

Abstract
BACKGROUND:
High circulating levels of lipoprotein(a) [Lp(a)], increase the risk of atherosclerosis and calcific aortic valve disease, affecting millions of patients worldwide. While atheroscleorosis is commonly treated with low-density liporprotein-targeting therapies these do not reduce Lp(a) or calcific aortic valve disease risk, a disease with no available drug therapies. Targeting Lp(a) production and catabolism may provide therapeutic benefit, but little is known about Lp(a) cellular uptake.
METHODS:
Here, unbiased ligand-receptor capture mass spectrometry was used to identify major facilitator superfamily domain containing 5 (MFSD5) as a novel receptor/co-factor involved in Lp(a) uptake.
RESULTS:
Reducing MFSD5 expression by a computationally-identified small molecule or siRNA supressed Lp(a) uptake and calcification in primary human valvular endothelial and interstitial cells. MFSD5 variants were associated with aortic stenosis (p=0.027 after multiple hypothesis testing) with suggestive evidence of an interaction with plasma Lp(a) levels.
CONCLUSIONS:
MFSD5 knockdown suppressing human valvular cell Lp(a) uptake and calcification along with meta-analysis of MFSD5 variants associating with aortic stenosis supports further preclinical assessment of MFSD5 in cardiovascular diseases, the leading cause of death worldwide.
Keywords: aortic valve stenosis, lipoprotein(a), MFSD5, lipoprotein receptors, cardiovascular calcification
Calcific aortic valve disease (CAVD) is the most common valve disease, with aortic sclerosis affecting 25% and aortic stenosis affecting 2% of individuals over age 65.1 Currently, the only treatment for symtomatic CAVD is invasive surgical or transcatheter valve replacements, which present some risk to patients and are not universaly available. With no approved pharmecutical therapies for CAVD, including statins that failed to provide benefit in clinical CAVD trials,2 there is urgent need for therapeutics which are targeted for early intervention against both the initiation and progression of CAVD.
A major risk factor of CAVD and atherosclerosis is lipoprotein(a) [Lp(a)], a low-density lipoprotein (LDL)-like molecule. Current estimates predict over 1.4 billion people worldwide have high Lp(a);3 however, there are no United States Food and Drug Administration-approved Lp(a)-lowering small-molecule therapies. Lp(a) abundance is controlled by genetics,4–6 is mostly independent of diet, and is not consistently reduced by LDL lowering drugs, such as statins. Like LDL, Lp(a) contains apolipoprotein B (ApoB), but differs from LDL in that it also contains Apo(a). Lp(a) also contains other pro-atherogenic components, including oxidized phospholids and apolipoprotein C III.7 Apolipoprotein C III on Lp(a) associates with Lp(a)-mediated valve disease and may promote human valvular intersititial cell (VIC) calcification through mitochondrial dysfunction, oxidative stress, and inflammatory pathways.8,9 While associated with increased cardiovascular disease risk,10,11 the normal physioloigcal functions of Lp(a) are unclear.12 Complicating investigative efforts, Lp(a) is not endogenously produced in most experimental animal models, with its expression largely restricted to humans and other primates. Lp(a) can be produced in transgenic mice,13 which have increased aorta lipid lesions, but there remains a paucity of animal models of Lp(a)-mediated CAVD pathology in the absence of other genetic modifications, necessisating the use of human studies to better understant Lp(a) biology.
Overall, there are insufficient means to reduce circulating Lp(a) amounts to lower cardiovascular disease risk.3 Strategies to limit Lp(a) effects include directly lowering Lp(a) levels and targeting Lp(a) uptake. Directly lowering Lp(a) levels can be achieved by lipoprotein aphresis, but this proceedure is costly, semi-invasive, and severly limits patient mobility, which leads to high patient refusal and low adherance rates.14 Proprotein convertase subtilisin/kexin type 9 inhibitors can reduce circulating Lp(a) abundance by 26.9%,15 but whether further reduction of circulating Lp(a) provides improvements in cardiovascular disease outcomes is unknown. Clinical trials demonstrated that concentrations of circulating Lp(a) can be consistently and potently reduced by Apo(a) antisense therapy, reducing Lp(a) abundance below the critical 50 mg/dL threshold in patients with established cardiovascular disease.16 Apo(a) antisense reduces circulating oxidized phospholipids in cardiovascular disease patients,16 but if antisense also affects other cardiovascular disease-promoting molecules found on Lp(a) particles is unclear. Whether Apo(a) antisense and other LPA RNA-targeting therapies improve patient cardiovascular outcomes beyond the extent of existing cardiovascular drugs is currently being investigated in clinical trials.17
Inhibiting Lp(a) uptake specifically in cardiovascular tissue where it acts, may be a clinically benefical approach. Several membrane proteins interact with Lp(a), and have been suggested to be putative Lp(a) receptors.18 It is possible that several receptors mediate Lp(a) clearance in the liver, the major route of Lp(a) catabolism, in addition to other tissue-specific mechanisms of Lp(a) catabolism. There are conflicting reports on Lp(a) uptake by many of these putative Lp(a) receptors, including LDL receptor (LDLR);19 as such, identification of definitive Lp(a) receptor(s) remains obscure in the liver and unknown in the aortic valve. In the aortic valve, Lp(a) has been observed in the extracellular matrix, and oxidized phospholipids on Lp(a), including lysophosphatidic acid, are metabolised and taken up by VICs, inducing calcification.7 Whereas Apo(a)-antisense may work to reduce production of Lp(a) through limiting Apo(a), combinatory therapies inhibiting Lp(a) production and receptor-mediated uptake in cardiovascular cells may further improve cardiovascular outcomes. As an example, combination therapy with ezetimibe, an inhibitor of the cholesterol absorption protein, Niemann-Pick C1 Like 1 protein with simvastatin that inhibits cholesterol synthesis, improves cardiovascular outcomes compared to statin therapy alone.20 Identifying receptors involved in Lp(a) catabolism would be adventageous, particularly in the context of CAVD, a disease with unmet therapuetic need. Therefore, we hypothesized that through an unbiased ligand-receptor capture mass spectrometry approach, Lp(a) receptor(s) controlling cellular Lp(a) uptake can be identified on VICs, a major cell type contibuting to aortic valve calcification.7,21
METHODS
Detailed methods and Major Resources Table provided in the Supplemental Material.
Cell Culture
Human aortic valve tissue was obtained with informed consent in accordance with the deceleration of Helsinki. Human aortic valve samples were handled under institutional review board protocol 2011P001703, approved by the Beth Israel Deaconess Medical Center and Partners Human Research Committee institutional review boards, and included written consent from all donors. Researchers were blinded to all clinical characteristics and patient data of the valve tissues used. Primary human VICs were obtained from human aortic valve tissues by collagenase digestion. Following removal of valvular endothelial cells (VECs) by scraping with a razor blade, valves were divided into small cubes (1–2mm) and digested in 1 mg/mL collagenase (Sigma Aldrich, St. Louis, MO) in Dulbecco’s Minimum Essential Medium (DMEM, Thermo Fisher Scientific) for 1 hour with agitation. Following removal of the 1-hour digest, valve pieces were redigested with 1 mg/mL collagenase for 3 hours. Resultant supernatant was then centrifuged (400g, 5 minutes, room temperature) and the VIC pellet resuspended in DMEM supplemented with 10% fetal bovine serum (unless otherwise stated, 1% penicillin/streptomycin (P/S), cultured (37°C 5% CO2, 90% humidity) and used between passages 1 and 5. VIC purity was validated by immunofluorescence staining for vimentin+ (abcam #ab92547) and negative for CD31− (abcam #ab9498), which was further verified by proteomics analysis. VECs (Factor VIII+, alpha-SMA−) were obtained from Lonza and cultured in the same manner as VICs. HepG2 cells were obtained from ATCC (Manassas, VA) and cultured in Eagle’s Minimum Essential Medium (Thermo Fisher Scientific) with 10% FBS (unless otherwise noted) and 1% P/S.
Lipoprotein(a) Uptake Assay
Uptake assays were performed as follows: briefly, VICs, VECs, and HepG2 cells were cultured in lipoprotein-deficient serum for 48 hours before incubation with Dil-labeled LDL (Thermo Fisher Scientific) or Lp(a) for 6 hours. To confirm removal of Lp(a) not taken up by cells, cells were stringently washed at room temperature (RT) as follows: three times in phosphate buffered saline (PBS) with 0.8% bovine serum albumin (BSA), twice in PBS with 0.8% BSA and 0.2 M ε-ACA for five minutes each, twice with 0.2 M acetic acid (pH 2.5) with 0.2 M NaCl for ten minutes each, and finally twice in PBS. Cells were stained with DAPI (4′,6-diamidino-2-phenylindole, Thermo Fisher Scientific), 488-conjugated wheat germ agglutinin (WGA) (Thermo Fisher Scientific) for the nuclei and membrane respectively, and imaged on a Nikon Confocal A1 microscope (Nikon, Tokyo, Japan). Fluorescence was quantified with an ImageXpress Pico digital fluorescence system (Molecular Devices, San Jose, CA), and assessed for percentage positive (Dil-Lp[a]/DAPI). Alternatively flurescence intensity was quantified using NIH ImageJ software from the Nikon Confocal A1 microscope images.
