

Abstract
Metalloradical catalysis (MRC) exploits the metal-centred radicals present in open-shell metal complexes as one-electron catalysts for the generation of metal-stabilized organic radicals–key intermediates that control subsequent one-electron homolytic reactions. Cobalt(II) complexes of porphyrins, as stable 15e-metalloradicals with a well-defined low-spin d7 configuration, have dominated the ongoing development of MRC. Here, to broaden MRC beyond the use of Co(II)-based metalloradical catalysts, we describe systematic studies that establish the operation of Fe(III)-based MRC and demonstrate an initial application for asymmetric radical transformations. Specifically, we report that five-coordinate iron(III) complexes of porphyrins with an axial ligand, which represent another family of stable 15e-metalloradicals with a d5 configuration, are potent metalloradical catalysts for olefin cyclopropanation with different classes of diazo compounds via a stepwise radical mechanism. This work lays a foundation and mechanistic blueprint for future exploration of Fe(III)-based MRC towards the discovery of diverse stereoselective radical reactions.
One-electron radical chemistry has attracted growing attention in organic synthesis due to its distinct reactivity compared to traditional two-electron polar chemistry1,2. Controlling the stereoselectivity, especially enantioselectivity, of radical reactions has long been a formidable challenge3,4. Among recent advances5–8, metalloradical catalysis (MRC) offers a conceptually different approach, achieving control over reactivity and stereoselectivity through the catalytic generation of metal-stabilized organic radicals as key intermediates governing subsequent homolytic radical reactions9–21. Co(II) complexes of porphyrins ([Co(Por)])–stable 15e-metalloradicals–have been demonstrated as effective MRC catalysts, involving fundamentally distinct α-Co(III)-alkyl and α-Co(III)-aminyl radical intermediates. Introducing D2-symmetric chiral amidoporphyrins as supporting ligands, Co(II)-based MRC has successfully enabled various asymmetric radical reactions, including C=C bond cyclopropanation and aziridination, and C–H bond alkylation and amination, with precise control over reactivity and stereoselectivity22–25. Expanding beyond Co(II)-based systems, we have explored other open-shell transition-metal complexes, in particular five-coordinate Fe(III) complexes of porphyrins ([Fe(Por)X]), as potential metalloradical catalysts for MRC. The low toxicity, abundance and cost-effectiveness of iron make Fe(III)-based metalloradical catalysts attractive, offering opportunities to explore diverse catalytic processes for stereoselective radical transformations.
Although the air-sensitive Fe(II) complexes of porphyrins have been studied extensively26–28, stable Fe(III) complexes of porphyrins have not been unambiguously established as genuine catalysts for catalytic transformations in metallocarbene systems, especially in radical pathways29,30. Although MRC has been primarily demonstrated using low-spin d7 [Co(Por)] (Fig. 1a), we questioned whether [Fe(Por)X], with the well-known spin-crossover of the d5 configuration, could also operate as potential metalloradical catalysts. Our focus was on investigating [Fe(Por)X] for olefin cyclopropanation with diazo compounds, a catalytic process previously presumed to proceed via in situ-reduced Fe(II) complexes and a concerted mechanism (Fig. 1b)29–44. We envisioned a stepwise radical mechanism for Fe(III)-catalysed cyclopropanation (Fig. 1c), generating trifluoromethyl-substituted cyclopropanes of great medicinal-chemistry interest (Supplementary Fig. 1)45–48. However, the ability of [Fe(Por)X] to homolytically activate diazo reagent 1′ and generate α-Fe(IV)-alkyl radical intermediate I remained unknown, along with crucial mechanistic considerations. Could α-Fe(IV)-alkyl radicals I serve as a catalytic intermediate for radical addition to substrate alkenes 2, and would the resulting γ-Fe(IV)-alkyl radicals II undergo intramolecular radical substitution via 3-exo-tet radical cyclization to deliver the cyclopropane products? Moreover, the diastereoselectivity and enantioselectivity controls of the radical transformation by Fe(III)-MRC have remained open questions. Drawing inspiration from Co(II)-MRC development, we anticipated D2-symmetric chiral amidoporphyrins to function as an effective ligand platform to support an Fe(III)-based metalloradical system and govern the stereochemical course of the proposed radical cyclopropanation.
Fig. 1 |. Proposed stepwise radical mechanism for cyclopropanation of alkenes with in situ-generated α-trifluoromethyldiazomethane via Fe(III)-based metalloradical catalysis.
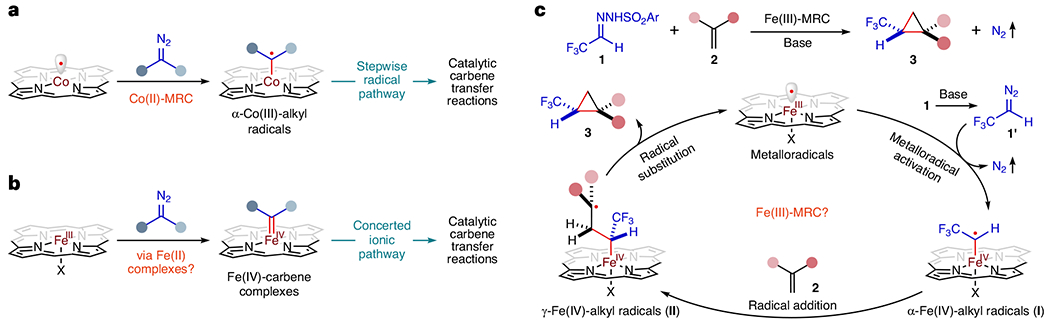
a, Co(II)-based metalloradical catalysis has emerged as a general catalytic approach for controlling the reactivity and selectivity of homolytic radical reactions. As stable 15e-metalloradicals, cobalt(II) complexes of porphyrins have been demonstrated with the capability of activating diazo compounds to generate α-Co(III)-alkyl radicals that can function as kinetically competent intermediates for catalytic carbene transfer reactions through a stepwise radical pathway. b, Iron(III) complexes of porphyrins have not been unambiguously established as genuine catalysts for catalytic transformations. They have been commonly assumed to be in situ-reduced to Fe(II) complexes as the actual catalysts for catalytic carbene transfer reactions through a concerted ionic pathway via metallocarbene intermediates. c, As another family of stable 15e-metalloradicals, Fe(III) complexes of porphyrins might have the potential to function as one-electron catalysts to homolytically activate diazo compounds such as in situ-generated α-trifluoromethyldiazomethane 1′ for catalytic radical cyclopropanation of alkenes 2 to form trifluoromethyl-substituted cyclopropanes 3. During this proposed Fe(III)-based metalloradical catalysis, the initially generated α-Fe(IV)-alkyl radical intermediates I could undergo radical addition to alkenes 2 to form γ-Fe(IV)-alkyl radicals II that would subsequently proceed via intramolecular radical substitution to deliver the cyclopropane products 3 while regenerating the Fe(III)-metalloradical catalysts.
Metal-catalysed cyclopropanation of alkenes with α-trifluoromethyldiazomethane allows for the synthesis of trifluoromethyl-substituted cyclopropanes49–52. Although some asymmetric catalytic systems have been reported53–61, their scope, yields and enantioselectivities were limited. Simonneaux and colleagues reported the first asymmetric catalytic systems based on Fe(III) and Ru(II) complexes of chiral porphyrins for the cyclopropanation of styrenes with pre-prepared α-trifluoromethyldiazomethane, furnishing three examples of trifluoromethyl-substituted cyclopropanes with excellent diastereoselectivities but only in low yields with moderate enantioselectivities40. In this report, the corresponding Fe(II) complex, which was in situ-generated from the reduction of the original Fe(III) complex with excess amounts of cobaltocene, was believed to be the real catalyst that catalyses the cyclopropanation process via a proposed addition mechanism involving a metallocarbene intermediate40. These previous systems utilized various protocols to handle α-trifluoromethyldiazomethane, including the use of the toxic and thermally unstable gas. To address these challenges, in this Article we report the successful development of an Fe(III)-based catalytic system using in situ generation of α-trifluoromethyldiazomethane from bench-stable hydrazone precursors under basic conditions. This one-pot operation provides a safe and convenient solution for the asymmetric radical cyclopropanation of alkenes. Our Fe(III)-based catalytic system, supported by an optimal D2-symmetric chiral amidoporphyrin ligand, effectively activates α-trifluoromethyldiazomethane under mild conditions, affording trifluoromethyl-substituted cyclopropanes in high yields with both excellent diastereoselectivities and enantioselectivities. Mechanistic insights from comprehensive experimental and computational studies support a stepwise radical mechanism involving α- and γ-Fe(IV)-alkyl radical intermediates, demonstrating the operation of Fe(III)-based metalloradical catalysis.
