

Abstract
Approximately one-third of endocrine-treated women with estrogen receptor-alpha positive (ER+) breast cancers (BC) are at risk of recurrence due to intrinsic or acquired resistance. Thus, it is vital to understand the mechanisms underlying endocrine therapy resistance in ER+ BC to improve patient treatment. Mitochondrial fatty acid β-oxidation (FAO) has been shown to be a major metabolic pathway in triple-negative BC (TNBC) that can activate Src signaling. Here, we found metabolic reprogramming that increases FAO in ER+ BC as a mechanism of resistance to endocrine therapy. A metabolically relevant, integrated gene signature was derived from transcriptomic, metabolomic, and lipidomic analyses in TNBC cells following inhibition of the FAO rate-limiting enzyme carnitine palmitoyl transferase 1 (CPT1), and this TNBC-derived signature was significantly associated with endocrine resistance in ER+ BC patients. Molecular, genetic, and metabolomic experiments identified activation of AMPK-FAO-oxidative phosphorylation (OXPHOS) signaling in endocrine-resistant ER+ BC. CPT1 knockdown or treatment with FAO inhibitors in vitro and in vivo significantly enhanced the response of ER+ BC cells to endocrine therapy. Consistent with the previous findings in TNBC, endocrine therapy-induced FAO activated the Src pathway in ER+ BC. Src inhibitors suppressed the growth of endocrine-resistant tumors, and the efficacy could be further enhanced by metabolic priming with CPT1 inhibition. Collectively, this study developed and applied a TNBC-derived signature to reveal that metabolic reprogramming to FAO activates the Src pathway to drive endocrine resistance in ER+ BC.
Introduction
Aromatase inhibitors, tamoxifen, and fulvestrant are the most common therapies for estrogen receptor-positive (ER+) breast cancer (BC). Endocrine-targeting drugs, such as tamoxifen, have been used clinically for several decades. However, almost half of patients with BC develop resistance to endocrine therapy, leading to tumor recurrence (1). Multiple mechanisms have been proposed for acquiring endocrine therapy resistance in ER+ BC, including ER alpha mutation/amplification (2,3), mTOR/AKT pathway activation (4,5), and cyclin D1 activation (5,6). Metabolic alterations have been proposed as an additional mechanism for endocrine resistance, including changes in amino acid or glucose metabolism and elevated lipid levels in endocrine-resistant ER+ BC cells (7,8). However, the mechanistic crosstalk between these metabolic alterations and endocrine pathways and their subsequent translational potential is not well understood.
Triple-negative BC (TNBC) is one of the most aggressive subtypes of BC. Using a trans-mitochondrial cybrid (cybrid) model, we previously discovered that cybrids with mitochondria from metastatic TNBC maintain high levels of mitochondrial fatty acid β-oxidation (FAO). We validated our finding of FAO as a major metabolic pathway in TNBC using parental BC cell lines, patient-derived xenograft (PDX) models, and clinical data (9). Our findings have been independently validated in a parallel study by Dr. Goga’s group (10).
We have also reported that FAO is critical for activating the Src pathway, one of the most frequently upregulated oncopathways in TNBC. Specifically, FAO induces autophosphorylation of c-Src at Y419 (pSrc Y419) (9). Interestingly, some studies, including our own study (11), reported that the Src inhibitor dasatinib is beneficial in ER+ BC when combined with endocrine therapy. However, the mechanisms underlying elevated phospho-Src levels in endocrine therapy-resistant BC remain unclear.
Carnitine palmitoyltransferase-1 (CPT1), the rate-limiting enzyme of FAO (12), is important for TNBC progression (9,10). Considering the critical role of FAO in TNBC, we genetically or pharmacologically inhibited CPT1 in three metastatic TNBC cell lines and performed transcriptomic analysis. We then developed a TNBC-derived metabolism-related gene signature by applying a multi-omics integration approach using metabolomic and lipidomic data from a CPT1-inhibited TNBC cell. We analyzed different BC databases to understand the significance of FAO-modulated TNBC-derived signature in other BC subgroups. Our TNBC-derived signature was significantly associated with endocrine therapy-resistant ER+ BC. Mechanistic studies confirmed the metabolic reprogramming of FAO and FAO-induced Src pathway in endocrine therapy-resistant ER+ BC. Our preclinical studies revealed a potential new therapeutic strategy targeting the FAO, oxidative phosphorylation (OXPHOS), or Src pathways to overcome endocrine resistance in ER+ BC patients.
Materials and Methods
Reagents
Etomoxir (ETX), tamoxifen (Tam) citrate (for in vivo), 4-hydroxy Tam (for in vitro), fulvestrant (Ful), dasatinib (Dasa), PP2, atovaquone (Ato), ranolazine (RNL), and 17ß-estradiol were purchased from Sigma-Aldrich. Metformin hydrochloride (Met) was purchased from TCI. ETX was purchased from Chemgood for in vivo studies. Details of the antibodies used are provided in the Supplementary Materials and Methods.
Cell models
TNBC cell lines MDA-MB-231, SUM159, SUM149, and ER+ BC cell lines MCF7 and T47D were maintained in the complete medium [high glucose DMEM supplemented with 5% fetal bovine serum (FBS) and 1% penicillin/streptomycin]. MDA-MB-231, MCF7 and T47D from ATCC were purchased via Molecular and Cellular Biology Tissue Culture Core at Baylor College of Medicine. SUM159 and SUM149 were from Asterand Bioscience kindly provided by Drs. Dave and Debeb respectively. All cells were verified by STR in the Cytogenetics and Cell Authentication Core facility at MD Anderson Cancer Center, Houston, TX. Cells were confirmed mycoplasma-free by using MycoAlert PLUS Mycoplasma detection Kit (Lonza, LT07–710) and/or Venor GeM Mycoplasma Detection Kit (Sigma, MP0025). Mostly, the experiments were performed within 20 passages after thawing the cells. Scrambled shRNA (shScrambled) and CPT1-specific shRNA (shCPT1)-infected TNBC cell lines MDA-MB-231 and SUM159 cells were generated as previously described (9). We have previously published the details of the generation of Tam-resistant (TR) MCF7 TR1 and TR2 cell lines (13). The details of the generation of MCF7 TRsa and T47D Ful-resistant (FulR) cell lines are described in the Supplementary Materials and Methods. Cells were maintained at 37 °C in 5% CO2.
Sulforhodamine B (SRB) assay
Cell proliferation was measured using an SRB-based colorimetric assay as described previously (14,15). Briefly, the cells were fixed with 5% trichloroacetic acid and stained with 1% SRB solution. The dye was dissolved in 10mM Tris buffer and the absorbance (510 nm) was measured using an Infinite M200 plate reader (Tecan).
Animal experiments
In vivo studies were performed following a protocol approved by the Institutional Animal Care and Use Committee of the Baylor College of Medicine (BCM). For animal experiments, 1 × 107 TR cells were injected subcutaneously into the mammary gland of 5–7 week-old SCID/Beige female mice. Water containing 8µg/mL 17β-estradiol (16–18) was provided to the mice from the day of cell line injection until their randomization to begin drug therapy. The mice were randomized into treatment groups of six mice per group. Power analysis was performed; assuming a variance of 33% in the population, with an n=6, we predicted to detect a fold change of 2 with a statistical power of 84%. Mice were treated with different drugs, with the dose and delivery method described in the figure legends. All the groups received low-dose Tam (2 mg/kg in corn oil) via oral gavage until the end of the experiment. Tumor size was measured using calipers, and tumor volume was calculated using the following equation: volume(mm3) = (width(mm))2 × length(mm).
Wound-healing assay
When cells reached confluence in a 96-well plate, a wound was created using the BioTek AutoScratch™ wound-making tool. The cells were then cultured in complete medium for an additional 24–72 hours. The wound distance was imaged at 10X magnification and analyzed using Cytation™ 5 at 0h, 24h, 48h, or 72h.
Targeted metabolomics analysis
shScrambled and shCPT1 MDA-MB-231 cells, as well as MCF7, TR1, and TRsa cells, were used for metabolomics analysis. Mass spectrometry (MS)-based metabolomics of carnitines was performed at the BCM Metabolomics core facility, as described previously (19). Differentially expressed metabolites were determined using a parametric t-test, with significance achieved at an FDR-adjusted p-value <0.25. Hierarchical clustering was performed using the R statistical system.
Unbiased lipidomics analysis
The shScrambled and shCPT1 MDA-MB-231 cells were used for lipidomic analysis at the BCM Metabolomics Core facility. Lipid extraction, MS data acquisition, MS raw data processing, and analysis were performed as previously described (20–22). Differentially expressed lipids were determined using a parametric t-test, with significance achieved at an FDR-adjusted p-value <0.25. Hierarchical clustering was performed using the R statistical system.
Transcriptomics analysis by microarray
Gene expression analysis in shScrambled and shCPT1 MDA-MB-231 TNBC cells was performed by microarray as we previously published (GSE72320) (9). Using this gene expression data, we have previously published that FAO regulate Src oncopathway in metastatic TNBC (9). Differential gene expression was computed using a parametric t-test, with significance achieved for a fold-change exceeding 1.25 × and an FDR-adjusted p-value<0.1. Total RNA from MCF7 parental cells (in charcoal-stripped medium) and TR cells (TR1 and TR2 cells with Tam) were used for microarray gene expression analysis. Affymetrix GeneChhip hybridization and quantification were performed as we previously described (23). The gene expression data was deposited to NCBI Gene Expression Omnibus (GEO) database (GSE241654). Differential gene expression was computed using a parametric t-test, with significance achieved for a fold-change exceeding 2× and an FDR-adjusted p-value<0.05.
RNA-Sequencing data analysis
RNA was extracted from SUM149 cell line treated with control or 100uM ETX for 7 days and SUM159 cell line stably infected with shScrambled or shCPT1. RNA-Sequencing was performed at Novogene Corporation Inc. (Sacramento, CA). Data was trimmed for low quality reads using trim_galore, then mapped to the human genome UCSC hg38 using STAR (24). Gene expression was quantified using featureCounts (25). Differentially expressed genes were determined using the R packages EdgeR (26) and RUVr (27). Significance was achieved for fold changes exceeding 1.25x and FDR<0.05. The RNA-Sequencing data was deposited to NCBI GEO (GSE240393).
