

Abstract
Charge detection mass spectrometry (CDMS) is a single-particle technique where the masses of individual ions are determined from simultaneous measurement of their mass-to-charge ratio (m/z) and charge. Masses are determined for thousands of individual ions, and then the results are binned to give a mass spectrum. Using this approach, accurate mass distributions can be measured for heterogeneous and high-molecular-weight samples that are usually not amenable to analysis by conventional mass spectrometry. Recent applications include heavily glycosylated proteins, protein complexes, protein aggregates such as amyloid fibers, infectious viruses, gene therapies, vaccines, and vesicles such as exosomes.
Graphical Abstract
1. INTRODUCTION
For the last 50 years there has been interest in extending mass spectrometry (MS) to measure accurate molecular weights (MWs) of larger and larger species. Initially, the challenge was placing large analytes into the gas phase where they could be measured. Early approaches included plasma desorption1 and fast atom bombardment.2 The development of MALDI (matrix-assisted laser desorption and ionization)3,4 and particularly electrospray ionization5 opened the flood gates and allowed the expansion of MS into the world of biological macromolecules. Once there, MS fueled the development of the omics revolution, including proteomics, glycomics, and metabalomics. At the same time there was growing interest in extending MS to the study of protein complexes and other large supramolecular assemblies. Such studies required that the assemblies remain intact when transferred into the gas phase for analysis. Most studies employed electrospray because electrospray offers the advantage of transferring analytes directly from solution. It was soon recognized that the solutions used for electrospray were too harsh,6,7 causing proteins to denature and complexes to disassemble. Switching to commonly used buffers was not viable because the presence of significant concentrations of involatile ions such as Na+ and Cl− suppressed ionization, and furthermore, the resulting peaks were broad because of adduct formation, where involatile salt molecules attach to the analyte. The solution to this problem was to buffer-exchange the sample into a solution of a volatile salt; ammonium acetate is now the most widely used.8,9 With this simple advance the field of native MS was born, and interest grew in using MS to determine accurate MWs for larger and larger complexes.10–12 Although, to realize this goal, it was still necessary to optimize conditions to transmit and detect high-MW ions.13,14
In conventional MS, the quantity that is measured is not the mass but the mass-to-charge ratio (m/z). Small ions are usually singly charged, so mass and m/z are synonymous. However, larger ions, particularly when they are generated by electrospray, are multiply charged, leading to a charge-state envelope—a distribution of peaks in the m/z spectrum due to ions with the same average MW but different charge. Multiple charging was often touted as an advantage of electrospray because it placed the m/z ratio of large proteins into the range accessible by existing mass spectrometers. However, when ions are multiply charged, it is necessary to determine their charge from the m/z spectrum so that the mass can be determined.15 If the resolving power is high enough, the charge can be determined from the natural isotopic distribution. This can be challenging for highly charged ions, where isotope peaks are closely spaced. In cases where the isotope distribution is not resolved, the charge can be determined from the separation between the peaks in the charge-state envelope. This is made difficult by heterogeneity. Heterogeneity can result from several sources, including post-translational modifications, counterions, residual salt, and trapped solvent. It broadens and shifts the peaks in the m/z spectrum, making it impossible to determine the MW from the m/z spectrum without prior knowledge of the MW. Heterogeneity usually gets worse with increasing MW and realistically restricts conventional MS measurements to ions with MWs less than ~1 megadalton. There have been some notable exceptions where charge-state distributions have been resolved for ions with MWs beyond a megadalton, but these are for samples that are highly homogeneous and extensively purified.16–21 There are also many examples of smaller analytes that cannot be analyzed by conventional MS because of heterogeneity. For example, many highly glycosylated proteins fall into this category.
One solution to the heterogeneity problem is to adopt a single-particle approach and measure the m/z and charge of individual ions. The product of an ion’s m/z and charge is its mass, and if the measurement is repeated many times for different ions, the resulting masses can be binned to give a mass distribution. This single-particle approach differs from the ensemble approach usually used in conventional mass spectrometry where the m/z spectrum is measured for many ions. The peaks in the m/z spectrum are assigned to charge states, and then the m/z values of the peaks are multiplied by the charge state to give the MW.
2. SINGLE-ION MS: MEASURING MASSES OF INDIVIDUAL IONS
Two basic approaches have been developed for determining the masses of individual ions by MS. The first involves charge stepping,22 where the m/z ratio of a single ion is measured, the charge on the ion is changed (see below), and then the m/z ratio is remeasured. The mass (and both charge states) can be deduced from two m/z measurements if the difference between the charge states is known. Usually, several charge-stepping cycles are performed to confirm the charge steps. Charge stepping has been performed in a quadrupole ion trap (QIT) and using Fourier transform ion cyclotron resonance (FTICR). The second approach to determining the MW of an individual ion involves simultaneous measurement of its m/z and charge. This includes charge detection mass spectrometry (CDMS) and direct charge detection in FTICR and Orbitrap instruments. Time-of-flight MS (TOF MS) with cryogenic detectors has also been used to measure m/z and charge simultaneously.
2.1. Charge Stepping
The charge-stepping approach was first implemented by Arnold and co-workers using a QIT.22 They irradiated the trapped particles with UV light to cause charge steps through photoemission;22 irradiation by an electron beam has also been used.23 The m/z of a charged particle in a 3D QIT can be determined by monitoring its Lissajous-like trajectory. In early work, the trajectories of μm-sized particles were monitored visually using a microscope to track them by their scattered light.24 An accurate value for the m/z ratio can be determined by adjusting the driving frequency so that the trajectory in the trap follows a stationary star pattern.25 The intensity of the scattered light decreases as d−6, and light scattering is unsuitable for particles <50 nm in diameter.26 The lower size limit can be overcome by studying fluorescent particles, either tagged27,28 or intrinsically fluorescent,29 or by the use of a bright probe particle to infer the behavior of a small dark particle.30 The QIT approach has been used by the Chang group to measure MWs for a variety of objects including dried E. coli cells,23 polystyrene spheres,31 vaccinia virus, and human red blood cells.32 Nie and co-workers measured MWs for adenovirus, iridovirus, and vaccinia virus.26,33 A resolving power (m/Δm) of 105 can be obtained using a novel light-scattering approach developed by Gerlich and co-workers.34,35 However, while the masses of trapped particles can be measured with high accuracy, this technology is not suited to the rapid analysis of many ions. Mass distributions determined using these methods usually contain tens of ions instead of the thousands needed to define a distribution.
Smith and co-workers used FTICR to perform single-molecule mass measurements in the mid-1990s.36–38 In these experiments, single ions were isolated, the trapped ion’s m/z was then measured with high precision, the charge was stepped by introducing a reagent, the reagent was removed, and the m/z was remeasured. Several charge-stepping cycles were performed because, in addition to proton transfer, adduction and elimination reactions can occur.38 This approach can achieve resolving powers in the 104–105 range. It avoids the size limitation due to the optical detection scheme used with the QIT, but as with the QIT approach, it is slow and has not been widely used. Another variation of the charge-stepping theme was recently described by McLuckey and co-workers.39 They used the attachment of multiply charged ions of the opposite charge (such as holo-myoglobin [hMb–13H]13−) to shift the charge. MWs measured for the 30S and 50S subunits of the E. coli ribosome (863.2 and 1497.7 kDa, respectively) were ~1.8% higher than expected. MWs determined for analytes electrosprayed from solutions of volatile salts are usually slightly higher than expected, but the difference is generally smaller, <1%.40
2.2. Simultaneous Measurement of the m/z Ratio and Charge
Simultaneous measurement of the m/z ratio and charge for individual ions provides a direct measure of the mass for each ion. The measurement is repeated a number of times, and then the masses are binned to give a mass spectrum. This approach can be accomplished in a few different ways, as described below.
2.2.1. Time-of-Flight MS with Cryogenic Detectors.
The m/z range of TOF MS is unlimited in theory. It is often coupled with MALDI. MALDI generates ions with a lower charge than electrospray and, hence, higher m/z values. In practice, the upper m/z limit of TOF MS is limited by the detector response.41 One solution to this problem is the use of a cryogenic detector,42–48 either a microcalorimeter or a super-conducting tunneling junction. In both cases, the response is related to the energy deposited by the ion as it strikes the detector. If the ions are accelerated through a fixed potential, each ion’s kinetic energy is proportional to its charge. Thus, in principle the charge can be deduced from the energy deposited into the detector. However, in practice the detector response is not linearly proportional to the ion charge above a few charges, so charge determination is not currently possible for ions with more than a few charges. This approach has not found widespread use.
2.2.2. Charge Detection Mass Spectrometry.
In CDMS, the ions pass through a detection cylinder. As the ion enters the cylinder, it induces a charge that is detected by a charge-sensitive amplifier. The induced charge dissipates when the ion leaves the cylinder. If the ion energy is known, the flight time through the cylinder provides the m/z ratio, and if the cylinder is long enough, the induced charge equals the charge on the ion.49,50 This approach was first used to measure the mass and velocities of micron-sized metal particles for impact studies to investigate the effect of dust-particle collisions on satellites.51 In 1995, Fuerstenau and Benner adapted it to analyze ions generated by electrospray.52 Because the charge carried by individual ions is small, the accuracy of the charge measurement is limited by electrical noise. In these early, ground-breaking measurements, the root mean square (RMS) noise was equivalent to 150 e (elementary charges), and the smallest charge that could be reliably detected was 425 e. This restricted the early CDMS measurements to highly charged ions, and the early studies of Benner and collaborators focused on measurements for DNA.52–54 In subsequent work, Benner and co-workers used the same approach to measure mass distributions for rice yellow mottle virus (RYMV) and tobacco mosaic virus (TMV).55 For RYMV they found a peak centered on ~6.5 MDa (close to the expected mass), but the peak was >10 MDa wide with a high mass tail that extended beyond 35 MDa. The results for TMV were similar. The poor accuracy of the charge measurement was probably the main contributor to the peak width in these early studies. In CDMS, the mass resolving power (m/Δm) depends on the uncertainties in both the m/z and charge, which are independent variables,
| (1) |
where Δm is the full width at half-maximum (fwhm) of the peak at mass m and σ(m/z) and σz are the root-mean-square deviations (RMSDs) for the m/z and charge determinations, respectively.
The main factor limiting the accuracy of the charge measurement is electrical noise. One way to improve the accuracy is to signal-average, either by embedding the detection tube in an electrostatic linear ion trap (ELIT),56,57 so that an ion oscillates back and forth through the tube many times,58,59 or by passing each ion through a series of detectors arranged one after the other in a linear array.60–64 When multiple measurements are performed, the uncertainty in the charge determination is , where σz1 is the RMSD in the charge measurement from a single pass through a detection cylinder and n is the number of times an ion passes through a cylinder. It follows that single-pass CDMS charge measurements are the least reliable, followed by a linear array, and then an ion trap where there can be thousands of oscillations. The main advantage of single-pass CDMS is that it has a much higher throughput than ion-trap CDMS so that a spectrum can be measured in <1 min. A linear array offers a compromise: a relatively high throughput with a charge accuracy better than that with single-pass CDMS, although not as good as with ion-trap CDMS. Most CDMS instruments now utilize the ion-trap approach, trading measurement time for charge accuracy and, hence, mass resolution.
2.2.2.1. Ion-Trap CDMS Instrumentation.
Figure 1a shows a schematic diagram of a state-of-the-art ion-trap CDMS instrument. Ions are generated by nanoelectrospray and carried into the instrument by ambient gas flow through a metal capillary. The ions are separated from the ambient gas as they travel through three stages of differential pumping. The first stage contains a FUNPET, an ion funnel–ion carpet hybrid that is optimized to thermalize high mass ions and provide ample time for them to desolvate.65 The following stages contain a radiofrequency (RF) hexapole and a segmented RF quadrupole. At the end of the quadrupole, ions are focused into a dual hemispherical deflection energy analyzer that transmits a narrow band of ion kinetic energies that are focused into the ELIT.
Figure 1.
