

SUMMARY
Abasic sites are common DNA lesions stalling polymerases and threatening genome stability. When located in single-stranded DNA (ssDNA), they are shielded from aberrant processing by 5-hydroxymethyl cytosine, embryonic stem cell (ESC)-specific (HMCES) via a DNA-protein crosslink (DPC) that prevents double-strand breaks. Nevertheless, HMCES-DPCs must be removed to complete DNA repair. Here, we find that DNA polymerase α inhibition generates ssDNA abasic sites and HMCES-DPCs. These DPCs are resolved with a half-life of approximately 1.5 h. HMCES can catalyze its own DPC self-reversal reaction, which is dependent on glutamate 127 and is favored when the ssDNA is converted to duplex DNA. When the self-reversal mechanism is inactivated in cells, HMCES-DPC removal is delayed, cell proliferation is slowed, and cells become hypersensitive to DNA damage agents that increase AP (apurinic/apyrimidinic) site formation. In these circumstances, proteolysis may become an important mechanism of HMCES-DPC resolution. Thus, HMCES-DPC formation followed by self-reversal is an important mechanism for ssDNA AP site management.
In brief
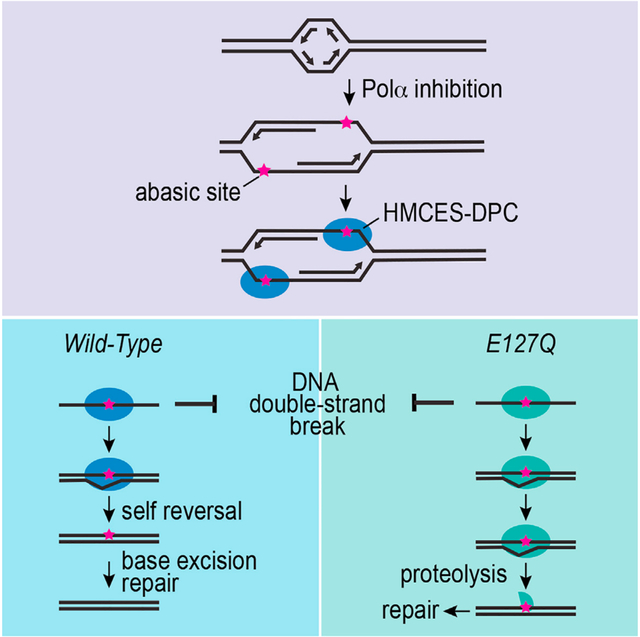
HMCES forms a reversible DNA-protein crosslink (DPC) to abasic sites in single-stranded DNA to prevent cleavage and double-strand break formation. Rua-Fernandez et al. demonstrate that an HMCES DPC self-reversal reaction is important to resolve the crosslink in cells. Inactivating self-reversal reduces cell fitness.
Graphical Abstract
INTRODUCTION
Apurinic/apyrimidinic (AP, or abasic) sites are one of the most common DNA lesions. They can be formed by spontaneous depurination/depyrimidination or as intermediates during the excision of damaged nucleobases by glycosylases.1–3 When AP sites are within double-stranded DNA (dsDNA), they can be repaired by base excision repair (BER). The endonuclease APE1 cleaves 5′ to the AP site, and Polβ uses the intact DNA strand as a template for synthesis to complete repair.4 AP sites can also be converted into strand breaks via a spontaneous β-elimination reaction.1,2 Spontaneous or enzymatic cleavage of AP sites located in ssDNA can lead to double-strand breaks (DSBs), which are highly toxic to cells. During DNA replication, AP sites are potent blocks to replicative polymerases, placing them at a dsDNA-ssDNA junction.5 Translesion DNA synthesis (TLS) polymerases can synthesize DNA across AP sites but will often cause mutations.
HMCES (5-hydroxymethyl cytosine, embryonic stem cell [ESC]-specific) was recently identified as a shield for ssDNA AP sites.6 HMCES is present at replication forks, interacts with PCNA (proliferating cell nuclear antigen), and covalently binds to ssDNA AP sites through an evolutionary conserved SOS response-associated peptidase (SRAP) domain, generating a DNA-protein crosslink (DPC).6 Unlike damaging DPC formation with other proteins, the HMCES-DPC is thought to be protective and beneficial to the cell. The HMCES-DPC prevents AP site cleavage, thereby reducing DSBs,6,7 and also decreases mutation frequency.6,8 HMCES and its bacterial ortholog, YedK, form a thiazolidine linkage between a ring-opened AP site and an N-terminal SRAP cysteine residue (C2).7,9,10 Inactivating HMCES through mutation of the cysteine, RNA interference, or gene disruption causes hypersensitivity to DNA-damaging agents that increase AP site frequency6,7 and synthetic lethality with nuclear expression of cytidine deaminase APOBEC3A,11,12 which also increases AP site formation. HMCES suppresses deletions during somatic hypermutation (SHM) by crosslinking to AP sites.13 Thus, HMCES acts by shielding ssDNA AP sites from inappropriate processing. Nevertheless, the HMCES-DPC itself is a bulky lesion that can interfere with replication and transcription.14 Persistence of DPCs in cells is associated with different diseases.15 Thus, the protective activity of HMCES-DPC should include its removal.
The HMCES-DPC is ubiquitylated, and proteasome inhibition may delay DPC removal after potassium bromate (KBrO3) treatment,6 suggesting a proteasome-dependent degradation of the DPC. In Xenopus egg extracts, the HMCES-DPC is removed by the SPRTN protease as an intermediate step in a DNA interstrand crosslink (AP-ICL) repair pathway that includes an ssDNA AP site.16 Proteasome inhibition had no effect on this system, despite HMCES-DPC being a target for ubiquitylation by the E3 ligase RFWD3 in Xenopus egg extracts.17 Other proteases have been reported to remove DPCs to maintain DNA replication and genome stability18–20; however, whether any act on HMCES-DPCs is unknown.
Recently, HMCES-DPC and YedK-DPC were shown to be reversible in biochemical reactions.14,21,22 Two residues in proximity to the thiazolidine linkage, His160 and Glu105, in YedK are important for this crosslink reversal process.21 Mutation of the human HMCES equivalent of Glu105 (Glu127) also impairs HMCES-DPC reversal in vitro.22 It is unknown whether this self-reversal activity is biologically important. Here, we developed a cellular system to detect, quantify, and track HMCES-DPC resolution. Our results provide evidence that self-reversal is an important mechanism in cells to remove the HMCES-DPC and promote cell fitness.
RESULTS
Polymerase alpha (POLα) inhibition generates ssDNA AP sites and HMCES-DPCs
Previous cellular studies of HMCES-DPC formation utilized DNA-damaging agents like KBrO3 to generate abasic sites. These approaches yield a modest increase in HMCES-DPC levels (approximately 3-fold).6 The generation of abasic sites in these circumstances occurs mostly in duplex DNA. Formation of the HMCES-DPC in response to this DNA damage likely requires unwinding of the DNA during DNA replication to move the AP site into ssDNA, where HMCES functions. Therefore, both the generation and the resolution of the HMCES-DPC in response to these types of DNA-damaging agents take hours. To better study DPC resolution, we looked for a cellular system in which AP site formation would be targeted to ssDNA and can be better separated from DPC resolution.
CD437 is a direct POLα inhibitor23 that prevents the synthesis of the lagging strand and rapidly generates large amounts of ssDNA compared to other replication stalling agents like hydroxyurea (Figure 1A).24 CD437 treatment dramatically slows DNA synthesis,25 and approximately half of the ongoing replication forks do not restart after its removal (Figure S1A). This acute, high-dose CD437 treatment is toxic to S-phase cells, but they remain viable for at least 8 h after exposure (Figure S1B). ssDNA is more vulnerable to chemical attack and spontaneous depurination,26 which leads to AP site formation. Indeed, we could detect AP sites with an aldehyde reactive probe (ARP) after the addition of CD437 (Figure 1B). Since HMCES crosslinks specifically to AP sites located in ssDNA,6 we hypothesized that CD437 would induce HMCES-DPC formation. As expected, cells treated with 5 μM CD437 for 30 min showed a large increase in the HMCES-DPC signal using the rapid approach to DNA adduct recovery (RADAR) assay (Figure 1C).27 The HMCES-DPCs generated by CD437 were greatly reduced in cells transduced with a plasmid expressing the uracil DNA glycosylase inhibitor (UGI)28 (Figure 1C), suggesting that HMCES reacted with AP sites largely created by glycosylase activity in these conditions. To confirm that CD437-induced HMCES-DPCs are formed via a C2 linkage, we tested DPC formation in cells in which endogenous HMCES was deleted by gene editing (HMCESΔ), and either wild-type (WT) or an HMCES C2A mutant protein that is unable to crosslink to the abasic site was expressed by retroviral integration. As expected, no HMCES-DPCs were detected in cells expressing HMCES C2A after CD437 treatment, whereas cells complemented with WT HMCES had an equivalent DPC level to the parental U2OS cells (Figure 1D). The HMCES C2A protein was expressed at similar levels to WT (Figure S2).
Figure 1. CD437 induces AP sites in ssDNA and HMCES-DPC formation in cells.