Integrated multi-omics analysis
For the differentially expressed metabolites and lipids, we determined the associated genes using the Human Metabolome Database (HMDB) (28) and then overlapped them with the differentially expressed genes identified by microarray and RNA-Seq. Metabolite- and lipid-related genes that overlapped with the microarray differential genes were combined to form the 3-model CPT1 suppression union signature.
Clinical association and survival prognosis using public transcriptomics datasets
We evaluated the association of our integrated signature using several cancer transcriptomics datasets including the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) ER-positive hormone treated dataset (29) and The Cancer Genome Atlas Program (TCGA) ER-positive dataset (30,31). We computed a gene signature activity score for each sample in each dataset as follows: first, all the genes were z-score normalized; next, z-scores of upregulated genes were added, and z-scores of downregulated genes were subtracted, generating a summed z-score for each patient for each signature used. To evaluate clinical association, we used an NCBI GEO dataset GSE74391 from Ful-resistant MCF7 cells. GSE74391 has gene expression data from 10 biological replicates of two Ful-resistant MCF7 cells (ICI182 and ICI164) and 6 replicates of Ful-sensitive MCF7 subline MCF7/S0.5 cells(32). Gene expression data for all 26 samples was used for the analysis. We determined association with Ful resistance (GSE74391) using a two-sided t-test, with significance achieved for FDR-adjusted p-value<0.05. To evaluate signature correlations, we derived a gene signature of Fulresistance in patient tumors by analyzing the NCBI GEO dataset GSE71791, which contains gene expression profiles from 38 ER+ breast carcinomas before and during presurgical treatment with Ful(33). We generated the Ful resistance signature using two-sided Student’s t-test, with significance achieved for FDR-adjusted p-value<0.1 and fold change exceeding 1.25x. We evaluated the correlation between the 3-model CPT1 suppression union gene signature and the Ful/Tam resistance signature in the METABRIC ER+ hormone treated dataset (29) and TCGA ER+ dataset(30,31). We first computed signature activity scores as described above, then evaluated signature correlation, with significance achieved at p<0.05. To assess survival prognosis capability, survival analysis was performed using the R package survival in the METABRIC BC cohort, focusing on ER positive patients receiving hormone therapy, with disease specific survival (DSS) as the outcome variable. We computed gene signature activity scores as described above and then selected the top and bottom 50% of the samples; association with DSS was determined using the log-rank test with significance achieved at p <0.05.
The Kaplan-Meier (KM) Plotter (34) protein dataset was used to generate an overall survival (OS) graph of ACAA2 protein expression in the tumor tissues of ER+ BC patients. KM Plotter microarray datasets were used to analyze the distant metastasis-free survival (DMFS) of ACAA2 and CPT1 mRNA expression in endocrine-treated ER+ BC patients. The median protein or mRNA expression levels were used for separation.
Statistics
Unless specifically mentioned in the figure legend, the statistical significance of different in vitro and in vivo experimental results was calculated by two-tailed Student’s t-test or two-way ANOVA using the GraphPad PRISM 7 software. Error bars represent the standard error of the mean (±SEM).
Additional materials and methods are described in the Supplementary Materials and Methods section.
Data Availability
The R and Python analysis codes used in the manuscript were uploaded to Zenodo repository, at https://doi.org/10.5281/zenodo.8207076 and https://doi.org/10.5281/zenodo.8253346. The RNA-Sequencing files have been deposited to NCBI GEO, accession GSE240393. The microarray data from MCF7 TR cells have been deposited into accession GSE241654. Other gene expression profile data analyzed in this study were obtained from GEO at GSE72320, GSE74391, and GSE71791. TCGA breast cancer data(31) was publicly available via http://firebrowse.org/. For METABRIC breast cancer data(29), the associated genotype and expression data have been deposited at the European Genome-Phenome Archive (http://www.ebi.ac.uk/ega/), which is hosted by the European Bioinformatics Institute, under accession number EGAS00000000083. Kaplan-Meier Plot (34) was generated from https://kmplot.com/. All other raw data generated in this study are available upon request from the corresponding author.
Results
A TNBC-derived gene signature correlated with the outcome of endocrine therapy in ER+ BC.
We developed in vitro TNBC cell models using well known metastatic TNBC cell lines MDA-MB-231, SUM159 and SUM149. These TNBC cell lines have been well studied for their tumor aggressiveness and metastatic character. We inhibited CPT1, the rate-limiting enzyme of FAO (12), in these TNBC cell lines using genetic and pharmacologic approaches. shScrambled or shCPT1 was stably infected in MDA-MB-231 (9) and SUM159 cells (supplementary Fig. S1A). SUM149 cells were treated with a CPT1 inhibitor, ETX, for 7 days. The MDA-MB-231 model was profiled using three omics platforms: microarray transcriptomics, targeted metabolomics, and unbiased lipidomics. SUM159 and SUM149 models were profiled using RNA-Seq analysis. We compared shCPT1 or ETX treatment with shScrambled or control to determine omics features associated with CPT1 suppression. First, we used a microarray transcriptomics dataset previously published by our group, GSE72320 (9). To ensure that the transcriptomic signature was relevant across multiple cell line models, we evaluated the results of CPT1 suppression in RNA-Sequencing data from SUM159 and SUM149 models. In SUM159, CPT1 suppression via shCPT1 led to 2735 increased and 2902 decreased differentially expressed genes (DEGs), whereas in SUM149, CPT1 inhibition via ETX led to 2537 increased and 3141 decreased DEGs (Fig. 1A). We further determined genes altered by CPT1 inhibition in either of the SUM149 or SUM159 models, or in common and in the same direction, leading to a signature of 7575 DEGs (Supplementary Fig. S1B). Overlapping the shCPT1 signature from the MDA-MB-231 microarray with the combined RNA-Sequencing DEGs, we obtained a 3-model CPT1 suppression signature with 547 gene (Fig. 1B). Finally, out of the 547 genes, integration with the metabolomics and lipidomics genes (Fig. 1C) led to a 3-model CPT1 suppression union signature, with 28 increased genes and 16 decreased genes (Fig. 1D, Supplementary Fig. S1C). Thus, we generated a robust transcriptomic signature of CPT1 suppression, and further determined a subset associated with a union of metabolic and lipidomic altered genes.
Figure 1. Multi-omics integration of transcriptomics, metabolomics, and lipidomics revealed a TNBC-derived gene signature associated with resistance to endocrine therapy in ER+ BC.
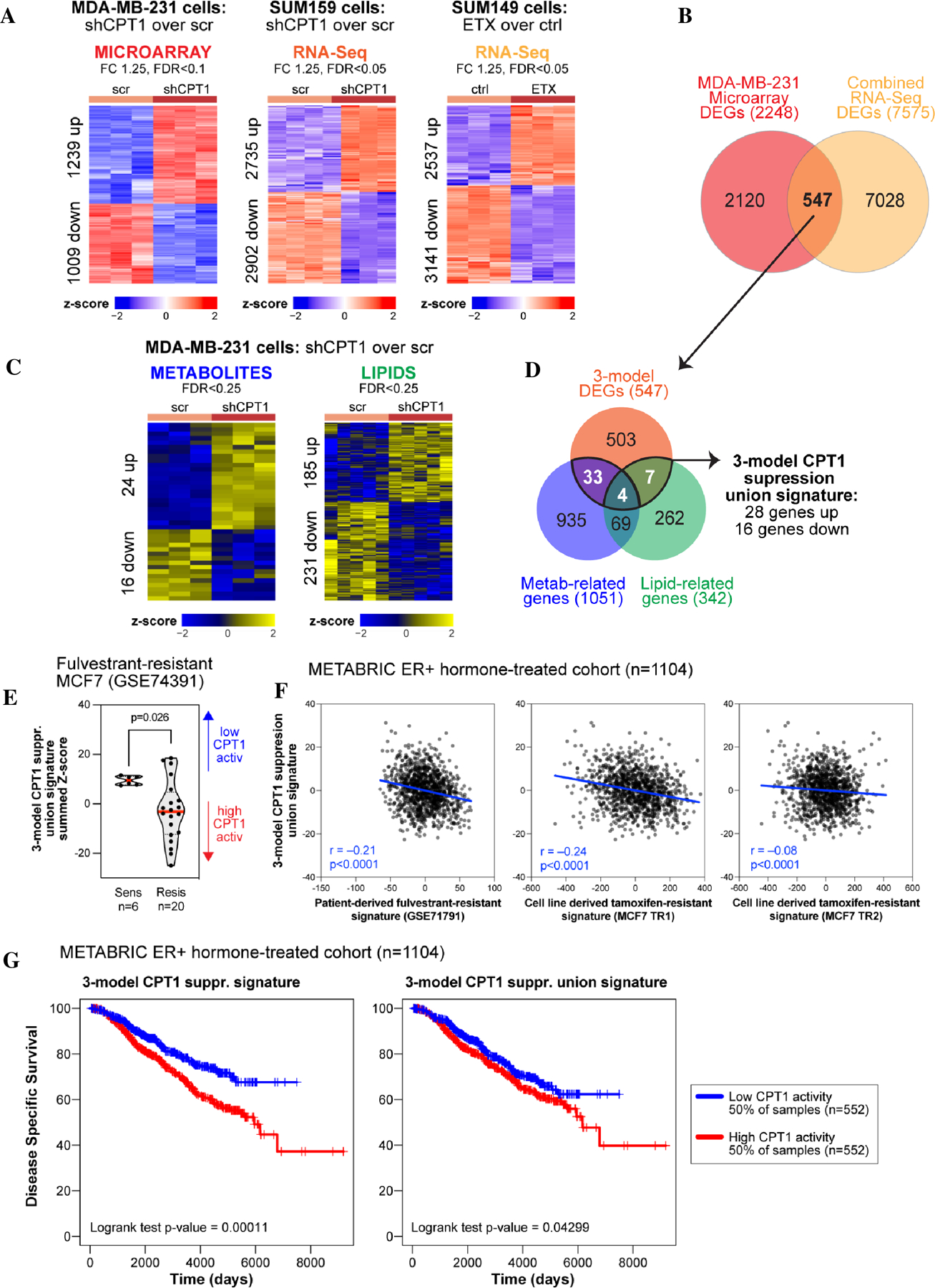
(A) MDA-MB-231 transfected with shScrambled (scr) or shCPT1 were profiled using microarray transcriptomics. Control (ctrl) and ETX treated SUM149 cells and SUM159 cells stably infected with scrambled or shCPT1 were profiled using RNA-Sequencing. Significant differentially expressed features shown as heatmaps. (B) A 3-model CPT1 suppression gene signature was derived, using MDA-MB-231, SUM159, and SUM149 gene expression data. (C) Scr or shCPT1 MDA-MB-231 cells were profiled using targeted metabolomics and unbiased lipidomics, with significant differentially expressed features shown as heatmaps. (D) A 3-model CPT1 suppression union signature was derived by overlapping 3-model CPT1 suppression gene signature with metabolic- and lipidomic- associated genes in MDA-BM-231 cells based on the CPT1 inhibition. (E) Distributions of the 3-model CPT1 suppression union signature activity in Ful-resistant MCF7 cells. (F) A Ful resistance signature was developed from BC patient tissue biopsies (GSE71791) and Tam resistance signatures were developed from two Tam resistant MCF7 cells (TR1 and TR2). Activity score correlation between the 3-model CPT1 suppression union signature and the three endocrine resistance signatures were computed over the patient tissue biopsies transcriptomic profiles from the METABRIC BC ER+ hormone-treated dataset. (G) Prognostic associations with disease-specific survival are shown for the 3-model CPT1 suppression gene signature and the 3-model CPT1 suppression union signature in the METABRIC BC ER+ hormone-treated dataset.