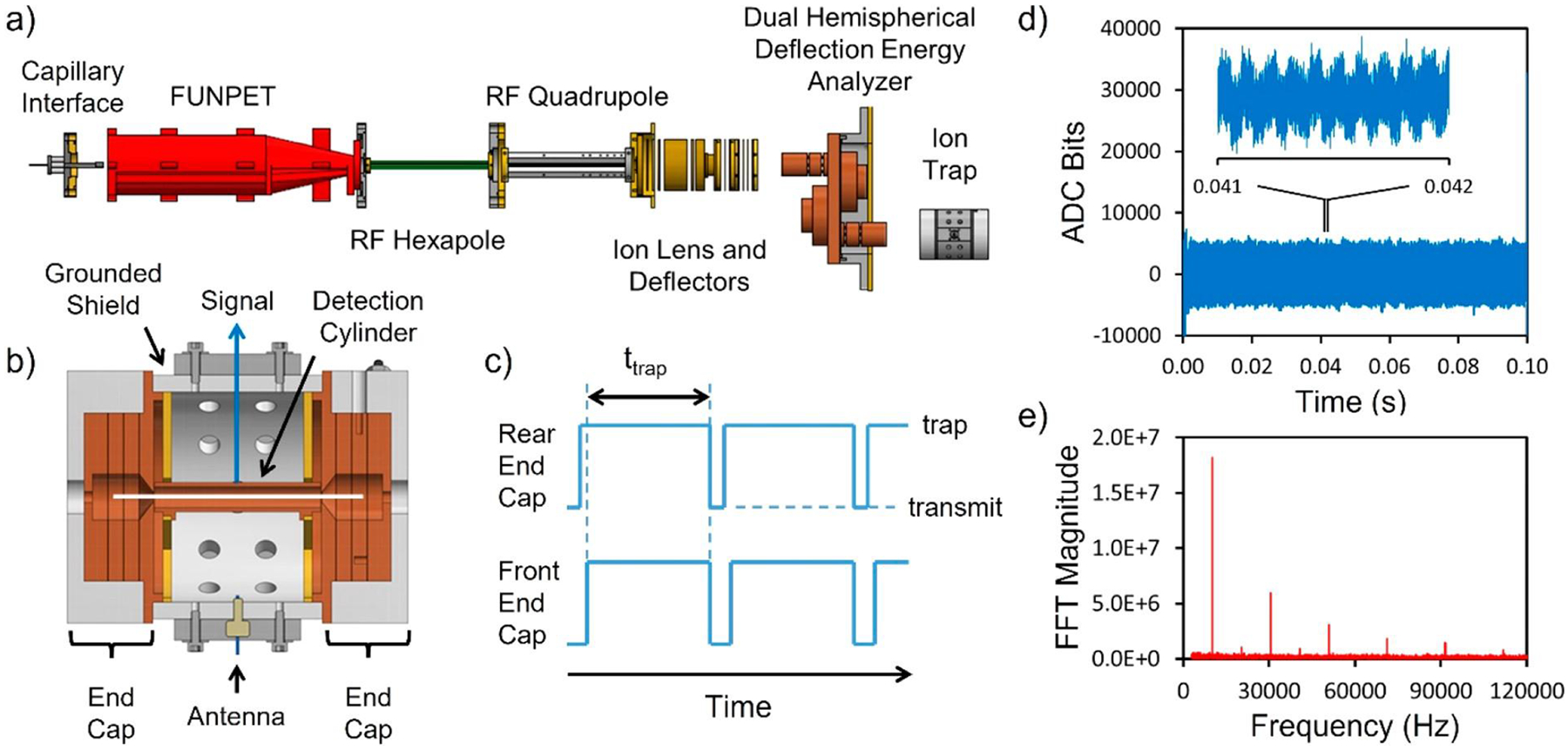
How CDMS measurements are performed. (a) Schematic diagram showing the main components of a state-of-the-art ion-trap CDMS instrument. (b) Cross section through the ELIT showing the detection cylinder between the two end-caps. The white line shows the trajectory of a trapped ion oscillating on the trap axis. (c) Time sequence of the potentials applied to the end-caps to trap ions. (d) Time domain signal resulting from a 0.1 s trapping event for a single ion. The inset shows an expanded view of the signal between 0.041 and 0.042 s (represented by the two closely spaced black vertical lines near the center of the plot). Oscillations in the signal due to the ion passing back and forth through the detection cylinder are apparent. (e) FFT of the full trapping event. The fundamental is at ~10 kHz, and the sequence of higher-frequency peaks is due to harmonics. The odd-numbered harmonics have much larger magnitudes than the even-numbered ones. (a) Adapted with permission from ref 73. Copyright 2019 American Chemical Society.
The ELIT consists of two end-caps that can be switched between transmission and trapping modes by changing the potentials on the end-cap electrodes. In our prototype instrument, we used a cone trap59,66 because of its simplicity (the end-cap consists of a single conical electrode), but simulations showed that better performance could be obtained from ELITs with multielectrode end-caps.67 Figure 1b shows a cross section through an ELIT optimized by trajectory simulations for CDMS measurements. In this example, both end-caps contain three electrodes and grounded shields separate the end-caps from the central detection cylinder. The potentials on the outer two electrodes are switched to trap ions.67 The time sequence of the end-cap potentials is shown schematically in Figure 1c. Initially, both end-caps are in transmission mode, and ions are focused through the trap. The potentials on the rear end-cap are switched to trapping mode, and ions are reflected back through the trap. Then, ~1 ms later, the front end-cap is switched to trapping mode, closing the trap. Trapped ions oscillate back and forth through the detection cylinder, inducing a periodic signal that is detected by the charge-sensitive amplifier. The white line in Figure 1b shows the trajectory of an ion oscillating along the trap axis. At the end of a predetermined trapping period (typically 0.1–1.5 s) the end-caps are switched to transmission mode to release the trapped ion. The trapping cycle is then repeated.
2.2.2.2. Data Analysis.
The signal from the oscillating ion is digitized and transferred to a computer. Figure 1d shows an example of the signal generated by a single ion trapped for 0.1 s. The inset shows the signal measured between 0.041 and 0.042 s, a time interval indicated by the two closely spaced vertical lines in the center of the plot. The oscillations visible in the signal in the inset result from the ion passing back and forth through the detection cylinder. The signal from the oscillating ion is a slightly rounded square wave. There are several high-frequency signals (100–400 kHz) superimposed on the ion signal. One is intentional: the 129 kHz reference signal applied to the antenna in Figure 1b. The reference signal is used to dynamically calibrate the charge measurement.68 Other high-frequency contributions include pick-up from the RF signals applied to the FUNPET, RF hexapole, and RF quadrupole. These are outside the frequency range of ion oscillation signals and do not interfere.
The time-domain signals are analyzed in real time using fast Fourier transforms (FFTs).59,69 The charge is obtained from the FFT magnitude, and the m/z ratio is determined from the oscillation frequency using the following equation,
| (2) |
where C is a constant determined from ion-trajectory simulations.59 Figure 1e shows a full-event FFT of the signal in Figure 1d. The fundamental is at ~10 kHz, and a series of odd harmonics are evident. The even harmonics are almost absent with this ELIT because it employs a 50% duty cycle (where the time spent in the end-cap equals the time in the detection cylinder).67 A 50% duty cycle optimizes the accuracy of the charge measurement and reduces the dependence of the oscillation frequency on the ion energy. In the program used to process the data, each trapping event is analyzed using a windowed FFT (where short overlapping sections of the time-domain signal are analyzed sequentially).59,69 Thus, the oscillation frequency and charge are monitored as a function of the trapping time. A gradual shift in the oscillation frequency during the trapping period can result from the loss of residual solvent, collisions with the background gas, and interactions between the ion and its image charge.70 Fragmentation or loss of a charge lead to a step in the oscillation frequency.70,71 If there is a step, or if the ion is not trapped to the end of the trapping period, the trapping event is discarded during data analysis.
2.2.2.3. Different Trapping Schemes.
To trap an ion in the ELIT, it is necessary to switch the end-cap potentials from transmission to trapping mode while an ion is in the trap. This can be accomplished in several ways. In early work, the end-caps were switched to trapping mode when the signal of an ion was sensed on the detection cylinder.58 This is called triggered trapping. The threshold was usually set to 5 times the RMS noise to avoid closing the trap on noise. This led to a high limit of detection (typically ~250 e), which restricted the early measurements to highly charged ions. Alternatively, the ion trap can be continuously opened and closed without prior knowledge of whether an ion is inside. The signal acquired during the trapping period is then analyzed to determine if an ion was trapped.59 This continuous-trapping mode is illustrated in Figure 1c. With this mode it is possible to detect ions with fewer charges than with triggered trapping. Even single, singly charged ions have been detected.72 However, continuous trapping is less sensitive than triggered trapping because the probability of trapping an ion is lower. In continuous trapping, the trap is open for a short time (a few milliseconds) during which ions may enter the trap, and then the trap is closed for the trapping period (typically 0.1–1.5 s). Any ions that arrive during the time when the trap is closed are lost. This loss can be avoided by accumulating the ions when the ELIT is closed and then pulsing them into the ELIT when it is opened to accept ions.73 This pulsed mode of operation increases the sensitivity of CDMS by >2 orders of magnitude, allowing much lower titer samples to be analyzed. A limit of detection of 1.3 μL of 3.3 × 108 particles/mL (7 × 10−19 mol) was obtained for hepatitis B virus with T = 4 capsids using pulsed-mode CDMS.73
2.2.2.4. Charge Uncertainty and Charge-State Resolution.
Charge is quantized. The measured charge can be used to assign the ion to its nearest charge state, and it is only necessary to measure the charge with enough accuracy to ensure that this assignment is correct.74 Figure 2a shows simulated charge spectra for three different charge RMSDs. For an RMSD of 0.1 e (the top charge spectrum in Figure 2a), the peaks corresponding to integer charges of 98, 99, 100, 101, and 102 e are well-resolved. Ions with a measured charge between 99.5 and 100.5 e can be assigned to an integer charge of 100 e with a very low error rate. The fraction of ions with an integer charge of 100 e that are assigned to higher or lower charge states is vanishingly small. For a charge RMSD of 0.4 e (bottom spectrum in Figure 2a), the charge-state distributions overlap, and the charge states are poorly resolved in the summed spectrum (black line). A substantial fraction of the ions with an integer charge of 100 e have measured charges that are <99.5 e or >100.5 e. These ions would be incorrectly assigned to lower or higher charge states if they are assigned to the nearest charge state. For a charge RMSD of 0.2 e (middle spectrum in Figure 2a), the charge states are well-resolved but just beginning to overlap. Figure 2b shows a plot of the quantized charge RMSD (i.e., the RMSD after the charge is assigned to the nearest charge state) plotted against the RMSD of the initial distribution (i.e., before quantization). For a charge RMSD >0.3 e, quantizing the charge increases the overall uncertainty in the charge, but for RMSDs <0.3 e, the uncertainty in the quantized charges drops rapidly. Figure 2c shows the fraction of ions assigned to the wrong charge state plotted against the initial RMSD (red line). The error rate can be substantially reduced by discarding ions that are close to the halfway point between two charge states (blue line). Taken together, these results show that a charge RMSD of ~0.2 e is required to have a well-resolved charge spectrum, where the number of ions assigned to the wrong charge state is small. The uncertainty in the charge no longer contributes significantly to the mass resolution, which becomes almost entirely determined by the uncertainty in the m/z.
Figure 2.
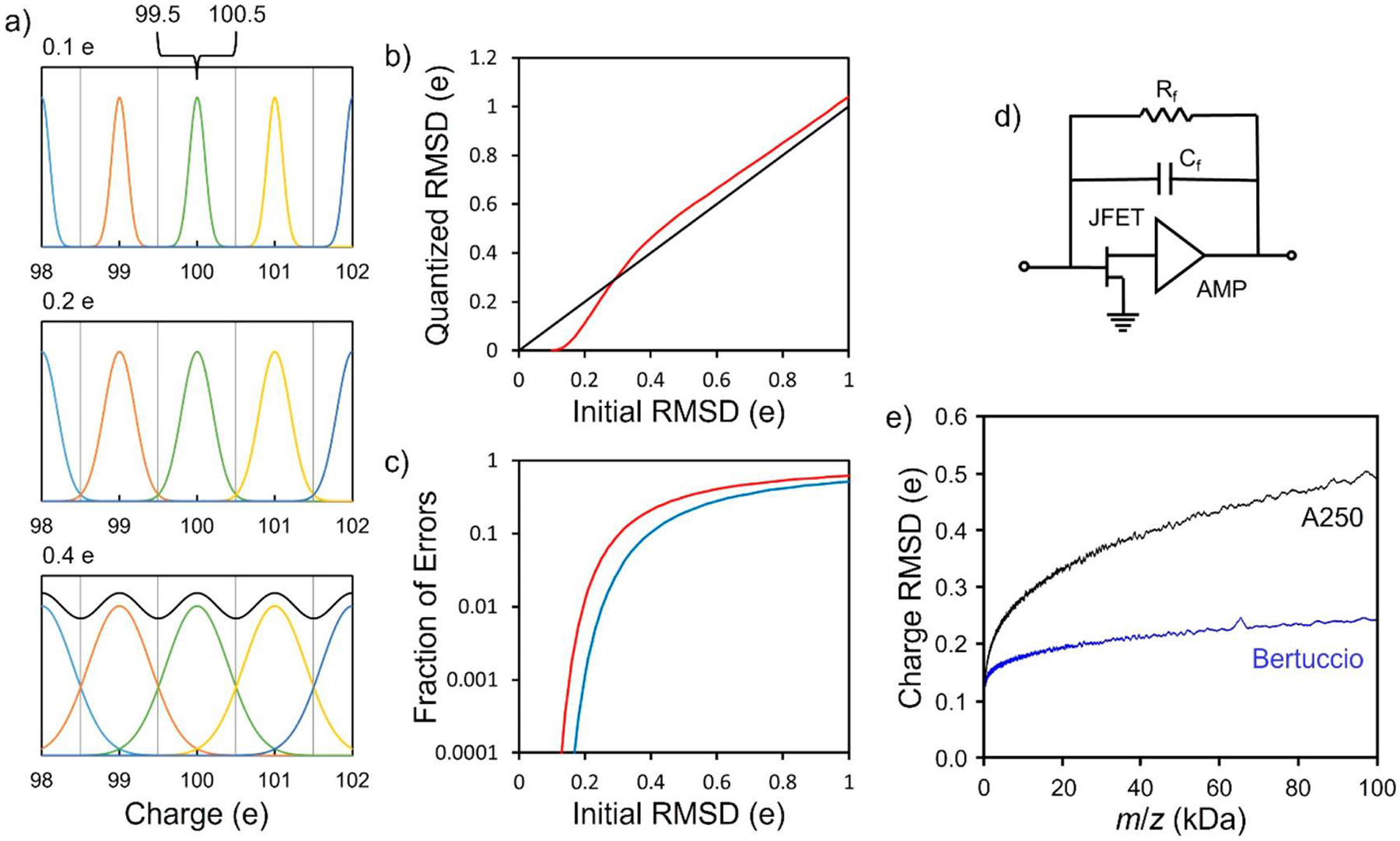
Charge-state resolution in CDMS. (a) Simulated charge spectra for three different charge RMSDs. The top spectrum has an RMSD of 0.1 e, the middle spectrum is 0.2 e, and the bottom spectrum is 0.4 e. The black line in the bottom spectrum is the sum. An RMSD of ~0.2 e is needed for good charge-state resolution. (b) RMSD after charge quantization (i.e., after assigning each ion to the nearest charge state) plotted against the RMSD of the initial distribution (red line). The black line is a guide showing the situation where the two RMSDs are equal. (c) Fraction of ions assigned to the wrong charge state plotted against the initial RMSD (red line). The blue line shows the reduction in the fraction misassigned when ions with charges near the midpoints between two integer charge states are discarded. In this case, ions within ±0.15 e of the midpoints were culled. (d) Schematic of a charge-sensitive amplifier configured for low charge signals. (e) Charge RMSD plotted against m/z. Results are shown for an Amptek A250 charge-sensitive amplifier with a 2SK152 JFET at the input (black line) and for an implementation of the Bertuccio design where the feedback resistor is removed (blue line). The input JFETs for both amplifiers were cryogenically cooled to 130–140 K, and in both cases the measurement time was 1.5 s. (b, c) Adapted with permission from ref 74. Copyright 2015 American Chemical Society. (d) Reproduced with permission from ref 72. Copyright 2020 American Chemical Society. (e) Adapted with permission from ref 72. Copyright 2020 American Chemical Society.
