

Abstract
Hematopoietic stem cells (HSC), which govern the production of all blood lineages, transition through a series of functional states characterized by expansion during fetal development, functional quiescence in adulthood, and decline upon aging. We describe central features of HSC regulation during ontogeny to contextualize how adaptive responses over the life of the organism ultimately form the basis for HSC functional degradation with age. We particularly focus on the role of cell cycle regulation, inflammatory response pathways, epigenetic changes, and metabolic regulation. We then explore how knowledge of age-related changes in HSC regulation can inform strategies for rejuvenation of old HSCs.
Introduction
Hematopoiesis is a constant, vital function by which all cells of the blood and immune system are produced throughout development, adulthood, and old age. Blood production by HSCs supports oxygen transport, wound healing, and immunological responses during different stages of life, with dynamic regulations tailoring the production of particular lineages of blood cells through a complex array of intrinsic and extrinsic mechanisms. HSCs are equipped with a unique network of stress-response mechanisms that enables them to rapidly respond to organismal demand while simultaneously protecting themselves from damage. However, these pro-survival mechanisms provoke specific genetic, epigenetic, and metabolic forms of damage which accumulate over time, leading to an expanded pool of indolent and functionally degraded old HSCs.
Here, we review the unique properties of HSCs that dictate their behavior, focusing on the fulfillment of distinct sets of organismal requirements during development and adult life. Through this lens, we examine how mechanisms used by HSCs during these periods engender features of aging, focusing on cell cycle regulation, inflammatory response pathways, epigenetics, and metabolism. Considering the changes affecting HSCs throughout the entire lifespan also provides the basis for strategies to forestall or reverse decline with age. As such, we conclude by reviewing current HSC rejuvenation strategies, with a focus on their mechanistic rationale for restoring youthful functionality.
Dynamic changes in HSC behavior throughout ontogeny
The hematopoietic system is a dynamic entity with HSC function evolving over the lifetime to support changing organismal demands. As the organism develops, functional changes in HSCs fulfill the distinct requirements of different stages of ontogeny. In the context of fetal and neonatal development, the prioritization of HSC expansion over blood production allows for the establishment of the hematopoietic compartment1,2. By contrast, during adulthood, balanced blood production and dynamic responses to stress take precedence to maintain peak efficiency during reproductive years3 (Figure 1). In this section, we review these changes in HSC behavior with a focus on the functional characteristics that allow hematopoiesis to be tailored to different stages of ontogeny.
Figure 1. Dynamic responses of HSCs to organismal demands throughout the lifespan.
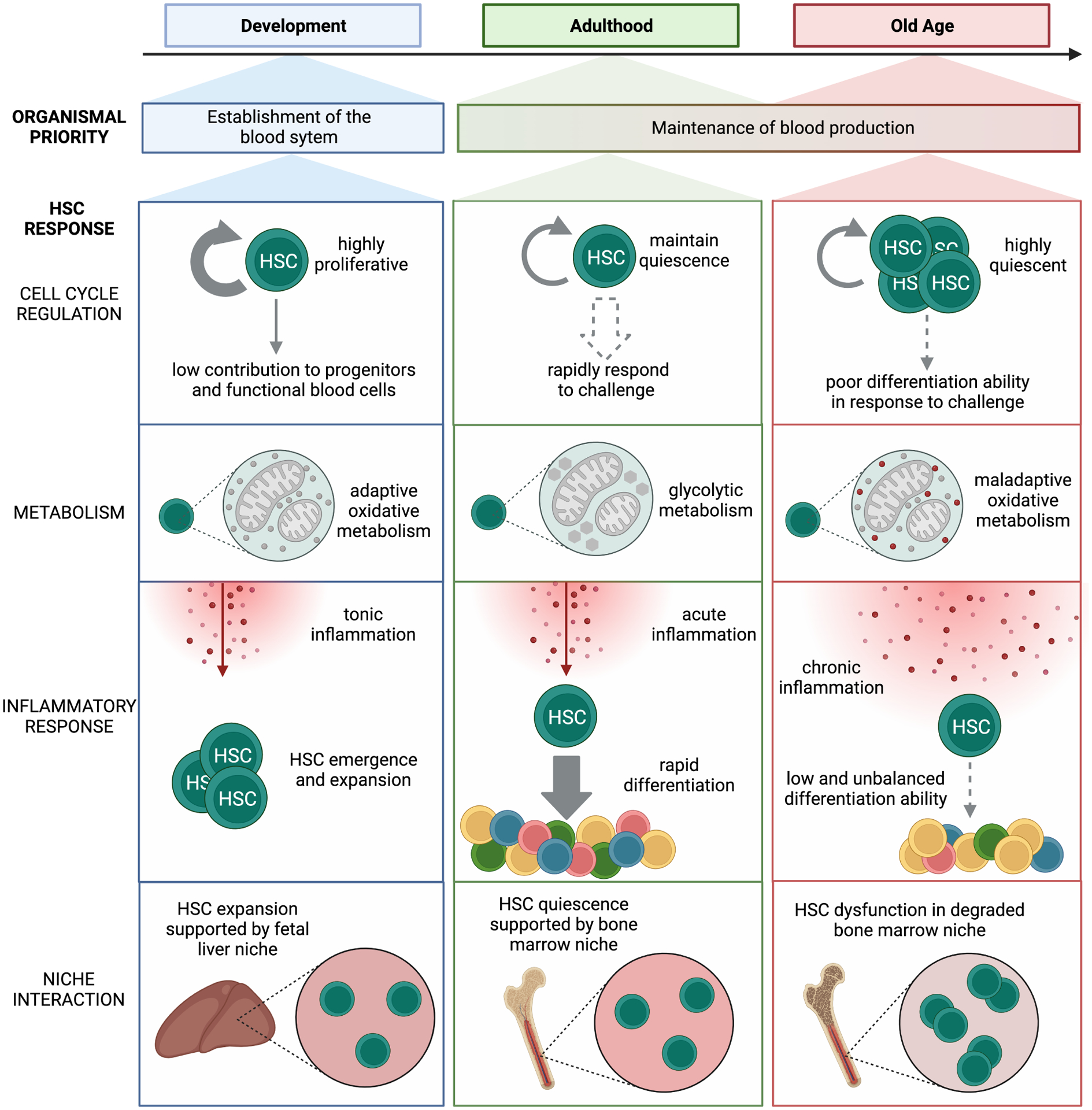
During development, HSCs primary function is to establish the adult blood system, with fetal HSCs residing in the fetal liver and engaging in oxidative phosphorylation to allow for rapid expansion. During adulthood, HSCs act to maintain organismal health and immunity via functional quiescence, a primarily glycolytic resting state that allows quick entry into the cell cycle in response to organismal demands. During aging, dysfunctional HSCs expand, survive, and maintain quiescence despite maladaptive oxidative metabolism and chronic inflammation in a degraded BM niche environment that promotes aberrant differentiation.
Fetal HSCs establish the hematopoietic compartment
In humans as well as zebrafish and murine model systems, hematopoiesis initiates during early embryonic development and occurs in successive waves that mark the transition from primitive hematopoiesis in the yolk sac to definitive hematopoiesis in the aorta-gonad-mesonephros (AGM) region and the fetal liver4. Primitive hematopoiesis has been extensively reviewed elsewhere5–9, and we focus here on definitive hematopoiesis, during which HSCs emerge from the hemogenic endothelium of the AGM region and migrate to the fetal liver before homing to the nascent bone marrow (BM) cavity10.
In the fetus, HSC behavior is driven by the fundamental requirement of establishing the hematopoietic compartment. In line with this objective, fetal HSCs demonstrate higher proliferative capacity and greater repopulation ability than adult HSCs11–16. This emphasis on proliferative capacity is enabled by numerous cell-intrinsic features, including active cell cycle engagement. Studies using murine models have shown that almost all HSCs from the fetal period until 3 weeks after birth are in the G1/S/G2-M phase of the cell cycle, with the G0 fraction estimated to be less than 0.02%17–19. Similarly, human HSC-enriched CD34+/CD38− cells from umbilical cord blood demonstrate increased propensity to transit through the cell cycle relative to their adult BM-derived counterparts20. To cope with the anabolic demands of cell division, murine fetal HSCs exhibit a greater reliance on oxidative metabolism compared to adult HSCs, with increased mitochondrial mass and oxygen consumption necessitating the buffering of reactive oxygen species by autophagic degradation21,22. Additionally, to mitigate the effects of elevated protein synthesis, murine fetal HSCs use bile acids from the maternal and fetal liver as chemical chaperones to suppress stress responses triggered by unfolded proteins23. These specific strategies allow fetal HSCs to maintain cellular integrity while supporting the proliferative demands of the rapidly developing organism.
Importantly, cell-extrinsic factors and inflammatory signals from the developing embryo and the maternal environment directly contribute to fetal HSC emergence, specification, and function24. Inflammatory signaling via tumor necrosis factor alpha (TNF-α) is required for the generation of HSCs in zebrafish, with Notch and NF-κB signaling pathways becoming activated to establish HSC fate25. Interferon (IFN) signaling has also been implicated in HSC emergence and maturation in both zebrafish and mouse models26,27. Converging evidence suggests that these inflammatory signals implicated in HSC emergence are largely sterile, with primitive neutrophils, and possibly primitive macrophages, contributing to the inflammatory milieu that drives HSPC expansion25,28–30. However, results in zebrafish indicate that inflammatory cytokine expression mediated by the commensal gut microbiome also promotes HSPC development31.
The expansion of murine fetal HSCs has classically been thought to occur in the vascularized fetal liver (FL) niche, with Nestin+NG2+ pericytes promoting HSC expansion32 and hepatic stellate and endothelial cells supporting HSC maintenance33. However, work in mice has challenged the model that fetal HSCs expand in the FL and progressively transition to the BM during the perinatal period, instead suggesting minimal contribution of FL HSCs to the adult pool, with most adult HSCs generated via postnatal expansion in the BM34,35. In this context, the placenta deserves further study as an extra-embryonic niche contributing to HSC expansion, given that phenotypic HSCs are known to expand in the murine placenta from E10.5 to E1136–38. The high frequency of HSPCs in the human placenta similarly suggests its role as an extra-embryonic niche for HSC expansion, an idea that is further supported by the ability of stromal cell lines from human placenta to support hematopoiesis in ex vivo co-culture39.
While the expansion of HSCs is critically important to the establishment of the adult HSC pool, the growing organism simultaneously has a vital need for erythroid cells to provide oxygen transport. Until recently, the mechanisms by which organisms balance these opposing requirements have been unclear. Lineage tracing experiments suggest that fetal HSCs only minimally contribute to the generation of progenitors and functional mature blood cells before birth, thereby prioritizing the growth of the HSC pool. Instead, a stem cell-independent, progenitor-based mechanism promotes the outburst of mature blood cells that support oxygen transport, stimulate rapid tissue growth, and establish the fetal immune system1,2. These stem cell-independent early embryonic multipotent progenitors (MPP) emerge as early as E10.5 in mice and produce multilineage progeny, with their contributions to blood production declining progressively postnatally but persisting up to 21 months of age1. The early embryonic MPPs also appear to be the dominant source of lifelong lymphoid output relative to HSC-derived MPPs, raising a possible connection to the decline in lymphoid production observed with age. Together, these findings start to map the numerous integrated mechanisms promoting the establishment of an HSC pool during fetal development and preparing the organism to support long-term blood production during adulthood. The extent to which these studies from mouse models are reflective of human HSC development remains to be investigated, with recent advances detailing the transcriptional evolution of human HSCs throughout the fetal period40 providing the foundation for further exploration.
The postnatal transition in HSC behavior revolves around establishing quiescence
Although fetal HSCs are primed to expand and generate the hematopoietic compartment, a significant transition occurs after birth. Postnatally, HSC behavior shifts from a constitutively proliferative state to one that prioritizes fitness during the reproductive lifespan by balancing long-term HSC survival with a demand-adapted response to stress. This abrupt transition in HSC behavior with adulthood is marked by the establishment of functional quiescence. Quiescence, a state of cell cycle and metabolic dormancy, protects HSCs from exhaustion. In mice, this marked functional change occurs between 3 and 4 weeks postnatally18. While HSCs from 1 to 3-week-old mice are still cycling, HSCs from 4-week-old mice and older are predominantly quiescent with more than 98% of HSCs in the G0 fraction17. Adult HSCs also have overall decreased self-renewal capacity compared to fetal HSCs, as evidenced by consistently lower levels of donor chimerism after transplantation into irradiated recipients18. This effect is likely a direct reflection of the changes in organismal needs for the HSC compartment, namely a shift from expansion to maintenance. In humans, this transition is suggested to occur at approximately 1 year of age, based on a marked decrease in the rate of telomere attrition at that time41. There may also be a role for inflammatory signaling in this transition, as the evolution from a fetal to an adult transcriptional state is associated with a prenatal spike in type I interferon (IFN) signaling42. However, the functional importance of this burst of inflammatory signaling remains unclear, with loss of the IFN-1 receptor in murine models resulting only in altered surface marker expression on HSPCs, and not affecting overall HSPC numbers or long-term mature blood cell production in neonates or adults43.
Although the transition to a quiescent phenotype occurs abruptly in the postnatal period, several murine studies using bulk and single-cell sequencing methods have indicated that the transcriptional underpinnings of this shift occur via progressive downregulation of fetal gene expression programs in conjunction with a gradual upregulation of adult gene expression42,44. While these transcriptional changes are likely connected with the differences in chromatin architecture between fetal and adult HSCs45,46, the functional consequences of perinatal epigenetic remodeling are just beginning to be explored. Advances in technology to examine chromatin organization and reorganization will herald more insights into how non-transcriptional changes affect the transition from fetal to adult HSC behavior.
The postnatal transition is accompanied by the migration of HSCs from the fetal liver to the adult BM niche, with interactions within the marrow microenvironment providing crucial signals reinforcing HSC dormancy. Mesenchymal stromal cells (MSC), endothelial cells, nerve cells, Schwann cells and various types of hematopoietic cells found in the BM niche all play intimate roles in governing HSC behavior during homeostasis and stress responses47. The endosteum, which lines the inner surface of the marrow cavity, seems particularly important for maintaining a highly quiescent subset of HSCs48,49. Bone-lining osteoblasts have been speculated to regulate HSC quiescence, but the relative importance of their various secreted factors, such as osteopontin, thrombopoietin and angiopoietin-1, remains unclear50. In the central marrow, megakaryocytes are critical for supporting HSC quiescence via the secretion of CXCL4 and TGFβ, both in terms of homeostatic maintenance and reinstatement following stress51–53. Similarly, sinusoidal endothelial cells, perivascular stromal cells, and non-myelinating Schwann cells are implicated in maintaining HSC quiescence through cytokines and chemokines, such as SCF and CXCL1254–57. These data show that the changes in the extrinsic environment following migration to the BM, along with cell-intrinsic transcriptional changes in the perinatal period, together reinforce the postnatal establishment of HSC quiescence.