Specifically, we first assessed the distributions of the 3-model CPT1 suppression union gene signature activity scores in a dataset of Ful resistance acquired in MCF7 cells (GSE74391) (Fig. 1E). The activity scores of the 3-model CPT1 suppression union signature were higher in the Ful-sensitive samples, suggesting that the loss of CPT1 activity best matched patients who were sensitive to endocrine therapy. To further assess the association between CPT1 inhibition and endocrine therapy resistance, we generated a patient-derived Ful resistance signature by analyzing the dataset GSE71791 using the same analytical approach as that used for our CPT1 suppression microarray. We also generated cell line-derived Tam resistance signatures from TR1 and TR2 cells and compared them with those of parental MCF7 cells using microarray data. Correlations between the 3-model CPT1 suppression union signature and these three signatures of endocrine therapy resistance were then computed across TCGA BC ER+ transcriptomic dataset (30,31) and METABRIC BC ER+ and hormone treated transcriptomic datasets (29). The two TR signatures were strongly correlated (r=0.93) and each TR signature was positively correlated with the patient-derived Ful resistance signature (Supplementary Fig. S1D). The 3-model CPT1 suppression union signature was negatively correlated with all three endocrine resistance signatures in METABRIC ER+ hormone-treated cohort (Fig. 1F) and TCGA ER+ cohort (Supplementary Fig. S1E), suggesting that targeted CPT1 modulation might be useful for overcoming Ful or Tam resistance in primary tumors. We further assessed the associations with disease-free survival for the 3-model CPT1 suppression with biopsies from the METABRIC BC cohort from ER+ hormone-treated BC patients. When comparing the bottom and top halves, we observed that both the 3-model CPT1 suppression DEG signature and 3-model CPT1 suppression union signature were associated with better prognosis (Fig. 1G) indicating a potential association with Tam resistance, which might be driven by a FAO metabolic phenotype.
Short-term endocrine therapy-induced FAO pathway in ER+ BC
To elucidate the mechanism underlying endocrine therapy resistance, we treated MCF7 and T47D cells with 1µM Tam or 1µM Ful for 24 h and assessed the expression of proteins related to the FAO pathway using Western Blot (WB) analysis. We observed a moderate increase in the expression levels of CPT1, CPT2, pACC (S79) and pAMPK (T172) in endocrine inhibitor-treated cells after the short-term treatment, indicating an increase in FAO metabolism after endocrine therapy (Fig. 2A; Supplementary Fig. S2A). Interestingly, we also observed an increase in pSrc (Y419) expression after endocrine therapy (Fig. 2A), which was previously shown to be activated by increased FAO in TNBC (9). We performed a similar experiment using the lower doses of Ful with charcoal stripped serum and confirmed the increase of pSrc (Y419) after Ful treatment (Supplementary Fig. S2B). Activation of the FAO pathway was more evident after three weeks of endocrine therapy (Fig. 2B, Supplementary Fig. S2C).
Figure 2. Activation of FAO signaling pathway after endocrine therapy in ER+ BC.
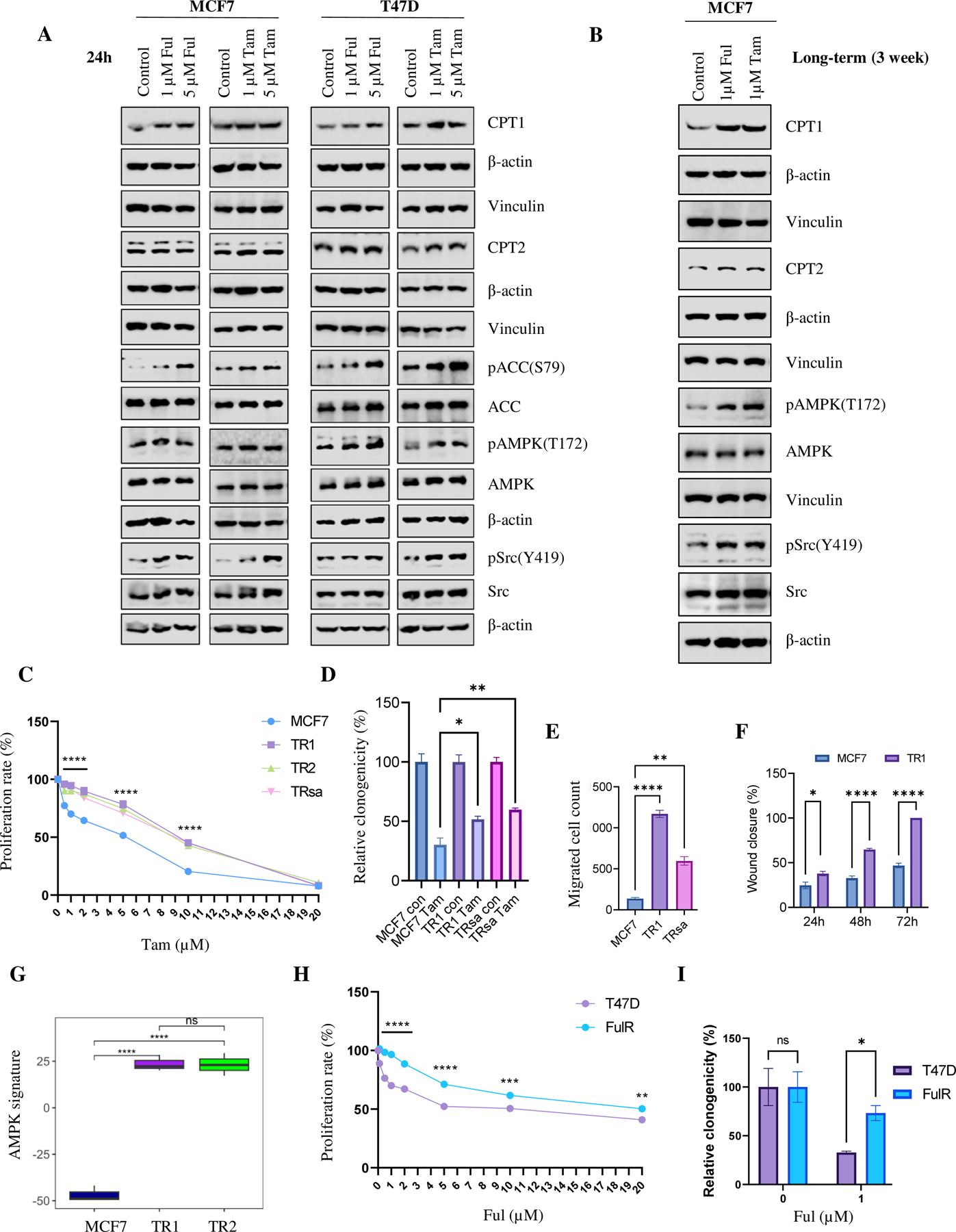
(A) WB analysis of FAO-related and Src proteins in MCF7 and T47D cells treated with Tam or Ful for 24h. β-actin and vinculin were used as loading controls. (B) WB of FAO-related and Src proteins in in MCF7 cells treated with Tam or Ful for three weeks. β-actin and vinculin were used as loading controls. (C) Quantification of SRB proliferation assay of MCF7, TR1, TR2, and TRsa cells after Tam therapy (n=3). (D) Quantification of clonogenic potential of MCF7 and TR cells after Tam (1 µM) therapy (n=3). (E) Quantification of migrated cells from MCF7 and TR cells in the transwell migration assay (n=3). (F) Quantification of wound healing distance in a scratch assay (n=4). (G) Microarray data analysis shows significant increase in the activity of AMPK signature in TR1 and TR2 cells compared to control MCF7 cells (n=3). (H) Quantification of SRB assay of T47D and FulR cells after Ful treatment (n=4). (I) Quantification of clonogenic potential of T47D and FulR cells after Ful (1 µM) treatment (n=3). Data are presented as mean S.E.M., *p < 0.05, ** p <0.01, *** p <0.001, **** p <0.0001, D-F, I: Two-tailed student t-test, C:two-way ANOVA compared to MCF7, H: two-way ANOVA compared to T47D.
Increased mitochondrial OXPHOS and FAO activity in endocrine therapy-resistant ER+ BC cells
To confirm the increased FAO activity after short-term endocrine therapy, we used Tam-resistant (TR) ER+ BC models derived from MCF7 cells. As shown in Figure 2C and 2D, TR cells (TR1, TR2, and TRsa) showed higher tolerance to Tam treatment in both SRB proliferation (Fig. 2C) and clonogenic assays (Fig. 2D; Supplementary Fig. S3A). The IC50 after Tam treatment was MCF7 =6.96 µM, TR1= 10.42 µM, TR2 = 10.25 µM, and TRsa = 10.00 µM. The TR cells also showed cross resistance to Ful treatment (Supplementary Fig. S3B). Transwell migration and wound healing assays suggested that the TR cells exhibited significantly higher migration (Fig. 2E, Supplementary Fig. S3C), and wound-healing motility potential (Fig. 2F; Supplementary Fig. S3D). Soft agar colony formation assays also showed an increased colony formation potential of TR1 cells compared to MCF7 cells (Supplementary Fig. S3E). These results confirmed that TR cells have increased endocrine resistance and tumor potential compared to their parental cells. Furthermore, using the MCF7-TR gene expression data, we analyzed the AMPK downstream gene signature, as we published earlier (15,35), to validate the increased mitochondrial OXPHOS activity in TR cells (Fig. 2G, Supplementary Fig. S4). As shown in Figure 2G, TR1 and TR2 cells showed significant induction of AMPK-related genes compared to MCF7 cells, confirming their increased mitochondrial OXPHOS activity. The T47D FulR cells showed significantly higher resistance to Ful treatment compared to parental T47D cells in SRB proliferation (Fig. 2H) and clonogenic (Fig. 2I) assays. The IC50 after Ful treatment was T47D = 12.71 µM and FulR = 17.19 µM. T47D FulR cells also showed cross resistance to Tam treatment in SRB assay (Supplementary Fig. S5A).