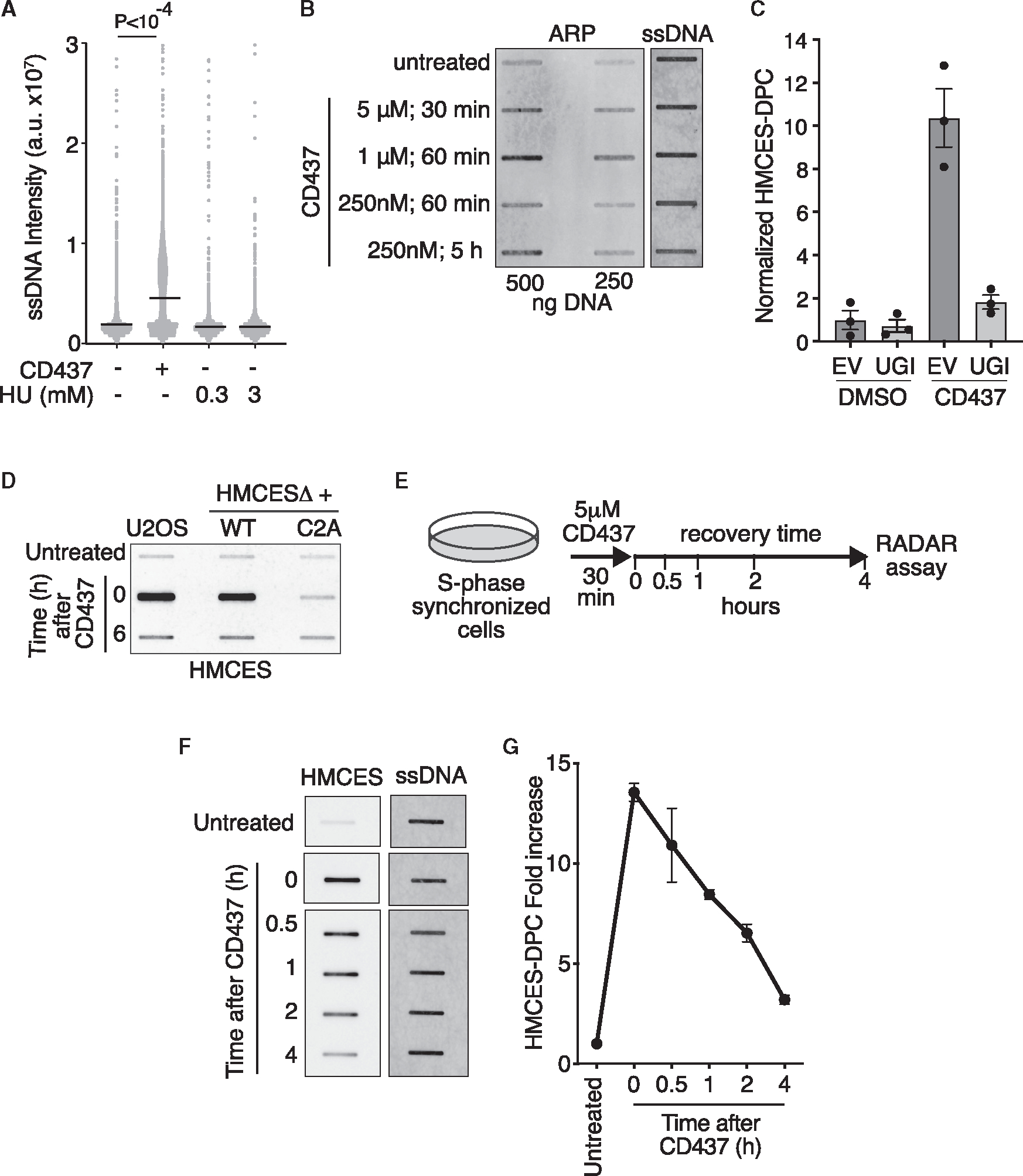
(A) Levels of ssDNA in HCT116 cells after treatment with 5 μncM CD437 or 0.3 or 3 mM HU for 30 min measured using the native BrdU staining assay. Intensity from individual nuclei and mean is shown; Kruskal-Wallis test.
(B) DNA was purified from cells treated as indicated and reacted with the aldehyde reactive probe (ARP) to measure AP sites. ARP-reacted DNA was blotted on membranes and probed with streptavidin-HRP to measure AP sites or with DNA antibody to measure DNA loading.
(C) Quantification of HMCES-DPC using RADAR assay. Cell expressing empty vector (EV) or UGI were treated with 5 μM CD437 or DMSO for 30 min (mean ± SEM, n = 3).
(D) Representative HMCES-DPC RADAR assay of parental U2OS or HMCESΔ cells complemented with WT or C2A HMCES and treated with 5 μM CD437 or DMSO (untreated) for 30 min. Cells were immediately harvested (T = 0) or the CD437 was removed, and cells were allowed to recover for 6 h prior to harvesting.
(E) Schematic of assay for detection of HMCES-DPC removal in cells after CD437 treatment used in all subsequent experiments. Cells were synchronized and released from thymidine for 2 h prior to treatment with 5 μM CD437 for 30 min. Samples were taken immediately after CD437 treatment (T = 0) and at the indicated time points.
(F) Representative image of HMCES-DPC resolution assay. HMCES-DPC was detected with an HMCES antibody. The samples were probed on the same blot, but the image was cut and reordered for the figure. The amount of DNA in each sample was quantified after deproteinization by blotting with a DNA antibody.
(G) RADAR assay of HMCES-DPC levels. Mean ± SEM, n = 3.
HMCES-DPC levels returned almost to the same level as untreated cells 6 h after removing CD437 (Figure 1D). To measure the rate of DPC resolution, S-phase synchronized cells were treated with CD437 for 30 min and then were allowed to recover in normal growth media and harvested at varying time points to analyze HMCES-DPC levels (Figure 1E). We observed a strong HMCES-DPC signal immediately after CD437 treatment that declined rapidly during recovery (Figure 1F). Quantification shows an HMCES-DPC half-life between 1 and 2 h, and approximately 80% of the HMCES-DPC is removed after 4 h (Figure 1G). We conclude that CD437 promotes HMCES-DPC formation in cells by increasing AP sites in ssDNA and provides a quantifiable system to analyze HMCES-DPC removal.
CD437-induced HMCES-DPC removal does not require the proteasome or SPRTN protease
Proteolysis is a major pathway to repair DPCs during DNA replication,29 and treatment with a proteasome inhibitor (MG132) appeared to delay HMCES-DPC resolution after KBrO3 treatment,6 suggesting a proteasome-dependent degradation of the HMCES-DPC. To test the activity of the proteasome in HMCES-DPC removal, we utilized our CD437 system to track DPC levels in the absence or presence of MG132. Treating cells with MG132 increased the total amount of HMCES-DPC formed after CD437 incubation by approximately 25% 30 min after CD437 removal (Figure 2A). However, MG132 did not prevent the resolution of HMCES-DPCs, which proceeded at least as quickly after the 30 min time point as vehicle-treated cells (Figures 2A and S3A). Incubating cells with MG132 alone in the absence of CD437 does not induce HMCES-DPC formation (Figure S3B); therefore, the increase within the first 30 min of release from CD437 required ssDNA formation. We also observed similar results by inhibiting the UBA1 E1 enzyme with TAK243,30 which blocks protein ubiquitylation (Figures 2B, S3A, and S3B). These results suggest that ubiquitylation and the proteasome are not essential for resolving the HMCES-DPC, although they may affect the amount of DPC formed or retained at early time points after CD437 treatment.
Figure 2. Inactivation of the proteasome or SPRTN does not prevent HMCES-DPC removal.
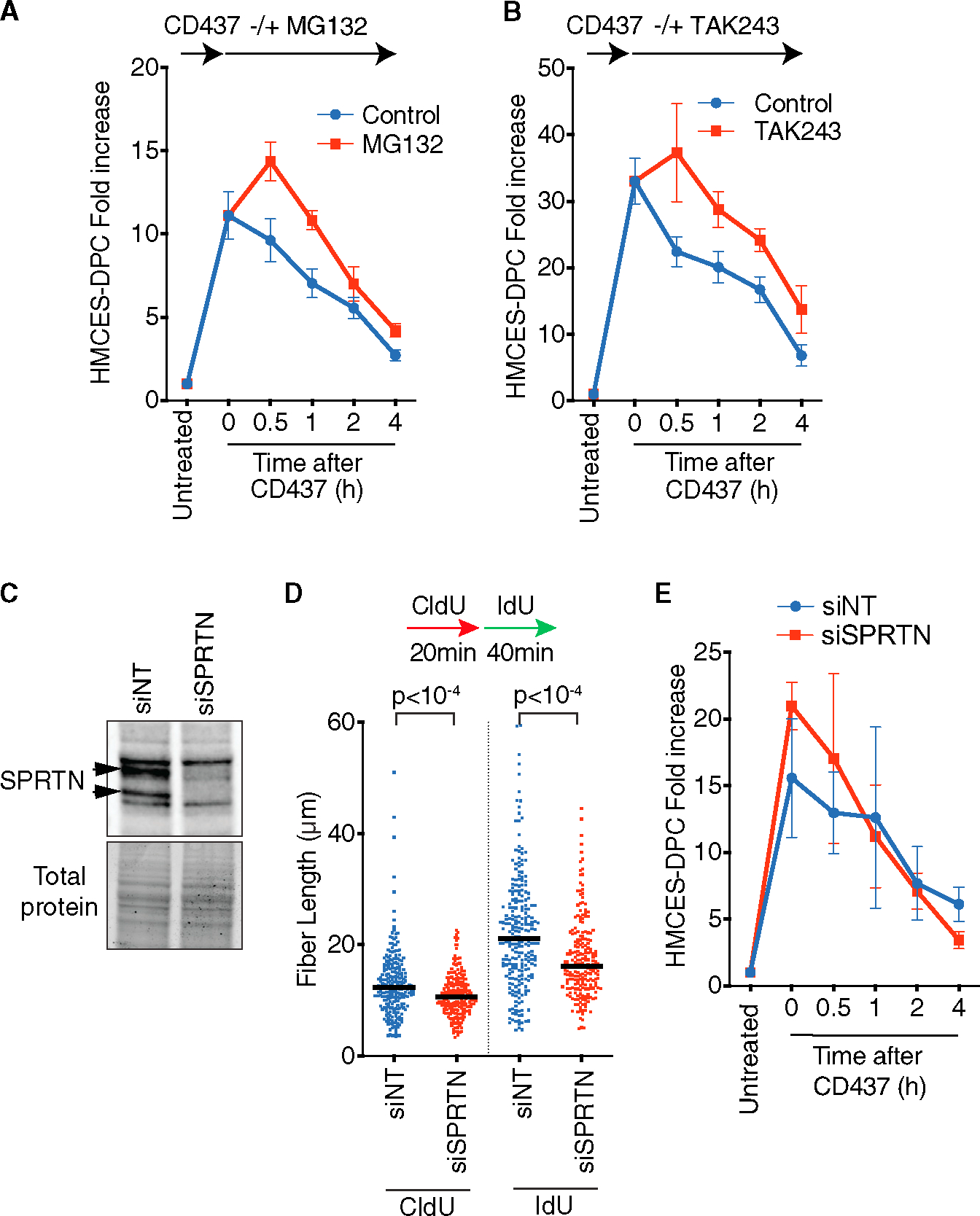
(A) RADAR assay of HMCES-DPC levels. MG132 was added immediately after CD437 treatment. Mean ± SEM, n = 5.