Results and discussion
Reaction development
We commenced our studies by evaluating the catalytic potential of [Fe(Por)Cl] in direct comparison with [Co(Por)] and [Mn(Por)Cl] for the cyclopropanation reaction of styrene with α-trifluoromethyldiazomethane (1′) through in situ generation from trifluoroacetaldehyde-derived hydrazone 1, a 2,4,6-triisopropylbenzenesulfonyl hydrazone whose structure was determined by X-ray crystallography, in the presence of Cs2CO3 as the base (Table 1). Although no reaction occurred without a catalyst (entry 1), Co(II)-metalloradical catalyst [Co(P1)] (P1 = 5,10,15,20-tetraphenylporphyrin) catalysed the reaction to form the desired trifluoromethyl-substituted cyclopropane 3a in 30% yield with a 99:1 diastereomeric ratio (d.r.) in favour of trans-diastereomer (entry 2). Although Mn(III)–metalloradical complex [Mn(P1)Cl] was ineffective (entry 3), Fe(III)–metalloradical complex [Fe(P1)Cl] outperformed [Co(P1)], delivering cyclopropane 3a in a considerably higher yield (88%) with the same level of diastereoselectivity (99:1 d.r.) (entry 4). These results exhibited a notable effect of metal ions on the catalytic reaction by metalloporphyrin-based catalysts, prompting us to investigate the difference in enantioselectivity with the support of D2-symmetric chiral amidoporphyrins. The Co(II) complex of first-generation D2-symmetric chiral amidoporphyrin [Co(P2)] (P2 = 3,5-DitBu-ChenPhyrin)62 furnished cyclopropane 3a in 47% yield with excellent diastereoselectivity (99:1 d.r.) but low enantioselectivity (10% e.e.) (entry 5). [Mn(P2)Cl], a Mn(III) complex of chiral porphyrin P2, was ineffective (entry 6). To our delight, the corresponding chiral Fe(III) complex [Fe(P2)Cl], the structure of which was determined by X-ray crystallography, was equally effective as [Co(P2)], generating cyclopropane 3a in a similar yield (45%) with the same excellent diastereoselectivity (98:2 d.r.) and slightly enhanced enantioselectivity (14% e.e.) (entry 7). Encouraged by this promising result, we further investigated the effect of ligand architecture on the catalytic reaction by using second-generation D2-symmetric chiral amidoporphyrin P3 (P3 = 3,5-DitBu-QingPhyrin)63. Ligand P3 differs from ligand P2 by replacement of one of the two methyl groups in each of the chiral amide units with a phenyl group, which could exert potential π-stacking interactions in stabilizing radical intermediates. [Co(P3)] notably improved the reaction, delivering cyclopropane 3a in high yield (92%) with excellent diastereoselectivity (99:1 d.r.) while increasing the enantioselectivity dramatically (−10% e.e. to 80% e.e.) (entry 8). Mn(III) complex [Mn(P3)Cl] remained unproductive (entry 9). Gratifyingly, Fe(III) complex [Fe(P3)Cl] was even more effective than [Co(P3)], producing cyclopropane 3a in excellent yield (96%) with excellent diastereoselectivity (98:2 d.r.) and even higher enantioselectivity (86% e.e.) (entry 10). These results illustrate a remarkable combined effect of the metal ion and supporting ligand on catalytic performance. [Fe(P3)Cl]-catalysed cyclopropanation was further improved by changing the solvent to hexanes (entry 11), and lowering the temperature to 4 °C further enhanced enantioselectivity to 91% e.e. without obviously affecting the high yield and excellent diastereoselectivity (entry 12).
Table 1 |.
Catalyst effect on asymmetric cyclopropanation with in situ-generated α-trifluoromethyldiazomethane
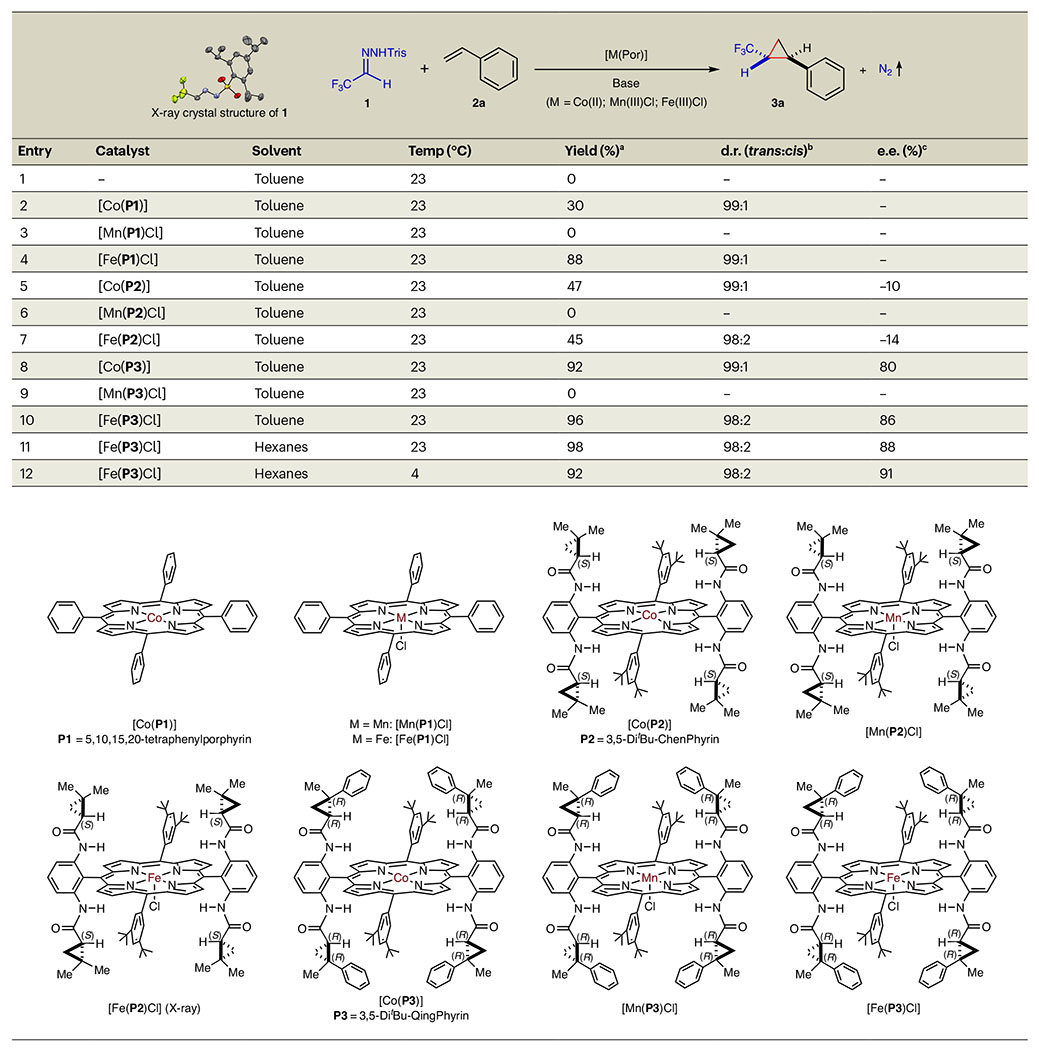
|
Conducted with 1 (0.12 mmol) and styrene (2a) (0.10 mmol) in the presence of Cs2CO3 (0.24 mmol) using [M(Por)] (M = Co(II); Mn(III)Cl; Fe(III)Cl) as catalyst (2 mol%) for 20 h.
isolated yields.
Diastereomeric ratio (d.r.) determined by 19F NMR analysis of reaction mixture.
Enantiomeric excess (e.e.) of trans-isomer determined by chiral HPLC. Tris = 2,4,6-triisopropylbenzenesulfonyl.
Substrate scope
Under optimized conditions, we evaluated the scope of [Fe(P3)Cl]-catalysed asymmetric cyclopropanation with in situ-generated trifluoromethyldiazomethane (1′) from hydrazone 1 in the presence of Cs2CO3, employing different alkenes as substrates (Table 2). Styrenes with different substituents, such as electron-neutral (–Me), electron-donating (–OMe) and electron-withdrawing groups (–CN), could be effectively cyclopropanated to generate trifluoromethyl-substituted cyclopropanes 3b–3d in high yields with excellent diastereoselectivities and high enantioselectivities. Halogenated styrenes with –Cl and –Br at different positions were also successfully transformed into cyclopropanes 3e–3h in excellent yields with high diastereoselectivities and enantioselectivities. The Fe(III)-catalysed asymmetric cyclopropanation tolerated functional groups, as exemplified for the formation of cyclopropanes 3i and 3j, containing formyl and benzodioxole functionalities, respectively. Additionally, extended aromatic olefins and heteroaromatic olefins were compatible, forming the corresponding cyclopropanes 3k–3m in excellent yields with high diastereoselectivities and enantioselectivities. The asymmetric catalytic reaction could be readily scaled up, as exemplified by the high-yielding synthesis of enantioenriched cyclopropane 3k on a 2.0 mmol scale.
Table 2 |.
