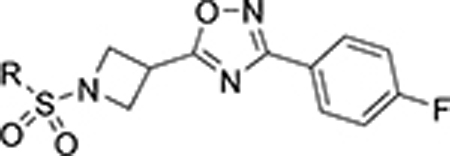
| ||||
|---|---|---|---|---|
|
| ||||
| No. | R | Modea | IC50 / EC50 (μM)b | Efficacy (%)b,c |
|
| ||||
| 111 d |
|
Inh | >10 | 46 |
| 109 |
|
Inh | 5.4 | 100 |
| 112 |
|
Act | ND | |
| 113 |
|
Inh | 8.8 | 91 |
| 114 |
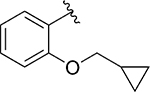
|
Inh | 6.0 | 95 |
| 110 |
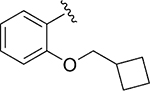
|
Inh | 9.3 | 88 |
| 115 |
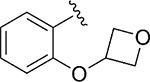
|
Act | 2.5 | 43 |
| 116 |
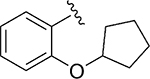
|
Inh | 5.8 | 93 |
| 117 |
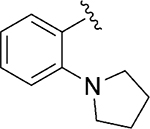
|
Inh | 8.9 | 92 |
| 118 |
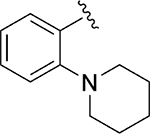
|
Inh | >10 | 28 |
Inh = inhibitor; Act = activator; NA = not active
Concentration-response curve (CRC) from Tl+ flux assay in HEK-293 cells expressing WT SLACK
Amplitude of response in the presence of 30 μM test compound as a percentage of the maximum response for VU0606170 (inhibitors) or loxapine (activators)
Synthesis described in Supplementary Material
Finally, a heterobicyclic ring library at the ring A region was pursued, maintaining what we postulate as critical structural features for SLACK inhibitory activity: a hydrogen bond acceptor directly attached to position 2 of the phenyl ring and a medium-length alkyl chain (Table 7). 8-Chromane 119 retained SLACK inhibitory activity, while 7-benzofuran 120 exhibited mode switching into an activator. Next, we turned our attention to nitrogen-containing heterocycles, such as 8-quinoline 121, which retained SLACK inhibitory activity. Installation of a methyl group at the 2-position of the 8-quinoline ring (122) led to enhanced potency and was expected to block a potential site for certain types of metabolism while simultaneously lowering the risk for mutagenicity.56, 57 Finally, given our objective to generate brain-penetrant analogs, we attempted to lower the lipophilicity of our compounds, which could also potentially lead to higher metabolic stability.42 Different nitrogen-containing heterocycles were prepared and evaluated as part of this exercise; unfortunately, both unsubstituted (125, 129, and 132) and methylated (126, 130, and 133) analogs proved detrimental for SLACK inhibitory activity (Table 7).
Table 7.
Ring A bicyclic analogs
| ||||
|---|---|---|---|---|
|
| ||||
| No. | R | Modea | IC50 / EC50 (μM)b | Efficacy (%)b,c |
|
| ||||
| 119 |
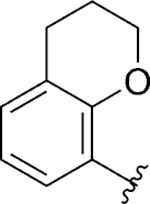
|
Inh | 8.7 | 93 |
| 120 |
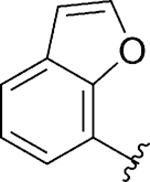
|
Act | 2.3 | 37 |
| 121 |
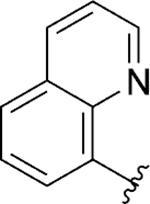
|
Inh | 7.4 | 98 |
| 122 |
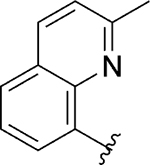
|
Inh | 1.1 | 99 |
| 125 |
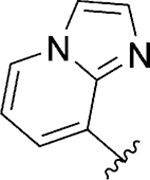
|
Act | >10 | 48 |
| 126 |
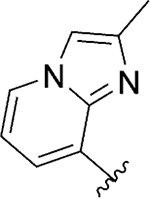
|
Act | >10 | 19 |
| 129 |
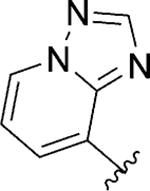
|
Act | >10 | 16 |
| 130 |
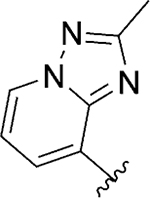
|
Act | >10 | 16 |
| 132 |
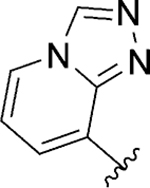
|
Act | >10 | 14 |
| 133 |
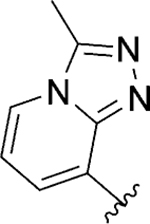
|
Inactive | ||
Inh = inhibitor; Act = activator; NA = not active
Concentration-response curve (CRC) from Tl+ flux assay in HEK-293 cells expressing WT SLACK
Amplitude of response in the presence of 30 μM test compound as a percentage of the maximum response for VU0606170 (inhibitors) or loxapine (activators)
2.2.2. Activity versus A934T SLACK
We decided to further evaluate the pharmacological profile of a selected set of analogs. In choosing this set, we of course selected some of our most potent analogs, e.g., 5, 43, 58, and 122. We also chose some more moderately potent analogs with unique structural elements. For example, we chose 27 to investigate the effect of branching on the 2-alkoxy group. We likewise chose 109 and 113 to investigate the impact of deuteration and fluorination, respectively, at the same position. We selected 60 as an example of a heteroaryl ring at the Ring D position and 76 as an example of piperidine at the Ring B position. Finally, we included 119 and 121 as additional examples of fused rings, one saturated and one unsaturated, at the Ring A position.
These 11 selected compounds were screened against A934T mutant SLACK, a GOF mutation commonly associated with MMPSI (Table 8).2, 3 All of the tested molecules showed inhibitory activity on the SLACK variant A934T, and were equipotent or slightly more potent against A934T compared to WT. Furthermore, two of our new analogs (60 and 122) showed submicromolar potency versus this mutant channel. This behavior matches that observed in an earlier hit optimization effort for another structurally distinct series of SLACK inhibitors,17 suggesting that our analogs exert their pharmacological action by the same mechanism. Moreover, it has been reported that SLACK mutants associated with epilepsy have an increased open channel probability compared to WT SLACK.15 Hence, it could be hypothesized that our analogs may inhibit an open conformation of the channel in a similar fashion to the binding of quinidine to other SLACK mutants associated with GOF behavior.14
Table 8.
Comparison of activity versus WT and A934T SLACK for selected inhibitors
| No. | WT | A934T | ||
|---|---|---|---|---|
| IC50 (μM)a | Efficacy (%)a,b | IC50 (μM)a | Efficacy (%)a,b | |
|
| ||||
| 5 | 2.1 | 102 | 0.82 | 72 |
| 27 | 3.7 | 103 | 1.1 | 109 |
| 43 | 3.2 | 105 | 1.0 | 110 |
| 58 | 2.1 | 95 | 2.0 | 95 |
| 60 | 4.8 | 105 | 0.97 | 103 |
| 76 | 4.9 | 105 | 4.3 | 129 |
| 109 | 5.4 | 100 | 2.3 | 83 |
| 113 | 8.8 | 91 | 2.5 | 72 |
| 119 | 8.7 | 93 | 2.6 | 104 |
| 121 | 7.4 | 98 | 1.5 | 112 |
| 122 | 1.1 | 99 | 0.67 | 72 |
CRC from Tl+ flux assay in HEK-293 cells expressing either WT or A934T SLACK
Amplitude of response in the presence of 30 μM test compound as a percentage of the maximum response for VU0606170
2.2.3. Selectivity Profile
We next assessed the selectivity profile for the same set of analogs against two additional members of the Slo family: SLICK (Slo2.1) and Maxi-K (Slo1 α1/β3) (Table 9). SLICK (Sequence like an intermediate conductance K+ channel) is another sodium-activated K+ channel, also known as Slo 2.1, encoded by the KCNT2 gene. SLICK shares almost 74% sequence homology with SLACK; furthermore, the RCK and transmembrane domains are almost identical. SLACK and SLICK mainly differ at the distal C-terminal region.13 Interestingly, SLICK and SLACK-B isoforms can exist as heteromeric channels with different properties than either channel expressed alone. For example, the time required to achieve 90% activation of the heteromeric channel is significantly longer than the time needed to activate either SLICK or SLACK-B homomeric currents.58 SLICK channels are implicated in epileptic encephalopathy and somatosensory processing.59–61 The selected analogs showed weak inhibition of SLICK (Table 10); however, analogs 43 and 121 showed an IC50 of 2.3 and 5.9 μM, and the efficacy was 50% and 56% of the positive control, respectively. The utility of analogs with SLICK/SLACK dual action is yet to be determined. On the other hand, Maxi-K or Slo1 (BK) channels are large-conductance Ca2+-activated potassium channels encoded by the KCNMA1 gene.62 Maxi-K channels share almost 7% identity with SLACK. It was demonstrated that SLACK and Maxi-K channels are similar at the pore-forming domain and the following S6 domain, in addition to the RCK domain.13 Satisfyingly, the selected analogs showed no activity versus Maxi-K at 30 μM.
Table 9.
Slo family selectivity for select SLACK inhibitors
| No. | SLICK (Slo2.1) | Maxi-K (Slo1 α1/β3) | ||
|---|---|---|---|---|
| IC50 (μM)a | Efficacy (%)a,b | IC50 (μM)a | Efficacy (%)a,b | |
|
| ||||
| 5 | >10c | 55 | inactive | |
| 27 | >10c | 47 | inactive | |
| 43 | 2.3 | 50 | inactive | |
| 58 | >10c | 44 | inactive | |
| 60 | >10c | 65 | inactive | |
| 76 | >10c | 35 | inactive | |
| 109 | >10c | 55 | inactive | |
| 113 | >10c | 27 | inactive | |
| 119 | >10c | 55 | inactive | |
| 121 | 5.9 | 56 | inactive | |
| 122 | >10c | 76 | inactive | |
CRC from Tl+ flux assay in HEK-293 cells expressing either SLICK (Slo2.1) or Maxi-K (Slo1 α1/β3)
Amplitude of response in the presence of 30 μM test compound as a percentage of the maximum response for SKF96365 (SLICK) or paxilline (Maxi-K)
CRC does not plateau
Table 10.
Whole-cell electrophysiology (EP) versus human WT SLACK for select inhibitors
| No. | IC50 (μM)a | Efficacy (%)b |
|---|---|---|
|
| ||
| VU0606170 | 2.28 | 95 |
| 5 | 0.64 | 100 |
| 27 | 1.09 | 101 |
| 43 | 1.00 | 101 |
| 58 | 0.43 | 100 |
| 60 | 0.70 | 100 |
| 76 | 1.76 | 100 |
| 109 | 1.04 | 101 |
| 113 | 1.76 | 100 |
| 119 | 1.63 | 100 |
| 121 | 1.58 | 100 |
| 122 | 0.32 | 100 |
CRC from whole-cell EP in CHO cells expressing WT SLACK utilizing an automated patch clamp system
Amplitude of response in the presence of 30 μM test compound as a percentage of the maximum response for 60 μM bepridil.
Bepridil and quinidine are both known to inhibit hERG channels and are associated with increased risk for cardiac arrhythmias due to QT prolongation.63, 64 Likewise, certain preclinical SLACK inhibitors demonstrated a propensity for hERG inhibition as well.38, 39 Thus, we were keen to evaluate the potential for hERG inhibition with our most potent compound. Gratifyingly, evaluation of a 10 μM concentration of analog 122 using our Tl+ flux assay in HEK-293 stably expressing hERG7 showed the compound to be inactive.
2.2.4. Electrophysiology
The SLACK inhibitory activity for the same set of analogs was assessed in whole-cell, voltage-clamp electrophysiology (EP) assays utilizing an automated patch clamp (APC) system (SyncroPatch 384) on Chinese Hamster Ovary (CHO) cell lines stably expressing human WT SLACK channels (Table 10). Recently, we have observed that on our Nanion SynchroPatch APC system, SLACK-expressing CHO cells demonstrated superior experimental success rates compared to SLACK-expressing HEK-293 cells. Thus, for our APC studies, SLACK-expressing CHO cells were used. The potency of VU0606170 is shown here and is similar across both cell lines.7 In this study, each of our analogs demonstrated a slightly higher potency in EP compared to those observed in the TI+ flux assay. We were likewise pleased to observe that three analogs have submicromolar potency (58, 60, and 122) besides hit compound VU245 (5). In fact, 2-methylquinoline 122 represents a notable advance, representing the most potent analog to emerge from our SAR efforts to date and will make another useful in vitro probe compound for studying SLACK channels.
2.3. In vitro Drug Metabolism and Pharmacokinetics (DMPK)
Our SAR studies around VU245 (5) only marginally improved upon the potency of the hit compound. Still, we were able to identify analogs that were equipotent or near equipotent to the original hit VU245 (5). Next, we advanced the same set of 11 analogs for further assessment in our frontline in vitro DMPK assays. Specifically, metabolic stability was determined by measuring the percent remaining of compounds after incubation for 10 minutes in mouse liver microsomes (MLMs). Likewise, the extent to which the analogs bind to the mouse plasma proteins was measured by equilibrium dialysis (Table 11). Branching the ethoxy into an isopropyl (27) was not a successful approach to improve the metabolic stability within this scaffold. However, replacing the thiophene (5) with 4-fluorophenyl (43) and 2-chloro-4-fluorophenyl (58) improved metabolic stability to some extent, supporting our hypothesis that the 3-thiophene ring in 5 contributed to its metabolic instability. 5-Fluoro-2-thiophene ring analog 60 proved no more metabolically stable than the unsubstituted 3-thiophene hit 5. Replacement of the azetidine (43) with a piperidine (76) ring led to increased metabolic clearance, possibly due to the introduction of additional sites for oxidative metabolism65 and higher lipophilicity compared to its azetidine ring counterpart. Gratifyingly, isosteric replacement of hydrogen (5) with deuterium (109) and fluorine (113) improved metabolic stability. Chromane 119 demonstrated a slight improvement in metabolic stability relative to 5; however, it is still a highly cleared analog. Interestingly, 8-quinoline 121 had notably improved metabolic stability to 74% remaining after 10 minutes. The introduction of a methyl group at position 2 of the quinoline ring (122) led to reduced metabolic stability, likely due to the introduction of a new metabolic soft spot to the molecule. We selected analog 121 for further assessment in mouse liver microsomes at multiple time points to enable the calculation of predicted hepatic clearance (CLHEP). These studies found the CLHEP to be 75 mL/min/kg, which is still considered high (> 2/3 hepatic blood flow). Overall, the results presented thus far provide evidence that both the ethoxy group and the 3-thiophene ring at VU245 (5) represent potential metabolic soft spots. Several approaches proved successful for marginally improving metabolic stability; however, the scaffold still suffers in that regard. The metabolic instability may be attributed to the sulfonyl azetidine, part of the molecule that was critical for optimal SLACK inhibitory activity.
Table 11.