Figure 2d shows a schematic of a charge-sensitive amplifier configured to detect small signals. There is a JFET (junction field-effect transistor) at the input and a feedback capacitor (Cf) and feedback resistor (Rf). The uncertainty in the charge measurement in CDMS is determined primarily by electrical noise. One way to reduce the uncertainty is to average for longer, i.e., to extend the trapping time.74,75 The charge RMSD scales as (trapping time)−1/2. However, a long trapping time is undesirable because it extends the acquisition time. The trapping time can be reduced by optimizing the design of the ELIT67 and minimizing the electrical noise by improving the design and implementation of the charge-sensitive amplifier. Several steps have been taken. The input FET is cryogenically cooled to 130–140 K to reduce noise.76 To eliminate thermal noise from the feedback resistor, it was removed in a recent implementation of the Bertuccio design77 and the JFET biased by its gate-leakage current.72 Finally, the charge measurement is dynamically calibrated using an internal standard generated by irradiating the detection cylinder with a 129 kHz signal by means of a small antenna (see Figure 1b).68 With dynamic calibration, the relative charge accuracy was reduced to 5 × 10−4, allowing charge-state resolution to be achieved for single ions with up to 500 charges. Dynamic calibration locks the charge calibration into a feedback loop so that it is stable for months. A trapping time of 1.5 s is currently needed to achieve a charge uncertainty of ~0.2 e.78
Because the uncertainty in the charge measurement is determined primarily by electrical noise, the absolute uncertainty is, to first order, independent of the charge. However, the charge uncertainty does depend on the oscillation frequency (and hence the m/z ratio). As m/z increases, the oscillation frequency decreases and the 1/f noise increases, which in turn causes the uncertainty in the charge to increase. Figure 2e shows a plot of the charge RMSD as a function of m/z for an Amptek A250 with a 2SK152 JFET (a widely used commercial package) and a recent implementation of the Bertuccio design76 without a feedback resistor.71 In both cases the input FET was cryogenically cooled to 130–140 K and the measurement time was 1.5 s. The contribution from the 1/f noise is substantially smaller for the Bertuccio design than for the A250, so the increase in the charge uncertainty with m/z is relatively weak, and the Bertuccio design has a lower charge uncertainty for all m/z values.
2.2.2.5. Reducing Measurement Time with Multiple-Ion Trapping.
Another way to reduce the measurement time is to trap more than one ion at a time, an approach that has been championed by Williams and co-workers.79,80 When multiple ions with similar oscillation frequencies are trapped, they may not be resolved in the frequency spectrum. Williams and co-workers have proposed that this problem can be ameliorated by increasing the breadth of the ions energy distribution, allowing enhanced multiplexing and a substantial reduction in measurement time.80 The ion energy also affects the oscillation frequency, and so increasing the energy distribution will affect the resolution. These studies were performed with a cone trap, which is capable of trapping ions with a relatively broad range of kinetic energies. Different ion energies lead to different end-cap penetration depths, which in turn lead to different duty cycles (the ratio of the time spent in the trap to the total cycle time). A change in the duty cycle is reflected in a change in the magnitudes of the fundamental and harmonics in the FFT of the signal. Williams and co-workers used the magnitudes to deduce the ion energy, which was in turn used with the oscillation frequency to determine the m/z.81 The charge measurement also depends on the magnitudes of the peaks in the FFT.74
While this approach offers reduced data-collection times, when multiple ions are trapped, ion–ion interactions can perturb the ion energies and ion trajectories, causing the oscillation frequencies to shift, which in turn degrades the resolution.82 The ion–ion interactions scale as z2 and hence become more important for larger ions. In some cases, the ion trajectories may be sufficiently perturbed that one or more ions are ejected from the trap. The best m/z resolution is obtained with ELITs designed to have the oscillation frequency independent of the ion kinetic energy. With these traps, it is feasible to trap up to 10 ions at a time without causing a significant degradation in performance. In the future, traps could be designed to optimize performance for multiple ions.82
2.2.2.6. m/z Resolution.
As noted earlier, the m/z ratio is determined from the oscillation frequency. In addition to the ion kinetic energy, the oscillation frequency also depends on ion-entry conditions (i.e., the radial offset and angular deviation of the entering ion). Ions that enter the trap off-axis undergo Lissajous-like trajectories with frequencies that differ slightly from ions that oscillate on-axis. Distributions in the ion energy and ion-entry conditions are primarily responsible for the m/z resolution. The most recent ELIT design (Figure 1b) reduces the dependence on the ion energy, and its m/z resolving power is ~330. Substantial improvements in the m/z resolving power are anticipated with better-optimized ELIT designs where the oscillation frequency dependencies on the ion energy and ion-entry conditions are reduced simultaneously. Resolving powers of >300 000 have been obtained in simulations.
2.2.2.7. High-Resolution CDMS Measurements: An Illustrative Example.
Figure 3 shows mass and charge distributions measured for hepatitis B virus (HBV) capsid ions assembled from truncated capsid protein (Cp149) dimers (see below). The charge distributions (Figure 3b) show good resolution of the charge states. The mass distribution shows intense peaks close to the masses expected for icosahedral capsids with 90 capsid protein (Cp) dimers (T = 3 capsids) and 120 Cp dimers (T = 4 capsids), as well as smaller peaks separated by the mass of a Cp dimer.
Figure 3.
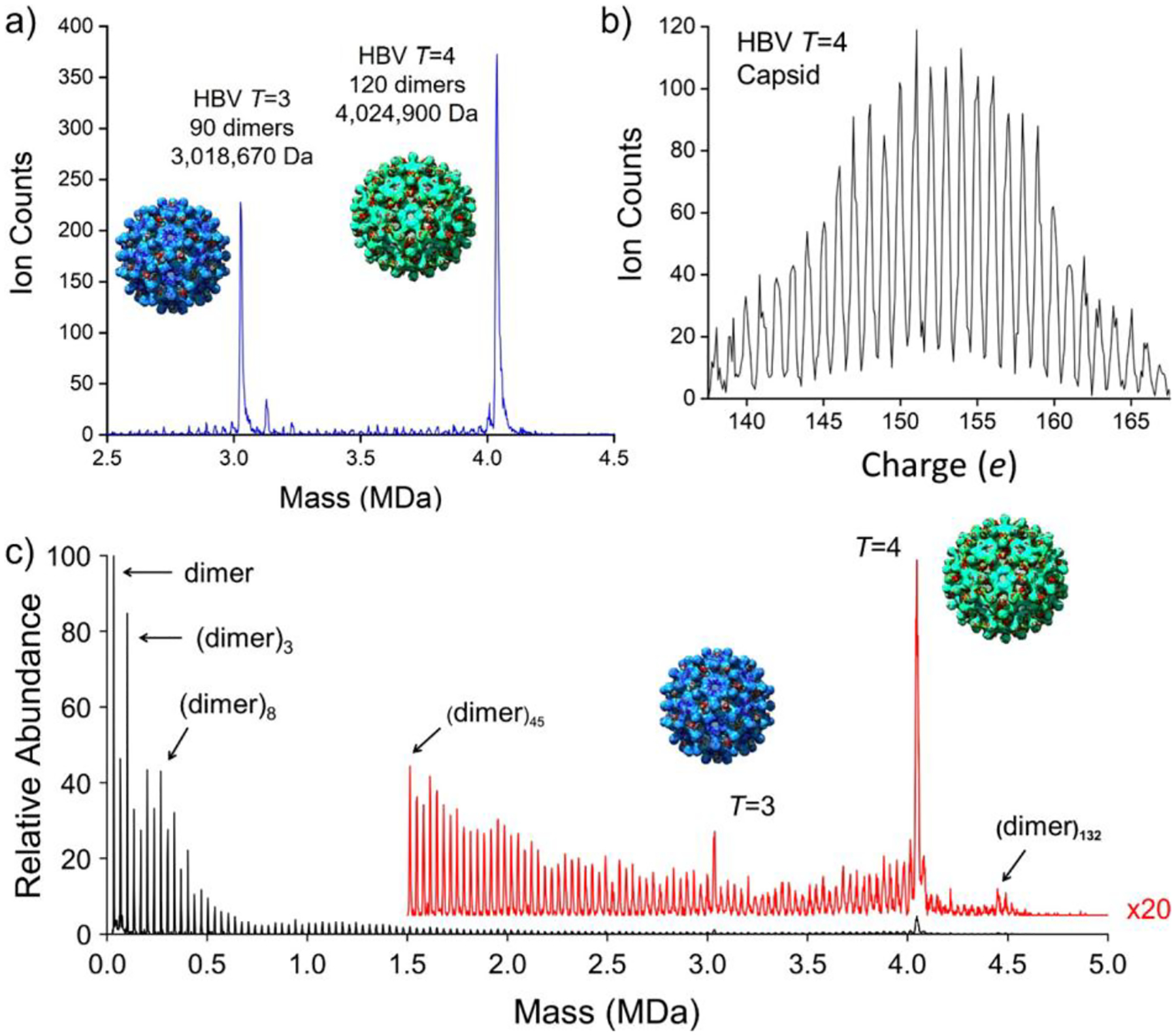
CDMS measurements for HBV capsids. (a) Mass distribution showing prominent peaks for icosahedral capsids with 90 Cp dimers (T = 3) at ~3 MDa and with 120 Cp dimers (T = 4) at ~4 MDa. The insets show the icosahedral geometries of the T = 3 and T = 4 capsids. (b) Charge spectrum measured for the T = 4 capsid showing good charge-state resolution. (c) Mass distribution for an HBV assembly reaction that stalled, leaving many trapped intermediates. (a, c) Adapted with permission from ref 78. Copyright 2020 American Chemical Society.
The measured peaks for the T = 3 and T = 4 capsids are at masses ~0.33% higher than expected from the known mass of the Cp149 dimer (33 540.8 Da). Such mass deviations are common for large protein complexes electrosprayed from volatile salt solutions.83,84 The deviation is attributed to counterions, salt adducts, and trapped solvent. Bearing in mind that the icosahedral HBV capsids are hollow and, when in solution, the inner cavity contains solvent (~1–2 × 105 water molecules), it is perhaps surprising that almost all of the solvent leaves the capsids as they are transported through the interface. On the other hand, the FUNPET interface employed here was designed to keep the ions in a buffer gas for a long time, allowing them plenty of time to desolvate.65 Furthermore, MD simulations have shown that virus capsids are quite porous.85,86 For example, according to simulations, all the water molecules inside an empty poliovirus capsid (~2 × 105) exchange in solution in ~25 μs.86
The peak widths in Figure 3a (9 375 Da fwhm for T = 3 and 13 377 Da fwhm for T = 4) are marginally larger that the values expected for the instrumental resolution, indicating that the underlying peak widths are substantially narrower. Ultimately, the peak width is limited by the isotope distribution. For a protein complex, the fwhm of the isotope distribution is given by approximately , which is ~109 Da for the T = 3 capsid and 126 Da for T = 4. Thus, with higher m/z resolving power, it would be possible to monitor small-molecule binding to viruses and other large complexes by CDMS. As described earlier, substantial improvements in the m/z resolving power are anticipated with better-optimized ELIT designs.
2.2.2.8. Deconstructing a Complex m/z Spectrum Using the Charge.
Figure 3c shows a mass distribution measured for an HBV assembly reaction that stalled, leaving behind trapped intermediates. All possible oligomers of the Cp are resolved, from the Cp dimer up to and beyond the icosahedral capsid with 120 Cp dimers (T = 4). Because of the heterogeneity of this sample, its mass distribution could not be determined by conventional MS; the m/z spectrum contains a charge-state envelope for each species, and the charge-state envelopes overlap so the spectrum is dense and uninterpretable. The m/z spectrum measured by CDMS for this sample is shown in Figure 4a. While a broad featureless m/z spectrum was anticipated, the measured spectrum contains resonances. The inset shows an expanded view of the resonance at an m/z of ~16 750. To deconstruct the m/z spectrum, Figure 4b shows a charge versus m/z scatter plot where each ion in the m/z spectrum in Figure 4a is represented by a point in Figure 4b. The points fall into tight groups. The inset shows an expanded view of the groups that have m/z values between 16 000 and 20 000 Da, and the green circle shows a further expansion of a portion of the inset. The ions are grouped because charge states are resolved in both the charge and m/z dimensions in this two-dimensional plot. Two of the groups in the Figure 4b inset are surrounded by red circles. The red circle on the right encompasses ions with m/z ≈ 16 822 and z = 76, which together yield an MW of 1.278 MDa. This corresponds to a (dimer)38 oligomer in the mass distribution in Figure 3c. The red circle on the left encompasses ions with m/z ≈ 16 604 and z = 77, which yields the same MW as the ions in the right circle (1.278 MDa). The ions in the two circles have the same nominal MW but differ by one elementary charge: they belong to the charge-state envelope of the (dimer)38 oligomer. The red lines in Figure 4b and its inset are lines of the same nominal MW—in this case, the MW of the (dimer)42 oligomer (1.412 MDa). Plotting out the intensity along these lines yields the charge-state envelope—the m/z spectrum—for this oligomer.
