


Abstract
Among the polymerases, DNA polymerase α-primase is involved in lagging strand DNA synthesis. A previous report indicated that DNA polymerase α-primase initiates primer RNA synthesis with purine bases on a single-stranded G-rich telomere repeat. In this study, we found that DNA polymerase α-primase precisely initiated with adenosine opposite the 3′-side thymidine in the G-rich telomere repeat 5′-(TTAGGG)n-3′ under rATP-rich conditions. Then, DNA polymerase α-primase synthesized the nascent DNA fragments by extending the primer. It was remarkable that DNA polymerase α-primase further expanded the product DNA far beyond the length of the template DNA, as ladders of multiple hexanucleotides on polyacrylamide gel electrophoresis. Using an oligomer duplex 5′-A(GGGTTA)5-3′/5′-(TAACCC)5T-3′ as a template–primer, we show that both the Klenow fragment of Escherichia coli DNA polymerase I and HIV reverse transcriptase could expand telomere DNA sequences as well, giving products greater than the size of the template DNA. The maximum product lengths with these polymerases were ∼40–90 nt longer than the template length. Our data imply that DNA polymerases have an intrinsic activity to expand the hexanucleotide repeats of the telomere sequence by a slippage mechanism and that DNA polymerase α uses both the repeat DNA primers and the de novo RNA primers for expansion. On the other hand, a plasmid harboring a eukaryotic telomere repeat showed remarkable genetic instability in E.coli. The telomere repeats exhibited either expansions or deletions by multiple hexanucleotide repeats during culture for a number of generations, suggesting involvement of the slippage mechanism in the instability of telomeric DNA in vivo.
INTRODUCTION
Instabilities in homopolymeric and dinucleotide repeat sequences are associated with biological functions and diseases, adaptive mutations in Escherichia coli genes (1,2) and the p53 tumor suppresser gene in human cancers (3) and in human hereditary colon cancers (4–6). These repeat instabilities may be caused by replication errors through a realignment of templates–primers (7,8). Trinucleotide repeat expansions are also associated with various human genetic diseases (9). The expansion of CAG/CTG, for example, is found in Huntington’s disease, spinal and bulbar muscular atrophy, spinocerebellar ataxia types 1, 2, 6 and 7, dentatorubral-pallido-luysian atrophy and Machado–Joseph disease in the coding regions and myotonic dystrophy in the non-coding regions (9–14). Trinucleotide repeat expansions appear to be byproducts of DNA replication rather than DNA recombination, because the expansions occur mainly towards the 3′-ends of the repeat tracts and are in linkage disequilibrium with franking markers (15). Consistent with this hypothesis, instability is detectable in in vitro experiments using synthetic oligonucleotide repeats and DNA polymerases (16,17). In order to elucidate what triggers these unusual DNA syntheses, we introduced one to six mismatches in CAG/CTG repeats flanked by non-repetitive sequence and observed a mismatch-dependent repeat expansion (18). Recently, mismatch-dependent repeat expansion has also been shown in AAT/ATT repeats (19) and to be affected by temperature, Mg2+ concentration (19), abasic sites (20) and hairpin stability (21,22).
In this work we have analyzed the extension and expansion of a telomere repeat sequence by DNA polymerases. In all vertebrates, including humans, the telomere sequences consist of hexanucleotide tandem repeats, such as d(TTAGGG/CCCTAA)n, each of which may extend to 2–30 kb (23). Telomerase is a unique enzyme that adds telomere repeats at the chromosomal terminus using an RNA subunit as template (24). In cells that are defective in telomerase, chromosomes become shorter by tens to hundreds of nucleotides per cell division (25,26). However, telomeres may not be maintained by a simple addition of telomere repeats, but be under a certain equilibrium of synthesis and degradation (27–30). Most of the telomere repeat sequences, which flank the chromosomal sequence, may be replicated by conventional replicative DNA polymerases, because it takes many generations to erase the telomere repeats in humans (25,31), mice (32,33) and yeast (34) that lack telomerase. Thus both telomerase and replicative DNA polymerases are employed in leading strand telomere replication. On the other hand, the replicative polymerases may be absolutely required for lagging strand synthesis on telomeres (35,36). In order to address telomere repeat stability during conventional DNA replication, we performed de novo DNA synthesis reactions and primer extension using sequences of hexanucleotide repeats and DNA polymerases. The stability of the eukaryotic telomere repeat was also measured in transformed E.coli as a model for the in vivo replication of telomere DNA without telomerase.
MATERIALS AND METHODS
Chemicals, enzymes and oligomers
The Klenow fragment of E.coli DNA polymerase I (Klenow fragment) was purchased from NEB Inc. (Beverly, MA) and HIV reverse transcriptase (HIV-RT) from Seikagaku Kogyo (Tokyo, Japan). Recombinant Taq DNA polymerase (Taq pol) was purchased from TaKaRa Corp. (Kyoto, Japan). Calf thymus DNA polymerase α-primase (pol α-primase) was purified as described previously (37). One unit of DNA polymerase was defined as the amount that catalyzes the incorporation of 1 nmol [3H]dTTP into an acid-insoluble product in 60 min at 37°C on activated calf thymus DNA. DNA polymerase ɛ (pol ɛ) was a gift from Dr Stuart Linn of the University of California, Berkeley. Oligomers were synthesized and gel purified by Amersham Pharmacia Biotech (Little Chalfont, UK). RP-A was isolated from HeLa cells according to Kenny et al. (38).
De novo DNA synthesis on the single-stranded G-rich telomere sequence by pol α-primase
The rATP-rich reaction mixture (25 µl), for the detection of DNA synthesis coupled with primer RNA synthesis (de novo DNA synthesis), contained 50 mM Tris–HCl, pH 7.5, 2 mM DTT, 5 mM MgCl2, 50 µM each dATP, dTTP, dCTP, rCTP and rUTP, 0.5 µM [α-32P]dGTP (800 Ci/mmol), 2 mM rATP and 100 ng 5′-C(TTAGGG)6-3′ (G-6 template). The rCTP-rich reaction mixture was the same as that for the rATP-rich conditions except that 50 µM rATP and 2 mM rCTP were used in place of 50 µM rCTP and 2 mM rATP, respectively. As a negative control, all four rNTPs were omitted (rNTPs minus conditions). Reactions took place at 37°C for 60 min and the reaction products were analyzed by 15% denaturing PAGE.