In vitro DMPK data for select SLACK inhibitors
| No. | cLogPa | % Remaining after 10 minb | Plasma-protein binding (fu)c |
|---|---|---|---|
|
| |||
| 5 | 2.32 | <10 | 0.022 |
| 27 | 2.63 | <10 | <0.01 |
| 43 | 2.55 | 58 | 0.012 |
| 58 | 3.02 | 59 | 0.014 |
| 60 | 2.69 | <10 | 0.024 |
| 76 | 3.52 | 21 | <0.01 |
| 109 | 2.55 | 63 | 0.012 |
| 113 | 2.82 | 57 | 0.012 |
| 119 | 3.05 | 19 | <0.01 |
| 121 | 2.34 | 71 | 0.014 |
| 122 | 2.84 | 26 | <0.01 |
Calculated using ChemDraw Professional. Version 16.0.
Liver microsomal stability was reported as percent of parent compound remaining after 10 min incubation with NADPH-supplemented mouse liver microsomes.
Determined from DMSO stock solution by equilibrium dialysis in mouse plasma.
We next turned our attention to evaluation of mouse plasma-protein binding, which is expressed here (Table 11) in terms of fraction unbound (fu). Interestingly, the 5-fluorothiophen-2-yl in 60 maintained the same level of plasma-protein binding as the hit compound VU245 (5). It seems possible that the thiophene ring is slightly more favorable in terms of protein binding compared to the halogenated phenyl ring counterparts (43 and 58); nonetheless, no significant difference in protein binding was observed across most of our analogs. All analogs showed high (fu = 0.01 – 0.024) protein binding except analogs 27, 76, 119, and 122 which demonstrated very high (fu < 0.01) protein binding. While plasma-protein binding is an important parameter to understand the DMPK and safety properties of our compounds, high plasma-protein binding is not always a liability.42
3. Conclusion
The present study was designed to determine the SAR around VU245 to optimize selective SLACK inhibitors and evaluate its potential as a lead series. We were able to identify critical structural features associated with optimal SLACK inhibitory activity, and an improvement in potency was achieved. Furthermore, we selected a set of analogs for further characterization in mouse plasma-protein binding and microsomal stability assays. Our analogs demonstrated high plasma-protein binding, and we were able to improve the metabolic stability in some cases; however, this series still suffers from high metabolic clearance in mouse liver microsomes. Notably, this series demonstrated a high degree of selectivity for SLACK channels versus other K+ channels from the same family: SLICK and Maxi-K. Furthermore, all selected analogs were active versus the A934T mutant SLACK and showed equivalent or increased potency versus the mutant compared to WT SLACK. Finally, select analogs were assessed for potency in whole-cell EP, and the results revealed submicromolar potency for multiple analogs, including an attractive new in vitro tool 122 (VU0935685) that also demonstrated selectivity versus hERG. Although the study has successfully characterized analogs with different structural features and improved some properties compared to the hit compound, the high metabolic clearance of this series likely precludes its use as an in vivo tool compound in mice. Hit evaluation and optimization continues in other distinct scaffolds identified through our HTS campaign in order to identify high-quality SLACK inhibitors tool compounds for use in vivo. Results from those ongoing studies will be reported in due course.
4. Experimental Section
Experimental procedures for synthesis and characterization of analogs 5, 11-28, 40-68, 109-110, and 112-122 are found below. Experimental procedures for pharmacology and in vitro DMPK studies as well as synthesis and characterization of all analogs, including 1H and 13C NMR spectra for all analogs submitted for biological testing, may be found in the Supplementary Material.
4.1. Synthesis and Purification.
Air-sensitive reactions were carried out under a nitrogen atmosphere. Starting materials, reagents, intermediates, and final compounds were weighed on a Mettler Toledo™ New Classic ME analytical balance or a Mettler Toledo™ New Classic ME toploader balance. Thin-layer chromatography (TLC) was conducted on glass plates coated with Silica Gel 60 F254 from Millipore Sigma. Normal-phase flash chromatography was carried out on either a CombiFlash® EZ Prep or CombiFlash® Rf+ automated flash chromatography system, both from Teledyne ISCO. Normal-phase flash chromatography was carried out using RediSep® Rf normal-phase, disposable flash columns from Teledyne ISCO or SiliaSep normal-phase, disposable flash columns (40–63 micron) from SiliCycle, Inc. Reverse-phase preparative chromatography was carried out on the CombiFlash® EZ Prep using a reusable RediSep® Rf C18 reverse-phase column. Microwave reactions were carried out an Anton Paar Monowave 200 automated microwave synthesizer. The Monowave 200 has an output power of 850W with a maximum temperature of 260 °C and a maximum pressure of 290 psi and is suitable for use with reaction volumes ranging from 0.5 to 20 mL.
4.2. Characterization.
All NMR spectra were recorded on a 300 MHz Bruker Fourier 300HD NMR spectrometer equipped with a dual 1H and 13C probe with Z-Gradient and automatic tuning and matching, full computer control of all shims with TopShim™, 24-sample SampleCase™ automation system, and TopSpin™ software. All NMR samples were prepared with either chloroform-d with 0.03% TMS (99.8+ atom % D, Acros Organics Catalog No. 209561000) or d6-dimethyl sulfoxide with 0.03% TMS (ACROS Organics Catalog No. 360000100). 1H and 13C chemical shifts are reported in δ values in ppm downfield. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), integration, coupling constant (Hz). High resolution mass spectrometry was conducted on an Agilent 6230 Accurate-Mass Time-of-Flight (TOF) LC/MS with ESI source equipped with MassHunter Walkup software. MS parameters were as follows: fragmentor: 175 V, capillary voltage: 3500 V, nebulizer pressure: 35 psig, drying gas flow: 11 L/min, drying gas temperature: 325 °C. Samples were introduced via an Agilent 1260 Infinity UHPLC comprised of a G4225A HiP Degasser, G1312B binary pump, G1367E ALS, G1316A TCC, and G1315C DAD VL+ with a 5 μL semi-micro flow cell with a 6 mm path length. UV absorption was observed at 220 nm and 254 nm with a 4 nm bandwidth. Column: Agilent Zorbax SB-C18, Rapid Resolution HT, 1.8 μm, 2.1 × 50 mm. Gradient conditions: Hold at 5% CH3CN in H2O (0.1% formic acid) for 1.0 min, 5% to 95% CH3CN in H2O (0.1% formic acid) over 5 min, hold at 95% CH3CN in H2O (0.1% formic acid) for 1.0 min, 0.5 mL/min. All samples submitted for biological testing were confirmed ≥ 95% pure by 1H NMR.
4.3. Chemical synthesis.
4.3.1. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(thiophen-3-yl)-1,2,4-oxadiazole (5).
(Step 1) N-hydroxythiophene-3-carboximidamide (7). Hydroxylamine hydrochloride (639 mg, 9.16 mmol, 2.0 eq) and sodium bicarbonate (773 mg, 9.16, 2.0 eq) were stirred in methanol (10 mL) for 30 minutes at room temperature. 3-Thiophenecarbonitrile (500 mg, 4.58 mmol, 1.0 eq) was added to the previous suspension and heated at 50 °C for 2 hours. The reaction was concentrated in vacuo, water was added, and extracted with ethyl acetate (2x). The combined organics were washed with brine, dried over sodium sulfate (Na2SO4), filtered, and concentrated in vacuo to afford 651 mg (100%) of the title compound as a white solid that was used further without purification. 1H NMR (300 MHz, CDCl3) δ 7.54 (t, J = 2.1 Hz, 1H), 7.34 (m, 2H), 4.86 (bs, 2H). (Step 2) tert-Butyl 3-(3-(thiophen-3-yl)-1,2,4-oxadiazol-5-yl)azetidine-1-carboxylate (9). Intermediate 7 (651 mg, 4.58, 1.0 eq), 1-boc-azetidine-3-carboxylic acid (8) (1.00 g, 5.04 mmol, 1.1 eq), hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU) (2.09 g, 5.50 mmol, 1.2 eq), and N,N-diisopropylethylamine (DIEA) (2.40 mL, 13.7 mmol, 3.0 eq) were dissolved in Dichloromethane (DCM) (15 mL) and allowed to react for 3 hours at room temperature. Water was added, and the reaction was extracted with ethyl acetate (2x), washed with brine, dried over sodium sulfate (Na2SO4), filtered, and concentrated in vacuo. The reaction was dissolved in N-Methyl-2-pyrrolidone (NMP) (1.0 mL) and heated at 150 °C for 1 hour. Water was added, and the reaction was extracted with ethyl acetate (2x). The combined organics were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel afforded 773 mg (55%) of the title compound. 1H NMR (300 MHz, CDCl3) δ 8.09 (dd, J = 3.0, 1.19 Hz, 1H), 7.64 (dd, J = 5.1, 1.2 Hz, 1H), 7.44 (dd, J = 5.1, 3.0 Hz, 1H), 4.35 (m, 4H), 4.04 (m, 1H), 1.47 (s, 9H). (Step 3) 3-(3-(Thiophen-3-yl)-1,2,4-oxadiazol-5-yl)azetidine hydrochloride (10). Intermediate 9 (773 mg, 2.51 mmol, 1.0 eq) was dissolved in 4M hydrochloric acid (HCl) in 1,4-dioxane (5.0 mL) and stirred at room temperature for 30 minutes. The reaction was concentrated in vacuo to afford 604 mg (99%) of the title compound as a white solid that was used further without purification. 1H NMR (300 MHz, DMSO-d6) δ 8.21 (dd, J = 2.8, 1.4 Hz, 1H), 7.63 (m, 2H), 4.64 – 4.42 (m, 5H). (Step 4) 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(thiophen-3-yl)-1,2,4-oxadiazole (5). Intermediate 10 (25 mg, 0.10 mmol, 1.0 eq) and triethylamine (TEA) (28 μL, 0.20 mmol, 2.0 eq) were dissolved in DCM (2.0 mL). 2-Ethoxybeznene sulfonyl chloride (33 mg, 0.15 mmol, 1.5 eq) was added, and the reaction was stirred for 1 hour at room temperature. Water was added, and the reaction was extracted with DCM (2x). The combined organics were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel afforded 15 mg (38%) of the title compound. 1H NMR (300 MHz, CDCl3) δ 8.02 (dd, J = 3.0, 1.2 Hz, 1H), 7.91 (dd, J = 7.9, 1.7 Hz, 1H), 7.60 (dd, J = 5.1, 1.2 Hz, 1H), 7.54 (m, 1H), 7.43 (dd, J = 3.0, 2.0 Hz, 1H), 7.06 (m, 2H), 4.46 (d, J = 2.7 Hz, 2H), 4.43 (d, J = 0.8 Hz, 2H), 4.20 (q, J = 7.0 Hz, 2H), 4.03 (m, 1H), 1.50 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 178.0, 165.0, 156.7, 135.0, 131.2, 128.0, 127.8, 127.2, 126.0, 125.9, 120.4, 113.5, 65.1, 54.5, 25.3, 14.7 ppm. LCMS RT = 5.09 min; HRMS, calc’d for C17H18N3O4S2+ [M+H], 392.0733; found 392.0740.
4.3.2. 5-(1-((2-Fluorophenyl)sulfonyl)azetidin-3-yl)-3-(thiophen-3-yl)-1,2,4-oxadiazole (11).
The title compound was prepared in 47% yield (17 mg) from intermediate 10 and 2-fluorobenzene sulfonyl chloride using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, CDCl3) δ 8.00 (dd, J = 3.0, 1.2 Hz, 1H), 7.91 (m, 1H), 7.64 (m, 1H), 7.58 (dd, J = 5.1, 1.2 Hz, 1H), 7.43 (q, J = 3.0 Hz, 1H), 7.32 (m, 2H), 4.43 (t, J = 8.3 Hz, 2H), 4.35 (t, J = 6.9 Hz, 2H), 4.05 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 167.5, 165.0, 159.3 (d, J(C,F) = 256.0 Hz), 135.7 (d, J(C,F) = 8.6 Hz), 131.2, 128.1, 127.7, 127.2, 126.0, 124.6 (d, J(C,F) = 3.78 Hz), 124.4 (d, J(C,F) = 14.7 Hz), 54.4 (d, J(C,F) = 2.1 Hz), 25.2 ppm. LCMS RT = 5.00 min; HRMS, calc’d for C15H13FN3O3S2+ [M+H], 366.0377; found 366.0378.
4.3.3. 5-(1-((3-Fluorophenyl)sulfonyl)azetidin-3-yl)-3-(thiophen-3-yl)-1,2,4-oxadiazole (12).
The title compound was prepared in 79% yield (29 mg) from intermediate 10 and 3-fluorobenzene sulfonyl chloride using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, CDCl3) δ 7.98 (dd, J = 3.02, 1.20 Hz, 1H), 7.69 (dt, J = 7.65, 1.38 Hz, 1H), 7.64 – 7.58 (m, 2H), 7.56 (dd, J = 5.08, 1.17 Hz, 1H), 7.43 (q, J = 3.16 Hz, 1H), 7.37 (m, 1H), 4.32 (t, J = 8.45 Hz, 2H), 4.17 (t, J = 6.32 Hz, 2H), 3.96 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 177.2, 165.0, 162.6 (d, J(C,F) = 252.3 Hz), 136.4 (d, J(C,F) = 6.6 Hz), 131.2 (d, J(C,F) = 7.7 Hz), 128.1, 127.6, 127.3, 125.9, 124.2 (d, J(C,F) = 3.4 Hz), 120.9 (d, J(C,F) = 21.1 Hz), 115.7 (d, J(C,F) = 24.3 Hz), 54.5, 25.2 ppm. LCMS RT = 5.04 min; HRMS, calc’d for C15H13FN3O3S2+ [M+H], 366.0377; found 366.0385.
4.3.4. 5-(1-((4-Fluorophenyl)sulfonyl)azetidin-3-yl)-3-(thiophen-3-yl)-1,2,4-oxadiazole (13).
The title compound was prepared in 68% (25 mg) yield from intermediate 10 and 4-fluorobenzene sulfonyl chloride using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, CDCl3) δ 7.97 (dd, J = 3.0, 1.2 Hz, 1H), 7.92 (m, 2H), 7.55 (dd, J = 5.0, 1.2 Hz, 1H), 7.43 (q, J = 3.0 Hz, 1H), 7.30 (m, 2H), 4.30 (t, J = 8.4 Hz, 2H), 4.13 (t, J = 6.3 Hz, 2H), 3.98 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 177.3, 165.8 (d, J(C,F) = 256 Hz), 165.0, 131.2 (d, J(C,F) = 9.4 Hz), 130.4 (d, J(C,F) = 3.3 Hz), 128.0, 127.6, 127.3, 125.9, 116.7 (d, J(C,F) = 22.6 Hz), 54.4, 25.2 ppm. LCMS RT = 5.00 min; HRMS, calc’d for C15H13FN3O3S2+ [M+H], 366.0377; found 366.0379.
4.3.5. 5-(1-((2-Chlorophenyl)sulfonyl)azetidin-3-yl)-3-(thiophen-3-yl)-1,2,4-oxadiazole (14).
The title compound was prepared in 45% (17 mg) yield from intermediate 10 and 2-chlorobenzene sulfonyl chloride using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, CDCl3) δ 8.1 – 8.0 (m, 2H), 7.62 (dd, J = 5.0, 1.2 Hz, 1H), 7.56 (m, 2H), 7.46 – 7.39 (m, 2H), 4.48 (m, 4H), 4.09 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 177.6, 165.1, 136.1, 134.1, 132.8, 132.1, 131.3, 128.1, 127.8, 127.2, 127.1, 126.0, 54.5, 25.1 ppm. LCMS RT = 5.20 min; HRMS, calc’d for C15H13ClN3O3S2+ [M+H], 382.0081; found 382.0089.