Calcification Assay
To induce calcification, 100% confluent VICs were cultured in DMEM for up to 21 days, in either normal medium (NM; DMEM supplemented with 10% FBS and 1% PS or in pro-calcifying medium (PM; NM with 10 nmol/L dexamethasone, 10 mmol/L β-glycerophosphate, and 50 μg/mL L-ascorbic acid). 10 μg/mL Lp(a) was included in PM when noted. VICs were stained with 2% Alizarin red (Lifeline Cell Technology, Frederick, MD) at day 14 and 21 of culture and quantified by extracting the stain with 100 mmol/L cetylpyridinium chloride (Thermo Fisher Scientific) at room temperature for 1 hour, and the absorbance was measured at 540 nm.
TriCEPS Mass Spectrometry
TriCEPS reagents were commercially obtained as Flow-TriCEPS and ligand-receptor capture-TriCEPs kits (Dual Systems, Zurich, Switzerland). Dot blots to validate TriCEPS reagent coupling to Lp(a) (20 μg) were performed according to the manufacturer’s instructions and blotted with three serial dilutions (1:10, 1:100, 1:1000). Transferrin was used as an active comparator and glycine as a negative control that quenches the amine-reactive moieties of TriCEPS. Fluorescence-activated cell sorting (FACS) of VICs incubated without Lp(a), with fluorescently labeled TriCEPS coupled to Lp(a), or fluorescently labeled TriCEPS coupled to Lp(a) that was quenched with glycine was assessed using an FACS Aria flow cytometer (BD Biosciences, Franklin Lakes, NJ; Harvard Immunology FACS core facility) and commercially obtained Flow-TriCEPS reagents according to manufacturer’s instructions. For TriCEPS experiments, six equally pooled VIC donors were used with a commercially obtained TriCEPS kit. VICs were plated at 100% confluency and 2 × 107 cells per sample were used. For ligand-receptor capture TriCEPS, the TriCEPS reagent was coupled to Lp(a) (300 μg ligand coupled to 150 μg TriCEPs) according to manufacturer’s instructions and incubated with VICs (4°C, 1 hour) to enable Lp(a) and receptor binding but not Lp(a) internalization. VICs were oxidized according to the TriCEPS kit instructions, allowing for crosslinking of TriCEPS-coupled Lp(a) to membrane proteins that were affinity purified and analyzed by LC-MS/MS.
MFSD5 siRNA
Knockdown studies in VICs and HepG2 cells were performed with siRNA targeting MFSD5 (20 nM, Assay IDs s39790, s224982 Thermo Fisher and Dharmacon Smartpool MFSD5 siRNA), LDLR (20 nM, Assay ID S4, Thermo Fisher) and non-targeting control siRNA (20 nM, Assay ID#4390843, Thermo Fisher) with Lipofectamine RNAi MAX transfection reagent (Thermo Fisher Scientific) according to manufacturer’s protocol. For VECs, 40 nM siRNAs were used. Three validated MFSD5 siRNAs were tested, and similar results were observed for all three with knockdown efficiency.
Lp(a) Interaction Analysis
The UK Biobank is a prospective cohort comprised of > 500,000 participants in the United Kingdom with broad phenotyping electronic health records, death registries, blood-based analyses, genotyping, and other methodologies.22 Genotyping had been performed using the Applied Biosystems UK Biobank Axiom Array and Applied Biosystems UK BiLEVE Axiom Array, followed by imputation using the 1000 Genomes phase 3, Haplotype Reference Consortium, and UK10K reference panels.23 Serum Lp(a) had been measured using an immuno-turbidimetric assay manufactured by Randox Bioscience, UK. Measures were set below or above the reportable range defined by the manufacturer (3.8 to 189 nmol/L) to 3.8 nmol/L or 189 nmol/L, respectively. The analysis included unrelated participants aged 55 years or older at baseline with genetically-confirmed White British ancestry. Individuals with congenital valvular heart disease in their hospital inpatient records (International Classification of Diseases, Ninth Revision [ICD-9] 746–747 or International Classification of Diseases, Tenth Revision [ICD-10] Q20-Q23) were excluded. Among the remaining 247,417 participants, aortic stenosis cases were identified through the presence of ICD-9 424.1, ICD-10 I35.0, or OPCS Classification of Surgical Operations and Procedures, Fourth Revision codes K26.1, K26.2, K26.3, K26.4, K31.2, K32.2, or K35.2 in the hospital inpatient records, with all other participants deemed to be controls. Associations with aortic stenosis were estimated in logistic regression models using PLINK version 2,24 adjusted for age, age squared, sex, genotype batch, and 20 principal components.
The Genetic Epidemiology Research on Adult Health and Aging cohort consists of > 100,000 individuals in northern California who are in the Kaiser Permanente healthcare delivery system.25 Genotyping had been performed using customized Affymetrix Axiom arrays,26 followed by imputation using the Michigan Imputation Server27 with the Haplotype Reference Consortium version r1.1 as the reference panel.28 Analysis included unrelated European ancestry participants aged 55 years or older. Using electronic health record data from January 1996 to December 2015, inclusively, individuals with congenital valvular heart disease were excluded (ICD-9 746–747) and identified aortic stenosis cases in the remaining 55,180 participants through the presence of ICD-9 424.1 or a procedure code for aortic valve replacement. All other participants were defined as controls. Associations with aortic stenosis were estimated in logistic regression models using PLINK version,24 adjusted for age, age squared, sex, and 10 principal components.
Firstly, analysis with a fixed effect model, inverse-variance weighted meta-analysis for aortic stenosis of the 291 variants within 50 kb of MFSD5 which had a minor allele frequency ≥ 0.001 and an imputation quality ≥ 0.3 in both cohorts was performed (consisted of 87 independent variants). For the independent (r2 < 0.1) variants with p < 0.01 in the meta-analysis (5 variants), the (i) direct effect on the natural logarithm of Lp(a) and (ii) the interaction with the natural logarithm of Lp(a) for the outcome of aortic stenosis were examined. For a variant which a significant interaction (p < 0.05), the prevalence of aortic stenosis stratified by copies of the minor allele and tertiles of Lp(a) was examined and performed logistic regression modelling the association of combinations of minor allele copies and Lp(a) tertiles with aortic stenosis. For all analyses, we adjusted p-values based on the number of independent variants tested by Bonferroni correction.
Data Availability
Data generated or analysed during the current study are indluded in the article and its Supplemental Data. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium by the PRIDE,29 partner repository with the dataset identifier PXD026861. Username: reviewer_pxd026861@ebi.ac.uk; password: uadLa8rZ.
Statistical Analysis
Analysis of variance (ANOVA) with Tukey’s multiple comparison testing and Welch’s t tests were performed with Prism 9 (GraphPad, La Jolla, CA). Data were analyzed for normality and equal variance to determine whether applied parametric tests were appropriate.
RESULTS
Ligand-receptor Capture Mass Spectrometry Identified a Previously Uncharacterized Lp(a) Receptor/Co-Factor on Human VICs
TriCEPS ligand-receptor capture mass spectrometry30 was used to identify membrane proteins that bind Lp(a) and mediate its uptake in human valves. For this experiment we used VICs isolated from human valves, validated by cells staining positive for an interstitial cell marker, vimentin+ and negative for an endothelial cell marker, CD31− (Figure S1A and S1B). We confirmed the presence and absence of these markers and further characterized these VICs using cell-fractionated proteomics for membrane, cytosolic, and nuclear proteins (Figure S2 and Excel File S1). TriCEPS reagent specifically couples to the N-terminus and lysines of ligands, with the whole complex then binding to plasma membrane receptors. Oxidization of cells allows for the TriCEPS reagent to crosslink and then be affinity purified, with the bound membrane proteins idenfied by liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) (Figure 1A). Dot blot analysis confirmed that Lp(a) could be coupled to the TriCEPS reagent using TriCEPS-transferrin plasma membrane receptor coupling as an active comparator and glycine that quenches the amine-reactive moieties of the TriCEPS reagent as a negative control (Figure S3A). Lp(a) was readily coupled to TriCEPS in a similar manner to that of transferrin supporting that the TriCEPS reagent is compatible with Lp(a) ligand (Figure S3A). This experimental process did not alter VIC viability, assessed by a live/dead cell assay (Figure S3B). Supporting that TriCEPS-coupled Lp(a) binds to VICs, TriCEPS-labeled Lp(a) increased VIC fluorescence that was partially suppressed by glycine quenching, as assessed by FACS (Figure S3C).