DNA primer extension reaction
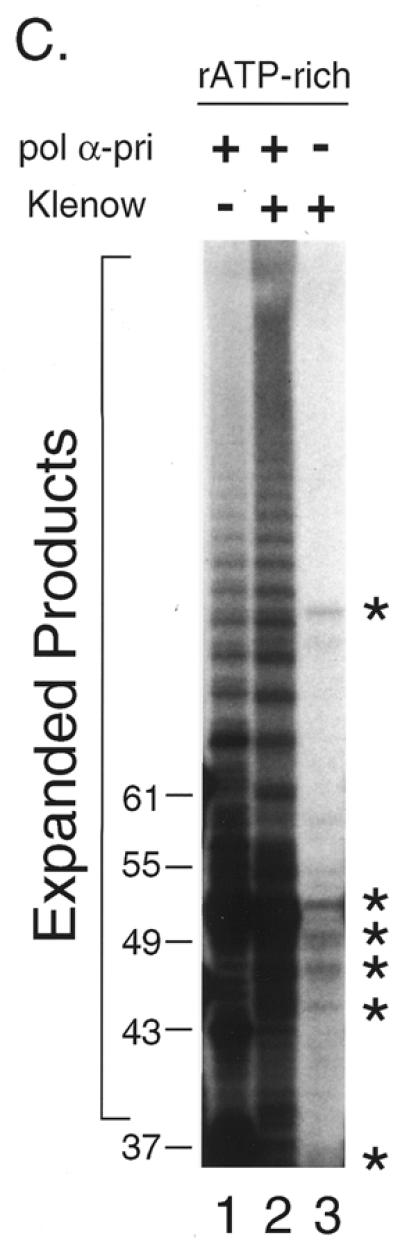
Oligomers 5′-A(GGGTTA)5-3′ (G-5) and 5′-(TAACCC)5T-3′ (C-5) were purified by PAGE and equal amounts of both strands were annealed by heating at 70°C for 15 min and cooling slowly at room temperature. The reaction mixture (25 µl) for HIV-RT contained 5 pmol annealed G-5/C-5, 100 µM each of three dNTPs, 10 µM [α-32P]dNTP, 10 mM Tris–HCl, pH 8.3, 50 mM KCl, 1.5 mM MgCl2 and the indicated amounts of HIV-RT. [α-32P]dGTP and [α-32P]dCTP were used for the extension of G-5 and C-5, respectively. Reaction mixtures for the Klenow fragment and pol α-primase were essentially the same as for HIV-RT except that 50 mM Tris–HCl, pH 7.5, 2 mM dithiothreitol (DTT) and 5 mM MgCl2 were used in place of the corresponding buffer, divalent cation and KCl, respectively. Reactions were performed at 37°C for 15 min for the Klenow fragment and HIV-RT and 60 min for pol α-primase, respectively. Reaction products were analyzed by electrophoresis on 12% polyacrylamide gels containing 8 M urea (denaturing PAGE). The bands at 67 nt or larger represent the expanded products. The frequency of annealing of the longest template–primer mediated by only one nucleotide (Fig. 2A, topmost panel) would be much lower than those by a hexanucleotide or a multiple of a hexanucleotide. Therefore, the product band at 61 nt may also represent the expansion of originally shorter products as well as the normal product on the 1 nt annealed template–primer.
Figure 2.

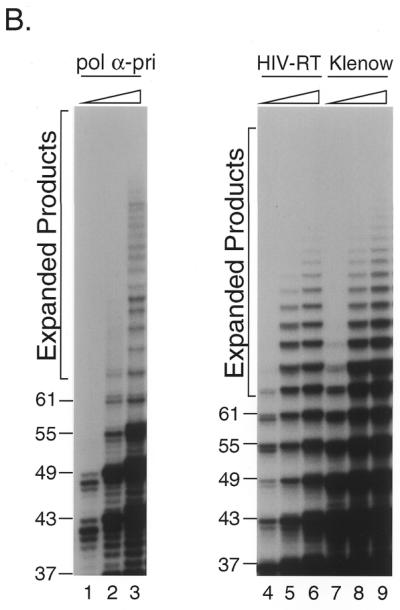
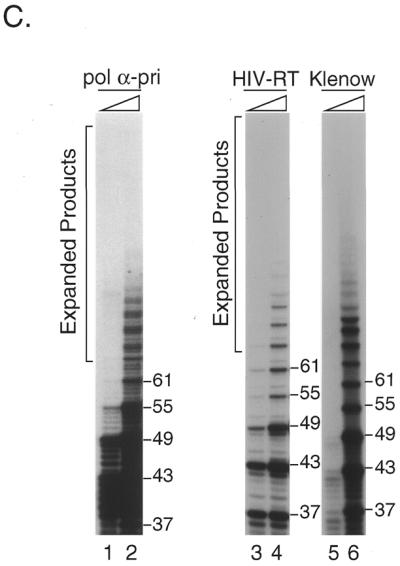
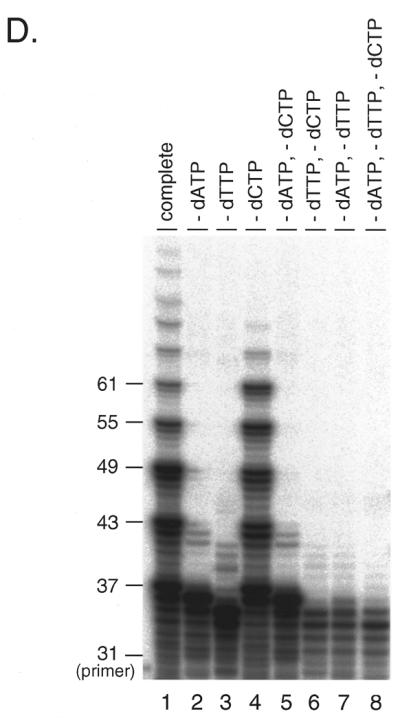
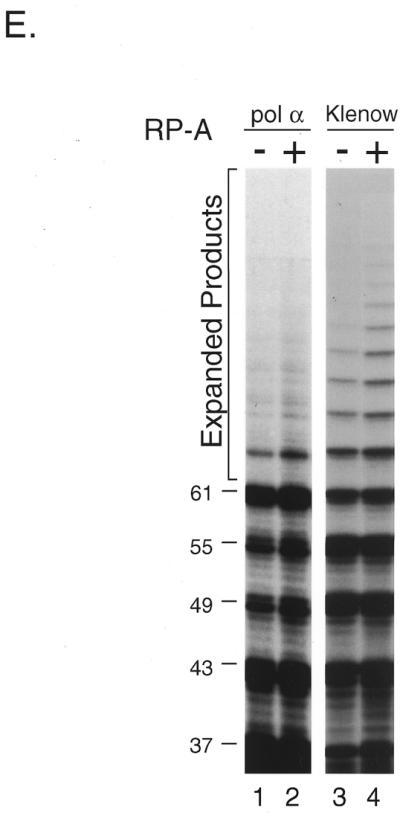
In vitro expansion of telomere repeats by DNA polymerases. (A) Templates–primers. Possible combinations of synthetic oligomers G-5 and C-5 are shown with expected product sizes without expansion. (B) In vitro expansion of the G-rich telomere strand. Reactions were carried out using 0.32, 0.8 and 1.6 U pol α-primase (lanes 1–3), 1.2, 2.4 and 6.0 U HIV-RT (lanes 4–6) and 1.0, 4.0 and 12.0 U Klenow fragment (lanes 7–9). The reaction products were resolved by denaturing PAGE and the incorporated [α-32P]dGMP was monitored with a BAS 2000 image analyzer (Fuji Film, Tokyo, Japan). Sizes of the reaction products were estimated in comparison with 32P-labeled marker oligonucleotides of 22, 31, 37, 49 and 69 nt length (not shown). Numbers on the left side of panels indicate products sizes. (C) In vitro expansion of the C-rich telomere strand. Reactions were carried out as for (B) except that [α-32P]dCTP was used instead of [α-32P]dGTP, with 0.32 and 1.6 U pol α-primase (lanes 1 and 2), 1.2 and 2.4 U HIV-RT (lanes 3 and 4) and 1.0 and 4.0 U Klenow fragment (lanes 5 and 6). Numbers on the left side of panels indicate product sizes. (D) Nucleotide requirements of the telomere repeat expansion by Klenow fragment (4.0 U). Reactions were carried out with all four dNTPs (lane 1), without dATP (lane 2), without dTTP (lane 3), without dCTP (lane 4), without dATP and dCTP (lane 5), without dTTP and dCTP (lane 6), without dATP and dTTP (lane 7) or without dATP, dTTP and dCTP (lane 8). Products were labeled with [α-32P]dGTP. (E) Effects of RP-A. In vitro expansion of the G-rich strand by pol α-primase (1.6 U; lanes 1 and 2) and Klenow fragment (4.0 U; lanes 3 and 4) were carried out with (lanes 2 and 4) or without (lanes 1 and 2) 1.0 µg RP-A purified from HeLa cells. Numbers on the left side of the figure indicate nucleotide sizes of products.