Young adult HSCs balance pro-survival and stress responses for functional maintenance
In contrast to the highly proliferative state of fetal HSCs, the enforcement of quiescence in young adult HSCs is a crucial characteristic protecting the established stem cell pool and allowing for lifelong blood production. The majority of adult HSCs exist in the G0 phase of the cell cycle17,58, and the potential for long-term HSC engraftment resides predominantly within this fraction59. Genetic mouse models that perturb cell cycle dormancy and induce HSCs to proliferate lead to exhaustion of the HSC compartment60. However, although quiescence enforcement protects the HSC pool, it can also hinder the elimination of damaged cells. To manage these two opposite requirements, HSCs engage a delicate balance of pro-survival and stress response mechanisms to maintain a functional HSC compartment.
Preserving HSC numbers.
In line with the goal of protecting the adult HSC pool from diminution, HSCs engage pro-survival mechanisms in the face of various forms of stress. For example, murine HSCs show enhanced resistance to apoptosis following irradiation as compared to downstream progenitors61. This finding is underscored by decreased pro-apoptotic gene expression and increased expression of pro-survival genes within the Bcl2 gene family61. Additional studies in mouse models further emphasize the importance of Bcl2 family members in regulating HSC number and survival, with inducible Mcl-1 deletion resulting in HSC loss62 versus constitutive expression of Bcl2 resulting in HSC accumulation63,64. These results contrast with studies showing that irradiated human cord blood-derived HSCs demonstrate a slower rate of double-stranded break repair and increased apoptosis relative to progenitors65, highlighting different priming towards preservation versus elimination in fetal versus adult HSCs. In addition to anti-apoptotic mechanisms, murine HSCs also engage gene programs that prevent necroptosis in the face of TNF-α induced inflammatory stress, emphasizing the close regulation of cell death pathways that are implicated in adult HSC pro-survival mechanisms66.
Protecting HSC functional integrity.
In parallel with the mechanisms preserving HSC number, other stress-response pathways are activated to protect adult HSC functionality. Induction of the cellular recycling processes of autophagy and mitophagy in response to stress are among these strategies, with loss of these mechanisms resulting in impaired self-renewal activity and diminished regenerative potential67–69. Autophagy serves as an essential gatekeeper of quiescence by degrading activated healthy mitochondria and terminating OXPHOS-driven pro-differentiation signals, thereby allowing HSCs to return to a quiescence-enforcing glycolytic metabolism following cell cycle activation69. Activation of mitophagy via the peroxisome proliferator-activated receptor and fatty acid oxidation pathways degrades damaged mitochondria, similarly reinforcing quiescence and protecting self-renewal activity68.
HSCs also rely on carefully tuned regulation of protein synthesis rates, with both increases or decreases in protein synthesis leading to impaired function70. As part of this regulatory framework, the process of protein degradation via aggrephagy plays a role in maintenance of the normal proteome and protection from accumulation of misfolded proteins specifically in HSCs71. Evidence also suggests that chaperone-mediated autophagy (CMA), a selective form of autophagy that regulates the proteome by targeting proteins with specific motifs for lysosomal degradation, protects HSC function in adult mice, with elimination of CMA leading to impaired self-renewal72. In addition, human HSCs have also been shown to activate the unfolded protein response (UPR) in response to stress-induced accumulation of misfolded proteins, which predisposes damaged cells to apoptosis73. However, HSCs must reach a pathologic threshold of ER stress to activate apoptotic UPR mechanisms74. As such, these pathways cannot fully protect from the accumulation of damage over time. We speculate that evolutionary pressure has tuned the threshold of damage needed for the clearance of adult HSCs, with an array of stress-response mechanisms mitigating low level insults to preserve HSC numbers, and only severe damage resulting in cell death, thereby optimizing the fidelity of the adult HSC pool to support effective lifelong hematopoiesis.
Regulating HSC metabolic activation.
The intrinsic wiring of adult HSCs within the hypoxic BM microenvironment serves to reinforce quiescence and protect HSCs from the cellular consequences of engaging active metabolism. Quiescent murine HSCs rely primarily on glycolysis as an energy source, limiting the mitochondrial respiration and reactive oxygen species generation associated with oxidative phosphorylation (OXPHOS)75–77. This quiescence-enforcing metabolic state of adult HSCs contrasts with the propensity of fetal HSCs to employ oxidative metabolism to support proliferation21,22.
The metabolic behavior of adult murine HSCs is further regulated via the inheritance of cellular degradative machinery, with asymmetric inheritance of lysosomes in HSC daughter cells predicting future patterns of activation78. The asymmetric inheritance of autophagic and mitophagic machinery may similarly regulate HSC integrity68. Growing evidence suggests an important interplay between the metabolic wiring and epigenetic state of adult HSCs. For instance, quiescent murine HSCs have high acetyl-CoA to NAD+ ratios, providing a substrate for histone acetyl transferases, such as CRB-binding protein, which play a role in preserving HSC self-renewal79 . Murine HSCs also show epigenetic remodeling through DNA demethylation when undergoing metabolic activation69. Regulatory pathways enforcing quiescence are also starting to be identified and often connect to amino acids or other metabolic cues such as vitamin C (ascorbate)80, vitamin A (retinoic acid)81,82, and vitamin B3 (nicotinamide riboside)83, which control HSC fate through epigenetic and transcriptional regulations. Ongoing work is actively mapping the cellular metabolism of quiescent and activated states84, with vital importance for understanding the mechanisms maintaining HSC function.
Controlling blood regeneration.
Although young adult HSCs maintain a quiescent state during steady-state hematopoiesis, they are responsible for orchestrating a dynamic response to stress, including regeneration following blood loss85, treatment with myeloablative agents53, and infectious challenges86. In this context, Type 1 IFN signaling directly activates dormant HSCs, leading to cell cycle entry and allowing for a robust proliferative response to stressors86,87. However, this proliferative response is transient, with the HSC pool rapidly returning to protective quiescence88. The inflammatory cytokine TNF-α also contributes to HSC pro-survival responses, while simultaneously inducing myeloid progenitor apoptosis66. The effect of TNF-α in protecting against HSC exhaustion in the face of inflammation may be mediated by activation of the transcription factor PU.1, which represses cell cycle and protein synthesis programs. Similarly, activation of PU.1 after stimulation with the pro-inflammatory cytokine interleukin (IL)-1 limits HSC expansion and mediates a return to quiescence89,90. Altogether, the ability of young adult HSCs to preserve their number and functional integrity while responding dynamically to inflammatory stress allows the hematopoietic system to maintain homeostasis during the reproductive lifespan.
The future of HSC ontogeny research
The tremendous progress in our understanding of dynamic changes in HSC function, supported by technological advances such as single-cell sequencing and in vivo lineage-tracing methods, have laid the foundation for further exploration into the regulators of HSC behavior in different developmental stages. In the fetal context, additional investigations to uncover the interconnected layers of genetic, epigenetic, and metabolic regulation that cue the transition toward HSC quiescence will be important. In the adult context, gaining a better understanding of the cell intrinsic programs controlling stress-response pathways and reinforcing quiescence, or allowing HSCs to return to quiescence upon activation, will help developing a working knowledge of functional quiescence and adult HSC maintenance. Additionally, developing a more detailed understanding of the three-dimensional spatial organization of the BM niche, which represents one of the most notable barriers in the field, will improve our assessment of the cell-extrinsic programs regulating adult HSC behavior. Importantly, such studies will provide the essential basis to explore how cellular programs become dysfunctional in aging and disease.
HSC aging: normal mechanisms of maintenance giving rise to dysfunction
Although young adult HSCs employ finely tuned mechanisms to balance pro-survival and stress responses, these maintenance strategies provoke vulnerabilities that manifest with continued age, leading to decline in function. In this section, we examine HSC biology through the lens of antagonistic pleiotropy, whereby the same mechanisms that are important for reproductive fitness form the basis for functional decline during aging91. We focus on the insults that HSCs face because of their unique job requirements, and how this contributes to the declining functionality of old HSCs (Figures 2 and 3). As in previous stages of life, we highlight the central role of cell cycle regulation, niche cell reliance, inflammatory responses, cellular memory, and distinct metabolic regulation in driving HSC aging features including expansion of the HSC pool, decreased self-renewal, myeloid-biased differentiation due to loss of lymphoid cell production, and genomic instability leading to clonal hematopoiesis (CH) and oncogenic transformation.
Figure 2. Cell-intrinsic and cell-extrinsic features of HSC aging.
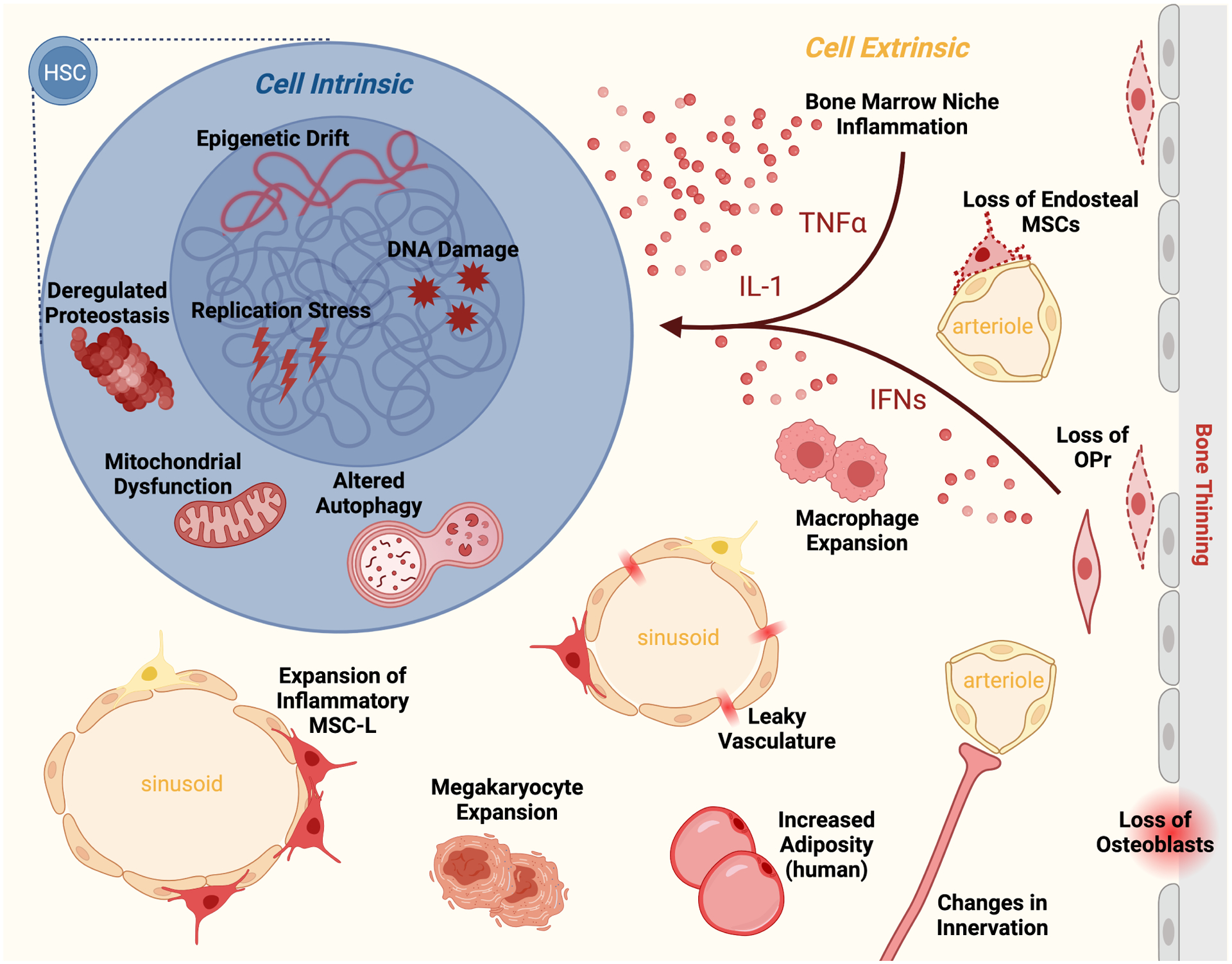
Old HSCs display many altered cell-intrinsic features including epigenetic drift, DNA damage, replication stress, deregulated proteostasis, mitochondrial dysfunction, and altered autophagy regulation. These cell-intrinsic changes are compounded by cell-extrinsic changes in the aged BM microenvironment including loss of specific niche cells (i.e., periarteriolar mesenchymal stromal cells (MSC), osteoblastic progenitors (OPr), osteoblasts), and accumulation of other cell types that contribute to an inflammatory milieu (i.e., inflammatory MSCs, macrophages, megakaryocytes) rich in pro-inflammatory factors (i.e., IL-1, IFNs, TNF-α, etc.). Old HSCs are also influenced by other niche changes, including altered innervation, increased adiposity, and leaky vasculature. Together, these features of HSC aging contribute to their functional decline.
Figure 3. Antagonistic pleiotropy of HSC features during young adulthood and old age.