Figure 1. MFSD5 in human aortic VICs binds to Lp(a).
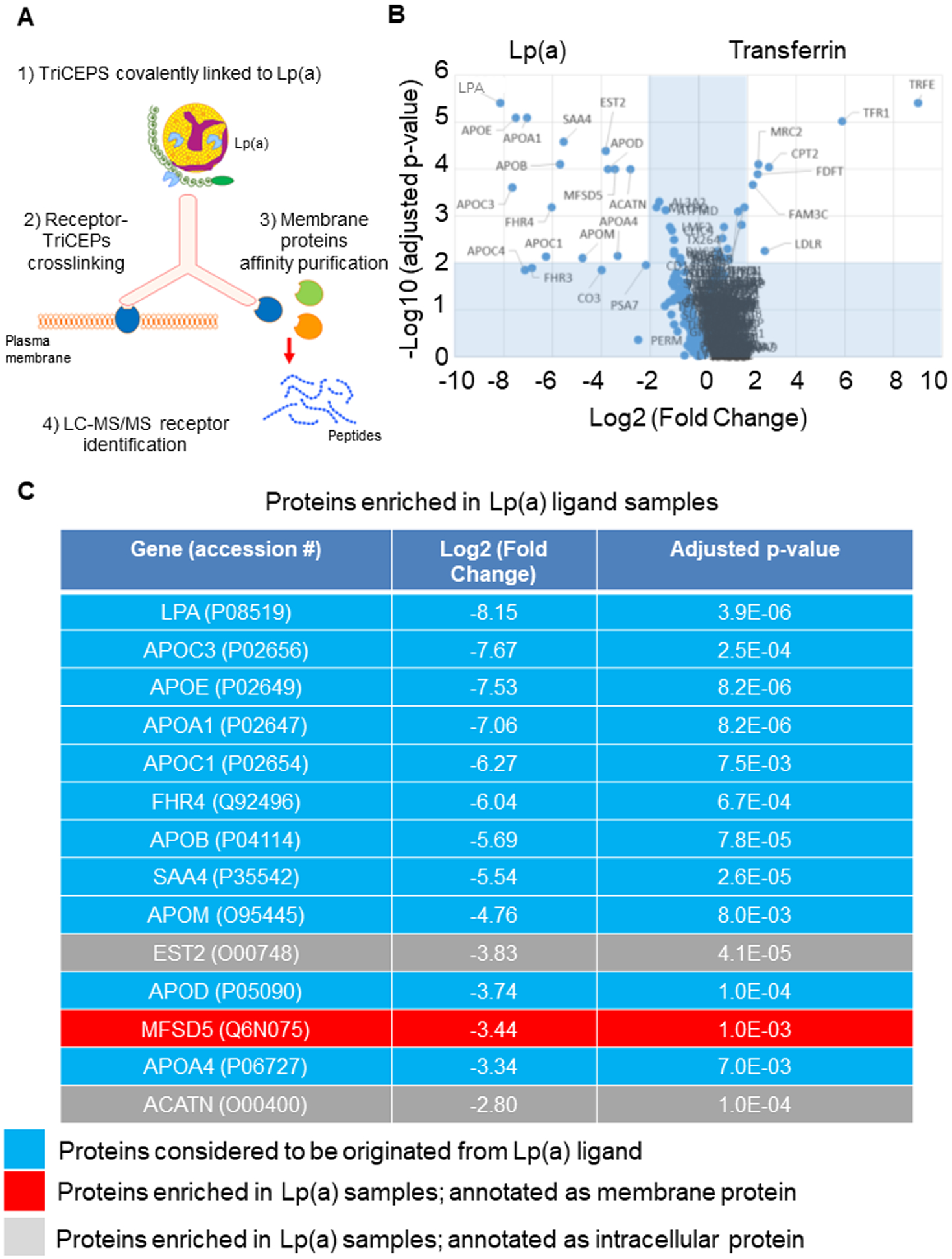
A, Cartoon showing the TriCEPS mass spectrometry experimental setup used to identify Lp(a) receptors on human aortic VICs. B, Volcano plot showing enrichment of mass spectrometry-identified proteins with TriCEPS-labeled Lp(a) incubation or TriCEPS-labeled transferrin used as an incubation control. C, Proteins purified and significantly enriched by TriCEPS-labeled Lp(a) capture in VICs shown. MFSD5 indicated in red; (n=3 replicate experiments with 6 pooled donors/experiment).
To identify which protein(s) TriCEPS-coupled Lp(a) bound to on VICs, ligand-receptor capture mass spectrometry was performed with Lp(a)-coupled TriCEPS and transferrin-coupled TriCEPS as a positive control. VICs took up Lp(a) in a time-dependent manner (Figure S3D), and incubation at 4°C inhibited interalization of Lp(a), enabling surface Lp(a) receptor identification using LC-MS/MS. Validating the experimental protocol, transferrin incubation lead to enrichment of the transferrin receptor (Figure 1B). Additional membrane proteins enriched in transferrin-incubated VICs included LDLR and mannose receptor C type 2 (Figure 1B). Rapid endocytosis in Lp(a) incubated cells could result in an apparent enrichment of Lp(a) receptors in the transferrin-incubated VIC group compared to the Lp(a)-incubated group, due to receptor internalization upon Lp(a) binding. However, since the TriCEPS incubation was carried out at 4°C, a condition that inhibits endocytosis therefore making this scenario less likely, we focused on receptors enriched with Lp(a) incubation in VICs. Most of the proteins enriched in the Lp(a)-incubated VICs were as expected, known components of Lp(a) particles,8 including Apo(a) and ApoB (Figure 1C). Given the expectation of an Lp(a) receptor being plasma membrane-localized enabling it to bind circulating Lp(a), we looked for enrichment of membrane-bound proteins. There was only one plasma membrane protein that was enriched in TriCEPS-coupled Lp(a)-incubated VICs, a ubiquitously expressed protein, major facilitator superfamily domain containing 5 (MFSD5) (Figure 1C). Additional Lp(a) receptors not identified by the TriCEPS capture method cannot be excluded from participating in Lp(a) uptake; however, identification of MFSD5 as the only markedly enriched receptor by TriCEPS-coupled Lp(a) immunopurification in VICs led us to focus on assessement of MFSD5 as a putative Lp(a) receptor.
Reducing MFSD5 Expression Inhibited Lp(a) Uptake
To determine whether MFSD5 participated in Lp(a) uptake in human cells, the effects of validated MFSD5 specific siRNA (confirmed by three separate siRNAs) on Lp(a) uptake in VICs was assessed. Purified Lp(a) was used throughout these experiments that was valdiated by apolipoprotein content and gel electrophoresis (Figure S4A and S4B). Lp(a) purity was additionally validated by Apo(a) immunogold electron microscopy, which showed that all lipoprotein particles were positively labeled with Apo(a), as well as demonstrating Lp(a) uptake by VICs (Figure S4C). Significant cellular uptake of Lp(a) was achieved at 10 ug/mL in human liver HepG2 cells (Figure S4D), a concentration previously associated with VIC calcificaiton,21 and was therefore used in our experiments. We also conducted a competition assay of Dil-labeled LDL and unlabeled Lp(a) (in excess; 10ug/mL) in HepG2 cells. With Lp(a) in excess, LDL was not taken up as readily in HepG2 cells as when incubated with Dil-LDL alone (Figure S4E). MFSD5 siRNA incubation with VICs was validated by quantitative PCR (Figure 2A) and reduced Lp(a) uptake assessed by immunofluorescence and Western blotting (Figure 2B; Figure S5). Given the similarity of Lp(a) to LDL, the ability of MFSD5 siRNA to alter Dil-LDL uptake by VICs was also assessed. Reduction of LDLR by siRNA did not reduce VIC MFSD5 mRNA (Figure 2A) or Lp(a) uptake in VICs (Figure 2B; Figure S5). As a control for LDLR knockdown, LDLR siRNA but not MFSD5 siRNA reduced LDLR mRNA (Figure 2C) and Dil-LDL uptake in VICs (Figure 2D). Similarly, MFSD5 siRNA reduced MFSD5 mRNA (Figure 3A) and Lp(a) uptake (Figure 3B) in primary human VECs. LDLR siRNA but not MFSD5 siRNA reduced VEC LDLR mRNA (Figure 3C) and Dil-LDL uptake (Figure 3D). These data support MFSD5 as a previously uncharacterized Lp(a) receptor or co-factor mediating Lp(a) uptake in human valvular cells.