Cloning of the expanded repeat
Reaction products with the Klenow fragment at 37°C for 60 min were separated by non-denaturing PAGE and visualized with SYBR™ Green I (Molecular Probes, Rockland ME). DNA fragments >66 nt were recovered from gels and ligated into a pGEM3Zf(+) vector cleaved by SmaI. Inserts in individual transformed clones were sequenced using a universal primer and an ABI model 373A DNA sequencer (Perkin-Elmer, Foster City, CA).
Insertion of mammalian telomere repeats into E.coli plasmids
The plasmids harboring 28 and 30 repeats of telomere units were constructed by ligating pGEM®-T Easy vector (Promega, Madison, WI) with the expanded reaction products using Taq pol I at 72°C for 60 min. They are named pGEM-G28 and pGEM-C30, because the G-rich repeats and the C-rich repeats are arranged as templates for leading strand DNA synthesis, respectively (Fig. 3). Orientations and sequences of inserts in individual transformed clones were confirmed with a DNA sequencer.
Figure 3.

Expansion and deletion of the eukaryotic telomere repeats in E.coli. (A) The 28 repeats of telomere units were inserted into pGEM plasmid DNA (pGEM-G28) in an orientation arranging the G-rich strand as the template for the leading strand (orientation I, illustrated at the top of the panel). The plasmid was introduced into E.coli JM109 and cultured for 40 (lanes 2–5) or 185 generations (lanes 7–10). Plasmid DNA was isolated from each culture, cut with EcoRI and subjected to 7.5% native PAGE as described in Materials and Methods. In lanes 1 and 6, the repeat sequence from the construct before culture is shown as a control. (B) The 30 repeats of telomere units were inserted into pGEM-C30 in an orientation reverse to that in (A), arranging the C-rich strand as the template for the leading strand (orientation II, see illustration). Other conditions were the same as described for (A). Lanes 1 and 6, controls; lanes 2–5, cultured for 40 generations; lanes 7–10, cultured for 185 generations. Arrowheads indicate the original size of the repeats. Closed boxes indicate the polycloning sites (PCS).
Expansion and deletion of telomere repeats in plasmids during long culture of E.coli
pGEM-G28 or pGEM-C30 was introduced into E.coli JM109 by means of electroporation. Escherichia coli was cultured on an agar plate and colonies were picked after incubation for 14 h at 37°C. Clones that had the correct lengths and orientations of the inserts were selected and grown in LB medium containing 100 µg/ml ampicillin. Genomic instability over the generations was measured according to Kang et al. (39). In brief, when cells grew until OD660 = 0.9 in liquid culture they were diluted 1–5 × 106 times with fresh medium. This process was repeated to maintain the cells in logarithmic phase. We estimated the cell generation by counting the colony number on an agar plate inoculated with an aliquot of culture at the time of dilution. The plasmid DNA, isolated by alkaline lysis from the colony on the plate, was digested with EcoRI and subjected to 7.5% native PAGE. The insert bands were visualized by ethidium bromide staining.
