

Abstract
Inflammation is a first responder against injury and infection and is also critical for the regeneration and repair of tissue after injury. The role of professional immune cells in tissue healing is well characterized. Professional immune cells respond to pathogens with humoral and cytotoxic responses; remove cellular debris through efferocytosis; secrete angiogenic cytokines and growth factors to repair the microvasculature and parenchyma. However, non-immune cells are also capable of responding to damage or pathogens. Non-immune somatic cells express pattern recognition receptors (PRRs) to detect pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). The PRRs activation leads to the release of inflammatory cytokines required for tissue defense and repair. Notably, the activation of PRRs also triggers epigenetic changes that promote DNA accessibility and cellular plasticity. Thus, non-immune cells directly respond to the local inflammatory cues and can undergo phenotypic modifications or even cell lineage transitions to facilitate tissue regeneration. This review will focus on the novel role of cell-autonomous inflammatory signaling in mediating cell plasticity, a process which is termed transflammation. We will discuss the regulation of this process by changes in the functions and expression levels of epigenetic modifiers, as well as metabolic and ROS/RNS-mediated epigenetic modulation of DNA accessibility during cell fate transition. We will highlight the recent technological developments in detecting cell plasticity and potential therapeutic applications of transflammation in tissue regeneration.
Keywords: Transflammation, innate immunity, DNA accessibility, epigenetics, metabolism, ROS, RNS, single-cell omics, regeneration
The Discovery of Transflammation
This story of transflammation has its origin in 2006 when Shinyi Yamanaka electrified the field of stem cell biology with his contribution to discovering the induced pluripotent stem cells (iPSCs)1. Dr. Yamanaka used a retroviral vector to forcibly express the transcriptional factors Oct4, Sox2, KLF4, and c-MYC in mouse fibroblasts to induce pluripotency. To avoid the use of an integrating vector in human cells (which could engender safety concerns for regenerative medicine applications), we generated cell-permeable peptides of the Yamanaka factors to enable nuclear reprogramming. Despite evidence that the cell-permeable peptides could enter the nuclei of human fibroblasts, no efficient induction of pluripotency genes was achieved, nor induced pluripotent stem cells were derived. Based on the earlier findings that empty virions could alter the phenotype of human cells2–4, we hypothesized that the retroviral vector used by Yamanaka played a role in reprogramming. To test this hypothesis, in addition to exposing human fibroblasts to our cell-permeant proteins, we added a retroviral vector encoding green fluorescent protein (GFP) and we were stunned to observe an efficient induction of pluripotency genes by this combination. We went on to demonstrate that the retroviral vector used to deliver the Yamanaka factors induced inflammatory signaling that was necessary for pluripotency induction. Specifically, stimulation of toll-like receptor 3 (TLR3) by the retroviral vector activated NFkB and IRF3, which then triggered global changes in the expression of epigenetic modifiers responsible for DNA accessibility5,6. This increase in DNA accessibility presumably gave the Yamanaka factors access to the promoter regions of the pluripotency genes required for iPSC formation. We could increase the generation of iPSCs with inflammatory activation and could abrogate iPSC production by blocking inflammatory activation. Thus, inflammatory signaling is necessary for efficient nuclear reprogramming of fibroblasts to iPSCs, as described in our Cell paper that was published the same month that Yamanaka garnered the Nobel Prize for his work6. The process whereby cell-autonomous inflammatory signaling enhances DNA accessibility to promote phenotypic plasticity has been termed “transflammation”7.
Transflammation has proven to be critical in other cell fate transitions besides nuclear reprogramming of somatic cells to iPSC. We have observed that this process is also involved in transdifferentiation. Transdifferentiation is defined as the process by which one somatic cell directly transforms into another somatic cell lineage without going through the stem-cell stage8.
Our group was the first to show that transdifferentiation of fibroblasts to endothelial cells (Mesenchymal-endothelial transition, MEndoT) can be induced in cell culture or mice in response to limb ischemia, which requires activation of inflammatory signaling6,9–11. In these studies, we focused on the transdifferentiation of fibroblasts to endothelial cells, but it seems likely that the transdifferentiation of any somatic cell to another lineage requires inflammatory signaling. In this conceptual framework, inflammatory signaling increases DNA accessibility to a broader genetic repertoire. This genetic repertoire may encode cell cycle proteins, growth factors, and cytoskeletal proteins, in the case of quiescent cells that are expected to become proliferative and migratory to heal a wound (e.g., keratinocytes at the edge of a cutaneous laceration). The genetic repertoire may include lineage factors to support transdifferentiation to another cell type (as when fibroblasts transdifferentiate into endothelial cells to support angiogenesis). During physiological conditions, the operational efficiency of a cell mandates the repression of genes that are not involved in cell identity or homeostatic processes of that cell lineage. However, when exposed to injury or invading pathogens, the ability of a somatic cell to rapidly change its genetic program and phenotype is adaptive. Thus, activation of PRRs induces cellular plasticity as well as cytokines for defense and regeneration, a parsimonious utilization of inflammatory signaling.
Since our first description of transflammation a decade ago, much has been learned about the process. Firstly, activation of cell surface PRRs such as TLRs or retinoic acid-inducible gene protein (RIG1)-like receptors5 has been confirmed to lead to NFKb and IRF-3 mediated alterations in expression and activities of epigenetic modifiers, thereby promoting DNA accessibility. The activation of transflammation is then associated with an upregulation of histone acetyltransferases (HATs), downregulation of histone deacetylases (HDACs)6, an increase in histone acetylation, and other epigenetic modifications associated with chromatin accessibility (e.g. H3K4me3). In addition, inducible nitric oxide synthase (iNOS) has been found to translocate to the nucleus during transdifferentiation. As described below, iNOS binds to repressive epigenetic modifiers, reducing their binding to and suppressive effect on the chromatin12,13. Metabolic regulation has also been found during transdifferentiation. The work of Lai et al. revealed a glycolytic shift during cell fate transitions that generate more of the substrate for histone acetylation, indicating that metabolism is coupled to epigenetic alterations14. These changes in epigenetic modifiers have the effect of globally altering histone markings to increase DNA accessibility and phenotypic fluidity. Thus, a cellular challenge, such as hypoxia, generates DAMPs which the cell senses, and responds to, with an increase in epigenetic plasticity that permits cellular plasticity and physiological adaptation. Furthermore, the most recent lineage tracing studies have shown evidence of this phenomenon in vivo, in the form of angiogenic transdifferentiation in a mouse hindlimb ischemic revascularization model10. The suppression of innate immune signaling by dexamethasone treatment or p65 conditional knockout in fibroblast cells impeded tissue regeneration which further supported the notion that transflammation is a key factor in cellular reprogramming.
The current understanding of the mechanisms underlying transflammation and cell plasticity is discussed in more detail below. (Figure1)
Figure1.
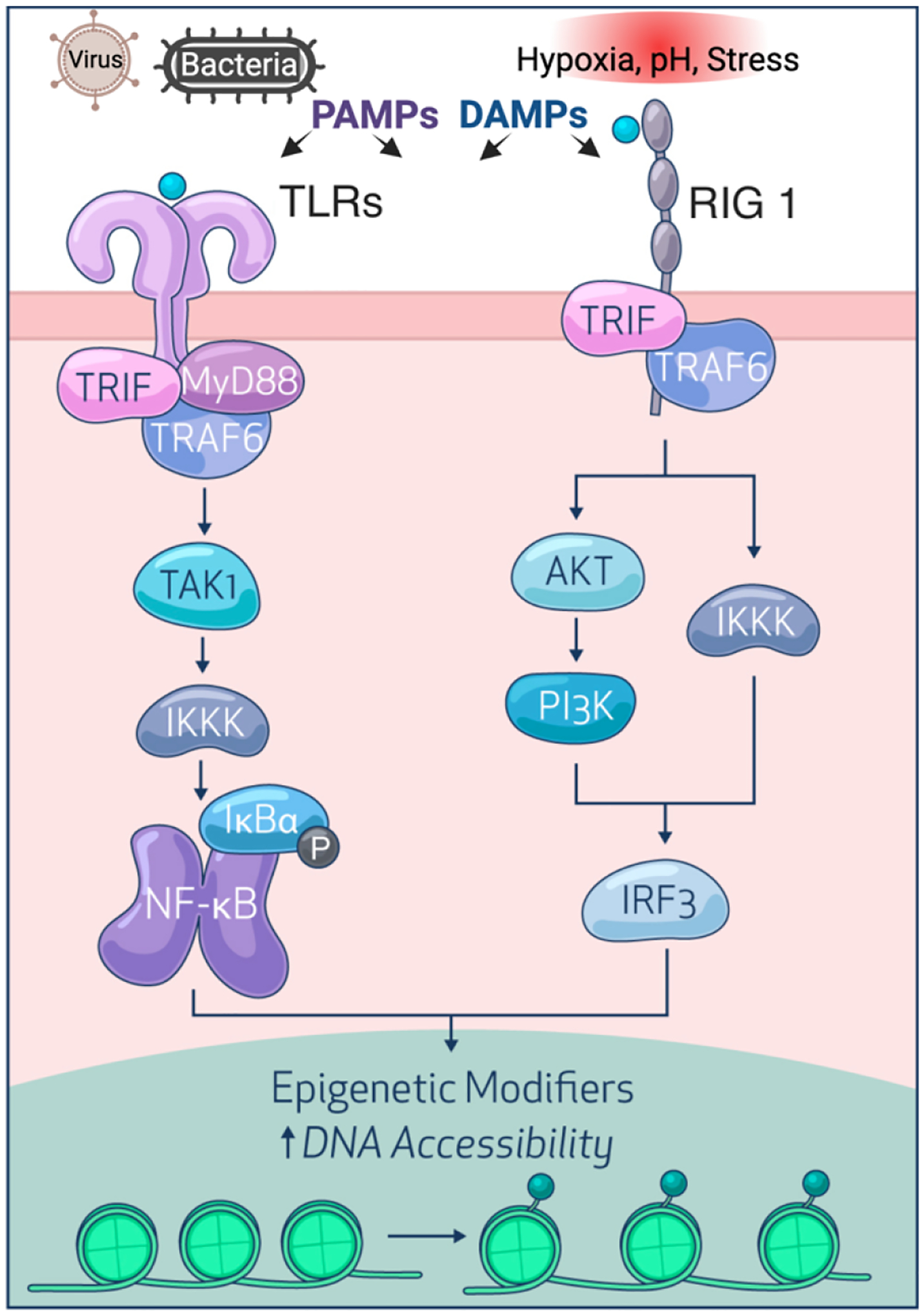
Transflammation. Viruses and bacteria produce PAMPs while hypoxia, pH, stress, and other injurious stimuli generate DAMPs, both of which trigger innate immune signaling by intracellular or transmembrane PRCs such as TLRs and RIG-I. Subsequently, NF-kB or IRF3 are activated and translocated to the nucleus to alter the expression of epigenetic modifiers, thereby increasing DNA accessibility to facilitate the cell fate transition.