4.3.6. 5-(1-((3-Chlorophenyl)sulfonyl)azetidin-3-yl)-3-(thiophen-3-yl)-1,2,4-oxadiazole (15).
The title compound was prepared in 60% (23 mg) yield from intermediate 10 and 3-chlorobenzene sulfonyl chloride using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, CDCl3) δ 7.99 (dd, J = 3.1, 1.2 Hz, 1H), 7.89 (t, J = 1.7 Hz, 1H), 7.77 (dt, J = 7.7, 1.3 Hz, 1H), 7.63 (m, 1H), 7.55 (m, 2H), 7.43 (q, J = 3.0 Hz, 1H), 4.32 (t, J = 8.5 Hz, 2H), 4.17 (t, J = 6.3 Hz, 2H), 3.99 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 177.3, 165.0, 136.2, 135.7, 133.7, 130.6, 128.3, 128.1, 127.6, 127.3, 126.4, 125.9, 54.5, 25.2 ppm. LCMS RT = 5.26 min; HRMS, calc’d for C15H13ClN3O3S2+ [M+H], 382.0081; found 382.0083.
4.3.7. 5-(1-((4-Chlorophenyl)sulfonyl)azetidin-3-yl)-3-(thiophen-3-yl)-1,2,4-oxadiazole (16).
The title compound was prepared in 63% (24 mg) yield from intermediate 10 and 4-chlorobenzene sulfonyl chloride using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, CDCl3) δ 7.95 (dd, J = 3.0, 1.2 Hz, 1H), 7.84 (m, 2H), 7.63 – 7.52 (m, 3H), 7.43 (q, J = 3.0 Hz, 1H), 4.31 (t, J = 8.4 Hz, 2H), 4.13 (t, J = 6.3 Hz, 2H), 3.98 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 177.4, 164.9, 140.3, 132.8, 129.9, 129.7, 128.1, 127.6, 127.3, 125.9, 54.6, 25.2 ppm. LCMS RT = 5.27 min; HRMS, calc’d for C15H13ClN3O3S2+ [M+H], 382.0081; found 382.0088.
4.3.8. 3-(Thiophen-3-yl)-5-(1-((2-(trifluoromethyl)phenyl)sulfonyl)azetidin-3-yl)-1,2,4-oxadiazole (17).
The title compound was prepared in 63% (26 mg) yield from intermediate 10 and 2-trifluoromethylbenzene sulfonyl chloride using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, CDCl3) δ 8.22 (m, 1H), 8.05 (dd, J = 3.0, 1.2 Hz, 1H), 7.92 (m, 1H), 7.74 (m, 2H), 7.61 (dd, J = 5.1, 1.2 Hz, 1H), 7.43 (q, J = 3.0 Hz, 1H), 4.42 (m, 4H), 4.07 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 177.5, 165.1, 137.0, 133.2, 132.5, 131.6, 128.5 (q, J(C,F) = 6.3 Hz), 128.4 (d, J(C,F) = 74.8 Hz), 128.1, 127.6, 127.2, 126.0, 122.5 (d, J(C,F) = 274 Hz), 54.5, 25.0 ppm. LCMS RT = 5.34 min; HRMS, calc’d for C16H13F3N3O3S2+ [M+H], 416.0345; found 416.0347.
4.3.9. 3-(Thiophen-3-yl)-5-(1-((3-(trifluoromethyl)phenyl)sulfonyl)azetidin-3-yl)-1,2,4-oxadiazole (18).
The title compound was prepared in 51% (21 mg) yield from intermediate 10 and 3-trifluoromethylbenzene sulfonyl chloride using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, CDCl3) δ 8.16 (m, 1H), 8.08 (m, 1H), 8.00 – 7.89 (m, 2H), 7.76 (m, 1H), 7.55 (dd, J = 5.1, 1.2 Hz, 1H), 7.43 (q, J = 3.0 Hz, 1H), 4.35 (t, J = 8.4 Hz, 2H), 4.19 (t, J = 6.3 Hz, 2H), 4.01 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 177.2, 165.0, 136.2, 132.1, 131.9, 131.5, 130.2 (m, J(C,F)), 128.1, 127.8, 127.2, 125.9, 125.3 (q, J(C,F) = 3.8 Hz), 123.2 (d, J(C,F) = 273.1 Hz), 54.5, 25.2 ppm. LCMS RT = 5.38 min; HRMS, calc’d for C16H13F3N3O3S2+ [M+H], 416.0345; found 416.0345.
4.3.10. 3-(Thiophen-3-yl)-5-(1-((4-(trifluoromethyl)phenyl)sulfonyl)azetidin-3-yl)-1,2,4-oxadiazole (19).
The title compound was prepared in 55% (23 mg) yield from intermediate 10 and 4-trifluoromethylbenzene sulfonyl chloride using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, CDCl3) δ 8.04 (m, 2H), 7.93 (dd, J = 3.0, 1.2 Hz, 1H), 7.89 (m, 2H), 7.52 (dd, J = 5.1, 1.2 Hz, 1H), 7.42 (q, J = 3.0 Hz, 1H), 4.35 (t, J = 8.4 Hz, 2H), 4.18 (t, J = 6.3 Hz, 2H), 4.00 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 177.3, 165.0, 136.9 (d, J(C,F) = 230 Hz), 135.0, 128.9, 128.0, 127.6, 127.3, 126.5 (q, J(C,F) = 3.7 Hz), 125.5, 123.2 (d, J(C,F) = 273.2 Hz), 54.6, 25.2 ppm. LCMS RT = 5.42 min; HRMS, calc’d for C16H13F3N3O3S2+ [M+H], 416.0345; found 416.0349.
4.3.11. 3-(Thiophen-3-yl)-5-(1-(o-tolylsulfonyl)azetidin-3-yl)-1,2,4-oxadiazole (20).
The title compound was prepared in 17% (6 mg) yield from intermediate 10 and 2-methylbenzene sulfonyl chloride using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, CDCl3) δ 8.06 (dd, J = 3.0, 1.2 Hz, 1H), 7.99 (m, 1H), 7.62 (dd, J = 6, 1.2 Hz, 1H), 7.51 (td, J = 6.9, 1.4 Hz, 1H), 7.43 (q, J = 3.0 Hz, 1H), 7.39 – 7.31 (m, 2H), 4.35 (t, J = 7.7 Hz, 2H), 4.26 (t, J = 6.0 Hz, 2H), 4.05 (m, 1H), 2.69 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 177.7, 165.1, 138.8, 134.8, 133.6, 132.8, 130.1, 128.1, 127.8, 127.2, 126.2, 126.0, 53.3, 25.3, 20.7 ppm. LCMS RT = 5.23 min; HRMS, calc’d for C16H16N3O3S2+ [M+H], 362.0628; found 362.0627.
4.3.12. 3-(Thiophen-3-yl)-5-(1-(m-tolylsulfonyl)azetidin-3-yl)-1,2,4-oxadiazole (21).
The title compound was prepared in 42% (15 mg) yield from intermediate 10 and 3-methylbenzene sulfonyl chloride using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, CDCl3) δ 7.97 (dd, J = 3.0, 1.2 Hz, 1H), 7.72 – 7.67 (m, 2H), 7.56 (dd, J = 5.1, 1.2 Hz, 1H), 7.53 – 7.40 (m, 3H), 4.28 (t, J = 8.4 Hz, 2H), 4.16 (t, J = 6.3 Hz, 2H), 3.95 (m, 1H), 2.46 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 177.4, 164.9, 139.7, 134.5, 134.0, 129.2, 128.7, 128.0, 127.7, 127.2, 125.9, 125.6, 54.3, 25.3, 21.5 ppm. LCMS RT = 5.12 min; HRMS, calc’d for C16H16N3O3S2+ [M+H], 362.0628; found 362.0634.
4.3.13. 3-(Thiophen-3-yl)-5-(1-tosylazetidin-3-yl)-1,2,4-oxadiazole (22).
The title compound was prepared in 28% yield (10 mg) from intermediate 10 and 4-methylbenzene sulfonyl chloride using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, CDCl3) δ 7.96 (dd, J = 3.0, 1.2 Hz, 1H), 7.78 (m, 2H), 7.55 (dd, J = 5.1, 1.2 Hz, 1H), 7.45 – 7.36 (m, 3H), 4.28 (t, J = 8.5 Hz, 2H), 4.13 (t, J = 6.3 Hz, 2H), 3.93 (m, 1H), 2.43 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 177.4, 164.9, 144.6, 131.0, 130.0, 128.6, 128.0, 127.7, 127.2, 125.9, 54.3, 25.3, 21.6 ppm. LCMS RT = 5.11 min; HRMS, calc’d for C16H16N3O3S2+ [M+H], 362.0628; found 362.0627.
4.3.14. 5-(1-((2-Methoxyphenyl)sulfonyl)azetidin-3-yl)-3-(thiophen-3-yl)-1,2,4-oxadiazole (23).
The title compound was prepared in 61% (23 mg) yield from intermediate 10 and 2-methoxybenzene sulfonyl chloride using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, CDCl3) δ 8.02 (dd, J = 3.0, 1.2 Hz, 1H), 7.92 (dd, J = 7.8, 1.7 Hz, 1H), 7.62 – 7.53 (m, 2H), 7.43 (dd, J = 5.1, 3.0 Hz, 1H), 7.08 (m, 2H), 4.42 (m, 4H), 4.05 (m, 1H), 3.96 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 177.9, 165.0, 157.1, 135.1, 131.3, 128.0, 127.8, 127.3, 126.0, 125.5, 120.6, 112.5, 56.2, 54.5, 25.2 ppm. LCMS RT = 4.85 min; HRMS, calc’d for C16H16N3O4S2+ [M+H], 378.0577; found 378.0578.
4.3.15. 5-(1-((3-Methoxyphenyl)sulfonyl)azetidin-3-yl)-3-(thiophen-3-yl)-1,2,4-oxadiazole (24).
The title compound was prepared in 61% (23 mg) yield from intermediate 10 and 3-methoxybenzene sulfonyl chloride using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, CDCl3) δ 7.98 (dd, J = 3.0, 1.2 Hz, 1H), 7.58 – 7.37 (m, 5H), 7.18 (m, 1H), 4.29 (t, J = 8.5 Hz, 2H), 4.17 (t, J = 6.3 Hz, 2H), 3.95 (m, 1H), 3.88 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 177.4, 164.9, 160.2, 135.1, 130.4, 128.0, 127.7, 127.2, 125.9, 120.6, 119.9, 113.2, 55.8, 54.4, 25.2 ppm. LCMS RT = 5.01 min; HRMS, calc’d for C16H16N3O4S2+ [M+H], 378.0577; found 378.0576.
4.3.16. 5-(1-((4-Methoxyphenyl)sulfonyl)azetidin-3-yl)-3-(thiophen-3-yl)-1,2,4-oxadiazole (25).
The title compound was prepared in 65% (25 mg) yield from intermediate 10 and 4-methoxybenzene sulfonyl chloride using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, DMSO-d6) δ 8.14 (dd, J = 3.0, 1.3 Hz, 1H), 7.84 – 7.76 (m, 3H), 7.49 (dd, J = 5.1, 1.2 Hz, 1H), 7.22 (m, 2H), 4.21–4.04 (m, 3H), 3.94 (m, 1H), 3.83 (s, 3H). LCMS RT = 4.93 min; HRMS, calc’d for C16H16N3O4S2+ [M+H], 378.0577; found 378.0581.
4.3.17. 5-(1-((2-Propoxyphenyl)sulfonyl)azetidin-3-yl)-3-(thiophen-3-yl)-1,2,4-oxadiazole (26).
Sodium hydride (NaH) (20 mg, 0.82 mmol, 10.0 eq) was added to n-propanol (2.0 mL) at 0 °C and was allowed to stir for 15 minutes. Analog 11 (30 mg, 0.082 mmol, 1.0 eq) was added afterwards, and the reaction was warmed to room temperature, then heated at 60 °C overnight. The reaction was cooled to room temperature and concentrated in vacuo. Water was added, and the reaction was extracted with DCM (2x). The combined organics were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel afforded 9 mg (28%) of the title compound. 1H NMR (300 MHz, CDCl3) δ 8.02 (dd, J = 3.0, 1.0 Hz, 1H), 7.91 (dd, J = 7.8, 1.7 Hz, 1H), 7.60 (dd, J = 5.1, 1.0 Hz, 1H), 7.54 (m, 1H), 7.43 (m, 1H), 7.05 (m, 2H), 4.43 (m, 4H), 4.12 – 3.96 (m, 3H), 4.03 (m, 1H), 1.89 (m, 2H), 1.08 (t, J = 7.4 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 177.9, 165.0, 156.9, 135.0, 131.3, 128.0, 127.8, 127.2, 126.0, 125.5, 120.3, 113.5, 70.9, 54.4, 25.3, 22.5, 10.4 ppm. LCMS RT = 5.37 min; HRMS, calc’d for C18H20N3O4S2+ [M+H], 406.0890; found 406.0887.
4.3.18. 5-(1-((2-Isopropoxyphenyl)sulfonyl)azetidin-3-yl)-3-(thiophen-3-yl)-1,2,4-oxadiazole (27).
The title compound was prepared in 26% (9 mg) yield from compound 11 and isopropanol using a method analogous to that described for the conversion of compound 11 into 26. 1H NMR (300 MHz, CDCl3) δ 8.03 (dd, J = 3.0, 1.2 Hz, 1H), 7.91 (dd, J = 7.8, 1.6 Hz, 1H), 7.60 (dd, J = 5.1, 1.2 Hz, 1H), 7.52 (m, 1H), 7.43 (m, 1H), 7.09 – 6.99 (m, 2H), 4.75 (m, 1H), 4.50 – 4.40 (m, 4H), 4.02 (m, 1H), 1.43 (d, J = 6.1 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 178.0, 165.0, 155.9, 134.8, 131.3, 128.0, 127.9, 127.2, 126.8, 126.0, 120.1, 114.7, 72.0, 54.4, 25.3, 22.0 ppm. LCMS RT = 5.33 min; HRMS, calc’d for C18H20N3O4S2+ [M+H], 406.0890; found 406.0889.
4.3.19. 5-(1-((2-Butoxyphenyl)sulfonyl)azetidin-3-yl)-3-(thiophen-3-yl)-1,2,4-oxadiazole (28).
The title compound was prepared in 17% (6 mg) yield from compound 11 and n-butanol using a method analogous to that described for the conversion of compound 11 into 26. 1H NMR (300 MHz, CDCl3) δ 8.03 (dd, J = 3.0, 1.2 Hz, 1H), 7.91 (m, 1H), 7.60 (dd, J = 5.1, 1.2 Hz, 1H), 7.54 (m, 1H), 7.43 (m, 1H), 7.05 (m, 2H), 4.49 – 4.37 (m, 4H), 4.15 – 3.96 (m, 3H), 1.84 (m, 2H), 1.53 (m, 2H), 0.97 (t, J = 7.4 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 178.0, 165.0, 156.9, 135.0, 131.3, 128.0, 127.8, 127.2, 126.0, 125.5, 120.3, 113.5, 69.2, 54.4, 31.2, 25.3, 19.1, 13.8 ppm. LCMS RT = 5.59 min; HRMS, calc’d for C19H22N3O4S2+ [M+H], 420.1046; found 420.1053.