Figure 2. MFSD5 specific siRNA reduced Lp(a) uptake by human VICs.
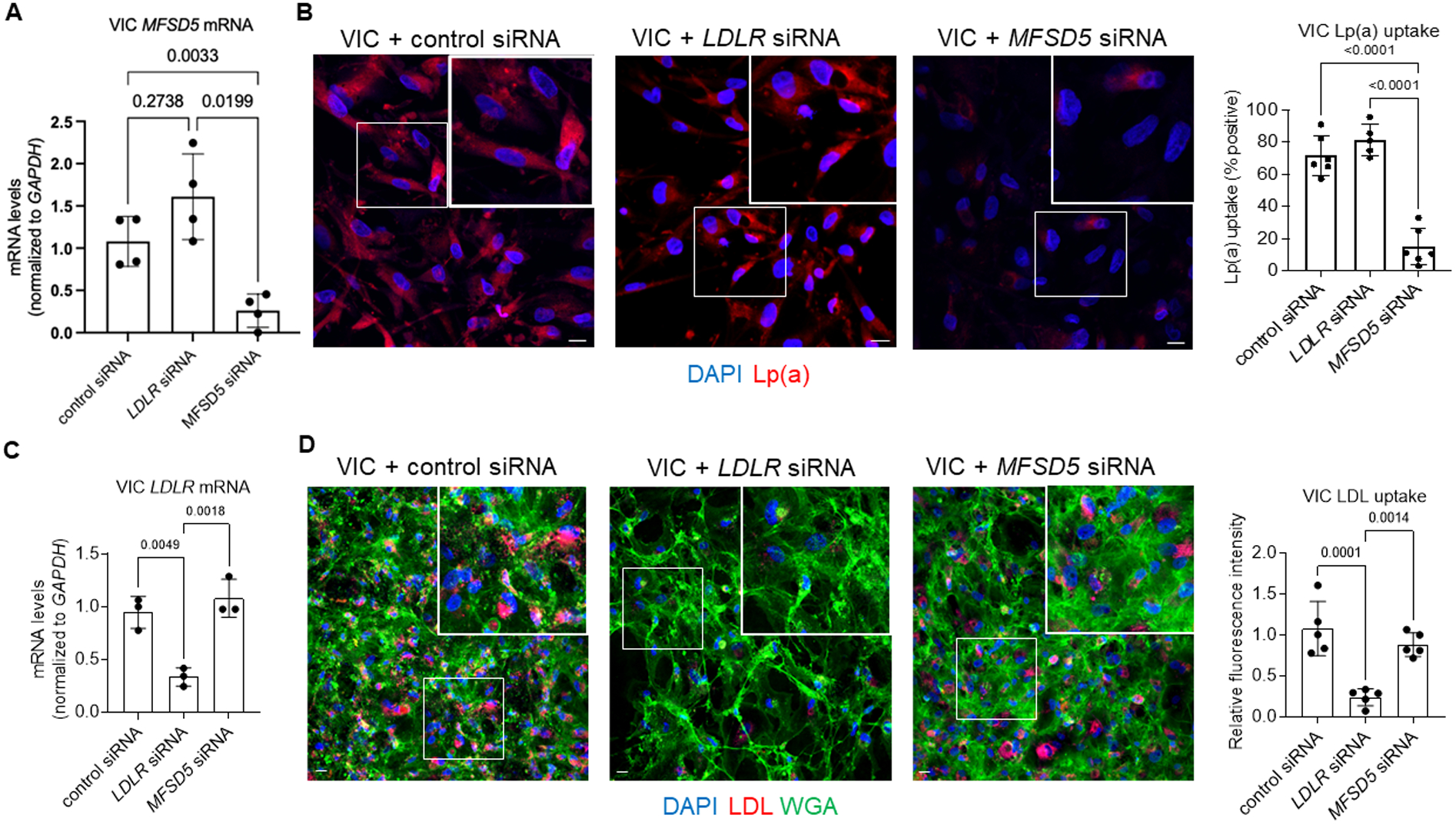
A, Analysis of MFSD5 mRNA abundance in VICs incubated with the indicated siRNAs (n=4 donors). B, Left: Confocal microscopy analysis of Dil-Lp(a) uptake by VICs incubated with the indicated siRNAs. Quantification of Lp(a) uptake by the indicated cells (n=5–6 donors). C, Analysis of LDLR mRNA abundance in VICs incubated with the indicated siRNAs (n=3 donors). D, Left: Confocal microscopy analysis of Dil-LDL uptake by VICs incubated with the indicated siRNAs. DAPI was used to stain nuclei and wheat germ agglutinin (WGA) was used to stain membranes. Right: Quantification of Dil-LDL uptake by VICs incubated with the indicated siRNAs (n=5 donors). Data are mean ± SD and were analyzed by ANOVA. Scale bars = 20 μm, higher magnification inset areas are indicated by white boxes.
Figure 3. MFSD5 specific siRNA reduced Lp(a) uptake by human VECs.
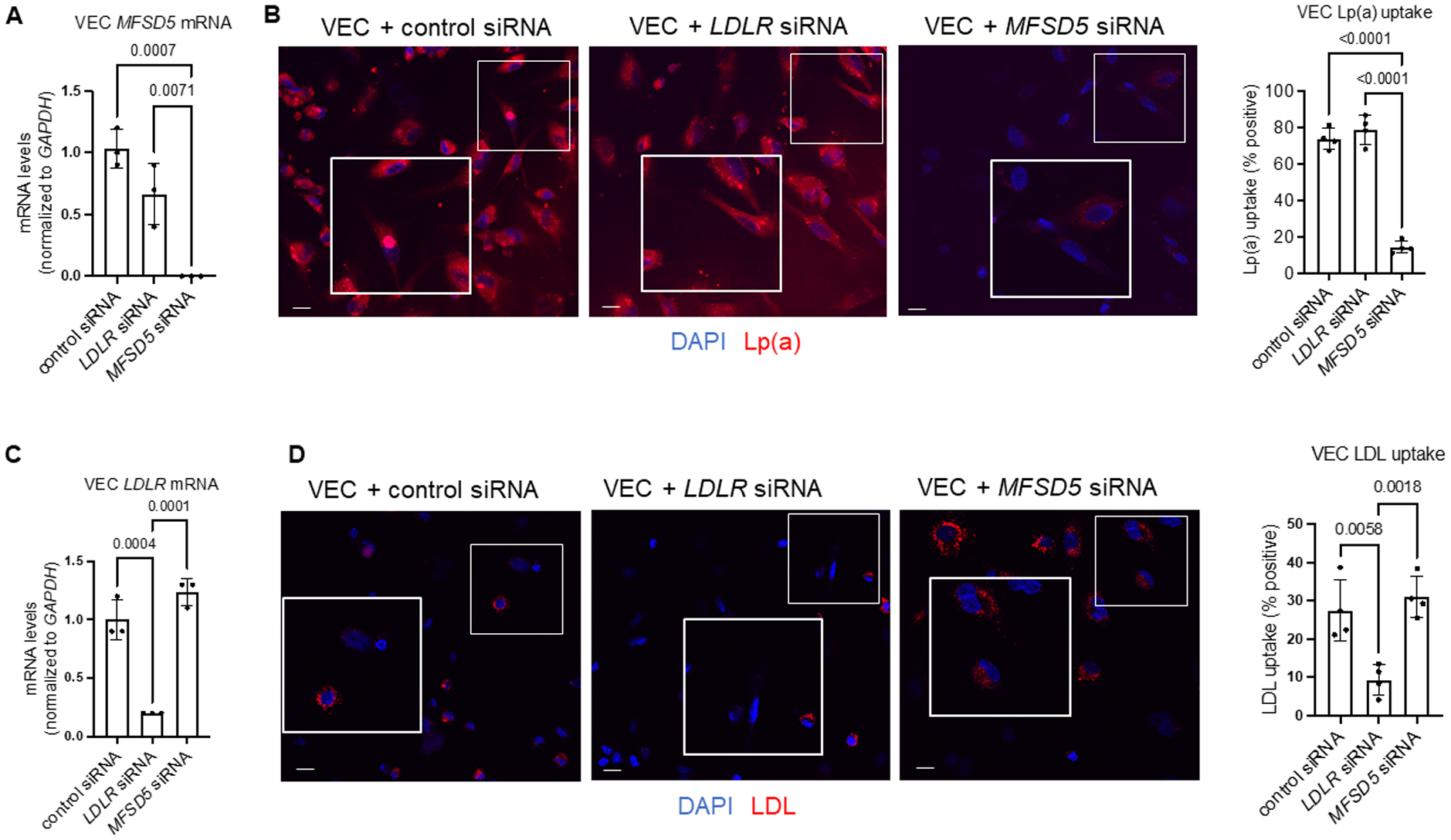
A, Analysis of MFSD5 mRNA abundance in VECs incubated with the indicated siRNAs (n=3 donors). B, Left: Confocal microscopy analysis of Dil-Lp(a) uptake by VECs incubated with the indicated siRNAs. Quantification of Lp(a) uptake by the indicated cells (n=4 donors). C, Analysis of LDLR mRNA abundance in VECs incubated with the indicated siRNAs (n=3 donors). D, Left: Confocal microscopy analysis of Dil-LDL uptake by VECs incubated with the indicated siRNAs. DAPI was used to stain nuclei. Right: Quantification of Dil-LDL uptake by VECs incubated with the indicated siRNAs (n=4 donors). Data are mean ± SD and were analyzed by ANOVA. Scale bars = 20 μm, higher magnification inset areas are indicated by white boxes.