Figure 4.
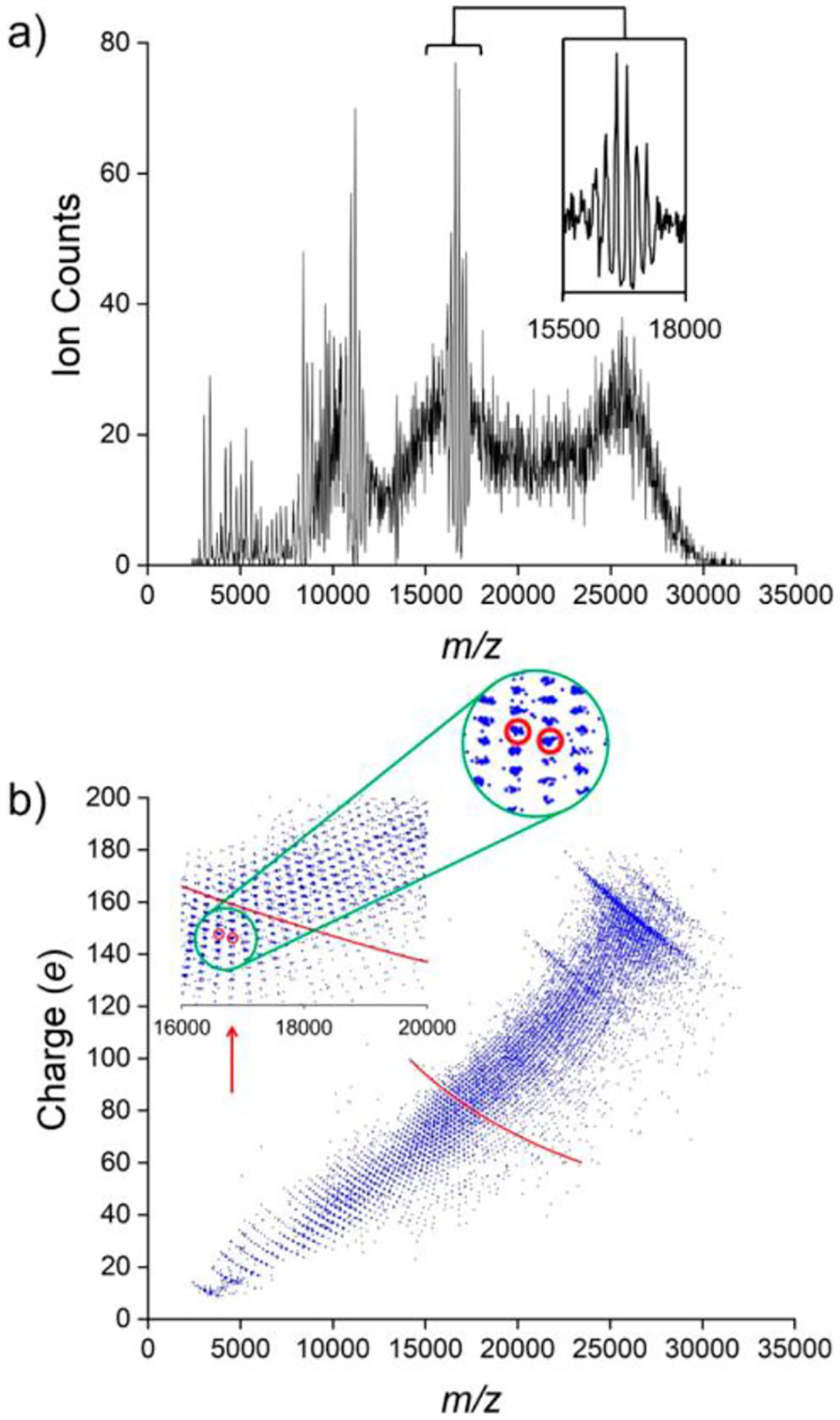
Resolution of charge states in both charge and m/z dimensions by CDMS allowing for deconstruction of the m/z spectrum. (a) m/z spectrum for the mass distribution in Figure 3c. The inset shows an expanded view of the resonance at an m/z of ~16 750 Da. (b) Charge versus m/z scatter plot for the ions in the m/z spectrum in (a). Each ion is represented by a point. The points are clustered into groups where ions with the same charge states are resolved in both the m/z and charge dimensions. The inset shows an expanded view of a portion of the scatter plot that shows the groups more clearly, and the green circle shows an expanded view of a portion of the inset. Two of the groups of ions are encompassed by red circles (see text). The red lines are lines of constant mass (1.412 MDa) that correspond to the mass of the (dimer)42 oligomer. The groups of points falling on the red line make up the m/z envelope for this oligomer mass. The red arrow in (b) shows the location in the inset of the resonance in (a) at an m/z of ~16 750. Adapted with permission from ref 78. Copyright 2020 American Chemical Society.
Finally, the charge versus m/z scatter plot provides an explanation for the resonances in the m/z spectrum. The resonances occur when the groups of ions are aligned vertically in the scatter plot. For example, the red arrow in the inset in Figure 4b points to the vertical alignment that leads to the resonance at m/z values of ~16 750. For the groups to align vertically, ions with different masses and charges must have almost identical m/z values, and this only occurs for certain combinations of oligomer size and charge.
2.2.2.9. CDMS for Light Ions: Analysis of Heterogeneous Mixtures.
While CDMS is usually viewed as a way to determine MWs for high-MW samples, it also can be used to measure MW distributions for lighter ions (<1 MDa). As noted earlier, the uncertainty in the charge determination improves slightly as the m/z decreases (and the oscillation frequency increases). Individual ions with a single charge have been detected by ion-trap CDMS.72 Angiotensin II, with an MW of 1046.2 Da, is the lightest ion that has been detected so far.72 For light ions, charge states can easily be resolved in the m/z spectrum. Ions associated with the different m/z charge states can be isolated, and then the average charges can be determined for the m/z separated ions from the charge measurements. A comparison of the average measured charges with the m/z charge states has been used to calibrate the charge.76 For low-heterogeneity samples of low-mass molecules, the relatively long measurement time of CDMS means that it is not competitive with ensemble measurements performed by conventional MS. However, CDMS can measure mass distributions for heterogeneous mixtures of lower-mass ions that are challenging to analyze by conventional MS and MS hyphenated with a separation technique, such as LC-MS. Some examples include polymers, micelles, nanoparticles, and proteins that have been extensively modified post-translation, as well as other heterogeneous mixtures of molecules that cannot be resolved by chromatography. The extensive sample cleanup that is necessary for conventional MS so that charge states can be resolved is not necessary for CDMS. For example, the analysis of serum and cell lysates with minimal sample processing is feasible.87
2.2.3. Single-Ion Mass Measurements by FTICR and an Orbitrap.
As described earlier, Smith and co-workers used FTICR to measure the masses of single high-mass ions using a charge-stepping approach where the mass was determined from multiple measurements of an ion’s m/z in different charge states.36–38 In subsequent work, the same group employed FTICR to perform a direct measurement of the charge.88,89 An FTICR cell is essentially a rectangular or cylindrical box with orthogonal pairs of excitation and detection electrodes arranged along the magnetic field axis. Excited ions undergo cyclotron motion perpendicular to the field axis and induce a charge on the detection electrodes that is detected by an amplifier.
In CDMS, the ion passes through a detection cylinder, and if the cylinder is long enough, the induced charge equals the charge on the ion. The induced charge does not depend on the ion’s trajectory through the cylinder. In an FTICR cell, the detection electrodes do not surround the ion, and hence, only a fraction of the ion’s charge is induced on them; the balance is induced on the excitation electrodes and the end-caps. The size of the induced charge depends on the distance between the electrode and the ion, which in turn depends on the radius of the cyclotron orbit, which is difficult to estimate. Chen and co-workers determined the charge by exciting each ion to a larger and larger cyclotron orbit until it was lost.88 For very large ions, where collisional relaxation was ineffective, a series of measurements were performed following random excitation or deexcitation of the ion.89 In both cases, the charge could be determined with an accuracy of ~10%. This approach was used to determine the mass of bacteriophage T4 DNA ions. Masses, ranging from 90.9 to 128.6 MDa, were reported for 6 ions. The sequence mass for the 168 kbp dsDNA T4 genome is 104.3 MDa. The small number of ions studied here speaks to the difficulty of the measurements.
There has recently been considerable excitement around single-ion measurement on an Orbitrap. An Orbitrap is an electrostatic ion trap where the ion circulates around a spindle electrode while it oscillates back and forth along the length of the spindle.90 The oscillation frequency depends on the ion’s m/z ratio, but it is relatively independent of the ion energy. The induced charge is picked up on two outer electrodes. Measurements are usually performed on packets of ions. Single-ion measurements were first performed by Makarov and Denisov more than a decade ago; they were able to detect single myoglobin ions with 20 charges.91 Only a single m/z measurement was made for each ion, so it was not possible to determine the mass without prior knowledge of the charge state. Interest in single-ion measurements has now been rekindled. Kelleher and co-workers recently reported that the analysis of single ions in an Orbitrap can lead to an increase in the m/z resolving power.92,93 For a packet of ions, field imperfections, either from space charge effects or geometric imperfections, cause dephasing, which reduces the resolving power. The dephasing is avoided for single ions, allowing an improvement in the m/z resolving power. This was first demonstrated by Smith and co-workers in FTICR measurements for single ions.94
Kelleher and Heck and their collaborators have now taken this work one step further and performed charge measurements for single ions using an Orbitrap mass spectrometer.95,96 This approach has been called individual-ion mass spectrometry or I2MS. For an ion trapped in an Orbitrap, only a fraction of its charge is induced on the detection electrodes, with most of the balance being induced on the center spindle. Thus, the size of the induced charge depends on the ion’s trajectory in the trap and specifically how close the ion’s path is to the detection electrodes. The same issue was discussed earlier for direct charge measurements using FTICR. For an Orbitrap, it appears that the uncertainty in the charge measurement is ~4% for a 2 s trapping time. Note that the uncertainty is expected to scale with the charge because it depends on the trajectory of the ion as well as amplifier noise. For heterogeneous samples, this relatively high charge uncertainty compromises the mass resolution. However, for less-heterogeneous samples where charge states are still resolved in the m/z spectrum, the average charge can be determined for ions in an m/z peak and used to assign the charge state to the peak (if there are enough ions in the peak that the average charge can be determined with enough precision). A similar approach has been used in CDMS to calibrate the charges.76 Orbitrap I2MS is a relatively recent development, and only a few demonstration results have been published so far.95,96
3. APPLICATIONS OF CDMS TO MOLECULAR BIOLOGY AND BIOTECHNOLOGY
CDMS can be used to analyze a wide range of heterogeneous and high-mass samples. While not the main focus of this Review, a considerable amount of work has also been done on nanoparticles,97,98 on polymers,99–108 and in the charging of, and charge separation in, droplets.62,109–115 The main focus here is on applications relevant to molecular biology and biotechnology, a number of which are discussed below.
3.1. DNA
In solution, DNA is negatively charged because at least some of the backbone phosphate groups are ionized. When electrosprayed, DNA acquires a substantial number of counterions that introduce heterogeneity and make it impossible to measure the mass of large DNA molecules by conventional electrospray MS. Benner and co-workers used CDMS to analyze single-stranded (ss) and double-stranded (ds) circular DNA in the range of 5.0–15.0 kb (kilobase).53 Electrospray can be used to generate both positive and negative ions, although negative-mode electrospray is considerably more difficult to perform because it is more susceptible to electric discharge. Negative-mode electrospray of plasmid pBR322 was found to result in a peak at close to the sequence (2.695 MDa).53 However, the charge distribution showed two peaks at around 980 and 2 220 e (elementary charges) that could be due to different three-dimensional structures. Large ions generated by electrospray are thought to be produced by the charge-residue mechanism,116,117 where a water droplet deposits its charge on the analyte as it evaporates away. The maximum charge deposited on a spherical ion can be predicted from the Rayleigh charge limit for a water droplet118 with the same diameter as the analyte. When assuming that a spherical ion has the same density as water, the Rayleigh charge limit is given by zR = 0.0778 m1/2.10 For a spherical ion with a mass of 2.7 MDa, the charge is expected to be ~130 e, which is much lower than those for both charge populations (980 and 2220 e) found in the experiments, indicating that for both populations the ions are far from spherical. The more highly charged population has roughly one net negative charge for every two base pairs.
For positively charged DNA ions, enough charge must be added during the electrospray process to neutralize the negative charge of the DNA in solution and provide a net positive charge. Ions generated by the positive-mode electrospray of pBR322 showed a single population with a charge of ~760 e. pBR322 is 4 361 bp. If all the backbone phosphate groups are ionized, the number of counterions should be (2 × 4 362) + 760 (for the charge) = 9 484. If the counterions are Na+, this would add 209 kDa to the mass of the positive ion, and for NH4+, 161 kDa would be added. The mass measured for the positively charged pBR322 ions is 2.76 MDa, which is 65 kDa (2.4%) higher than the sequence (2.695 MDa). An excess mass of 65 kDa is smaller than that anticipated, which suggests that many of the backbone phosphate groups are un-ionized.