4.3.20. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-phenyl-1,2,4-oxadiazole (40).
(Step 1) Methyl 1-((2-ethoxyphenyl)sulfonyl)azetidine-3-carboxylate (38). Methyl azetidine-3-carboxylate hydrochloride (37) (618 mg, 4.08 mmol, 1.2 eq) and N,N-diisopropylethylamine (1.78 mL, 10.2 mmol, 3.0 eq) were dissolved in DCM (10 mL), followed by the addition of 2-ethoxybenzene sulfonyl chloride (750 mg, 3.40 mmol, 1.0 eq). The reaction was stirred for 2 hours at room temperature. Water was added, and the reaction was extracted with DCM (2x). The combined organics were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel afforded 830 mg (82%) of the title compound. 1H NMR (300 MHz, CDCl3) δ 7.88 (dd, J = 8.1, 1.8 Hz, 1H), 7.53 (m, 1H), 7.08 – 6.98 (m, 2H), 4.27 – 4.12 (m, 6H), 3.70 (s, 3H), 3.35 (m, 1H), 1.51 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 172.2, 156.7, 134.8, 131.1, 126.1, 120.3, 113.5, 65.0, 53.1, 52.4, 31.5, 14.7 ppm. LCMS RT = 3.94 min; HRMS, calc’d for C13H18NO5S+ [M+H], 300.0900; found 300.0899. (Step 2) 1-((2-Ethoxyphenyl)sulfonyl)azetidine-3-carboxylic acid (39). Intermediate 38 (830 mg, 2.77 mmol, 1.0 eq) was dissolved in THF (5.0 mL) and H2O (5.0 mL), followed by the addition of lithium hydroxide (LiOH) monohydrate (233 mg, 5.54 mmol, 2.0 eq). The reaction was stirred at room temperature for 2 hours. The solvents were removed in vacuo, and the reaction was acidified with 1N HCl. The aqueous layer was extracted with ethyl acetate (2x), the combined organics were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo to afford 720 mg (91%) as a white solid that was used further without purification. 1H NMR (300 MHz, CDCl3) δ 7.88 (dd, J = 8.2, 1.7 Hz, 1H), 7.53 (m, 1H), 7.08 – 6.99 (m, 2H), 4.30 – 4.12 (m, 6H), 3.38 (m, 1H), 1.51 (t, J = 7.0 Hz, 3H). (Step 3) 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-phenyl-1,2,4-oxadiazole (40). Intermediate 39 (50 mg, 0.18 mmol, 1.0 eq), N,N-diisopropylethylamine (94 μL, 0.54 mmol, 3.0 eq), and HATU (103 mg, 0.27 mmol, 1.5 eq) were dissolved in DMF (2.0 mL) and allowed to stir for 15 minutes, followed by the addition of benzamidoxime (49 mg, 0.36 mmol, 2.0 eq). The reaction was allowed to stir for 1 hour at room temperature, afterwards the reaction temperature was brought to 140 °C and stirred for 2 hours. Water was added, and the reaction was extracted with ethyl acetate (2x). The combined organics were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel afforded 28 mg (40%) of the title compound. 1H NMR (300 MHz, CDCl3) δ 8.03 (m, 2H), 7.91 (dd, J = 7.8, 1.8 Hz, 1H), 7.59 – 7.43 (m, 4H), 7.10 – 6.99 (m, 2H), 4.46 (m, 4H), 4.20 (q, J = 7.0 Hz, 2H), 4.05 (m, 1H), 1.50 (t, J = 7. 0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 178.2, 168.6, 156.7, 135.0, 131.5, 131.2, 128.9, 127.5, 126.4, 125.9, 120.4, 113.5, 65.1, 54.5, 25.3, 14.7 ppm. LCMS RT = 5.27 min; HRMS, calc’d for C19H20N3O4S+ [M+H], 386.1169; found 386.1177.
4.3.21. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(2-fluorophenyl)-1,2,4-oxadiazole (41).
Intermediate 39 (50 mg, 0.18 mmol, 1.0 eq), DIEA (63 μL, 0.36 mmol, 2.0 eq), 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (52 mg, 0.27 mmol, 1.5 eq), and Hydroxybenzotriazole (HOBt) (41 mg, 0.27 mmol, 1.5 eq) were dissolved in 1,4-dioxane (2.0 mL). The reaction was allowed to stir for 30 minutes at room temperature, followed by the addition of 2-fluorobenzamidoxime (41 mg, 0.27 mmol, 1.5 eq). The reaction was stirred at room temperature for 2 hours, afterwards the reaction temperature was brought to 90 °C and stirred overnight. Water was added, and the reaction was extracted with ethyl acetate (2x). The combined organics were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel afforded 4 mg (6%) of the title compound. 1H NMR (300 MHz, CDCl3) δ 8.00 (td, J = 7.3, 1.8 Hz, 1H), 7.91 (dd, J = 8.0, 1.6 Hz, 1H), 7.58 – 7.46 (m, 2H), 7.34 – 7.19 (m, 2H), 7.10 – 6.99 (m, 2H), 4.46 (m, 4H), 4.20 (q, J = 7.0 Hz, 2H), 4.08 (m, 1H), 1.50 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 177.8, 163.9 (d, J(C,F) = 228 Hz), 159.0, 156.7, 135.0, 133.0 (d, J(C,F) = 8.6 Hz), 131.2, 130.7 (d, J(C,F) = 2.2 Hz), 125.9, 124.5 (d, J(C,F) = 3.8 Hz), 120.4, 116.8 (d, J(C,F) = 21.1 Hz), 114.7 (d, J(C,F) = 12.4 Hz), 113.5, 65.1, 54.6, 25.3, 14.7 ppm. LCMS RT = 5.15 min; HRMS, calc’d for C19H19FN3O4S+ [M+H], 404.1075; found 404.1080.
4.3.22. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(3-fluorophenyl)-1,2,4-oxadiazole (42).
The title compound was prepared in 11% yield (8 mg) from intermediate 39 and 3-fluorobenzamidoxime using a method analogous to that described for the conversion of compound 39 into 41. 1H NMR (300 MHz, CDCl3) δ 7.92 (dd, J = 7.8, 1.7 Hz, 1H), 7.84 (dt, J = 8.0, 1.2 Hz, 1H), 7.73 (m, 1H), 7.60 – 7.41 (m, 2H), 7.22 (m, 2H), 7.11 – 7.01 (m, 2H), 4.46 (m, 4H), 4.20 (q, J = 7.0 Hz, 2H), 4.05 (m, 1H), 1.50 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 178.6, 166.2 (d, J(C,F) = 244.4 Hz), 161.2, 156.7, 135.0, 131.2, 130.7 (d, J(C,F) = 8.0 Hz), 128.4 (d, J(C,F) = 8.6 Hz), 125.9, 123.2 (d, J(C,F) = 3.2 Hz), 120.4, 118.5 (d, J(C,F) = 21.3 Hz), 114.6 (d, J(C,F) = 23.8 Hz), 113.6, 65.1, 54.5, 25.3, 14.7 ppm. LCMS RT = 5.38 min; HRMS, calc’d for C19H19FN3O4S+ [M+H], 404.1075; found 404.1076.
4.3.23. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(4-fluorophenyl)-1,2,4-oxadiazole (43).
The title compound was prepared in 10% yield (7 mg) from intermediate 39 and 4-fluorobenzamidoxime using a method analogous to that described for the conversion of compound 39 into 41. 1H NMR (300 MHz, CDCl3) δ 8.04 (m, 2H), 7.92 (dd, J = 7.9, 1.7 Hz, 1H), 7.54 (m, 1H), 7.17 (m, 2H), 7.10 – 6.99 (m, 2H), 4.46 (m, 4H), 4.20 (q, J = 7.0 Hz, 2H), 4.04 (m, 1H), 1.50 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 178.3, 167.8, 164.7 (d, J(C,F) = 252 Hz), 156.7, 135.0, 131.1, 129.6 (d, J(C,F) = 8.8 Hz), 126.0, 122.6 (d, J(C,F) = 3.4 Hz), 120.4, 116.2 (d, J(C,F) = 22.1 Hz), 113.5, 65.1, 54.5, 25.3, 14.7 ppm. LCMS RT = 5.35 min; HRMS, calc’d for C19H19FN3O4S+ [M+H], 404.1075; found 404.1084.
4.3.24. 3-(2-Chlorophenyl)-5-(1-((2-ethoxyphenyl)sulfonyl)azetidin-3-yl)-1,2,4-oxadiazole (44).
The title compound was prepared in 5% yield (4 mg) from intermediate 39 and 2-chlorobenzamidoxime using a method analogous to that described for the conversion of compound 39 into 41. 1H NMR (300 MHz, CDCl3) δ 7.90 (m, 2H), 7.59 – 7.33 (m, 4H), 7.10 – 6.99 (m, 2H), 4.47 (m, 4H), 4.20 (q, J = 7.0 Hz, 2H), 4.09 (m, 1H), 1.50 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 177.7, 167.4, 156.7, 135.0, 133.5, 131.9, 131.7, 131.2, 131.0, 126.9, 125.9, 125.6, 120.4, 113.6, 65.1, 54.5, 25.3, 14.8 ppm. LCMS RT = 5.34 min; HRMS, calc’d for C19H19ClN3O4S+ [M+H], 420.0779; found 420.0788.
4.3.25. 3-(3-Chlorophenyl)-5-(1-((2-ethoxyphenyl)sulfonyl)azetidin-3-yl)-1,2,4-oxadiazole (45).
The title compound was prepared in 7% yield (5 mg) from intermediate 39 and 3-chlorobenzamidoxime using a method analogous to that described for the conversion of compound 39 into 41. 1H NMR (300 MHz, CDCl3) δ 8.02 (t, J = 1.6 Hz, 1H), 7.93 (m, 2H), 7.61 – 7.38 (m, 3H), 7.12 – 7.01 (m, 2H), 4.46 (m, 4H), 4.20 (q, J = 7.0 Hz, 2H), 4.05 (m, 1H), 1.50 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 178.6, 167.6, 156.8, 155.0, 135.1, 131.5, 131.3, 130.3, 128.1, 127.6, 125.8, 125.5, 120.4, 113.6, 65.1, 54.5, 25.3, 14.7 ppm. LCMS RT = 5.34 min; HRMS, calc’d for C19H19ClN3O4S+ [M+H], 420.0779; found 420.0787.
4.3.26. 3-(4-Chlorophenyl)-5-(1-((2-ethoxyphenyl)sulfonyl)azetidin-3-yl)-1,2,4-oxadiazole (46).
The title compound was prepared in 5% yield (4 mg) from intermediate 39 and 4-chlorobenzamidoxime using a method analogous to that described for the conversion of compound 39 into 41. 1H NMR (300 MHz, CDCl3) δ 7.98 (m, 2H), 7.91 (dd, J = 7.8, 1.8 Hz, 1H), 7.54 (m, 1H), 7.47 (m, 2H), 7.11 – 6.99 (m, 2H), 4.46 (m, 4H), 4.20 (q, J = 7.0 Hz, 2H), 4.05 (m, 1H), 1.50 (t, J = 7. 0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 178.5, 167.8, 156.7, 137.7, 135.0, 131.1, 129.3, 128.8, 126.0, 124.9, 120.4, 113.5, 65.1, 54.5, 25.3, 14.7 ppm. LCMS RT = 5.34 min; HRMS, calc’d for C19H19ClN3O4S+ [M+H], 420.0779; found 420.0788.
4.3.27. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(2-(trifluoromethyl)phenyl)-1,2,4-oxadiazole (47).
The title compound was prepared in 11% yield (9 mg) from intermediate 39 and 2-trifluoromethylbenzamidoxime using a method analogous to that described for the conversion of compound 39 into 41. 1H NMR (300 MHz, CDCl3) δ 7.90 (m, 1H), 7.87 – 7.81 (m, 1H), 7.79 – 7.73 (m, 1H), 7.71 – 7.63 (m, 2H), 7.53 (m, 1H), 7.05 (m, 2H), 4.46 (m, 4H), 4.25 – 4.02 (m, 3H), 1.49 (t, J = 7. 0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 178.2, 167.8, 156.7, 135.0, 131.9, 131.8, 131.1, 131.0, 129.6, 129.2, 127.0 (q, J(C,F) = 5.3 Hz), 125.8, 125.2 (q, J(C,F) = 3.4 Hz), 120.4, 113.5, 65.1, 54.5, 25.3, 14.7 ppm. LCMS RT = 5.38 min; HRMS, calc’d for C20H19F3N3O4S+ [M+H], 454.1043; found 454.1044.
4.3.28. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(3-(trifluoromethyl)phenyl)-1,2,4-oxadiazole (48).
The title compound was prepared in 17% yield (14 mg) from intermediate 39 and 3-trifluoromethylbenzamidoxime using a method analogous to that described for the conversion of compound 39 into 41. 1H NMR (300 MHz, CDCl3) δ 8.33 – 8.19 (m, 2H), 7.92 (dd, J = 7.8, 1.6 Hz, 1H), 7.79 (m, 1H), 7.63 (m, 1H), 7.55 (m, 1H), 7.06 (m, 2H), 4.47 (m, 4H), 4.21 (q, J = 7.0 Hz, 2H), 4.07 (m, 1H), 1.51 (t, J = 7. 0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 178.9, 167.6, 156.8, 135.1, 131.8, 131.3, 130.5, 129.6, 128.0 (q, J(C,F) = 3.7 Hz), 127.3, 125.8, 125.5, 124.5 (q, J(C,F) = 3.9 Hz), 120.4, 113.6, 65.1, 54.5, 25.2, 14.7 ppm. LCMS RT = 5.69 min; HRMS, calc’d for C20H19F3N3O4S+ [M+H], 454.1043; found 454.1043.
4.3.29. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(4-(trifluoromethyl)phenyl)-1,2,4-oxadiazole (49).
The title compound was prepared in 11% yield (9 mg) from intermediate 39 and 4-trifluoromethylbenzamidoxime using a method analogous to that described for the conversion of compound 39 into 41. 1H NMR (300 MHz, CDCl3) δ 8.17 (d, J = 8.1 Hz, 2H), 7.92 (dd, J = 7.8, 1.7 Hz, 1H), 7.57 (d, J = 8.2 Hz, 2H), 7.55 (m, 1H), 7.06 (m, 2H), 4.47 (m, 4H), 4.20 (q, J = 7.0 Hz, 2H), 4.07 (m, 1H), 1.51 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 178.8, 167.6, 156.6, 135.0, 133.1 (q, J(C,F) = 32.8 Hz), 131.1, 129.8, 125.8, 125.9 (q, J(C,F) = 3.5 Hz), 125.5, 121.9, 120.5, 113.6, 65.1, 54.5, 25.3, 14.7 ppm. LCMS RT = 5.71 min; HRMS, calc’d for C20H19F3N3O4S+ [M+H], 454.1043; found 454.1042.
4.3.30. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(o-tolyl)-1,2,4-oxadiazole (50).