Asymmetric cyclopropanation of alkenes by [Fe(P3)Cl] with in situ-generated α-trifluoromethyldiazomethane and various diazo compounds
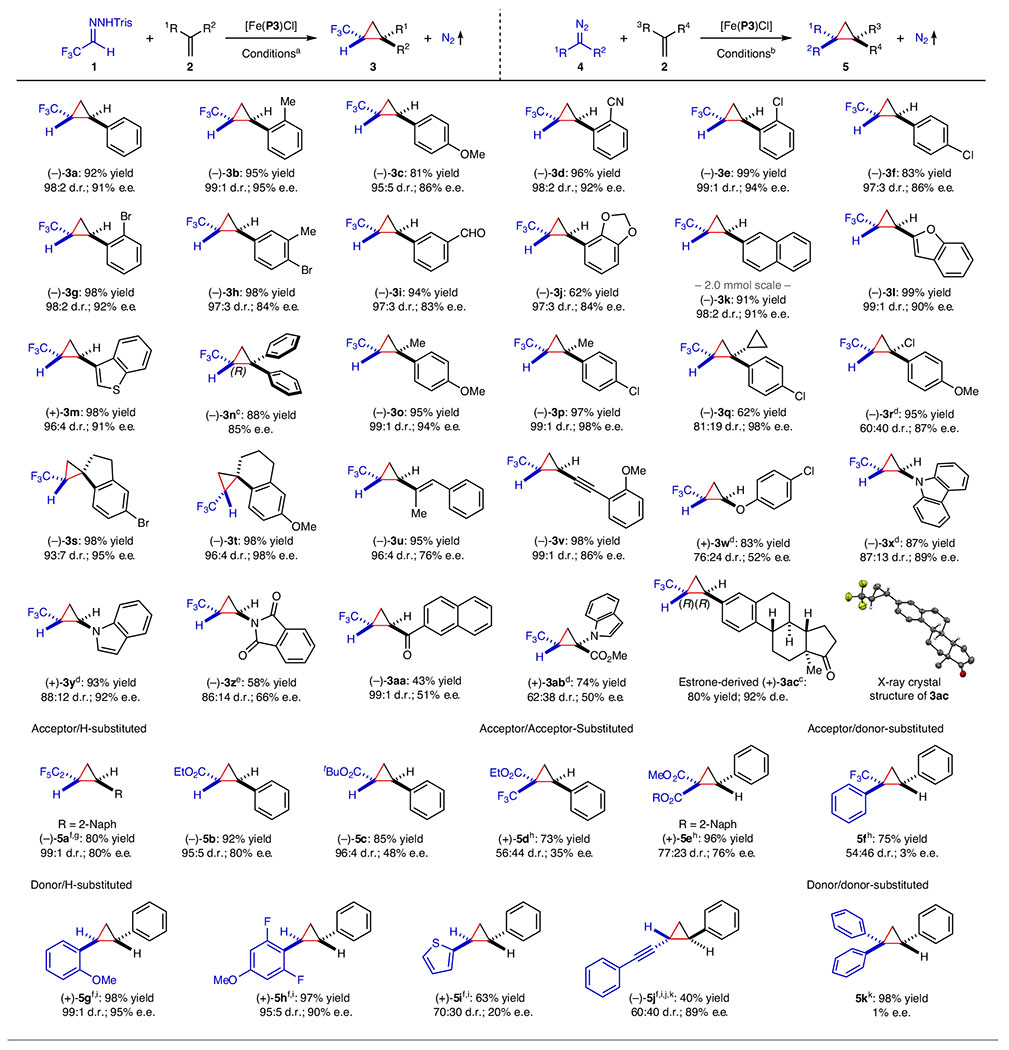
|
Conducted with hydrazone 1 (0.12 mmol) and alkene 2 (0.10 mmol) in the presence of Cs2CO3 (0.24 mmol) using [Fe(P3)Cl] as catalyst (2 mol%) in hexanes at 4 °C for 20 h; isolated yields; diastereomeric ratio (d.r.) determined by 19F NMR analysis of reaction mixture; enantiomeric excess (e.e.) of trans-isomer determined by chiral HPLC.
Conducted with diazo compound 4 (0.10 mmol) and alkene 2 (0.60 mmol) using [Fe(P3)Cl] as catalyst (2 mol%) in toluene at 23 °C for 24 h; isolated yields; d.r. determined by 1H NMR analysis of the reaction mixture; e.e. of the major isomer determined by chiral HPLC.
Absolute configuration determined by X-ray crystallography.
In toluene at 23 °C.
With 1 (0.20 mmol).
Diazo compound in situ-generated from trisylhydrazone in the presence of Cs2CO3 (0.20 mmol).
With 2 (0.12 mmol) in hexanes at 4 °C.
At 80 °C.
With 2 (0.20 mmol).
In ethyl acetate.
At 40 °C.
1,1-Disubstituted alkenes also served as suitable substrates for the construction of trifluoromethyl-substituted cyclopropanes using the [Fe(P3)Cl]-based catalytic system. For example, 1,1-diphenylethylene was successfully cyclopropanated to furnish cyclopropane 3n in high yield with high enantioselectivity as the (R)-configuration by X-ray crystallography. Encouraged by this result, we explored different 1,1-disubstituted alkenes as substrates. α-Methylstyrenes bearing p-OMe and p-Cl groups were both asymmetrically transformed to cyclopropanes 3o and 3p, with exceptional stereocontrol of the all-carbon quaternary stereogenic centre on the three-membered ring. With α-cyclopropylstyrene containing a p-Cl atom, cyclopropane 3q resulted, containing two cyclopropane rings linked at the all-carbon quaternary stereogenic centre, with excellent enantioselectivity but moderate diastereoselectivity. The α-halogen styrenes enabled stereoselective construction of cyclopropanes with chiral α-tertiary halides, as exemplified by cyclopropane 3r. The Fe(III)-catalysed cyclopropanation was further applied for asymmetric construction of spiro-structures, leading to spiro-cyclopropanes 3s and 3t in almost quantitative yields, with excellent diastereoselectivities and enantioselectivities. Additionally, conjugated dienes and enynes were regio- and chemo-selectively cyclopropanated at the terminal alkene units, affording cyclopropanes 3u and 3v bearing alkenyl and alkynyl functionalities, respectively.
The Fe(III)-catalysed cyclopropanation was also applicable to electron-rich olefins such as vinyl aryl ether, N-vinylcarbazole, N-vinylindole and N-vinylphthalimide, producing cyclopropanes 3w–3z in good to high yields with varied stereoselectivities. Electron-deficient olefins like vinyl ketones and dehydroaminocarboxylates, known as challenging substrates for catalytic systems involving electrophilic metallocarbene intermediates, were also successfully employed, delivering trifluoromethyl-substituted cyclopropanes 3aa and 3ab containing additional functionalities. The Fe(III)-based catalytic system was amenable to late-stage cyclopropanation of complex molecules, as exemplified by the high-yielding synthesis of estrone-derived cyclopropane 3ac with excellent stereoselectivity as (1R,2R) by X-ray crystallography. For aliphatic and internal olefins under standard conditions, only small amounts of the desired cyclopropanes were generated (Supplementary Figs. 2–4), highlighting the need for a more effective chiral porphyrin ligand in supporting Fe(III)-catalysed cyclopropanation for these challenging substrates.
The versatility of the Fe(III)-based metalloradical system was explored using different classes of diazo compounds (4) as potential metalloradicophiles for asymmetric cyclopropanation (Table 2). The fluorinated diazo compound pentafluoroethyldiazomethane (4a′), in situ-generated from the corresponding hydrazone, could also be effectively activated by [Fe(P3)Cl] for asymmetric cyclopropanation of 2-vinylnaphthalene, delivering cyclopropane 5a in high yield with excellent diastereoselectivity and high enantioselectivity. Other acceptor/H-substituted diazo compounds, such as ethyl diazoacetate (4b) and tert-butyl diazoacetate (4c), were also successfully activated by [Fe(P3)Cl] for asymmetric cyclopropanation of styrene, affording cyclopropanecarboxylates 5b and 5c, respectively, with high diastereoselectivities and moderate enantioselectivities. Additionally, [Fe(P3)Cl] exhibited the ability to activate acceptor/acceptor-substituted diazo compounds, as shown with α-trifluoromethyldiazoacetate 4d and diazomalonate 4e, providing trifluoromethylcyclopropaneester 5d and 1,1-cyclopropanediester 5e, respectively, in high yields with moderate stereoselectivities. Furthermore, acceptor/donor-substituted diazo compounds could be applied as metalloradicophiles for [Fe(P3)Cl)]-catalysed asymmetric cyclopropanation, as exemplified by the formation of cyclopropane 5f, albeit with inferior control of stereoselectivities. Moreover, various donor/H-substituted diazo compounds (for example, α-aryldiazomethanes 4g′ and 4h′, α-heteroaryldiazomethane 4i′ and α-alkynyldiazomethane 4j′) were effectively activated by the Fe(III)-based metalloradical system, resulting in cyclopropanes 5g–5j in good to high yields with generally high stereoselectivities. For donor/donor-substituted diazo compound diphenyldiazomethane (4k), the desired triphenylcyclopropane 5k was obtained in excellent yield but without controlling the challenging enantioselectivity. These results demonstrate the potential of Fe(III)-based metalloradical catalysis for broad synthetic applications.