Antoine and co-workers have reported several studies using CDMS to investigate the multiphoton dissociation of DNA ions. In these studies, the ions were trapped in an ELIT and irradiated with a CO2 laser.119–121 In principle, this approach allows one to follow the fragmentation pathways at a single-molecule level. Three different processes were identified: sudden loss of the signal due to the trapped ion, a gradual loss of signal, and a multistep process where the signal disappears in steps. Finally, Antoine and co-workers have recently used CDMS to measure the MW distribution of calf thymus DNA.122 They found two broad distributions centered around 3 and 10 MDa. The lower mass component was thought to be degradation products.
3.2. Glycosylated Proteins
Glycosylation is one of the most common forms of protein post-translational modification, and around half of cellular proteins are glycosylated. Glycosylation affects protein structural stability and plays an important role in recognition. Hence, it plays a central role in almost all biological processes. Glycosylation increases diversity in the proteome. At any given glycosylation site, a variety of glycans can be attached, and when there are multiple glycosylation sites, the number of different glycoforms increases exponentially. For example, if a protein has 20 glycosylation sites that can be randomly populated with 3 different glycans, the number of glycoforms is 320 or 3.5 × 109. Protein glycosylation is usually studied using a bottom-up approach where the glycoprotein is enzymatically digested and the fragments are analyzed by LC-MS.123 Using this approach, it is possible to determine the glycan population at specific sites within the protein. However, it is not possible to measure the MW distribution of the intact glycoprotein by conventional MS if there are a significant number of glycosylation sites because of heterogeneity. For example, the spike protein trimer of SARS-CoV-2 has 66 N-glycosylation sites (22 per spike protein). SARS-CoV-2 is responsible for the COVID-19 pandemic that emerged in 2020.124–128 Trimers of the spike protein decorate the virus surface and are the primary target for vaccine development.129,130 The glycans provide a shield that frustrates the immune response. Bottom-up glycoproteomics studies have been used to determine the glycan composition at the N-glycan sites; high mannose, hybrid, and complex glycans were found.131,132 The blue line in Figure 5 shows the mass distribution measured for the spike protein trimer by CDMS.133 The orange inset is the measured charge distribution, which shows good resolution of the charge states (RMSD = 0.191 e). The gray line shows the mass distribution measured for β-galactosidase under identical conditions. The measured mass (467.6 kDa) is <0.5% larger than the expected mass (465.4 kDa). The difference, attributed to counterions, salt adducts, and trapped solvent, is in line with deviations usually found for protein complexes electrosprayed from volatile salt solutions. The dashed vertical line indicates the expected mass of unglycosylated spike protein trimer (414.2 kDa) calculated from its sequence. The red line shows the mass distribution expected for the N-glycosylated trimer assuming the glycoforms are populated randomly according to the glycan distribution determined in the glycoproteomics studies.131 This means that glycans populating each site are unaffected by the glycans populating other sites (i.e., the sites are uncorrelated). The average glycoprotein mass determined from the CDMS measurement of the intact mass (145.3 kDa) is substantially larger than the average from the bottom-up glycoproteomics studies (107.5 kDa). One possible explanation for this difference is that the glycoproteomics studies underestimate the abundances of larger, more complex glycoforms. The measured glycan distribution is also much broader than expected for a random glycan distribution. It has been reported that the S protein is mannosylated before it assembles into trimers in the ER and acquires complex N-glycans in the Golgi.134 The broad glycan mass distribution observed in the experiments suggests that glycan processing in the Golgi is correlated, so that, for some trimers, many of the glycan sites are lightly processed, while for others, many are heavily processed. A random distribution would lead to a narrower mass distribution.
Figure 5.
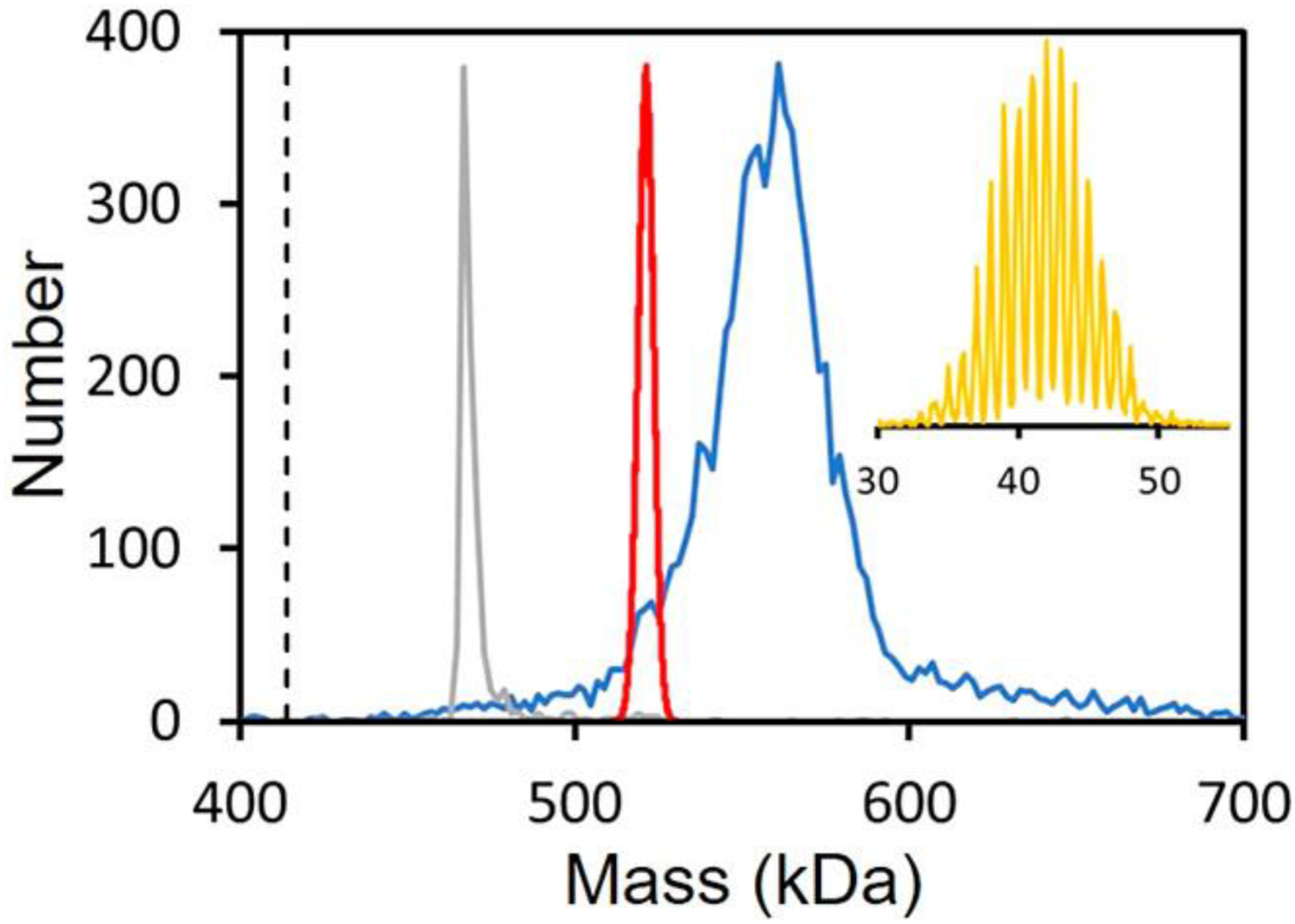
CDMS spectrum measured for the heavily glycosylated SARS-CoV-2 S protein trimer (blue line). The gray line shows the mass distribution measured for β-galactosidase under identical conditions. The dashed vertical line shows the sequence mass of the unglycosylated trimer (414.2 kDa). The red line shows the mass distribution predicted for a statistical distribution using the glycan populations determined from glycoproteomic measurements (see text). The orange inset shows the measured charge distribution, which shows good resolution of the charge states (RMSD = 0.191 e). Adapted with permission from ref 133. Copyright 2021 American Chemical Society.
3.3. Protein Complexes
A number of CDMS measurements have been performed for protein complexes with masses in the half megadalton range, such as pyruvate kinase,135 β-galactosidase,67,133 and RuBis-CO.80,79 However, these studies were directed at using the complexes to characterize CDMS rather than learning new information about the complexes. Accurate MWs determined by CDMS can provide information about the stoichiometry of protein complexes. A good illustration is provided by studies performed on the yeast nuclear pore complex (NPC).136 The NPC mediates transport between the cytoplasm and the nucleus and consequently plays an important regulatory role. The dynamic nature of the NPC makes it difficult to characterize. The structure was deduced through a combination of cross-linking studies using MS and cryogenic electron microscopy (cryo-EM) measurements. The NPC contains ~550 copies of ~30 different proteins. The expected mass of the native NPC is 52 MDa, which increases to ~87 MDa when the membrane, cargo, and nuclear transport factors are included. The CDMS mass distribution showed a broad peak centered on ~80 MDa, which confirmed the overall stoichiometry of the NPC.136
3.4. Protein Aggregation
Protein aggregation is associated with several diseases,137 and it is a significant issue with high-MW therapeutics such as antibody formulations. IgG oligomers up to the hexamer and IgM oligomers containing 4–6 monomers have been resolved using Orbitrap I2MS.96 Pyruvate kinase (PK) oligomers containing up to 10 PK tetramers have been detected by CDMS.135 It is usually possible to track the early stages of protein aggregation using conventional MS,138–140 and the end point can be determined using a variety of particle-analysis methods such as dynamic light scattering or multiangle light scattering. CDMS offers the advantage of being able to cover a size range from monomers to aggregates with MWs into the gigadalton range. Thus, it should be possible to track aggregation over the entire size range of interest using CDMS.
MW distributions have been measured for amyloid fibers from several different sources by single-pass CDMS.141–143 In the case of fibers generated from α-lactalbumin,141 the average MW of the fibers was found to be 395 MDa. The average length of the fibers from EM images was 2.01 μm. Both the MW distributions and the length distributions were log-normal. Polydispersity indices for both distributions were similar (1.62 for mass and 1.65 for length), suggesting that the main feature driving the MW distribution was the length distribution and not heterogeneity in the number of filaments in each fiber. The average charge on the fibers is ~1 180 e. This is almost the same as the charge found for adenovirus, which has an MW of ~150 MDa and a roughly spherical shape with diameter of ~165 nm (see below). It is surprising that the much larger fibers have such a low charge relative to that of adenovirus. This suggests that the fibers are wrapped up tightly during the electrospray process; a much higher charge would be expected for an elongated structure.
Assembly of HET-s (a fungal prion protein) yielded such long fibers that it was difficult to determine the length distribution from atomic force microscopy (AFM) and EM images.141 The mean mass determined by CDMS was 112 MDa. The mass distribution was again log-normal, although the polydispersity index (1.19) was smaller than that found for α-lactalbumin fibers (see earlier). In the AFM and EM images, the fibers had a uniform diameter of ~3 nm. This value for the diameter, an assumed density, and the average mass provided an estimate of the average length of the fibers of 12.2 μm. The average charge carried by the fibers was 650 e, which was around half the value for the α-lactalbumin fibers, while the HET-s fibers were ~6 times longer. The low charge was not consistent with an elongated conformation in the gas phase.
For fibers generated from two polypeptides involved in neurodegenerative diseases, Aβ1–42 and tau, two coexisting species were detected in the CDMS measurements.143 Results for the tau amyloid fibers are shown in Figure 6. Two populations are evident in the CDMS mass distribution (Figure 6a) and charge versus mass heat map (Figure 6b). The low-mass population was attributed to the spherical oligomers indicated by the white arrows in the EM image (Figure 6c). The low-mass population was ~5% of the total. In the charge versus mass plot, the charge increases much more rapidly with mass for the lower-mass population than for the higher-mass one (see dashed lines in Figure 6b).
Figure 6.