Next, we assessed the expression of FAO pathway proteins in TR cells by WB analysis. As observed with short-term endocrine therapy in ER+ BC cells (Fig. 2), TR cells also demonstrated increased FAO pathway signaling, indicated by higher expression of pAMPK (T172), pACC (S79), CPT1, and CPT2, compared to parental MCF7 cells (Fig. 3A). Seahorse flux analysis showed increased mitochondrial respiration in the TRsa cells (Fig. 3B). The activity of the ETC complex-1 OXPHOS enzyme in the mitochondria was also higher in TR cells than in parental MCF7 cells (Fig. 3C), whereas the activity of citrate synthase, one of the enzymes in the TCA cycle of mitochondria, did not show major difference (Supplementary Fig. S5B). While most of the intermediate carnitine metabolites of the FAO pathway were decreased in TR cells by targeted metabolomics, acetyl carnitine, the final metabolite of FAO, was significantly increased in both TR1 and TRsa cells (Fig. 3D and E). This indicates active FAO metabolism in TR cells. We then analyzed the mRNA and protein expression levels of ACAA2, the last enzyme in the FAO pathway responsible for generating acetyl-carnitine. ACAA2 mRNA and protein levels were increased in TR cells (Fig. 3F and G). Moreover, KM survival analysis suggested that increased ACAA2 protein expression in primary tumors resulted in worse overall survival in patients with ER+ BC (Fig. 3H). KM survival analysis of mRNA data in tumors of endocrine therapy-treated ER+ BC patients suggested that the higher expression of ACAA2 or CPT1 mRNA in tumors resulted in significantly reduced distant metastasis-free survival (Fig. 3I and J). Since CPT1 activity is a major rate-limiting step of FAO, we analyzed the CPT1 enzymatic activity of TR cells in lipid depleted (Fig. 3K) and complete (Supplementary Fig. S5C) serum media. In both media, significantly increased CPT1 activity was observed in TR cells compared to that in parental MCF7 cells. Together, these findings confirmed increased FAO activity in endocrine therapy-resistant ER+ BC cells.
Figure 3. Increased FAO metabolism in TR cells.
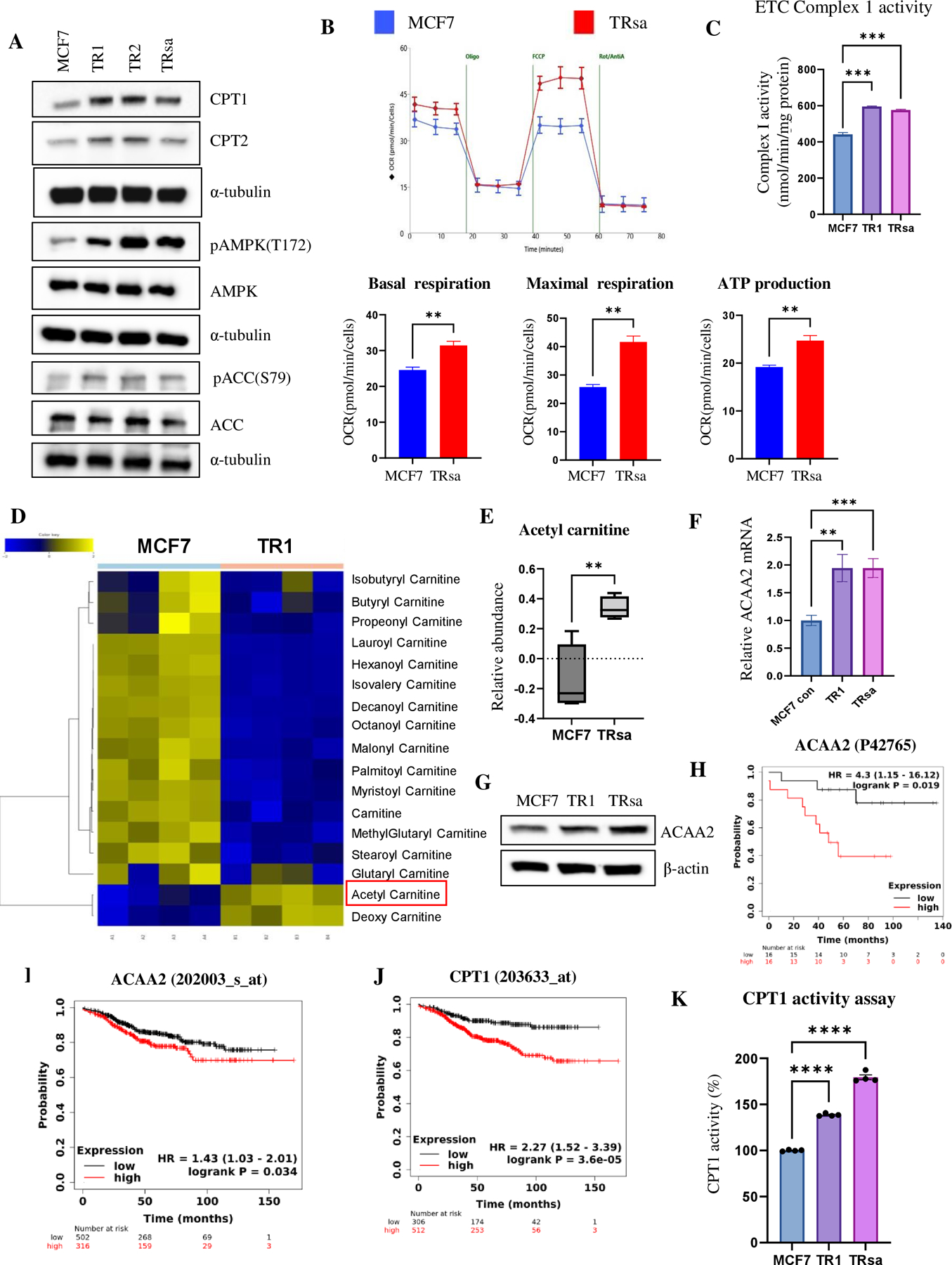
(A) WB analysis of FAO-related proteins in MCF7 and TR cells. α-tubulin was used as a loading control. (B) Seahorse analysis of MCF7 and TR cells to determine the oxygen consumption rate (OCR). Basal respiration, maximal respiration, and ATP production were calculated from the Seahorse assay (n=3). (C) Complex1 activity of MCF7 and TR cells was measured using the ETC assay (n=3). (D) Metabolomics analysis of carnitines in MCF7 and TR1 cells. Acetyl carnitine is marked by a red box. (E) Boxplot of acetyl carnitine levels from the metabolomic analysis of MCF7 and TRsa cells (n=4). (F) qPCR analysis of relative mRNA levels of ACAA2 in MCF7 and TR cells (n=6). (G) WB analysis of ACAA2 protein in MCF7 and TR cells. β-actin was used as a loading control. (H) KM overall survival plot of ER+ BC patients (n=32) separated by the median ACAA2 protein expression in tumors. (I-J) KM distant metastasis-free survival plots of endocrine therapy treated ER+ BC patients (n=818), separated by median ACAA2 (I) or CPT1 (J) mRNA expression in the primary tumors. (K) CPT1 enzymatic activity in MCF7 and TR cells in lipid-depleted serum medium. Data are presented as mean S.E.M. *p < 0.05, ** p <0.01, *** p <0.001 using a two-tailed student t-test.
TR cells were sensitive to FAO and OXPHOS inhibition
Considering the increased FAO activity observed in TR cells, we analyzed their sensitivity to FAO inhibitors. Cell proliferation by SRB assay showed that TR cells were more sensitive to the FAO inhibitor ETX than parental MCF7 cells, as evidenced by the decreased proliferation rate (Fig. 4A). Clonogenic assays using ETX and an FDA-approved partial FAO inhibitor RNL also showed increased sensitivity in TR cells to these FAO inhibitors compared to parental cells (Fig. 4B and C). We then analyzed the role of FAO inhibitors in the migratory and anchored-dependent growth potential of TR cells. As expected, FAO inhibition suppressed the wound healing properties (Fig. 4D) and soft agar colony formation potential of TR cells (Fig. 4E; Supplementary Fig. S3E). To confirm our observations, we generated shRNA-mediated CPT1 knockdown cells using both MCF7 and T47D cell lines. Knockdown was verified by WB (Fig. 4F). In both cell line models, the loss of CPT1 showed significantly higher sensitivity to Tam and Ful therapy in the MTT cell proliferation (Fig. 4G) and clonogenic (Fig. 4H) assays.
Figure 4. TR cells have increased sensitivity to FAO inhibition.