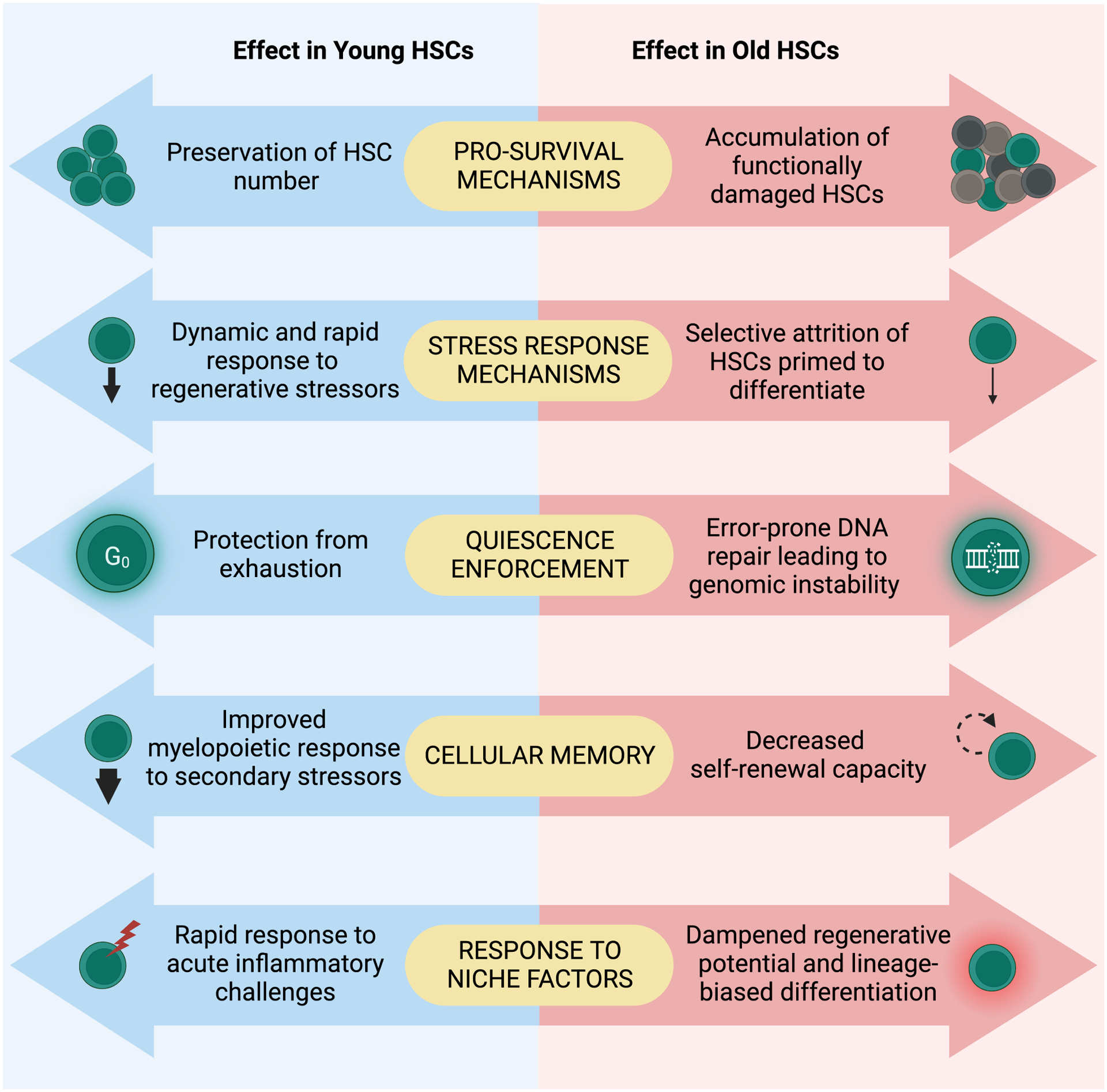
Many features of HSCs, such as pro-survival mechanisms, stress response mechanisms, quiescence, cellular memory, and response to niche factors, are beneficial for optimal HSC function and response to organismal demands in young adulthood but become detrimental and maladaptive during aging.
Cell cycle regulation and DNA damage repair
The dynamic regulation of cell cycling that predominantly maintains a state of quiescence is crucial for protecting adult HSCs from age-dependent exhaustion due to over-proliferation, as well as from the genetic and metabolic insults associated with activation. However, residence in G0 also restricts murine HSCs from engaging homologous recombination and repairing DNA with high fidelity, instead limiting them to fallible end-joining repair mechanisms61. HSC aging is also associated with downregulation of mini-chromosome maintenance genes and a loss of dormant origins, which cause old murine HSCs to experience delayed cell cycling and severe replication stress upon cycle entry, leading to further mutational accumulation and an inability to mount appropriate regenerative responses92. The repression of cell cycling and cell cycle gene expression is shared in human HSCs, and has been connected to reduced mitogenic signaling93. Nevertheless, intact DNA damage repair machinery94 and reduced apoptotic priming95 allow old HSCs to survive in the face of physiological levels of genotoxic stressors, which allows for the persistence of the compartment while also preventing clearance of mutated stem cells. Thus, although repression of cell cycling and specific mechanisms of DNA damage repair are necessary to ensure lifelong HSC survival, they inevitably lead to genomic instability during aging. This is particularly relevant to understanding the mechanistic basis for human genomic studies showing that somatic mutations associated with CH and leukemic transformation are increasingly prevalent with advanced age96. Indeed, some recurrent CH mutations are directly attributable to end-joining based repair mechanisms97. Still, CH-associated mutations are nearly ubiquitous at low variant allele frequencies (VAF) in healthy humans over 50 years old and do not uniformly expand to higher VAFs, implying that environmental factors are also crucial in this respect98,99. Given emerging evidence from mouse models that genetic lesions cooperate with external signals, in particular heightened inflammation, to drive CH100–103, we speculate that both are needed to drive clonal expansion.
Niche cell alterations and inflammaging
Cellular and molecular changes to BM niche cells are already apparent at mid-life, well before the emergence of the most striking features of HSC aging, such as numerical expansion and defective transplantation104,105. Although the order of events is not yet fully detangled, the specific relationships between niche alterations and hematopoietic aging features are beginning to be dissected. Changes in the cellular composition and functionality of the BM vasculature and lymphatic system with age play a direct role in driving defective hematopoietic regeneration of old murine HSCs104,106,107. Similarly, the nervous and mesenchymal components of aged murine BM contribute to myeloid-biased hematopoiesis and HSC dysfunction via localized production of pro-inflammatory cytokines104,108,109. Recent work has added mechanistic understanding to the crosstalk between mesenchymal and hematopoietic cells in aging. For example, an osteolectin-expressing subset of MSCs that is responsible for supporting osteogenesis and lymphopoiesis declines with age110, whereas the remaining osteoprogenitor cells secrete IL-1 that reprograms central marrow MSCs, causes defective hematopoietic regeneration, and impairs HSC function104. Furthermore, reduced insulin-like growth factor (IGF)-1 production by murine BM MSCs during middle age drives an age-related, myeloid-biased HSC phenotype105. These interweaved dependencies illustrate the complex influence of BM niche cell ecology to HSC aging, and future work will uncover additional cellular and molecular mediators of this crosstalk. For one, BM-resident T cells directly regulate HSCs111 and themselves decline in function with age112. We therefore speculate that alterations to BM T cells are an important unstudied driver of hematopoietic aging.
Chronic inflammation is a well-appreciated driver of organismal aging113 and is implicated in impairing HSC self-renewal activity66,87,89,114. Recent work shows that temporally discrete inflammatory events accelerate murine HSC aging and lead to irreversible attrition of self-renewal capacity115. Age-related inflammation in the BM microenvironment was first reported over a decade ago116. Since then, different niche cell types, including macrophages117, MSCs108 and osteoprogenitors104, as well as adrenergic innervation118 and dysbiosis of the gut microbiome119, have all been implicated as important sources of chronic inflammation promoting age-related HSC dysfunction. To return to the lens of antagonistic pleiotropy, whereas the ability of HSCs to respond to inflammatory signals enables rapid responses to infection and injury, it also renders HSCs susceptible to maladaptive, low-grade inflammation within the aged marrow cavity. Heterogeneity within the HSC compartment regarding the potential of an individual HSC to engage in differentiation versus self-renewal leads to variable responses to inflammation in different HSC subsets120,121. This adaptive behavior allows the HSC compartment en masse to mount a response to inflammatory stress while also retaining self-renewal capacity long-term. However, it likely leads to the selective attrition of those HSCs that are primed to differentiate122, resulting in a compartment that is less conducive to activation with age. It therefore seems likely that HSC responses to acute inflammatory episodes over the lifetime of the organism, as well as to chronic inflammation in the BM niche that progressively worsens with age, will favor the retention of a biased subset of HSCs.
Replicative history, epigenetic drift, and trained immunity
HSC aging is associated with the accumulation of epigenetic changes. Studies in both mice and humans indicate that these age-related epigenetic changes include shifts in histone marks, such as global increases in the active mark H3K4me3, global decreases in the repressive mark H3K27me3123,124, and loss of H4K16ac polarity125. Although global DNA methylation in HSCs is mostly stable during aging, site-specific alterations in DNA methylation are observed in old HSCs and correlate with methylome changes induced by proliferation126. Consistently, HSCs seem to carry memory of their activation history, with progressive cell divisions causing loss of self-renewal127.
Perhaps unsurprisingly, clonal tracking experiments have revealed that epigenetic heterogeneity plays a role in the disparate response of genetically normal murine HSCs to various stressors128. The functional consequences of epigenetic changes encompass an effect on HSC self-renewal capacity123 and may also account for the predominance of old HSC subsets that are responsible for altered responses to inflammatory challenges129. Additionally, studies have provided evidence for trained immunity occurring at the level of HSCs and their immediate downstream progeny130–132, where epigenetic changes following inflammatory challenges reinforce and expedite the transcriptional response to subsequent insults. These findings are again consistent with the theme of antagonistic pleiotropy, in that epigenetic memories of previous inflammatory challenges enable organismal fitness during adulthood by providing improved myelopoietic responses to pathogens, but also likely engender aging features by encoding chromatin states that bias toward the myeloid lineage.
HSC metabolism, proteostasis, and autophagy in aging
Mirroring the context-dependent regulation of cell cycling, response to inflammatory signals, and epigenetics, HSCs depend on discrete metabolic requirements that undergo marked alterations upon activation and during aging22. In particular, HSCs necessarily switch from glycolysis to OXPHOS to achieve the anabolic demands of activation and differentiation133. Notably, genetic models that interfere with transcriptional enforcement of glycolysis and activate OXPHOS lead to loss of HSC quiescence and self-renewal, paralleling functional features of aging134,135. Furthermore, there is a close coupling between HSC cell cycle activity and metabolic requirements22. This interlinked relationship is relevant to aging in part because asymmetric mitochondrial segregation imbues daughter cells with an additional form of memory associated with declining regenerative potential136. Given the intimate connection between metabolism and epigenetics, an important interplay likely exists between these two HSC regulatory mechanisms. Thus far, the metabolic-epigenetic axis, as it pertains to HSC aging, has been examined mainly through investigations of sirtuin (SIRT) genes, which are NAD+-dependent deacetylases regulating both histone architecture and cell signaling pathways with well-known connections to aging biology137. SIRT1 is involved in the activation of FOXO3A, which enforces HSC quiescence and stress resilience throughout life67, and SIRT1 deletion promotes an accelerated HSC aging phenotype138. SIRT2 is required to repress the NLRP3 inflammasome downstream of mitochondrial stress139. SIRT3 and SIRT7 both suppress mitochondrial metabolism, with SIRT7 specifically tuning protein translation and inducing a mitochondrial UPR that maintains HSC metabolism during aging140,141. Proteostasis more generally is also important for HSC aging, with evidence suggesting that middle-aged murine HSCs depend on the stress response gene HSF1 to maintain low translation rate and engraftment capability142, and that mechanisms of proteostasis are altered in old HSCs71.
Autophagy has long been associated with organismal aging, with interventions that increase lifespan, such as calorie restriction or rapamycin treatment, directly linked to autophagy induction137. Work over the past decade has demonstrated the specific relevance of autophagy to HSC aging, with disruption of macroautophagy69 as well as CMA resulting in premature aging phenotypes72. Still, a subset of old HSCs retains the ability to engage autophagy via a FOXO3A dependent pro-survival circuit67. Mouse studies using autophagy reporter systems have confirmed the functional importance of this process, showing that those HSCs with robust autophagy engagement are the most fit69 . In contrast, old HSCs that fail to engage autophagy display metabolic deregulation and impaired self-renewal activity69,71. Future work will indicate what triggers specific subsets of old HSCs to engage autophagy. Old HSCs also show reduced CMA activity, with genetic mouse models demonstrating impaired glycolysis and loss of proteostasis in CMA-deficient HSCs72. Collectively, this work illustrates that the mechanisms that regulate HSC metabolism undergo marked alterations during aging. We speculate that metabolically protective processes such as autophagy and CMA engagement become increasingly important upon aging to compensate for increased cellular stressors.
The future of HSC aging research
Studying HSC aging is critical because an accurate understanding of the underlying mechanisms will pave the way for root-cause therapies for age-related disease. The implications of such studies extend beyond the blood system, as hematopoietic aging contributes to solid tissue senescence143. Accordingly, understanding and targeting the defects arising in HSCs will likely have important consequences in other aging tissues. Although much has been uncovered regarding individual drivers of HSC aging, these processes are often studied in isolation, and future work will begin to examine their relationships to each other. HSC aging is a consequence of many cellular alterations, and such integrated approaches are needed to develop a comprehensive understanding of the process as a whole. Application of advanced technologies will also provide fresh insight into important aspects of HSC aging. For instance, clonal barcoding technologies have revealed unique features of fetal and adult HSC biology1,144, and it will be exciting to see what they will uncover in the aging context. Spatial transcriptomics applied to the aging BM niche is similarly promising in its potential to resolve the relationship between specific niche cell alterations and transcriptional features of aged HSCs. Additionally, CRISPR-enabled genome engineering broadens the feasibility of genetic studies on HSC aging in model organisms, providing the opportunity for more mechanistic insight. Lastly, much of the groundwork for understanding the connection between aging and the development of CH and hematological malignancies has been established in mouse models, which will guide translation into human hematopoietic systems with the goal of improving outcomes for individuals suffering from age-related blood diseases.
Intervention strategies for rejuvenating aged HSCs
Given that age-related changes in HSCs are thought to be key contributing factors to infectious susceptibility, reduced vaccination efficacy, leukemic emergence, and even solid tissue senescence, a focus has been placed on mitigating or reversing these changes for systemic functional benefit145. In general, studies have predominantly been performed in mouse models and have focused on restoring the engraftment potential and reverting the lineage-biased differentiation of old HSCs. Attempts at HSC rejuvenation have employed a diverse array of environmental, metabolic, and epigenetic strategies targeting mechanisms associated with HSC functional decline. These rejuvenation attempts can be broadly categorized into two types of approaches: 1) preventing HSC functional decline by diminishing the effect of aging, for example by reducing inflammation, preventing epigenetic alteration, or forcing metabolic reprogramming; and 2) restoring overall HSC functionality, for example by removing damaged HSCs. Here, we summarize the state of these efforts and their potential for translational applications aimed at improving immune responses in older individuals and mitigating the development of age-associated blood disorders (Table 1).