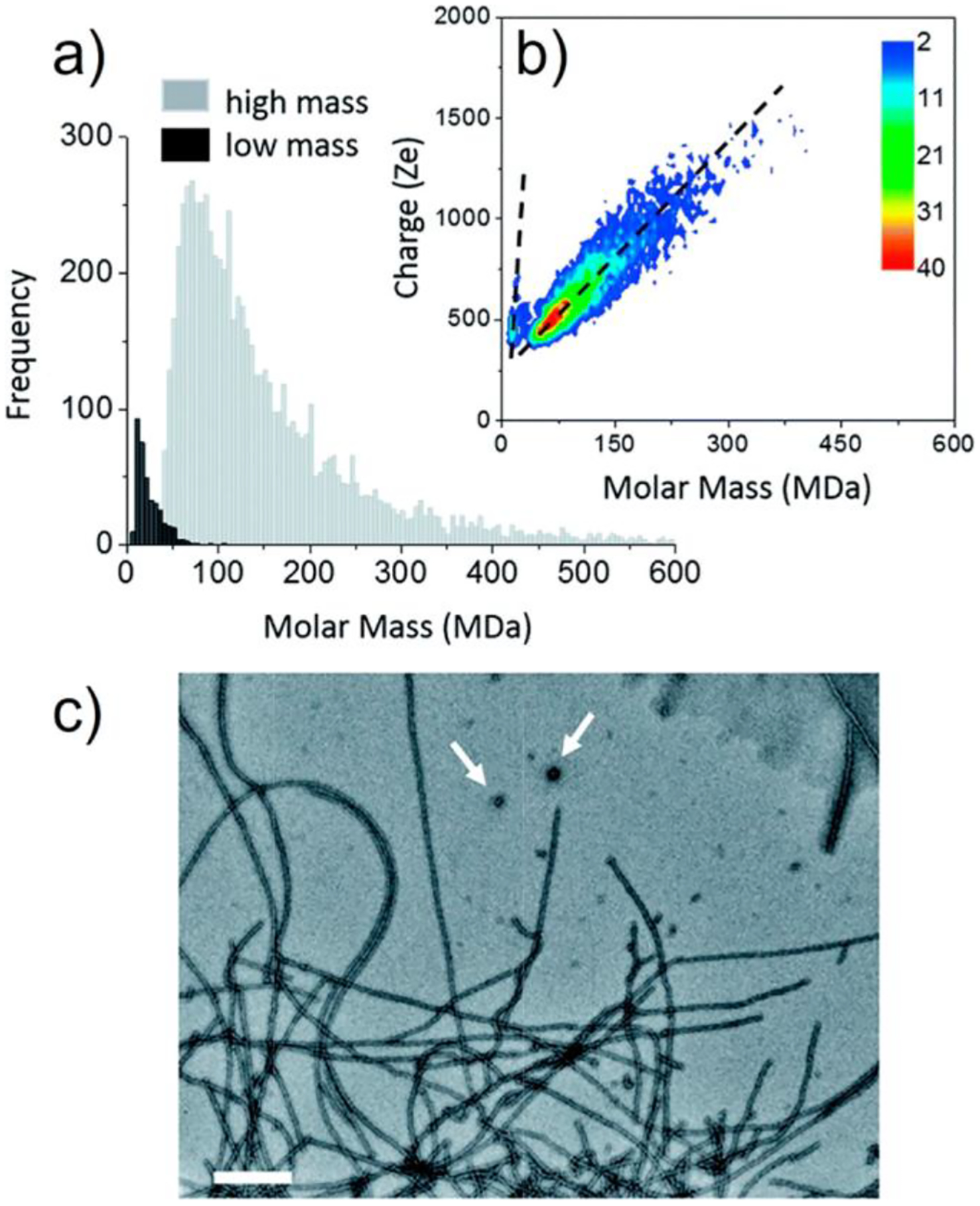
Tau amyloid fibers. (a) CDMS mass distribution for tau amyloid fibers plotted using 5 MDa bins. Low-mass and high-mass populations are distinguished by dark and light shading, respectively. (b) Charge versus mass heat map for tau amyloid fibers. The dashed lines are guides indicating how the charge depends on mass for the two populations. (c) Transmission electron microscopy (TEM) image of tau amyloid fibers. Scale bar: 200 nm. Features assigned to spherical oligomers are shown with white arrows. Adapted with permission from ref 143. Copyright 2018 Royal Society of Chemistry under Creative Commons Attribution 3.0 Unported License (CC By 3.0), https://creativecommons.org/licenses/by/3.0/legalcode.
3.5. Viruses
A capsid, the protein shell that surrounds the genetic material of a virus, is assembled from many, often hundreds, capsid proteins. Around half of known virus families have icosahedral capsids. The structures of icosahedral capsids are defined by a triangulation number (T) where 60T is the number of capsid proteins present.144 The capsid proteins are arranged into 12 pentamers plus 10(T – 1) hexamers. The allowed T values can be obtained from T = h2 + hk + k2, where h and k are positive integers. The first few allowed T values are 1, 3, 4, 7, 9, 12, etc. The T = 3 capsid has the same basic geometry as a soccer ball, with the pentagons separated by a single hexagon. Some viruses are enveloped, where the capsid is encased in a lipid membrane derived from the host cells. In addition to providing a remarkable example of self-assembly, there is also the possibility of exploiting capsid assembly to develop novel viromimetic particles that could act as nanocontainers and templates.145,146
3.5.1. Hepatitis B Virus Capsid Assembly.
Hepatitis B virus (HBV) has emerged as a model system to study virus capsid assembly. It is also a destructive pathogen; worldwide, ~250 million people suffer from chronic HBV infection and ~900 000 die annually. The virion consists of an outer lipid envelope with an icosahedral nucleocapsid core. The assembly of the HBV nucleocapsid is an attractive target for the development of assembly-directed antivirals,147–151 making information about HBV capsid assembly particularly pertinent. HBV self-assembly is usually studied with the core protein assembly domain (Cp149), a 149-residue protein that lacks the arginine-rich, C-terminal DNA-binding domain. The building block is the Cp149 dimer, and it spontaneously assembles into icosahedral capsids with 90 dimers (in a T = 3 structure) and 120 dimers (in a T = 4 structure).152,153 The assembly reaction, which can be triggered by raising the ionic strength, is thought to occur through a series of coupled equilibria, with each one corresponding to the addition of a dimer in a bimolecular reaction.154–156 The equilibrium constant for the overall reaction,
| (3) |
for the T = 4 capsid is
| (4) |
The extraordinarily high dependence on the dimer concentration leads to pseudocritical behavior if we define the total dimer concentration as the sum of the free dimer and the dimer in capsids. For total dimer concentrations below the pseudocritical concentration, the amount of capsid present is vanishingly small; for those above the pseudocritical concentration, the capsid concentration increases linearly, while the dimer concentration remains fixed at the pseudocritical value.156 Reversibility during assembly is thought to be critical because it provides a mechanism for error correction.157 Accelerating assembly provides a way to overwhelm self-correction, leading to defective capsids. The concentrations of intermediates are expected to be low during assembly.156 CDMS has been used to investigate HBV assembly in several studies. In the first study,158 the assembly reaction was performed in sodium chloride, and then the products were dialyzed into ammonium acetate solution before electrospray. Kinetically trapped, late-assembly intermediates were observed. While the mass resolution used in these early studies was insufficient to resolve specific intermediates, a prominent feature was observed with ~104–105 dimers (i.e., missing 15–16 dimers). Cryo-EM studies performed in tandem suggested that the trapped intermediates were incomplete capsids with a single hole due to the missing dimers.158
By performing the assembly reactions in ammonium acetate, the dialysis step could be avoided, and the assembly reactions could be monitored in real time.159,160 Examples of time-resolved CDMS spectra are shown in Figure 7. With a dimer concentration of 5 μM and a salt concentration of 210 mM, the assembly reaction is slow enough that the T = 4 capsid grows in during the 90 min time period that was monitored (Figure 7a). Increasing the dimer concentration to 10 μM causes the reaction rate to increase so that the bulk of the assembly occurs in the first-time window (Figure 7b). Note that the concentration of the intermediates is relatively low for assembly in 210 mM ammonium acetate. However, when the salt concentration is increased to 510 mM (Figure 7c), trapped intermediates are observed between 3 and 4 MDa. These intermediates are thought to be off-pathway—the result of assembly errors that prevent completion of an icosahedral structure and require partial disassembly to correct. The higher salt concentration strengthens dimer–dimer interactions and reduces the rate of disassembly. Note that the intermediates first appear around 3 MDa, close to the expected mass of the T = 3 capsid with 90 dimers. An intermediate with a mass of ~3 MDa is also observed in the disassembly of T = 4 capsids. However, the 3 MDa intermediate is not thought to be the T = 3 capsid. Instead, it was proposed that the intermediates result from hole collapse,161 where the hole in an intermediate on the road to a T = 4 capsid closes up to form a strained nonspherical shell. Hole collapse is favorable if the energy gained by the additional dimer–dimer contacts exceeds the strain energy for distorting to a closed structure.
Figure 7.
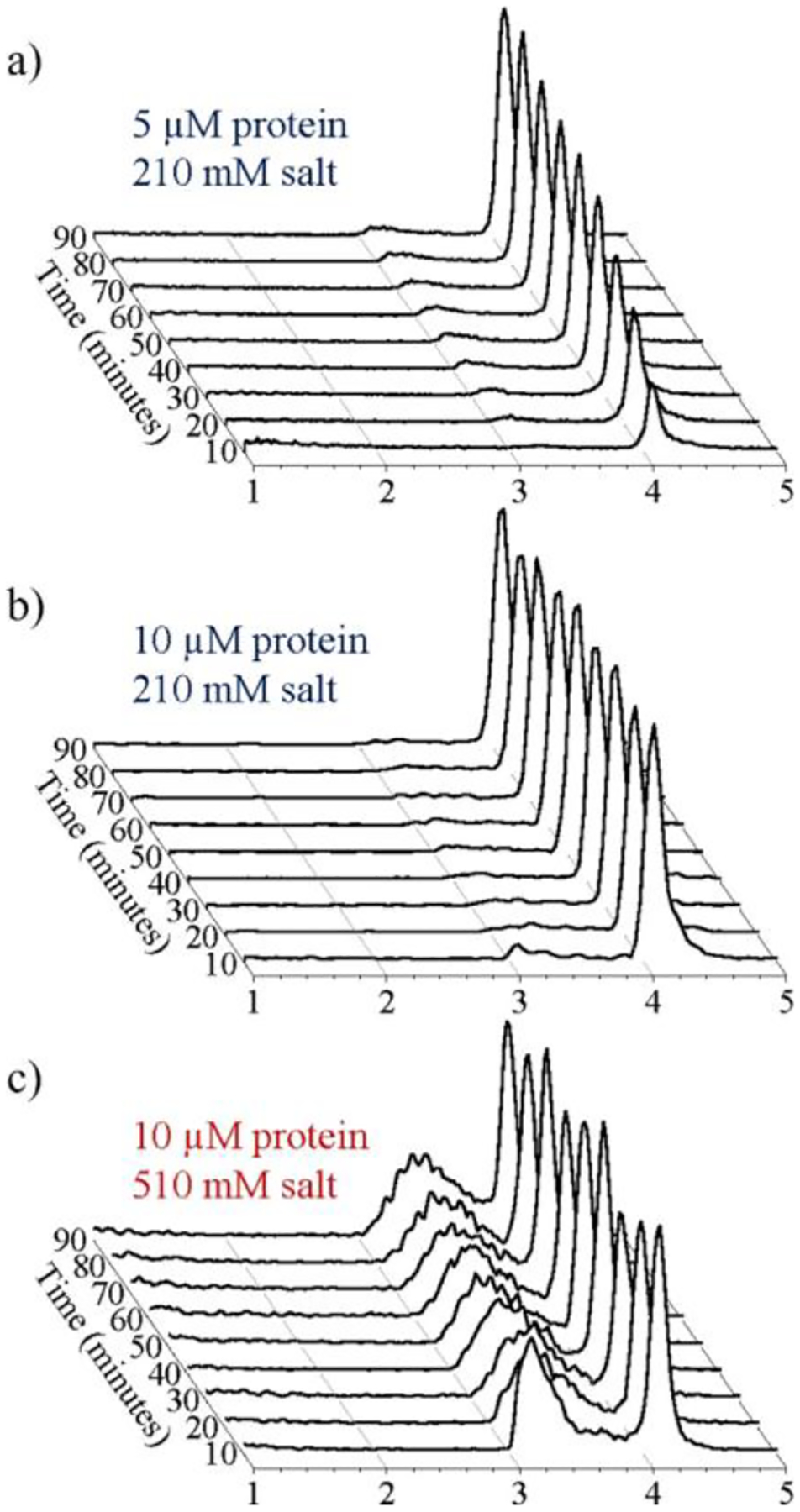
Time-resolved CDMS spectra showing the progression of HBV capsid assembly over the first 90 min: (a) for a reaction mixture containing an initial dimer concentration of 5 μM in 210 mM ammonium acetate; (b) for a 10 μM dimer in 210 mM ammonium acetate; and (c) for a 10 μM dimer in 510 mM ammonium acetate. The earliest time interval (0–10 min) is at the front. The spectra were generated using 20 kDa bins. Each spectrum is normalized by the sum of all ions measured for each time point. Adapted with permission from ref 159. Copyright 2017 American Chemical Society.
The final step in the assembly reaction is thought to be insertion of the last capsid building block to complete the icosahedron. It is accepted that the rate of subunit addition should decrease as the capsid nears completion,162 and it has been suggested that the stiffening of the capsid that occurs on completion could even make completion thermodynamically unfavorable.163 CDMS studies of HBV T = 4 capsid assembly have shown that some of the capsids formed in the initial assembly reaction have slightly more than the expected number of dimers (120). The overgrown capsids slowly relax back to the mass expected for the perfect T = 4 icosahedron.159 This provides a proof-reading mechanism where the accumulation of a few imperfections during assembly leads to defective and overgrown particles that are more labile than perfect capsids and subsequently self-correct over time.
Woodchuck hepatitis virus (WHV) has been used as a model for HBV infection in humans. Sequence alignments of the WHV and HBV assembly domains show 65% identity. The T = 4 capsids are structurally similar; however, T = 3 capsids are much less abundant for WHV than for HBV.164 There have been several studies of WHV assembly that have utilized CDMS.164,165 While overgrowth and self-correction were observed for HBV, for WHV the overgrowth was much more extensive and mostly does not self-correct to the T = 4 icosahedron. In particular, there are prominent peaks for capsids with ~132 dimers and 150 dimers.165 The peak at 150 dimers can be attributed to elongation of a T = 4 capsid along its 5-fold axis by the addition of a ring of hexamers. Such a geometry has precedent; prolate capsids such at bacteriophage T4 and rice tungro bacilliform virus have capsids that are elongated along the 5-fold axis in discrete steps of hexamers.166–172
During infection, mature cores (the HBV T = 4 capsid with a packaged dsDNA genome) are transported to the nucleus through the nuclear pore complex. Transport is mediated by a nuclear transport receptor (a complex of importin α and importin β) that binds to the C-terminal domain (CTD) of the HBV capsid protein.173 The CTD is a basic, 34-residue, nucleic acid binding domain that is localized to the capsid interior in mature cores but has at least transient external exposure. While binding between the nuclear transport receptor and the mature cores occurs through importin α, CDMS studies indicate that importin β can also bind directly to empty capsids.174 Cryo-EM image reconstruction show that the C-terminal domains of empty capsids can extrude through holes in the capsid surface near the quasi-6-fold vertices. The importin β density in the vicinity suggests a maximum of 30 importin β per capsid. However, CDMS measurements indicate that up to 90 importin β molecules bind to some capsids. Cryo-EM images for HBV incubated in excess importin β show that importin β had been internalized, suggesting that the capsids transiently open and close, possibly because they are destabilized by importin β.