As Lp(a) catabolism occurs primarily in the liver and MFSD5 has ubiquitous expression, the functional role of MFSD5 in human liver HepG2 cells was also assessed. Reduced MFSD5 with MFSD5-specific siRNA incubation was validated by quantitative PCR analysis in HepG2 cells (Figure S6A). MFSD5 siRNA reduced Lp(a) uptake in HepG2 cells (Figure S6B). No change in MFSD5 levels were observed with LDLR specific siRNA in HepG2 cells (Figure S6A). Supression of HepG2 cells LDLR mRNA (Figure S6C) by LDLR siRNA did not reduce Lp(a) uptake (Figure S6B), but did reduce Dil-LDL uptake (Figure S6D) in our experimental conditions. Together these data demonstrate that MFSD5 is unlikely to interact with ApoB protein present on both Lp(a) and LDL.
Next, small molecule inhibition of MFSD5 Lp(a) uptake was conducted through the L1000 platform,31,32 in which genes and drugs are connected by common gene-expression signatures enabling prediction of compounds that modulate genes of interest. L1000 analysis supported aminopurvalanol A, a cyclin-dependent kinase inhibitor, as a top, commercially available compound that could modulate MFSD5 expression. Quantitative PCR demonstrated that aminopurvanol A could suppress MFSD5 mRNA levels in VICs (Figure S7A). VICs incubated with aminopurvalanol A were then incubated with Lp(a), to confirm a reduction in uptake. In agreement with the MFSD5 siRNA data, Lp(a) uptake was reduced by aminopurvalanol A incubation (Figure S7B). These data provide an initial in vitro proof-of-concept that MFSD5 and associated Lp(a) uptake can be regulated by small molecules, in addition to providing an independent validation of our MFSD5 siRNA data.
Reducing MFSD5 Expression Suppressed VIC Calcification
To assess the potential of reducing VIC Lp(a) uptake in the etiology of CAVD through reduced MFSD5, we first used siRNA to suppress MFSD5 expression. The presence of MFSD5 in human aortic valve tissues from which primary VICs were obtained was shown with RNAscope (Figure 4A). After isolation, purified VICs were pre-incubated with MFSD5 or control siRNA and then incubated in pro-calcifying media (PM) containing inorganic phosphate and ascorbic acid, with or without Lp(a). Lp(a) incubation led to earlier calcification compared to that induced by PM alone (Figure 4B). In agreement with a role of MFSD5 in mediating Lp(a) uptake that promotes calcification, MFSD5 siRNA suppressed calcification in VICs, assessed by Alizarin red staining (Figure 4C). Similarly, aminopurvalanol A incubation inhibited primary human VIC calcification by Lp(a) (Figure S7C).
Figure 4. MFSD5 specific siRNA reduced Lp(a)-mediated calcification of human VICs.
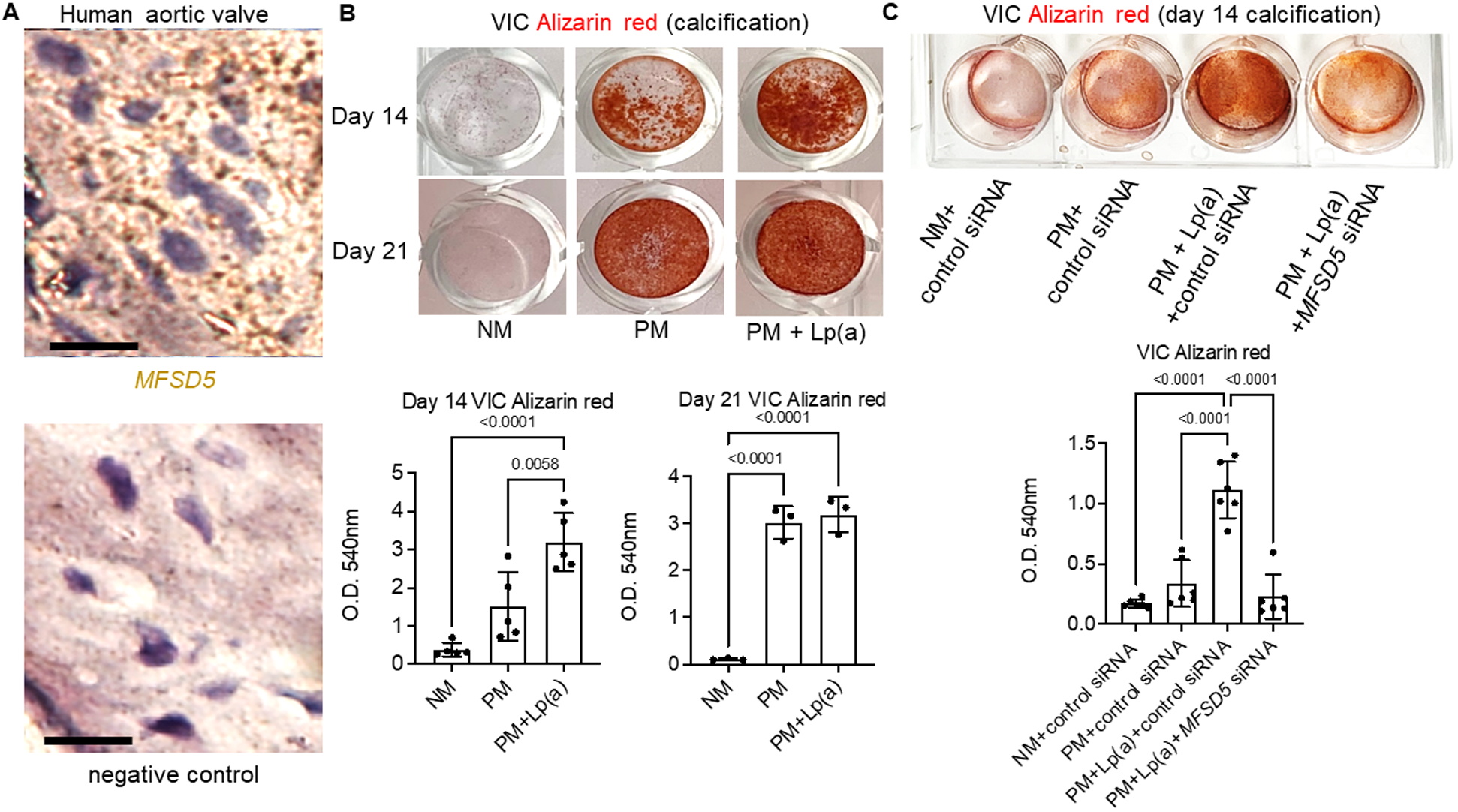
A, MFSD5 RNAscope in human aortic valves (n=5 donors with an example donor image shown stained with an MFSD5 probe (brown stain) and a negative control probe in the same region of a serial section). Scale bars = 20 μm. B, VIC calcification (Alizarin red) images (top) and quantification (bottom) for VICs incubated in normal control media (NM) or pro-calcifying media (PM) with or without Lp(a) (n=5 donors for day 14, and 3 donors for day 21; analyzed by ANOVA). C, VICs incubated with the indicated siRNAs (n=6 donors; analyzed by ANOVA). Data are mean ± SD.