RESULTS
Lagging strand synthesis by pol α-primase on the G-rich strand
Lagging strand DNA synthesis is required to complete the double-stranded telomere DNA (40). To examine whether the telomere repeat is used as a template for nascent DNA synthesis, we prepared a new 37 nt oligonucleotide, 5′-C(TTAGGG)6, and performed de novo DNA synthesis using pol α-primase. This template was designed such that the initiation nucleotide is known from the product length detected with [32P]dGMP incorporated at the 3′-terminus (41). Pol α-primase initiated ribonucleotide-dependent DNA synthesis on 5′-C(TTAGGG)6 and gave a product ladder with an interval of 6 nt (Fig. 1A and B), which were degraded by several nucleotides on treatment with RNase (data not shown).
Figure 1.
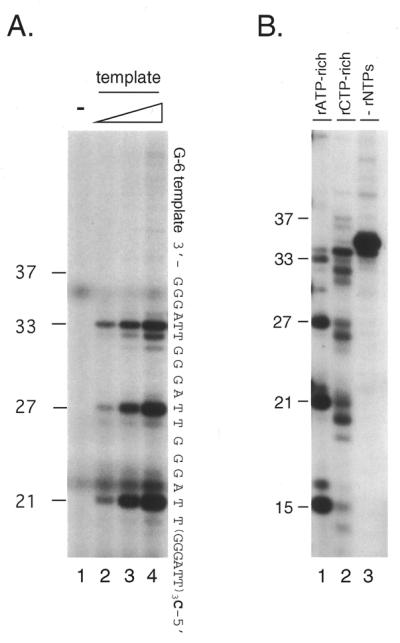
Lagging strand synthesis by pol α-primase on the telomere G-rich strand. (A) Initiation of RNA primer synthesis at a specific sequence. Reactions were performed with four dNTPs and four rNTPs using 0.08 U pol α-primase as described in Materials and Methods (rATP-rich conditions). As templates, 0, 10, 50 and 100 ng G-6 template were used (lanes 1–4, respectively). The reaction products, primer RNA–DNA conjugates, labeled with [α-32P]dGMP at the 3′-end of the DNA, were resolved by denaturing PAGE to show the initiation sites. The sequence of the G-6 template, constructed to incorporate dGMP at the 3′-end, as well as the positions of size markers are indicated. Numbers on the left side of panels indicate products sizes. (B) Shift of initiation sites by changing the ratio of rNTPs. Reactions were performed as described for (A) except that, in addition to rATP-rich conditions (lane 1), rCTP-rich conditions were employed (lane 2) using 100 ng G-6 template and 0.16 (lane 1) or 0.32 U (lane 2 and 3) pol α-primase (Materials and Methods). Reactions without rNTPs were performed as a negative control (lane 3). Sizes of the products are indicated on the left side of the figure. (C) Expansion of RNA-primed DNA products on the G-rich telomere strand by pol α-primase. The rNTPs-dependent DNA synthesis reaction was performed under rATP-rich conditions with 100 ng G-6 template and 1.6 U pol α-primase (lane 1). As a positive control for expansion, 1.0 U Klenow fragment was further added (lane 2). As a negative control (lane 3), primase (pol α-primase) was omitted from the reaction mixture used for lane 2. Products were labeled with [α-32P]dGTP at the 3′-ends of the the DNA that was extended from RNA primers and their sizes were estimated from positions of 32P-labeled oligonucleotides. The asterisks indicate non-specific products.
It has been reported that the RNA primer exclusively initiates with purine and that the highest concentration of ribonucleotide defines the eventual second nucleotide of the RNA sequence (41–43). Consistently, the initiation sites on the telomere repeats were also influenced by the concentration of ribonucleoside triphosphate. Under rATP-rich conditions, the major products were 21, 27 and 33 nt long, which initiated opposite the second (3′-side) thymine in the 5′-TTAGGG-3′ repeat unit (Fig. 1A). When the reaction was performed with an increased concentration of rCTP (40-fold rATP), new product bands appeared at 20, 26 and 32 nt, corresponding to a new initiation site at the first thymine (5′-site) in the repeats (Fig. 1B).
Expansion beyond the template size was not seen when the molar ratio template:pol α-primase was 1:1.5 (Fig. 1A). We increased the amount of pol α-primase by 20-fold. Interestingly, under the new conditions expanded reaction products beyond 37 nt long were observed. Additional Klenow fragment further increased the products beyond 37 nt long (Fig. 1C). In both cases the reaction products migrated with an interval of 6 nt. These reaction products were not observed when rNTPs were omitted (Fig. 1B, lane 3) and are considered to be primed by pol α-primase.
Primer extension by DNA polymerases
To determine whether other DNA polymerases can also expand the products beyond the template length, we performed the primer extension assay using various DNA polymerases. We prepared two synthetic oligonucleotides, 5′-A(GGGTTA)5 and 5′-(TAACCC)5T, which were annealed and used as the template–primer. Since each strand was of 31 nt containing five repeat units plus one nucleotide, the length of reaction products was expected to be 61 nt or shorter unless the primer slipped on the template oligomer during DNA synthesis (Fig. 2A). Reactions were carried out at 37°C with either pol α-primase, HIV-RT or Klenow fragment. The polymerases extended primer 5′-A(GGGTTA)5 up to 61 nt and then further elongated it as long as 67 nt or more (Fig. 2B and C). The reaction products migrated at intervals of 6 nt, although some of them were found to be doublets (17,18). The maximum product lengths with pol α-primase, Klenow fragment and HIV-RT were ∼100, 150 and 130 nt, respectively (Fig. 2B). We also analyzed Taq pol I and pol ɛ under the same conditions. Taq pol I did not support efficient expansion at 37°C, which was far below the optimum temperature, nor did pol ɛ extend the primer more than the template length (data not shown).
If slippage of the template–primer caused the expansion of the primer DNA, the reaction products should consist of the repeat sequences. We purified and cloned the 67 nt and longer reaction products produced by Klenow fragment. All the clones exclusively contained 11–14 repeat units (data not shown). From these results we concluded that the telomere repeat sequence is capable of being expanded by slippage DNA synthesis in vitro.
Orientation-dependent expansion and deletion have been reported in CTG and GGC repeats (39,44). To study if the C-rich telomere strand is also expanded during the reaction, we used [32P]dCTP to label the products. Roughly the same amount of 5′-(TAACCC)5T primer was expanded as 5′-A(GGGTTA)5 primer by pol α-primase, Klenow fragment and HIV-RT (Fig. 2C).