Table 1.
Attempts to rejuvenate aged HSCs.
| Intervention: | Methodology: | Result: | Reference: | |
|---|---|---|---|---|
| Systemic | Rapamycin treatment | 4mg/kg rapamycin injection every other day for 6 weeks in 22-month-old mice | More youthful HSC numbers and HSC cell cycling in vivo; improved engraftment and lymphoid output upon HSC transplantation; increased B cell progenitors and vaccine response in primary mice | Chen et al, 2009174 |
| Prolonged fasting | 8 cycles of 48 hour fasting in 18-month-old mice | Decreased frequency of myeloid-biased HSCs and increased peripheral blood lymphocytes in primary mice | Cheng et al, 2014183 | |
| Exposure to young bloodborne factors | Heterochronic parabiosis (1 month) or injection of young plasma (8x over 1 month) in 24-month-old mice | No effect on old HSC function (engraftment or myeloid bias upon HSC transplantation, division time in vitro) | Ho et al, 2021152 | |
| Exercise | 7-week long free access to running wheel in 18-month-old-mice | No effect on HSC function (engraftment or myeloid bias upon HSC transplantation) | Ho et al, 2021152 | |
| Calorie restriction | 30% calorie restriction for 9 months, analysis of 12-month-old mice | Decreased HSC number and decreased frequency of myeloid-biased HSCs in primary mice; improved engraftment and lymphoid output upon HSC transplantation | Tang et al, 2016184 | |
| 40% reduction in calorie intake in 3.5- to24-month-old mice | No effect on HSC function (engraftment or myeloid bias upon HSC transplantation) | Ho et al, 2021152 | ||
| Microenvironmental | Endothelial cell transplantation | Young endothelial cells infusion for 4 consecutive days in 24-month-old-mice followed by sublethal irradiation and whole BM transplantation 1 month later | Improved peripheral blood mature cell production following irradiation; improved engraftment and lymphoid output upon whole BM transplantation | Poulos et al, 2017155 |
| Small molecule mimetic of β3-adrenergic signaling | 12-week delivery with osmotic pumps in 20- to 24-month-old-mice | Improved engraftment and lymphoid output upon primary HSC transplantation and secondary BM transplantation | Maryanovich et al, 2018109 | |
| 8 weeks daily injection in progeroid mice | Decreased myeloid and increased lymphoid cells in peripheral blood | Ho et al, 2019118 | ||
| IGF1 stimulation | In vitro treatment of 14-month-old HSCs with 100ng/ml IGF1 | Rescue of γH2AX foci, Cdc42 polarization, mitochondrial membrane potential, and morphology | Young et al, 2021105 | |
| IL1R antagonism using Anakinra | 3 weeks daily injection in 24-month-old mice before HSC transplantation | Increased lymphoid output in HSC transplantation; no difference in engraftment | Kovtonyuk et al, 2022119 | |
| 2 weeks daily injection in 24-month-old mice before immunophenotyping and HSC transplantation | Decreased HSC numbers in primary mice; no effect on HSC function (engraftment or myeloid bias upon HSC transplantation) | Mitchell et al, 2023104 | ||
| 2 weeks daily injection starting 2 days prior to 5-FU myeloablation in 24-month-old mice | Improved regenerative response; Increased B cell and red blood cell recovery in peripheral blood; increased lymphoid-biased progenitors and erythroid progenitors in BM | Mitchell et al, 2023104 | ||
| Netrin-1 supplementation | 10 injections over 2-week period of 4 μg of recombinant murine Netrin-1 in 18-month-old mice | Improved engraftment and lymphoid output in primary mice | Ramalingam et al, 2023185 | |
| Microbiome modulation | 1-year-old germ-free mice or 8-week antibiotic treatment in 2-year-old mice | Improved lymphoid output upon HSC transplantation; no difference in engraftment | Kovtonyuk et al, 2022119 | |
| Oral gavage in 20- to 24-month mice with fecal slurry from 7- to 8-week-old mice | Improved B cell numbers in primary mice, improved chimerism and HSC engraftment in transplantation | Zeng et al., 2023160 | ||
| Epigenetic | Cdc42 inhibition | In vitro treatment with small molecule inhibitor (CASIN) in 20- to 26-month-old mice | Rescue of Cdc42 and H4K16 acetylation polarization; improved myeloid bias upon primary HSC transplantation; improved engraftment and myeloid bias upon secondary BM transplantation | Florian et al, 2012165 |
| Satb1 overexpression | Retroviral transduction of LSK cells from 2-year-old mice In vitro | Improved growth of T cells, B cells and NK cells in co-culture with lymphopoiesis supporting stromal cells | Satoh et al, 2013166 | |
| Yamanaka factor reprogramming | iPSC generation from HSC clones from 23-month-old mice followed by redifferentiation into HSCs (“iHSC”) | T cell chimerism following transplantation of iHSC derived from aged HSC clones which previously did not exhibit T cell output | Wahlestedt et al, 2017167 | |
| Metabolic/mitochondrial | Rapamycin treatment | 4 mg/kg every other day for 6 weeks in 22-month-old mice | Decreased HSC numbers; improved BrdU incorporation; decreased p16 and Arf expression; improved engraftment and lymphoid output upon HSC transplantation; improved vaccine response in vivo | Chen et al, 2009174 |
| Sirt2 overexpression | Lentiviral transduction of HSCs from 20- to 24-month-old mice in vitro | Decreased caspase-1 activation; improved engraftment and lymphoid output upon HSC transplantation | Luo et al, 2019139 | |
| Sirt3 overexpression | Lentiviral transduction of lineage-depleted BM cells from 18- 24-month-old mice in vitro | Improved engraftment upon HSC transplantation | Brown et al, 2013140 | |
| Sirt7 overexpression | Lentiviral transduction of murine HSCs in vitro | Reduced mitochondrial protein folding stress; improved engraftment and lymphoid output upon HSC transplantation | Mohrin et al, 2015141 | |
| NLRP3/Caspase-1 knockdown | Lentiviral shRNA knockdown of HSCs from 20- to 24-month-old mice in vitro | Improved engraftment and lymphoid output upon HSC transplantation | Luo et al, 2019139 | |
| Nicotinamide riboside supplementation | 3mg/mL in drinking water for 8 weeks in 20- to 24-month-old mice | Decreased HSC and GMP frequency and number in primary mice; more youthful HSC transcriptome; improved metabolic parameters; improved engraftment in HSC transplantation | Sun et al, 2021176 | |
Many groups have applied interventions to old HSCs, aimed at returning them to a more youthful state. These attempts span a wide range of techniques and biological mechanisms and have had mixed success. GMP, granulocyte-macrophage progenitor, iPSC: induced pluripotent stem cell, LSK: lineage−/Sca-1+/c-Kit+, shRNA: small hairpin RNA, 5-FU: 5-fluorouracil.
Unique challenges to HSC rejuvenation
Experiments suggesting that the rate of aging can be modulated and delayed in model organisms via dietary, pharmacological, and genetic means have resulted in growing scientific and public interest in interventions aimed at functional rejuvenation of aged tissues146. Some approaches have been described as “universal” rejuvenation strategies, due to broad successes in rejuvenation across multiple tissues. For example, heterochronic parabiosis rejuvenates muscle function, bone healing, and neurogenesis in aged mice147–149. Additionally, life-long caloric restriction improves neural stem cell activity and reduces cardiovascular disease in aged mice and primates150. Exercise has also been described as an anti-aging strategy, with effects spanning multiple tissues and leading to reduced risk of cardiovascular disease as well as improved muscle function, cognition, and neurogenesis151. However, these bloodborne systemic rejuvenation approaches have not been successful in functionally rejuvenating old HSCs152,153 or improving other aspects of the aging blood system, such as diminished thymic function154. For instance, the lodging and long-term residence of old HSCs in the young BM cavity, which results from cross-over observed in heterochronic parabiosis pairs, does not restore old HSC regenerative potential152. Additionally, transplantation of old murine HSCs into young recipients does not revert age-associated DNA methylation changes153, and young plasma injection does not dampen chronic inflammation in the aged BM microenvironment152. This lack of success likely reflects unique aspects of the biology of HSCs and their niche compared to other tissues and stem cell populations and demonstrates the interdependency of cell-intrinsic deregulations and cell-extrinsic environmental changes in damaging old HSC function. Given the limited effectiveness of broad, systemic rejuvenation approaches, strategies that take advantage of our growing knowledge of age-related changes in HSC properties and in the BM niche, such as the metabolic-epigenetic axis, will be important to continue developing promising interventions. Meanwhile, the field will continue to grapple with the cost and time required to age relevant mouse models, which limits mechanistic insight into the genetics of the aging process, and the challenge of translating findings in model organisms to humans, which age on a different timescale.
Targeting niche cell alterations and inflammaging
Targeting specific cellular, molecular, and structural changes occurring in the aged BM niche has shown promise in restoring HSC function and more youthful blood production. Transplantation of young BM endothelial cells into aged recipients can improve hematopoietic regeneration, presumably by compensating for age-related decline in the number and functionality of these vascular cells155. BM MSCs also undergo alteration with age and perhaps could be similarly harnessed, but the translational potential is currently limited by the same barriers encountered by all cell-based therapies in clinical applications156. From the structural perspective, murine BM stiffens with age, and a study suggests that modulating BM stiffness ex vivo may be a potential rejuvenation strategy157. Pharmacological modulation of microenvironmental alterations has also shown some success, with a chemical agonist mimicking adaptive β3-adrenergic signaling in the aging BM niche improving old HSC functionality and regenerative potential in transplantation assays109. The same compound has been effective at rebalancing lineage output in a progeroid model of aging118, and future work will determine whether boosting β3-adrenergic signaling may also be beneficial in physiological aging. Artificial restoration of secreted factors has also shown promise, with recombinant osteopontin treatment ex vivo improving old HSC function158 and ex vivo treatment with IGF-1 improving the myeloid bias of 12-month-old HSCs105. However, given that increased signaling through IGF-1 promotes organismal aging in mice and other mammalian models146, artificial activation of this pathway may be challenging to implement in humans, unless employed in a highly targeted manner.
The low-grade, chronic inflammation of the aged BM niche provides another attractive opportunity for intervention159. For instance, in aged mice, normalizing overactive IL-1 signaling via the FDA-approved IL-1 antagonist, Anakinra, improves the hematopoietic response to a regenerative challenge imposed by the chemotherapeutic agent 5-fluorouracil104. Lifelong reduction of IL-1 signaling in mice lacking the IL-1 receptor also prevents the emergence of inflammatory MSC subsets in the central marrow and improves aged blood production and old HSC engraftment potential104. In addition, antibiotic suppression of the microbiota or pharmacological blockade of IL-1 signaling are able to reverse the myeloid-biased output of old HSCs119. Further support for modulation of the commensal microbiome as a tractable HSC rejuvenation strategy comes from a study showing that fecal transplantation from young mice improves HSC function by suppressing age-related inflammation160. Moreover, both IL-1100 and microbiome-driven IL-6161,162 signaling have been implicated in the progression of Tet2 mutant CH in mouse models, suggesting a potential role for targeted interventions to minimize the negative health consequences associated with CH163. In fact, clinical trial data suggest that treatment of patients harboring TET2-mutant CH with the anti-IL-1β antibody canakinumab decreases the incidence of cardiovascular events162. Collectively, these studies suggest that targeting either specific pro-inflammatory molecules or broad cellular changes in the BM niche can maintain a more youthful blood production and forestall the progression of age-related disease. Still, such strategies do not correct the intrinsic changes occurring in old HSCs that are implicated in age-related dysfunction.
Targeting HSC epigenetic drift
Correcting the epigenetic and transcriptional alterations occurring in aged HSCs is another window for therapeutic intervention, especially given the range of small molecules available for epigenetic tuning164. Both genetic and pharmacological approaches have been used to alter the epigenome of aged HSCs. Inhibition of the small RhoGTPase Cdc42 can restore the spatial distribution of H4K16 acetylation in old HSCs, improving their engraftment potential and lineage bias in transplantation assays165. Overexpression of Satb1, a lymphoid differentiation transcriptional regulator, also improves the lineage output of old HSCs in vitro166. Growing evidence suggests that epigenetic reprogramming instead of epigenetic correction might be a better avenue for functional rejuvenation of old HSCs. Notably, HSCs derived from induced pluripotent stem cells (iPSC) that originated from the mature cell progeny of lineage-biased old HSCs have successfully rejuvenated lineage-balanced blood production and engraftment potential in vivo167. This finding has coincided with broad interest in the aging field in using epigenetic reprogramming via Yamanaka transcription factor (Oct4, Sox2, Klf4, and c-Myc) overexpression to push aged cells and tissues towards an embryonic state168. Some success has been achieved with this reprogramming strategy in various organs169–172, although most of these studies rely on settings of progeria or inflammatory challenge and report epigenetic “clock”-based assessments of rejuvenation in lieu of functional measurements. It therefore remains to be seen whether Yamanaka factor induction in vivo can delay hematopoietic aging or rejuvenate old HSC function. Future implementation of such reprogramming strategies must also include consideration of inherent associated risks, such as teratoma formation171. Nevertheless, there is great promise for the development of successful interventions that target epigenetic drift, particularly as our knowledge of the functional implications of these changes continues to grow.
Metabolic-targeted rejuvenation approaches
Given the important role of metabolic deregulation in HSC aging, it is not surprising that this has been a prominent target for rejuvenation approaches. Modulation of the mTOR pathway through calorie restriction or small molecule inhibition is the most well-studied anti-aging strategy and is effective in an array of model organisms173. In the context of HSC aging, rapamycin treatment improves engraftment in transplantation assays as well as the vaccination response of old mice to influenza virus174. Modulation of the mTOR pathway has also been extended to human clinical trials, with data suggesting that the elderly patients’ response to seasonal influenza vaccines can be improved after treatment with an mTOR inhibitor175. Given the immunosuppressive effects of mTOR inhibitors, intermittent low-dose treatments will likely provide the most favorable risk-benefit ratio, if large-scale trials support their widespread implementation. Targeting of sirtuins has also garnered interest as a method of improving HSC functionality with aging. In particular, overexpressing SIRT2 improves the engraftment potential of old HSCs by dampening mitochondrial stress and NLRP3 inflammasome activation139. Overexpression of SIRT3 or SIRT7 also improves old HSC function in transplantation assays, by reducing oxidative stress140 and mitochondrial protein folding stress141, respectively. Sirtuins are NAD+-dependent enzymes, and several studies have suggested that a systemic decline in NAD+ levels with aging could contribute to a broad decrease in SIRT activity. To combat this, nicotinamide riboside (NR), which is endogenously converted to NAD+, has been studied as a method to restore NAD+ levels and promote youthful metabolic function in aged HSCs. Indeed, continuously treating old mice with NR for 8 weeks shifts old HSCs toward a more youthful transcriptional state and improves their reconstitution potential176. However, NR treatment has not demonstrated a significant impact on other functional features of HSC aging, such as lineage-bias, and old HSCs revert to decayed metabolic activity and function upon treatment discontinuation. However, despite current limitations, rejuvenation approaches that target metabolism, such as dietary strategies, are often among the most accessible interventions, and therefore represent a critical area of investigation.