Intermediate 39 (30 mg, 0.11 mmol, 1.0 eq) and 1,1’-carbonyldiimidazole (CDI) (27 mg, 0.17 mmol, 1.5 eq) were dissolved in anhydrous DMF (2.0 mL), and the reaction was allowed to stir for 30 minutes at room temperature followed by the addition of 2-methylbenzamidoxime (25 mg, 0.17 mmol, 1.5 eq), and another 1.5 eq of CDI. The reaction was stirred overnight at 80 °C. Water was added, and the reaction was extracted with DCM (2x). The combined organics were washed with water, brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel afforded 19 mg (43%) of the title compound. 1H NMR (300 MHz, CDCl3) δ 7.92 (dd, J = 7.8, 1.7 Hz, 1H), 7.88 – 7.79 (m, 2H), 7.53 (m, 1H), 7.41 – 7.29 (m, 2H), 7.11 – 6.98 (m, 2H), 4.46 (m, 4H), 4.20 (q, J = 7.0 Hz, 2H), 4.04 (m, 1H), 2.43 (s, 3H), 1.50 (t, J = 7.0 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 178.1, 168.7, 156.7, 138.7, 135.0, 132.2, 131.2, 128.8, 128.0, 126.2, 126.0, 124.6, 120.4, 113.6, 65.1, 54.5, 25.4, 21.3, 14.7 ppm. LCMS RT = 5.54 min; HRMS, calc’d for C20H22N3O4S+ [M+H], 400.1326; found 400.1327.
4.3.31. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(m-tolyl)-1,2,4-oxadiazole (51).
The title compound was prepared in 43% yield (29 mg) from intermediate 39 and 3-methylbenzamidoxime using a method analogous to that described for the conversion of compound 39 into 50. 1H NMR (300 MHz, CDCl3) δ 7.97 – 7.87 (m, 2H), 7.53 (m, 1H), 7.44 – 7.35 (m, 1H), 7.34 – 7.27 (m, 2H), 7.09 – 6.99 (m, 2H), 4.46 (m, 4H), 4.19 (q, J = 7.0 Hz, 2H), 4.06 (m, 1H), 2.60 (s, 3H), 1.49 (t, J = 7.0 Hz, 3H). LCMS RT = 5.50 min; 13C NMR (75 MHz, CDCl3) δ 177.1, 169.1, 156.7, 138.3, 135.0, 131.5, 131.2, 130.8, 130.1, 126.0, 125.9, 125.6, 120.4, 113.6, 65.1, 54.6, 25.3, 22.2, 14.7 ppm. LCMS RT = 5.50 min; HRMS, calc’d for C20H22N3O4S+ [M+H], 400.1326; found 400.1326.
4.3.32. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(p-tolyl)-1,2,4-oxadiazole (52).
The title compound was prepared in 39% yield (26 mg) from intermediate 39 and 4-methylbenzamidoxime using a method analogous to that described for the conversion of compound 39 into 50. 1H NMR (300 MHz, CDCl3) δ 7.91 (d, J = 8.3 Hz, 3H), 7.53 (m, 1H), 7.28 (m, 2H), 7.09 – 6.99 (m, 2H), 4.45 (m, 4H), 4.19 (q, J = 7.0 Hz, 2H), 4.03 (m, 1H), 2.42 (s, 3H), 1.49 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 178.0, 168.6, 156.7, 141.8, 135.0, 131.2, 129.6, 127.4, 125.9, 123.5, 120.4, 113.6, 65.1, 54.5, 25.3, 21.6, 14.7 ppm. LCMS RT = 5.53 min; HRMS, calc’d for C20H22N3O4S+ [M+H], 400.1326; found 400.1327.
4.3.33. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(2-methoxyphenyl)-1,2,4-oxadiazole (53).
The title compound was prepared in 11% yield (8 mg) from intermediate 39 and 2-methoxyamidoxime using a method analogous to that described for the conversion of compound 39 into 41. 1H NMR (300 MHz, CDCl3) δ 7.92 (m, 2H), 7.58 – 7.44 (m, 2H), 7.12 – 6.98 (m, 4H), 4.45 (m, 4H), 4.19 (q, J = 7.0 Hz, 2H), 4.07 (m, 1H), 3.97 (s, 3H), 1.49 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 176.9, 167.1, 158.1, 156.7, 135.0, 132.6, 131.4, 131.1, 125.8, 120.7, 120.4, 115.3, 113.5, 111.7, 65.1, 56.0, 54.5, 25.3, 14.7 ppm. LCMS RT = 5.00 min; HRMS, calc’d for C20H22N3O5S+ [M+H], 416.1275; found 416.1284.
4.3.34. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(3-methoxyphenyl)-1,2,4-oxadiazole (54).
The title compound was prepared in 17% yield (12 mg) from intermediate 39 and 3-methoxybenzamidoxime using a method analogous to that described for the conversion of compound 39 into 41. 1H NMR (300 MHz, CDCl3) δ 7.92 (dd, J = 7.8, 1.7 Hz, 1H), 7.63 (dt, J = 1.2 Hz, 1H), 7.59 – 7.49 (m, 2H), 7.39 (t, J = 8.0 Hz, 1H), 7.11 – 6.99 (m, 3H), 4.46 (m, 4H), 4.20 (q, J = 7.0 Hz, 2H), 4.05 (m, 1H), 3.88 (s, 3H), 1.50 (t, J = 7. 0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 178.2, 168.5, 159.9, 156.7, 135.0, 131.2, 130.1, 127.5, 125.9, 120.4, 119.9, 117.9, 113.5, 112.1, 65.1, 56.0, 54.5, 25.3, 14.7 ppm. LCMS RT = 5.30 min; HRMS, calc’d for C20H22N3O5S+ [M+H], 416.1275; found 416.1278.
4.3.35. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(4-methoxyphenyl)-1,2,4-oxadiazole (55).
The title compound was prepared in 15% yield (11 mg) from intermediate 39 and 4-methoxybenzamidoxime using a method analogous to that described for the conversion of compound 39 into 41. 1H NMR (300 MHz, CDCl3) δ 8.01 – 7.87 (m, 3H), 7.54 (m, 1H), 7.11 – 6.92 (m, 4H), 4.45 (m, 4H), 4.19 (q, J = 7.0 Hz, 2H), 4.03 (m, 1H), 3.88 (s, 3H), 1.50 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 177.9, 168.3, 162.1, 156.7, 135.0, 131.2, 129.1, 125.9, 120.4, 118.8, 114.3, 113.5, 65.1, 55.4, 54.5, 25.3, 14.7 ppm. LCMS RT = 5.25 min; HRMS, calc’d for C20H22N3O5S+ [M+H], 416.1275; found 416.1284.
4.3.36. 3-(2,4-Difluorophenyl)-5-(1-((2-ethoxyphenyl)sulfonyl)azetidin-3-yl)-1,2,4-oxadiazole (56).
The title compound was prepared in 18% yield (13 mg) from intermediate 39 and 2,4-difluorobenzamidoxime using a method analogous to that described for the conversion of compound 39 into 41. 1H NMR (300 MHz, CDCl3) δ 8.02 (m, 1H), 7.91 (dd, J = 7.8, 1.6 Hz, 1H), 7.54 (m, 1H), 7.12 – 6.94 (m, 4H), 4.46 (m, 4H), 4.20 (q, J = 7.0 Hz, 2H), 4.07 (m, 1H), 1.50 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 178.0, 164.8 (d, J(C,F) = 260 Hz), 164.9, 164.8, 164.8 (d, J(C,F) = 253 Hz), 159.6, 169.5, 156.7, 135.0, 132.1 (d, J(C,F) = 10.5 Hz), 132.02 (d, J(C,F) = 10.1 Hz), 131.1, 126.0, 120.4, 113.6, 112.2 (d, J(C,F) = 21.6 Hz), 112.1 (d, J(C,F) = 21.5 Hz), 111.3 (d, J(C,F) = 4.4 Hz), 111.1 (d, J(C,F) = 3.8 Hz), 105.3 (d, J(C,F) = 25.2 Hz), 65.1, 54.4, 25.2, 14.7 ppm. LCMS RT = 5.27 min; HRMS, calc’d for C19H18F2N3O4S+ [M+H], 422.0981; found 422.0985.
4.3.37. 3-(4-Chloro-2-fluorophenyl)-5-(1-((2-ethoxyphenyl)sulfonyl)azetidin-3-yl)-1,2,4-oxadiazole (57).
The title compound was prepared in 8% yield (6 mg) from intermediate 39 and 4-chloro-2-fluorobenzamidoxime using a method analogous to that described for the conversion of compound 39 into 41. 1H NMR (300 MHz, CDCl3) δ 8.02 – 7.86 (m, 2H), 7.54 (m, 1H), 7.34 – 7.23 (m, 2H), 7.11 – 6.99 (m, 2H), 4.46 (m, 4H), 4.20 (q, J = 7.0 Hz, 2H), 4.07 (m, 1H), 1.50 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 177.1, 164.9, 163.5 (d, J(C,F) = 192.4 Hz), 158.7, 156.7, 138.4 (d, J(C,F) = 9.9 Hz), 135.0, 131.4 (d, J(C,F) = 3.1), 131.1, 126.0, 125.1 (d, J(C,F) = 3.7 Hz), 120.4, 117.6 (d, J(C,F) = 24.4 Hz), 113.5, 65.1, 54.4, 25.2, 14.7 ppm. LCMS RT = 5.56 min; HRMS, calc’d for C19H18ClFN3O4S+ [M+H], 438.0685; found 438.0689.
4.3.38. 3-(2-Chloro-4-fluorophenyl)-5-(1-((2-ethoxyphenyl)sulfonyl)azetidin-3-yl)-1,2,4-oxadiazole (58).
The title compound was prepared in 11% yield (8 mg) from intermediate 39 and 2-chloro-4-fluorobenzamidoxime using a method analogous to that described for the conversion of compound 39 into 41. 1H NMR (300 MHz, CDCl3) δ 7.98 – 7.86 (m, 2H), 7.53 (m, 1H), 7.29 (dd, J = 8.5, 2.5 Hz, 1H), 7.18 – 6.98 (m, 3H), 4.46 (m, 4H), 4.20 (q, J = 7. 0 Hz, 2H), 4.08 (m, 1H), 1.50 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 177.8, 166.7, 163.7 (d, J(C,F) = 255.5 Hz), 156.7, 135.0, 134.8 (d, J(C,F) = 10.6 Hz), 133.3, 131.1, 126.0, 122.0 (d, J(C,F) = 5.5 Hz), 120.4, 118.6 (d, J(C,F) = 25.0 Hz), 114.6 (d, J(C,F) = 21.5 Hz), 113.6, 65.1, 54.5, 25.3, 14.8 ppm. LCMS RT = 5.47 min; HRMS, calc’d for C19H18ClFN3O4S+ [M+H], 438.0685; found 438.0688.
4.3.39. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(thiophen-2-yl)-1,2,4-oxadiazole (59).
The title compound was prepared in 6% yield (4 mg) from intermediate 39 and thiophene-2-amidoxime using a method analogous to that described for the conversion of compound 39 into 40. 1H NMR (300 MHz, CDCl3) δ 7.91 (dd, J = 8.3, 1.6 Hz, 1H), 7.76 (dd, J = 3.7, 1.2 Hz, 1H), 7.59 – 7.49 (m, 2H), 7.16 (m, 1H), 7.06 (m, 2H), 4.44 (m, 4H), 4.21 (q, J = 7.0 Hz, 2H), 4.04 (m, 1H), 1.51 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 178.1, 164.6, 156.7, 135.1, 131.2, 129.8, 129.6, 128.1, 127.8, 125.8, 120.4, 113.5, 65.1, 54.4, 25.3, 14.8 ppm. LCMS RT = 5.10 min; HRMS, calc’d for C17H18N3O4S2+ [M+H], 392.0733; found 392.0753.
4.3.40. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(5-fluorothiophen-2-yl)-1,2,4-oxadiazole (60).
CDI (24 mg, 0.15 mmol, 1.2 eq) was added to a solution of intermediate 39 (35 mg, 0.12 mmol, 1.1 eq) in dimethyl sulfoxide (DMSO) (0.5 mL). The reaction was stirred for 30 minutes at room temperature, followed by the addition of 5-fluorothiophene-2-amidoxime (18 mg, 0.11, 1.0 eq). The reaction was stirred at room temperature overnight, followed by the addition of sodium hydroxide (NaOH) (6 mg, 0.15 mmol, 1.2 eq), and was stirred for additional 2 hours at room temperature. The reaction was diluted with ice-cold water and extracted with DCM (2x). The combined organics were washed with water, brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel afforded 12 mg (27%) of the title compound. 1H NMR (300 MHz, CDCl3) δ 7.90 (dd, J = 8.3, 1.8 Hz, 1H), 7.55 (m, 1H), 7.42 (t, J = 4.0 Hz, 1H), 7.06 (m, 2H), 6.56 (dd, J = 4.2, 1.6 Hz, 1H), 4.43 (m, 4H), 4.21 (q, J = 7. 0 Hz, 2H), 4.01 (m, 1H), 1.51 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 178.2, 168.1, 164.2, 156.7, 135.0, 131.2, 127.1 (d, J(C,F) = 4.3 Hz), 125.9, 120.4, 116.5 (d, J(C,F) = 4.7 Hz), 113.5, 108.8 (d, J(C,F) = 11.2 Hz), 65.1, 54.4, 25.2, 14.7 ppm. LCMS RT = 5.40 min; HRMS, calc’d for C17H17FN3O4S2+ [M+H], 410.0639; found 410.0645.
4.3.41. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(thiazol-2-yl)-1,2,4-oxadiazole (61).
The title compound was prepared in 65% yield (43 mg) from intermediate 39 and thiazole-2-amidoxime using a method analogous to that described for the conversion of compound 39 into 60. 1H NMR (300 MHz, CDCl3) δ 8.07 (d, J = 3.2 Hz, 1H), 7.90 (dd, J = 8.3, 1.8 Hz, 1H), 7.6 (d, J = 3.1 Hz, 1H), 7.5 (m, 1H), 7.1 (m, 2H), 4.5 (m, 4H), 4.21 (q, J = 7.0 Hz, 2H), 4.10 (m, 1H), 1.50 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 179.2, 164.2, 156.7, 153.8, 145.2, 135.0, 131.1, 126.0, 122.8, 120.4, 113.6, 65.1, 54.3, 25.4, 14.8 ppm. LCMS RT = 4.57 min; HRMS, calc’d for C16H17N4O4S2+ [M+H], 393.0686; found 393.0689.
4.3.42. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(isothiazol-4-yl)-1,2,4-oxadiazole (62).
The title compound was prepared in 39% yield (26 mg) from intermediate 39 and isothiazole-4-amidoxime using a method analogous to that described for the conversion of compound 39 into 60. 1H NMR (300 MHz, CDCl3) δ 9.26 (s, 1H), 9.00 (s, 1H), 7.91 (dd, J = 7.9, 1. 6 Hz, 1H), 7.55 (m, 1H), 7.06 (m, 2H), 4.46 (m, 4H), 4.20 (q, J = 7.0 Hz, 2H), 4.06 (m, 1H), 1.51 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 178.7, 163.2, 156.7, 156.1, 149.0, 135.0, 131.1, 126.0, 126.0, 120.4, 113.6, 65.1, 54.4, 25.2, 14.7 ppm. LCMS RT = 4.76 min; HRMS, calc’d for C16H17N4O4S2+ [M+H], 393.0686; found 393.0691.