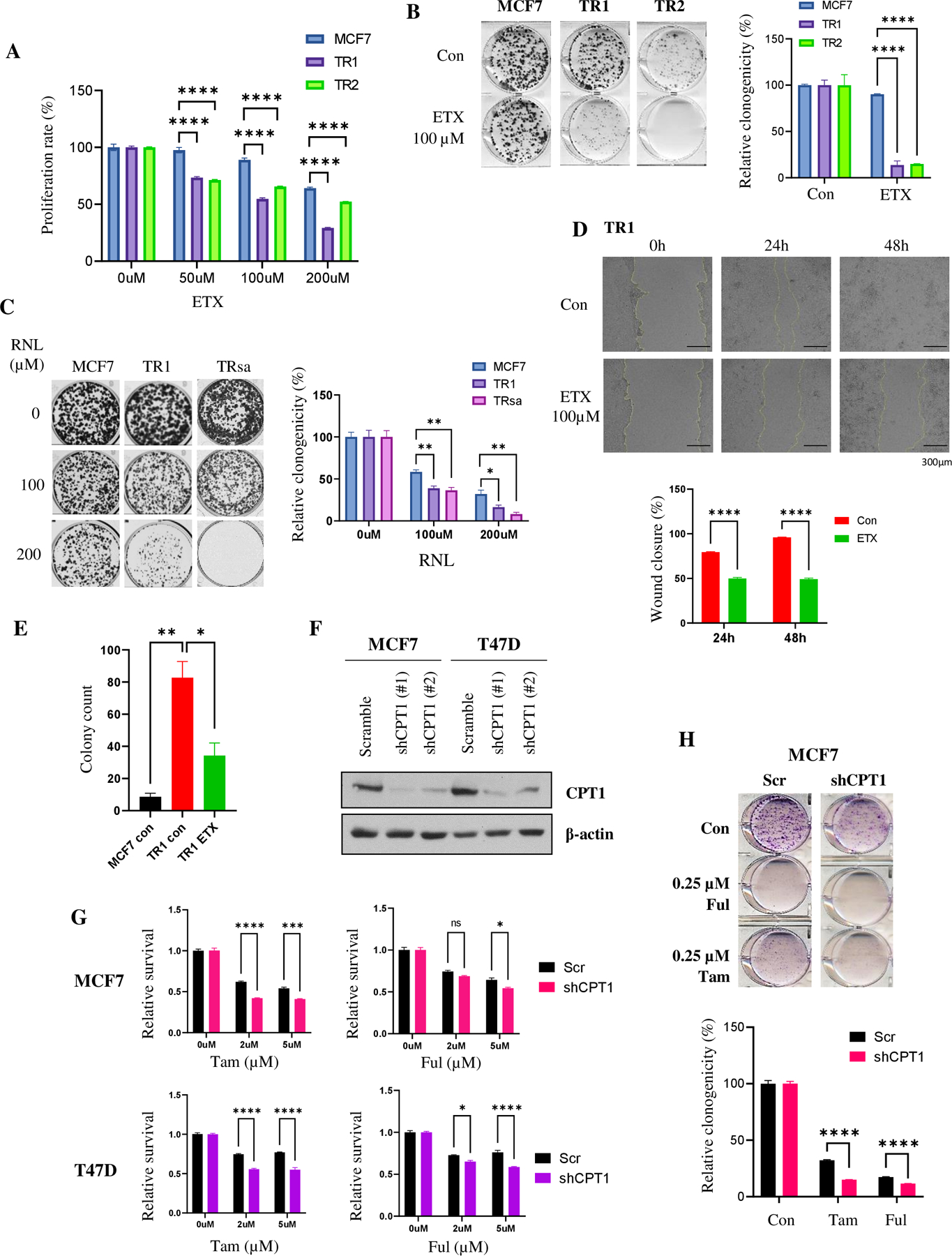
(A) SRB proliferation assay of MCF7 and TR cells treated with ETX (n=6). (B) & (C) Clonogenic assay of MCF7 and TR cells treated with ETX (B) or RNL (C) (n=3). (D) Wound healing analysis of TR cells after treatment with ETX (n=4). (E) Quantification of anchorage-independent growth analysis using soft agar colony formation assay (n=3). (F) WB confirmation of decreased CPT1 protein expression in shCPT1 knockdown in MCF7 and T47D cells. β-actin was used as a loading control. (G) & (H) Analysis of cell viability using MTT assay (G) and clonogenicity using clonogenic assay (H) after Tam and Ful treatment in shScrambled or shCPT1 ER+ BC cells (n=3). Data are presented as mean S.E.M., *p < 0.05, ** p <0.01, *** p <0.001, **** p <0.0001, A, B, D, and G: two-way ANOVA, C, E and H: two-tailed student t-test.
It is well known that FAO activation leads to increased mitochondrial OXPHOS (36,37). Using the OXPHOS inhibitors, Ato and Met, we tested whether the increased sensitivity of TR cells to FAO inhibitors was also true with OXPHOS inhibition. Clonogenic (Fig. 5A and B) assays showed a significantly increased sensitivity of TR cells to Ato and Met therapy compared to that of parental MCF7 cells, confirming their increased dependency on mitochondrial ETC. Wound healing assay also showed increased sensitivity of TR cells to Ato (Fig. 5C). Similar to TR cells, T47D FulR cells also showed higher sensitivity to Ato (Fig. 5D) and Met (Fig. 5E) compared to T47D parental cells. Furthermore, FulR cells were more sensitive to FAO inhibitors ETX (Supplementary Fig. S5D) and RNL (Supplementary Fig. S5E).
Figure 5. FAO and OXPHOS inhibitors suppress in vivo growth potential of TR cells.
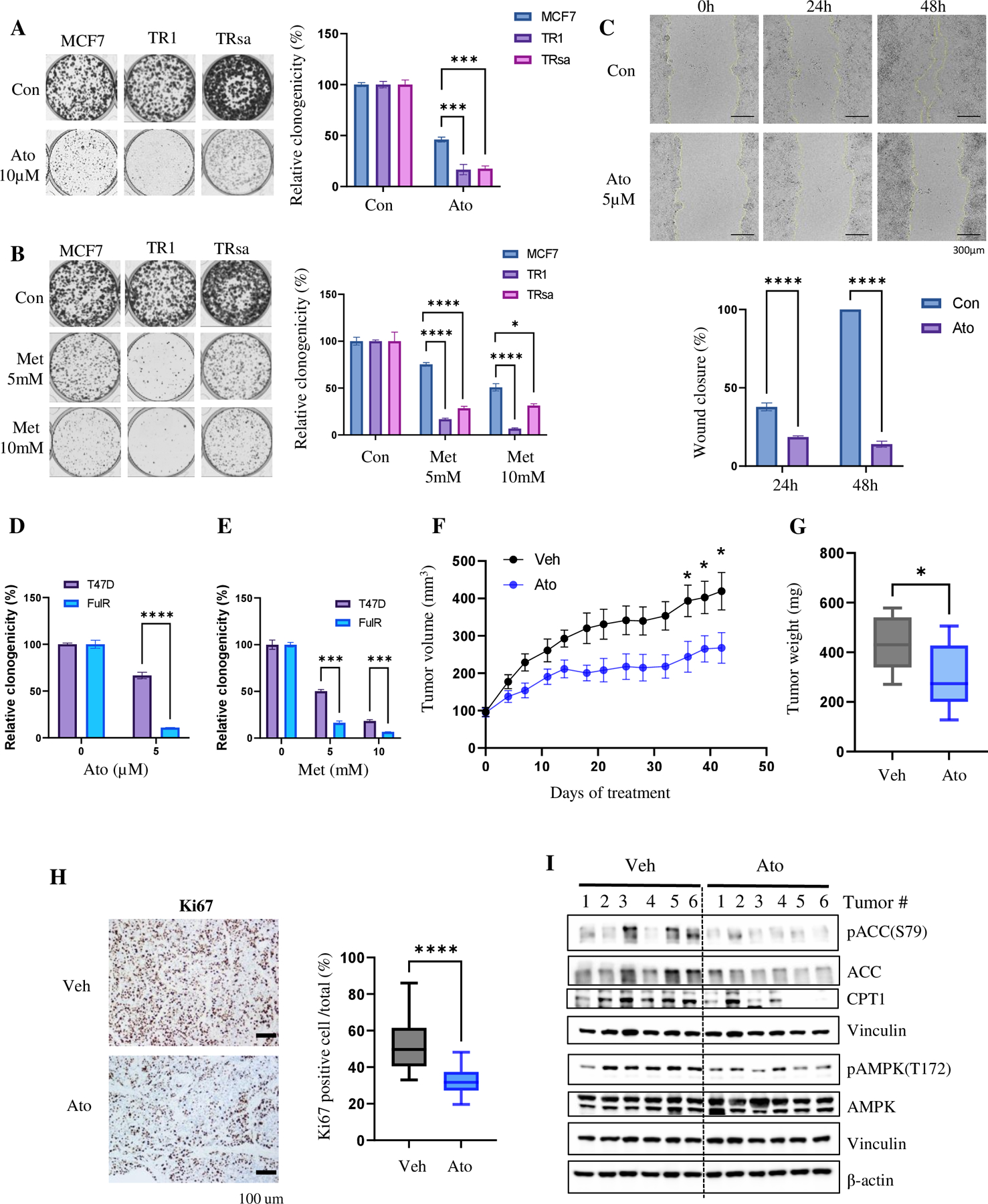
(A & B) Clonogenic assay of MCF7 and TR cells after treatment with OXPHOS inhibitors Ato (A) and Met (B) (n=3). (C) Wound healing assay of TR1 cells after treatment with Ato (n=4). (D & E) Clonogenic assay of T47D and FulR cells after treatment with OXPHOS inhibitors Ato (D; control n=6, Ato n=3) and Met (E; control: n=6, Met: n=3). (F-H) In vivo Ato therapy in TRsa cells. (F) Tumor growth curve of mice with TR cell xenografts treated with vehicle or Ato (50mg/kg, 5 times/week, p.o., n=6). All mice received Tam (2mg/kg, 5 times/week, p.o.). (G) Tumor weight at the end point (n=6). (H) IHC analysis of Ki67 in tumor tissues and quantification of Ki67 positive cells (n=6 tumors x 3 representative areas). (I) WB analysis of the tumor tissues from Fig. 5F. Vinculin and ß-actin are loading controls. Data are presented as mean S.E.M., *p < 0.05, ** p <0.01, *** p <0.001, **** p <0.0001, A-C & F =Two -way ANOVA; D-E & H = two-tailed student t-test, G = one-tailed student t-test.
To confirm the in vitro findings, we performed in vivo experiments using TR cells. TRsa cells were injected into the mammary glands of female SCID mice and treated with vehicle (corn oil) or ETC inhibitor Ato in the presence of Tam. The Ato treatment significantly reduced the tumor growth (Fig. 5F) and tumor weight (Fig. 5G) compared to vehicle-treated mice. No considerable changes were observed in body weight between vehicle- and Ato-treated mice (Supplementary Fig. S6A). IHC analysis of Ki67 revealed significantly decreased Ki67 positive cells in Ato-treated tumors, suggesting reduced cell proliferation after OXPHOS inhibitor therapy (Fig. 5H). WB analysis of the tumors showed significantly decreased expression of pACC (S79), CPT1, pAMPK (T172) proteins in the Ato-treated tumors (Fig. 5I, Supplementary Fig. S6B). These data confirm that Tam-resistant cells have increased FAO and OXPHOS activity in vivo and are sensitive to FAO or OXPHOS inhibition.