Killing dysfunctional old HSCs
Another method to improve the overall function of the aged HSC pool is to selectively ablate dysfunctional cells. Several features of old HSCs, including altered metabolism, mutations, deregulated cell cycling, and neo-antigen presentation, can provide potential targets for removal. The metabolic heterogeneity of the aged HSC compartment, wherein a subset of autophagy-engaging HSCs retains the highest functionality upon transplantation69, may provide one such avenue for selectively ablating dysfunctional old HSCs. Such metabolic targeting has shown promise in the context of acute myeloid leukemia (AML), where leukemic stem cells (LSCs) have different metabolic properties from healthy HSCs. For example, LSCs demonstrate greater dependence on mitochondrial respiration for their survival, leaving them vulnerable to therapies that reduce OXPHOS. Methods for selectively killing LSCs by targeting their altered metabolism include inhibiting amino acid metabolism177, inducing cell cycle entry and exhaustion by inhibiting fatty acid oxidation178, and BCL-2 inhibition179. BCL-2 and BCL-xL inhibition using the compound ABT263 attenuates age-related changes of non-malignant old murine HSCs via senescent cell clearence180. These targeting strategies may provide a future avenue for selective removal of dysfunctional aged HSCs with altered metabolism to improve the fitness of the remaining aged HSC pool.
Another promising method to distinguish the most dysfunctional aged HSCs might be found in the antigen presentation ability of HSCs. Both murine and human HSCs constitutively express MHC-II antigen presentation machinery111 and, in AML, antigen-presentation correlates with stemness of LSCs. The presentation of immunogenic neoantigens by aberrant HSCs activates antigen specific CD4+ T cells that drive HSC differentiation and subsequent exhaustion111, suggesting a mechanism for selective elimination of HSCs undergoing malignant transformation. The use of PARP inhibitors for AML treatment has also been successful in inducing expression of the C-type lectin-like receptor NKG2D on LSCs, making them vulnerable to killing by NK cells181. These few examples highlight the therapeutic potential of killing LSCs to prevent AML development and progression and provide a roadmap on how clearance of dysfunctional old HSCs could be used for rejuvenating the aged HSC compartment. In HSC aging, enhancement of the immune response to neoantigens or induction of NKG2D ligands may allow for the selective elimination of old HSCs harboring mutations, thereby improving the functionality of the aged HSC compartment en masse. However, further progress in this area will rely on the identification of strategies to selectively induce expression of either neoantigens or NKG2D ligands in old HSCs, which has thus far been challenging.
The future of HSC rejuvenation
The development of translatable rejuvenation approaches for the hematopoietic system will be considerably helped by continued advances in our methods for assessing HSC function, a more rigorous definition of HSC aging, and the establishment of better criteria to claim successful rejuvenation of aged HSCs. Choosing effective intervention strategies will necessarily rely on distinguishing indispensable adaptive survival responses from targetable maladaptive features of aging. For decades, assessments of HSC function have relied primarily on transplantation, a method which is constrained by its reliance on homing and engraftment processes, despite the emergence of many new cellular and functional attributes of young and old HSC states. Other methods for assessing age, such as ‘epigenetic clocks’ for determination of epigenetic age, are limited by a lack of specificity to HSCs and an uncertain correlation to functionality and age-related phenotypes. We anticipate that multi-dimensional assessments that include regeneration challenges, immune response to infection and vaccination, and assessments of the epigenetic and metabolic cell state will be important in validating improvements in rejuvenation approaches. With our growing understanding of HSC aging, the potential applications of HSC rejuvenation strategies to human health and disease will continue to be explored. Notably, HSCs are uniquely poised for ex vivo interventions, due to their ability to be isolated via pharmacological mobilization into the peripheral blood. Technological advances that allow human HSCs to be genetically edited ex vivo and subsequently transplanted back into the BM niche provide a mechanism for genetically driven rejuvenation strategies. Such gene therapy strategies are already being applied in other clinical areas182, heralding new opportunities for translational approaches. Although strategies for HSC rejuvenation remain in early stages, these methods open the door to designing rational methods for future geromedical treatments.
Conclusions: toward an integrated understanding of HSC aging
The HSC compartment is governed by a complex network of cell-intrinsic and cell-extrinsic factors that enable a balance between self-renewal and differentiation to maintain long-term blood production. Here, we reviewed the features governing HSC behavior throughout ontogeny that reinforce this balance, highlighting how pro-survival mechanisms lead to maladaptation with age. As additional studies continue to dissect the genetic, epigenetic, metabolic, and inflammatory drivers of aging, new strategies to combat aging phenotypes via functional rejuvenation approaches will follow. Importantly, these future studies must account for heterogeneity in the kinetics of aging, which in mouse models may be obscured by a selection bias inherent to the prominent use of 24-month-old mice from the C57Bl6 genetic background that are fit to survive into old age. Alternative models including other strains of mice, other species, and improved human cell systems will aid in confirming the applicability of insights from established models. Longitudinal studies that encompass the full spectrum of aging - from birth, through different stages of adulthood, and into old age – have the potential to contextualize this heterogeneity, particularly given advances in sequencing technologies for in-depth profiling of genetic and epigenetic landscapes. Integration of these genetic and epigenetic profiles at different stages of life with functional features, such as repopulation ability and response to inflammation, may open the door to an era of personalized medicine strategies for HSC rejuvenation.
Acknowledgments
M.K. discloses support for this work from ASH RTAF Award, C.A.M. from NIH F31HL160207, M.A.P. from NIH TL1DK136048, and E.P. from NIH R01AG073599 and CU21-0225 from the Milky Way Research Foundation. The funders had no role in the preparation of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
E.P. is a member of Cell Stem Cell advisory board. The authors declare no other competing interests.
References
- 1.Patel SH, Christodoulou C, Weinreb C, Yu Q, da Rocha EL, Pepe-Mooney BJ, Bowling S, Li L, Osorio FG, Daley GQ, et al. (2022). Lifelong multilineage contribution by embryonic-born blood progenitors. Nature 606, 747–753. 10.1038/s41586-022-04804-z. [DOI] [PubMed] [Google Scholar]
- 2.Yokomizo T, Ideue T, Morino-Koga S, Tham CY, Sato T, Takeda N, Kubota Y, Kurokawa M, Komatsu N, Ogawa M, et al. (2022). Independent origins of fetal liver haematopoietic stem and progenitor cells. Nature 609, 779–784. 10.1038/s41586-022-05203-0. [DOI] [PubMed] [Google Scholar]
- 3.Pietras EM, Warr MR, and Passegué E (2011). Cell cycle regulation in hematopoietic stem cells. J. Cell Biol 195, 709–720. 10.1083/jcb.201102131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orkin SH, and Zon LI (2008). Hematopoiesis: An Evolving Paradigm for Stem Cell Biology. Cell 132, 631–644. 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyd KL, and Bolon B (2022). Embryonic and Fetal Hematopoiesis. In Schalm’s Veterinary Hematology (John Wiley & Sons, Ltd; ), pp. 1–8. 10.1002/9781119500537.ch1. [DOI] [Google Scholar]
- 6.Dzierzak E, and Bigas A (2018). Blood Development: Hematopoietic Stem Cell Dependence and Independence. Cell Stem Cell 22, 639–651. 10.1016/j.stem.2018.04.015. [DOI] [PubMed] [Google Scholar]
- 7.McGrath KE, and Palis J (2005). Hematopoiesis in the yolk sac: more than meets the eye. Exp. Hematol 33, 1021–1028. 10.1016/j.exphem.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 8.Wittamer V, and Bertrand JY (2020). Yolk sac hematopoiesis: does it contribute to the adult hematopoietic system? Cell. Mol. Life Sci 77, 4081–4091. 10.1007/s00018-020-03527-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamane T (2018). Mouse Yolk Sac Hematopoiesis. Front. Cell Dev. Biol 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ciriza J, Thompson H, Petrosian R, Manilay JO, and García-Ojeda ME (2013). The migration of hematopoietic progenitors from the fetal liver to the fetal bone marrow: Lessons learned and possible clinical applications. Exp. Hematol 41, 411–423. 10.1016/j.exphem.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Hao Q, Shah A, Thiemann F, Smogorzewska E, and Crooks G (1995). A functional comparison of CD34 + CD38− cells in cord blood and bone marrow. Blood 86, 3745–3753. 10.1182/blood.V86.10.3745.bloodjournal86103745. [DOI] [PubMed] [Google Scholar]
- 12.Holyoake TL, Nicolini FE, and Eaves CJ (1999). Functional differences between transplantable human hematopoietic stem cells from fetal liver, cord blood, and adult marrow. Exp. Hematol 27, 1418–1427. 10.1016/s0301-472x(99)00078-8. [DOI] [PubMed] [Google Scholar]
- 13.Lansdorp PM, Dragowska W, and Mayani H (1993). Ontogeny-related changes in proliferative potential of human hematopoietic cells. J. Exp. Med 178, 787–791. 10.1084/jem.178.3.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Micklem HS, Ford CE, Evans EP, Ogden DA, and Papworth DS (1972). Competitive in vivo proliferation of foetal and adult haematopoietic cells in lethally irradiated mice. J. Cell. Physiol 79, 293–298. 10.1002/jcp.1040790214. [DOI] [PubMed] [Google Scholar]
- 15.Morrison SJ, Hemmati HD, Wandycz AM, and Weissman IL (1995). The purification and characterization of fetal liver hematopoietic stem cells. Proc. Natl. Acad. Sci 92, 10302–10306. 10.1073/pnas.92.22.10302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pawliuk R, Eaves C, and Humphries RK (1996). Evidence of Both Ontogeny and Transplant Dose-Regulated Expansion of Hematopoietic Stem Cells In Vivo. Blood 88, 2852–2858. 10.1182/blood.V88.8.2852.bloodjournal8882852. [DOI] [PubMed] [Google Scholar]
- 17.Bowie MB, McKnight KD, Kent DG, McCaffrey L, Hoodless PA, and Eaves CJ (2006). Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. J. Clin. Invest 116, 2808–2816. 10.1172/JCI28310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bowie MB, Kent DG, Dykstra B, McKnight KD, McCaffrey L, Hoodless PA, and Eaves CJ (2007). Identification of a new intrinsically timed developmental checkpoint that reprograms key hematopoietic stem cell properties. Proc. Natl. Acad. Sci 104, 5878–5882. 10.1073/pnas.0700460104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nygren JM, Bryder D, and Jacobsen SEW (2006). Prolonged cell cycle transit is a defining and developmentally conserved hemopoietic stem cell property. J. Immunol. Baltim. Md 1950 177, 201–208. 10.4049/jimmunol.177.1.201. [DOI] [PubMed] [Google Scholar]
- 20.da Silva CL, Gonçalves R, Porada CD, Ascensão JL, Zanjani ED, Cabral JMS, and Almeida-Porada G (2009). Differences amid bone marrow and cord blood hematopoietic stem/progenitor cell division kinetics. J. Cell. Physiol 220, 102–111. 10.1002/jcp.21736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manesia JK, Xu Z, Broekaert D, Boon R, van Vliet A, Eelen G, Vanwelden T, Stegen S, Van Gastel N, Pascual-Montano A, et al. (2015). Highly proliferative primitive fetal liver hematopoietic stem cells are fueled by oxidative metabolic pathways. Stem Cell Res. 15, 715–721. 10.1016/j.scr.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 22.Nakamura-Ishizu A, Ito K, and Suda T (2020). Hematopoietic Stem Cell Metabolism during Development and Aging. Dev. Cell 54, 239–255. 10.1016/j.devcel.2020.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sigurdsson V, Takei H, Soboleva S, Radulovic V, Galeev R, Siva K, Leeb-Lundberg LMF, Iida T, Nittono H, and Miharada K (2016). Bile Acids Protect Expanding Hematopoietic Stem Cells from Unfolded Protein Stress in Fetal Liver. Cell Stem Cell 18, 522–532. 10.1016/j.stem.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 24.Collins A, Mitchell CA, and Passegué E (2021). Inflammatory signaling regulates hematopoietic stem and progenitor cell development and homeostasis. J. Exp. Med 218, e20201545. 10.1084/jem.20201545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Espín-Palazón R, Stachura DL, Campbell CA, García-Moreno D, Del Cid N, Kim AD, Candel S, Meseguer J, Mulero V, and Traver D (2014). Proinflammatory signaling regulates hematopoietic stem cell emergence. Cell 159, 1070–1085. 10.1016/j.cell.2014.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sawamiphak S, Kontarakis Z, and Stainier DYR (2014). Interferon gamma signaling positively regulates hematopoietic stem cell emergence. Dev. Cell 31, 640–653. 10.1016/j.devcel.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim PG, Canver MC, Rhee C, Ross SJ, Harriss JV, Tu H-C, Orkin SH, Tucker HO, and Daley GQ (2016). Interferon-α signaling promotes embryonic HSC maturation. Blood 128, 204–216. 10.1182/blood-2016-01-689281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frame JM, Kubaczka C, Long TL, Esain V, Soto RA, Hachimi M, Jing R, Shwartz A, Goessling W, Daley GQ, et al. (2020). Metabolic Regulation of Inflammasome Activity Controls Embryonic Hematopoietic Stem and Progenitor Cell Production. Dev. Cell 55, 133–149.e6. 10.1016/j.devcel.2020.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Esain V, Teng L, Xu J, Kwan W, Frost IM, Yzaguirre AD, Cai X, Cortes M, Maijenburg MW, et al. (2014). Inflammatory signaling regulates embryonic hematopoietic stem and progenitor cell production. Genes Dev. 28, 2597–2612. 10.1101/gad.253302.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tie R, Li H, Cai S, Liang Z, Shan W, Wang B, Tan Y, Zheng W, and Huang H (2019). Interleukin-6 signaling regulates hematopoietic stem cell emergence. Exp. Mol. Med 51, 1–12. 10.1038/s12276-019-0320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong D, Jiang H, Zhou C, Ahmed A, Li H, Wei X, Lian Q, Tastemel M, Xin H, Ge M, et al. (2023). The microbiota regulates hematopoietic stem and progenitor cell development by mediating inflammatory signals in the niche. Cell Rep. 42, 112116. 10.1016/j.celrep.2023.112116. [DOI] [PubMed] [Google Scholar]