4.3.43. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(furan-2-yl)-1,2,4-oxadiazole (63).
The title compound was prepared in 28% yield (18 mg) from intermediate 39 and furan-2-amidoxime using a method analogous to that described for the conversion of compound 39 into 41. 1H NMR (300 MHz, CDCl3) δ 7.90 (dd, J = 8.3, 1.8 Hz, 1H), 7.62 (dd, J = 1.8, 0.8 Hz, 1H), 7.54 (m, 1H), 7.11 (dd, J = 3.5, 0.8 Hz, 1H), 7.05 (m, 2H), 6.58 (dd, J = 3.5, 1.8 Hz, 1H), 4.44 (m, 4H), 4.20 (q, J = 7. 0 Hz, 2H), 4.04 (m, 1H), 1.50 (t, J = 7.0 Hz, 3H). LCMS RT = 4.82 min; 13C NMR (75 MHz, CDCl3) δ 178.2, 161.4, 156.7, 145.5, 141.9, 135.0, 131.1, 125.9, 120.4, 114.2, 113.5, 111.9, 65.1, 54.4, 25.3, 14.7 ppm. HRMS, calc’d for C17H18N3O5S+ [M+H], 376.0962; found 376.0963.
4.3.44. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(pyridin-2-yl)-1,2,4-oxadiazole (64).
The title compound was prepared in 31% yield (20 mg) from intermediate 39 and pyridine-2-amidoxime using a method analogous to that described for the conversion of compound 39 into 41. 1H NMR (300 MHz, CDCl3) δ 8.80 (dq, J = 4.8, 0.9 Hz, 1H), 8.10 (dt, J = 7.9, 1.0 Hz, 1H), 7.96 – 7.82 (m, 2H), 7.58 – 7.39 (m, 2H), 7.05 (m, 2H), 4.48 (m, 4H), 4.26 – 4.04 (m, 3H), 1.49 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 178.9, 168.4, 156.7, 150.5, 145.9, 137.2, 135.0, 131.0, 126.0, 125.8, 123.3, 120.4,113.5, 65.1, 54.3, 25.5, 14.7 ppm. LCMS RT = 4.45 min; HRMS, calc’d for C18H19N4O4S+ [M+H], 387.1122; found 387.1124.
4.3.45. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(pyridin-3-yl)-1,2,4-oxadiazole (65).
The title compound was prepared in 24% yield (16 mg) from intermediate 39 and pyridine-3-amidoxime using a method analogous to that described for the conversion of compound 39 into 41. 1H NMR (300 MHz, CDCl3) δ 9.26 (s, 1H), 8.76 (d, J = 3.8 Hz, 1H), 8.32 (dt, J = 8.0, 1.9 Hz, 1H), 7.92 (m, 1H), 7.55 (m, 1H), 7.44 (m, 1H), 7.06 (m, 2H), 4.47 (m, 4H), 4.21 (q, J = 7.0 Hz, 2H), 4.08 (m, 1H), 1.51 (t, J = 7.0 Hz, 3H). LCMS RT = 4.36 min; HRMS, calc’d for C18H19N4O4S+ [M+H], 387.1122; found 387.1128.
4.3.46. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(pyridin-4-yl)-1,2,4-oxadiazole (66).
The title compound was prepared in 40% yield (26 mg) from intermediate 39 and pyridine-4-amidoxime using a method analogous to that described for the conversion of compound 39 into 60. 1H NMR (300 MHz, CDCl3) δ 8.79 (d, J = 5.9 Hz, 2H), 7.96 – 7.84 (m, 3H), 7.55 (m, 1H), 7.06 (m, 2H), 4.47 (m, 4H), 4.20 (q, J = 7.0 Hz, 2H), 4.08 (m, 1H), 1.51 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 179.2, 167.1, 156.7, 150.8, 135.0, 133.8, 131.1, 126.0, 121.2, 120.5, 113.6, 65.1, 54.4, 25.3, 14.7 ppm. LCMS RT = 4.29 min; HRMS, calc’d for C18H19N4O4S+ [M+H], 387.1122; found 387.1129.
4.3.47. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(5-fluoropyridin-2-yl)-1,2,4-oxadiazole (67).
The title compound was prepared in 29% yield (20 mg) from intermediate 39 and 4-fluoropyridine-2-amidoxime using a method analogous to that described for the conversion of compound 39 into 60. 1H NMR (300 MHz, CDCl3) δ 8.64 (d, J = 2.8 Hz, 1H), 8.14 (m, 1H), 7.90 (m, 1H), 7.63 – 7.47 (m, 2H), 7.05 (m, 2H), 4.48 (m, 4H), 4.28 – 4.03 (m, 3H), 1.50 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 179.1, 167.6, 160.4 (d, J(C,F) = 261.9 Hz), 156.7, 142.1 (d, J(C,F) = 4.2 Hz), 139.2 (d, J(C,F) = 24.8 Hz), 135.0, 131.0, 126.1, 124.7 (d, J(C,F) = 5.2 Hz), 123.9 (d, J(C,F) = 18.8 Hz), 120.4, 113.5, 65.1, 54.3, 25.5, 14.7 ppm. LCMS RT = 4.69 min; HRMS, calc’d for C18H18FN4O4S+ [M+H], 405.1027; found 405.1031.
4.3.48. 5-(1-((2-Ethoxyphenyl)sulfonyl)azetidin-3-yl)-3-(pyrimidin-2-yl)-1,2,4-oxadiazole (68).
The title compound was prepared in 29% yield (19 mg) from intermediate 39 and pyrimidine-2-amidoxime using a method analogous to that described for the conversion of compound 39 into 60. 1H NMR (300 MHz, CDCl3) δ 8.98 (d, J = 4.9 Hz, 1H), 7.89 (dd, J = 8.1, 1.8 Hz, 1H), 7.58 – 7.43 (m, 2H), 7.09 – 6.97 (m, 2H), 4.50 (m, 4H), 4.28 – 4.08 (m, 3H), 1.49 (t, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 179.6, 167.9, 158.1, 156.6, 155.8, 135.0, 131.0, 126.0, 122.4, 120.4, 113.5, 65.1, 54.2, 25.7, 14.7 ppm. LCMS RT = 4.04 min; HRMS, calc’d for C17H18N5O4S+ [M+H], 388.1074; found 388.1077.
4.3.49. 5-(1-((2-(Ethoxy-d5)phenyl)sulfonyl)azetidin-3-yl)-3-(4-fluorophenyl)-1,2,4-oxadiazole (109).
(Step 1) 3-(4-Fluorophenyl)-5-(1-((2-fluorophenyl)sulfonyl)azetidin-3-yl)-1,2,4-oxadiazole (104). The title compound was prepared in 81% yield (1.10 g) from intermediate 167 and 2-fluorobenzene-sulfonyl chloride, using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, CDCl3) δ 8.00 (m, 2H), 7.90 (m, 1H), 7.64 (m, 1H), 7.31 (m, 2H), 7.16 (m, 2H), 4.40 (m, 4H), 4.07 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 177.9, 164.7 (d, J(C,F) = 252 Hz), 159.4 (d, J(C,F) = 256 Hz), 135.7 (d, J(C,F) = 8.4 Hz), 131.1, 129.6 (d, J(C,F) = 8.8 Hz), 124.6 (d, J(C,F) = 3.8 Hz), 123.5 (d, J(C,F) = 153.8 Hz), 123.4 (d, J(C,F) = 135.9 Hz), 117.5 (d, J(C,F) = 21.8 Hz), 116.1 (d, J(C,F) = 22.1 Hz), 54.4 (d, J(C,F) = 2.1 Hz), 25.2 ppm. (Step 2) 5-(1-((2-(Allyloxy)phenyl)sulfonyl)azetidin-3-yl)-3-(4-fluorophenyl)-1,2,4-oxadiazole (105). Intermediate 104 (1.10 g, 2.91 mmol, 1.0 eq), Cs2CO3 (2.84 g, 8.73 mmol, 3.0 eq), and allyl alcohol (10 mL) were heated at 70 °C overnight. The reaction was cooled to room temperature, water was added, and the reaction was extracted with DCM (2x). The combined organics were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel afforded 630 mg (52%) of the title compound. 1H NMR (300 MHz, CDCl3) δ 8.04 (m, 2H), 7.93 (dd, J = 7.8, 1.7 Hz, 1H), 7.54 (m, 1H), 7.17 (m, 2H), 7.12 – 7.00 (m, 2H), 6.08 (m, 1H), 5.54 (m, 1H), 5.32 (m, 1H), 4.69 (dt, J = 5.2, 1.5 Hz, 2H), 4.44 (m, 4H), 4.03 (m, 1H). LCMS RT = 5.44 min; HRMS, calc’d for C20H19FN3O4S+ [M+H], 416.1075; found 416.1081. (Step 3) 2-((3-(3-(4-Fluorophenyl)-1,2,4-oxadiazol-5-yl)azetidin-1-yl)sulfonyl)phenol (106). Tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4) (70 mg, 0.06 mmol, 0.01 eq) was added to a solution of intermediate 105 (630 mg, 1.52 mmol, 1.0 eq) and K2CO3 (840 mg, 6.08 mmol, 4.0 eq) in methanol (5.0 mL), and stirred overnight at room temperature. The reaction was concentrated in vacuo, followed by the addition of water, and extraction with DCM (2x). The combined organics were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel afforded 435 mg (77%) of the title compound as white solid. 1H NMR (300 MHz, CDCl3) δ 7.98 (m, 2H), 7.67 (dd, J = 9. 0, 1.5 Hz, 1H), 7.52 (t, J = 7.8 Hz, 1H), 7.23 – 6.96 (m, 4H), 4.26 (m, 4H), 3.98 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 177.5, 167.7, 164.7 (d, J(C,F) = 252.2 Hz), 136.3, 129.7 (d, J(C,F) = 8.8 Hz), 129.6, 122.4 (d, J(C,F) = 3.2 Hz), 120.1, 119.5, 117.0, 116.1 (d, J(C,F) = 22.1 Hz), 54.3, 25.3 ppm. LCMS RT = 4.98 min; HRMS, calc’d for C17H15FN3O4S+ [M+H], 376.0762; found 376.0769. (Step 4) Ethyl-d5 4-methylbenzenesulfonate (107). Ethanol-d6 (534 mg, 10.5 mmol, 1.0 eq), and triethylamine (2.86 mL, 21.0 mmol, 2.0 eq) were dissolved in DCM (15 mL), followed by the addition of p-toluenesulfonyl chloride (1.80 g, 9.45 mmol, 0.9 eq), and DMAP (12 mg, 0.10 mmol, 0.01 eq). The reaction was stirred at room temperature overnight. Water was added and the reaction was extracted with DCM (2x). The combined organics were washed with 1N HCl, saturated NaHCO3, and brine; dried over Na2SO4, filtered, and concentrated in vacuo to afford 1.60 g (83%) of the title compound that was used further without purification. 1H NMR (300 MHz, CDCl3) δ 7.80 (m, 2H), 7.35 (m, 2H), 2.45 (s, 3H). (Step 5) 5-(1-((2-(Ethoxy-d5)phenyl)sulfonyl)azetidin-3-yl)-3-(4-fluorophenyl)-1,2,4-oxadiazole (109). Intermediate 106 (30 mg, 0.08 mmol, 1.0 eq), intermediate 107 (33 mg, 0.16 mol, 2.0 eq), Cs2CO3 (78 mg, 0.24 mmol, 3.0 eq), and DMF (0.5 mL) were added to a microwave vial and heated in a microwave reactor at 120 °C for 20 minutes. Water was added, and the reaction was extracted with DCM (2x). The combined organics were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel afforded 23 mg (70%) of the title compound. 1H NMR (300 MHz, CDCl3) δ 8.04 (m, 2H), 7.91 (dd, J = 7.8, 1.8 Hz, 1H), 7.54 (m, 1H), 7.17 (m, 2H), 7.09 – 7.00 (m, 2H), 4.45 (m, 4H), 4.05 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 178.4, 167.8, 164.7 (d, J(C,F) = 252.0 Hz), 156.7, 135.0, 131.1, 129.6 (d, J(C,F) = 8.7 Hz), 125.9, 122.6 (d, J(C,F) = 3.3 Hz), 122.4, 116.1 (d, J(C,F) = 22.1 Hz), 113.5, 54.5, 25.3 ppm. LCMS RT = 5.35 min; HRMS, calc’d for C19H14D5FN3O4S+ [M+H], 409.1389; found 409.1400.
4.3.50. 5-(1-((2-(Cyclobutylmethoxy)phenyl)sulfonyl)azetidin-3-yl)-3-(4-fluorophenyl)-1,2,4-oxadiazole (110).
The title compound was prepared in 56% yield (12 mg) from intermediate 106 and cyclobutane methanol using a method analogous to that described for the conversion of compound 106 into 109. 1H NMR (300 MHz, CDCl3) δ 8.04 (m, 2H), 7.91 (m, 1H), 7.54 (m, 1H), 7.17 (m, 2H), 7.05 (m, 2H), 4.48 – 4.36 (m, 4H), 4.08 (d, J = 6.8 Hz, 2H), 4.02 (m, 1H), 2.86 (m, 1H), 2.23 – 2.09 (m, 2H), 2.04 – 1.83 (m, 4H); 13C NMR (75 MHz, CDCl3) δ 178.3, 165.4 (d, J(C,F) = 356.4 Hz), 157.1, 133.2 (d, J(C,F) = 279 Hz), 130 (d, J(C,F) = 8.8 Hz), 125.5, 122.6 (d, J(C,F) = 3.1 Hz), 120.3, 116.3, 116.0, 113.7, 73.6, 54.2, 34.4, 25.3, 25.0, 18.5 ppm. LCMS RT = 5.87 min; HRMS, calc’d for C22H23FN3O4S+ [M+H], 444.1388; found 444.1392.
4.3.51. 5-(1-((2-(2,2-Difluoroethoxy)phenyl)sulfonyl)azetidin-3-yl)-3-(4-fluorophenyl)-1,2,4-oxadiazole (112).
The title compound was prepared in 52% yield (11 mg) from intermediate 104 and difluoroethanol using a method analogous to that described for the conversion of compound 104 into 105. The title compound was prepared in 35% yield (8 mg) from intermediate 104 and trifluoroethanol. 1H NMR (300 MHz, CDCl3) δ 8.04 (m, 2H), 7.96 (dd, J = 7.9 Hz, 1H), 7.61 (m, 1H), 7.27 – 7.07 (m, 4H), 4.59 – 4.36 (m, 6H), 4.04 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 178.1, 167.8, 164.7 (d, J(C,F) = 251.8 Hz), 155.0, 133.2 (d, J(C,F) = 285.3 Hz), 129.7 (d, J(C,F) = 8.8 Hz), 128.0, 124.8, 123.2, 122.6 (d, J(C,F) = 3.3 Hz), 121.1, 116.1 (d, J(C,F) = 22.1 Hz), 115.8, 67.8 (q, J(C,F) = 36.2 Hz), 54.3, 25.2 ppm. LCMS RT = 5.43 min; HRMS, calc’d for C19H16F4N3O4S+ [M+H], 458.0792; found 458.0795.
4.3.52. 3-(4-Fluorophenyl)-5-(1-((2-(2,2,2-trifluoroethoxy)phenyl)sulfonyl)azetidin-3-yl)-1,2,4-oxadiazole (113).