Mechanistic investigations
To elucidate the underlying mechanism of the Fe(III)-based metalloradical system for olefin cyclopropanation, we performed experimental studies and density functional theory (DFT) computations. Comparative studies were conducted to probe the oxidation state of the active iron catalyst (Fig. 2). First, we compared the cyclopropanation reaction of alkene 2c by [Fe(P3)Cl] in air and under an inert atmosphere64. Both reactions proceeded equally well, providing cyclopropane 3c in similar yields (95% versus 93%) with identical diastereoselectivities (93:7 d.r.) and enantioselectivities (80% e.e.) (entries 1 and 2). These results suggest that the original Fe(III) complex [Fe(P3)Cl] is the true catalyst, challenging the assumption of in situ-reduced Fe(II) complex [Fe(P3)]. Next, we compared the cyclopropanation of alkene 2k by [Fe(P3)Cl] with and without 4-(dimethylamino)pyridine (DMAP). Both reactions afforded cyclopropane 3k with identical diastereoselectivities (98:2 d.r.) and enantioselectivities (86% e.e.), despite different yields (99% versus 82%) (entries 3 and 4). When [Fe(P3)Cl] was replaced by [Co(P3)] as the catalyst, the reactions produced cyclopropane 3k with similar but non-identical diastereoselectivities (99:1 versus 98:2 d.r.) and notably different enantioselectivities (75% versus 64% e.e.), along with different yields (99% versus 93%) (entries 5 and 6). The decrease of product yields in the Fe(III)- and Co(II)-catalysed reactions with DMAP was attributed to the partial generation of the catalytically inactive six-coordinate metalloporphyrin complexes [(DMAP) Fe(P3)Cl] and [(DMAP)2Co(P3)]. The difference in stereoselectivities in the Co(II)-catalysed reaction is probably due to the formation of the five-coordinate Co(II) complex [(DMAP)Co(P3)] (Fig. 2a), which remains catalytically active but exhibits different reactivity and selectivity due to a potential trans-effect exerted by the additional DMAP ligand41. In the Fe(III)-catalysed reaction, the analogous six-coordinate complex [(DMAP)Fe(P3)Cl] is coordinately saturated and does not contribute to an erosion in stereoselectivities. Similarly, the five-coordinate Fe(II) complex [(DMAP)Fe(P3)] should exhibit different reactivity and selectivity compared to the four-coordinate Fe(II) complex [Fe(P3)]. The observation that DMAP has no effect on the stereoselectivities of the Fe(III)-catalysed reaction provides additional evidence supporting the original Fe(III) complex [Fe(P3)Cl] as the active catalyst. To provide further evidence that the Fe–Cl bond remains intact during catalysis, we made considerable efforts to detect the proposed α-Fe(IV)-alkyl radical key intermediate I by high-resolution mass spectrometry (HRMS). The analysis of the reaction mixture of ethyl diazoacetate (4b) with [Fe(P3)Cl] using HRMS with electrospray ionization (ESI) exhibited a clear signal corresponding to the carbene radical complex of [Fe(P3)Cl] I[Fe(P3Cl)]/4b (Fig. 2b and Supplementary Fig. 8). Both the exact mass and the isotope distribution pattern match acceptably with those calculated from the formula of [C108H110ClFeN8O6 + Na]+, resulting from the sodium adduct of the molecular ion I[Fe(P3)Cl]/4b. Similar HRMS analysis of the reaction mixture of α-phenyldiazomethane (41′) with [Fe(P3)Cl] detected a signal corresponding to the carbene radical complex of [Fe(P3)Cl] I[Fe(P3)Cl]/4l (Fig. 2c and Supplementary Fig. 9), resulting from the potassium adduct of the molecular ion. The direct detections of α-Fe(IV)-alkyl radicals I by HRMS, together with comparative studies, strongly support Fe(III) complex [Fe(Por)Cl] as the genuine catalyst for the catalytic transformation.
Fig. 2 |. Comparative studies on catalytic cyclopropanation and detection of intermediates by HRMS to probe the oxidation state of iron porphyrin catalysts.
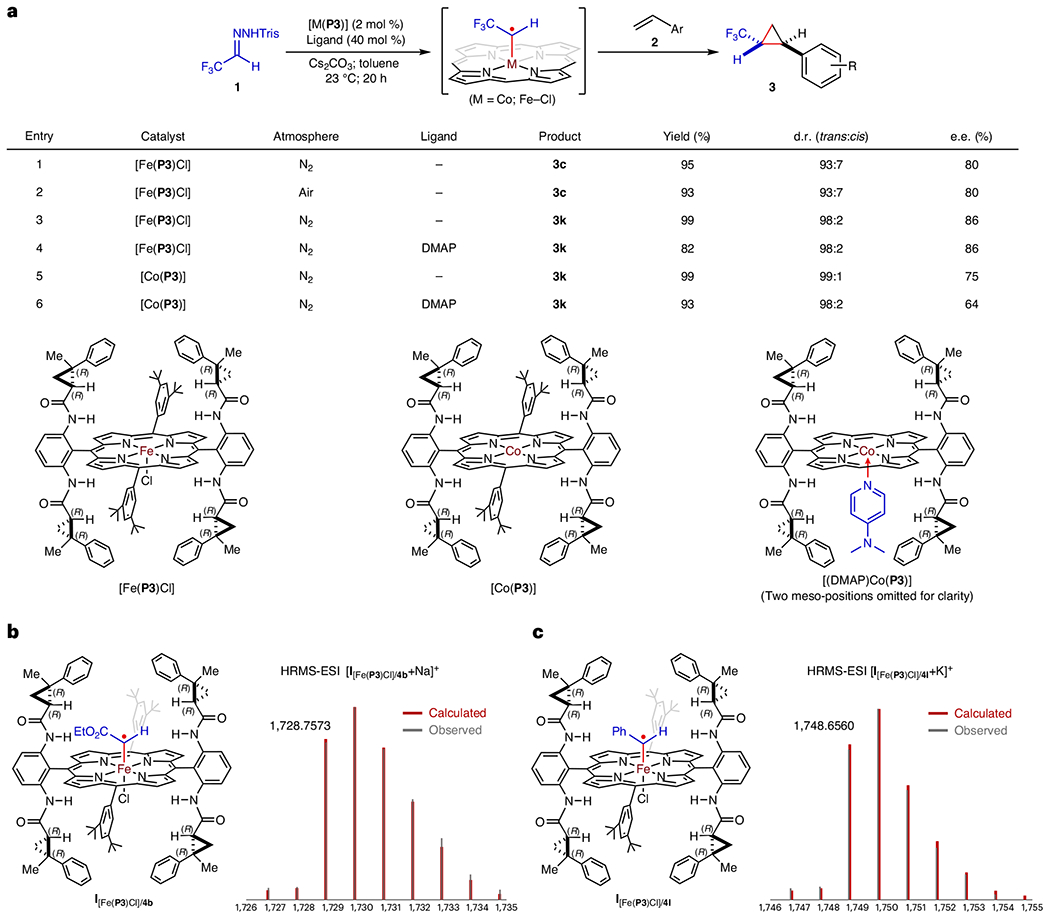
a, To probe the oxidation state of the active iron catalyst, comparative studies on catalytic cyclopropanation of alkenes 2 with in situ-generated α-trifluoromethyldiazomethane (1′) by [Fe(P3)Cl] were performed under a nitrogen atmosphere and in air, as well as in the absence and presence of DMAP. As a parallel comparison, the catalytic cyclopropanation reaction was also carried out with the [Co(P3)] as the catalyst in the absence and presence of DMAP. b, HRMS-ESI detection of α-Fe(IV)-alkyl radical I[Fe(p3)Cl]/4b from a reaction mixture of ethyl diazoacetate (4b) and [Fe(P3)Cl]. c, HRMS-ESI detection of α-Fe(IV)-alkyl radical I[Fe(P3)Cl]/4l from a reaction mixture of α-phenyldiazomethane (4l′) and [Fe(P3)Cl].
DFT calculations were conducted to examine the details of the catalytic pathways and associated energetics for the asymmetric cyclopropanation reaction of styrene (2a) with trifluoromethyldiazomethane (1′) by catalyst [Fe(P3)Cl] (A) (Fig. 3a and Supplementary Figs. 36–39). The five-coordinate d5-Fe(III)-metalloradical catalyst A exhibits three distinct spin states with comparable energy: high-spin sextet 6A (S = 5/2; 0.0 kcal mol−1), intermediate-spin quartet 4A (S = 3/2; 3.2 kcal mol−1) and low-spin doublet 2A (S = 1/2; 4.1 kcal mol−1). The DFT calculations reveal the initial formation of intermediate B, arising from the interaction between diazomethane 1′ and catalyst A through a network of attractive non-covalent forces. This interaction precisely positions the α-carbon atom of trifluoromethyldiazomethane 1′ in close proximity to the Fe(III)-metalloradical centre, priming it for further interaction. The three spin states of intermediate B (6B, 0.9 kcal mol−1; 4B, 2.1 kcal mol−1; 2B, 6.4 kcal mol−1) manifest substantial variations in the energies of their corresponding transition states (6TS1, 41.6 kcal mol−1; 4TS1, 36.8 kcal mol−1; 2TS1, 19.2 kcal mol−1), implying a spin-crossover phenomenon from the sextet state to the doublet state during the activation process. Following the most favourable pathway, the metalloradical activation exhibits a spin-crossover from 6B to 2TS1, leading to the generation of α-Fe(IV)-alkyl radical intermediate 2C and simultaneous elimination of dinitrogen as a by-product. This activation pathway, while slightly exergonic by 1.8 kcal mol−1, was identified as the rate-determining step due to its relatively high, yet accessible, activation barrier (2TS1, ΔG‡ = 18.3 kcal mol−1). Detailed analysis, presented in the spin plot of intermediate 2C (Fig. 3a), demonstrates a substantial redistribution of spin density from the iron centre to the α-carbon atom, indicating a critical role of spin changes during the metalloradical activation process. The subsequent radical addition of α-Fe(IV)-alkyl radical 2C to alkene 2a, being exergonic by 18.9 kcal mol−1, proceeds through an exceedingly low activation barrier (2TS2, ΔG‡ = 1.3 kcal mol−1), leading to the formation of γ-Fe(IV)-alkyl radical intermediate 2D. As illustrated in the DFT-optimized structure of 2TS2 (Fig. 3a), it is evident that the network of non-covalent attractions, including multiple hydrogen-bonding interactions between the substrates and the catalyst, work synergistically to lower the activation barrier of the transition state. As shown in the spin plot of intermediate 2D, the radical addition results in the translocation of the spin density from the α-carbon atom to the γ-carbon atom (Fig. 3a). To decipher the origin of the stereoselectivities, the DFT calculations probed the associated energetics governing the formation of corresponding major and minor diastereomers as well as enantiomers during the radical addition step (Supplementary Figs. 37 and 39). These calculations unveiled a preference for the production of trans-(R,R)-3a, with γ-Fe(IV)-alkyl radical intermediate 2D(R,R) being energetically favoured (ΔG‡ = 1.3 kcal mol−1) over 2D(S,S) (ΔG‡ = 4.3 kcal mol−1), both kinetically (ΔΔG‡ = 3.0 kcal mol−1) and thermodynamically (ΔΔG° = 3.1 kcal mol−1) (Fig. 3b). However, the calculated differentiation between the two transition states exhibited a quantitative overestimation of the ΔΔG‡ value compared to the experimental observation of trans-(R,R)-3a as the major product. This differentiation was further elucidated through the non-covalent interaction (NCI) plots of the DFT-optimized transition-state structures (Fig. 3b), revealing that 2TS2(R,R) experiences a stronger network of non-covalent interactions, including two-point hydrogen bonding, C–H⋯Cl and N–H/C–H⋯π interactions, between the substrates and ligand compared to 2TS2(S,S). The difference in non-covalent interactions was responsible for the observed enantioselectivity. The DFT calculations unravelled that the final step of intramolecular radical substitution (3-exo-tet radical cyclization) of intermediate 2D is highly exergonic by 34.2 kcal mol−1 and virtually barrierless, delivering cyclopropane 3a as the product while regenerating catalyst [Fe(P3)Cl]. Comparative energetics were calculated for the alternative concerted pathway for [Fe(P3)Cl]-catalysed cyclopropanation of styrene (2a) with diazomethane 1′, revealing a higher activation barrier than the stepwise mechanism (Supplementary Figs. 38 and 39). Additionally, further DFT computations were performed to investigate the asymmetric cyclopropanation reactions of styrene (2a) with ethyl diazoacetate (4b) and α-phenyldiazomethane (4l′) (Supplementary Figs. 40–43).