(B) RADAR assay of HMCES-DPC levels. TAK243 was added immediately after CD437 treatment. Mean ± SEM, n = 4.
(C) Immunoblot of cells transfected with non-targeting siRNA (siNT) or siRNA against SPRTN (siSPRTN).
(D) DNA combing assay analysis of replication fork speed in cells transfected with siNT or siSPRTN. Bar represents the median, and p values were derived from Mann-Whitney test.
(E) RADAR assay of HMCES-DPC levels in cells transfected with indicated siRNAs. Mean ± SEM, n = 3.
We next tested whether the SPRTN protease is important for HMCES-DPC resolution. SPRTN is essential for mammalian cell survival31; therefore, we used small interfering RNA (siRNA) to deplete SPRTN acutely and test its activity. Knockdown efficiency was confirmed by immunoblotting (Figure 2C). We also verified that SPRTN was functionally inactivated by measuring replication fork speed, which was previously shown to be slowed by SPRTN inactivation (Figure 2D).32 Depletion of SPRTN did not prevent CD437-induced HMCES-DPC removal (Figure 2E). Additionally, we tested a possible redundancy between SPRTN and the proteasome; however, MG132 did not prevent HMCES-DPC removal in SPRTN knockdown cells (Figure S3C). Therefore, our data suggest that neither the proteasome nor SPRTN activity is critical to remove HMCES-DPCs induced by POLα inhibition.
HMCES Glu127 mediates a self-reversal reaction, which is stimulated by a duplex-forming oligonucleotide
Although the HMCES-DPC thiazolidine linkage appears to be stable and resistant to repair enzymes like AP endonucleases, biochemical experiments using a second ssDNA oligonucleotide containing an AP site as a trap showed that it is reversible.21 In the HMCES bacterial ortholog YedK, the reversal reaction was disrupted by E105Q and H160Q mutations, with the former having a stronger effect. To test if the equivalent glutamic acid residue is also important for the self-reversal of human HMCES, we took a similar experimental approach. The HMCES-DPC was formed by incubation of the SRAP domain with a 20-nucleotide DNA oligo containing an AP site (DPC-20). Next, we added a 40-nucleotide AP DNA oligo in 50-fold excess to trap any self-reversed protein during incubation at 37°C (Figure 3A). The DPC-20 slowly decreased over time as a DPC-40 formed, in accordance with self-reversal regenerating an intact and active HMCES protein capable of crosslinking again to another available ssDNA AP site (Figures 3B and 3C). In contrast, the E127Q HMCES-DPC is completely unable to reverse even after 24 h of incubation. This result indicates that E127 is necessary for human HMCES-DPC self-reversal.
Figure 3. HMCES-DPC self-reversal is dependent on E127 and is stimulated by duplex DNA.
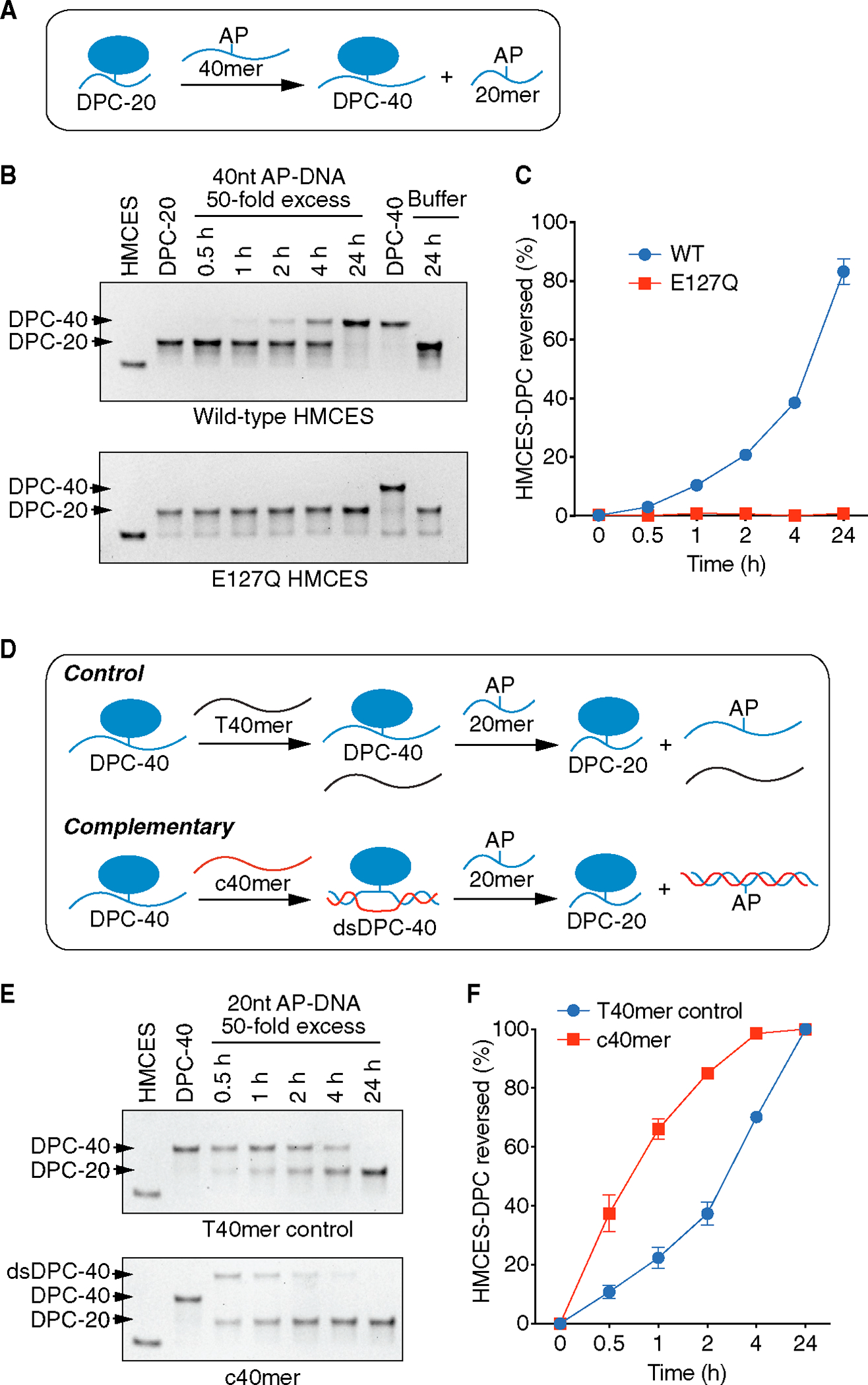
(A) Schematic of HMCES-DPC biochemical self-reversal assay. The AP site containing 40-nucleotide ssDNA oligonucleotide (40-mer) was added at 50× excess.
(B) Representative time course experiment of HMCES-DPC reversal with purified WT or E127Q HMCES. The first lane is HMCES protein without any DNA. The free protein and DPC-20 (DPC with 20-nucleotide oligonucleotide) or DPC-40 (DPC with 40-nucleotide oligonucleotide) were visualized by Coomassie-stained SDS-PAGE.
(C) Quantification of HMCES-DPC reversal. Percent reversed is the amount of DPC-40 product compared to the total. Mean ± SEM, n = 3.
(D) Schematic of HMCES-DPC reversal assay comparing ssDNA vs. duplex DNA. The HMCES DPC-40 was incubated with a complementary (c40mer) or non-complementary (T40mer control) oligonucleotide with a 20-nucleotide ssDNA trap containing an AP site.
(E) Representative time course of HMCES-DPC reversal. The higher migrating band is the product of the hybridization of c40mer with ssDNA HMCES-DPC (dsDPC-40).
(F) Quantification of HMCES-DPC reversal from ssDNA and dsDNA. Percent reversed is the amount of the DPC-20 product compared to the total. Mean ± SEM, n = 3.
The half-life of the HMCES-DPC in vitro is greater than the half-life of the DPC seen after CD437 treatment in cells. HMCES movement from one AP-ssDNA site to another requires not only the reversal of the thiazolidine linkage but also disengagement of DNA binding. Our previous studies showed that even without crosslinking, HMCES has a very high affinity for ssDNA.6 In contrast, HMCES cannot bind dsDNA. Thus, we reasoned that if a complementary oligonucleotide capable of forming a duplex with the ssDNA to which HMCES is crosslinked was included in the reversal reaction, we may be able to increase the speed at which we could observe the reversal. Indeed, this is the case. After generating HMCES-DPC-40, we added a complemented oligo (c40mer), which shifted the DPC-40 band in an SDS-PAGE gel (dsDPC-40). Next, we added a 50-fold excess of 20-mer containing an AP site to trap any HMCES that undergoes self-reversal (Figure 3D). As expected, the control sample containing a non-duplex-forming 40-mer oligonucleotide (T40mer control) showed a reduction of DPC-40 along with the formation of DPC-20. However, the reversal of DPC-40 in the presence of the duplex-forming oligonucleotide is faster compared to the control (Figure 3E). After 1 h incubation, the dsDNA structure yielded more than 60% reversal compared to HMCES-DPC in ssDNA, which reached a little more than 20% (Figure 3F). This result is consistent with two recent reports that found that DNA duplex formation caused an apparent accelerated self-reversal rate14,22 and indicates that the intrinsic rate of self-reversal is comparable with the reversal rate observed in cells.