The title compound was prepared in 35% yield (8 mg) from intermediate 104 and trifluoroethanol using a method analogous to that described for the conversion of compound 104 into 105. 1H NMR (300 MHz, CDCl3) δ 8.04 (m, 2H), 7.96 (dd, J = 7.9 Hz, 1H), 7.61 (m, 1H), 7.27 – 7.07 (m, 4H), 4.59 – 4.36 (m, 6H), 4.04 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 178.1, 167.8, 164.7 (d, J(C,F) = 251.8 Hz), 155.0, 133.2 (d, J(C,F) = 285.3 Hz), 129.7 (d, J(C,F) = 8.8 Hz), 128.0, 124.8, 123.2, 122.6 (d, J(C,F) = 3.3 Hz), 121.1, 116.1 (d, J(C,F) = 22.1 Hz), 115.8, 67.8 (q, J(C,F) = 36.2 Hz), 54.3, 25.2 ppm. LCMS RT = 5.43 min; HRMS, calc’d for C19H16F4N3O4S+ [M+H], 458.0792; found 458.0795.
4.3.53. 5-(1-((2-(Cyclopropylmethoxy)phenyl)sulfonyl)azetidin-3-yl)-3-(4-fluorophenyl)-1,2,4-oxadiazole (114).
The title compound was prepared in 28% yield (6 mg) from intermediate 104 and cyclopropane methanol using a method analogous to that described for the conversion of compound 104 into 105. 1H NMR (300 MHz, CDCl3) δ 8.04 (m, 2H), 7.92 (dd, J = 7.9, 1.7 Hz, 1H), 7.52 (m, 1H), 7.17 (m, 2H), 7.10 – 6.95 (m, 2H), 4.52 (d, J = 7.7 Hz, 4H), 4.06 (m, 1H), 3.95 (d, J = 7.0 Hz, 2H), 1.35 (m, 1H), 0.66 (m, 2H), 0.39 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 178.4, 167.8, 164.7 (d, J(C,F) = 251.9 Hz), 156.7, 133.0 (d, J(C,F) = 288.7 Hz), 129.6 (d, J(C,F) = 8.7 Hz), 126.3, 122.6 (d, J(C,F) = 3.3 Hz), 120.4, 116.3, 116.0, 113.7, 74.4, 54.5, 25.3, 10.1, 3.6 ppm. LCMS RT = 5.59 min; HRMS, calc’d for C21H21FN3O4S+ [M+H], 430.1231; found 430.1238.
4.3.54. 3-(4-Fluorophenyl)-5-(1-((2-(oxetan-3-yloxy)phenyl)sulfonyl)azetidin-3-yl)-1,2,4-oxadiazole (115).
The title compound was prepared in 47% yield (10 mg) from intermediate 104 and oxetan-3-ol using a method analogous to that described for the conversion of compound 104 into 105. 1H NMR (300 MHz, CDCl3) δ 8.03 (m, 2H), 7.96 (dd, J = 7.9, 1.7 Hz, 1H), 7.53 (m, 1H), 7.22 – 7.09 (m, 3H), 6.60 (dd, J = 8.3, 0.7 Hz, 1H), 5.33 (m, 1H), 5.01 (m, 2H), 4.85 (m, 2H), 4.56 – 4.42 (m, 4H), 4.09 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 178.2, 167.8, 164.7 (d, J(C,F) = 252.0 Hz), 154.4, 133.4 (d, J(C,F) = 253.4 Hz), 129.7 (d, J(C,F) = 8.7 Hz), 126.4, 122.5 (d, J(C,F) = 3.3 Hz), 121.6, 116.3, 116.0, 113.5, 71.7, 54.5, 25.3 ppm. LCMS RT = 4.96 min; HRMS, calc’d for C20H19FN3O5S+ [M+H], 432.1024; found 432.1027.
4.3.55. 5-(1-((2-(cyclopentyloxy)phenyl)sulfonyl)azetidin-3-yl)-3-(4-fluorophenyl)-1,2,4-oxadiazole (116).
The title compound was prepared in 34% yield (7 mg) from intermediate 104 and cyclopentanol using a method analogous to that described for the conversion of compound 104 into 105. 1H NMR (300 MHz, CDCl3) δ 8.03 (m, 2H), 7.90 (dd, J = 7.8, 1.7 Hz, 1H), 7.52 (m, 1H), 7.17 (m, 2H), 7.08 – 6.97 (m, 2H), 4.93 (m, 1H), 4.48 – 4.35 (m, 4H), 4.02 (m, 1H), 2.00 – 1.76 (m, 6H), 1.71 – 1.55 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 178.3, 167.7, 164.7 (d, J(C,F) = 252 Hz), 156.1, 133.1 (d, J(C,F) = 251 Hz), 129.6 (d, J(C,F) = 8.8 Hz), 125.9, 122.6 (d, J(C,F) = 3.2 Hz), 119.9, 116.3, 116.0, 114.6, 81.1, 54.3, 32.8, 25.3, 24.0 ppm. LCMS RT = 5.81 min; HRMS, calc’d for C22H23FN3O4S+ [M+H], 444.1388; found 444.1400.
4.3.56. 3-(4-Fluorophenyl)-5-(1-((2-(pyrrolidin-1-yl)phenyl)sulfonyl)azetidin-3-yl)-1,2,4-oxadiazole (117).
NaH (26 mg, 1.1 mmol, 10.0 eq) was added to pyrrolidine (0.5 mL) at 0 °C. Intermediate 104 (40 mg, 0.11 mmol, 1.0 eq) was added afterwards, and the reaction was warmed to room temperature, then heated at 80 °C overnight. The reaction was cooled to room temperature and concentrated in vacuo. Water was added, and the reaction was extracted with DCM (2x). The combined organics were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel afforded 7 mg (16%) of the title compound. 1H NMR (300 MHz, CDCl3) δ 8.06 (m, 2H), 7.98 (d, J = 8.07, 1.7 Hz, 1H), 7.45 (m, 1H), 7.23 – 7.12 (m, 3H), 7.01 (m, 1H), 4.36 (m, 4H), 4.03 (m, 1H), 3.38 (m, 4H), 1.94 (m, 4H); 13C NMR (75 MHz, CDCl3) δ 178.5, 167.8, 164.7 (d, J(C,F) = 252.0 Hz), 150.0, 133.9, 131.5, 129.7 (d, J(C,F) = 8.8 Hz), 128.6, 122.7 (d, J(C,F) = 3.3 Hz), 120.6 (d, J(C,F) = 5.1 Hz), 116.3, 116.0, 53.9, 53.5, 25.4, 25.1 ppm. LCMS RT = 5.84 min; HRMS, calc’d for C21H22FN4O3S+ [M+H], 429.1391; found 429.1397.
4.3.57. 3-(4-Fluorophenyl)-5-(1-((2-(piperidin-1-yl)phenyl)sulfonyl)azetidin-3-yl)-1,2,4-oxadiazole (118).
The title compound was prepared in 18% yield (9 mg) from intermediate 104 and piperidine using a method analogous to that described for the conversion of compound 104 into 117. 1H NMR (300 MHz, CDCl3) δ 8.05 – 7.95 (m, 3H), 7.56 (m, 1H), 7.37 (dd, J = 8.1, 1.1 Hz, 1H), 7.29 – 7.12 (m, 3H), 4.38 (m, 4H), 3.97 (m, 1H), 2.98 (m, 4H), 1.72 (m, 4H), 1.57 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 178.3, 167.0 (d, J(C,F) = 101 Hz), 154.6, 134.3, 132.2, 132.0, 129.6 (d, J(C,F) = 8.7 Hz), 124.4, 123.4, 122.6 (d, J(C,F) = 3.3 Hz), 116.3, 116.0, 55.8, 54.6, 26.1, 25.3, 24.0 ppm. LCMS RT = 6.08 min; HRMS, calc’d for C22H24FN4O3S+ [M+H], 443.1548; found 443.1554.
4.3.58. 5-(1-(chroman-8-ylsulfonyl)azetidin-3-yl)-3-(4-fluorophenyl)-1,2,4-oxadiazole (119).
The title compound was prepared in 72% yield (32 mg) from 3-(3-(4-fluorophenyl)-1,2,4-oxadiazol-5-yl)azetidin-1-hydrochloride (prepared using a method analogous to that described for 10) and chroman-8-sulfonyl chloride using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, CDCl3) δ 8.03 (m, 2H), 7.72 (m, 1H), 7.26 (m, 1H), 7.17 (m, 2H), 6.94 (t, J = 7.7 Hz, 1H), 4.49 – 4.39 (m, 4H), 4.33 (t, J = 5.2 Hz, 2H), 4.06 (m, 1H), 2.83 (t, J = 6.5 Hz, 2H), 2.06 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 177.4, 167.7, 164.7 (d, J(C,F) = 252.1 Hz), 152.8, 135.4, 129.6 (d, J(C,F) = 8.8 Hz), 129.2, 124.6, 124.3, 122.6 (d, J(C,F) = 3.3 Hz), 119.7, 116.2 (d, J(C,F) = 22.1 Hz), 67.3, 54.5, 25.3, 24.8, 21.4 ppm. LCMS RT = 5.33 min; HRMS, calc’d for C20H19FN3O4S+ [M+H], 416.1075; found 416.1076.
4.3.59. 5-(1-(Benzofuran-7-ylsulfonyl)azetidin-3-yl)-3-(4-fluorophenyl)-1,2,4-oxadiazole (120)
The title compound was prepared in 63% yield (28 mg) from 3-(3-(4-fluorophenyl)-1,2,4-oxadiazol-5-yl)azetidin-1-hydrochloride (prepared using a method analogous to that described for 10) and benzofuran-7-sulfonyl chloride using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, CDCl3) δ 7.96 – 7.76 (m, 4H), 7.79 (d, J = 2.2 Hz, 1H), 7.42 (t, J = 7.8 Hz, 1H), 7.15 (m, 2H), 6.89 (d, J = 2.2 Hz, 1H), 4.40 (m, 4H), 3.97 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 177.9, 167.6, 164.7 (d, J(C,F) = 252 Hz), 150.5, 146.6, 129.9, 129.6 (d, J(C,F) = 8.8 Hz), 127.3, 126.3, 123.0, 122.4 (d, J(C,F) = 3.3 Hz), 119.4, 116.1 (d, J(C,F) = 22.1 Hz), 106.9, 54.4, 25.2 ppm. LCMS RT = 5.23 min; HRMS, calc’d for C19H15FN3O4S+ [M+H], 400.0762; found 400.0762.
4.3.60. 3-(4-Fluorophenyl)-5-(1-(quinolin-8-ylsulfonyl)azetidin-3-yl)-1,2,4-oxadiazole (121).
The title compound was prepared in 57% yield (26 mg) from 3-(3-(4-fluorophenyl)-1,2,4-oxadiazol-5-yl)azetidin-1-hydrochloride (prepared using a method analogous to that described for 10) and 8-quinolinesulfonyl chloride using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, CDCl3) δ 9.05 (dd, J = 4.2, 1.8 Hz, 1H), 8.49 (dd, J = 7.4, 1.4 Hz, 1H), 8.23 (dd, J = 8.5, 1.8 Hz, 1H), 8.07 (dd, J = 8.2, 1.4 Hz, 1H), 7.95 (m, 2H), 7.66 (dd, J = 8.1, 7.5 Hz, 1H), 7.51 (q, J = 4.3 Hz, 1H), 7.15 (m, 2H), 4.71 (m, 4H), 4.04 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 178.5, 167.6, 164.6 (d, J(C,F) = 252 Hz), 151.4, 144.2, 136.7, 136.4, 133.8, 132.4, 129.6 (d, J(C,F) = 8.8 Hz), 129.1, 125.6, 122.6 (d, J(C,F) = 3.3 Hz), 122.2, 116.1 (d, J(C,F) = 22.1 Hz), 55.0, 25.4 ppm. LCMS RT = 5.09 min; HRMS, calc’d for C20H16FN4O3S+ [M+H], 411.0922; found 411.0927.
4.3.61. 3-(4-Fluorophenyl)-5-(1-((2-methylquinolin-8-yl)sulfonyl)azetidin-3-yl)-1,2,4-oxadiazole (122).
The title compound was prepared in 81% yield (38 mg) from 3-(3-(4-fluorophenyl)-1,2,4-oxadiazol-5-yl)azetidin-1-hydrochloride (prepared using a method analogous to that described for 10) and 2-methylquinoline-8-sulfonyl chloride using a method analogous to that described for the conversion of compound 10 into 5. 1H NMR (300 MHz, CDCl3) δ 8.45 (dd, J = 7.4, 1.43 Hz, 1H), 8.08 (d, J = 8.5, 1H), 8.04 – 7.90 (m, 3H), 7.58 (t, J = 7.8 Hz, 1H), 7.36 (d, J = 8.5 Hz, 1H), 7.15 (m, 2H), 4.74 (m, 4H), 4.05 (m, 1H), 2.76 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 178.6, 167.6, 164.7 (d, J(C,F) = 252 Hz), 160.6, 143.8, 136.4, 136.3, 133.4, 132.1, 129.6 (d, J(C,F) = 8.8 Hz), 127.4, 124.6, 123.1, 122.6 (d, J(C,F) = 3.3 Hz), 116.1 (d, J(C,F) = 22.1 Hz), 54.9, 25.5, 25.4 ppm. LCMS RT = 5.22 min; HRMS, calc’d for C21H18FN4O3S+ [M+H], 425.1078; found 425.1074.
Supplementary Material
Acknowledgements
We thank the National Institute of Neurological Disorders and Stroke (R33NS109521 to C.D.W. and K.A.E.) for their support of our program in the development of small molecule inhibitors of Slack channels. Portions of this work has been supported by certain funds managed by Deerfield Management Company, L.P. Funding for the WaveFront Biosciences Panoptic and the Syncropatch 768 PE platform was provided by the Office of The Director (OD) of the National Institutes of Health under the award numbers 1S10OD021734 and 1S10OD021734, respectively. These instruments are housed and maintained in the Vanderbilt Institute of Chemical Biology’s High-throughput Screening Center.
Footnotes
Declaration of Competing Interest
C.D.W. is an owner of WaveFront Biosciences and ION biosciences, makers of the Panoptic plate reader and Thallos, Tl+-sensitive fluorescent indicators used in the these studies, respectively.
Data Availability
Data will be made available on request.