Fig. 3 |. DFT calculations on the catalytic mechanism for olefin cyclopropanation by [Fe(P3)CI].
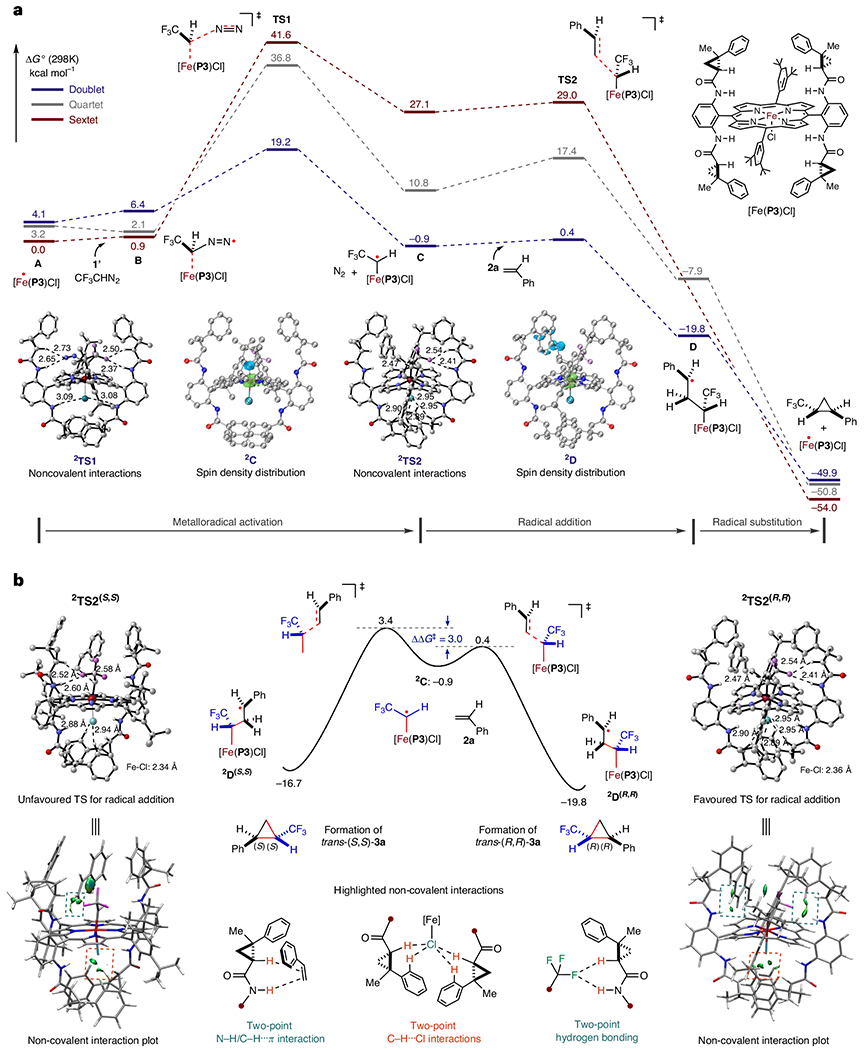
a, DFT calculations on the catalytic pathway and associated energies for the asymmetric cyclopropanation of styrene with α-trifluoromethyldiazomethane (1′) by [Fe(P3)Cl] in three different spin states. b, DFT study on the origin of the enantioselectivity of the asymmetric cyclopropanation of styrene with α-trifluoromethyldiazomethane (1′) by [Fe(P3)Cl].
To determine the spin states of the Fe(III)-centre in [Fe(P3)Cl]65,66, the magnetic moment was measured using a superconducting quantum interference device (SQUID) over a temperature range of 6–300 K (Fig. 4a). The effective magnetic moment remained constant at ~5.68 μB per ion after an initial sharp increase before ~50 K. In solution (CDCl3) at 298 K, the magnetic moment of [Fe(P3)Cl] was found to be 5.62 μB, similar to the solid state. These smaller values compared to the high-spin complex [Fe(P1)Cl] (~5.90 μB)67 indicate a quantum mechanical spin admixture of high spin (S = 5/2) and intermediate spin (S = 3/2) rather than a purely high-spin (S = 5/2) state68. The different spin states of the Fe(III) centres in [Fe(P3)Cl] and [Fe(P1)Cl] are probably attributed to potential hydrogen-bonding interactions between the axial chloride and the two amides in the P3 ligand, which are absent in the P1 ligand69. Additionally, X-band electron paramagnetic resonance (EPR) measurements on [Fe(P3)Cl] at different temperatures ranging from 298 K to 13 K (Fig. 4b) display two notable signals (g⊥ = 5.81 and g|| = 1.99) that increase in intensity as the temperature decreases. In comparison with the EPR signals of purely high-spin complex [Fe(P1)Cl] from the literature (g⊥ = 6.00 and g|| = 2.00)67, the pair of peaks at g|| = 5.81 and g|| = 1.99 demonstrate the predominant contribution of high-spin (S = 5/2) character and the minor contribution of intermediate-spin (S = 3/2) character67–69. The EPR studies, along with the magnetic moment measurements, suggest that [Fe(P3)Cl] is a spin-admixed (S = 5/2, 3/2) complex adopting more high-spin (S = 5/2) character, consistent with DFT calculations. Furthermore, the X-ray structural parameters of analogous [Fe(P2)Cl] (Supplementary Tables 1–7) also support the assignment of an admixed spin state (S = 5/2 and 3/2)67, in line with the experimental findings for [Fe(P3)Cl].
Fig. 4 |. Determination of iron spin states in [Fe(Por)Cl].
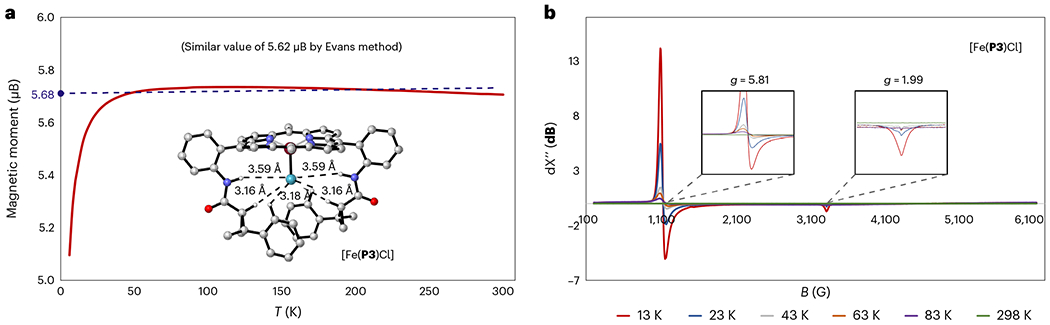
a, Measurement of the magnetic moment of [Fe(P3)Cl] by variable-temperature SQUID and Evans methods. b, X-band EPR measurement for [Fe(P3)Cl] at different temperatures.