HMCES-DPC self-reversal is an important resolution pathway in human cells
To test if HMCES-DPC self-reversal is an important resolution pathway in cells, we made use of the E127Q HMCES protein. First, we complemented the HMCESΔ cells to create stable cell lines expressing only the E127Q or WT HMCES at near-endogenous levels (Figure 4A). Analysis of HMCES-DPC levels in untreated, synchronized cells showed more DPC in E127Q cells compared to WT cells (Figure 4B), suggesting that the crosslinked state is increased in cells expressing the E127Q mutant even in the absence of added genotoxic stress. We next treated the E127Q- or WT-HMCES-expressing cells with CD437 to induce HMCES-DPC formation and tracked DPC resolution over time. The E127Q HMCES-DPC formed a similar total level of DPC as WT cells 30 min after CD437 treatment (Figure 4C), although the fold increase, when compared to the untreated cell control, was less. This may be because the baseline levels of E127Q HMCES-DPCs are higher, and the mutation modestly affects DPC formation as a result of the role of E127 in ribose ring opening.21 Strikingly, after removing CD437, the resolution kinetics of the E127Q was significantly delayed by at least 1 h compared to WT HMCES (Figures 4D and 4E), suggesting that E127-dependent self-reversal is an important process in cells.
Figure 4. Inactivating HMCES self-reversal delays DPC resolution in cells.
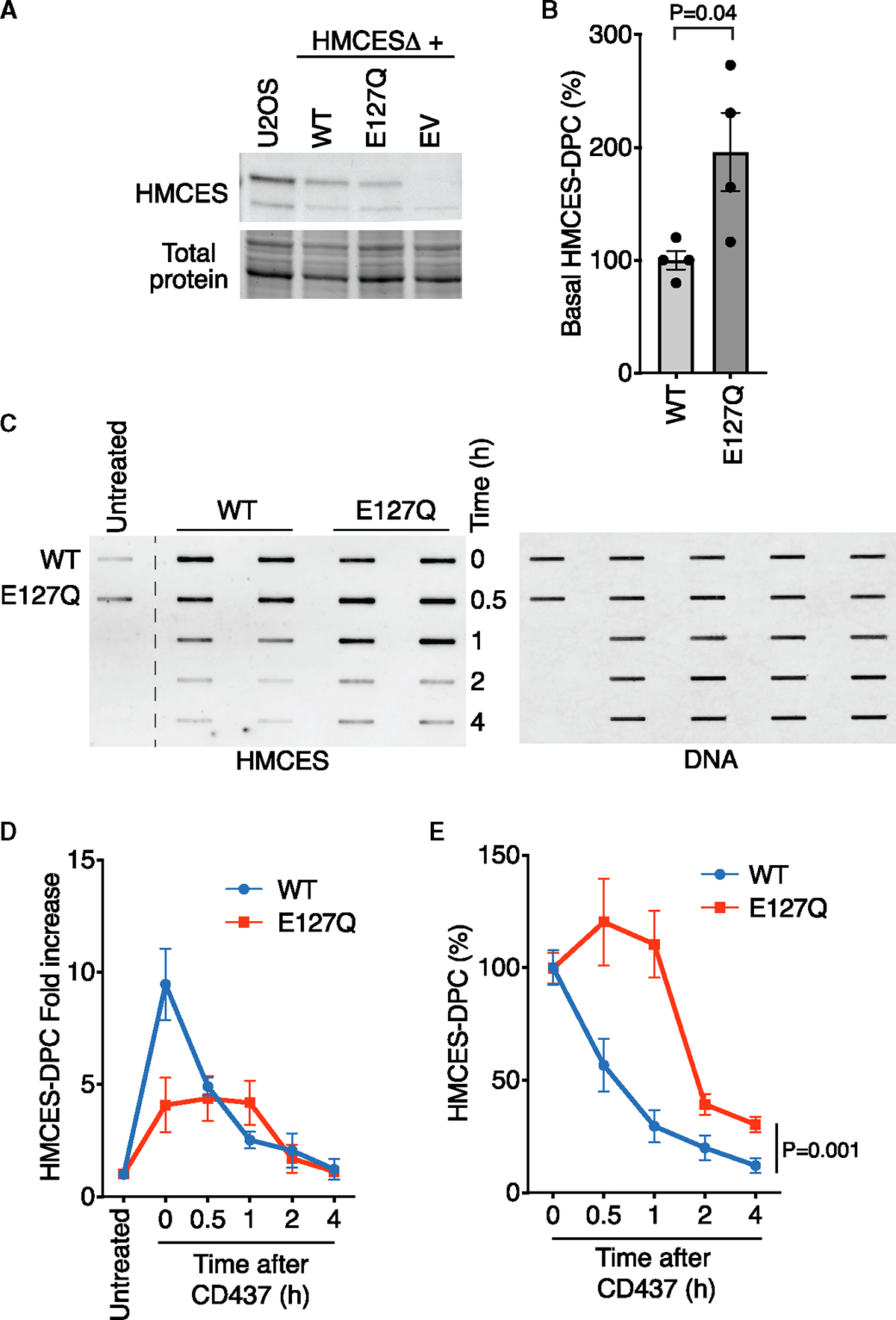
(A) Immunoblot of U2OS or HMCESΔ cells complemented with WT HMCES, E127Q HMCES, or EV.
(B) Quantification of HMCES-DPC levels in untreated cells that express WT or E127Q HMCES. RADAR assay was performed with samples from S-phase synchronized cells. Mean ± SEM, n = 4, two-tailed t test.
(C) Representative image of RADAR assay to detect HMCES-DPC from WT- or E127Q-HMCES-expressing cells.
(D) RADAR assay of HMCES-DPC levels from WT- or E127Q-HMCES-expressing cells. Mean ± SEM, n = 4.
(E) Quantification of HMCES-DPC levels with time zero immediately after CD437 set at 100%. Mean ± SEM, n = 4, two-way ANOVA.
After 2 h recovery, the E127Q HMCES-DPC level was reduced almost to approximately the same amount as the WT HMCES-DPC, suggesting alternative mechanisms to complete removal in the absence of self-reversal. We tested whether DPC proteolysis was involved. Indeed, TAK243 treatment or SPRTN siRNA increased the levels of the E127Q HMCES-DPC in the first hour after CD437 treatment (Figures S4A and S4B). However, neither completely prevented removal.
Effects of expressing the self-reversal-deficient E127Q HMCES protein
HMCESΔ cells accumulate DSBs.6 Expressing endogenous levels of either the WT or E127Q HMCES protein reduced the DSBs as measured by a neutral comet assay, indicating that the E127Q protein can at least partly protect abasic sites from cleavage (Figure S5A).
DPC accumulation is detrimental for cells. Hence, we analyzed how the expression of the E127Q HMCES protein affects cell fitness. Without the addition of exogenous DNA-damaging agents, cells expressing near-endogenous levels of E127Q HMCES exhibited 30% less growth/viability as measured by an alamarBlue assay than HMCESΔ cells containing an empty vector (EV) or HMCESΔ cells expressing WT HMCES (Figure 5A). We noticed that the growth defect of the E127Q-expressing cells diminished over time as they were maintained in culture, suggesting an adaptation mechanism.
Figure 5. HMCES self-reversal is important for cell fitness and responses to DNA damage.