MFSD5 Variants Associated with Aortic Stenosis and may Modify Lp(a) Mediated Risk in Human Aortic Stenosis
To further evaluate the contribution of MFSD5 to Lp(a)-related phenotypes in humans, the role of MFSD5-region variants on Lp(a) driven CAVD was assessed. Five independent (r2 < 0.1) variants (Table S1) were selected within 50 kb of MFSD5 associated with aortic stenosis at p < 0.01 in an inverse-variance weighted, fixed effects meta-analysis of 247,417 UK Biobank White British participants (3,443 cases) and 55,180 Genetic Epidemiology Research on Adult Health and Aging European-ancestry participants (3,469 cases) for 291 variants. One variant, rs61760168, was found to be significantly associated with AS in meta-analysis after adjustment for multiple hypothesis testing (OR 2.03, 95% CI 1.38,2.97; p = 3×10−4, padj= 0.027 for multiple comparisons). No significant direct effect of MFSD5 variants on Lp(a) levels in the UK Biobank was found (Table S1). We observed a differential effect of Lp(a) on aortic stenosis by the MFSD5 rs73101935 genotype (interaction p=0.031; padj=0.155 for multiple comparisons, Figure 5). Among individuals in the highest tertile of Lp(a), the prevalence of aortic stenosis was 1.8% in individuals with no copies of rs73101935-T, compared to 4.7% in individuals with one copy (Figure 5). In regression analysis, compared to individuals in the lowest tertile of Lp(a) and with zero copies of rs73101935-T, the odds of aortic stenosis were 54% higher for individuals in the highest tertile of Lp(a) but with zero copies of rs73101935-T (odds ratio, 1.54; 95% CI, 1.41 to 1.67; p = 2.0 × 10−23), and 341% higher for individuals in the highest Lp(a) tertile with one copy of rs73101935-T (odds ratio, 4.41; 95% CI, 2.64 to 7.35; p = 1.3 × 10−8) (Table S2).
Figure 5. Prevalence of aortic stenosis in the UK Biobank, stratified by the number of MFSD5 rs73101935-T alleles and tertiles of Lp(a).
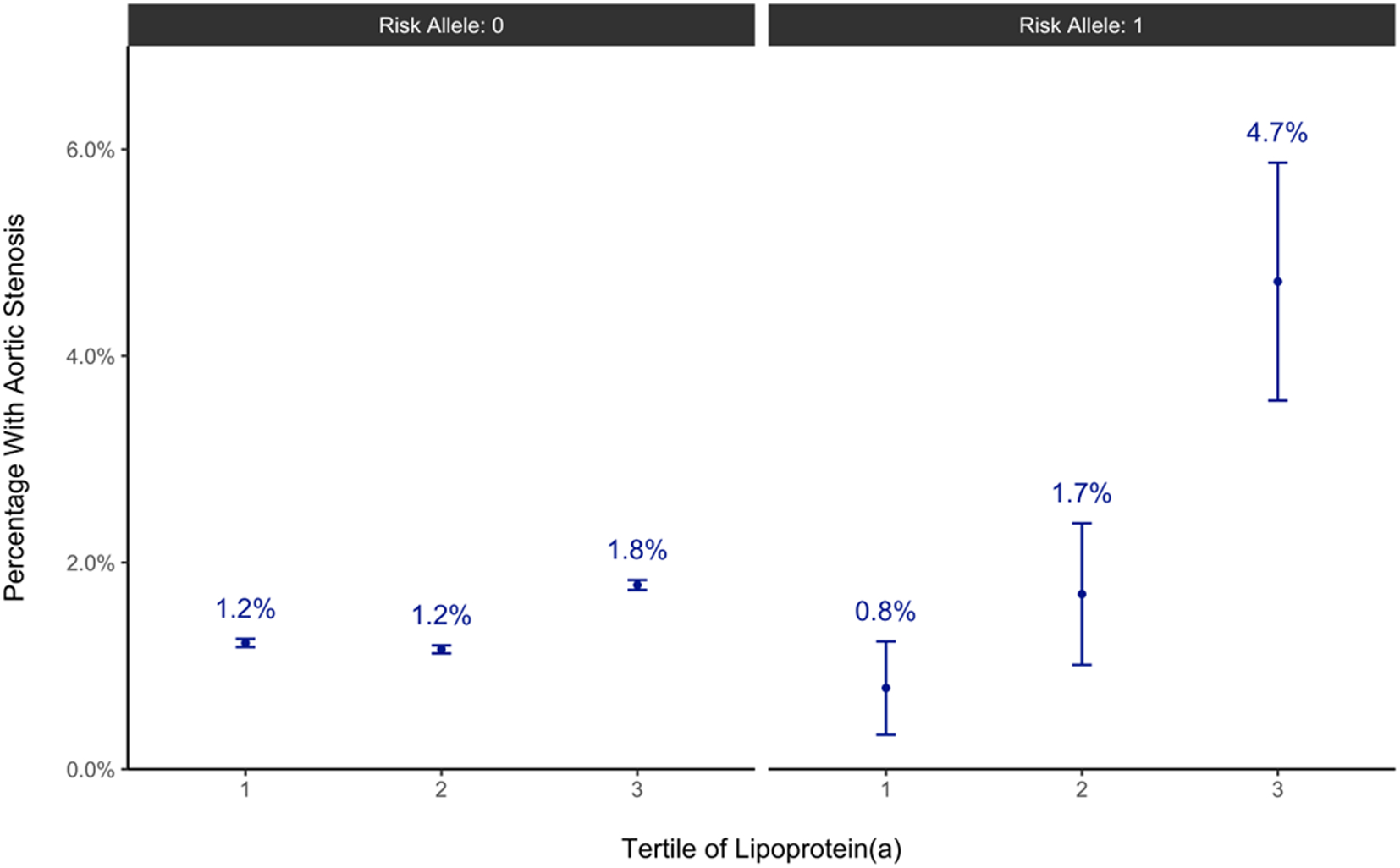
Prevalence is expressed as mean ± SE (n=75,774, 75,792, and 75,808 participants for the 1, 2, and 3 respective Lp(a) tertiles with Risk allele: 0, and 382, 354, and 339 participants for the 1, 2, and 3 respective Lp(a) tertiles with Risk allele: 1). A table format of the data is included the Supplemental Material, Tables S1 and S2.
DISCUSSION
The current study demonstrates a role of MFSD5 as a new receptor/co-factor involved in Lp(a) uptake and aortic stenosis. Moreover, the data reveals MFSD5 binding of Lp(a) through a crosslinking affinity purification assay. MFSD5 contribution to Lp(a) uptake and VIC calcification was further demonstration by reducing MFSD5 expression. An MFSD5 variant, which enhanced the effect of Lp(a) on aortic stenosis greater than 4-fold was identified in the UK Biobank genetic dataset that further supports a role for MFSD5 in Lp(a)-mediated cardiovascular disease. While we observed a reduction of Lp(a) uptake in HepG2 cells with MFSD5 siRNA, albeit less than detected in valvular cells, we did not find a direct effect on Lp(a) plasma concentrations in the UK Biobank dataset. Lp(a) production is largely impacted by the LPA locus and a lack of direct effect on Lp(a) levels by variants in MFSD5 does not preclude a proposed role of MFSD5 as an Lp(a) receptor or co-factor.
Despite the clinical importance of Lp(a), the cellular and biochemical pathways involved in Lp(a) catabolism remain unclear. Reducing Lp(a) mediated pathology might be achieved by altering its uptake, like statins in reducing the effects of LDL in part via increasing LDL receptor abundance. However, unlike LDL, the receptors involved in Lp(a) catabolism remain unclear. Several types of receptors have been suggested to be involved in Lp(a) uptake including lipoprotein receptors, scavenger receptors, toll-like receptors, lectins, and plasminogen receptors, and Lp(a) uptake may involve multiple receptors.18 In our experimental conditions, LDLR specific siRNA did not reduce Lp(a) uptake in human valvular or HepG2 cells; however, we cannot exclude a role of LDLR in Lp(a) catabolism in other conditions. Lp(a) contains Apo(a) that may impair the ability of LDLR to bind ApoB on Lp(a) particles, resulting in slower Lp(a) uptake than LDL, which was reduced by LDLR specific siRNA in human valvular and HepG2 cells. Whether MFSD5 interacts with oxidized lipids is unknown. MFSD5 and lysophosphatidic acid receptor, a receptor associated with aortic stenosis mediated by oxidative transformation of low-density lipoprotein,33 was suggested as one of 56,000 candidate interactions identified by an affinity purification-mass spectrometry approach.34 As such, if MFSD5 acts as a co-factor with other receptors associated with Lp(a) and oxidized lipids could be further explored, but is beyond the scope of the present study.