Additional experimental studies were performed to detect and probe the radical intermediates involved in the catalytic cycle (Fig. 5). To trap the proposed α-Fe(IV)-alkyl radical I for EPR spectroscopy, the spin-trapping reagent PBN (N-tert-butyl-α-phenylnitrone) was added to a mixture of [Fe(P1)Cl] with hydrazone 1 in the presence of base in benzene without an olefin substrate (Fig. 5a). The isotropic X-band EPR spectrum displays the characteristic triplet of doublet signal at a g value of ~2.00, providing evidence for the formation of nitroxyl radical 6 resulting from PBN trapping of α-Fe(IV)-trifluoroethyl radical I[Fe(P1)Cl]/1. The experimental spectrum could be fittingly simulated based on the hyperfine couplings by 14N (I = 1) and 1H (I = 1/2) at a g value of 2.00641 with A(N) = 41.1 MHz and A(H) = 7.5 MHz. Similarly, when DMPO (5,5-dimethyl-1-pyrroline N-oxide) was used, the corresponding DMPO-trapped nitroxyl radical 7 from I[Fe(P1)Cl]/1 was detected by EPR, exhibiting the characteristic doublet of triplet signal. Simulation of the EPR spectrum generated a g value of 2.00640 with A(N) = 39.9 MHz and A(H) = 60.5 MHz. Additionally, the corresponding α-Fe(IV)-alkyl radicals I from the activation of other diazo compounds such as diazoacetate 4b, α-trifluoromethyldiazoacetate 4d, α-cyanodiazoacetate 4n and α-aryldiazomethane 41′ by [Fe(P1)Cl] were successfully trapped by PBN and DMPO. The EPR spectra display characteristic signals at a g value of ~2.00, which were adequately simulated, showing differences in fine details depending on the nature of the specific substituents (Supplementary Figs. 15–20). However, efforts to isolate products resulting from the trapping of α-Fe(IV)-trifluoroethyl radical I by H-atom sources and stable TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) radical were not successful, presumably due to the volatility or instability of the resulting fluorine compounds. Accordingly, α-aryldiazomethanes were employed as surrogates for trifluoromethyldiazomethane (1′) in the trapping experiments. Gratifyingly, when α-phenyldiazomethane, generated in situ from hydrazone 4l, was reacted with [Fe(P1)Cl] in the presence of triphenylsilane as a H-atom source, compound 8, identified as benzyltriphenylsilane, could be isolated in 31% yield (Fig. 5b). Formation of silane 8 was seen to proceed through a sequence of radical H-atom abstraction from triphenylsilane by the corresponding α-Fe(IV)-benzyl radical intermediate I[Fe(P1)Cl]/4l, followed by subsequent radical substitution of the resulting Fe(IV)-benzyl complex III[Fe(P1)Cl]/4l with triphenylsilyl radical. In a similar reaction, benzyltriphenylstannane (8′) was formed in 85% yield when triphenyltin hydride was employed as the H-atom source (Supplementary Table 11). Moreover, it was shown that α-Fe(IV)-benzyl radical I[Fe(P1)Cl]/4m, resulting from metalloradical activation of α-(p-cyano) phenyldiazomethane, generated in situ from hydrazone 4m, by [Fe(P1)Cl] could be successfully trapped by TEMPO to form product 9 in 70% yield (Fig. 5b). As confirmed by X-ray crystallography, compound 9 is a bis(TEMPO)methane derivative bearing two geminal TEMPO units at the α-position of p-cyanotoluene. Evidently, α-Fe(IV)-benzyl radical I[Fe(P1)Cl]/4m was first trapped by TEMPO to form Fe(IV)–benzyl complex IV[Fe(P1)Cl]/4m, which then underwent radical substitution with another molecule of TEMPO to give the final product 9.
Fig. 5 |. Experimental studies on the catalytic mechanism of olefin cyclopropanation by the Fe(III)-based metalloradical system.
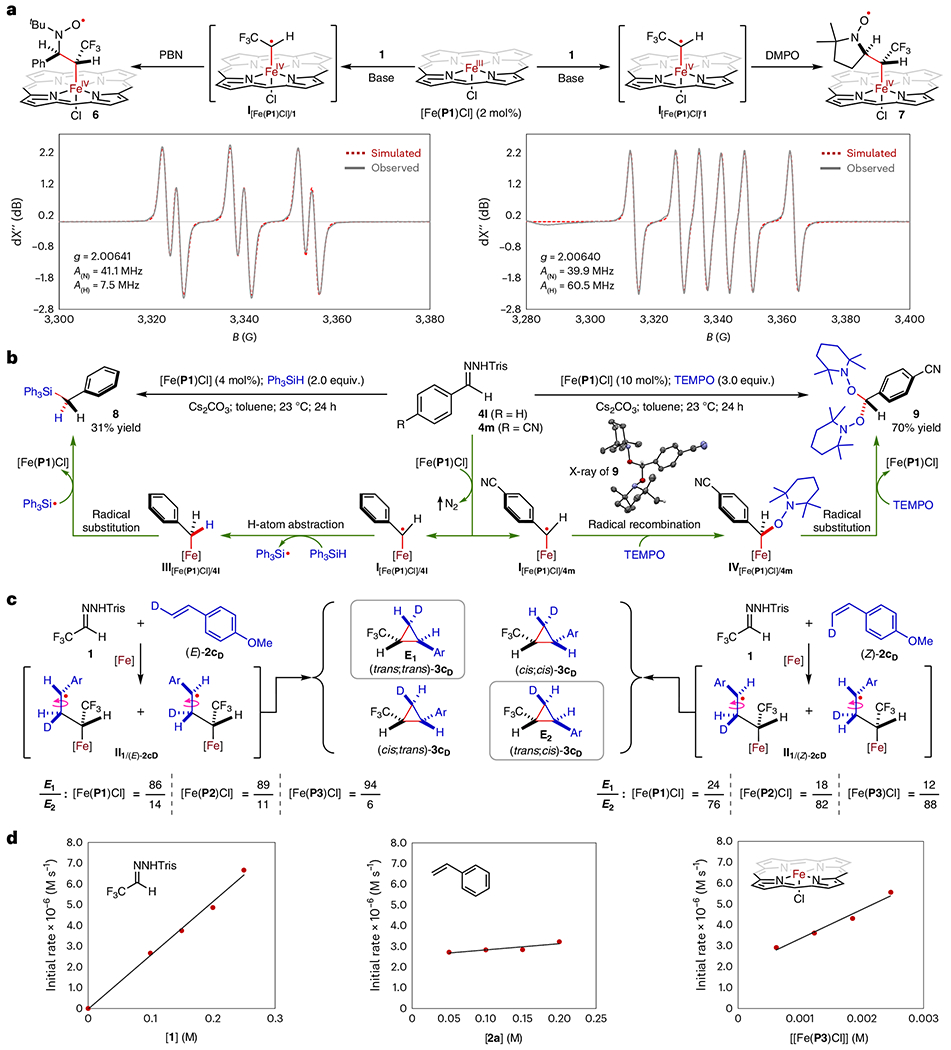
a, Trapping of the α-Fe(IV)-alkyl radical intermediate by spin traps PBN and DMPO for EPR detection. b, Trapping of α-Fe(IV)-benzyl radicals from metalloradical activation of aryldiazomethanes by Ph3SiH and by TEMPO. c, Probing of the γ-Fe(IV)-alkyl radical intermediate by cyclopropanation reactions of (E)- and (Z)-β-deutero-p-methoxystyrene. d, Dependence of the cyclopropanation reaction rate on concentrations of substrates and catalyst through kinetic studies.
To probe the involvement of δ-Fe(IV)-alkyl radical II as an intermediate in the proposed mechanism, both isotopomers of β-deutero-p-methoxystyrene (E)-2cD and (Z)-2cD were applied as substrates for Fe(III)-catalysed cyclopropanation with hydrazone 1 in the presence of Cs2CO3 (Fig. 5c). Unlike the stereospecific formation of cyclopropanes in the well-known concerted mechanism70, the stepwise radical mechanism is anticipated to generate four diastereomers of cyclopropanes, owing to potential β-C–C bond rotation in intermediate II before 3-exo-tet cyclization (Fig. 1). As anticipated, the reactions of (E)-2cD and (Z)-2cD with 1 produced a mixture of four diastereomers (trans;trans)-3cD, (cis;cis)-3cD, (cis;trans)-3cD and (trans;cis)-3cD. Isotopomers (trans;trans)-3cD (E1) and (trans;cis)-3cD (E2) were the predominated products due to the high diastereoselectivity (Fig. 5c and Supplementary Figs. 21–25). The isotopomeric ratio of E1 to E2 could be accurately determined by a combination of 1H- and 2H NMR analysis. With [Fe(P1)Cl] as the catalyst, the isotopomeric ratio of E1 to E2 was 86:14 for the cyclopropanation of (E)-2cD and 24:76 for the cyclopropanation of (Z)-2cD. The observation of both trans- and cis-isotopomers of trans-3c in the reactions indicates free rotation of the β-C–C bond in the corresponding γ-Fe(IV)-alkyl radical intermediates II1/(E)-2cD and II1/(Z)-2cD. Switching the catalyst to [Fe(P2)Cl] changed the isotopomeric ratio of E1 to E2 to 89:11 in the reactions of (E)-2cD and 18:82 in the reaction of (Z)-2cD, indicating a lower degree of rotation freedom in a more sterically hindered catalyst environment. The use of [Fe(P3)Cl], with four phenyl groups in place of the methyl groups, further decreased the degree of the β-C–C bond rotation, changing the isotopomeric ratio of E1 to E2 to 94:6 and 12:88 for the cyclopropanation reactions of (E)-2cD and (Z)-2cD, respectively. These findings support the involvement of γ-Fe(IV)-alkyl radical intermediate II, exhibiting a varying degree of β-C–C bond rotation, in contrast to the non-existence of such a feature in concerted pathways. Similar experiments were performed with isotopomers (E)-2aD and (Z)-2aD to probe the involvement of γ-Fe(IV)-alkyl radical II in cyclopropanation reactions by [Fe(P2)Cl] with other classes of diazo compounds such as diazoacetate 4b, diazomalonate 4e and α-aryldiazomethane 4o′. In all cases, evidence of free rotations of the β-C–C bond in the corresponding γ-Fe(IV)-alkyl radical intermediates were observed, occurring to varying degree depending on the specific substituents (Supplementary Figs. 26–28). Together, these results are in full agreement with the proposed stepwise radical mechanism for Fe(III)-catalysed cyclopropanation.