- 32.Khan JA, Mendelson A, Kunisaki Y, Birbrair A, Kou Y, Arnal-Estapé A, Pinho S, Ciero P, Nakahara F, Ma’ayan A, et al. (2016). Fetal liver hematopoietic stem cell niches associate with portal vessels. Science 351, 176–180. 10.1126/science.aad0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee Y, Leslie J, Yang Y, and Ding L (2020). Hepatic stellate and endothelial cells maintain hematopoietic stem cells in the developing liver. J. Exp. Med 218, e20200882. 10.1084/jem.20200882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hall TD, Kim H, Dabbah M, Myers JA, Crawford JC, Morales-Hernandez A, Caprio CE, Sriram P, Kooienga E, Derecka M, et al. (2022). Murine fetal bone marrow does not support functional hematopoietic stem and progenitor cells until birth. Nat. Commun 13, 5403. 10.1038/s41467-022-33092-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ganuza M, Hall T, Myers J, Nevitt C, Sánchez-Lanzas R, Chabot A, Ding J, Kooienga E, Caprio C, Finkelstein D, et al. (2022). Murine foetal liver supports limited detectable expansion of life-long haematopoietic progenitors. Nat. Cell Biol 24, 1475–1486. 10.1038/s41556-022-00999-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao X, Xu C, Asada N, and Frenette PS (2018). The hematopoietic stem cell niche: from embryo to adult. Dev. Camb. Engl 145, dev139691. 10.1242/dev.139691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gekas C, Dieterlen-Lièvre F, Orkin SH, and Mikkola HKA (2005). The placenta is a niche for hematopoietic stem cells. Dev. Cell 8, 365–375. 10.1016/j.devcel.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 38.Ottersbach K, and Dzierzak E (2005). The murine placenta contains hematopoietic stem cells within the vascular labyrinth region. Dev. Cell 8, 377–387. 10.1016/j.devcel.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 39.Robin C, Bollerot K, Mendes S, Haak E, Crisan M, Cerisoli F, Lauw I, Kaimakis P, Jorna R, Vermeulen M, et al. (2009). Human placenta is a potent hematopoietic niche containing hematopoietic stem and progenitor cells throughout development. Cell Stem Cell 5, 385–395. 10.1016/j.stem.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calvanese V, Capellera-Garcia S, Ma F, Fares I, Liebscher S, Ng ES, Ekstrand S, Aguadé-Gorgorió J, Vavilina A, Lefaudeux D, et al. (2022). Mapping human haematopoietic stem cells from haemogenic endothelium to birth. Nature 604, 534–540. 10.1038/s41586-022-04571-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rufer N, Brümmendorf TH, Kolvraa S, Bischoff C, Christensen K, Wadsworth L, Schulzer M, and Lansdorp PM (1999). Telomere Fluorescence Measurements in Granulocytes and T Lymphocyte Subsets Point to a High Turnover of Hematopoietic Stem Cells and Memory T Cells in Early Childhood. J. Exp. Med 190, 157–168. 10.1084/jem.190.2.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Y, Kong W, Yang W, Patel RM, Casey EB, Okeyo-Owuor T, White JM, Porter SN, Morris SA, and Magee JA (2020). Single-Cell Analysis of Neonatal HSC Ontogeny Reveals Gradual and Uncoordinated Transcriptional Reprogramming that Begins before Birth. Cell Stem Cell 27, 732–747.e7. 10.1016/j.stem.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y, Yang W, Wang HC, Patel RM, Casey EB, Denby ED, and Magee JA (2023). Basal type I interferon signaling has only modest effects on neonatal and juvenile hematopoiesis. Blood Adv, bloodadvances.2022008595. 10.1182/bloodadvances.2022008595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Copley MR, Babovic S, Benz C, Knapp DJHF, Beer PA, Kent DG, Wohrer S, Treloar DQ, Day C, Rowe K, et al. (2013). The Lin28b–let-7–Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nat. Cell Biol 15, 916–925. 10.1038/ncb2783. [DOI] [PubMed] [Google Scholar]
- 45.Chen C, Yu W, Tober J, Gao P, He B, Lee K, Trieu T, Blobel GA, Speck NA, and Tan K (2019). Spatial Genome Re-organization between Fetal and Adult Hematopoietic Stem Cells. Cell Rep. 29, 4200–4211.e7. 10.1016/j.celrep.2019.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao P, Chen C, Howell ED, Li Y, Tober J, Uzun Y, He B, Gao L, Zhu Q, Siekmann AF, et al. (2020). Transcriptional regulatory network controlling the ontogeny of hematopoietic stem cells. Genes Dev. 34, 950–964. 10.1101/gad.338202.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schepers K, Campbell TB, and Passegué E (2015). Normal and Leukemic Stem Cell Niches: Insights and Therapeutic Opportunities. Cell Stem Cell 16, 254–267. 10.1016/j.stem.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Itkin T, Gur-Cohen S, Spencer JA, Schajnovitz A, Ramasamy SK, Kusumbe AP, Ledergor G, Jung Y, Milo I, Poulos MG, et al. (2016). Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 532, 323–328. 10.1038/nature17624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, Mizoguchi T, Wei Q, Lucas D, Ito K, et al. (2013). Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643. 10.1038/nature12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pinho S, and Frenette PS (2019). Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol 20, 303–320. 10.1038/s41580-019-0103-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, Scheiermann C, Schiff L, Poncz M, Bergman A, et al. (2014). Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat. Med 20, 1315–1320. 10.1038/nm.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao M, Perry JM, Marshall H, Venkatraman A, Qian P, He XC, Ahamed J, and Li L (2014). Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat. Med 20, 1321–1326. 10.1038/nm.3706. [DOI] [PubMed] [Google Scholar]
- 53.Hérault A, Binnewies M, Leong S, Calero-Nieto FJ, Zhang SY, Kang Y-A, Wang X, Pietras EM, Chu SH, Barry-Holson K, et al. (2017). Myeloid progenitor cluster formation drives emergency and leukaemic myelopoiesis. Nature 544, 53–58. 10.1038/nature21693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ding L, and Morrison SJ (2013). Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495, 231–235. 10.1038/nature11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ding L, Saunders TL, Enikolopov G, and Morrison SJ (2012). Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481, 457–462. 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Greenbaum A, Hsu Y-MS, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, Nagasawa T, and Link DC (2013). CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 495, 227–230. 10.1038/nature11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, Taketo MM, Karlsson S, Iwama A, and Nakauchi H (2011). Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell 147, 1146–1158. 10.1016/j.cell.2011.09.053. [DOI] [PubMed] [Google Scholar]
- 58.Cheshier SH, Morrison SJ, Liao X, and Weissman IL (1999). In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc. Natl. Acad. Sci 96, 3120–3125. 10.1073/pnas.96.6.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Passegué E, Wagers AJ, Giuriato S, Anderson WC, and Weissman IL (2005). Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J. Exp. Med 202, 1599–1611. 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Orford KW, and Scadden DT (2008). Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat. Rev. Genet 9, 115–128. 10.1038/nrg2269. [DOI] [PubMed] [Google Scholar]
- 61.Mohrin M, Bourke E, Alexander D, Warr MR, Barry-Holson K, Le Beau MM, Morrison CG, and Passegué E (2010). Hematopoietic Stem Cell Quiescence Promotes Error-Prone DNA Repair and Mutagenesis. Cell Stem Cell 7, 174–185. 10.1016/j.stem.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, and Korsmeyer SJ (2005). Obligate Role of Anti-Apoptotic MCL-1 in the Survival of Hematopoietic Stem Cells. Science 307, 1101–1104. 10.1126/science.1106114. [DOI] [PubMed] [Google Scholar]
- 63.Domen J, Gandy KL, and Weissman IL (1998). Systemic Overexpression of BCL-2 in the Hematopoietic System Protects Transgenic Mice From the Consequences of Lethal Irradiation. Blood 91, 2272–2282. 10.1182/blood.V91.7.2272. [DOI] [PubMed] [Google Scholar]
- 64.Domen J, Cheshier SH, and Weissman IL (2000). The Role of Apoptosis in the Regulation of Hematopoietic Stem Cells: Overexpression of BCL-2 Increases Both Their Number and Repopulation Potential. J. Exp. Med 191, 253–264. 10.1084/jem.191.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Milyavsky M, Gan OI, Trottier M, Komosa M, Tabach O, Notta F, Lechman E, Hermans KG, Eppert K, Konovalova Z, et al. (2010). A distinctive DNA damage response in human hematopoietic stem cells reveals an apoptosis-independent role for p53 in self-renewal. Cell Stem Cell 7, 186–197. 10.1016/j.stem.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 66.Yamashita M, and Passegué E (2019). TNF-α Coordinates Hematopoietic Stem Cell Survival and Myeloid Regeneration. Cell Stem Cell 25, 357–372.e7. 10.1016/j.stem.2019.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, Debnath J, and Passegué E (2013). FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 494, 323–327. 10.1038/nature11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ito K, Turcotte R, Cui J, Zimmerman SE, Pinho S, Mizoguchi T, Arai F, Runnels JM, Alt C, Teruya-Feldstein J, et al. (2016). Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science 354, 1156–1160. 10.1126/science.aaf5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, Figueroa ME, and Passegué E (2017). Autophagy maintains the metabolism and function of young and old stem cells. Nature 543, 205–210. 10.1038/nature21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Signer RAJ, Magee JA, Salic A, and Morrison SJ (2014). Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509, 49–54. 10.1038/nature13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chua BA, Lennan CJ, Sunshine MJ, Dreifke D, Chawla A, Bennett EJ, and Signer RAJ (2023). Hematopoietic stem cells preferentially traffic misfolded proteins to aggresomes and depend on aggrephagy to maintain protein homeostasis. Cell Stem Cell 0. 10.1016/j.stem.2023.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dong S, Wang Q, Kao Y-R, Diaz A, Tasset I, Kaushik S, Thiruthuvanathan V, Zintiridou A, Nieves E, Dzieciatkowska M, et al. (2021). Chaperone-mediated autophagy sustains haematopoietic stem-cell function. Nature 591, 117–123. 10.1038/s41586-020-03129-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van Galen P, Kreso A, Mbong N, Kent DG, Fitzmaurice T, Chambers JE, Xie S, Laurenti E, Hermans K, Eppert K, et al. (2014). The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature 510, 268–272. 10.1038/nature13228. [DOI] [PubMed] [Google Scholar]
- 74.van Galen P, Mbong N, Kreso A, Schoof EM, Wagenblast E, Ng SWK, Krivdova G, Jin L, Nakauchi H, and Dick JE (2018). Integrated Stress Response Activity Marks Stem Cells in Normal Hematopoiesis and Leukemia. Cell Rep. 25, 1109–1117.e5. 10.1016/j.celrep.2018.10.021. [DOI] [PubMed] [Google Scholar]
- 75.Chandel NS, Jasper H, Ho TT, and Passegué E (2016). Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat. Cell Biol 18, 823–832. 10.1038/ncb3385. [DOI] [PubMed] [Google Scholar]
- 76.Kohli L, and Passegué E (2014). Surviving Change: The Metabolic Journey of Hematopoietic Stem Cells. Trends Cell Biol. 24, 479–487. 10.1016/j.tcb.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Suda T, Takubo K, and Semenza GL (2011). Metabolic Regulation of Hematopoietic Stem Cells in the Hypoxic Niche. Cell Stem Cell 9, 298–310. 10.1016/j.stem.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 78.Loeffler D, Schneiter F, and Schroeder T (2020). Pitfalls and requirements in quantifying asymmetric mitotic segregation. Ann. N. Y. Acad. Sci 1466, 73–82. 10.1111/nyas.14284. [DOI] [PubMed] [Google Scholar]
- 79.Chan W-I, Hannah RL, Dawson MA, Pridans C, Foster D, Joshi A, Göttgens B, Van Deursen JM, and Huntly BJP (2011). The Transcriptional Coactivator Cbp Regulates Self-Renewal and Differentiation in Adult Hematopoietic Stem Cells. Mol. Cell. Biol 31, 5046–5060. 10.1128/MCB.05830-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Agathocleous M, Meacham CE, Burgess RJ, Piskounova E, Zhao Z, Crane GM, Cowin BL, Bruner E, Murphy MM, Chen W, et al. (2017). Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476–481. 10.1038/nature23876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cabezas-Wallscheid N, Buettner F, Sommerkamp P, Klimmeck D, Ladel L, Thalheimer FB, Pastor-Flores D, Roma LP, Renders S, Zeisberger P, et al. (2017). Vitamin A-Retinoic Acid Signaling Regulates Hematopoietic Stem Cell Dormancy. Cell 169, 807–823.e19. 10.1016/j.cell.2017.04.018. [DOI] [PubMed] [Google Scholar]
- 82.Schönberger K, Obier N, Romero-Mulero MC, Cauchy P, Mess J, Pavlovich PV, Zhang YW, Mitterer M, Rettkowski J, Lalioti M-E, et al. (2022). Multilayer omics analysis reveals a non-classical retinoic acid signaling axis that regulates hematopoietic stem cell identity. Cell Stem Cell 29, 131–148.e10. 10.1016/j.stem.2021.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vannini N, Campos V, Girotra M, Trachsel V, Rojas-Sutterlin S, Tratwal J, Ragusa S, Stefanidis E, Ryu D, Rainer PY, et al. (2019). The NAD-Booster Nicotinamide Riboside Potently Stimulates Hematopoiesis through Increased Mitochondrial Clearance. Cell Stem Cell 24, 405–418.e7. 10.1016/j.stem.2019.02.012. [DOI] [PubMed] [Google Scholar]