3.5.2. Brome Mosaic Virus Assembly and Disassembly.
The viral genome plays an active role in the assembly of small, nonenveloped, icosahedral RNA viruses.17,162,175,176 It recruits capsid proteins to form a complex that self-organizes to give a protective protein shell around the genome. The capsid proteins can also self-assemble around nongenomic cargo (including nanoparticles), forming capsids identical to those formed around genomic RNA.177 In the host-cell cytoplasm, assembly leads overwhelmingly to the packaging of viral RNA, even though there are many other polyanionic species present. Brome mosaic virus (BMV) assembly around the wt (wild-type) genome leads to an icosahedral capsid with T = 3 symmetry. BMV is a plant virus that has emerged as a useful model for ssRNA virus replication.178 Studies of the assembly of brome mosaic virus capsid proteins around ssDNA oligomers using a combination of CDMS and cryo-EM showed that assembly resulted in protein shells with nonicosahedral structures.179
Figure 8 shows the CDMS spectrum measured for BMV capsid proteins assembled with 52 nt polyadenine oligonucleotides. There are prominent peaks at 2.5, 3.0, and 3.4 MDa. An empty T = 3 capsid has an expected mass of 3.65 MDa. The N-terminus of the BMV capsid protein has a basic arginine-rich motif with 9 positive charges that interact with RNA.180 For a ratio of ~1 nucleotide/coat protein arginine,181 a T = 3 particle mass of ~4.15 MDa is expected. A ratio closer to 1.6 is usually observed with wt viruses.182 EM studies showed particles with two distinct sizes (22 and 25 nm) with the smaller particles being dominant. The smaller particles have previously been attributed to pseudo-T = 2 symmetry.183–185 However, this would lead to an expected mass of at least 2.75 MDa, considerably higher than the most prominent peak in the CDMS mass spectrum at ~2.5 MDa (Figure 8). The only peak that could be due to a pseudo-T = 2 particle is the one at ~3.0 MDa, leaving the peak at ~2.5 MDa unassigned. Structural characterization of cryo-EM images using unbiased 2D classification revealed that many of the 22 and 25 nm particles showed 4-fold rotational symmetry, indicating a structural organization that is not icosahedral. Further analysis revealed the geometries labeled H8 and H15 in the insets to Figure 8. H8 is a D6 hexagonal barrel containing 12 pentameric and 8 hexameric capsomers. It contains 108 capsid proteins and has a total protein mass of 2.19 MDa. Nineteen polyadenine oligonucleotides are needed to attain a nucleotide/arginine ratio of ~1, which gives a total mass of 2.5 MDa, in good agreement with the main peak in the CDMS spectrum (Figure 8). H15 has a geometry consisting of 12 pentameric and 15 hexameric capsomeres. It has 150 capsid proteins and a protein mass of 3.05 MDa. Twenty-six oligomers are needed to achieve a nucleotide/arginine ratio of ~1, leading to a total mass of 3.46 MDa, in good agreement with the CDMS peak at 3.4 MDa (see Figure 8). As noted earlier, the larger (25 nm) particles are in the minority and the smaller (22 nm) particles were dominant in the EM images, which is consistent with the relative abundances in Figure 8.
Figure 8.
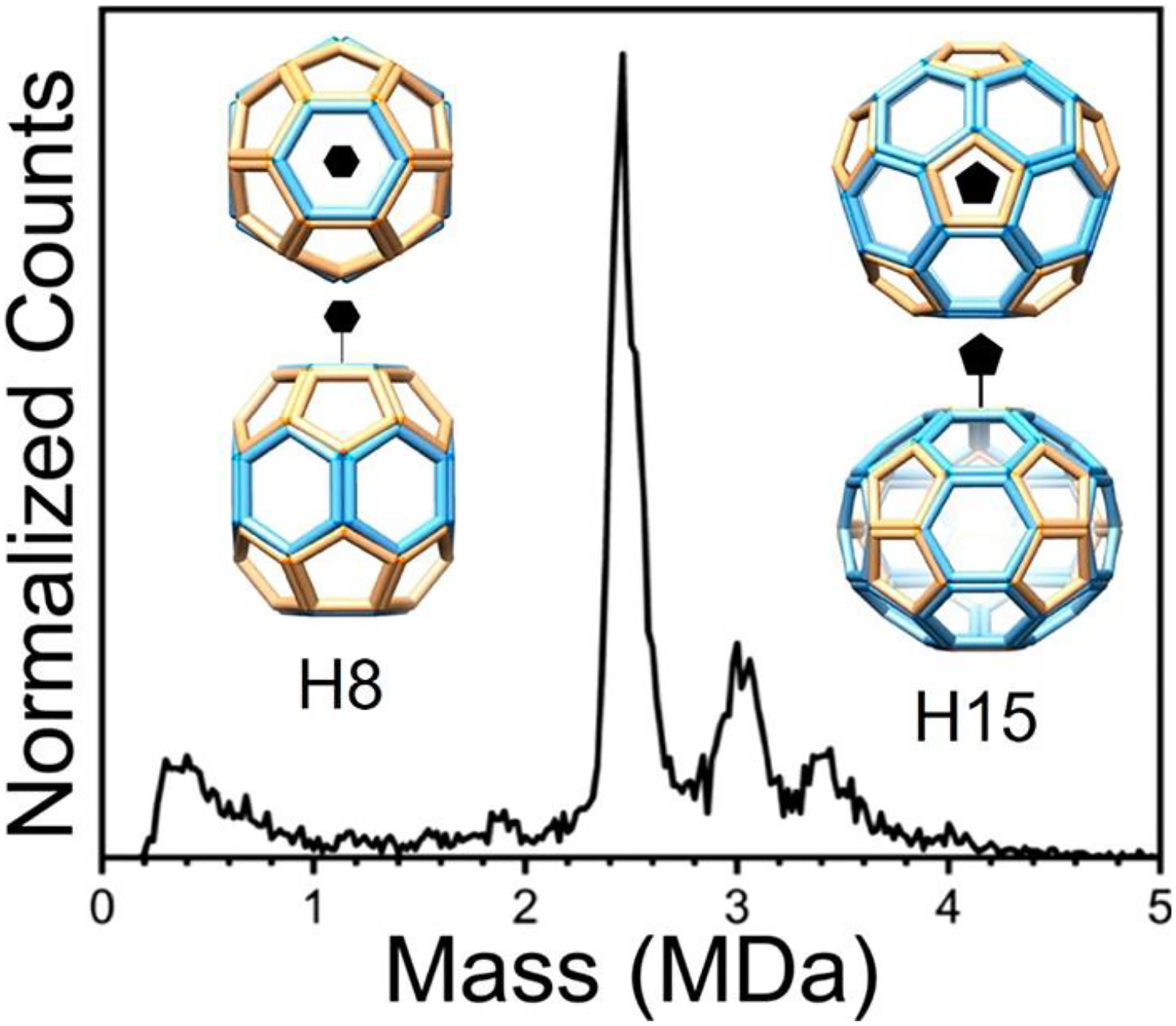
CDMS spectrum measured for BMV capsid proteins assembled with 52 nt polyadenine oligonucleotides. The insets show the H8 and H15 geometries determined from the CDMS and cryo-EM studies. The peak at 2.5 MDa is assigned to the H8 structure, and the one at 3.4 MDa is assigned to the H15. Adapted with permission from ref 179. Copyright 2020 John Wiley and Sons.
Capsid disassembly and genome release are critical steps in the lifecycle of a virus. The capsid must be stable enough to persist in the cellular and extracellular environments and then dissociate to allow release of the viral genome at the right time during infection. How this happens is poorly understood. CDMS and EM were used to investigate the disassembly of BMV under two situations: a pH jump to a basic environment at low ionic strength and under slow buffer exchange to remove magnesium cations that stabilize interactions at capsid protein interfaces.186 In the case of a pH jump, disassembly occurred through a global cleavage event to give broad mass distributions at around 3.33 and 1.42 MDa. The sum of these masses is close to the mass of the intact virion (4.71 MDa). The higher mass fragment was attributed to an almost-intact empty capsid (the expected mass of an intact empty capsid is 3.65 MDa), and the lower mass fragment was attributed to the RNA associated with some capsid proteins. In the case of disassembly by removal of magnesium ions, low-mass capsid fragments (<1 MDa) were found to accumulate over time, and at the same time the peak attributed to the virion broadened and shifted to higher mass. When the magnesium ions were removed at neutral pH, ionized glutamic acid side chains at the capsid protein–protein interfaces repelled each other, causing the BMV capsid to swell, which in turn opened pores that exposed the RNA to the surrounding solution.187 The RNA could recruit more capsid proteins, causing the observed increase in the mass. It is likely that this disassembly pathway is more closely related to what happens in vivo. It matches the cotranslational disassembly model originally proposed by Wilson and co-workers where capsid proteins are not stripped from the RNA but continue to protect it as the host machinery binds to the RNA to begin translation.188,189
3.5.3. Bacteriophage P22 Assembly.
P22 is a tailed dsDNA bacteriophage that infects Salmonella. Assembly begins with the formation of an icosahedral precursor (called a procapsid) from capsid and scaffolding proteins. P22 normally assembles into procapsids with T = 7 symmetry that contain 420 copies of the capsid protein. The scaffolding protein is an assembly chaperone that facilitates assembly of procapsids with the correct structure, and after assembly some remain trapped inside and leave later in the lifecycle.190 CDMS measurements of procapsids produced in E. coli showed a prominent peak at ~23.60 MDa and a smaller peak at 19.84 M Da.191 The lower-mass peak was 1.3% above the mass expected for a procapsid without a scaffolding protein. The higher-mass peak could be accounted for by the addition of ~112 scaffolding proteins. Procapsids could assemble in the absence of scaffolding proteins, but that process is very slow and leads to the formation of both T = 4 and T = 7 particles.192,193 Thus, the procapsids without scaffolding protein detected in the CDMS measurements (the peak at 19.84 MDa) are presumably due to procapsids that have lost the scaffolding protein after assembly.
Single amino acid substitutions can dramatically affect procapsid assembly. Substitutions at A285 are known to lead to “petite” particles.194 The assembly products of variants A285Y and A285T were analyzed by CDMS. In both cases, broad mass distributions extending from 5 to 15 MDa were observed. The charge of the ions was found to be valuable in distinguishing between hollow-shell structures (i.e., capsids) and more compact protein aggregates; the hollow capsids acquire a higher charge than the compact aggregates. For the A285T variant, T = 4 and T = 7 capsids were found. A285Y made T = 3 and T = 4 capsids.195
During maturation, the dsDNA genome of P22 is packaged through a portal complex that occupies a single vertex on the icosahedral capsid, replacing five capsid proteins. How the portal complex is incorporated into the icosahedral lattice is not fully understood. CDMS measurements showed that the portal proteins assembled exclusively into a dodecamer.196 Further studies suggested that the scaffolding protein played a role in triggering the portal ring formation and helped to recruit them into the growing procapsid.
The main components of mature P22 are shown in Figure 9a.197,198 It is believed to contain 521 copies of 9 different proteins and a 42 724 bp genome. Summing the masses of all of the components197–200 (see Table 1) leads to an expected mass of 51.614 MDa. The CDMS mass distribution for mature P22 shows a single Gaussian peak centered on 52.18 MDa (Figure 9b).198 The measured peak is at a mass 1.1% larger than the expected mass. The difference is attributable to counterions, salt adducts, and trapped solvent. The peak is 1.33 MDa wide (fwhm). After accounting for the instrumental resolution, the underlying peak width is ~750 kDa. This heterogeneity can be entirely accounted for by the distribution of packaged DNA. The DNA is packaged by the headful mechanism,197,201 where DNA is actively spooled into the capsid through the tail machine until the capsid is full and then the DNA is cleaved. Around 1600 bp more than a full genome is packaged, so the ends are redundant. In addition, headful packaging leads to a distribution in the length of the packaged DNA, which accounts for the underlying peak width.198 Because the width can be accounted for by the heterogeneity in the DNA, there can be little heterogeneity in the copy numbers of the protein constituents. The average charge on mature P22 is 490 e. For the procapsid, the average charge is lower, ~425 e. The increase in charge on going from the procapsid to the mature virion probably results from two structural changes. First, the capsid is known to expand slightly when the DNA is packaged, and second, the procapsids studied earlier do not incorporate the tail machine that protrudes from the capsid surface (see Figure 9a).
Figure 9.