Inflammatory signaling in other types of transdifferentiation.
Whereas our work has focused on the role of transflammation in the nuclear reprogramming of fibroblasts to pluripotency, or to their transdifferentiation to endothelial cells or MEndoT, it is likely that the phenomenon of transflammation also applies to other cell fate transitions. Indeed, Dzau and colleagues have shown that activation of innate immunity is required for fibroblast to cardiomyocyte transdifferentiation15. In addition, the maturation of the induced cardiomyocytes requires TLR3-mediated NFKb activity16. These studies suggested that the transdifferentiation of any somatic cell to another lineage may require inflammatory signaling. This response to inflammatory signaling may be a primordial mechanism that provides cells with the ability to respond to a challenge with increased adaptability.
The association of inflammatory signaling with pathological transdifferentiation, i.e. epithelial-mesenchymal transition (EMT) or endothelial-mesenchymal transition (EndoMT), has been described in chronic diseases like cancer17–20 and organ fibrosis21–27. In cancers, the chronic inflammation in the tumor microenvironment causes sustained activation of immune-responsive transcriptional factors (TF)s, including TGF-β and NF-KB, which collaboratively initiate the epithelial transformation and facilitate the dissemination of different cancers18,28–30 by activating the expression of EMT-TFs, such as TWIST, SLUG31, and SNAIL17. Similar mechanisms are reported in EndoMT32–34 which contributes to the production of Cancer-Associated-Fibroblasts (CAFs)35 to further supports tumor growth and metastasis36. Tissue fibrosis is also characterized as a common feature of a variety of chronic pathological processes, affecting major organs including the heart37,38, kidney39,40, liver41–43, and lung44,45, etc. In all cases, inflammation response is considered to play a role in the disease progression46,47. However, the cellular origins of the activated fibroblasts remain controversial48,49. Seminal works have connected pathological transdifferentiation with cardiovascular fibrotic events. Specifically, EMT50,51 and EndoMT52,53 are reported to be involved in the response to cardiac injury, mediated by inflammatory signaling and TGF-β54–56. These mechanisms may also underlie cardiac fibrosis in diabetic57,58 and hypertrophic59,60 conditions. Some forms of cardiac fibrosis may occur in the absence of EndoMT such as post-transverse aortic constriction (TAC)61,62 or myocardial infarction (MI) surgery63–65. Moreover, other forms of transdifferentiation including macrophages to myofibroblasts transition (MMT) are also reported in fibrotic disorders66–68. So, more lineage tracing work in vivo in combine with fate mapping using single-cell sequencing analysis will be needed to confirm the role of pathological transdifferentiation under different disease conditions.
Immunometabolism in cell fate transition
Metabolic pathways provide the energy and building blocks through catabolism and anabolism to meet the diverse demands of cellular processes69,70. Cellular metabolism also provides intermediates as the substrate for epigenetic events that are required for cell fate transition71–75. Several of the tricarboxylic acid metabolites mediate the activities of the chromatin-modifying proteins. An example of a metabolite with an epigenetic role is acetyl-coA76,77. It is generated through the metabolism of several precursors, including fatty acids, acetate, pyruvate, and glutamine. Acetyl-CoA can then be used for lipid synthesis and protein acetylation. In the nucleus, acetyl-CoA is utilized in histone acetylation, a major regulatory process in chromatin configuration and gene expression. Histone acetylation neutralizes the positive charge of the lysine on the histone tail and decreases the interaction between histone and DNA, which makes DNA more accessible for the transcriptional machinary78,79. The enzymes that are responsible for acetyl-CoA synthesis, including ATP-citrate lyase (ACL)80 and acetyl-CoA synthetase (ACSS)81, have become therapeutic targets82 in diseases including cancer83–85 and atherosclerosis86–89 to reduce histone acetylation and cellular activation in those conditions.
Immunometabolism describes the intricate interplay between metabolism and immune cell activation and differentiation, also termed intrinsic immunometabolism. For example, the M2 macrophage relies on β-oxidation. Inhibition of fatty acid oxidation is sufficient to alter macrophage polarization, switching the immune-repressive M2 phenotype to the pro-inflammatory M1 phenotype90–92. Furthermore, the metabolic-epigenetic axis is known to be involved in T cell fate determination, including effector93,94, Treg95–97, memory98,99, and exhausted100,101 T cells. Additionally, studies performed to explore the role of immune cells in systemic metabolism, including obesity102 and diabetes103,104, termed the extrinsic immunometabolism, have further unveiled a crucial role of chronic inflammation in insulin resistance105,106 and other metabolic diseases107–109. However, although many studies have characterized the molecular circuits traversing the reciprocal relationship between inflammation and metabolism, much less attention has been focused on metabolism and inflammatory signaling in non-immune cells. Here, the interaction between inflammatory activation and metabolism appears to be crucial for cell function, cell identity, and cell fate transition.
One example of inflammatory signaling and cell plasticity in non-professional immune cells is the endothelial-to-mesenchymal transition (EndoMT) that contributes to atherosclerosis, pulmonary hypertension, and cardiac fibrosis110,111. In those pathological conditions, the endothelial cells persistently express a high level of pro-inflammatory leukocyte adhesion molecules and growth factors that induce endothelial dysfunction, and which promote a mesenchymal phenotypic switch. Recent studies highlight a role for metabolism, in that fatty acid metabolism provides an essential pool of acetyl-CoA to maintain endothelial cell identity112.
In the transdifferentiation of fibroblasts to endothelial cells11, metabolic-epigenetic coupling plays a critical role14. Specifically, upon TLR3 activation, a rapid Warburg effect is triggered, in which glycolysis exceeds oxidative phosphorylation, coupled with a non-canonical tricarboxylic acid cycle (TCA cycle) in which glucose-derived citrate accumulates and is exported out of the mitochondria through citrate transporter, Slc25a1. Concurrently there is an increase in the expression of ATP-citrate lyase (ACL) in the nucleus, which converts citrate to acetyl-CoA. Acetyl-CoA is the substrate for histone acetylation which increases DNA accessibility to facilitate cellular reprogramming. These observations represented a novel metabolism-driven signaling cascade across mitochondria, cytoplasm, and nucleus, linking metabolism with cell-autonomous inflammatory signaling, epigenetic regulation, and cell plasticity which may serve as a general mechanism in many inflammation-induced cell fate transitions113–116 (Figure2). A similar non-canonical TCA cycle phenomenon was later observed in embryonic stem cells (ESCs) with [U-13C] glucose tracing assay117. In this study, Arnold et al. observed ESCs prefer a non-canonical TCA cycle in which the mitochondrial citrate tends to be shunted into the cytoplasm where it is converted to oxaloacetate and malate. Malate is then transported back to mitochondria through Slc25a1, the citrate/malate antiporter, to replenish mitochondrial oxaloacetate. They documented that the utilization of the non-canonical TCA cycle in naïve ES cells was increased when the cells exited the naïve state. Similarly, during the differentiation from muscle myoblast to myotubes, the utilization of the canonical pathway increased, suggesting a dynamic regulation of metabolism during cell fate transition. Together with the findings from earlier paper14, this non-canonical TCA cycle may play an important role in cell fate transitions.
Figure 2.