To study the kinetics of the Fe(III)-based catalytic system, the cyclopropanation reaction of alkene 2a with hydrazone 1 in the presence of Cs2CO3 by [Fe(P3)Cl] was monitored by 19F NMR and 1H NMR at regular intervals over the course of 24 h at room temperature (Fig. 5d and Supplementary Figs. 29–31). Within 5h, hydrazone 1 was nearly completely converted to diazomethane 1′, while the conversion of alkene 2a and diazomethane 1′ to cyclopropane 3a was completed in ~15 h. The effectiveness of the catalytic reaction at room temperature aligns with the low overall activation barrier predicted by DFT calculations. To gain insights into how the catalyst and reactants influence the catalytic process, NMR monitoring experiments were performed for a series of reactions by systematically varying the concentrations of hydrazone 1, alkene 2a and catalyst [Fe(P3)Cl] (Supplementary Figs. 32–35). The plots of the initial rate for 3a formation against concentration (Fig. 5d) revealed that the rate of the catalytic process increased proportionally with the concentration of 1 (first-order) but remained independent of the concentration of 2a (zero-order). Interestingly, the catalytic process was found to be a fractional order in catalyst [Fe(P3)Cl] (0.5 order), indicating its coexistence with an inactive resting state. Overall, these kinetic orders determined from these experimental studies are fully consistent with the DFT calculations, which identified the metalloradical activation of diazomethane 1′ by catalyst [Fe(P3)Cl] as the rate-determining step.
Conclusion
In summary, we have explicitly demonstrated the operation of Fe(III)-based MRC in the context of its application for asymmetric olefin cyclopropanation. Supported by the D2-symmetric chiral amidoporphyrin 3,5-DitBu-QingPhyrin as the optimal ligand, the Fe(III)-based metalloradical system, which operates under mild conditions in a one-pot fashion with in situ-generated α-trifluoromethyldiazomethane, is highly effective for radical cyclopropanation of a wide range of alkenes, affording trifluoromethyl-substituted cyclopropanes in high yields with both high diastereoselectivity and enantioselectivity. Additionally, we have shown that the Fe(III)-based metalloradical system is broadly effective in activating different classes of diazo compounds for asymmetric radical cyclopropanation, offering a potentially general and practically sustainable approach for stereoselective synthesis of valuable three-membered carbocycles. Our systematic experimental studies, in combination with detailed DFT computations, provide multiple lines of convincing evidence supporting the genuine action of Fe(III)-based metalloradical catalysis and shed light on the underlying stepwise radical mechanism. Specifically, we have established that the five-coordinate Fe(III) complexes of porphyrins ([Fe(Por)X]), a class of stable 15e-metalloradical complexes, can function as potent metalloradical catalysts that have the ability to homolytically activate diazo compounds and generate α-Fe(IV)-alkyl and γ-Fe(IV)-alkyl radicals as catalytic intermediates. Beyond cyclopropanation, our ongoing research aims to explore the reactivity of α-Fe(IV)-alkyl radicals, in H-atom and X-atom abstraction reactions. We believe that the outcomes presented here will lay a solid foundation for Fe(III)-based metalloradical catalysis and pave the way for the development of novel stereoselective radical transformations. This work represents an important step towards advancing the field of metalloradical catalysis and holds promise for the design of efficient radical methods for stereoselective organic synthesis.
Methods
In a general procedure for Fe(III)-catalysed olefin cyclopropanation, an oven-dried Schlenk tube was charged with Fe(III)-metalloradical catalyst (2 mol%), hydrazone (0.12 mmol, 1.2 equiv.) and Cs2CO3 (0.24 mmol, 2.4 equiv.). The Schlenk tube was then evacuated and backfilled with nitrogen three times. Alkene 2 (0.10 mmol, 1.0 equiv.) and solvent (1.0 ml) were added under nitrogen. The tube was purged with nitrogen and then stirred at 4 °C for 20 h. After completion of the reaction, the reaction mixture was filtered through a plug of silica gel and then concentrated by rotary evaporation. The residue was then subject to purification by flash chromatography to afford the pure cyclopropane product.
Supplementary Material
Acknowledgements
We are grateful for financial support by the NSF (CHE-2154885) and in part by NIH (R01-GM102554). W.-C.C.L. was supported by a LaMattina Graduate Fellowship and Dean’s Dissertation Fellowship. We thank B. Li (Boston College) for X-ray structure determination, J. Jin (Boston College) for EPR measurements, M. Graf (Boston College) for SQUID measurements and M. Kumar (Massachusetts Institute of Technology) for HRMS measurements. We thank J. Zhang (Johns Hopkins University) for helpful discussions and valuable suggestions. We also acknowledge financial support by NIH (S10-OD026910) and NSF (CHE-2117246) for the purchase of NMR spectrometers at the Magnetic Resonance Center of Boston College.
Footnotes
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41557-023-01317-8.
Competing interests
The authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41557-023-01317-8.
Data availability
All data are available in the main text or the Supplementary Information. The crystal structure data of compounds [Fe(P2)Cl], 1, (R)-3n, (1R,2R)-3ac, 8′ and 9 have been deposited in the Cambridge structural database under reference nos. CCDC 2128685, 2128686, 2128688, 2128687, 2128689 and 2043165, respectively. Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/.
References
- 1.Zard SZ Radical Reactions in Organic Synthesis (Oxford Chemistry Masters, 2003). [Google Scholar]
- 2.Curran DP, Porter NA & Giese B Stereochemistry of Radical Reactions: Concepts, Guidelines and Synthetic Applications (Wiley, 2008). [Google Scholar]
- 3.Bar G & Parsons AF Stereoselective radical reactions. Chem. Soc. Rev 32, 251–263 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Mondal S. et al. Enantioselective radical reactions using chiral catalysts. Chem. Rev 122, 5842–5976 (2022). [DOI] [PubMed] [Google Scholar]
- 5.Kern N, Plesniak MP, McDouall JJW & Procter DJ Enantioselective cyclizations and cyclization cascades of samarium ketyl radicals. Nat. Chem 9, 1198–1204 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Wang F, Chen PH & Liu GS Copper-catalyzed radical relay for asymmetric radical transformations. Acc. Chem. Res 51, 2036–2046 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Gu QS, Li ZL & Liu XY Copper(I)-catalyzed asymmetric reactions involving radicals. Acc. Chem. Res 53, 170–181 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Li ZL, Fang GC, Gu QS & Liu XY Recent advances in copper-catalyzed radical-involved asymmetric 1,2-difunctionalization of alkenes. Chem. Soc. Rev 49, 32–48 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Lee WCC & Zhang XP Asymmetric radical cyclopropanation of alkenes. Trends Chem. 4, 850–851 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Leest NP, de Zwart FJ, Zhou M & de Bruin B Controlling radical-type single-electron elementary steps in catalysis with redox-active ligands and substrates. JACS Au 1, 1101–1115 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajanbabu TV & Nugent WA Selective generation of free-radicals from epoxides using a transition-metal radical. A powerful new tool for organic synthesis. J. Am. Chem. Soc 116, 986–997 (1994). [Google Scholar]
- 12.Funken N, Muhlhaus F & Gansauer A General, highly selective synthesis of 1,3-and 1,4 difunctionalized building blocks by regiodivergent epoxide opening. Angew. Chem. Int. Ed 55, 12030–12034 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Yao CB, Dahmen T, Gansauer A & Norton J Anti-Markovnikov alcohols via epoxide hydrogenation through cooperative catalysis. Science 364, 764–767 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Ye KY, McCallum T & Lin S Bimetallic radical redox-relay catalysis for the isomerization of epoxides to allylic alcohols. J. Am. Chem. Soc 141, 9548–9554 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Chan YW & Chan KS Metalloradical-catalyzed aliphatic carbon-carbon activation of cyclooctane. J. Am. Chem. Soc 132, 6920–6922 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Roy S, Das SK & Chattopadhyay B Cobalt(II)-based metalloradical activation of 2-(diazomethyl)-pyridines for radical transannulation and cyclopropanation. Angew. Chem. Int. Ed 57, 2238–2243 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Roy S, Khatua H, Das SK & Chattopadhyay B Iron(II)-based metalloradical activation: switch from traditional click chemistry to denitrogenative annulation. Angew. Chem. Int. Ed 58, 11439–11443 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Das SK, Roy S, Khatua H & Chattopadhyay B Iron-catalyzed amination of strong aliphatic C(sp3)-H bonds. J. Am. Chem. Soc 142, 16211–16217 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Roy S. et al. Road map for the construction of high-valued N-heterocycles via denitrogenative annulation. Acc. Chem. Res 54, 4395–4409 (2021). [DOI] [PubMed] [Google Scholar]