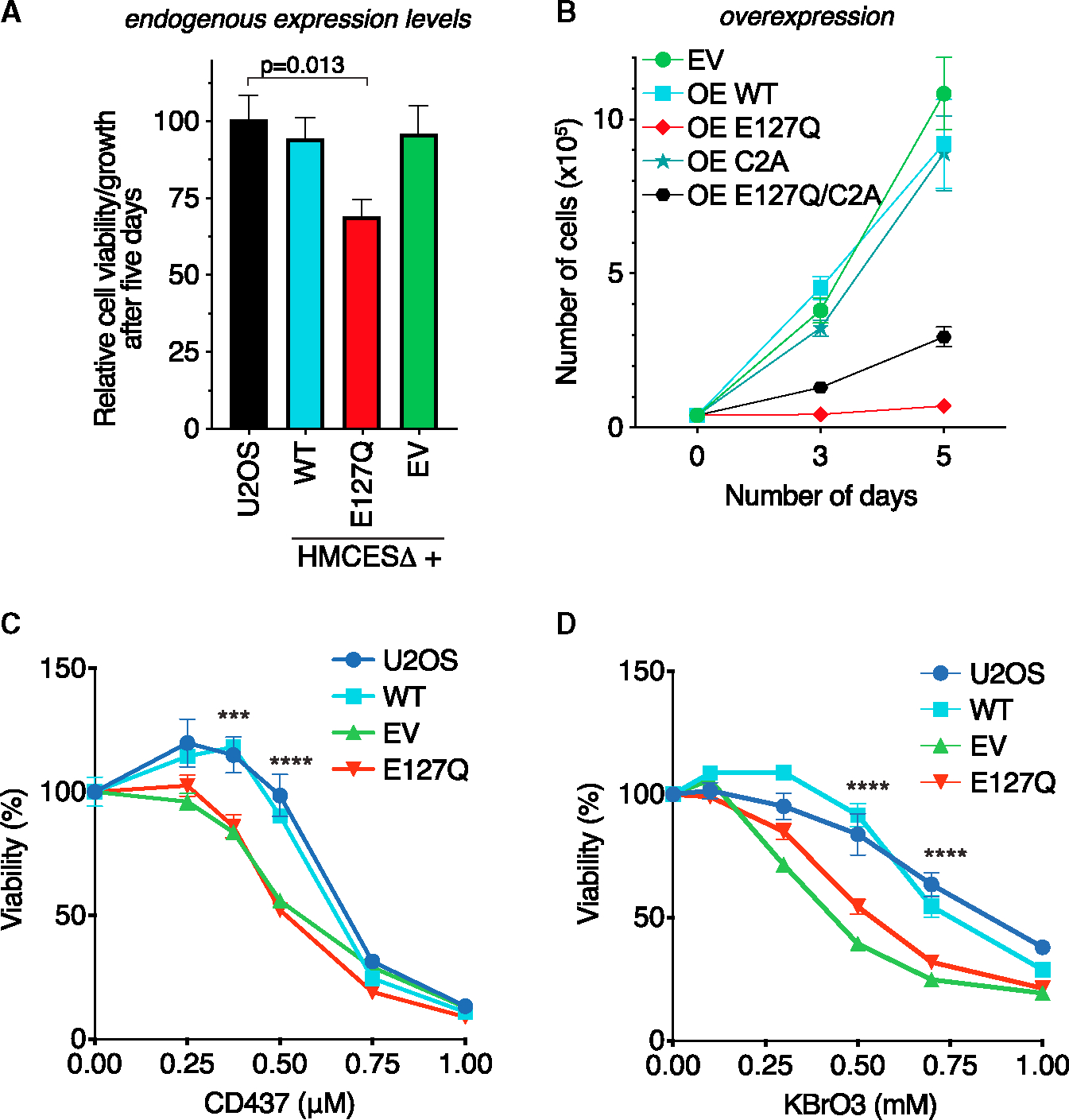
(A) U2OS cells and HMCESΔ cells expressing an EV or near-endogenous levels of WT or E127Q HMCES were plated at equal cell numbers. Cell proliferation/viability was measured using alamarBlue 5 days later. Mean ± SEM, n = 6, one-way ANOVA with Dunnett post-test.
(B) HMCESΔ cells were infected with retroviruses to overexpress the indicated HMCES WT and mutant proteins. Cells were selected for 3 days with puromycin and then plated at equal cell numbers. Viable cells were counted using trypan blue staining at each time point. Mean ± SEM, n = 3.
(C) Percentage of the viability of the indicated cells treated with CD437 measured using alamarBlue 4 days after a 24 h exposure to drug. Two-way ANOVA, n = 3. WT vs. E127Q, ***p < 0.0004, ****p < 0.0001.
(D) Percentage of viability of the indicated cells treated with KBrO3 as measured using alamarBlue 3 days after a 48 h exposure to drug. Two-way ANOVA, n = 3. WT vs. E127Q, ****p < 0.0001.
We next infected HMCESΔ cells with retrovirus containing HMCES WT and mutant cDNAs expressed from a strong promoter to cause overexpression. Overexpressing E127Q HMCES from a strong promoter (OE E127Q) had a more deleterious effect leading to substantially slower cell growth (Figures 5B and S5B). In contrast, cells overexpressing WT HMCES (OE WT) or HMCESΔ cells infected with an EV proliferated normally (Figure 5B). Combining a C2A mutation with the E127Q mutation partially mitigated the growth defect of the E127Q overexpression, consistent with the idea that persistent thiazolidine linkages in the E127Q-HMCES-overexpressing cells contribute to the reduced cell fitness (Figure 5B).
HMCESΔ cells and cells that express crosslink-deficient HMCES mutants are hypersensitive to DNA-damaging agents that generate AP sites.6,11,12 To test whether inactivating self-reversal also causes hypersensitivity, we exposed cells expressing near-endogenous levels of WT HMCES or the E127Q mutant to increasing doses of CD437 and let them recover in normal growth media before measuring viability. As expected, HMCESΔ cells transduced with an EV had reduced viability in response to CD437 treatment. E127Q cells also showed the same detrimental effect; meanwhile, WT HMCES recapitulated the viability of control U2OS cells (Figure 5C). Similarly, E127Q cells were hypersensitive to KBrO3 (Figure 5D). These results further support the idea that HMCES self-reversal is an important mechanism for resolving the HMCES-DPC.
DISCUSSION
Abasic sites are frequent DNA lesions that threaten genome stability, especially when they are present in ssDNA, where BER cannot be used for repair. HMCES provides an evolutionarily conserved mechanism to recognize and shield these ssDNA lesions from inappropriate processing that can generate DSBs. However, repair requires the removal of the HMCES-DPC. Our results demonstrate that there is a self-reversal mechanism in human cells. The reversibility of the crosslink depends on E127, which is positioned adjacent to the thiazolidine linkage created by the N-terminal cysteine residue.7,9,10 Mutation of E127 largely prevents the reversal reaction without preventing crosslink formation in biochemical assays and delays the resolution of the HMCES-DPC in cells. Furthermore, cells expressing only the E127Q HMCES protein proliferate slowly, accumulate HMCES-DPCs even in the absence of added genotoxic stress, and are hypersensitive to agents that increase AP site formation.
Some of the effects of E127Q HMCES in cells could be because of a reduction in the rate of crosslink formation or other effects of the mutation other than lack of self-reversal. Although not essential to form the DPC, the E127 residue promotes ring opening of the AP site deoxyribose from the furan to aldehyde form to facilitate crosslink formation.21 The E127Q HMCES-DPC levels do not increase as much as the WT HMCES-DPC in response to CD437. This difference could be due to the increase in the steady-state level of the E127Q HMCES-DPC compared to WT in the untreated cells making the fold change look smaller, but it also likely reflects a reduced amount of crosslink formation. Nonetheless, the E127Q HMCES protein can at least partly protect AP sites from forming DSBs, and E127A HMCES was also previously shown to protect AP sites during SHM.13
Endogenous levels of E127Q expression modestly reduce cell growth, but this effect disappears over increasing passages, suggesting the cells adapt. Overexpression of E127Q from a strong promoter causes severe toxicity. The overexpression toxicity is substantially, but not completely, mitigated by combining E127Q with a C2A mutation, which blocks thiazolidine DPC formation. The lack of complete rescue suggests the E127Q mutation also has another effect other than preventing reversal of the thiazolidine DPC linkage. Mutation of E127 or the equivalent site in YedK is reported to increase its ssDNA binding affinity compared to WT.9,22 In addition, the C2A HMCES protein can form an unstable Schiff base with the AP site and promote DNA strand cleavage via a β-elimination reaction.7,21 These other effects of these mutations (or a combination of them in the double mutant) may contribute to the cellular phenotypes generated by expressing these proteins.
Inhibiting HMCES-DPC self-reversal delayed, but did not prevent, DPC removal. Previous studies on DPCs in Xenopus egg extracts found two major repair mechanisms involving the SPRTN protease and the proteasome.33 Inhibiting the ubiquitin-dependent or SPRTN-dependent pathways yielded a modestly higher level of the E127Q HMCES-DPC after CD437 treatment, suggesting they may act in in parallel or as backup pathways to the self-resolution mechanism. The increase in the amount of the HMCES-DPC at early time points in these circumstances could also be due to unknown indirect effects on DPC formation. SPRTN removes DPCs at replication forks,34,35 showing full activity when these bulky lesions are located at ssDNA/dsDNA junctions.36,37 Furthermore, SPRTN can remove HMCES-DPCs formed as an intermediate in ICL repair in Xenopus egg extracts.16 The lack of a strong requirement for SPRTN in HMCES-DPC removal in the CD437-treated human cells could be because we did not achieve sufficient SPRTN inactivation with siRNA. It also could be because an appropriate substrate is not generated in the CD437 system. Since many forks do not restart, these CD437-generated ssDNA AP sites may not be present at ssDNA/dsDNA junctions. In addition to SPRTN and the proteasome, proteases including GCNA,18 FAM111A,19 and DDI120 can participate in DPC repair. Further studies will be needed to understand which, if any, of these mechanisms contribute to the resolution of the HMCES-DPC in cells.
The DNA-binding cleft in HMCES and YedK only accommodates ssDNA on one side of the AP site.7,9,10,38 This creates specificity for either AP sites in ssDNA or the ones that exist at a dsDNA/ssDNA junction such as what would form if a POL stalls at the lesion. The apparent rate of self-reversal is greatly increased when a complementary oligonucleotide is added to the HMCES-DPC to generate duplex DNA. This increased reversal rate is likely due to the inability of HMCES to rebind the duplex DNA that forms as HMCES releases from the ssDNA. Coupling DPC resolution to the generation of duplex DNA in cells would allow HMCES to shield the AP site until it can be properly repaired by BER (Figure 6). Duplex DNA formation in cells could be generated by TLS synthesis across from the HMCES-DPC. Recent studies showed that TLS across from the DPC could be facilitated either by HMCES proteolysis14,16 or by the action of FANCJ on the intact DPC.39 In either case, the outcome of TLS would include mutations. However, HMCES-deficient cells have increased mutation rates and increased recruitment of TLS POLs to replication forks and exhibit synthetic lethality with TLS POL inactivation, suggesting that another mechanism might operate normally.6,8,11 Alternatively, template switching could be utilized to generate the duplex DNA, providing an error-free repair mechanism (Figure 6). Further studies will be needed to determine if duplex DNA formation in cells is important and which of these mechanisms is preferred.
Figure 6. Model of CD437-induced HMCES-DPC reversal and repair.