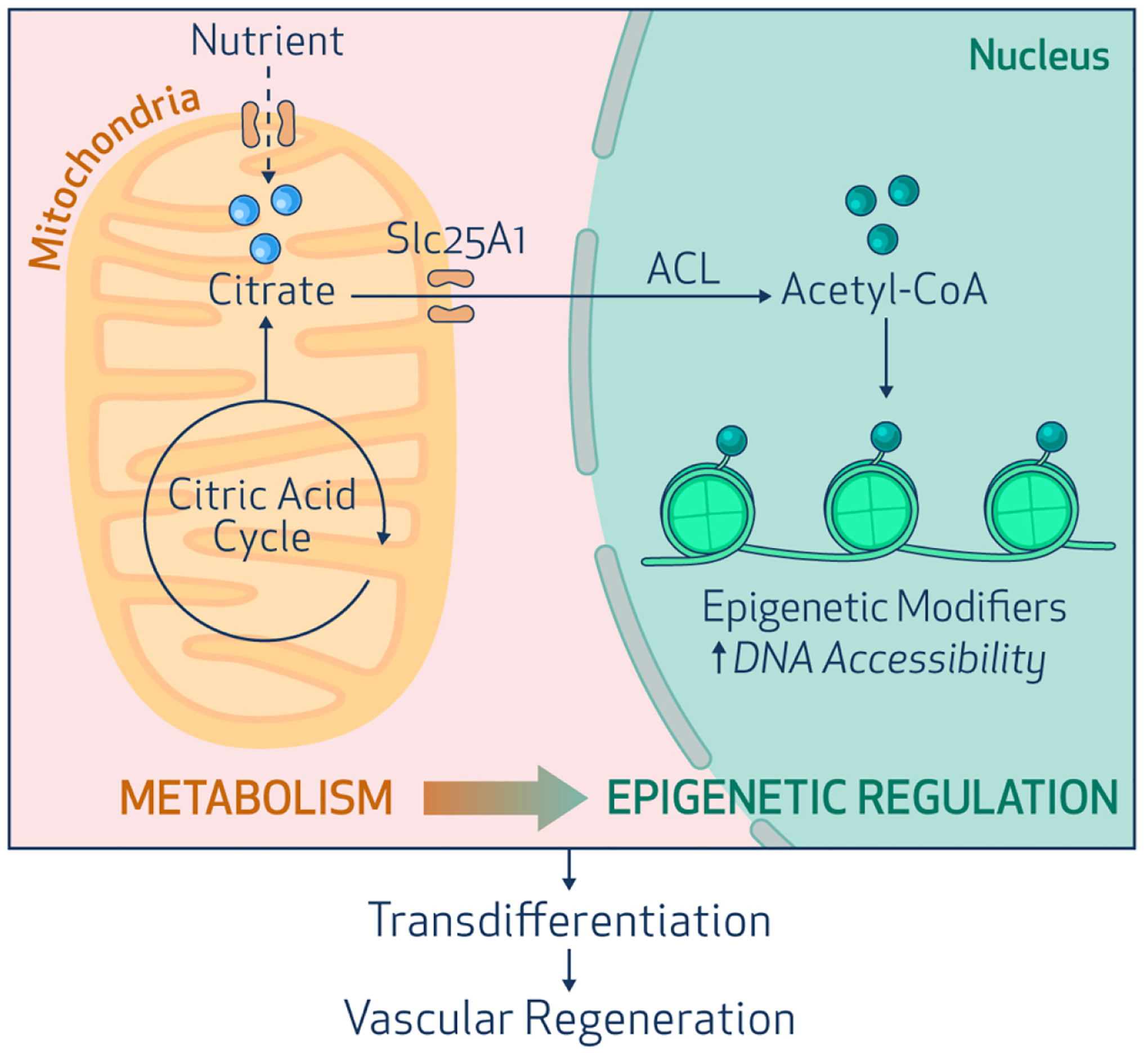
A metabolic-epigenetic axis controls cell fate transition. Innate immune activation induces glycolysis and facilitates mitochondrial export of citrate to the nucleus. In the nucleus, ATP-Citrate Lyase (ACL) converts citrate to acetyl-CoA. There it serves as the substrate for histone acetylation to increase DNA accessibility, and facilitate cell fate transitions, as in vascular regeneration.
The role of ROS/RNS in the cell plasticity
It is well-known that inflammation induces the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS)118–120. These free radicals are key signaling molecules that drive many homeostatic as well as pathophysiological processes121. They also have a prominent impact on the cell fate transition122,123.
ROS plays essential roles in signaling to maintain cellular homeostasis124–126. A low and modest level of ROS is known to be beneficial for survival and proliferation127 in many cells including T cells128,129 and cancer cells130 which normally rely on glycolysis for energy expenditure. Similarly, iPSCs are known to be better cultured under conditions that favor glycolysis131. A study from Gang et. al has observed that ROS production is transiently increased during nuclear reprogramming to generate iPSCs132 using mice embryonic fibroblasts carrying the doxycycline-inducible Yamanaka cassette (Oct4, Sox2, Klf4, and c-Myc). Upon the initiation of nuclear reprogramming, there is a transient spike in ROS generation which subsides at a later phase of the process. Inhibition of ROS generation by knockdown of NADPH oxidase 2 (Nox2), or the use of ROS scavengers, at the onset of nuclear reprogramming, abrogated iPSC formation. Furthermore, this phenomenon is mediated by NF-KB signaling which is in line with our initial finding about the function of innate immune activation during nuclear reprogramming6. The inhibition of NFKB phosphorylation by BAY117085 decreases the early upregulation of Nox and iPSC formation. Later in the process, the generation of ROS subsides, and here the administration of antioxidants enhances reprogramming. Interestingly, the overproduction of ROS by Nox overexpression or a high dose of hydrogen peroxide, even at the initial stage of reprogramming, impairs iPSC formation. These observations suggest that there is an optimal range of ROS generation for effective nuclear reprogramming and are consistent with the Goldilocks’ zone for inflammatory signaling and cell fate plasticity described below.
Reactive nitrogen species are another form of free radical. They are generated through 3 major nitric oxide synthases (NOS), including neuronal isoform (nNOS), endothelial isoform (eNOS), and the inducible isoform, iNOS133. The activation of iNOS is a notable feature of inflammation134. The nitric oxide (NO) generated by NOS enzymes is important for many cellular processes maintaining vascular homeostasis135–137. The reduced expression of eNOS and reduced bioavailability of NO are often associated with cardiovascular diseases including atherosclerosis, hypertension, and aging138,139. The NOS enzymes exert their influence in part by post-translational modification of proteins through S-Nitrosylation140 which was found to occur on epigenetic modifiers during cell fate transitions. Specifically, during the induction of pluripotency through nuclear reprogramming, an increased iNOS expression was observed. Furthermore, iNOS translocated to the nucleus to bind and S-nitrosylates MTA3, which is a component of the nucleosome remodeling and deacetylase (NuRD) complex141. The MTA3 S-nitrosylation is associated with reduced association of the NuRD complex with chromatin; reduced HDAC activity; increased coverage of the chromatin with the active marker H3K27ac and decreased coverage with the repressive marker H3K27me3 on promoter regions of pluripotency genes12. Similarly, iNOS is induced and NO generation is increased, during transdifferentiation of fibroblasts to endothelial cells. Concurrently, S-nitrosylation of polycomb repressive complex protein, RING1A142, was observed, which causes dissociation of the repressive polycomb complex from chromatin to increase epigenetic plasticity13.
Goldilocks zone of inflammatory signaling in regeneration
In view of the many facets of immune signaling in physiological and pathological conditions, it is easy to speculate that the timing and the intensity of inflammatory signaling modulate the downstream effects of immune activation. Indeed, the observations during the induction of pluripotency, or transdifferentiation, strongly suggest a Goldilocks zone for innate immune signaling to activate cellular plasticity and tissue regeneration143. In these studies, a range of doses of TLR3 agonist Poly I: C was used to activate inflammatory signaling during cell fate transition. Distinct outcomes are observed for different doses of the TLR3 agonist, and intermediate doses of Poly I: C (10 to 100 ng/ml) enhances cell fate transitions, whereas doses above this level impair reprogramming efficiency12. This discordance may be partially explained by an overproduction of ROS which was previously observed to impair reprogramming to iPSCs132. Furthermore, during nuclear reprogramming, the repressive epigenetic factor, MTA3, and NuRD complex activity are downregulated with 30 ng/ml Poly I: C, while upregulated with 1000ng/ml Poly I: C. Also, the MNase digestion assay which reflects the DNA accessibility showed the highest mononucleosome to dinucleosome and mononucleosome to trinucleosome ratios in the optimal range of innate immune signaling. These data indicate that there is an optimal dose of inflammatory signaling that can increase DNA accessibility for phenotypic fluidity.
It is also possible that sub-optimal inflammatory signaling fails to orchestrate the energy supply/expenditure as well as the metabolic-epigenetic coupled regulation during cellular reprogramming144. For example, under chronic inflammatory conditions, e.g., atherosclerosis, ACL is highly expressed, and its knockdown generates a more favorable plaque phenotype145. The Goldilocks zone of inflammatory signaling may be reflected in the apparently contradictory observations about the role of inflammation in tissue regeneration. For example, the poor wound healing in the patient who receives intensive steroid therapy146,147 may be due to impaired inflammatory activation of DNA accessibility and regenerative cellular plasticity. In other words, these patients have inadequate stimulation of the epigenetic and metabolic mechanisms required for an open chromatin state and transcriptional activation of regenerative pathways. On the contrary, the patient with a diabetic foot ulcer148,149 may have failed to heal due to excessive inflammatory activation, which may also impair DNA accessibility and cellular plasticity (Figure 3). Understanding how to manipulate inflammatory signaling, with attention to spatiotemporal control, may provide a new avenue in the management of non-healing wounds. In this regard, novel anti-inflammatory drugs have exciting applications for cardiovascular disease150, but there should be a heightened level of concern in using such agents in patients in the setting of surgery or trauma150,151.