References
- 1.Coppola G, Plouin P, Chiron C, Robain O, Dulac O. Migrating Partial Seizures in Infancy: A Malignant Disorder with Developmental Arrest. Epilepsia. 1995;36(10): 1017–1024. [DOI] [PubMed] [Google Scholar]
- 2.McTague A, Nair U, Malhotra S, et al. Clinical and Molecular Characterization of KCNT1-Related Severe Early-Onset Epilepsy. Neurology. 2018;90(1): e55–e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barcia G, Fleming MR, Deligniere A, et al. De novo Gain-of-Function KCNT1 Channel Mutations Cause Malignant Migrating Partial Seizures of Infancy. Nature Genetics. 2012;44(11): 1255–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cole BA, Clapcote SJ, Muench SP, Lippiat JD. Targeting K(Na)1.1 Channels in KCNT1-Associated Epilepsy. Trends in Pharmacol Sciences. 2021;42(8): 700–713. [DOI] [PubMed] [Google Scholar]
- 5.Yuan A, Santi CM, Wei A, et al. The Sodium-Activated Potassium Channel is Encoded by a Member of the Slo Gene Family. Neuron. 2003;37(5): 765–773. [DOI] [PubMed] [Google Scholar]
- 6.Joiner WJ, Tang MD, Wang LY, et al. Formation of Intermediate-Conductance Calcium-Activated Potassium Channels by Interaction of Slack and Slo subunits. Nature Neuroscience. 1998;1(6): 462–469. [DOI] [PubMed] [Google Scholar]
- 7.Spitznagel BD, Mishra NM, Qunies AM, et al. VU0606170, a Selective Slack Channels Inhibitor, Decreases Calcium Oscillations in Cultured Cortical Neurons. ACS Chemical Neuroscience. 2020;11(21): 3658–3671. [DOI] [PubMed] [Google Scholar]
- 8.Barcia G, Chemaly N, Kuchenbuch M, et al. Epilepsy with Migrating Focal Seizures: KCNT1 Mutation Hotspots and Phenotype Variability. Neurology Genetics. 2019;5(6): e363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhattacharjee A, Gan L, Kaczmarek LK. Localization of the Slack Potassium Channel in the Rat Central Nervous System. Journal of Comparitive Neurology 2002;454(3): 241–254. [DOI] [PubMed] [Google Scholar]
- 10.The Human Protein Atlas. KCNT1.; https://www.proteinatlas.org/ENSG00000107147-KCNT1/tissue. Accessed 2/18/2021.
- 11.Bhattacharjee A, Joiner WJ, Wu M, Yang Y, Sigworth FJ, Kaczmarek LK. Slick (Slo2.1), A Rapidly-Gating Sodium-Activated Potassium Channel Inhibited by ATP. Journal of Neuroscience 2003;23(37): 11681–11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Z, Rosenhouse-Dantsker A, Tang QY, Noskov S, Logothetis DE. The RCK2 Domain Uses a Coordination Site Present in Kir Channels to Confer Sodium Sensitivity to Slo2.2 Channels. Journal of Neuroscience. 2010;30(22): 7554–7562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaczmarek LK. Slack, Slick and Sodium-activated Potassium Channels. ISRN Neuroscience. 2013;2013(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rizzo F, Ambrosino P, Guacci A, et al. Characterization of Two De Novo KCNT1 Mutations in Children with Malignant Migrating Partial Seizures in Infancy. Molecular and Cellular Neurosciences. 2016;72: 54–63. [DOI] [PubMed] [Google Scholar]
- 15.Tang QY, Zhang FF, Xu J, et al. Epilepsy-Related Slack Channel Mutants Lead to Channel Over-Activity by Two Different Mechanisms. Cell Reports. 2016;14(1): 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nuwer MO, Picchione KE, Bhattacharjee A. cAMP-Dependent Kinase Does Not Modulate the Slack Sodium-Activated Potassium Channel. Neuropharmacology. 2009;57(3): 219–226. [DOI] [PubMed] [Google Scholar]
- 17.Qunies AM, Mishra NM, Spitznagel BD, et al. Structure-Activity Relationship Studies in a New Series of 2-Amino-N-Phenylacetamide Inhibitors of Slack Potassium Channels. Bioorganic & Medicinal Chemistry Letters. 2022;76: 129013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quraishi IH, Mercier MR, McClure H, et al. Impaired Motor Skill Learning and Altered Seizure Susceptibility in Mice with Loss or Gain of Function of the Kcnt1 Gene Encoding Slack (KNa1.1) Na(+)-Activated K(+) Channels. Scientific Reports. 2020;10(1): 3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim Grace E, Kronengold J, Barcia G, et al. Human Slack Potassium Channel Mutations Increase Positive Cooperativity between Individual Channels. Cell Reports. 2014;9(5): 1661–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Milligan CJ, Li M, Gazina EV, et al. KCNT1 Gain of Function in 2 Epilepsy Phenotypes is Reversed by Quinidine. Annals of Neurology. 2014;75(4): 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin HC, Kim GE, Pagnamenta AT, et al. Clinical Whole-Genome Sequencing in Severe Early-Onset Epilepsy Reveals New Genes and Improves Molecular Diagnosis. Human Molecular Genetics. 2014;23(12): 3200–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heron SE, Smith KR, Bahlo M, et al. Missense Mutations in the Sodium-Gated Potassium Channel Gene KCNT1 Cause Severe Autosomal Dominant Nocturnal Frontal Lobe Epilepsy. Nature Genetics. 2012;44(11): 1188–1190. [DOI] [PubMed] [Google Scholar]
- 23.Ohba C, Kato M, Takahashi N, et al. De Novo KCNT1 Mutations in Early-Onset Epileptic Encephalopathy. Epilepsia. 2015;56(9): e121–128. [DOI] [PubMed] [Google Scholar]
- 24.Moller RS, Heron SE, Larsen LH, et al. Mutations in KCNT1 Cause a Spectrum of Focal Epilepsies. Epilepsia. 2015;56(9): e114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Borlot F, Abushama A, Morrison-Levy N, et al. KCNT1-Related Epilepsy: An International Multicenter Cohort of 27 Pediatric Cases. Epilepsia. 2020. [DOI] [PubMed] [Google Scholar]
- 26.Routier L, Verny F, Barcia G, et al. Exome Sequencing Findings in 27 Patients with Myoclonic-Atonic Epilepsy: Is There a Major Genetic Factor? Clinical Genetics. 2019;96(3): 254–260. [DOI] [PubMed] [Google Scholar]
- 27.Hansen N, Widman G, Hattingen E, Elger CE, Kunz WS. Mesial Temporal Lobe Epilepsy Associated with KCNT1 Mutation. Seizure. 2017;45: 181–183. [DOI] [PubMed] [Google Scholar]
- 28.Quraishi IH, Stern S, Mangan KP, et al. An Epilepsy-Associated KCNT1 Mutation Enhances Excitability of Human iPSC-Derived Neurons by Increasing Slack KNa Currents. Journal of Neuroscience. 2019;39(37): 7438–7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown MR, Kronengold J, Gazula VR, et al. Fragile X Mental Retardation Protein Controls Gating of the Sodium-Activated Potassium Channel Slack. Nature Neuroscience. 2010;13(7): 819–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fleming MR, Brown MR, Kronengold J, et al. Stimulation of Slack K(+) Channels Alters Mass at the Plasma Membrane by Triggering Dissociation of a Phosphatase-Regulatory Complex. Cell Reports. 2016;16(9): 2281–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bearden D, Strong A, Ehnot J, DiGiovine M, Dlugos D, Goldberg EM. Targeted Treatment of Migrating Partial Seizures of Infancy with Quinidine. Annals of Neurology. 2014;76(3): 457–461. [DOI] [PubMed] [Google Scholar]
- 32.Dilena R, DiFrancesco JC, Soldovieri MV, et al. Early Treatment with Quinidine in 2 Patients with Epilepsy of Infancy with Migrating Focal Seizures (EIMFS) Due to Gain-of-Function KCNT1 Mutations: Functional Studies, Clinical Responses, and Critical Issues for Personalized Therapy. Neurotherapeutics: The Journal of the American Society for Experimental NeuroTherapeutics. 2018;15(4): 1112–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jia Y, Lin Y, Li J, et al. Quinidine Therapy for Lennox-Gastaut Syndrome with KCNT1 Mutation. A Case Report and Literature Review. Frontiers in Neurology. 2019;10: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patil AA, Vinayan KP, Roy AG. Two South Indian Children with KCNT1-Related Malignant Migrating Focal Seizures of Infancy - Clinical Characteristics and Outcome of Targeted Treatment with Quinidine. Annals of Indian Academy of Neurology. 2019;22(3): 311–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Passey CC, Erramouspe J, Castellanos P, O’Donnell EC, Denton DM. Concurrent Quinidine and Phenobarbital in the Treatment of a Patient with 2 KCNT1 Mutations. Current Therapeutic Research, Clinical and Experimental. 2019;90: 106–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.El Kosseifi C, Cornet M-C, Cilio MR. Neonatal Developmental and Epileptic Encephalopathies. Seminars in Pediatric Neurology. 2019;32: 100770. [DOI] [PubMed] [Google Scholar]
- 37.Yoshitomi S, Takahashi Y, Yamaguchi T, et al. Quinidine Therapy and Therapeutic Drug Monitoring in Four Patients with KCNT1 Mutations. Epileptic Disorders: International Epilepsy Journal with Videotape. 2019;21(1): 48–54. [DOI] [PubMed] [Google Scholar]
- 38.Cole BA, Johnson RM, Dejakaisaya H, et al. Structure-Based Identification and Characterization of Inhibitors of the Epilepsy-Associated K(Na)1.1 (KCNT1) Potassium Channel. iScience. 2020;23(5): 101100–101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Griffin AM, Kahlig KM, Hatch RJ, et al. Discovery of the First Orally Available, Selective KNa1.1 Inhibitor: In Vitro and In Vivo Activity of an Oxadiazole Series. ACS Medicinal Chemistry Letters. 2021;12(4): 593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qunies AM, Emmitte KA. Small-Molecule Inhibitors of Slack Potassium Channels as Potential Therapeutics for Childhood Epilepsies. Pharmaceutical Patent Analyst. 2022;11(2): 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Treiman DM. GABAergic Mechanisms in Epilepsy. Epilepsia. 2001;42 Suppl 3: 8–12. [DOI] [PubMed] [Google Scholar]
- 42.Rankovic Z CNS Drug Design: Balancing Physicochemical Properties for Optimal Brain Exposure. Journal of Medicinal Chemistry. 2015;58(6): 2584–2608. [DOI] [PubMed] [Google Scholar]
- 43.Pajouhesh H, Lenz GR. Medicinal Chemical Properties of Successful Central Nervous System Drugs. NeuroRx. 2005;2(4): 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.cLogP and tPSA calculated using ChemDraw® Professional v. 16.0.
- 45.Grone BP, Baraban SC. Animal Models in Epilepsy Research: Legacies and New Directions. Nature Neuroscience. 2015;18(3): 339–343. [DOI] [PubMed] [Google Scholar]
- 46.Felts AS, Rodriguez AL, Morrison RD, et al. Discovery of Imidazo[1,2-a]-, [1,2,4]Triazolo[4,3-a]-, and [1,2,4]Triazolo[1,5-a]Pyridine-8-Carboxamide Negative Allosteric Modulators of Metabotropic Glutamate Receptor Subtype 5. Bioorganic & Medicinal Chemistry Letters. 2017;27(21): 4858–4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baldauf C, Günther R, Hofmann H-J. Conformational Properties of Sulfonamido Peptides. Journal of Molecular Structure: THEOCHEM. 2004;675(1): 19–28. [Google Scholar]
- 48.Gramec D, Peterlin Mašič L, Sollner Dolenc M. Bioactivation Potential of Thiophene-Containing Drugs. Chemical Research in Toxicology. 2014;27(8): 1344–1358. [DOI] [PubMed] [Google Scholar]
- 49.Oberdorf C, Schepmann D, Vela JM, Diaz JL, Holenz J, Wünsch B. Thiophene Bioisosteres of Spirocyclic σ Receptor Ligands. 1. N-Substituted Spiro[piperidine-4,4′-thieno[3,2-c]pyrans]. Journal of Medicinal Chemistry. 2008;51(20): 6531–6537. [DOI] [PubMed] [Google Scholar]
- 50.Zhao Z, Koeplinger KA, Peterson T, et al. Mechanism, Structure-Activity Studies, and Potential Applications of Glutathione S-Transferase-Catalyzed Cleavage of Sulfonamides. Drug Metabolism and Disposition. 1999;27(9): 992–998. [PubMed] [Google Scholar]
- 51.Meunier B, de Visser SP, Shaik S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chemical Reviews. 2004;104(9): 3947–3980. [DOI] [PubMed] [Google Scholar]
- 52.Subbaiah MAM, Meanwell NA. Bioisosteres of the Phenyl Ring: Recent Strategic Applications in Lead Optimization and Drug Design. Journal of Medicinal Chemistry. 2021;64(19): 14046–14128. [DOI] [PubMed] [Google Scholar]
- 53.Pirali T, Serafini M, Cargnin S, Genazzani AA. Applications of Deuterium in Medicinal Chemistry. Journal of Medicinal Chemistry. 2019;62(11): 5276–5297. [DOI] [PubMed] [Google Scholar]
- 54.Meanwell NA. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. Journal of Medicinal Chemistry. 2018;61(14): 5822–5880. [DOI] [PubMed] [Google Scholar]
- 55.Naumovich V, Grishina M, Novak J, et al. Electronic Properties Investigation of Human Dihydrofolate Reductase Complexes with Ligands. Journal of Biomolecular Structure & Dynamics. 2022;40(11): 4775–4790. [DOI] [PubMed] [Google Scholar]
- 56.Barreiro EJ, Kümmerle AE, Fraga CAM. The Methylation Effect in Medicinal Chemistry. Chemical Reviews. 2011;111(9): 5215–5246. [DOI] [PubMed] [Google Scholar]
- 57.Kato T-a, Hakura A, Mizutani T, Saeki K-i. Anti-Mutagenic Structural Modification by Fluorine-Substitution in Highly Mutagenic 4-Methylquinoline Derivatives. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 2000;465(1): 173–182. [DOI] [PubMed] [Google Scholar]
- 58.Chen H, Kronengold J, Yan Y, et al. The N-Terminal Domain of Slack Determines the Formation and Trafficking of Slick/Slack Heteromeric Sodium-Activated Potassium Channels. The Journal of Neuroscience. 2009;29(17): 5654–5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Flauaus C, Engel P, Zhou F, et al. Slick Potassium Channels Control Pain and Itch in Distinct Populations of Sensory and Spinal Neurons in Mice. Anesthesiology. 2022;136(5): 802–822. [DOI] [PubMed] [Google Scholar]
- 60.Gong P, Jiao X, Yu D, Yang Z. Case Report: Causative De novo Variants of KCNT2 for Developmental and Epileptic Encephalopathy. Frontiers in Genetics. 2021;12: 649556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gururaj S, Palmer EE, Sheehan GD, et al. A De Novo Mutation in the Sodium-Activated Potassium Channel KCNT2 Alters Ion Selectivity and Causes Epileptic Encephalopathy. Cell Rep. 2017;21(4): 926–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Toro L, Li M, Zhang Z, Singh H, Wu Y, Stefani E. MaxiK Channel and Cell Signalling. Pflugers Archive. 2014;466(5): 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stork D, Timin EN, Berjukow S, et al. State Dependent Dissociation of HERG Channel Inhibitors. British Journal of Pharmacology. 2007;151(8): 1368–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsujimae K, Suzuki S, Yamada M, Kurachi Y. Comparison of Kinetic Properties of Quinidine and Dofetilide Block of HERG Channels. European Journal of Pharmacology. 2004;493(1–3): 29–40. [DOI] [PubMed] [Google Scholar]
- 65.Obach RS, LaChapelle EA, Brodney MA, Vanase-Frawley M, Kauffman GW, Sawant-Basak A. Strategies Toward Optimization of the Metabolism of a Series of Serotonin-4 Partial Agonists: Investigation of Azetidines as Piperidine Isosteres. Xenobiotica. 2016;46(12): 1112–1121. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.