TR cells have FAO-induced Src activation
Src is activated in endocrine-resistant BC cells (38). However, the underlying mechanisms have not been elucidated. Short-term (Fig. 2A) and long-term (Fig. 2B and 6A) endocrine treatments increased pSrc (Y419) expression in ER+ BC cells. Thus, we treated MCF7 and TR cells with FAO inhibitors for 24 hours and analyzed Src phosphorylation. The basal levels of pSrc (Y419) were higher in TR cells than in parental MCF7 cells. As expected, the FAO inhibitor ETX suppressed the increase in Src activation in the TR cells (Fig. 6B). This suggests that Tam-induced FAO leads to Src activation in ER+ BC cells, which can be reversed by an FAO inhibitor.
Figure 6. TR cells depend on FAO-induced Src activation.
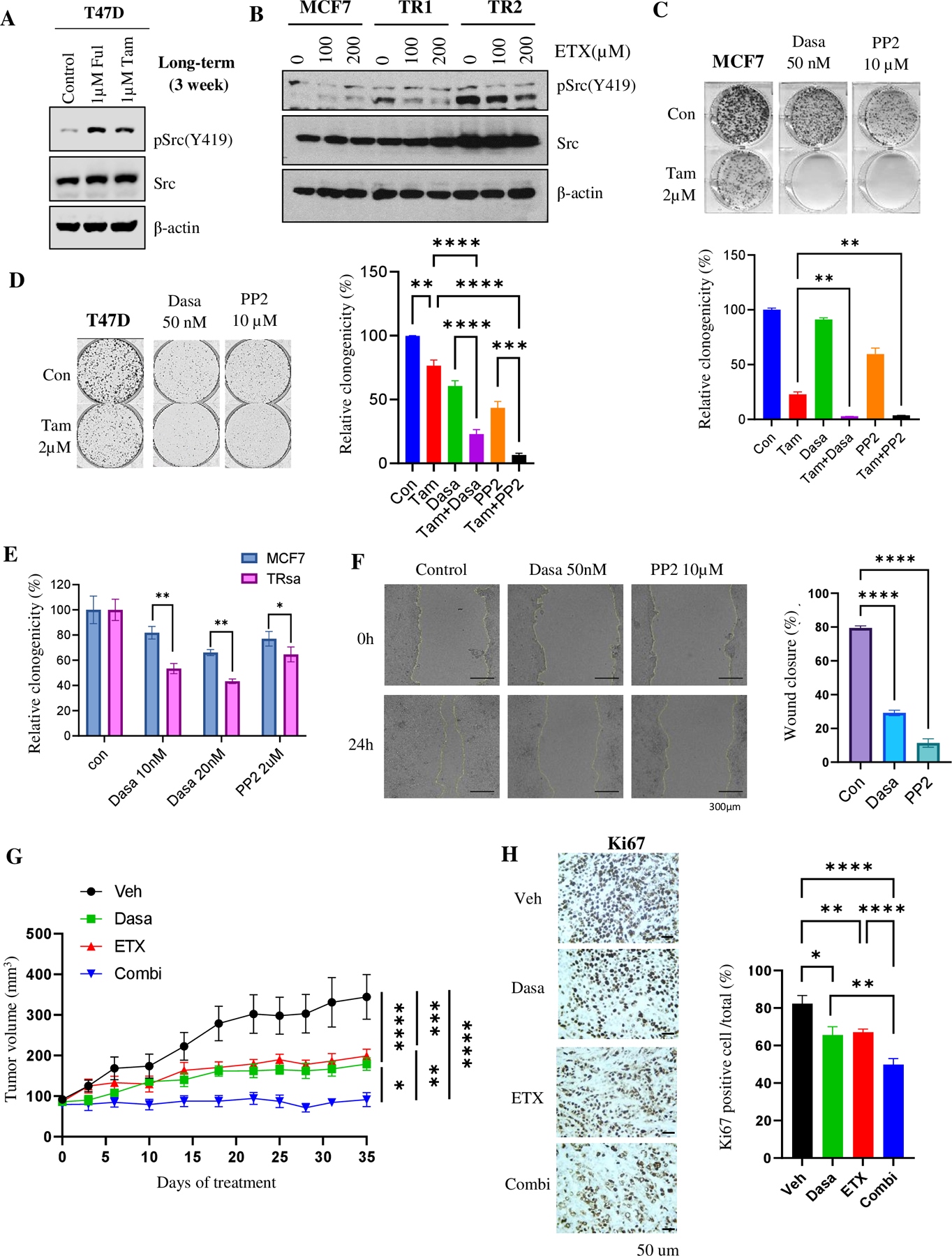
(A) WB of pSrc (Y419) and Src in MCF7 cells treated with Tam for 2-weeks. Vinculin was used as a loading control. (B) WB of pSrc (Y419) and Src levels in MCF7 and TR cells treated with ETX for 24 hours. β-actin was used as a loading control. (C & D) Clonogenic assay of MCF7 (C) and T47D (D) cells after treatment with Tam and Src inhibitors alone or in combination (n=3). (E) Quantification of the clonogenic assay in MCF7 or TRsa cells after treatment with Src inhibitors (Dasa or PP2) (n=3). (F) Wound healing assay of TR1 cells after treatment with Src inhibitors (n=4). (G) Tumor growth curve of TRsa cell xenografts treated with Dasa (20mg/kg, 3 times/week, p.o., n=6), ETX (50mg/kg, 3 times/week, i.p., n=6) or combination of Dasa + ETX (n=6). All mice received Tam (2mg/kg, 5 times/week, p.o.). (H) IHC analysis of Ki67 in tumor tissue from Fig 5G (n=4 tumors x 3 representative areas). Data are presented as mean S.E.M., *p < 0.05, ** p <0.01, *** p <0.001, **** p <0.0001, C, D, F & H= One-way ANOVA, E= Two-tailed student t-test, G= Two -way ANOVA
Using a colony formation assay, we analyzed the antitumor activity of short-term Tam therapy in MCF7 and T47D cells in the presence of Src inhibitors Dasa and PP2. Src inhibitors enhanced the anticancer effects of Tam and significantly decreased the colony formation potential of endocrine therapy-sensitive cells (Fig. 6C and D). We then analyzed the role of Src inhibitors in TR cells. The colony formation assay results suggested that TR cells were significantly more sensitive to Src inhibitors than their parental MCF7 cells (Fig. 6E; Supplementary Fig. S7A). Src inhibitors also significantly inhibited the migration potential of TR cells, as shown in the wound healing scratch assay (Fig. 6F). T47D FulR cells also showed increased sensitivity to PP2 and Dasa treatment in colony formation assay (Supplementary Fig. S7B).
Considering FAO induction and increased sensitivity to Src inhibitors in TR cells, we hypothesized that the combination with an upstream FAO pathway inhibitor might increase the sensitivity of TR cells to Src inhibitors. Thus, we generated tumors from TRsa cells and performed a 4-arm study with vehicle, ETX, Dasa and ETX+Dasa. All tumors, including the vehicle group, were maintained under a low dose of Tam. As shown in Fig. 6G and Supplementary Fig. S7C, compared to vehicle-treated mice, the tumor volumes were significantly reduced after ETX or Dasa therapy alone. Interestingly, a combination of ETX and Dasa significantly prevented the tumor growth compared to the single drug therapy. Ki67 staining also showed significant reduction of tumor cell proliferation in ETX and Dasa alone treated group. As expected, tumors from the combination-treated mice showed significantly reduced the Ki67 positive cells compared to the single drug treated tumors (Fig. 6H). Treatment did not cause any significant differences in the body weights of the mice (Supplementary Fig. S7D). Furthermore, WB analysis of the tumors from ETX-treated mice showed considerable decrease of pSrc expression after FAO inhibitor therapy (Supplementary Fig. S7E). We then hypothesized that an initial short-term pre-treatment with ETX might increase the sensitivity of TR cells to Src inhibitors. Therefore, we adopted a sequential treatment approach in this study as illustrated in the Supplementary Fig. S8A. Once the TR tumors were palpable, the mice were treated with vehicle or the FAO inhibitor ETX for three weeks. Low-dose Tam (2mg/kg) was administered to both groups. After three weeks, we stopped ETX therapy and replaced it with the Src inhibitor, Dasa. Sequential treatment with FAO followed by Src inhibitor significantly reduced the tumor growth (Supplementary Fig. S8B). However, no significant differences were observed in the body weights of the mice (Supplementary Fig. S8C). These data suggest that pretreatment with metabolic inhibitors may reduce endocrine resistance and enhance the activity of Src inhibitors in a subgroup of tumors with Tam resistance.
Overall, as shown in Figure 7, the results of our study suggest that endocrine therapy-induced mitochondrial metabolic reprogramming activated the FAO-OXPHOS-Src resistance pathway in ER+ BC cells.
Fig7. Hypothetical model of metabolic reprogramming of FAO-driven endocrine therapy resistance mechanism in ER+ BC.