Figure 3.
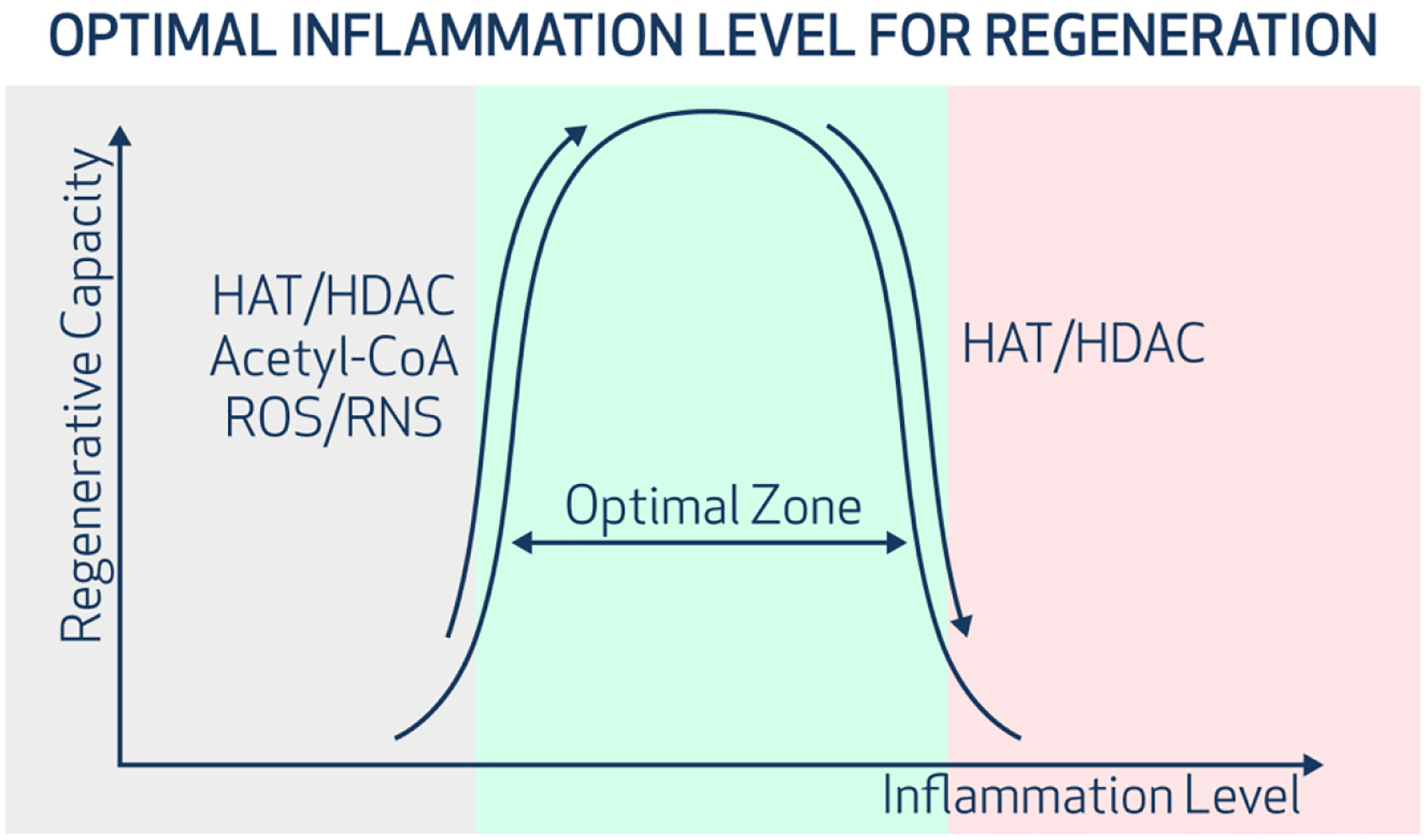
The Goldilocks zone of inflammatory signaling in regeneration. An optimal intensity of inflammatory signaling increases DNA accessibility and cellular plasticity. Insufficient or excessive inflammation reduces DNA accessibility and limits cellular adaptability to injury or stress. HAT/HDAC represents the ratio of gene expression of histone acetyltransferases (HAT) to histone deacetylases (HDAC). A higher HAT/HDAC ratio suggested a more open chromatin and higher DNA accessibility. ROS=reactive oxygen species. RNS = reactive nitrogen species.
New technologies for characterizing mechanisms of cell plasticity
Cell plasticity in response to a pathogenic challenge or injury is required for tissue defense and regeneration. Elucidation of the mechanisms undergirding cell plasticity should lead to major advances in regenerative medicine. The technology of single-cell multi-omics has transformed the field of regenerative medicine. The traditional approaches to identifying cell subpopulations are largely dependent on the low throughput and low-resolution flow cytometry or immunostaining with known cell identity markers. Furthermore, bulk RNA sequencing may dilute rare transcriptional events and cellular subpopulations that are important in regeneration. With the rapid development of single-cell multi-omics and computational analysis tools, genetic and epigenetic information can be profiled for the same cell152, and databases from different experiments can be further integrated153. Now spatiotemporal transcriptomics can profile genetic information on tissue sections with the resolution of a few cells. In combination with single-cell RNA sequencing datasets, single-cell resolution transcriptomics can be generated from tissue slides154.
These advances in technology have led to fruitful discoveries. For example, a single cell RNA seq study in a murine model of experimental myocardial infarction identified an endothelial subpopulation that transiently expresses mesenchymal signatures early after the surgery155. This observation supports the concept of an endothelial-mesenchymal transition which contributes to tissue remodeling and fibrosis post-cardiac ischemia, a concept that has been debated for years155. Another single-cell RNA seq study of endothelial zonation in the mouse brain also finds evidence for the transdifferentiation of endothelial cells to other cell types and the metabolic underpinnings which can be potentially harnessed for therapeutic strategies156. As another example, eight divergent subpopulations of fibroblasts in a model of hindlimb ischemia mouse model are discovered when combining single-cell RNA seq with lineage tracing strategy. Furthermore, with experimental induction of limb ischemia, two of these fibroblast subpopulations (clusters 5 and 8) increased significantly and appeared to contribute to angiogenesis. These clusters were then isolated using specific surface markers, and cultured ex vivo. Cluster 8 generated angiogenic cytokines, whereas cluster 5 expressed some endothelial identity genes, and in Matrigel, formed tubes and expressed endothelial surface markers suggestive of transdifferentiation to an angiogenic phenotype. In the murine ischemic hindlimb model, inhibition of inflammatory signaling markedly reduced the number of these “angiogenic fibroblasts”; impaired wound healing in the ischemic limb; and reduced perfusion recovery10. This study may explain the different responses of fibroblasts to the same stimuli. The ability of a fibroblast to transdifferentiate may be pre-determined by the epigenetic and transcriptional heterogeneity of the fibroblasts. With the unprecedented development of single cell-resolution transcriptome and epitranscriptome profiling and analysis methods157, it will be possible to predict and target the specific sub-clusters that drive the pathophysiological processes.
Single-cell proteomics and metabolomics are the newer members of the single-cell omics field. The single-cell proteomics that characterizes the amount, the post-translational modification, and the kinetics of the thousands of proteins at the same time are complementary to and synergistic with, transcriptomic studies158. This approach mainly includes single-cell barcoding, which is similar to single-cell RNA seq but with a mass spectrometry version (isobaric tags), in a nanoliter-scale reaction system called nanoPOTS for protein lysis, followed by ultra-high-resolution mass spectrometry159. Advances have been made by combining nanoPOTS with laser-capture microdissection, mass spectrometry, and a newly developed computational algorithm, HIT-MAP, which adds spatial information in proteomics studies160. The single-cell metabolomics is the newest single-cell omics technology and provides additional single-cell phenotypic information161. Unlike the previously described omics, single-cell metabolomics doesn’t involve a barcoding process. Instead, matrix-assisted laser desorption/ionization mass spectrometry (MALDI–MS) is integrated with an imaging system to identify single cells. Then the identified cell is irradiated with a UV laser beam to ionize analytes for assessment by mass spectrometry162. These advances in single-cell omics, together with new bioinformatic approaches (such as RNA velocity algorithms) will provide more comprehensive transcriptional, epigenetic, and metabolic profiles to characterize the determinants of cell identity and plasticity. These fundamental insights will no doubt contribute to the development of novel therapeutic avenues.