- 84.Morganti C, Cabezas-Wallscheid N, and Ito K (2022). Metabolic Regulation of Hematopoietic Stem Cells. HemaSphere 6, e740. 10.1097/HS9.0000000000000740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cheshier SH, Prohaska SS, and Weissman IL (2007). The effect of bleeding on hematopoietic stem cell cycling and self-renewal. Stem Cells Dev. 16, 707–717. 10.1089/scd.2007.0017. [DOI] [PubMed] [Google Scholar]
- 86.Baldridge MT, King KY, Boles NC, Weksberg DC, and Goodell MA (2010). Quiescent haematopoietic stem cells are activated by IFN-γ in response to chronic infection. Nature 465, 793–797. 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Essers MAG, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, and Trumpp A (2009). IFNα activates dormant haematopoietic stem cells in vivo. Nature 458, 904–908. 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- 88.Pietras EM, Lakshminarasimhan R, Techner J-M, Fong S, Flach J, Binnewies M, and Passegué E (2014). Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J. Exp. Med 211, 245–262. 10.1084/jem.20131043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, Lakshminarasimhan R, Chin CP, Techner J-M, Will B, et al. (2016). Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol 18, 607–618. 10.1038/ncb3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chavez JS, Rabe JL, Loeffler D, Higa KC, Hernandez G, Mills TS, Ahmed N, Gessner RL, Ke Z, Idler BM, et al. (2021). PU.1 enforces quiescence and limits hematopoietic stem cell expansion during inflammatory stress. J. Exp. Med 218, e20201169. 10.1084/jem.20201169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Williams GC (1957). Pleiotropy, Natural Selection, and the Evolution of Senescence. Evolution 11, 398–411. 10.2307/2406060. [DOI] [Google Scholar]
- 92.Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM, Reynaud D, Alvarez S, Diolaiti ME, Ugarte F, Forsberg EC, et al. (2014). Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 512, 198–202. 10.1038/nature13619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hammond CA, Wu SW, Wang F, MacAldaz ME, and Eaves CJ (2023). Aging alters the cell cycle control and mitogenic signaling responses of human hematopoietic stem cells. Blood 141, 1990–2002. 10.1182/blood.2022017174. [DOI] [PubMed] [Google Scholar]
- 94.Beerman I, Seita J, Inlay MA, Weissman IL, and Rossi DJ (2014). Quiescent Hematopoietic Stem Cells Accumulate DNA Damage during Aging that Is Repaired upon Entry into Cell Cycle. Cell Stem Cell 15, 37–50. 10.1016/j.stem.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gutierrez-Martinez P, Hogdal L, Nagai M, Kruta M, Singh R, Sarosiek K, Nussenzweig A, Beerman I, Letai A, and Rossi DJ (2018). Diminished apoptotic priming and ATM signalling confer a survival advantage onto aged haematopoietic stem cells in response to DNA damage. Nat. Cell Biol 20, 413–421. 10.1038/s41556-018-0054-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. (2014). Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med 371, 2488–2498. 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Feldman T, Bercovich A, Moskovitz Y, Chapal-Ilani N, Mitchell A, Medeiros JJF, Biezuner T, Kaushansky N, Minden MD, Gupta V, et al. (2021). Recurrent deletions in clonal hematopoiesis are driven by microhomology-mediated end joining. Nat. Commun 12, 2455. 10.1038/s41467-021-22803-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guermouche H, Ravalet N, Gallay N, Deswarte C, Foucault A, Beaud J, Rault E, Saindoy E, Lachot S, Martignoles J-A, et al. (2020). High prevalence of clonal hematopoiesis in the blood and bone marrow of healthy volunteers. Blood Adv. 4, 3550–3557. 10.1182/bloodadvances.2020001582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Young AL, Challen GA, Birmann BM, and Druley TE (2016). Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun 7, 12484. 10.1038/ncomms12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Caiado F, Kovtonyuk LV, Gonullu NG, Fullin J, Boettcher S, and Manz MG (2023). Aging drives Tet2+/− clonal hematopoiesis via IL-1 signaling. Blood 141, 886–903. 10.1182/blood.2022016835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Heyde A, Rohde D, McAlpine CS, Zhang S, Hoyer FF, Gerold JM, Cheek D, Iwamoto Y, Schloss MJ, Vandoorne K, et al. (2021). Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell 184, 1348–1361.e22. 10.1016/j.cell.2021.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hormaechea-Agulla D, Matatall KA, Le DT, Kain B, Long X, Kus P, Jaksik R, Challen GA, Kimmel M, and King KY (2021). Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNγ signaling. Cell Stem Cell 28, 1428–1442.e6. 10.1016/j.stem.2021.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Avagyan S, Henninger JE, Mannherz WP, Mistry M, Yoon J, Yang S, Weber MC, Moore JL, and Zon LI (2021). Resistance to inflammation underlies enhanced fitness in clonal hematopoiesis. Science 374, 768–772. 10.1126/science.aba9304. [DOI] [PubMed] [Google Scholar]
- 104.Mitchell CA, Verovskaya EV, Calero-Nieto FJ, Olson OC, Swann JW, Wang X, Hérault A, Dellorusso PV, Zhang SY, Svendsen AF, et al. (2023). Stromal niche inflammation mediated by IL-1 signalling is a targetable driver of haematopoietic ageing. Nat. Cell Biol 25, 30–41. 10.1038/s41556-022-01053-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Young K, Eudy E, Bell R, Loberg MA, Stearns T, Sharma D, Velten L, Haas S, Filippi M-D, and Trowbridge JJ (2021). Decline in IGF1 in the bone marrow microenvironment initiates hematopoietic stem cell aging. Cell Stem Cell 28, 1473–1482.e7. 10.1016/j.stem.2021.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kusumbe AP, Ramasamy SK, Itkin T, Mäe MA, Langen UH, Betsholtz C, Lapidot T, and Adams RH (2016). Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature 532, 380–384. 10.1038/nature17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Biswas L, Chen J, De Angelis J, Singh A, Owen-Woods C, Ding Z, Pujol JM, Kumar N, Zeng F, Ramasamy SK, et al. (2023). Lymphatic vessels in bone support regeneration after injury. Cell 186, 382–397.e24. 10.1016/j.cell.2022.12.031. [DOI] [PubMed] [Google Scholar]
- 108.Ambrosi TH, Marecic O, McArdle A, Sinha R, Gulati GS, Tong X, Wang Y, Steininger HM, Hoover MY, Koepke LS, et al. (2021). Aged skeletal stem cells generate an inflammatory degenerative niche. Nature 597, 256–262. 10.1038/s41586-021-03795-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Maryanovich M, Zahalka AH, Pierce H, Pinho S, Nakahara F, Asada N, Wei Q, Wang X, Ciero P, Xu J, et al. (2018). Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat. Med 24, 782–791. 10.1038/s41591-018-0030-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shen B, Tasdogan A, Ubellacker JM, Zhang J, Nosyreva ED, Du L, Murphy MM, Hu S, Yi Y, Kara N, et al. (2021). A mechanosensitive peri-arteriolar niche for osteogenesis and lymphopoiesis. Nature 591, 438–444. 10.1038/s41586-021-03298-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hernández-Malmierca P, Vonficht D, Schnell A, Uckelmann HJ, Bollhagen A, Mahmoud MAA, Landua S-L, van der Salm E, Trautmann CL, Raffel S, et al. (2022). Antigen presentation safeguards the integrity of the hematopoietic stem cell pool. Cell Stem Cell 29, 760–775.e10. 10.1016/j.stem.2022.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mittelbrunn M, and Kroemer G (2021). Hallmarks of T cell aging. Nat. Immunol 22, 687–698. 10.1038/s41590-021-00927-z. [DOI] [PubMed] [Google Scholar]
- 113.Franceschi C, Garagnani P, Morsiani C, Conte M, Santoro A, Grignolio A, Monti D, Capri M, and Salvioli S (2018). The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front. Med 5, 61. 10.3389/fmed.2018.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Takizawa H, Fritsch K, Kovtonyuk LV, Saito Y, Yakkala C, Jacobs K, Ahuja AK, Lopes M, Hausmann A, Hardt W-D, et al. (2017). Pathogen-Induced TLR4-TRIF Innate Immune Signaling in Hematopoietic Stem Cells Promotes Proliferation but Reduces Competitive Fitness. Cell Stem Cell 21, 225–240.e5. 10.1016/j.stem.2017.06.013. [DOI] [PubMed] [Google Scholar]
- 115.Bogeska R, Mikecin A-M, Kaschutnig P, Fawaz M, Büchler-Schäff M, Le D, Ganuza M, Vollmer A, Paffenholz SV, Asada N, et al. (2022). Inflammatory exposure drives long-lived impairment of hematopoietic stem cell self-renewal activity and accelerated aging. Cell Stem Cell 29, 1273–1284.e8. 10.1016/j.stem.2022.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ergen AV, Boles NC, and Goodell MA (2012). Rantes/Ccl5 influences hematopoietic stem cell subtypes and causes myeloid skewing. Blood 119, 2500–2509. 10.1182/blood-2011-11-391730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Frisch BJ, Hoffman CM, Latchney SE, LaMere MW, Myers J, Ashton J, Li AJ, Saunders J, Palis J, Perkins AS, et al. (2019). Aged marrow macrophages expand platelet-biased hematopoietic stem cells via interleukin-1B. JCI Insight 4. 10.1172/jci.insight.124213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ho Y-H, del Toro R, Rivera-Torres J, Rak J, Korn C, García-García A, Macías D, González-Gómez C, del Monte A, Wittner M, et al. (2019). Remodeling of Bone Marrow Hematopoietic Stem Cell Niches Promotes Myeloid Cell Expansion during Premature or Physiological Aging. Cell Stem Cell 25, 407–418.e6. 10.1016/j.stem.2019.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kovtonyuk LV, Caiado F, Garcia-Martin S, Manz E-M, Helbling P, Takizawa H, Boettcher S, Al-Shahrour F, Nombela-Arrieta C, Slack E, et al. (2022). IL-1 mediates microbiome-induced inflammaging of hematopoietic stem cells in mice. Blood 139, 44–58. 10.1182/blood.2021011570. [DOI] [PubMed] [Google Scholar]
- 120.Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, et al. (2008). Hematopoietic Stem Cells Reversibly Switch from Dormancy to Self-Renewal during Homeostasis and Repair. Cell 135, 1118–1129. 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 121.Zhao M, Tao F, Venkatraman A, Li Z, Smith SE, Unruh J, Chen S, Ward C, Qian P, Perry JM, et al. (2019). N-Cadherin-Expressing Bone and Marrow Stromal Progenitor Cells Maintain Reserve Hematopoietic Stem Cells. Cell Rep. 26, 652–669.e6. 10.1016/j.celrep.2018.12.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Higa KC, Goodspeed A, Chavez JS, De Dominici M, Danis E, Zaberezhnyy V, Rabe JL, Tenen DG, Pietras EM, and DeGregori J (2021). Chronic interleukin-1 exposure triggers selection for Cebpa-knockout multipotent hematopoietic progenitors. J. Exp. Med 218, e20200560. 10.1084/jem.20200560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, Wang H, Le T, Faull KF, Chen R, et al. (2014). Epigenomic Profiling of Young and Aged HSCs Reveals Concerted Changes during Aging that Reinforce Self-Renewal. Cell Stem Cell 14, 673–688. 10.1016/j.stem.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Adelman ER, Huang H-T, Roisman A, Olsson A, Colaprico A, Qin T, Lindsley RC, Bejar R, Salomonis N, Grimes HL, et al. (2019). Aging Human Hematopoietic Stem Cells Manifest Profound Epigenetic Reprogramming of Enhancers That May Predispose to Leukemia. Cancer Discov. 9, 1080–1101. 10.1158/2159-8290.CD-18-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Grigoryan A, Guidi N, Senger K, Liehr T, Soller K, Marka G, Vollmer A, Markaki Y, Leonhardt H, Buske C, et al. (2018). LaminA/C regulates epigenetic and chromatin architecture changes upon aging of hematopoietic stem cells. Genome Biol. 19, 189. 10.1186/s13059-018-1557-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Beerman I, Bock C, Garrison BS, Smith ZD, Gu H, Meissner A, and Rossi DJ (2013). Proliferation-Dependent Alterations of the DNA Methylation Landscape Underlie Hematopoietic Stem Cell Aging. Cell Stem Cell 12, 413–425. 10.1016/j.stem.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 127.Bernitz JM, Kim H-S, MacArthur B, Sieburg H, and Moore K (2016). Hematopoietic Stem Cells Count and Remember Self-Renewal Divisions. Cell 167, 1296–1309.e10. 10.1016/j.cell.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yu VWC, Yusuf RZ, Oki T, Wu J, Saez B, Wang X, Cook C, Baryawno N, Ziller MJ, Lee E, et al. (2016). Epigenetic Memory Underlies Cell-Autonomous Heterogeneous Behavior of Hematopoietic Stem Cells. Cell 167, 1310–1322.e17. 10.1016/j.cell.2016.10.045. [DOI] [PubMed] [Google Scholar]
- 129.Mann M, Mehta A, de Boer CG, Kowalczyk MS, Lee K, Haldeman P, Rogel N, Knecht AR, Farouq D, Regev A, et al. (2018). Heterogeneous Responses of Hematopoietic Stem Cells to Inflammatory Stimuli Are Altered with Age. Cell Rep. 25, 2992–3005.e5. 10.1016/j.celrep.2018.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.de Laval B, Maurizio J, Kandalla PK, Brisou G, Simonnet L, Huber C, Gimenez G, Matcovitch-Natan O, Reinhardt S, David E, et al. (2020). C/EBPβ-Dependent Epigenetic Memory Induces Trained Immunity in Hematopoietic Stem Cells. Cell Stem Cell 26, 657–674.e8. 10.1016/j.stem.2020.01.017. [DOI] [PubMed] [Google Scholar]
- 131.Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonça LE, Pacis A, Tzelepis F, Pernet E, Dumaine A, Grenier J-C, et al. (2018). BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell 172, 176–190.e19. 10.1016/j.cell.2017.12.031. [DOI] [PubMed] [Google Scholar]