Primer extension was also performed in reaction mixtures lacking one or more dNTPs (minus conditions). Under either minus dATP or minus dTTP conditions, primer 5′-A(GGGTTA)5 was extended, but this ceased 1 nt before the sites where dAMP or dTMP would be incorporated and was then degraded by a few nucleotides due to the 3′→5′ exonuclease associated with the Klenow fragment (Fig. 2D, lanes 2 and 3). Under the conditions where two or more nucleotides were omitted, DNA synthesis was severely impaired (Fig. 2D).
Interestingly, amounts of extension/expansion products of 55 nt and longer under the minus dCTP conditions were not the same as under the complete conditions (Fig. 2D, lanes 1 and 4), although dCTP is not required for extension of 5′-A(GGGTTA)5. Under the complete conditions expanded products over 61 nt were more frequent than under the minus dCTP conditions. In contrast, reaction products of 55 and 61 nt were less frequent than under minus dCTP conditions. These data indicate that extension of the 5′-(TAACCC)5T strand is required for efficient expansion of the 5′-A(GGGTTA)5 strand and argues against possible contamination by any templates longer than 61 nt. We observed essentially the same results using 5′-(TAACCC)5T as the primer (data not shown).
Effects of RP-A
Replication protein A (RP-A) is one of the components comprising the eukaryotic DNA replication machinery. This single-stranded DNA-binding protein is essential for SV40 DNA replication (38,45). To determine whether this single-stranded DNA-binding protein may accelerate or stabilize loop formation in slippage expansion, we performed DNA synthesis on telomere repeats in the presence of human RP-A. RP-A markedly enhanced telomere repeat expansion by Klenow fragment (Fig. 2E, lanes 2 and 4), but affected the expansion by pol α to a lesser extent. Although stimulation of expansion by pol α was observed at 67 nt, it was not evident at greater sizes. RP-A enhanced the 61 nt band (Fig. 2E, lanes 1 and 2), which is presumably composed of the expanded products of originally shorter sequences (see Materials and Methods).
Expansion and deletion of the mammalian telomere repeat in E.coli during culture
The involvement of DNA replication in telomere repeat expansion in vivo was examined with proliferating E.coli cells transformed with pGEM plasmids harboring mammalian telomere sequences. The 28 and 30 telomere unit repeats were inserted into pGEM plasmids in different orientations, so as to measure expansion of the repeat sequence in both leading and lagging strand synthesis (Fig. 3A and B; pGEM28G and pGEM30C in orientations I and II, respectively). pGEM is a derivative of pUC that has a unidirectional replication origin of ColE1 (the sense strand of the β-lactamase gene corresponds to the leading strand in replication). A recombination-deficient strain of E.coli, JM109 (recA1), was transformed with the plasmids and then cultured for a large number of generations. At 40 generations (Fig. 3) the sizes of inserts had changed in only a small fraction, <10% of the total colonies. After 185 generations (Fig. 3), both deletion and the expansion of the inserts by multiples of 6 nt appeared as ladders at high frequencies, in >80% of total colonies. These results indicate that expansions and deletions of the telomere repeat sequence occur frequently in vivo in E.coli cells, presumably by the slippage synthesis mechanism. Besides the 6 nt ladders of expansion or deletion, larger deletions (Fig 3A, lane 8) or expansions (data not shown) were also detected. The large changes occurred in four of 30 clones at 185 generations. These changes were observed with both pGEM28G and pGEM30C and no significant differences were seen between these two orientations.
DISCUSSION
De novo DNA synthesis and repeat expansion
We analyzed replication of the G-rich telomere repeat using pol α-primase, a unique complex that initiates lagging strand synthesis. A previous study has shown that α-primase may initiate RNA priming on pyrimidines in the telomere G-rich strand (40). The G-rich strand, (GGGTTA)n, contains a putative preferred initiation site for primase, which is the dT adjacent to a dA (42,46). We performed de novo DNA synthesis reactions under rATP- and rCTP-rich conditions and observed two distinct initiation products that migrated at the predicted positions (Fig. 1A). Consistent with the previously known characteristics of α-primase (40), a unique RNA priming was preferentially initiated from the position opposite the 3′-side thymine in the G-rich telomere repeat under the rATP-rich conditions, and the initiation site shifted 1 nt under the rCTP-rich conditions. It has been shown that the three contiguous guanines next to TTA in the telomere repeat may not be favored by primase as priming sites (43). In cells, where RP-A acts as a molecular tether between G-rich strands and pol α-primase (47), priming efficiency would be higher than in the in vitro system.
DNA synthesis took place by successively extending the RNA primer. Expansion of the telomere repeats beyond the template sizes was clearly observed when an excess of pol α-primase was used (Fig. 1A). The expanded products migrated as ladders on PAGE with an interval of 6 nt. Therefore, the expansion that follows de novo DNA synthesis may operate by the slippage mechanism, as in the expansion of mono-, di- and trinucleotides.
Primer extension analysis
To determine whether other DNA polymerases could also expand telomere hexanucleotide repeats beyond the template length, we performed the extension assay using E.coli pol I Klenow fragment and HIV-RT. They were capable of extending both 5′-A(GGGTTA)5 and 5′-(TAACCC)5T strands more than the template length (Fig. 2B and C). These expanded products were shown to be composed exclusively of the telomere repeat sequences (data not shown). It is likely that the products shorter than the template may be generated by hairpin formation of the template strand, though this cannot be measured precisely in our detection system. These results imply that these DNA polymerases have an intrinsic activity for expansion of the hexanucleotide repeats such as telomere repeats as well as for simpler tandem repeats.
Expansion was observed when the reaction was carried out in an excess of polymerase over template (Fig. 2B and C). The polymerases that showed expansions are all distributive. A processive DNA polymerase, human pol ɛ, in contrast, did not produce expansion beyond the template length (data not shown). These results suggest that frequent dissociation and re-association of DNA polymerases with the primer terminus are essential for expansion, in agreement with the model that expansion is mediated by a slippage-based template–primer realignment (7,8,16,17,48,49).