Conclusion and Discussion
Whereas the role of professional immune cells in tissue repair and regeneration has been well-characterized, non-immune cells are also capable of a response to injury, in part by sensing the molecular patterns presented by pathogens or damaged tissue. Essentially, the stimulation of pattern recognition receptors by damage-associated or pathogen-associated molecular patterns activates inflammatory signaling which triggers a cascade of cellular signaling (mediated by RNS, ROS, and metabolites) to cause changes in epigenetic modifiers that increase DNA accessibility. The cell is now in a state of epigenetic plasticity that permits phenotypic fluidity. However, the trajectory of the transition is dependent upon the milieu. In a setting where ischemia triggers the generation of angiogenic cytokines, a subset of fibroblasts may become endothelial cells. However, it is also possible that, if the ischemia is too severe, or the inflammatory signaling is too profound, the transdifferentiation of fibroblasts to endothelial cells may be abrogated. Indeed, we have shown that there is a Goldilocks zone for inflammatory signaling in cell fate transitions. With excessive inflammatory signaling, the generation of induced pluripotent stem cells from fibroblasts is attenuated. With excessive production of ROS, fibroblast reprogramming to pluripotency is reduced12,132. Furthermore, other factors in the milieu, such as intercellular or tissue-derived signals generated by cytokines, neurohormonal factors, exosomes, or alterations in the composition of the extracellular matrix will contribute to modifying the trajectory and direction of cell fate transitions. For example, in a murine model of myocardial ischemia, activation of the TLR3 pathway by mechanical stimulation releases exosomes containing angiogenic microRNA from endothelial cells to stimulate angiogenesis163,164.
Based on our studies and others, we believe that inflammatory signaling has epigenetic effects to increase DNA accessibility, whereas cell fate is determined by the microenvironment. Differences in the microenvironment may play a critical role in the different responses observed in myocardial versus limb ischemia. Whereas transdifferentiation of cardiac fibroblasts to endothelial cells in the setting of myocardial ischemia is controversial63,64,155,165,166, the contribution of fibroblast transdifferentiation to endothelial cells in the recovery from limb ischemia has been convincingly shown10. This difference may be due to the fact that there is a greater capacity for arteriogenesis and collateralization in the limb in comparison to the heart. In C57BL6J mice, femoral artery ligation reduces limb blood flow by 80%. However, this ischemic challenge triggers a robust arteriogenic and angiogenic response that largely recovers perfusion within 2 to 3 weeks. Conversely, with ligation of the left anterior descending (LAD) coronary artery, there is extensive necrosis and scarring of the myocardium, with little perfusion recovery167 168. The profound ischemic insult of LAD ligation, combined with the ongoing metabolic demand of the working heart, may limit the degree of recovery of the microvasculature.
Ours and others’ recent findings about the role of metabolism and ROS/RNS in the process of transflammation also suggested a new path toward developing therapeutic strategies targeting these pathways to enhance DNA accessibility, cellular plasticity, and tissue repair. For example, pharmacotherapeutics that increase ACL activity and histone acetylation may be useful for tissue regeneration. As another example, spatiotemporal control of iNOS activity and S-nitrosylation may have regenerative medicine applications. As we begin to identify subpopulations of regenerative cells by single-cell omics and the determinants of their fate and function, better markers of tissue health and regeneration will be developed to orchestrate a regenerative response. The single-cell characterization of these subpopulations by transcriptional, epigenetic, proteomic, and metabolomic approaches will lead to novel strategies to target and manipulate those populations to enhance tissue repair and regeneration.
Acknowledgment
We thank Ms. Rachael Whitehead of Houston Methodist Academic Institute for her elegant illustrations.
Funding
This work is supported in part by grants to JPC from the National Institutes of Health (NIH) R01s HL133254, HL157790, and HL148338; as well as the Cancer Prevention and Research Institute of Texas CPRIT RP150611). This work is supported in part by a grant to LL from the National Institutes of Health (NIH) R56HL169204.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
J.P.C. and L.L. are inventors on a pending patent assigned to Houston Methodist Hospital on the use of modulators of O-GlcNAcylation for the treatment of vascular diseases.
References
- 1.Takahashi K & Yamanaka S Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676, doi: 10.1016/j.cell.2006.07.024 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Pant A, Dsouza L & Yang Z Alteration in Cellular Signaling and Metabolic Reprogramming during Viral Infection. mBio 12, e0063521, doi: 10.1128/mBio.00635-21 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thaker SK, Ch’ng J & Christofk HR Viral hijacking of cellular metabolism. BMC Biol 17, 59, doi: 10.1186/s12915-019-0678-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oldstone MB Viral alteration of cell function. Sci Am 261, 42–48, doi: 10.1038/scientificamerican0889-42 (1989). [DOI] [PubMed] [Google Scholar]
- 5.Sayed N et al. Retinoic Acid Inducible Gene 1 Protein (RIG1)-Like Receptor Pathway Is Required for Efficient Nuclear Reprogramming. Stem Cells 35, 1197–1207, doi: 10.1002/stem.2607 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee J et al. Activation of innate immunity is required for efficient nuclear reprogramming. Cell 151, 547–558, doi: 10.1016/j.cell.2012.09.034 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooke JP Therapeutic transdifferentiation: a novel approach for vascular disease. Circ Res 112, 748–750, doi: 10.1161/CIRCRESAHA.113.301053 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jopling C, Boue S & Izpisua Belmonte JC Dedifferentiation, transdifferentiation and reprogramming: three routes to regeneration. Nat Rev Mol Cell Biol 12, 79–89, doi: 10.1038/nrm3043 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Liu C et al. HIF1alpha Regulates Early Metabolic Changes due to Activation of Innate Immunity in Nuclear Reprogramming. Stem Cell Reports 14, 192–200, doi: 10.1016/j.stemcr.2020.01.006 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meng S et al. Reservoir of Fibroblasts Promotes Recovery From Limb Ischemia. Circulation 142, 1647–1662, doi: 10.1161/CIRCULATIONAHA.120.046872 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sayed N et al. Transdifferentiation of human fibroblasts to endothelial cells: role of innate immunity. Circulation 131, 300–309, doi: 10.1161/CIRCULATIONAHA.113.007394 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chanda PK et al. Nuclear S-Nitrosylation Defines an Optimal Zone for Inducing Pluripotency. Circulation 140, 1081–1099, doi: 10.1161/CIRCULATIONAHA.119.042371 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meng S et al. Transdifferentiation Requires iNOS Activation: Role of RING1A S-Nitrosylation. Circ Res 119, e129–e138, doi: 10.1161/CIRCRESAHA.116.308263 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai L, Reineke E, Hamilton DJ & Cooke JP Glycolytic Switch Is Required for Transdifferentiation to Endothelial Lineage. Circulation 139, 119–133, doi: 10.1161/CIRCULATIONAHA.118.035741 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baksh SS, Hu J, Pratt RE, Dzau VJ & Hodgkinson CP Rig1 receptor plays a critical role in cardiac reprogramming via YY1 signaling. Am J Physiol Cell Physiol 324, C843–C855, doi: 10.1152/ajpcell.00402.2022 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hodgkinson CP et al. Cardiomyocyte Maturation Requires TLR3 Activated Nuclear Factor Kappa B. Stem Cells 36, 1198–1209, doi: 10.1002/stem.2833 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stanisavljevic J, Porta-de-la-Riva M, Batlle R, de Herreros AG & Baulida J The p65 subunit of NF-kappaB and PARP1 assist Snail1 in activating fibronectin transcription. J Cell Sci 124, 4161–4171, doi: 10.1242/jcs.078824 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Huber MA et al. NF-kappa B is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. Journal of Clinical Investigation 114, 569–581, doi: 10.1172/Jci200421358 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Q & Verma IM NF-kappaB regulation in the immune system. Nat Rev Immunol 2, 725–734, doi: 10.1038/nri910 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Platel V, Faure S, Corre I & Clere N Endothelial-to-Mesenchymal Transition (EndoMT): Roles in Tumorigenesis, Metastatic Extravasation and Therapy Resistance. J Oncol 2019, 8361945, doi: 10.1155/2019/8361945 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kalluri R & Weinberg RA The basics of epithelial-mesenchymal transition. J Clin Invest 119, 1420–1428, doi: 10.1172/JCI39104 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang S & Susztak K Epithelial Plasticity versus EMT in Kidney Fibrosis. Trends Mol Med 22, 4–6, doi: 10.1016/j.molmed.2015.11.009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu L et al. Epithelial-mesenchymal transition in organ fibrosis development: current understanding and treatment strategies. Burns Trauma 10, tkac011, doi: 10.1093/burnst/tkac011 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rieder F et al. Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am J Pathol 179, 2660–2673, doi: 10.1016/j.ajpath.2011.07.042 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshimatsu Y & Watabe T Emerging roles of inflammation-mediated endothelial-mesenchymal transition in health and disease. Inflamm Regen 42, 9, doi: 10.1186/s41232-021-00186-3 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piera-Velazquez S, Li Z & Jimenez SA Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am J Pathol 179, 1074–1080, doi: 10.1016/j.ajpath.2011.06.001 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nieto MA, Huang RY, Jackson RA & Thiery JP Emt: 2016. Cell 166, 21–45, doi: 10.1016/j.cell.2016.06.028 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Brabletz T, Kalluri R, Nieto MA & Weinberg RA EMT in cancer. Nat Rev Cancer 18, 128–134, doi: 10.1038/nrc.2017.118 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Zavadil J, Haley J, Kalluri R, Muthuswamy SK & Thompson E Epithelial-mesenchymal transition. Cancer Res 68, 9574–9577, doi: 10.1158/0008-5472.CAN-08-2316 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Zhang N et al. Novel therapeutic strategies: targeting epithelial-mesenchymal transition in colorectal cancer. Lancet Oncol 22, e358–e368, doi: 10.1016/S1470-2045(21)00343-0 (2021). [DOI] [PubMed] [Google Scholar]