MFSD5 is a largely unstudied protein and the physiologic function of MFSD5, a plasma membrane protein, is unknown, but it may act in transport and metabolic functions. To our knowledge, this is the first report of MFSD5 function using human cells and tissues. MFSD5 overexpression experiments in yeast cells suggest that it is a solute transporter that transfers molybdate anions.35 MFSD5 interacts with overexpressed glucagon-like peptide-1 receptor in Chinese hamster ovary cells,36 a receptor involved in controling blood glucose levels through increasing insulin secretion. MFSD5 is a member of the mammalian solute carrier family, a group with at least 395 members in humans and which translocate a broad range of substrates across membranes.37 Given that MFSD5 is evolutionarily conserved, MFSD5 likely has additional functions unrelated to Lp(a). Whereas rodents do not endogenously produce Lp(a), Mfsd5 mRNA is reduced by either starvation or high-fat diet in mice, suggesting a possible additional function in energy regulation.37 MFSD5 involvement in metabolism may be related to cardiovascular disease pathology. We demonstrated inhibition of MFSD5 expression suppressed Lp(a) uptake in VICs and VECs. Lp(a) enhances glycolysis that induces endothelial cell inflammation associated with transendothelial migration of leukocytes in the arterial wall.38 In addition to regulating recruitment of immune cells and expression of pro-calcific proteins, VECs can undergo an endothelial-to-mesenchymal transformation into osteogenic cells,39 and high glucose stimulates endothelial-to-mesenchymal transformation of VECs.40
Our current TriCEPS ligand-receptor capture affinity purification results support MFSD5 binding to Lp(a) and implicate a functional role in a lipid metabolism. It remains unclear what portion of Lp(a) that MFSD5 interacts with, but it is unlikely to be ApoB given that MFSD5 siRNA did not reduce LDL uptake in vitro. How increased Lp(a) uptake promotes VIC calcification is an active area of research. We and others have shown the oxidized phospholipid component of Lp(a) increases inflammatory and osteogenic gene transcription in VICs.7,21 Similar VIC phenotype changes were also observed with a proinflammatory component of Lp(a), apolipoprotein C III.9 Additionally, Lp(a) promotes the release of calcifying extracellular vesicles in human VICs.41 While these inflammatory and osteogenic changes would be expected to be suppressed by reduced MFSD5 expression, as this condition reduced Lp(a) uptake and calcification, the key molecular pathways through which MFSD5 regulates calcification with high Lp(a) requires further study. Given the association we observed with the MFSD5 variant rs73101935 and increased odds of aortic stenosis in patients with high Lp(a), understanding the molecular changes associated with this variant may help elicudate mechanisms through which high Lp(a) increases aortic stenosis risk.
A limit of our study is a need for further demonstration of direct MFSD5 and Lp(a) binding. Our TriCEPS ligand-receptor capture experimental approach suggests an interaction of MFSD5 with Lp(a) but does not exclude other nearby proteins directly binding to Lp(a). A more direct biochemical means could be explored in future studies to show specific binding of Lp(a) and MFSD5 that could mechanistically explain why Lp(a) uptake was suppressed in vitro when we inhibited MFSD5 expression. In human VICs, a computationally identified MFSD5 suppressing small molecule, aminopurvalanol A, inhibited calcification, supporting our siRNA data and the potential druggability of MFSD5. However, aminopurvalanol A is not a specific MFSD5 inhibitor, and we cannot exclude other mechanisms contributing to its anti-calcification and Lp(a) uptake suppression effects. Another limitation of the present study is the lack of an in vivo animal model supporting our in vitro and human genetics data. While not trivial, development of in vivo MFSD5-deficient models with high Lp(a)-driven CAVD pathology could provide a means to gain further mechanistic insight into the role of MFSD5 in this disease.
In conclusion, this study identifies a previously unknown biological function of MFSD5 in lipid metabolism and provides initial proof-of-concept and support for further preclinical assessement of MFSD5 in cardiovascular disease, including through perturbation of Lp(a) metabolism.
Supplementary Material
Clinical Perspective.
What is new?
First identification of MFSD5 as a receptor/co-factor involved in Lp(a) uptake and calcific aortic valve disease.
MFSD5 variant rs73101935 magnified the effect of high Lp(a) on aortic stenosis.
What are the clinical implications?
MFSD5 is a novel and potentially druggable target with a computationally identified small molecule and MFSD5 siRNA suppressing MFSD5 expression, Lp(a) uptake and calcification in primary human valvular cells.
Acknowledgments
Analyses conducted using the UK Biobank Resource were performed under Application Number 41025.
Funding
National Institutes of Health grants R01HL136431, R01HL147095, R01HL141917 to E.A.; National Institutes of Health grant RO1HL127564 to P.N.; Fonds de Recherche Santé - Québec to G.T.; Canadian Institutes for Health Research to J.C.E., G.T.; Heart and Stroke Foundation of Canada to G.T.; National Institutes of Health grant R01HL128550 to G.T.; Kowa Grant to M.A.; the funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosures
C.J.O’D. is employed by Novartis Institutes for Biomedical Research. M.A.R. is employed by Intellia Therapeutics, but completed all work related to this study prior to this employment, while employed by Brigham and Women’s Hospital. P.N. reports grants from Amgen, Apple, AstraZeneca, Boston Scientific, and Novartis, personal fees from Apple, AstraZeneca, Blackstone Life Sciences, Foresite Labs, Genentech/Roche, Novartis, and TenSixteen Bio, equity in geneXwell, TenSixteen Bio, and Zizi, co-founder of TenSixteen Bio, and spousal employment at Vertex, all unrelated to the present work. E.A. is on advisory board for Elastrin Therapeutics Inc. G.T. has received consulting fees from Ionis Pharmaceuticals and has participated in advisory boards for Amgen and Sanofi. The authors declare no other conflicts of interests.
Non-standard Abbreviations and Acronyms:
- DAPI
4′,6-diamidino-2-phenylindole
- Apo(a)
apolipoprotein(a)
- ApoB
apolipoprotein B
- CAVD
calcific aortic valve disease
- DMEM
Dulbecco’s Minimum Essential Medium
- FACS
fluorescence-activated cell sorting
- Lp(a)
lipoprotein(a)
- LC-MS/MS
liquid chromatography-mass spectrometry/mass spectrometry
- LDL
low-density lipoprotein
- LDLR
low-density lipoprotein receptor
- MFSD5
major facilitator superfamily domain containing 5
- NM
normal media
- P/S
penicillin/streptomycin
- PheWAS
phenome-wide association studies
- PBS
phosphate buffered saline
- PM
pro-calcifiying media
- VIC
valvular intersititial cell
- VEC
valvular endothelial cell
- WGA
wheat germ agglutinin
Footnotes
SUPPLEMENTAL MATERIAL
Tables S1, S2, and Major Resources Table
REFERENCES
- 1.Blaser MC, Kraler S, Lüscher TF, Aikawa E. Multi-Omics Approaches to Define Calcific Aortic Valve Disease Pathogenesis. Circulation Research. 2021;128:1371–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yadgir S, Johnson CO, Aboyans V, Adebayo OM, Adedoyin RA, Afarideh M, Alahdab F, Alashi A, Alipour V, Arabloo J. Global Burden of Disease Study 2017 Nonrheumatic Valve Disease Collaborators. Global, Regional, and National Burden of Calcific Aortic Valve and Degenerative Mitral Valve Diseases, 1990–2017. Circulation. 2020;141:1670–1680. [DOI] [PubMed] [Google Scholar]
- 3.Tsimikas S, Stroes ESG. The dedicated “Lp(a) clinic”: a concept whose time has arrived? Atherosclerosis. 2020;300:1–9. [DOI] [PubMed] [Google Scholar]
- 4.Zekavat SM, Ruotsalainen S, Handsaker RE, Alver M, Bloom J, Poterba T, Seed C, Ernst J, Chaffin M, Engreitz J, et al. Deep coverage whole genome sequences and plasma lipoprotein(a) in individuals of European and African ancestries. Nat Commun. 2018;9:2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoekstra M, Chen HY, Rong J, Dufresne L, Yao J, Guo X, Tsai MY, Tsimikas S, Post WS, Vasan RS, Rotter JI, Larson M, Thanassoulis G, Engert JC. Genome-wide association study highlights APOH as a novel locus for lipoprotein(a) levels-brief report. Aterioscler Thromb Vasc Biol. 2021;41:458–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trinder M, Uddin M, Finneran P, Aragam KG, Natarajan P. Clinical utility of lipoprotein(a) and LPA genetic risk score in risk prediction of incident atherosclerotic cardiovascular disease. JAMA Cardiol. 2021;6:287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouchareb R, Mahmut A, Nsaibia MJ, Boulanger MC, Dahou A, Lépine JL, Laflamme MH, Hadji F, Couture C, Trahan S, et al. Autotaxin derived from lipoprotein(a) and valve interstitial cells promotes inflammation and mineralization of the aortic valve. Circulation. 2015;132:677–690. [DOI] [PubMed] [Google Scholar]