Orientation-dependent repeat instability has been reported using CTG/CAG repeats in E.coli (39) and in yeast (50). In an in vitro experiment using the GGC/GCC triplet repeat in Fragile X syndrome, active expansion was found in the GCC strand and addition to the GGC strand was passive (16). At first we expected that the G-rich strand would be preferentially expanded by hairpin structure formation, as occurs with other G-rich trinucleotide repeats. However, approximately the same number of 5′-(TAACCC)5T primers were expanded as the 5′-A(GGGTTA)5 primer by pol α-primase, Klenow fragment and HIV-RT (Fig. 2C). Thus, preferential G-rich strand expansion was not apparent with any DNA polymerase (Fig. 2B and C).
In this context, symmetrical expansion was confirmed by the experiments under the minus one nucleotide conditions. Because the telomere repeat sequences were composed of three kinds of deoxynucleotides, each of the two primers was selectively extended while another was forced to cease extension (Fig. 2D). This ‘forced’ asymmetrical reaction produced less expanded product than the symmetrical reaction (Fig. 2D). These results show that both strand extensions are required for efficient primer expansion.
Effects of single-stranded DNA-binding protein
Various protein factors are involved in DNA replication. Among them, single-stranded DNA-binding proteins, such as eukaryotic RP-A and E.coli SSB, play important roles in DNA metabolism, including DNA replication, repair and recombination (38,45,51). Here we have examined the effects of RP-A on the expansion of telomere repeats.
As shown in Figure 2E, RP-A markedly enhanced the reaction, accompanied by enhanced expansion of repeats by both pol α-primase and Klenow fragment (Fig. 2E, lanes 2 and 4). On the other hand, SSB did not enhance but suppressed synthesis of not only the expanded products, but also the products less than 61 nt long in both DNA polymerase reactions (data not shown).
It has been shown that RP-A is essential for the formation of replication foci and the initiation of cellular chromosomal DNA replication (52,53). RP-A reduces pausing by DNA pol α-primase at specific sites in the single-stranded templates (54,55) and increases the accuracy of DNA pol α-primase (51,54). Besides the interaction with DNA, RP-A stimulates DNA polymerases by a protein–protein interaction and this direct interaction may also result in the stimulation of telomere repeat expansion.
These results indicate that DNA-binding proteins may affect slippage expansion in telomere DNA replication. In telomere DNA replication, telomerase may work in concert with DNA polymerases and primase, in the presence of telomere-binding proteins such as TRF1, TRF2 and TIN2. These telomere-specific proteins might modulate the secondary or tertiary structures of telomere DNA and regulate telomere maintenance.
Instability of the telomere repeat sequence in E.coli
Expansion and deletion of the CTG trinucleotide repeat in E.coli have been reported by Kang et al. (39). They proposed the possibility of orientation-, length- and generation-dependent instability. To determine if the telomere hexanucleotide repeat is also deleted and expanded in vivo, we also used E.coli cells as a model for in vitro telomere DNA replication. We constructed plasmids in which 28 and 30 telomere repeats were inserted in both orientations into pGEM plasmids and introduced them into a recombination-deficient E.coli strain, which was cultured for a number of generations (Fig. 3). It can clearly be seen that the inserted telomere repeats underwent deletion or expansion, giving a ladder of product bands with an interval of 6 nt. These results support the involvement of DNA replication in the instability of telomere repeats in vivo. The instability was dependent on the number of generations (Fig. 3) and on the length of inserted telomere repeats (10–100 repeats; data not shown), as was observed with the CTG repeat. On the other hand, the orientation-dependent difference was not obvious in expansion or deletion of telomere repeats either in vivo (Fig. 3) or in vitro (Fig. 2). This suggests that, unlike the trinucloetide repeats, both strands of mammalian telomere repeats form loops at similar frequency.
Furthermore, the telomere sequences in the plasmids showed much larger deletions and expansions in four of 30 clones after culture for 185 generations (Fig. 3A, lane 8, and data not shown). Recently, Morag et al. showed that difficulties in DNA replication may trigger recombinational expansions and deletions of two 787 bp repeats (56). The large changes in telomere repeats in the plasmids, therefore, might be produced either by forming a large hairpin structure or by a recombination process. Furthermore, conditions such as length of repeat, copy number of plasmid, generation of the cell, genetic make-up of host cell and site of insertion in the vector may influence the frequency of deletions and/or expansions.
Slippage DNA synthesis in telomere maintenance
At present we are not able to assess precisely the frequency of single template–primer slippage or a single repeat expansion, because of the absence of strict kinetic studies. Since relatively enzyme-rich conditions were required to achieve visualization of the expansions (Fig. 1C), the probability of repeat expansion may not be as high under the usual assay conditions. Obviously, slippage-mediated expansion by pol α-primase would not normally compensate for telomere maintenance in vivo, since chromosomes become shorter by tens to hundreds of nucleotides per cell division in cells that lack telomerase (25,31). Nevertheless, the intrinsic potential of slippage expansion by pol α-primase may be important in genomic instability. Genetic analyses have indicated that the lagging strand is more susceptible to genome instability than the leading strand in E.coli (39,49,57) and in yeast (50), presumably due to the asymmetrical nature of DNA synthesis. Pol α mutants of Schizosaccharomyces pombe, as well as mutants of other protein factors involved in lagging strand DNA synthesis, show genome instability due to aberrant DNA synthesis at the semi-permissive temperature (58). Abnormal telomere elongation has been observed in a pol α mutant cdc17-1 strain of Saccharomyces cerevisiae (59). Diede and Gottshling (36) have recently shown, in contrast, that telomerase is unable to add nucleotides onto the 3′-end in the absence of pol α and pol δ, while it does so without pol ɛ, suggesting that extension of the TG1–3 strand of the yeast telomere by telomerase is closely coordinated with replication of the lagging C1–3A strand. All of these results strongly suggest that pol α-primase plays a part in telomere DNA replication.
Telomerase is found in immortalized cells as well as in cancer cells, which can add telomere repeats at the telomere ends to preserve telomere length (60). However, some immortalized cell lines maintain their telomere length without detectable telomerase activity (61,62), which suggests that one or more unknown mechanisms other than via telomerase exist in these telomerase-negative cells to preserve telomere length. Our present results, showing that the telomere hexanucleotide repeats are expanded beyond the template length by pol α-primase and other DNA polymerases, indicate the possible involvement of this intrinsic activity in telomere maintenance under certain conditions.