- 31.Pires BR et al. NF-kappaB Is Involved in the Regulation of EMT Genes in Breast Cancer Cells. PLoS One 12, e0169622, doi: 10.1371/journal.pone.0169622 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adjuto-Saccone M et al. TNF-alpha induces endothelial-mesenchymal transition promoting stromal development of pancreatic adenocarcinoma. Cell Death Dis 12, 649, doi: 10.1038/s41419-021-03920-4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi KJ, Nam JK, Kim JH, Choi SH & Lee YJ Endothelial-to-mesenchymal transition in anticancer therapy and normal tissue damage. Exp Mol Med 52, 781–792, doi: 10.1038/s12276-020-0439-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calon A, Tauriello DV & Batlle E TGF-beta in CAF-mediated tumor growth and metastasis. Semin Cancer Biol 25, 15–22, doi: 10.1016/j.semcancer.2013.12.008 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Yang D, Liu J, Qian H & Zhuang Q Cancer-associated fibroblasts: from basic science to anticancer therapy. Exp Mol Med, doi: 10.1038/s12276-023-01013-0 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Z et al. Metastasis-associated fibroblasts: an emerging target for metastatic cancer. Biomark Res 9, 47, doi: 10.1186/s40364-021-00305-9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Travers JG, Kamal FA, Robbins J, Yutzey KE & Blaxall BC Cardiac Fibrosis: The Fibroblast Awakens. Circ Res 118, 1021–1040, doi: 10.1161/CIRCRESAHA.115.306565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hinderer S & Schenke-Layland K Cardiac fibrosis - A short review of causes and therapeutic strategies. Adv Drug Deliv Rev 146, 77–82, doi: 10.1016/j.addr.2019.05.011 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Huang R, Fu P & Ma L Kidney fibrosis: from mechanisms to therapeutic medicines. Signal Transduct Target Ther 8, 129, doi: 10.1038/s41392-023-01379-7 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li L, Fu H & Liu Y The fibrogenic niche in kidney fibrosis: components and mechanisms. Nat Rev Nephrol 18, 545–557, doi: 10.1038/s41581-022-00590-z (2022). [DOI] [PubMed] [Google Scholar]
- 41.Kisseleva T & Brenner D Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol 18, 151–166, doi: 10.1038/s41575-020-00372-7 (2021). [DOI] [PubMed] [Google Scholar]
- 42.Ramachandran P et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575, 512–+, doi: 10.1038/s41586-019-1631-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hammerich L & Tacke F Hepatic inflammatory responses in liver fibrosis. Nat Rev Gastroenterol Hepatol, doi: 10.1038/s41575-023-00807-x (2023). [DOI] [PubMed] [Google Scholar]
- 44.Wilson MS & Wynn TA Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol 2, 103–121, doi: 10.1038/mi.2008.85 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martinez FJ et al. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers 3, 17074, doi: 10.1038/nrdp.2017.74 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Bhattacharya M & Ramachandran P Immunology of human fibrosis. Nat Immunol, doi: 10.1038/s41590-023-01551-9 (2023). [DOI] [PubMed] [Google Scholar]
- 47.Wynn TA & Ramalingam TR Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nature Medicine 18, 1028–1040, doi: 10.1038/nm.2807 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krenning G, Zeisberg EM & Kalluri R The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol 225, 631–637, doi: 10.1002/jcp.22322 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim J & Braun T Targeting the cellular origin of organ fibrosis. Cell stem cell 16, 3–4, doi: 10.1016/j.stem.2014.12.008 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Lepilina A et al. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell 127, 607–619, doi: 10.1016/j.cell.2006.08.052 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Russell JL et al. A dynamic notch injury response activates epicardium and contributes to fibrosis repair. Circ Res 108, 51–59, doi: 10.1161/CIRCRESAHA.110.233262 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zeisberg EM et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 13, 952–961, doi: 10.1038/nm1613 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Cheng W, Li X, Liu D, Cui C & Wang X Endothelial-to-Mesenchymal Transition: Role in Cardiac Fibrosis. J Cardiovasc Pharmacol Ther 26, 3–11, doi: 10.1177/1074248420952233 (2021). [DOI] [PubMed] [Google Scholar]
- 54.Sanchez-Duffhues G et al. Inflammation induces endothelial-to-mesenchymal transition and promotes vascular calcification through downregulation of BMPR2. J Pathol 247, 333–346, doi: 10.1002/path.5193 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang L et al. Knockout RAGE alleviates cardiac fibrosis through repressing endothelial-to-mesenchymal transition (EndMT) mediated by autophagy. Cell Death Dis 12, 470, doi: 10.1038/s41419-021-03750-4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yoshimatsu Y & Watabe T Roles of TGF-beta signals in endothelial-mesenchymal transition during cardiac fibrosis. Int J Inflam 2011, 724080, doi: 10.4061/2011/724080 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Widyantoro B et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation 121, 2407–2418, doi: 10.1161/CIRCULATIONAHA.110.938217 (2010). [DOI] [PubMed] [Google Scholar]
- 58.Wang M, Li Y, Li S & Lv J Endothelial Dysfunction and Diabetic Cardiomyopathy. Front Endocrinol (Lausanne) 13, 851941, doi: 10.3389/fendo.2022.851941 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teekakirikul P et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. The Journal of clinical investigation 120, 3520–3529, doi: 10.1172/JCI42028 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Illigens BM et al. Vascular Endothelial Growth Factor Prevents Endothelial-to-Mesenchymal Transition in Hypertrophy. Ann Thorac Surg 104, 932–939, doi: 10.1016/j.athoracsur.2017.01.112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peisker F et al. Mapping the cardiac vascular niche in heart failure. Nat Commun 13, 3027, doi: 10.1038/s41467-022-30682-0 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moore-Morris T et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. The Journal of clinical investigation 124, 2921–2934, doi: 10.1172/JCI74783 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li Y, Lui KO & Zhou B Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat Rev Cardiol 15, 445–456, doi: 10.1038/s41569-018-0023-y (2018). [DOI] [PubMed] [Google Scholar]
- 64.Zhang S et al. Seamless Genetic Recording of Transiently Activated Mesenchymal Gene Expression in Endothelial Cells During Cardiac Fibrosis. Circulation 144, 2004–2020, doi: 10.1161/CIRCULATIONAHA.121.055417 (2021). [DOI] [PubMed] [Google Scholar]
- 65.He L et al. Preexisting endothelial cells mediate cardiac neovascularization after injury. The Journal of clinical investigation 127, 2968–2981, doi: 10.1172/JCI93868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Little K et al. Macrophage to myofibroblast transition contributes to subretinal fibrosis secondary to neovascular age-related macular degeneration. J Neuroinflammation 17, 355, doi: 10.1186/s12974-020-02033-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wei J, Xu Z & Yan X The role of the macrophage-to-myofibroblast transition in renal fibrosis. Front Immunol 13, 934377, doi: 10.3389/fimmu.2022.934377 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu H, Guan Q, Zhao P & Li J TGF-beta-induced CCR8 promoted macrophage transdifferentiation into myofibroblast-like cells. Exp Lung Res, 1–14, doi: 10.1080/01902148.2022.2055227 (2022). [DOI] [PubMed] [Google Scholar]
- 69.Ly CH, Lynch GS & Ryall JG A Metabolic Roadmap for Somatic Stem Cell Fate. Cell Metab 31, 1052–1067, doi: 10.1016/j.cmet.2020.04.022 (2020). [DOI] [PubMed] [Google Scholar]
- 70.Zhu J & Thompson CB Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Biol 20, 436–450, doi: 10.1038/s41580-019-0123-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Keating ST & El-Osta A Epigenetics and metabolism. Circ Res 116, 715–736, doi: 10.1161/CIRCRESAHA.116.303936 (2015). [DOI] [PubMed] [Google Scholar]
- 72.Lu C & Thompson CB Metabolic regulation of epigenetics. Cell Metab 16, 9–17, doi: 10.1016/j.cmet.2012.06.001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tatapudy S, Aloisio F, Barber D & Nystul T Cell fate decisions: emerging roles for metabolic signals and cell morphology. EMBO Rep 18, 2105–2118, doi: 10.15252/embr.201744816 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ye C & Tu BP Sink into the Epigenome: Histones as Repositories That Influence Cellular Metabolism. Trends Endocrinol Metab 29, 626–637, doi: 10.1016/j.tem.2018.06.002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baker SA & Rutter J Metabolites as signalling molecules. Nat Rev Mol Cell Bio 24, 355–374, doi: 10.1038/s41580-022-00572-w (2023). [DOI] [PubMed] [Google Scholar]