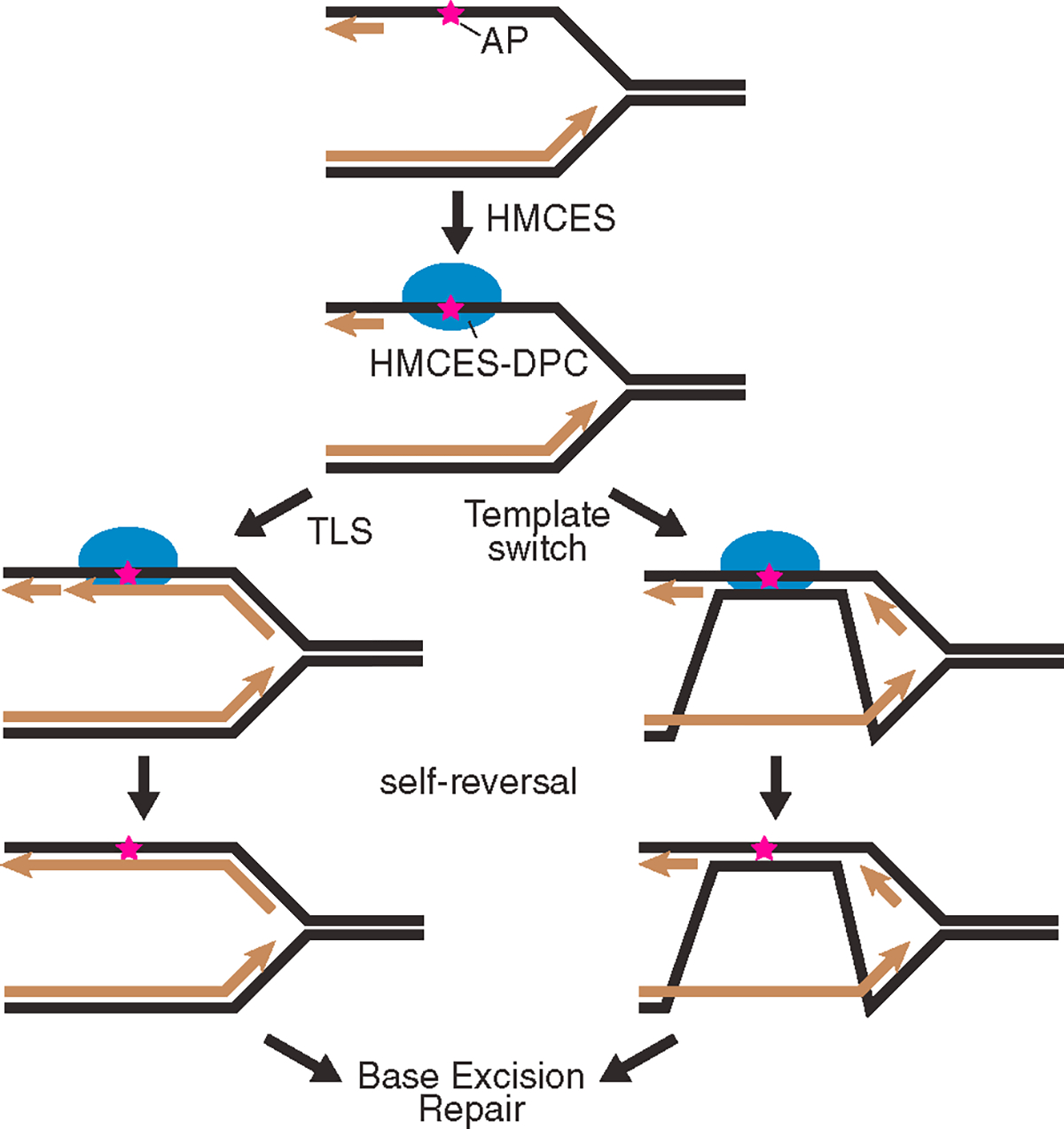
Our data are consistent with recent biochemical studies showing that the thiazolidine linkage is reversible.14,21,22 This linkage was initially thought to be highly stable because it appeared unchanged in biochemical reactions even days after formation, could be observed by crystallography, and blocked the action of AP endonucleases.7 However, the addition of a second ssDNA-AP oligonucleotide trap revealed that HMCES could move to another substrate,21 indicating the DPC formation is reversible and regenerates an intact HMCES protein.
Finally, to analyze HMCES-DPC resolution kinetics in cells, we utilized POLα inhibition by CD437. Inhibiting POLα rapidly generates large amounts of ssDNA and HMCES-DPC formation. These results suggest that ssDNA formation in other contexts could generate a need for HMCES. Although abasic sites form much more rapidly in ssDNA than in dsDNA, further studies are needed to understand why CD437 is such an efficient generator of glycosylase-dependent HMCES-DPCs. In addition, the mechanisms that operate to remove the HMCES-DPC may differ depending on the context in which they are formed. Nonetheless, the synchrony in the formation and resolution of the HMCES-DPC after CD437 treatment provided a useful system to study how the HMCES-DPC is removed.
Limitations of the study
HMCES-DPC resolution mechanisms may depend on the context or cell type. The E127Q HMCES mutation reduces DPC formation efficiency and may affect DNA binding or other HMCES functions in addition to preventing self-reversal. These changes could contribute to the phenotypes observed in cells expressing this mutant protein. Finally, further studies will be needed to determine if duplex DNA formation in cells is important to the resolution mechanism and how the duplex DNA is generated.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David Cortez (david.cortez@vanderbilt.edu).
Materials availability
Plasmids and cell lines generated in this study are available upon request from the lead contact.
Data and code availability
The published article includes all datasets generated or analyzed during this study.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell lines
HCT116 cells (male) were cultured in 7.5% McCoy’s 5A supplemented with 7.5% FBS. U2OS cells (female) were cultured in DMEM supplemented with 7.5% FBS. Cells were cultured at 37°C and 5% CO2 with humidity.
METHOD DETAILS
Generation of stable cell lines
U2OS HMCESΔ cells were generated previously.6 Stable cell lines expressing WT-HMCES, E127Q, or empty vector (EV) were generated by infection of U2OS HMCESΔ cells with a pLPG backbone lentivirus. Stable cell lines overexpressing WT HMCES or HMCES mutants were generated by infection of U2OS HMCESΔ cells with pLPCX backbone retrovirus.
Cell transfections
siRNA transfections were performed using Lipofectamine RNAimax according to manufacturer’s instructions.
Plasmids
HMCES E127Q plasmid was generated by Gibson assembly of gene block containing the point mutation into the pLPG plasmid with gD promoter. Overexpression plasmids were created by PCR subcloning HMCES WT, E127Q, C2A, or C2A/E127Q cDNA into pLPCX plasmid containing CMV promoter. Plasmids were corroborated by sequencing. pUGI-NLS UDG Inhibitor (UGI) plasmid was purchased from Addgene (Cat#101091).
Analysis of parental ssDNA with native BrdU staining
Cells were plated in a 96-well glass-bottom, poly-L-lysine coated plate and pulsed with 2mM BrdU for 18 h. BrdU was washed off for 2 h before drug treatment. Cells were treated with CD437 (5μM) or HU (0.3mM, 3mM) for 30 min and subsequently fixed with 3% paraformaldehyde, 2% sucrose for 10 min. Cells were permeabilized in PBT (0.5% Triton X-100 in PBS) for 10 min, blocked for 1 h in 10% normal goat serum, and probed with anti-BrdU antibody (AbCam Cat#ab6326), followed by Goat anti-Rat IgG (Thermo Cat# A11007). The plate was imaged and analyzed directly using a Molecular Devices ImageXpress high-content imager.
AP site detection
Genomic DNA was purified as described in the RADAR method, quantified, and diluted to 100 ng/μL in dH20. Abasic sites were labeled by incubation of 2.5 μg DNA with 5 mM biotinylated aldehyde reactive probe (ARP; Dojindo Laboratories, A305) for 1 h at 37°C. The DNA was ethanol precipitated, washed twice with 70% ethanol, resuspended in dH20, and quantified. For the loading control, 50 ng DNA was diluted in 6X SSC, denatured, and dot blotted onto a nylon membrane. The membrane was treated with 1.5 M NaCl, 0.5 N NaOH for 10 min, followed by 1 M NaCl, 0.5M Tris-HCl pH 7.0 for 10 min, and DNA was crosslinked to the membrane using a Stratalinker. The membrane was blocked with 5% milk in TBST immunoblotted for ssDNA (Millipore Sigma, MAB3034). For ARP detection, 500 ng or 250 ng DNA was diluted in 6X SSC, denatured, and applied to a nylon membrane, as above. The membrane was blocked with 5% bovine serum albumin and biotin was detected with Streptavidin-HRP (ThermoFisher).
Immunoblotting
Cell lysates were extracted using Igepal lysis buffer (1% Igepal, 150mM NaCl, 50mM Tris pH 7.4) enriched with protease inhibitor cocktail (Roche). Proteins were analyzed by SDS-PAGE and immunoblotting.
DNA combing
Cells were labeled for 20 min with 20 μM CldU (Sigma, C6891) followed by 40 min with 100 μM IdU (Sigma, l7125) and approximately 300,000 cells were collected by trypsinization and embedded in agarose plugs. DNA combing was performed according to the manufacturer’s instructions (Genomic Vision) with minor modifications using a combing machine. DNA-combed coverslips were baked for 2 h at 65°C and stored at −20°C. The DNA-coated coverslips were denatured with freshly prepared 0.5M NaOH, 1M NaCl solution, washed with PBS, and dehydrated consecutively in 70%, 90%, and 100% ethanol before air drying. Coverslips were blocked with 10% goat serum, 0.1% Triton X-100 in 1X PBS and immunostaining was performed with antibodies that recognize CldU (Abcam, ab6326) and IdU (BD Biosciences, 347580) for 1 h at room temperature. Coverslips were then washed in PBS, probed with secondary antibodies for 30 min at room temperature, washed with PBS and mounted using ProLong Gold (ThermoFisher). Images were captured using a 40X oil objective (Nikon Eclipse Ti) and fiber length analysis was performed using Nikon Elements software.