- 20.Roy S. et al. Iron-catalyzed radical activation mechanism for denitrogenative rearrangement over C(sp3)-H amination. Angew. Chem. Int. Ed 60, 8772–8780 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Das SK et al. An iron(II)-based metalloradical system for intramolecular amination of C(sp2)-H and C(sp3)-H bonds: synthetic applications and mechanistic studies. Chem. Sci 13, 11817–11828 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee W-CC et al. Asymmetric radical cyclopropanation of dehydroaminocarboxylates: stereoselective synthesis of cyclopropyl α-amino acids. Chem 7, 1588–1601 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riart-Ferrer X. et al. Metalloradical activation of carbonyl azides for enantioselective radical aziridination. Chem 7, 1120–1134 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie JJ et al. New catalytic radical process involving 1,4-hydrogen atom abstraction: asymmetric construction of cyclobutanones. J. Am. Chem. Soc 143, 11670–11678 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lang K, Hu Y, Lee W-CC & Zhang XP Combined radical and ionic approach for the enantioselective synthesis of β-functionalized amines from alcohols. Nat. Synth 1, 548–557 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woggon WD Metalloporphyrines as active site analogues—lessons from enzymes and enzyme models. Acc. Chem. Res 38, 127–136 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Oohora K, Onoda A & Hayashi T Hemoproteins reconstituted with artificial metal complexes as biohybrid catalysts. Acc. Chem. Res 52, 945–954 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Yang Y & Arnold FH Navigating the unnatural reaction space: directed evolution of heme proteins for selective carbene and nitrene transfer. Acc. Chem. Res 54, 1209–1225 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolf JR et al. Shape and stereoselective cyclopropanation of alkenes catalyzed by iron porphyrins. J. Am. Chem. Soc 117, 9194–9199 (1995). [Google Scholar]
- 30.Lai TS et al. Alkene cyclopropanation catalyzed by Halterman iron porphyrin: participation of organic bases as axial ligands. Dalton Trans. 2006, 4845–4851 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Aggarwal VK, de Vicente J & Bonnert RV Catalytic cyclopropanation of alkenes using diazo compounds generated in situ. A novel route to 2-arylcyclopropylamines. Org. Lett 3, 2785–2788 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Li Y. et al. Remarkably stable iron porphyrins bearing nonheteroatom-stabilized carbene or (alkoxycarbonyl)carbenes: isolation, X-ray crystal structures, and carbon atom transfer reactions with hydrocarbons. J. Am. Chem. Soc 124, 13185–13193 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Adams LA et al. Diastereoselective synthesis of cyclopropane amino acids using diazo compounds generated in situ. J. Org. Chem 68, 9433–9440 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Morandi B & Carreira EM Iron-catalyzed cyclopropanation in 6 M KOH with in situ generation of diazomethane. Science 335, 1471–1474 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Morandi B, Dolva A & Carreira EM Iron-catalyzed cyclopropanation with glycine ethyl ester hydrochloride in water. Org. Lett 14, 2162–2163 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Zhu SF & Zhou QL Iron-catalyzed transformations of diazo compounds. Natl Sci. Rev 1, 580–603 (2014). [Google Scholar]
- 37.Allouche EMD, Al-Saleh A & Charette AB Iron-catalyzed synthesis of cyclopropanes by in situ generation and decomposition of electronically diversified diazo compounds. Chem. Commun 54, 13256–13259 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Ning YQ et al. Difluoroacetaldehyde N-triftosylhydrazone (DFHZ-Tfs) as a bench-stable crystalline diazo surrogate for diazoacetaldehyde and difluorodiazoethane. Angew. Chem. Int. Ed 59, 6473–6481 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Damiano C, Sonzini P & Gallo E Iron catalysts with N-ligands for carbene transfer of diazo reagents. Chem. Soc. Rev 49, 4867–4905 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Le Maux P, Juillard S & Simonneaux G Asymmetric synthesis of trifluoromethylphenyl cyclopropanes catalyzed by chiral metalloporphyrins. Synthesis 2006, 1701–1704 (2006). [Google Scholar]
- 41.Chen Y & Zhang XP Asymmetric cyclopropanation of styrenes catalyzed by metal complexes of D2-symmetrical chiral porphyrin: superiority of cobalt over iron. J. Org. Chem 72, 5931–5934 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Intrieri D. et al. Highly diastereoselective cyclopropanation of α-methylstyrene catalysed by a C2-symmetrical chiral iron porphyrin complex. Chem. Commun 50, 1811–1813 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Carminati DM et al. Designing ‘totem’ C2-symmetrical iron porphyrin catalysts for stereoselective cyclopropanations. Chem. Eur. J 22, 13599–13612 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Carminati DM et al. Synthesis, characterisation and catalytic use of iron porphyrin amino ester conjugates. New J. Chem 41, 5950–5959 (2017). [Google Scholar]
- 45.Bos M. et al. Recent progress toward the synthesis of trifluoromethyl-and difluoromethyl-substituted cyclopropanes. Chem. Eur. J 23, 4950–4961 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Mykhailiuk PK 2,2,2-Trifluorodiazoethane (CF3CHN2): a long journey since 1943. Chem. Rev 120, 12718–12755 (2020). [DOI] [PubMed] [Google Scholar]
- 47.Decaens J. et al. Synthesis of fluoro-, monofluoromethyl-, difluoromethyl-and trifluoromethyl-substituted three-membered rings. Chem. Eur. J 27, 2935–2962 (2021). [DOI] [PubMed] [Google Scholar]
- 48.Pons A. et al. Asymmetric synthesis of fluoro, fluoromethyl, difluoromethyl and trifluoromethylcyclopropanes. Acc. Chem. Res 54, 2969–2990 (2021). [DOI] [PubMed] [Google Scholar]
- 49.Morandi B & Carreira EM Iron-catalyzed cyclopropanation with trifluoroethylamine hydrochloride and olefins in aqueous media: in situ generation of trifluoromethyl diazomethane. Angew. Chem. Int. Ed 49, 938–941 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Morandi B, Cheang J & Carreira EM Iron-catalyzed preparation of trifluoromethyl substituted vinyl-and alkynylcyclopropane. Org. Lett 13, 3080–3081 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Phelan JP et al. Redox-neutral photocatalytic cyclopropanation via radical/polar crossover. J. Am. Chem. Soc 140, 8037–8047 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang XY et al. Use of trifluoroacetaldehyde N-tfsylhydrazone as a trifluorodiazoethane surrogate and its synthetic applications. Nat. Commun 10, 284 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Denton JR, Sukumaran D & Davies HML Enantioselective synthesis of trifluoromethyl-substituted cyclopropanes. Org. Lett 9, 2625–2628 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Zhang XY et al. Fluoroalkyl N-triftosylhydrazones as easily decomposable diazo surrogates for asymmetric [2 + 1] cycloaddition: synthesis of chiral fluoroalkyl cyclopropenes and cyclopropanes. ACS Catal. 11, 8527–8537 (2021). [Google Scholar]
- 55.Huang WS et al. General catalytic enantioselective access to monohalomethyl and trifluoromethyl cyclopropanes. Chem. Eur. J 24, 10339–10343 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Pagire SK, Kumagai N & Shibasaki M Highly enantio-and diastereoselective synthesis of 1,2,3-trisubstituted cyclopropanes from α,β-unsaturated amides and stabilized sulfur ylides catalyzed by a chiral copper(I) complex. ACS Catal. 11, 11597–11606 (2021). [Google Scholar]
- 57.Morandi B, Mariampillai B & Carreira EM Enantioselective cobalt-catalyzed preparation of trifluoromethyl-substituted cyclopropanes. Angew. Chem. Int. Ed 50, 1101–1104 (2011). [DOI] [PubMed] [Google Scholar]
- 58.Tinoco A, Steck V, Tyagi V & Fasan R Highly diastereo- and enantioselective synthesis of trifluoromethyl-substituted cyclopropanes via myoglobin-catalyzed transfer of trifluoromethylcarbene. J. Am. Chem. Soc 139, 5293–5296 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carminati DM et al. Biocatalytic strategy for the highly stereoselective synthesis of CHF2-containing trisubstituted cyclopropanes. Angew. Chem. Int. Ed 60, 7072–7076 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kotozaki M. et al. Highly enantioselective synthesis of trifluoromethyl cyclopropanes by using Ru(II)-Pheox catalysts. Chem. Commun 54, 5110–5113 (2018). [DOI] [PubMed] [Google Scholar]
- 61.Altarejos J, Sucunza D, Vaquero JJ & Carreras J Enantioselective copper-catalyzed synthesis of trifluoromethyl-cyclopropylboronates. Org. Lett 23, 6174–6178 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen Y, Fields KB & Zhang XP Bromoporphyrins as versatile synthons for modular construction of chiral porphyrins: cobalt-catalyzed highly enantioselective and diastereoselective cyclopropanation. J. Am. Chem. Soc 126, 14718–14719 (2004). [DOI] [PubMed] [Google Scholar]
- 63.Xu X. et al. Highly asymmetric intramolecular cyclopropanation of acceptor-substituted diazoacetates by Co(II)-based metalloradical catalysis: iterative approach for development of new-generation catalysts. J. Am. Chem. Soc 133, 15292–15295 (2011). [DOI] [PubMed] [Google Scholar]
- 64.Kweon J & Chang S Highly robust iron catalyst system for intramolecular C(sp3)-H amidation leading to γ-lactams. Angew. Chem. Int. Ed 60, 2909–2914 (2021). [DOI] [PubMed] [Google Scholar]
- 65.Nakamura M. Electronic structures of highly deformed Iron(III) porphyrin complexes. Coord. Chem. Rev 250, 2271–2294 (2006). [Google Scholar]
- 66.Nakamura M. in Fundamentals of Porphyrin Chemistry: A 21st Century Approach (eds Brothers PJ & Senge MO) 631–659 (Wiley, 2022). [Google Scholar]
- 67.Cheng RJ et al. Control of spin state by ring conformation of Iron(III) porphyrins. A novel model for the quantum-mixed intermediate spin state of ferric cytochrome c′ from photosynthetic bacteria. J. Am. Chem. Soc 119, 2563–2569 (1997). [Google Scholar]
- 68.Stuzhin PA et al. Effects of solvation on the spin state of Iron(III) in 2,8,12,18-tetrabutyl-3,7,13,17-tetramethyl-5,10-diazaporphyrinatoiron(III) chloride. Inorg. Chem 49, 4802–4813 (2010). [DOI] [PubMed] [Google Scholar]
- 69.Sahoo D, Quesne MG, de Visser SP & Rath SP Hydrogen-bonding interactions trigger a spin-flip in Iron(III) porphyrin complexes. Angew. Chem. Int. Ed 54, 4796–4800 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wei Y. et al. Cyclopropanations via heme carbenes: basic mechanism and effects of carbene substituent, protein axial ligand, and porphyrin substitution. J. Am. Chem. Soc 140, 1649–1662 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the Supplementary Information. The crystal structure data of compounds [Fe(P2)Cl], 1, (R)-3n, (1R,2R)-3ac, 8′ and 9 have been deposited in the Cambridge structural database under reference nos. CCDC 2128685, 2128686, 2128688, 2128687, 2128689 and 2043165, respectively. Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/.