- 76.Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F & Kroemer G Acetyl coenzyme A: a central metabolite and second messenger. Cell metabolism 21, 805–821, doi: 10.1016/j.cmet.2015.05.014 (2015). [DOI] [PubMed] [Google Scholar]
- 77.Sivanand S, Viney I & Wellen KE Spatiotemporal Control of Acetyl-CoA Metabolism in Chromatin Regulation. Trends Biochem Sci 43, 61–74, doi: 10.1016/j.tibs.2017.11.004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Morgan MAJ & Shilatifard A Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nature genetics 52, 1271–1281, doi: 10.1038/s41588-020-00736-4 (2020). [DOI] [PubMed] [Google Scholar]
- 79.Bannister AJ & Kouzarides T Regulation of chromatin by histone modifications. Cell research 21, 381–395, doi: 10.1038/cr.2011.22 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wellen KE et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080, doi: 10.1126/science.1164097 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mews P et al. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 546, 381–386, doi: 10.1038/nature22405 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Palsson-McDermott EM & O’Neill LAJ Targeting immunometabolism as an anti-inflammatory strategy. Cell research 30, 300–314, doi: 10.1038/s41422-020-0291-z (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Miller KD et al. Targeting ACSS2 with a Transition-State Mimetic Inhibits Triple-Negative Breast Cancer Growth. Cancer Res 81, 1252–1264, doi: 10.1158/0008-5472.CAN-20-1847 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schug ZT et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27, 57–71, doi: 10.1016/j.ccell.2014.12.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zaidi N, Swinnen JV & Smans K ATP-citrate lyase: a key player in cancer metabolism. Cancer Res 72, 3709–3714, doi: 10.1158/0008-5472.CAN-11-4112 (2012). [DOI] [PubMed] [Google Scholar]
- 86.Feng X, Zhang L, Xu S & Shen AZ ATP-citrate lyase (ACLY) in lipid metabolism and atherosclerosis: An updated review. Prog Lipid Res 77, 101006, doi: 10.1016/j.plipres.2019.101006 (2020). [DOI] [PubMed] [Google Scholar]
- 87.Pinkosky SL et al. Liver-specific ATP-citrate lyase inhibition by bempedoic acid decreases LDL-C and attenuates atherosclerosis. Nat Commun 7, 13457, doi: 10.1038/ncomms13457 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pinkosky SL, Groot PHE, Lalwani ND & Steinberg GR Targeting ATP-Citrate Lyase in Hyperlipidemia and Metabolic Disorders. Trends Mol Med 23, 1047–1063, doi: 10.1016/j.molmed.2017.09.001 (2017). [DOI] [PubMed] [Google Scholar]
- 89.Holm H et al. Mendelian Randomization Study of ACLY and Cardiovascular Disease. The New England journal of medicine 383, e50, doi: 10.1056/NEJMc1908496 (2020). [DOI] [PubMed] [Google Scholar]
- 90.Johnson AR et al. Metabolic reprogramming through fatty acid transport protein 1 (FATP1) regulates macrophage inflammatory potential and adipose inflammation. Mol Metab 5, 506–526, doi: 10.1016/j.molmet.2016.04.005 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nomura M et al. Fatty acid oxidation in macrophage polarization. Nat Immunol 17, 216–217, doi: 10.1038/ni.3366 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wculek SK, Dunphy G, Heras-Murillo I, Mastrangelo A & Sancho D Metabolism of tissue macrophages in homeostasis and pathology. Cell Mol Immunol 19, 384–408, doi: 10.1038/s41423-021-00791-9 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Desdin-Mico G et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 368, 1371–1376, doi: 10.1126/science.aax0860 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chapman NM, Boothby MR & Chi H Metabolic coordination of T cell quiescence and activation. Nat Rev Immunol 20, 55–70, doi: 10.1038/s41577-019-0203-y (2020). [DOI] [PubMed] [Google Scholar]
- 95.Kishore M et al. Regulatory T Cell Migration Is Dependent on Glucokinase-Mediated Glycolysis. Immunity 48, 831–832, doi: 10.1016/j.immuni.2018.03.034 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.van Loosdregt J et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood 115, 965–974, doi: 10.1182/blood-2009-02-207118 (2010). [DOI] [PubMed] [Google Scholar]
- 97.Peng M et al. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 354, 481–484, doi: 10.1126/science.aaf6284 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gubser PM et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol 14, 1064–1072, doi: 10.1038/ni.2687 (2013). [DOI] [PubMed] [Google Scholar]
- 99.Raud B et al. Etomoxir Actions on Regulatory and Memory T Cells Are Independent of Cpt1a-Mediated Fatty Acid Oxidation. Cell metabolism 28, 504–515 e507, doi: 10.1016/j.cmet.2018.06.002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fisicaro P et al. Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nature medicine 23, 327–336, doi: 10.1038/nm.4275 (2017). [DOI] [PubMed] [Google Scholar]
- 101.Scharping NE et al. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 45, 374–388, doi: 10.1016/j.immuni.2016.07.009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Russo L & Lumeng CN Properties and functions of adipose tissue macrophages in obesity. Immunology 155, 407–417, doi: 10.1111/imm.13002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Buzzetti R, Zampetti S & Maddaloni E Adult-onset autoimmune diabetes: current knowledge and implications for management. Nature reviews. Endocrinology 13, 674–686, doi: 10.1038/nrendo.2017.99 (2017). [DOI] [PubMed] [Google Scholar]
- 104.Velloso LA, Eizirik DL & Cnop M Type 2 diabetes mellitus--an autoimmune disease? Nature reviews. Endocrinology 9, 750–755, doi: 10.1038/nrendo.2013.131 (2013). [DOI] [PubMed] [Google Scholar]
- 105.Sam S & Mazzone T Adipose tissue changes in obesity and the impact on metabolic function. Translational research : the journal of laboratory and clinical medicine 164, 284–292, doi: 10.1016/j.trsl.2014.05.008 (2014). [DOI] [PubMed] [Google Scholar]
- 106.Brestoff JR et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature 519, 242–246, doi: 10.1038/nature14115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Palomino-Segura M & Hidalgo A Circadian immune circuits. J Exp Med 218, doi: 10.1084/jem.20200798 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Keller M et al. A circadian clock in macrophages controls inflammatory immune responses. Proc Natl Acad Sci U S A 106, 21407–21412, doi: 10.1073/pnas.0906361106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Downton P et al. Chronic inflammatory arthritis drives systemic changes in circadian energy metabolism. Proceedings of the National Academy of Sciences of the United States of America 119, e2112781119, doi: 10.1073/pnas.2112781119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Alvandi Z & Bischoff J Endothelial-Mesenchymal Transition in Cardiovascular Disease. Arteriosclerosis, thrombosis, and vascular biology 41, 2357–2369, doi: 10.1161/ATVBAHA.121.313788 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kovacic JC et al. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. Journal of the American College of Cardiology 73, 190–209, doi: 10.1016/j.jacc.2018.09.089 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xiong J et al. A Metabolic Basis for Endothelial-to-Mesenchymal Transition. Mol Cell 69, 689–698 e687, doi: 10.1016/j.molcel.2018.01.010 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fanucchi S, Dominguez-Andres J, Joosten LAB, Netea MG & Mhlanga MM The Intersection of Epigenetics and Metabolism in Trained Immunity. Immunity 54, 32–43, doi: 10.1016/j.immuni.2020.10.011 (2021). [DOI] [PubMed] [Google Scholar]
- 114.Zhang X, Liu J & Cao X Metabolic control of T-cell immunity via epigenetic mechanisms. Cell Mol Immunol 15, 203–205, doi: 10.1038/cmi.2017.115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Noe JT et al. Lactate supports a metabolic-epigenetic link in macrophage polarization. Sci Adv 7, eabi8602, doi: 10.1126/sciadv.abi8602 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lauterbach MA et al. Toll-like Receptor Signaling Rewires Macrophage Metabolism and Promotes Histone Acetylation via ATP-Citrate Lyase. Immunity 51, 997–1011 e1017, doi: 10.1016/j.immuni.2019.11.009 (2019). [DOI] [PubMed] [Google Scholar]
- 117.Arnold PK et al. A non-canonical tricarboxylic acid cycle underlies cellular identity. Nature 603, 477–481, doi: 10.1038/s41586-022-04475-w (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q & Griendling KK Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circulation research 122, 877–902, doi: 10.1161/CIRCRESAHA.117.311401 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li Y, Deng SL, Lian ZX & Yu K Roles of Toll-Like Receptors in Nitroxidative Stress in Mammals. Cells 8, doi: 10.3390/cells8060576 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yang CS et al. TLR3-triggered reactive oxygen species contribute to inflammatory responses by activating signal transducer and activator of transcription-1. J Immunol 190, 6368–6377, doi: 10.4049/jimmunol.1202574 (2013). [DOI] [PubMed] [Google Scholar]