RADAR assay
DPC levels were measured using the RADAR assay,27 and all resolution experiments were performed using the same conditions. Briefly, cells were synchronized with 2mM thymidine overnight. Then, thymidine was removed, and cells recovered in normal growth medium for 2h prior to treatment with 5μM CD437 for 30 min. Cells were washed twice with PBS and recovered in normal growth medium. Cells were lysed in RADAR buffer (RLT plus buffer supplemented with 1% Sarkosyl). Genomic DNA was ethanol precipitated by the addition of ½ volume 100% ethanol and incubation at −20°C for 5 min. After full-speed centrifugation, the DNA pellet was washed twice with 70% ethanol and resuspended in 8mM NaOH at 65°C at 800rpm for 3 h. DNA concentration was determined by spectrophotometry. DNA sample (20μg) was digested with Pierce universal nuclease in 1X TBS with 2mM MgCl2 at 37°C at 300rpm for 1 h. Samples were boiled for 5 min and applied to a nitrocellulose membrane with a slot blot apparatus. The membrane was blocked for 1 h with 5% non-fat dry milk in TBST and immunoblotted for HMCES. For the DNA blot, DNA sample (1ug) was added in 1mL of 6X SSC buffer. The sample was boiled and then on ice for 10 min each and added to a nylon membrane with a slot apparatus. The membrane was placed face up on Whatman paper soaked with solution A (1.5M NaCl, 0.5M NaOH) for 10 min and in solution B (1.5M NaCl, 0.5M Tris pH7.5) for 5 min. After air drying, the membrane is crosslinked with UV 1200J/m2 and blocked for 1 h with 5% non-fat dry milk in TBST and immunoblotted for ssDNA.
Preparation of AP-DNA
Sequences of oligonucleotides used in the biochemical assays are listed in key resources table. AP-DNA was prepared by incubation of 200μM uracil-containing oligonucleotide (70μM of the trap oligo) and 8 U UDG in UDG Buffer (supplemented with 1mM DTT) at 37°C for 20 min. AP-DNA was prepared fresh for each reaction.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit polyclonal anti HMCES | Sigma | Cat#HPA044968; RRID:AB_2679160 |
| Mouse anti-DVC1 (SPRTN) | John Rouse lab | N/A |
| Mouse anti ssDNA | DSHB | AB_10805144 |
| Goat Anti-Rabbit IgG (H + L) | Fisher Scientific | Cat#111-035-144; RRID:AB_2307391 |
| StarBright Blue 700 Goat Anti-Mouse IgG | Bio-Rad | Cat#12004158; RRID:AB_2884948 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| RLT plus buffer | Qiagen | Cat#1053393 |
| IdU | Sigma Aldrich | Cat#I7125 |
| KBrO3 | Acros | Cat#446941000 |
| MG132 | Sigma Aldrich | Cat#M7449 |
| TAK243 | Chemietek | Cat#AOB 87172 |
| CD437 | Tocris bioscience | Cat#1549 |
| Uracil-DNA Glycosylase (UDG) | NEB | Cat#M0280S |
| alamarBlue | ThermoFisher | Cat#DAL1100 |
| Aldehyde reactive probe | Dojindo | Cat#A305 |
| Wild-type HMCES SRAP protein | This study | N/A |
| E127Q HMCES SRAP protein | This study | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| Comet assay kit | Tevigen | Cat#4250-050-K |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| U2OS | ATCC | Cat#HTB-96; RRID:CVCL_0042 |
| HCT-116 | ATCC | Cat#CRL-11268; RRID:CVCL_1926 |
|
| ||
| Oligonucleotides | ||
|
| ||
| SPRTN siRNA | Dharmacon | J-015442-19-0002 |
| SPRTN siRNA | Dharmacon | J-015442-20-0002 |
| SPRTN siRNA | Dharmacon | J-015442-21-0002 |
| SPRTN siRNA | Dharmacon | J-015442-22-0002 |
| 20mer = d(TCTTCTGGTCUGGATGGTAGT) | IDT | N/A |
| 40mer = d(GGAATCTGACTCTTCTGGTCUGGATGGTAGTTAAGTCTTGT) | IDT | N/A |
| C40mer = d(ACAAGACTTAACTACCATCCAGACCAGAAGAGTCAGATTCC) | IDT | N/A |
| T40mer control = d(GGAATCTGACTCTTCTGGTCTGGATGGTAGTTAAGTCTTGT) | IDT | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pLPG-HMCES (pKM354) | This manuscript | N/A |
| pLPG-E127Q HMCES (pJRF27) | This manuscript | N/A |
| pLPCX-EV (pJRF32) | This manuscript | N/A |
| pLPCX-OE WT HMCES (pJRF01) | This manuscript | N/A |
| pLPCX-OE E127Q HMCES (pJRF33) | This manuscript | N/A |
| pLPCX-OE C2A HMCES (pDC1351) | This manuscript | N/A |
| pLPCX-OE C2A/E127Q HMCES (pDC1352) | This manuscript | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Graphpad Prism | Graphpad Software | https://www.graphpad.com/scientific-software/prism/; RRID: SCR_000306 |
| Image Lab | Bio-Rad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
Biochemical crosslink reversal assay
WT and E127Q HMCES were purified as previously described.21 HMCES-DPC was formed by incubation of 10μM 20mer AP-DNA with 2μM HMCES (WT or E127Q) in DPC buffer (20μM HEPES, 10mM NaCl, 1mM EDT, pH 8) overnight at 37°C. DPC-20 was incubated with a 50-fold excess of 40mer AP-DNA to trap any reversed HMCES. Reactions were stopped by adding an equal volume of 2X SDS buffer. Each time point was initiated in reverse so that all reactions were quenched for the same length of time. Reaction products were resolved on 4 to 12% Bis-Tris gels and Coomassie stained for detection.
Reversal trapping in duplex DNA required incubation of 10μM 40mer AP-DNA with 2μM WT HMCES in DPC buffer overnight at 37°C. DPC-40 was incubated with a 1.4-fold excess of non-complementary oligo (40mer control) or the complementary oligo (c40mer). Simultaneously, a 50-fold excess of 20mer AP-DNA was added to trap any reversed HMCES. Each time point was initiated in reverse, so all reactions were quenched for the same length of time and by adding an equal volume of 2X SDS buffer. Reactions products were resolved on 4 to 12% Bis-tris gels, 1X MES running buffer, and Coomassie stained for protein. The reversal percentage was calculated as the percentage of DPC-20 compared to total at each time point.
Viability assays
For the endogenous level expression analysis, 5×103 cells/well were plated in a 96-well dish. Cells were incubated in a DMEM growth medium for 5 days prior to performing an alamarBlue assay according to the manufacturer’s instructions. Analysis of cell growth after HMCES overexpression was done infecting cells with pLPCX retroviruses, selection with puromycin for three days, then seeding in a 6 well dish at equal cell number. Viable cells were then counted by trypan blue staining 3 and 5 days after plating. For drug hypersensitivity assays, cells were treated with CD437 for 24 h, or KBrO3 for 48hoursand then recovered in normal growth medium for 4 and 3 days, respectively. The alamarBlue assay was performed using a BioTek multimode reader. These viability measurements are presented as a percentage of the untreated control for each cell line.
Neutral comet assay
The neutral comet assay was performed according to the manufacturer’s instructions.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were completed with PRISM v.9. Descriptions of statistical tests can be found in the figure legends.
Supplementary Material
Highlights.
ssDNA generation by Polα inhibition generates abasic sites protected by an HMCES-DPC
HMCES-DPC self-reversal is favored by duplex DNA formation and requires the residue E127
Mutation of E127 delays HMCES-DPC resolution in cells and reduces cell proliferation
ACKNOWLEDGMENTS
We thank Dr. John Rouse for providing the SPRTN antibody. We thank Vanderbilt Antibody and Protein Resource for purification of the ssDNA antibody. The Vanderbilt Antibody and Protein Resource is supported by the Vanderbilt Institute of Chemical Biology and the Vanderbilt-Ingram Cancer Center (P30CA68485). This work was supported by NIH grants R01ES030575 to D.C., R35GM136401 to B.F.E., K99ES034058 to K.P.M.M., and F31ES032334 to K.A.P.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as living with a disability. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location.
Footnotes
DECLARATION OF INTERESTS
D.C. is a member of the Cell Reports advisory board.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113427.