Overexpression of pol α and/or other events causing replication errors leading to instability of the double-strandedness of telomere DNA would induce expansion of the telomere sequences by slippage, followed by telomere instability.
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank Drs Keiko Tamiya-Koizumi and Nobuki Hayakawa in our laboratory for helpful discussions. We are grateful to Mr Yasutomo Ito, Ms Miwa Takahashi and Mrs Tazuko Tomita for expert technical assistance. K.N. is a Research Fellow of the Japan Society for Promotion of Science. This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan
REFERENCES
- 1.Foster P.L. and Trimarchi,J.M. (1994) Science, 265, 407–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg S.M., Longerich,S., Gee,P. and Harris,R.S. (1994) Science, 265, 405–407. [DOI] [PubMed] [Google Scholar]
- 3.Greenblatt M.S., Grollman,A.P. and Harris,C.C. (1996) Cancer Res., 56, 2130–2136. [PubMed] [Google Scholar]
- 4.Aquilina G., Hess,P., MacGeoch,C., Casciano,I., Karran,P. and Bignami,M. (1994) Proc. Natl Acad. Sci. USA, 91, 8905–8909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aaltonen L.A., Peltomaki,P., Mecklin,J.-P., Jarvinen,H., Jass,J.R., Green,J.S., Lynch,H.T., Watson,R., Tallqvist,G., Juhola,M., Sistonen,P., Hamilton,S.R., Kinzler,K.W., Vogelstein,B. and de la Chapelle,A. (1994) Cancer Res., 54, 1645–1648. [PubMed] [Google Scholar]
- 6.Umar A., Boyer,J.C., Thomas,D.C., Nguyen,D.C., Risinger,J.I., Boyd,J., Ionov,Y., Perucho,M. and Kunkel,T.A. (1994) J. Biol. Chem., 269, 14367–14370. [PubMed] [Google Scholar]
- 7.Kornberg A. and Baker,T.A. (1992) DNA Replication, 2nd edn. Freeman, New York, NY.
- 8.Kunkel T.A. (1992) J. Biol. Chem., 267, 18251–18254. [PubMed] [Google Scholar]
- 9.Ashley C. Jr and Warren,S.T. (1995) Annu. Rev. Genet., 29, 703–728. [DOI] [PubMed] [Google Scholar]
- 10.Pulst S.M., Nechiporuk,A., Nechiporuk,T., Gispert,S., Chen,X.N., Lopes-Cendes,I., Pearlman,S., Starkman,S., Orozco-Diaz,G., Lunkes,A., DeJong,P., Rouleau,G.A., Auburger,G., Korenberg,J.R., Figueroa,C. and Sahba,S. (1996) Nature Genet., 14, 269–276. [DOI] [PubMed] [Google Scholar]
- 11.Sanpei K., Takano,H., Igarashi,S., Sato,T., Oyake,M., Sasaki,H., Wakisaka,A., Tashiro,K., Ishida,Y., Ikeuchi,T., Koide,R., Saito,M., Sato,A., Tanaka,T., Hanyu,S., Takiyama,Y., Nishizawa,M., Shimizu,N., Nomura,Y., Segawa,M., Iwabuchi,K., Eguchi,I., Tanaka,H., Takahashi,H. and Tsuji,S. (1996) Nature Genet., 14, 277–284. [DOI] [PubMed] [Google Scholar]
- 12.David G., Abbas,N., Stevanin,G., Durr,A., Yvert,G., Cancel,G., Weber,C., Imbert,G., Saudou,F., Antoniou,E., Drabkin,H., Gemmill,R., Giunti,P., Benomar,A., Wood,N., Ruberg,M., Agid,Y., Mandel,J.L. and Brice,A. (1997) Nature Genet., 17, 65–70. [DOI] [PubMed] [Google Scholar]
- 13.Zhuchenko O., Bailey,J., Bonnen,P., Ashizawa,T., Stockton,D.W., Amos,C., Dobyns,W.B., Subramony,S.H., Zoghbi,H.Y. and Lee,C.C. (1997) Nature Genet., 15, 62–69. [DOI] [PubMed] [Google Scholar]
- 14.Imbert G., Saudou,F., Yvert,G., Devys,D., Trottier,Y., Garnier,J.M., Weber,C., Mandel,J.L., Cancel,G., Abbas,N., Durr,A., Didierjean,O., Stevanin,G., Agid,Y. and Brice,A. (1996) Nature Genet., 14, 285–291. [DOI] [PubMed] [Google Scholar]
- 15.Kunst C.B. and Warren,S.T. (1994) Cell, 77, 853–861. [DOI] [PubMed] [Google Scholar]
- 16.Ji J., Clegg,N.J., Peterson,K.R., Jackson,A.L., Laird,C.D. and Loeb,L.A. (1996) Nucleic Acids Res., 24, 2835–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schlotterer C. and Tautz,D. (1992) Nucleic Acids Res., 20, 211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakayabu M., Miwa,S., Suzuki,M., Izuta,S., Sobue,G. and Yoshida,S. (1998) Nucleic Acids Res., 26, 1980–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lyons-Darden T. and Topal,M.D. (1999) Nucleic Acids Res., 27, 2235–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lyons-Darden T. and Topal,M.D. (1999) J. Biol. Chem., 274, 25975–25978. [DOI] [PubMed] [Google Scholar]
- 21.Gacy A.M. and McMurray,C.T. (1998) Biochemistry, 37, 9426–9434. [DOI] [PubMed] [Google Scholar]
- 22.Petruska J., Hartenstine,M.J. and Goodman,M.F. (1998) J. Biol. Chem., 273, 5204–5210. [DOI] [PubMed] [Google Scholar]
- 23.Blackburn E.H. (1991) Nature, 350, 569–573. [DOI] [PubMed] [Google Scholar]
- 24.Greider C.W. (1996) Annu. Rev. Biochem., 65, 337–365. [DOI] [PubMed] [Google Scholar]
- 25.Levy M.Z., Allsopp,R.C., Futcher,A.B., Greider,C.W. and Harley,C.B. (1992) J. Mol. Biol., 225, 951–960. [DOI] [PubMed] [Google Scholar]
- 26.Harley C.B. (1991) Mutat. Res., 256, 271–282. [DOI] [PubMed] [Google Scholar]