- 121.Zarkovic N Roles and Functions of ROS and RNS in Cellular Physiology and Pathology. Cells 9, doi: 10.3390/cells9030767 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yermolaieva O, Brot N, Weissbach H, Heinemann SH & Hoshi T Reactive oxygen species and nitric oxide mediate plasticity of neuronal calcium signaling. Proceedings of the National Academy of Sciences of the United States of America 97, 448–453, doi: 10.1073/pnas.97.1.448 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Matrone G et al. Nuclear S-nitrosylation impacts tissue regeneration in zebrafish. Nat Commun 12, 6282, doi: 10.1038/s41467-021-26621-0 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Morris G, Gevezova M, Sarafian V & Maes M Redox regulation of the immune response. Cell Mol Immunol 19, 1079–1101, doi: 10.1038/s41423-022-00902-0 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang K et al. Redox homeostasis: the linchpin in stem cell self-renewal and differentiation. Cell Death Dis 4, e537, doi: 10.1038/cddis.2013.50 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Panieri E & Santoro MM ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Dis 7, e2253, doi: 10.1038/cddis.2016.105 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sena LA et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236, doi: 10.1016/j.immuni.2012.10.020 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Belikov AV, Schraven B & Simeoni L T cells and reactive oxygen species. J Biomed Sci 22, 85, doi: 10.1186/s12929-015-0194-3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Peng HY et al. Metabolic Reprogramming and Reactive Oxygen Species in T Cell Immunity. Front Immunol 12, 652687, doi: 10.3389/fimmu.2021.652687 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liou GY & Storz P Reactive oxygen species in cancer. Free Radic Res 44, 479–496, doi: 10.3109/10715761003667554 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wu J, Ocampo A & Belmonte JCI Cellular Metabolism and Induced Pluripotency. Cell 166, 1371–1385, doi: 10.1016/j.cell.2016.08.008 (2016). [DOI] [PubMed] [Google Scholar]
- 132.Zhou G, Meng S, Li Y, Ghebre YT & Cooke JP Optimal ROS Signaling Is Critical for Nuclear Reprogramming. Cell Rep 15, 919–925, doi: 10.1016/j.celrep.2016.03.084 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Griendling KK et al. Measurement of Reactive Oxygen Species, Reactive Nitrogen Species, and Redox-Dependent Signaling in the Cardiovascular System: A Scientific Statement From the American Heart Association. Circulation research 119, e39–75, doi: 10.1161/RES.0000000000000110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tripathi P, Tripathi P, Kashyap L & Singh V The role of nitric oxide in inflammatory reactions. FEMS Immunol Med Microbiol 51, 443–452, doi: 10.1111/j.1574-695X.2007.00329.x (2007). [DOI] [PubMed] [Google Scholar]
- 135.Huwiler A & Pfeilschifter J Recuperation of Vascular Homeostasis. Circulation research 129, 237–239, doi: 10.1161/CIRCRESAHA.121.319558 (2021). [DOI] [PubMed] [Google Scholar]
- 136.Gheibi S, Jeddi S, Kashfi K & Ghasemi A Regulation of vascular tone homeostasis by NO and H2S: Implications in hypertension. Biochem Pharmacol 149, 42–59, doi: 10.1016/j.bcp.2018.01.017 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tousoulis D, Kampoli AM, Tentolouris C, Papageorgiou N & Stefanadis C The role of nitric oxide on endothelial function. Curr Vasc Pharmacol 10, 4–18, doi: 10.2174/157016112798829760 (2012). [DOI] [PubMed] [Google Scholar]
- 138.Chen JY et al. Nitric oxide bioavailability dysfunction involves in atherosclerosis. Biomed Pharmacother 97, 423–428, doi: 10.1016/j.biopha.2017.10.122 (2018). [DOI] [PubMed] [Google Scholar]
- 139.Walsh T, Donnelly T & Lyons D Impaired endothelial nitric oxide bioavailability: a common link between aging, hypertension, and atherogenesis? J Am Geriatr Soc 57, 140–145, doi: 10.1111/j.1532-5415.2008.02051.x (2009). [DOI] [PubMed] [Google Scholar]
- 140.Hess DT & Stamler JS Regulation by S-nitrosylation of protein post-translational modification. The Journal of biological chemistry 287, 4411–4418, doi: 10.1074/jbc.R111.285742 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Burgold T et al. The Nucleosome Remodelling and Deacetylation complex suppresses transcriptional noise during lineage commitment. The EMBO journal 38, doi: 10.15252/embj.2018100788 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.van Kruijsbergen I, Hontelez S & Veenstra GJ Recruiting polycomb to chromatin. Int J Biochem Cell Biol 67, 177–187, doi: 10.1016/j.biocel.2015.05.006 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Abnave P & Ghigo E Role of the immune system in regeneration and its dynamic interplay with adult stem cells. Semin Cell Dev Biol 87, 160–168, doi: 10.1016/j.semcdb.2018.04.002 (2019). [DOI] [PubMed] [Google Scholar]
- 144.Aloysius A, Saxena S & Seifert AW Metabolic regulation of innate immune cell phenotypes during wound repair and regeneration. Curr Opin Immunol 68, 72–82, doi: 10.1016/j.coi.2020.10.012 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Baardman J et al. Macrophage ATP citrate lyase deficiency stabilizes atherosclerotic plaques. Nat Commun 11, 6296, doi: 10.1038/s41467-020-20141-z (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wang AS, Armstrong EJ & Armstrong AW Corticosteroids and wound healing: clinical considerations in the perioperative period. Am J Surg 206, 410–417, doi: 10.1016/j.amjsurg.2012.11.018 (2013). [DOI] [PubMed] [Google Scholar]
- 147.Barnard AR, Regan M, Burke FD, Chung KC & Wilgis EF Wound healing with medications for rheumatoid arthritis in hand surgery. ISRN Rheumatol 2012, 251962, doi: 10.5402/2012/251962 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rehak L et al. The Immune-Centric Revolution in the Diabetic Foot: Monocytes and Lymphocytes Role in Wound Healing and Tissue Regeneration-A Narrative Review. J Clin Med 11, doi: 10.3390/jcm11030889 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tuttolomondo A, Maida C & Pinto A Diabetic foot syndrome: Immune-inflammatory features as possible cardiovascular markers in diabetes. World J Orthop 6, 62–76, doi: 10.5312/wjo.v6.i1.62 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ridker PM et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. The New England journal of medicine 377, 1119–1131, doi: 10.1056/NEJMoa1707914 (2017). [DOI] [PubMed] [Google Scholar]
- 151.Cooke JP Inflammation and Its Role in Regeneration and Repair. Circulation research 124, 1166–1168, doi: 10.1161/CIRCRESAHA.118.314669 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ogbeide S, Giannese F, Mincarelli L & Macaulay IC Into the multiverse: advances in single-cell multiomic profiling. Trends in genetics : TIG 38, 831–843, doi: 10.1016/j.tig.2022.03.015 (2022). [DOI] [PubMed] [Google Scholar]
- 153.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821, doi: 10.1016/j.cell.2019.05.031 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wei R et al. Spatial charting of single-cell transcriptomes in tissues. Nat Biotechnol, doi: 10.1038/s41587-022-01233-1 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tombor LS et al. Single cell sequencing reveals endothelial plasticity with transient mesenchymal activation after myocardial infarction. Nature communications 12, 681, doi: 10.1038/s41467-021-20905-1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pasut A, Becker LM, Cuypers A & Carmeliet P Endothelial cell plasticity at the single-cell level. Angiogenesis 24, 311–326, doi: 10.1007/s10456-021-09797-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Li S et al. A relay velocity model infers cell-dependent RNA velocity. Nat Biotechnol, doi: 10.1038/s41587-023-01728-5 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Perkel JM Single-cell proteomics takes centre stage. Nature 597, 580–582, doi: 10.1038/d41586-021-02530-6 (2021). [DOI] [PubMed] [Google Scholar]
- 159.Cong Y et al. Ultrasensitive single-cell proteomics workflow identifies >1000 protein groups per mammalian cell. Chem Sci 12, 1001–1006, doi: 10.1039/d0sc03636f (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Guo G et al. Automated annotation and visualisation of high-resolution spatial proteomic mass spectrometry imaging data using HIT-MAP. Nature communications 12, 3241, doi: 10.1038/s41467-021-23461-w (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Seydel C Single-cell metabolomics hits its stride. Nature methods 18, 1452–1456, doi: 10.1038/s41592-021-01333-x (2021). [DOI] [PubMed] [Google Scholar]
- 162.Guo S, Zhang C & Le A The limitless applications of single-cell metabolomics. Curr Opin Biotechnol 71, 115–122, doi: 10.1016/j.copbio.2021.07.015 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Graber M et al. Cardiac Shockwave Therapy - A Novel Therapy for Ischemic Cardiomyopathy? Front Cardiovasc Med 9, 875965, doi: 10.3389/fcvm.2022.875965 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Holfeld J et al. Toll-like receptor 3 signalling mediates angiogenic response upon shock wave treatment of ischaemic muscle. Cardiovascular research 109, 331–343, doi: 10.1093/cvr/cvv272 (2016). [DOI] [PubMed] [Google Scholar]
- 165.Ubil E et al. Mesenchymal-endothelial transition contributes to cardiac neovascularization. Nature 514, 585–590, doi: 10.1038/nature13839 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Dong W et al. Mesenchymal-endothelial transition-derived cells as a potential new regulatory target for cardiac hypertrophy. Sci Rep 10, 6652, doi: 10.1038/s41598-020-63671-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wang G et al. Role of Endothelial and Mesenchymal Cell Transitions in Heart Failure and Recovery Thereafter. Front Genet 11, 609262, doi: 10.3389/fgene.2020.609262 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Muthuramu I, Lox M, Jacobs F & De Geest B Permanent ligation of the left anterior descending coronary artery in mice: a model of post-myocardial infarction remodelling and heart failure. J Vis Exp, doi: 10.3791/52206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]