REFERENCES
- 1.Nakamura J, and Nakamura M (2020). DNA-protein crosslink formation by endogenous aldehydes and AP sites. DNA Repair 88, 102806. 10.1016/j.dnarep.2020.102806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thompson PS, and Cortez D (2020). New insights into abasic site repair and tolerance. DNA Repair 90, 102866. 10.1016/j.dnarep.2020.102866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, and Ellenberger T (2006). DNA Repair and Mutagenesis, Second Edition (American Society for Microbiology; ). [Google Scholar]
- 4.Krokan HE, and Bjørås M (2013). Base excision repair. Cold Spring Harb. Perspect. Biol. 5, a012583. 10.1101/cshperspect.a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi JY, Lim S, Kim EJ, Jo A, and Guengerich FP (2010). Translesion synthesis across abasic lesions by human B-family and Y-family DNA polymerases alpha, delta, eta, iota, kappa, and REV1. J. Mol. Biol. 404, 34–44. 10.1016/j.jmb.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mohni KN, Wessel SR, Zhao R, Wojciechowski AC, Luzwick JW, Layden H, Eichman BF, Thompson PS, Mehta KPM, and Cortez D (2019). HMCES Maintains Genome Integrity by Shielding Abasic Sites in Single-Strand DNA. Cell 176, 144–153.e13. 10.1016/j.cell.2018.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thompson PS, Amidon KM, Mohni KN, Cortez D, and Eichman BF (2019). Protection of abasic sites during DNA replication by a stable thiazolidine protein-DNA cross-link. Nat. Struct. Mol. Biol. 26, 613–618. 10.1038/s41594-019-0255-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Srivastava M, Su D, Zhang H, Chen Z, Tang M, Nie L, and Chen J (2020). HMCES safeguards replication from oxidative stress and ensures error-free repair. EMBO Rep. 21, e49123. 10.15252/embr.201949123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang N, Bao H, Chen L, Liu Y, Li Y, Wu B, and Huang H (2019). Molecular basis of abasic site sensing in single-stranded DNA by the SRAP domain of E. coli yedK. Nucleic Acids Res. 47, 10388–10399. 10.1093/nar/gkz744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halabelian L, Ravichandran M, Li Y, Zeng H, Rao A, Aravind L, and Arrowsmith CH (2019). Structural basis of HMCES interactions with abasic DNA and multivalent substrate recognition. Nat. Struct. Mol. Biol. 26, 607–612. 10.1038/s41594-019-0246-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mehta KPM, Lovejoy CA, Zhao R, Heintzman DR, and Cortez D (2020). HMCES Maintains Replication Fork Progression and Prevents Double-Strand Breaks in Response to APOBEC Deamination and Abasic Site Formation. Cell Rep. 31, 107705. 10.1016/j.celrep.2020.107705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biayna J, Garcia-Cao I, Álvarez MM, Salvadores M, Espinosa-Carrasco J, McCullough M, Supek F, and Stracker TH (2021). Loss of the abasic site sensor HMCES is synthetic lethal with the activity of the APOBEC3A cytosine deaminase in cancer cells. PLoS Biol. 19, e3001176. 10.1371/journal.pbio.3001176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu L, Shukla V, Yadavalli AD, Dinesh RK, Xu D, Rao A, and Schatz DG (2022). HMCES protects immunoglobulin genes specifically from deletions during somatic hypermutation. Genes Dev. 36, 433–450. 10.1101/gad.349438.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sugimoto Y, Masuda Y, Iwai S, Miyake Y, Kanao R, and Masutani C (2023). Novel mechanisms for the removal of strong replication-blocking HMCES- and thiazolidine-DNA adducts in humans. Nucleic Acids Res. 51, 4959–4981. 10.1093/nar/gkad246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruggiano A, Vaz B, Kilgas S, Popović M, Rodriguez-Berriguete G, Singh AN, Higgins GS, Kiltie AE, and Ramadan K (2021). The protease SPRTN and SUMOylation coordinate DNA-protein crosslink repair to prevent genome instability. Cell Rep. 37, 110080. 10.1016/j.celrep.2021.110080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Semlow DR, MacKrell VA, and Walter JC (2022). The HMCES DNA-protein cross-link functions as an intermediate in DNA interstrand cross-link repair. Nat. Struct. Mol. Biol. 29, 451–462. 10.1038/s41594-022-00764-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gallina I, Hendriks IA, Hoffmann S, Larsen NB, Johansen J, Colding-Christensen CS, Schubert L, Sellés-Baiget S, Fábián Z, Kühbacher U, et al. (2021). The ubiquitin ligase RFWD3 is required for translesion DNA synthesis. Mol. Cell 81, 442–458.e9. 10.1016/j.molcel.2020.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borgermann N, Ackermann L, Schwertman P, Hendriks IA, Thijssen K, Liu JC, Lans H, Nielsen ML, and Mailand N (2019). SUMOylation promotes protective responses to DNA-protein crosslinks. The EMBO journal 38, e101496. 10.15252/embj.2019101496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kojima Y, Machida Y, Palani S, Caulfield TR, Radisky ES, Kaufmann SH, and Machida YJ (2020). FAM111A protects replication forks from protein obstacles via its trypsin-like domain. Nat. Commun. 11, 1318. 10.1038/s41467-020-15170-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yip MCJ, Bodnar NO, and Rapoport TA (2020). Ddi1 is a ubiquitin-dependent protease. Proc. Natl. Acad. Sci. USA 117, 7776–7781. 10.1073/pnas.1902298117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paulin KA, Cortez D, and Eichman BF (2022). The SOS response-associated peptidase (SRAP) domain of YedK catalyzes ring opening of abasic sites and reversal of its DNA-protein cross-link. J. Biol. Chem. 298, 102307. 10.1016/j.jbc.2022.102307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donsbach M, Dürauer S, Grünert F, Nguyen KT, Nigam R, Yaneva D, Weickert P, Bezalel-Buch R, Semlow DR, and Stingele J (2023). A non-proteolytic release mechanism for HMCES-DNA-protein crosslinks. EMBO J. 42, e113360. 10.15252/embj.2022113360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han T, Goralski M, Capota E, Padrick SB, Kim J, Xie Y, and Nijhawan D (2016). The antitumor toxin CD437 is a direct inhibitor of DNA polymerase alpha. Nat. Chem. Biol. 12, 511–515. 10.1038/nchembio.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ercilla A, Benada J, Amitash S, Zonderland G, Baldi G, Somyajit K, Ochs F, Costanzo V, Lukas J, and Toledo L (2020). Physiological Tolerance to ssDNA Enables Strand Uncoupling during DNA Replication. Cell Rep. 30, 2416–2429.e7. 10.1016/j.celrep.2020.01.067. [DOI] [PubMed] [Google Scholar]
- 25.Mehta KPM, Thada V, Zhao R, Krishnamoorthy A, Leser M, Lindsey Rose K, and Cortez D (2022). CHK1 phosphorylates PRIMPOL to promote replication stress tolerance. Sci. Adv. 8, eabm0314. 10.1126/sciadv.abm0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan K, Sterling JF, Roberts SA, Bhagwat AS, Resnick MA, and Gordenin DA (2012). Base damage within single-strand DNA underlies in vivo hypermutability induced by a ubiquitous environmental agent. PLoS Genet. 8, e1003149. 10.1371/journal.pgen.1003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kiianitsa K, and Maizels N (2013). A rapid and sensitive assay for DNA-protein covalent complexes in living cells. Nucleic Acids Res. 41, e104. 10.1093/nar/gkt171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cone R, Bonura T, and Friedberg EC (1980). Inhibitor of uracil-DNA glycosylase induced by bacteriophage PBS2. Purification and preliminary characterization. J. Biol. Chem. 255, 10354–10358. [PubMed] [Google Scholar]
- 29.Kühbacher U, and Duxin JP (2020). How to fix DNA-protein crosslinks. DNA Repair 94, 102924. 10.1016/j.dnarep.2020.102924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barghout SH, Patel PS, Wang X, Xu GW, Kavanagh S, Halgas O, Zarabi SF, Gronda M, Hurren R, Jeyaraju DV, et al. (2019). Preclinical evaluation of the selective small-molecule UBA1 inhibitor, TAK-243, in acute myeloid leukemia. Leukemia 33, 37–51. 10.1038/s41375-018-0167-0. [DOI] [PubMed] [Google Scholar]
- 31.Maskey RS, Kim MS, Baker DJ, Childs B, Malureanu LA, Jeganathan KB, Machida Y, van Deursen JM, and Machida YJ (2014). Spartan deficiency causes genomic instability and progeroid phenotypes. Nat. Commun. 5, 5744. 10.1038/ncomms6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halder S, Torrecilla I, Burkhalter MD, Popović M, Fielden J, Vaz B, Oehler J, Pilger D, Lessel D, Wiseman K, et al. (2019). SPRTN protease and checkpoint kinase 1 cross-activation loop safeguards DNA replication. Nat. Commun. 10, 3142. 10.1038/s41467-019-11095-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larsen NB, Gao AO, Sparks JL, Gallina I, Wu RA, Mann M, Räschle M, Walter JC, and Duxin JP (2019). Replication-Coupled DNA-Protein Crosslink Repair by SPRTN and the Proteasome in Xenopus Egg Extracts. Mol. Cell 73, 574–588.e7. 10.1016/j.molcel.2018.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vaz B, Popovic M, Newman JA, Fielden J, Aitkenhead H, Halder S, Singh AN, Vendrell I, Fischer R, Torrecilla I, et al. (2016). Metalloprotease SPRTN/DVC1 Orchestrates Replication-Coupled DNA-Protein Crosslink Repair. Mol. Cell 64, 704–719. 10.1016/j.molcel.2016.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stingele J, Bellelli R, Alte F, Hewitt G, Sarek G, Maslen SL, Tsutakawa SE, Borg A, Kjær S, Tainer JA, et al. (2016). Mechanism and Regulation of DNA-Protein Crosslink Repair by the DNA-Dependent Metalloprotease SPRTN. Mol. Cell 64, 688–703. 10.1016/j.molcel.2016.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reinking HK, Kang HS, Götz MJ, Li HY, Kieser A, Zhao S, Acampora AC, Weickert P, Fessler E, Jae LT, et al. (2020). DNA Structure-Specific Cleavage of DNA-Protein Crosslinks by the SPRTN Protease. Mol. Cell 80, 102–113.e6. 10.1016/j.molcel.2020.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li F, Raczynska JE, Chen Z, and Yu H (2019). Structural Insight into DNA-Dependent Activation of Human Metalloprotease Spartan. Cell Rep. 26, 3336–3346.e4. 10.1016/j.celrep.2019.02.082. [DOI] [PubMed] [Google Scholar]
- 38.Amidon KM, and Eichman BF (2020). Structural biology of DNA abasic site protection by SRAP proteins. DNA Repair 94, 102903. 10.1016/j.dnarep.2020.102903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yaneva D, Sparks JL, Donsbach M, Zhao S, Weickert P, Bezalel-Buch R, Stingele J, and Walter JC (2023). The FANCJ helicase unfolds DNA-protein crosslinks to promote their repair. Mol. Cell 83, 43–56.e10. 10.1016/j.molcel.2022.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.