- 27.Shore D. (1997) Trends Biochem. Sci., 22, 233–235. [DOI] [PubMed] [Google Scholar]
- 28.Fan X. and Price,C.M. (1997) Mol. Biol. Cell, 8, 2145–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bryan T.M., Englezou,A., Dunham,M.A. and Reddel,R.R. (1998) Exp. Cell Res., 239, 370–378. [DOI] [PubMed] [Google Scholar]
- 30.Sprung C.N., Sabatier,L. and Murnane,J.P. (1999) Exp. Cell Res., 247, 29–37. [DOI] [PubMed] [Google Scholar]
- 31.Harley C.B., Futcher,A.B. and Greider,C.W. (1990) Nature, 345, 458–460. [DOI] [PubMed] [Google Scholar]
- 32.Niida H., Matsumoto,T., Satoh,H., Shiwa,M., Tokutake,Y., Furuichi,Y. and Shinkai,Y. (1998) Nature Genet., 19, 203–206. [DOI] [PubMed] [Google Scholar]
- 33.Blasco M.A., Lee,H.W., Hande,M.P., Samper,E., Lansdorp,P.M., DePinho,R.A. and Greider,C.W. (1997) Cell, 91, 25–34. [DOI] [PubMed] [Google Scholar]
- 34.Lundblad V. and Blackburn,E.H. (1993) Cell, 73, 347–360. [DOI] [PubMed] [Google Scholar]
- 35.Adams Martin A., Dionne,I., Wellinger,R.J. and Holm,C. (2000) Mol. Cell Biol., 20, 786–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diede S.J. and Gottschling,D.E. (1999) Cell, 99, 723–733. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki M., Izuta,S., Savoysky,E., Sakurai,T., Simbulan,C., Tatebe,M., Kojima,K. and Yoshida,S. (1993) Biochem. Mol. Biol. Int., 29, 645–652. [PubMed] [Google Scholar]
- 38.Kenny M.K., Schlegel,U., Furneaux,H. and Hurwitz,J. (1990) J. Biol. Chem., 265, 7693–7700. [PubMed] [Google Scholar]
- 39.Kang S., Jaworski,A., Ohshima,K. and Wells,R.D. (1995) Nature Genet., 10, 213–218. [DOI] [PubMed] [Google Scholar]
- 40.Reveal P.M., Henkels,K.M. and Turchi,J.J. (1997) J. Biol. Chem., 272, 11678–11681. [DOI] [PubMed] [Google Scholar]
- 41.Suzuki M., Savoysky,E., Izuta,S., Tatebe,M., Okajima,T. and Yoshida,S. (1993) Biochemistry, 32, 12782–12792. [DOI] [PubMed] [Google Scholar]
- 42.Yamaguchi M., Hendrickson,E.A. and DePamphilis,M.L. (1985) Mol. Cell. Biol., 5, 1170–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheaff R.J. and Kuchta,R.D. (1993) Biochemistry, 32, 3027–3037. [DOI] [PubMed] [Google Scholar]
- 44.Chen X., Mariappan,S.V., Catasti,P., Ratliff,R., Moyzis,R.K., Laayoun,A., Smith,S.S., Bradbury,E.M. and Gupta,G. (1995) Proc. Natl Acad. Sci. USA, 92, 5199–5203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wold M.S. (1997) Annu. Rev. Biochem., 66, 61–92. [DOI] [PubMed] [Google Scholar]
- 46.Suzuki M., Savoysky,E., Izuta,S., Tatebe,M., Okajima,T. and Yoshida,S. (1993) Biochemistry, 32, 12782–12792. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki M., Tamiya-Koizumi,K., Takemura,M., Furuta,K., Izuta,S., Savoysky,E., Miura,A. and Yoshida,S. (1996) J. Biochem. Tokyo, 120, 766–772. [DOI] [PubMed] [Google Scholar]
- 48.Tran H.T., Keen,J.D., Kricker,M., Resnick,M.A. and Gordenin,D.A. (1997) Mol. Cell. Biol., 17, 2859–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trinh T.Q. and Sinden,R.R. (1991) Nature, 352, 544–547. [DOI] [PubMed] [Google Scholar]
- 50.Freudenreich C.H., Stavenhagen,J.B. and Zakian,V.A. (1997) Mol. Cell. Biol., 17, 2090–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suzuki M., Izuta,S. and Yoshida,S. (1994) J. Biol. Chem., 269, 10225–10228. [PubMed] [Google Scholar]
- 52.Adachi Y. and Laemmli,U.K. (1992) J. Cell Biol., 119, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adachi Y. and Laemmli,U.K. (1994) EMBO J., 13, 4153–4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carty M.P., Levine,A.S. and Dixon,K. (1992) Mutat. Res., 274, 29–43. [DOI] [PubMed] [Google Scholar]
- 55.Podust V.N. and Hubscher,U. (1993) Nucleic Acids Res., 21, 841–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morag A.S., Saveson,C.J. and Lovett,S.T. (1999) J. Mol. Biol., 289, 21–27. [DOI] [PubMed] [Google Scholar]
- 57.Veaute X. and Fuchs,R.P. (1993) Science, 261, 598–600. [DOI] [PubMed] [Google Scholar]
- 58.Liu V.F., Bhaumik,D. and Wang,T.S. (1999) Mol. Cell. Biol., 19, 1126–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adams A.K. and Holm,C. (1996) Mol. Cell. Biol., 16, 4614–4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim N.W., Piatyszek,M.A., Prowse,K.R., Harley,C.B., West,M.D., Ho,P.L., Coviello,G.M., Wright,W.E., Weinrich,S.L. and Shay,J.W. (1994) Science, 266, 2011–2015. [DOI] [PubMed] [Google Scholar]
- 61.Bryan T.M., Englezou,A., DallaPozza,L., Dunham,M.A. and Reddel,R.R. (1997) Nature Med., 3, 1271–1274. [DOI] [PubMed] [Google Scholar]
- 62.Strahl C. and Blackburn,E.H. (1996) Mol. Cell. Biol., 16, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]