- 8.Capoulade R, Torzewski M, Mayr M, Chan KL, Mathieu P, Bossé Y, Dumesnil JG, Tam J, Teo KK, Burnap SA, et al. ApoCIII-Lp(a) complexes in conjunction with Lp(a)-OxPL predict rapid progression of aortic stenosis. Heart. 2020;106:738–745. [DOI] [PubMed] [Google Scholar]
- 9.Schlotter F, de Freitas RCC, Rogers MA, Blaser MC, Wu PJ, Higashi H, Halu A, Iqbal F, Andraski AB, Rodia CN, et al. ApoC-III is a novel inducer of calcification in human aortic valves. J Biol Chem. 2020;296:100193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bartoli-Leonard F, Turner ME, Zimmer J, Chapurlat R, Pham T, Aikawa M, Pradhan AD, Szulc P, Aikawa E. Elevated lipoprotein(a) as a predictor for coronary events in older men. J Lipid Res. 2022;63:100242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reyes-Soffer G, Ginsber HN, Berglund L, Duell PB, Heffron SP, Kamstrip PR, Lloyd-Jones DM, Marcovina SM, Yeang C, Koschinsky ML, American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Radiology and Intervention; and Council on Peripheral Vascular Disease. Lipoprotein(a): a genetically determined, causal, and prevalent risk factor for atherosclerotic cardiovascular disease: a scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol. 2022;42:e48–e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lippi G, Guidi G. Lipoprotein(a): from ancestral benefit to modern pathogen? QJM. 2000;93:75–84. [DOI] [PubMed] [Google Scholar]
- 13.Schneider M, Witztum JL, Young SG, Ludwig EH, Miller ER, Tsimikas S, Curtiss LK, Marcovina SM, Taylor JM, Lawn RM, Innerarity TL, Pitas RE. High-level lipoprotein [a] expression in transgenic mice: evidence for oxidized phospholipids in lipoprotein [a] but not in low density lipoproteins. J Lipid Res. 2005;46:769–78. [DOI] [PubMed] [Google Scholar]
- 14.Kayikcioglu M LDL apheresis and Lp(a) apheresis: a clinician’s perspective. Curr Atheroscler Rep. 2021;23:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Donoghue ML, Fazio S, Giugliano RP, Stroes ESG, Kanevsky E, Gouni-Berthold I, Im K, Pineda AL, Wasserman SM, Ceska R, et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation. 2019;139:1483–1492. [DOI] [PubMed] [Google Scholar]
- 16.Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, Tardif JC, Baum SJ, Steinhagen-Thiessen E, Shapiro MD, Stroes ESG, Moriarty PM, Nordestgaard BG, et al. Lipoprotein(a) reduction in persons with cardiovascular disease. N Engl J Med. 2020;382:244–255. [DOI] [PubMed] [Google Scholar]
- 17.Tsimikas S, Moriarty PM, Stroes ES, Emerging RNA Therapeutics to lower blood levels of Lp(a): JACC focus seminar 2/4. J Am Coll Cardiol. 2021;77:1576–1589. [DOI] [PubMed] [Google Scholar]
- 18.McCormick SPA, Schneider WJ. Lipoprotein(a) catabolism: a case of multiple receptors. Pathology. 2019;51:155–164. [DOI] [PubMed] [Google Scholar]
- 19.Kraft HG, Lingenhel A, Raal FJ, Hohenegger M, Utermann G. Lipoprotein(a) in homozygous familial hypercholesterolemia. Aterioscler Thromb Vasc Biol. 2000;20:522–528. [DOI] [PubMed] [Google Scholar]
- 20.Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, Darius H, Lewis BS, Ophuis TO, Jukema JW, et al. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387–2397. [DOI] [PubMed] [Google Scholar]
- 21.Zheng KH, Tsimikas S, Pawade T, Kroon J, Jenkins WSA, Doris MK, White AC, Timmers NKLM, Hjortnaes J, Rogers MA, et al. Lipoprotein(a) and oxidized phospholipids promote valve calcification in patients with aortic stenosis. J Am Coll Cardiol. 2019;73:2150–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, et al. UK Biobank: An Open Access Resource for Identifying the Causes of a Wide Range of Complex Diseases of Middle and Old Age. PLOS Medicine. 2015;12:e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang CC, Carson CC, Tellier LC, Vattikuti S, Purcell SM, JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Banda Y, Kvale MN, Hoffmann TJ, Hesselson SE, Ranatunga D, Tang H, Sabatti C, Croen LA, Dispensa BP, Hendersen M, et al. Characterizing race/ethnicity and genetic ancestry for 100,000 subjects in the genetic epidemiology research on adult health and aging (GERA) cohort. Genetics. 2015;200:1285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffmann TJ, Kvale MN, Hesselson SE, Zhan Y, Aquino C, Cao Y, Cawley S, Chung E, Connell S, Eshragh J, et al. Next generation genome-wide association tool: design and coverage of a high-throughput European-optimized SNP array. Genomics. 2011;98:79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48:1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K, et al. Haplotype Reference Consortium. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frei AP, Jeon OY, Kilcher S, Moest H, Henning LM, Jost C, Plückthun A, Mercer J, Aebersold R, Carreira EM, Wollscheid B. Direct identification of ligand-receptor interactions on living cells and tissues. Nat Biotechnol. 2012;30:997–1001. [DOI] [PubMed] [Google Scholar]
- 31.Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK, et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017;171:1437–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asano T, Chelvanambi S, Decano JL, Whelan MC, Aikawa E, Aikawa M. In silico drug screening approach using L1000-based connectivity map and its application to COVID-19. Front Cardiovasc Med. 2022;9:842641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nsaibia MH, Boulanger MC, Bouchared R, Mkannez G, Quang KL, Hadji F, Argaud D, Dahou A, Bossé Y, Koschinsky ML, Pibarot P, Arsenault BJ, Marette A, Mathieu P. OxLDL-derived lysophosphatidic acid promotes the progression of aortic valve stenosis through a LPAR1-RhoA-NF-κB. Cardiovasc Res. 2017;113:1351–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017;545:505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tejada-Jimenez M, Galvan A, Fernandez E. Algae and humans share a molybdate transporter. Proc Natl Acad Sci U S A. 2011;108:6420–6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang X, Dai FF, Gaisano G, Giglou K, Han J, Zhang M, Kittanakom S, VWong V, Wei L, Showalter AD, et al. The identification of novel proteins that interact with the GLP-1 receptor and restrain its activity. Mol Endocrinol. 2013;27:1550–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perland E, Lekholm E, Eriksson MM, Bagchi S, Arapi V, Fredriksson R, The putative SLC transporters Mfsd5 and Mfsd11 are abundantly expressed in the mouse brain and have a potential role in energy homeostasis. PloS One. 2016;11:e0156912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schnitzler JG, Hoogeveen RM, Ali L, Prange KHM, Waissi F, van Weeghel M, Bachmann JC, Versloot M, Borrelli MJ, Yeang C, et al. Atherogenic Lipoprotein(a) Increases Vascular Glycolysis, Thereby Facilitating Inflammation and Leukocyte Extravasation. Cir Res. 2020;126:1346–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hjortnaes J, Shapero K, Goettsch C, Hutcheson JD, Keegan J, Kluin J, Mayer JE, Bischoff J, Aikawa E. Valvular interstitial cells suppress calcification of valvular endothelial cells. Atherosclerosis. 2015;242:251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cecoltan S, Ciortan L, Macarie RD, Vadana M, Mihaila AC, Tucureanu M, Vlad ML, Droc I, Gherghiceanu M, Simionescu A, et al. High Glucose Induced Changes in Human VEC Phenotype in a 3D Hydrogel Derived From Cell-Free Native Aortic Root. Front Cardiovasc Med. 2021;8:714573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rogers MA, Atkins SK, Zheng KH, Singh SA, Chelvanambi S, Pham TH, Kuraoka S, Stroes ESG, Aikawa M, Aikawa E, Lipoprotein(a) induces vesicular cardiovascular calcification revealed with single-extracellular vesicle analysis. Front Cardiovasc Med. 2022;9:778919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated or analysed during the current study are indluded in the article and its Supplemental Data. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium by the PRIDE,29 partner repository with the dataset identifier PXD026861. Username: reviewer_pxd026861@ebi.ac.uk; password: uadLa8rZ.