- 132.Mitroulis I, Ruppova K, Wang B, Chen L-S, Grzybek M, Grinenko T, Eugster A, Troullinaki M, Palladini A, Kourtzelis I, et al. (2018). Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 172, 147–161.e12. 10.1016/j.cell.2017.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yu W-M, Liu X, Shen J, Jovanovic O, Pohl EE, Gerson SL, Finkel T, Broxmeyer HE, and Qu C-K (2013). Metabolic Regulation by the Mitochondrial Phosphatase PTPMT1 Is Required for Hematopoietic Stem Cell Differentiation. Cell Stem Cell 12, 62–74. 10.1016/j.stem.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, Shima H, Johnson RS, Hirao A, Suematsu M, et al. (2010). Regulation of the HIF-1α Level Is Essential for Hematopoietic Stem Cells. Cell Stem Cell 7, 391–402. 10.1016/j.stem.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 135.Takubo K, Nagamatsu G, Kobayashi CI, Nakamura-Ishizu A, Kobayashi H, Ikeda E, Goda N, Rahimi Y, Johnson RS, Soga T, et al. (2013). Regulation of Glycolysis by Pdk Functions as a Metabolic Checkpoint for Cell Cycle Quiescence in Hematopoietic Stem Cells. Cell Stem Cell 12, 49–61. 10.1016/j.stem.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hinge A, He J, Bartram J, Javier J, Xu J, Fjellman E, Sesaki H, Li T, Yu J, Wunderlich M, et al. (2020). Asymmetrically Segregated Mitochondria Provide Cellular Memory of Hematopoietic Stem Cell Replicative History and Drive HSC Attrition. Cell Stem Cell 26, 420–430.e6. 10.1016/j.stem.2020.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rubinsztein DC, Mariño G, and Kroemer G (2011). Autophagy and Aging. Cell 146, 682–695. 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 138.Rimmelé P, Bigarella CL, Liang R, Izac B, Dieguez-Gonzalez R, Barbet G, Donovan M, Brugnara C, Blander JM, Sinclair DA, et al. (2014). Aging-like Phenotype and Defective Lineage Specification in SIRT1-Deleted Hematopoietic Stem and Progenitor Cells. Stem Cell Rep. 3, 44–59. 10.1016/j.stemcr.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Luo H, Mu W-C, Karki R, Chiang H-H, Mohrin M, Shin JJ, Ohkubo R, Ito K, Kanneganti T-D, and Chen D (2019). Mitochondrial Stress-Initiated Aberrant Activation of the NLRP3 Inflammasome Regulates the Functional Deterioration of Hematopoietic Stem Cell Aging. Cell Rep. 26, 945–954.e4. 10.1016/j.celrep.2018.12.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Brown K, Xie S, Qiu X, Mohrin M, Shin J, Liu Y, Zhang D, Scadden DT, and Chen D (2013). SIRT3 Reverses Aging-Associated Degeneration. Cell Rep. 3, 319–327. 10.1016/j.celrep.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mohrin M, Shin J, Liu Y, Brown K, Luo H, Xi Y, Haynes CM, and Chen D (2015). A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 347, 1374–1377. 10.1126/science.aaa2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kruta M, Sunshine MJ, Chua BA, Fu Y, Chawla A, Dillingham CH, Hidalgo San Jose L, De Jong B, Zhou FJ, and Signer RAJ (2021). Hsf1 promotes hematopoietic stem cell fitness and proteostasis in response to ex vivo culture stress and aging. Cell Stem Cell. 10.1016/j.stem.2021.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yousefzadeh MJ, Flores RR, Zhu Y, Schmiechen ZC, Brooks RW, Trussoni CE, Cui Y, Angelini L, Lee K-A, McGowan SJ, et al. (2021). An aged immune system drives senescence and ageing of solid organs. Nature 594, 100–105. 10.1038/s41586-021-03547-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bowling S, Sritharan D, Osorio FG, Nguyen M, Cheung P, Rodriguez-Fraticelli A, Patel S, Yuan W-C, Fujiwara Y, Li BE, et al. (2020). An Engineered CRISPR-Cas9 Mouse Line for Simultaneous Readout of Lineage Histories and Gene Expression Profiles in Single Cells. Cell 181, 1410–1422.e27. 10.1016/j.cell.2020.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.de Haan G, and Lazare SS (2018). Aging of hematopoietic stem cells. Blood 131, 479–487. 10.1182/blood-2017-06-746412. [DOI] [PubMed] [Google Scholar]
- 146.de Cabo R, Carmona-Gutierrez D, Bernier M, Hall MN, and Madeo F (2014). The Search for Antiaging Interventions: From Elixirs to Fasting Regimens. Cell 157, 1515–1526. 10.1016/j.cell.2014.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, and Rando TA (2005). Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 433, 760–764. 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- 148.Loffredo FS, Steinhauser ML, Jay SM, Gannon J, Pancoast JR, Yalamanchi P, Sinha M, Dall’Osso C, Khong D, Shadrach JL, et al. (2013). Growth Differentiation Factor 11 Is a Circulating Factor that Reverses Age-Related Cardiac Hypertrophy. Cell 153, 828–839. 10.1016/j.cell.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Villeda SA, Plambeck KE, Middeldorp J, Castellano JM, Mosher KI, Luo J, Smith LK, Bieri G, Lin K, Berdnik D, et al. (2014). Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat. Med 20, 659–663. 10.1038/nm.3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Horowitz AM, and Villeda SA (2017). Therapeutic potential of systemic brain rejuvenation strategies for neurodegenerative disease. F1000Research 6, 1291. 10.12688/f1000research.11437.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Garatachea N, Pareja-Galeano H, Sanchis-Gomar F, Santos-Lozano A, Fiuza-Luces C, Morán M, Emanuele E, Joyner MJ, and Lucia A (2015). Exercise attenuates the major hallmarks of aging. Rejuvenation Res. 18, 57–89. 10.1089/rej.2014.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ho TT, Dellorusso PV, Verovskaya EV, Bakker ST, Flach J, Smith LK, Ventura PB, Lansinger OM, Hérault A, Zhang SY, et al. (2021). Aged hematopoietic stem cells are refractory to bloodborne systemic rejuvenation interventions. J. Exp. Med 218, e20210223. 10.1084/jem.20210223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kuribayashi W, Oshima M, Itokawa N, Koide S, Nakajima-Takagi Y, Yamashita M, Yamazaki S, Rahmutulla B, Miura F, Ito T, et al. (2021). Limited rejuvenation of aged hematopoietic stem cells in young bone marrow niche. J. Exp. Med 218, e20192283. 10.1084/jem.20192283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kim M-J, Miller CM, Shadrach JL, Wagers AJ, and Serwold T (2015). Young, proliferative thymic epithelial cells engraft and function in aging thymuses. J. Immunol. Baltim. Md 1950 194, 4784–4795. 10.4049/jimmunol.1403158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Poulos MG, Ramalingam P, Gutkin MC, Llanos P, Gilleran K, Rabbany SY, and Butler JM (2017). Endothelial transplantation rejuvenates aged hematopoietic stem cell function. J. Clin. Invest 127, 4163–4178. 10.1172/JCI93940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bashor CJ, Hilton IB, Bandukwala H, Smith DM, and Veiseh O (2022). Engineering the next generation of cell-based therapeutics. Nat. Rev. Drug Discov 21, 655–675. 10.1038/s41573-022-00476-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zhang X, Cao D, Xu L, Xu Y, Gao Z, Pan Y, Jiang M, Wei Y, Wang L, Liao Y, et al. (2023). Harnessing matrix stiffness to engineer a bone marrow niche for hematopoietic stem cell rejuvenation. Cell Stem Cell 30, 378–395.e8. 10.1016/j.stem.2023.03.005. [DOI] [PubMed] [Google Scholar]
- 158.Guidi N, Sacma M, Ständker L, Soller K, Marka G, Eiwen K, Weiss JM, Kirchhoff F, Weil T, Cancelas JA, et al. (2017). Osteopontin attenuates aging-associated phenotypes of hematopoietic stem cells. EMBO J. 36, 840–853. 10.15252/embj.201694969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Verovskaya EV, Dellorusso PV, and Passegué E (2019). Losing Sense of Self and Surroundings: Hematopoietic Stem Cell Aging and Leukemic Transformation. Trends Mol. Med 25, 494–515. 10.1016/j.molmed.2019.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zeng X, Li X, Li X, Wei C, Shi C, Hu K, Kong D, Luo Q, Xu Y, Shan W, et al. (2023). Fecal microbiota transplantation from young mice rejuvenates aged hematopoietic stem cells by suppressing inflammation. Blood 141, 1691–1707. 10.1182/blood.2022017514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Meisel M, Hinterleitner R, Pacis A, Chen L, Earley ZM, Mayassi T, Pierre JF, Ernest JD, Galipeau HJ, Thuille N, et al. (2018). Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 557, 580–584. 10.1038/s41586-018-0125-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cai Z, Kotzin JJ, Ramdas B, Chen S, Nelanuthala S, Palam LR, Pandey R, Mali RS, Liu Y, Kelley MR, et al. (2018). Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell 23, 833–849.e5. 10.1016/j.stem.2018.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, et al. (2017). Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med 377, 111–121. 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hauser A-T, Robaa D, and Jung M (2018). Epigenetic small molecule modulators of histone and DNA methylation. Curr. Opin. Chem. Biol 45, 73–85. 10.1016/j.cbpa.2018.03.003. [DOI] [PubMed] [Google Scholar]
- 165.Florian MC, Dörr K, Niebel A, Daria D, Schrezenmeier H, Rojewski M, Filippi M-D, Hasenberg A, Gunzer M, Scharffetter-Kochanek K, et al. (2012). Cdc42 Activity Regulates Hematopoietic Stem Cell Aging and Rejuvenation. Cell Stem Cell 10, 520–530. 10.1016/j.stem.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Satoh Y, Yokota T, Sudo T, Kondo M, Lai A, Kincade PW, Kouro T, Iida R, Kokame K, Miyata T, et al. (2013). The Satb1 Protein Directs Hematopoietic Stem Cell Differentiation toward Lymphoid Lineages. Immunity 38, 1105–1115. 10.1016/j.immuni.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wahlestedt M, Erlandsson E, Kristiansen T, Lu R, Brakebusch C, Weissman IL, Yuan J, Martin-Gonzalez J, and Bryder D (2017). Clonal reversal of ageing-associated stem cell lineage bias via a pluripotent intermediate. Nat. Commun 8, 14533. 10.1038/ncomms14533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Takahashi K, and Yamanaka S (2016). A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol 17, 183–193. 10.1038/nrm.2016.8. [DOI] [PubMed] [Google Scholar]
- 169.Browder KC, Reddy P, Yamamoto M, Haghani A, Guillen IG, Sahu S, Wang C, Luque Y, Prieto J, Shi L, et al. (2022). In vivo partial reprogramming alters age-associated molecular changes during physiological aging in mice. Nat. Aging 2, 243–253. 10.1038/s43587-022-00183-2. [DOI] [PubMed] [Google Scholar]
- 170.Lu JY, Simon M, Zhao Y, Ablaeva J, Corson N, Choi Y, Yamada KYH, Schork NJ, Hood WR, Hill GE, et al. (2022). Comparative transcriptomics reveals circadian and pluripotency networks as two pillars of longevity regulation. Cell Metab. 34, 836–856.e5. 10.1016/j.cmet.2022.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ocampo A, Reddy P, Martinez-Redondo P, Platero-Luengo A, Hatanaka F, Hishida T, Li M, Lam D, Kurita M, Beyret E, et al. (2016). In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 167, 1719–1733.e12. 10.1016/j.cell.2016.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rodríguez-Matellán A, Alcazar N, Hernández F, Serrano M, and Ávila J (2020). In Vivo Reprogramming Ameliorates Aging Features in Dentate Gyrus Cells and Improves Memory in Mice. Stem Cell Rep. 15, 1056–1066. 10.1016/j.stemcr.2020.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, et al. (2009). Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395. 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chen C, Liu Y, Liu Y, and Zheng P (2009). mTOR Regulation and Therapeutic Rejuvenation of Aging Hematopoietic Stem Cells. Sci. Signal 2, ra75–ra75. 10.1126/scisignal.2000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, Lonetto MA, Maecker HT, Kovarik J, Carson S, et al. (2014). mTOR inhibition improves immune function in the elderly. Sci. Transl. Med 6, 268ra179–268ra179. 10.1126/scitranslmed.3009892. [DOI] [PubMed] [Google Scholar]
- 176.Sun X, Cao B, Naval-Sanchez M, Pham T, Sun YBY, Williams B, Heazlewood SY, Deshpande N, Li J, Kraus F, et al. (2021). Nicotinamide riboside attenuates age-associated metabolic and functional changes in hematopoietic stem cells. Nat. Commun 12, 2665. 10.1038/s41467-021-22863-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, Pei S, Khan N, Adane B, Ye H, et al. (2018). Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 34, 724–740.e4. 10.1016/j.ccell.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, Ikeda Y, Rosenblatt J, Avigan DE, Teruya-Feldstein J, and Pandolfi PP (2008). PML targeting eradicates quiescent leukaemia-initiating cells. Nature 453, 1072–1078. 10.1038/nature07016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O’Dwyer KM, et al. (2013). BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell Stem Cell 12, 329–341. 10.1016/j.stem.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chang J, Wang Y, Shao L, Laberge R-M, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, et al. (2016). Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med 22, 78–83. 10.1038/nm.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Paczulla AM, Rothfelder K, Raffel S, Konantz M, Steinbacher J, Wang H, Tandler C, Mbarga M, Schaefer T, Falcone M, et al. (2019). Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 572, 254–259. 10.1038/s41586-019-1410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Orkin SH, and Bauer DE (2019). Emerging Genetic Therapy for Sickle Cell Disease. Annu. Rev. Med 70, 257–271. 10.1146/annurev-med-041817-125507. [DOI] [PubMed] [Google Scholar]
- 183.Cheng C-W, Adams GB, Perin L, Wei M, Zhou X, Lam BS, Da Sacco S, Mirisola M, Quinn DI, Dorff TB, et al. (2014). Prolonged Fasting Reduces IGF-1/PKA to Promote Hematopoietic-Stem-Cell-Based Regeneration and Reverse Immunosuppression. Cell Stem Cell 14, 810–823. 10.1016/j.stem.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tang D, Tao S, Chen Z, Koliesnik IO, Calmes PG, Hoerr V, Han B, Gebert N, Zörnig M, Löffler B, et al. (2016). Dietary restriction improves repopulation but impairs lymphoid differentiation capacity of hematopoietic stem cells in early aging. J. Exp. Med 213, 535–553. 10.1084/jem.20151100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ramalingam P, Gutkin MC, Poulos MG, Tillery T, Doughty C, Winiarski A, Freire AG, Rafii S, Redmond D, and Butler JM (2023). Restoring bone marrow niche function rejuvenates aged hematopoietic stem cells by reactivating the DNA Damage Response. Nat. Commun 14, 2018. 10.1038/s41467-023-37783-4. [DOI] [PMC free article] [PubMed] [Google Scholar]