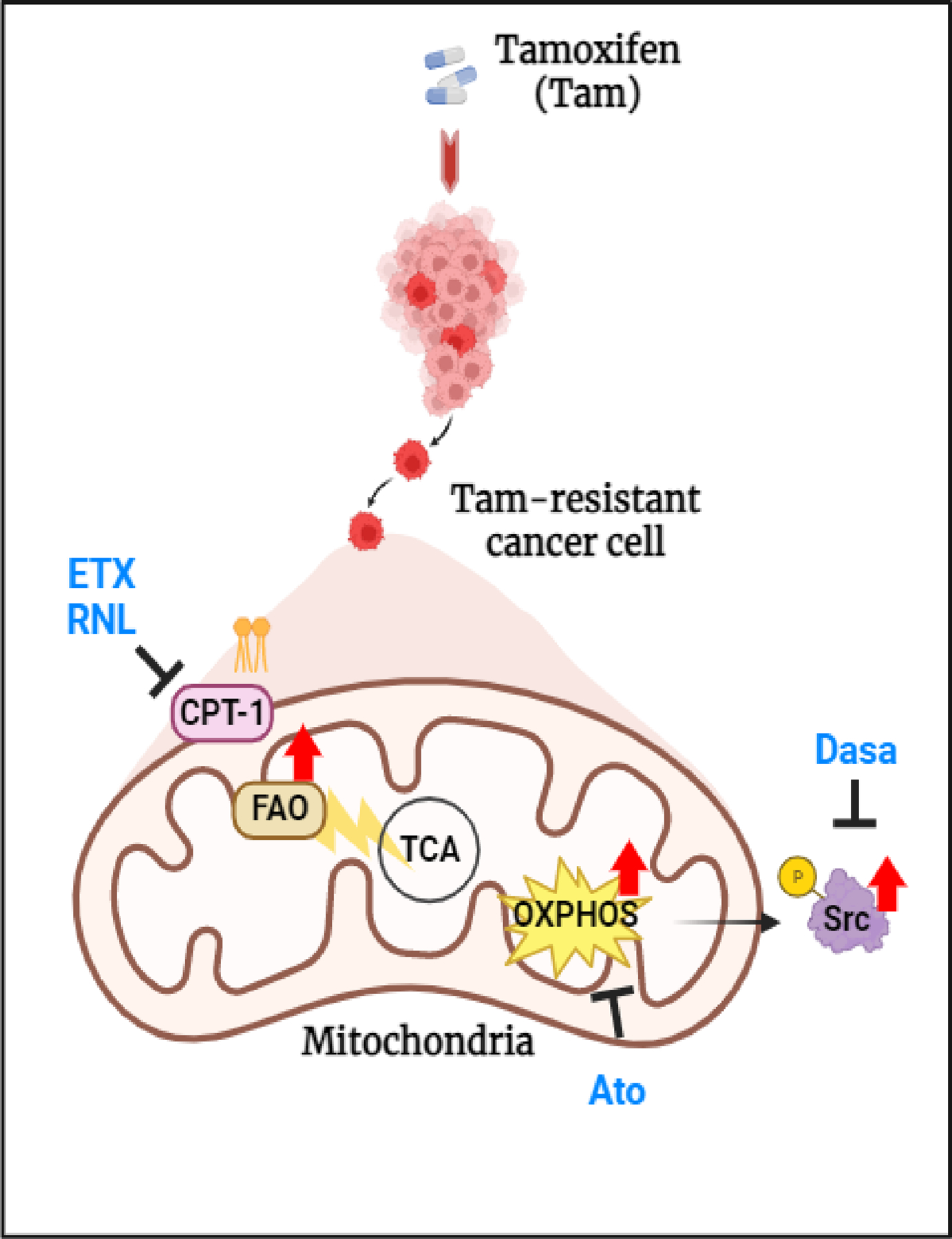
(Created with BioRender.com).
Discussion
The recurrence of ER+ BC after hormonal therapy is a major obstacle limiting the success of cancer treatment (39). This endocrine resistance cannot be explained merely by altered gene expression, common mutations, or simple changes in molecular signaling. Recently, several reports have demonstrated metabolic reprogramming and functional changes in the mitochondria of endocrine-resistant BC cells (7,40,41). Unlike other BC subtypes, ER+ BC has a higher dependency on glycolysis (42–44), and menopause onset or endocrine therapy inhibits estrogen secretion, leading to the initial inhibition of glycolysis (45). However, multiple studies have reported changes in amino acid and lipid metabolism in endocrine-resistant cells (40,46–48). Moreover, Hultsch et al. reported a striking increase in lipid and cholesterol accumulation in Tam-resistant cells (48). Fatty acid synthase (FASN) is highly expressed in the ER+/HER2+ BC subtype in the Cancer Proteome Atlas and FASN inhibitors sensitize Tam resistance in vivo (47). However, the association between endocrine therapy and mitochondrial metabolism-mediated regulation of oncopathways and the underlying mechanisms remain unclear. In this study, we showed that endocrine therapy activates the FAO and Src signaling pathways, which contribute to the therapeutic resistance of ER+ BC. We further demonstrated that ETX, a direct inhibitor of CPT1, inhibited the growth of Tam-resistant cells in vitro and in vivo. By mining public data, we found that the signature of high FAO activity was highly correlated with Tam- or Ful-treated patient transcriptomics. Clinical data showed that ER+ BC patients with high CPT1 activity tended to have poor prognosis (Fig. 1G and 3J).
Integrating multiple omics, we developed a TNBC-derived gene signature with metabolic significance. We focused on CPT1, the key modulator of FAO that is involved in metastatic progression (9,49,50). Interestingly, lipid metabolism and CPT1 activity have been reported to potentially affect resistance to other therapeutic interventions such as kinase inhibitors (51) or radiation therapy (52). After knockdown of CPT1 in TNBC cells, we profiled our cell model using transcriptomics, metabolomics, and lipidomics assays, and derived a 3-model CPT1 suppression union signature by intersecting the CPT1 suppression gene signatures across three cell models with genes associated with altered metabolites or lipids, resulting in a final signature comprised of 28 induced and 16 suppressed genes in shCPT1 cells. This TNBC-derived union signature achieved prognostic significance in endocrine-resistant ER+ BC patients.
Increased FAO metabolism leads to the activation of OXPHOS, which is more dependent on the mitochondrial respiration process because FAO can provide more reducing equivalents to the mitochondria that accelerate OXPHOS, followed by ATP synthesis. We observed that mitochondrial activity was higher in endocrine-resistant cells than in the parental cells. We also showed that inhibiting mitochondrial OXPHOS using Met or Ato, FDA-approved OXPHOS inhibitors, abrogated endocrine resistance in ER+ BC. Thus, these drugs can be considered attractive therapeutic targets for endocrine resistance.
Furthermore, our mechanistic studies showed that FAO inhibitors reduced pSrc (Y419) and Src oncogenic signaling pathways in TR cells, as previously observed in TNBC tumors (9). Interestingly, the FDA-approved Src inhibitor Dasa caused tumor regression in ETX-pretreated TR cells. Previous studies have shown that Tam-resistant cells are more sensitive to Dasa (53). However, the mechanism by which Src signaling is increased in endocrine-resistant cells remains to be elucidated. In this study, we confirmed that mitochondrial metabolic reprogramming to FAO is a major driving factor for activation of the Src oncopathway in endocrine-resistant tumors. Thus, endocrine-resistant tumors also exhibit an activated FAO-OXPHOS-Src axis, as observed in metastatic TNBC.
Metabolic inhibitors alone may not be sufficient to control tumor progression due to their low cytotoxicity. However, they can prime tumors for better therapeutic response, as shown in the supplementary Fig. S8B. Moreover, several studies have shown that metabolic alterations are a common phenomenon in the tumor microenvironment (54,55). Thus, understanding the metabolic reprogramming of a tumor can have a significant impact on designing suitable combination therapies to sensitize and target currently non-manageable drug-resistant tumors. Further studies are necessary to understand the significance of metabolic inhibitors in enhancing the effects of chemotherapy, checkpoint inhibitors, and other targeted therapies in endocrine-resistant tumors.
In conclusion, our data suggest that endocrine-resistant BC depends on FAO, which plays a critical role in activating oncopathways to develop therapeutic resistance (Fig. 7). Additionally, metabolic priming may sensitize endocrine-resistant tumors to targeted therapies in patients with ER+ BC.
Supplementary Material
Significance:
Increased fatty acid oxidation induced by endocrine therapy activates Src signaling to promote endocrine resistance in breast cancer, which can be overcome using clinically-approved therapies targeting FAO and Src.
Acknowledgements
The authors acknowledge the Metabolomics Core facility at BCM for providing expertise in metabolic profiling and Mr. Divya Vella in the BCM Multi Omics Data Analysis Core (MODAC) for processing RNA-Seq data. S.A, J.H.P, and B.A.K. were partially supported by National Institutes of Health (NIH) grants (R01CA253445, R01CA234479, W81XWH-18-1-0714, and R03CA277079). S. L. G. and C.C. were partially supported by The Cancer Prevention Institute of Texas (CPRIT) grants RP210227, and RP200504; NIH P30 shared resource grant CA125123; NIEHS grants P30 ES030285 and P42 ES027725; NIMHD grant P50 MD015496, and NIMH grant R01 MH134392. SAWF was supported by the BCRF 19-055, NIH R01 CA207270, NIH R01CA072038, and CPRIT MIRA RP180712 (Kelly Hunt).
Footnotes
Conflicts of interest
The authors of this manuscript have no conflict of interest to declare
References
- 1.Anurag M, Ellis MJ, Haricharan S. DNA damage repair defects as a new class of endocrine treatment resistance driver. Oncotarget 2018;9:36252–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clarke R, Liu MC, Bouker KB, Gu Z, Lee RY, Zhu Y, et al. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene 2003;22:7316–39 [DOI] [PubMed] [Google Scholar]
- 3.Herynk MH, Fuqua SA. Estrogen receptor mutations in human disease. Endocr Rev 2004;25:869–98 [DOI] [PubMed] [Google Scholar]
- 4.deGraffenried LA, Friedrichs WE, Russell DH, Donzis EJ, Middleton AK, Silva JM, et al. Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt Activity. Clin Cancer Res 2004;10:8059–67 [DOI] [PubMed] [Google Scholar]
- 5.Mills JN, Rutkovsky AC, Giordano A. Mechanisms of resistance in estrogen receptor positive breast cancer: overcoming resistance to tamoxifen/aromatase inhibitors. Curr Opin Pharmacol 2018;41:59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butt AJ, McNeil CM, Musgrove EA, Sutherland RL. Downstream targets of growth factor and oestrogen signalling and endocrine resistance: the potential roles of c-Myc, cyclin D1 and cyclin E. Endocr Relat Cancer 2005;12 Suppl 1:S47–59 [DOI] [PubMed] [Google Scholar]
- 7.Hussein S, Khanna P, Yunus N, Gatza ML. Nuclear Receptor-Mediated Metabolic Reprogramming and the Impact on HR+ Breast Cancer. Cancers (Basel) 2021;13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mishra A, Srivastava A, Pateriya A, Tomar MS, Mishra AK, Shrivastava A. Metabolic reprograming confers tamoxifen resistance in breast cancer. Chem Biol Interact 2021;347:109602. [DOI] [PubMed] [Google Scholar]
- 9.Park JH, Vithayathil S, Kumar S, Sung PL, Dobrolecki LE, Putluri V, et al. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer. Cell Rep 2016;14:2154–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, Anderton B, et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med 2016;22:427–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herynk MH, Beyer AR, Cui Y, Weiss H, Anderson E, Green TP, et al. Cooperative action of tamoxifen and c-Src inhibition in preventing the growth of estrogen receptor-positive human breast cancer cells. Mol Cancer Ther 2006;5:3023–31 [DOI] [PubMed] [Google Scholar]
- 12.Schlaepfer IR, Joshi M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020;161 [DOI] [PubMed] [Google Scholar]
- 13.Barone I, Brusco L, Gu G, Selever J, Beyer A, Covington KR, et al. Loss of Rho GDIalpha and resistance to tamoxifen via effects on estrogen receptor alpha. J Natl Cancer Inst 2011;103:538–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc 2006;1:1112–6 [DOI] [PubMed] [Google Scholar]
- 15.Jia D, Lu M, Jung KH, Park JH, Yu L, Onuchic JN, et al. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc Natl Acad Sci U S A 2019;116:3909–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gordon MN, Osterburg HH, May PC, Finch CE. Effective oral administration of 17 beta-estradiol to female C57BL/6J mice through the drinking water. Biol Reprod 1986;35:1088–95 [DOI] [PubMed] [Google Scholar]
- 17.Levin-Allerhand JA, Sokol K, Smith JD. Safe and effective method for chronic 17beta-estradiol administration to mice. Contemp Top Lab Anim Sci 2003;42:33–5 [PubMed] [Google Scholar]
- 18.Scherer SD, Riggio AI, Haroun F, DeRose YS, Ekiz HA, Fujita M, et al. An immune-humanized patient-derived xenograft model of estrogen-independent, hormone receptor positive metastatic breast cancer. Breast Cancer Res 2021;23:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gohlke JH, Lloyd SM, Basu S, Putluri V, Vareed SK, Rasaily U, et al. Methionine-Homocysteine Pathway in African-American Prostate Cancer. JNCI Cancer Spectr 2019;3:pkz019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Purwaha P, Gu F, Piyarathna DWB, Rajendiran T, Ravindran A, Omilian AR, et al. Unbiased Lipidomic Profiling of Triple-Negative Breast Cancer Tissues Reveals the Association of Sphingomyelin Levels with Patient Disease-Free Survival. Metabolites 2018;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vantaku V, Dong J, Ambati CR, Perera D, Donepudi SR, Amara CS, et al. Multi-omics Integration Analysis Robustly Predicts High-Grade Patient Survival and Identifies CPT1B Effect on Fatty Acid Metabolism in Bladder Cancer. Clin Cancer Res 2019;25:3689–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piyarathna DWB, Rajendiran TM, Putluri V, Vantaku V, Soni T, von Rundstedt FC, et al. Distinct Lipidomic Landscapes Associated with Clinical Stages of Urothelial Cancer of the Bladder. Eur Urol Focus 2018;4:907–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cui Y, Parra I, Zhang M, Hilsenbeck SG, Tsimelzon A, Furukawa T, et al. Elevated expression of mitogen-activated protein kinase phosphatase 3 in breast tumors: a mechanism of tamoxifen resistance. Cancer Res 2006;66:5950–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30:923–30 [DOI] [PubMed] [Google Scholar]
- 26.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Risso D, Ngai J, Speed TP, Dudoit S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol 2014;32:896–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, et al. HMDB 3.0--The Human Metabolome Database in 2013. Nucleic Acids Res 2013;41:D801–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012;486:346–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thennavan A, Beca F, Xia Y, Recio SG, Allison K, Collins LC, et al. Molecular analysis of TCGA breast cancer histologic types. Cell Genom 2021;1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alves CL, Elias D, Lyng M, Bak M, Kirkegaard T, Lykkesfeldt AE, et al. High CDK6 Protects Cells from Fulvestrant-Mediated Apoptosis and is a Predictor of Resistance to Fulvestrant in Estrogen Receptor-Positive Metastatic Breast Cancer. Clin Cancer Res 2016;22:5514–26 [DOI] [PubMed] [Google Scholar]
- 33.Patani N, Dunbier AK, Anderson H, Ghazoui Z, Ribas R, Anderson E, et al. Differences in the transcriptional response to fulvestrant and estrogen deprivation in ER-positive breast cancer. Clin Cancer Res 2014;20:3962–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat 2010;123:725–31 [DOI] [PubMed] [Google Scholar]
- 35.Yu L, Lu M, Jia D, Ma J, Ben-Jacob E, Levine H, et al. Modeling the Genetic Regulation of Cancer Metabolism: Interplay between Glycolysis and Oxidative Phosphorylation. Cancer Res 2017;77:1564–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin Z, Liu F, Shi P, Song A, Huang Z, Zou D, et al. Fatty acid oxidation promotes reprogramming by enhancing oxidative phosphorylation and inhibiting protein kinase C. Stem Cell Res Ther 2018;9:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Mohsen AW, Mihalik SJ, Goetzman ES, Vockley J. Evidence for physical association of mitochondrial fatty acid oxidation and oxidative phosphorylation complexes. J Biol Chem 2010;285:29834–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo J, Zou H, Guo Y, Tong T, Ye L, Zhu C, et al. SRC kinase-mediated signaling pathways and targeted therapies in breast cancer. Breast Cancer Res 2022;24:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salvo EM, Ramirez AO, Cueto J, Law EH, Situ A, Cameron C, et al. Risk of recurrence among patients with HR-positive, HER2-negative, early breast cancer receiving adjuvant endocrine therapy: A systematic review and meta-analysis. Breast 2021;57:5–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steifensand F, Gallwas J, Bauerschmitz G, Grundker C. Inhibition of Metabolism as a Therapeutic Option for Tamoxifen-Resistant Breast Cancer Cells. Cells 2021;10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tomkova V, Sandoval-Acuna C, Torrealba N, Truksa J. Mitochondrial fragmentation, elevated mitochondrial superoxide and respiratory supercomplexes disassembly is connected with the tamoxifen-resistant phenotype of breast cancer cells. Free Radic Biol Med 2019;143:510–21 [DOI] [PubMed] [Google Scholar]
- 42.Kostanyan A, Nazaryan K. Rat brain glycolysis regulation by estradiol-17 beta. Biochim Biophys Acta 1992;1133:301–6 [DOI] [PubMed] [Google Scholar]
- 43.Imbert-Fernandez Y, Clem BF, O’Neal J, Kerr DA, Spaulding R, Lanceta L, et al. Estradiol stimulates glucose metabolism via 6-phosphofructo-2-kinase (PFKFB3). J Biol Chem 2014;289:9440–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Minchenko OH, Ochiai A, Opentanova IL, Ogura T, Minchenko DO, Caro J, et al. Overexpression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-4 in the human breast and colon malignant tumors. Biochimie 2005;87:1005–10 [DOI] [PubMed] [Google Scholar]
- 45.Alemany M Estrogens and the regulation of glucose metabolism. World J Diabetes 2021;12:1622–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Demas DM, Demo S, Fallah Y, Clarke R, Nephew KP, Althouse S, et al. Glutamine Metabolism Drives Growth in Advanced Hormone Receptor Positive Breast Cancer. Front Oncol 2019;9:686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Menendez JA, Papadimitropoulou A, Vander Steen T, Cuyas E, Oza-Gajera BP, Verdura S, et al. Fatty Acid Synthase Confers Tamoxifen Resistance to ER+/HER2+ Breast Cancer. Cancers (Basel) 2021;13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hultsch S, Kankainen M, Paavolainen L, Kovanen RM, Ikonen E, Kangaspeska S, et al. Association of tamoxifen resistance and lipid reprogramming in breast cancer. BMC Cancer 2018;18:850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiong Y, Liu Z, Zhao X, Ruan S, Zhang X, Wang S, et al. CPT1A regulates breast cancer-associated lymphangiogenesis via VEGF signaling. Biomed Pharmacother 2018;106:1–7 [DOI] [PubMed] [Google Scholar]
- 50.Ruidas B, Sur TK, Das Mukhopadhyay C, Sinha K, Som Chaudhury S, Sharma P, et al. Quercetin: a silent retarder of fatty acid oxidation in breast cancer metastasis through steering of mitochondrial CPT1. Breast Cancer 2022;29:748–60 [DOI] [PubMed] [Google Scholar]
- 51.Feng WW, Kurokawa M. Lipid metabolic reprogramming as an emerging mechanism of resistance to kinase inhibitors in breast cancer. Cancer Drug Resist 2020;3:1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Han S, Wei R, Zhang X, Jiang N, Fan M, Huang JH, et al. CPT1A/2-Mediated FAO Enhancement-A Metabolic Target in Radioresistant Breast Cancer. Front Oncol 2019;9:1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Larsen SL, Laenkholm AV, Duun-Henriksen AK, Bak M, Lykkesfeldt AE, Kirkegaard T. SRC drives growth of antiestrogen resistant breast cancer cell lines and is a marker for reduced benefit of tamoxifen treatment. PLoS One 2015;10:e0118346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garcia-Canaveras JC, Chen L, Rabinowitz JD. The Tumor Metabolic Microenvironment: Lessons from Lactate. Cancer Res 2019;79:3155–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eisenberg L, Eisenberg-Bord M, Eisenberg-Lerner A, Sagi-Eisenberg R. Metabolic alterations in the tumor microenvironment and their role in oncogenesis. Cancer Lett 2020;484:65–71 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The R and Python analysis codes used in the manuscript were uploaded to Zenodo repository, at https://doi.org/10.5281/zenodo.8207076 and https://doi.org/10.5281/zenodo.8253346. The RNA-Sequencing files have been deposited to NCBI GEO, accession GSE240393. The microarray data from MCF7 TR cells have been deposited into accession GSE241654. Other gene expression profile data analyzed in this study were obtained from GEO at GSE72320, GSE74391, and GSE71791. TCGA breast cancer data(31) was publicly available via http://firebrowse.org/. For METABRIC breast cancer data(29), the associated genotype and expression data have been deposited at the European Genome-Phenome Archive (http://www.ebi.ac.uk/ega/), which is hosted by the European Bioinformatics Institute, under accession number EGAS00000000083. Kaplan-Meier Plot (34) was generated from https://kmplot.com/. All other raw data generated in this study are available upon request from the corresponding author.