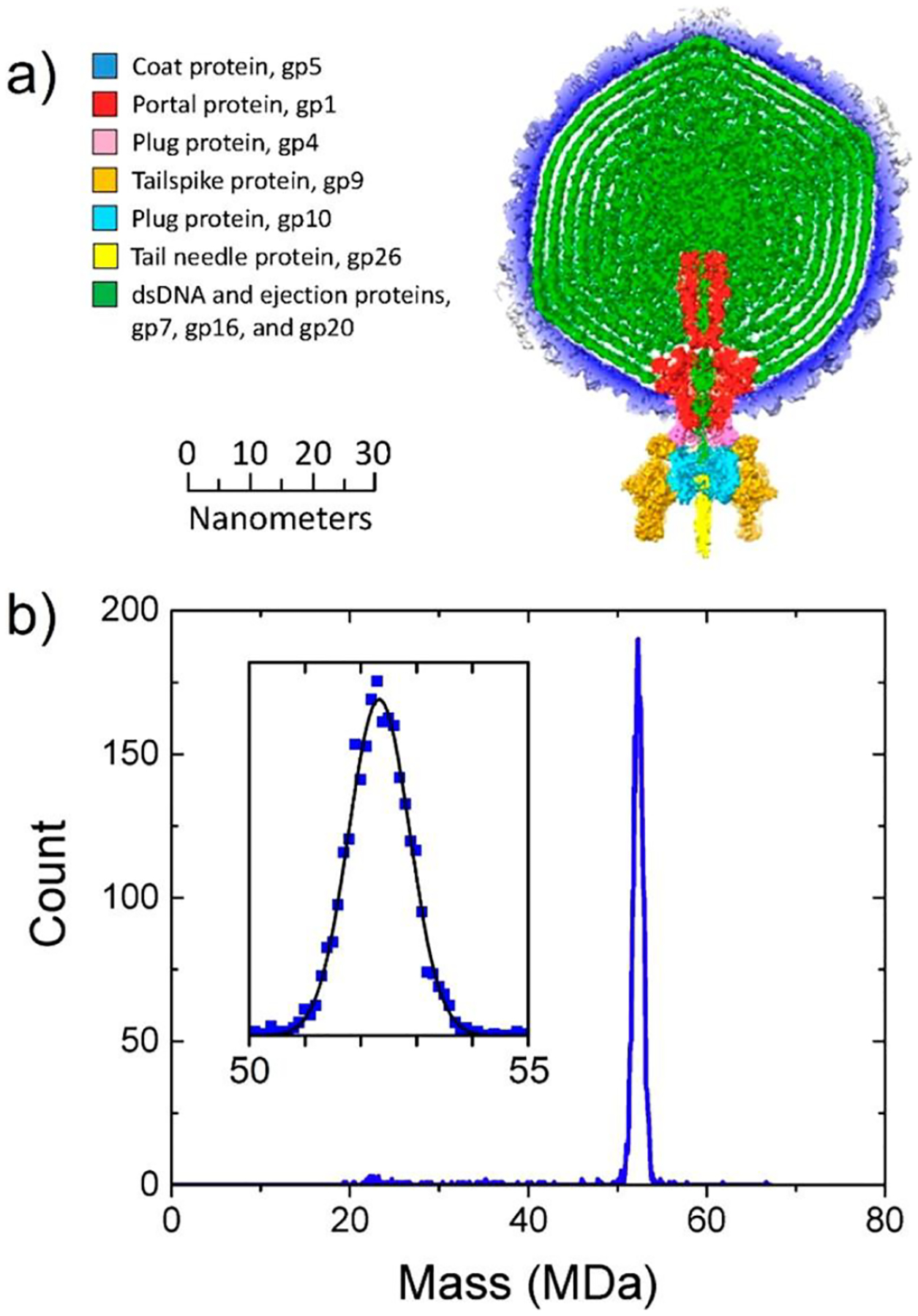
CDMS measurements for mature bacteriophage P22. (a) Main components of P22. (b) CDMS mass distribution. The inset shows an expanded view of the main peak in the spectrum. Adapted with permission from ref 198. Copyright 2016 John Wiley and Sons.
Table 1.
Components of Bacteriophage P22 and Their Massesa
| component | mass (Da) | copies | total mass (Da) | |
|---|---|---|---|---|
| dsDNA | genome + 1600 bp | 26 768 698 | 1 | 26 768 698 |
| capsid | coat protein, gp5 | 46 621 | 415 | 19 347 715 |
| tail machine | portal protein, gp1 | 82 612 | 12 | 2 889 621 |
| plug protein, gp4 | 18 025 | 12 | ||
| tailspike protein, gp9 | 71 857 | 18 | ||
| plug protein, gp10 | 52 457 | 6 | ||
| tail needle protein, gp26 | 24 603 | 3 | ||
| ejection proteins | gp7 | 21 093 | (11) | 2 607 551 |
| gp16 | 64 358 | (12) | ||
| gp20 | 50 101 | (32) | ||
| total mass of intact virion (Da) | 51 613 585 | |||
Reproduced with permission from ref 198. Copyright 2016 John Wiley and Sons.
3.5.4. Sindbis.
Sindbis virus (SINV) is an enveloped, single-stranded RNA virus, ~70 nm in diameter.202,203 It consists of a nucleocapsid core surrounded by a lipid bilayer. The particle surface has 80 trimeric glycoprotein spikes arranged with T = 4 icosahedral symmetry. SINV is an alphavirus with arthropod (primarily mosquito) and vertebrate hosts. There is a large difference in the infectivity of SINV particles from different hosts: those produced in mosquito cells are >1 order of magnitude more infectious than those produced in mammalian cells.204,205 CDMS measurements of SINV from mammalian (BHK) and insect (C6/36) cell lines were performed to shed light on the infectivity difference.206 BHK-derived SINV was found to have a substantially higher mass than SINV from C6/36 cells, 52.88 versus 50.69 MDa. From an MS analysis of the lipids extracted from the lipid bilayer, it was found that the mass difference was due to differences in the nature of the lipids. BHK-derived SINV had a higher proportion of lipids with longer-chain fatty acids. The difference in the lipid composition may affect the organization of the lipid bilayer, which could affect multiple phases of the viral lifecycle and thus influence the infectivity. The peaks in the CDMS mass distributions were ~2 MDa wide fwhm. This is broader than those measured for other viruses of a similar size. There are two potential sources of heterogeneity that deserve mention: heterogeneity in the glycans on the glycoprotein spikes and, probably more important, heterogeneity in the lipid bilayer.
3.6. Analysis of Gene-Therapy Vectors
Gene therapy (where genetic material is introduced into cells to correct defective genes) has had some notable successes in the last 5 years. Luxturna (which treats RPE65 mutation-induced retinal dystrophy) was approved by the FDA in 2017, and Zolgensma (which treats SMN1 mutation-induced spinal muscular atrophy, the most common genetic cause of infant death) was approved in 2019. Both drugs use recombinant adeno-associated virus (rAAV) to transport a DNA payload.207,208 AAV is a small, nonenveloped, single-stranded DNA virus, ~25 nm in diameter (see inset in Figure 10a).209,210 The AAV capsid contains three proteins (VP1–3). All three share the VP3 sequence, VP2 adds an N-terminal domain, and VP1 adds another N-terminal domain to VP2.211 The AAV capsid has a total of 60 copies of VP1–3 arranged in quasi-icosahedral symmetry.212–214 However, the number of VP1, VP2, and VP3 in each capsid is random. For capsids derived from HEK293 cells, the average ratios are around 1:1:10,215–218 which yields an expected mass of ~3.732 MDa (for serotype 8). Wild-type AAV packages ~4.7 kilobases (kb),211 which has a mass of ~1.4 MDa. So, a fully loaded AAV is expected to have a mass of ~5.1 MDa.
Figure 10.
CDMS mass distribution for AAV gene-therapy vectors. (a) CDMS mass distribution measured for a sample of empty AAV8 particles. The red dashed line shows the calculated mass distribution. An external view of the 25 nm AAV8 capsid is shown in the inset. (b) CDMS mass spectrum for the ATCC AAV8 Reference Standard (blue line). The yellow line shows the spectrum × 10 and offset vertically. The dashed red line shows the distribution expected when the sample heterogeneity and experimental resolution are included. The red arrow shows the location of the empty AAV8 particle. Adapted with permission from ref 227. Copyright 2021 Elsevier under Creative Commons Attribution 4.0 International Public License (CC BY 4.0) http://creativecommons.org/licenses/by/4.0/legalcode.
Characterizing rAAV vectors is challenging. Along with capsids that have packaged the full genome of interest, empty capsids are generated,219 and some capsids package a partial genome.220–222 The encapsidation of DNA impurities, such as heterogeneous fragments of host cellular, helper, or plasmid DNAs, also occurs.223,224 Finally, aggregation can reduce bioavailability and influence biodistribution and may also cause an immune response.225 CDMS has emerged as a valuable tool to analyze the DNA content of rAAV vectors.222,226 It can go beyond characterization of the empty/full ratios and provide information about particle heterogeneity including packaging of heterogeneous DNA.227 Figure 10a shows a CDMS mass distribution measured for empty AAV8 particles separated by ultracentrifugation with a CsCl gradient. The red dashed line shows the expected peak for AAV8 with a 1:1:10 ratio of VP1–3, accounting for the experimental resolution. There is a high-mass tail that was attributed to packaging of small DNA fragments from the host cells. A CDMS mass distribution recorded for the ATCC AAV8 reference standard is shown in Figure 10b. The main peak at ~5.25 MDa is assigned to AAV8 particles that have packaged a full genome. The charge carried by the ions that make up the peak at 5.25 MDa (~170 e) is very similar to the charge carried by the peak due to empty particles in Figure 10a. This indicates that the DNA is packaged inside the capsid. If the DNA was external, a substantially higher charge would be expected.
The dashed red line in Figure 10b shows the expected mass distribution calculated for the full particle considering sample heterogeneity and instrument resolution. The yellow line shows the counts × 10 and offset vertically. The red arrow shows the expected mass of empty particles. Only a few ions were detected with masses in the range expected for empty AAV8, reflecting expectations that this was a highly purified sample. However, ~14% of the ions had masses that fell below the mass of the main peak and were attributed to particles that packaged less than the full genome.222 The difference between the masses of empty and full particles can be used to determine the genome mass. This difference was determined as a function of the genome length, and a plot of the measured difference against the DNA sequence mass yielded a straight line with an intercept of 0.016 MDa and a slope of 1.041.227 The non-zero intercept and the reason the slope is not exactly 1.000 are both attributed to counterions. ssDNA and ssRNA viruses often have basic sites inside the capsid to neutralize some of the charge associated with the backbone phosphates of the packaged genome.182,228 When no genome is present, these basic sites will have counterions that are lost when the genome is packaged. The negative intercept is attributed to these counterions. Its small size (0.016 MDa) indicates that the number of excess basic sites per capsid protein is small (~3), which is consistent with the known structure of the AAV8 capsid.229 The slope (1.041) is attributed to the packaging of counterions along with the negatively charged DNA. A slope of 1.075 is expected if all the counterions are Na+, 1.059 for NH4+, 1.039 for Mg2+, and 1.003 for H+ (i.e., unionized). While it is not possible to draw a conclusion about the nature of the counterions, the measured slope (1.041) is in the expected range.
One of the main limitations of AAV as a gene-therapy vector is its small packaging capacity (~5 kb).230,231 Adenovirus is much larger than AAV and overcomes this limitation; human adenovirus 5 (HAdV5) has a 36 kbp dsDNA genome. Recombinant adenovirus (AdV) has been used as a gene-therapy vector and as a vaccine vector. Its use as a vaccine vector has come to the fore recently as it has been employed in some COVID-19 vaccines (for example, Oxford/AstraZeneca, Sputnick 5, and Johnson & Johnson). AdV has a pseudo-T = 25 icosahedral capsid made of 720 copies of the hexon protein and 60 copies of the penton protein. Groups of five penton proteins, arranged into pentagons, occupy the 12 vertices of the capsid. The pentons are decorated with fibers that are trimers of the fiber protein (see inset in Figure 11a). There are 36 copies of the fiber proteins, so with the hexon and penton proteins there are 816 structural proteins in the adenovirus capsid. The capsid is stabilized by four cement proteins that are located inside and outside of the capsid. There is some uncertainty in their copy numbers, but in HAdV5 there are thought to be ~785 copies altogether.232 Finally, there are another 6 proteins in mature AdV located in the core. Together there are ~998 copies of the core proteins in HAdV5.233,234 One important function of the core proteins is condensation of the genomic DNA, and the three proteins involved in this task have basic domains and are closely associated with the genome. Including the structural proteins (816 copies), the cement proteins (~785 copies), and the core proteins (~998 copies), there are expected to be ~2 599 proteins in HAdV5. From the sequences and copy numbers, the expected mass of HAdV5 is 142.5 MDa, including the dsDNA genome.235
Figure 11.
CDMS measurements for adenovirus. (a) CDMS mass spectrum measured for HAdV5. An external view of HAdV5 is shown in the inset. The hexons are purple, the pentons are blue, and the fibers are green. (b) Charge versus mass heat map for the data in (a). Warmer colors indicate higher intensity. Adapted with permission ref 235. Copyright 2021 American Chemical Society.
CDMS measurements of HAdV5 show a sharp peak at 156.1 MDa that is attributed to the mature virus and a smaller peak at 129.6 MDa (see Figure 11a).235 The 129.6 MDa peak is due to empty or light particles that do not contain the genome but may contain packaging and capsid-assembly proteins that are not present in the mature virion.236,237 Light and heavy particles are separable by gradient centrifugation. Figure 11b shows a heat map of charge versus mass for the data in Figure 11a. The ions that make up the 129.6 MDa peak have a significantly lower charge than the ions in the 156.1 MDa peak (930 versus 1120 e). The lower charge may result because the light particles lack some or all of the fibers that decorate the surface of the mature virion.236 The fibers that protrude from the pentagonal vertices of the icosahedral capsid (see inset in Figure 11a) significantly increase the size of the virus, allowing it to carry much more charge.
The peak attributed to mature HAdV5 at 156.1 MDa is at a significantly higher mass than expected from sequence masses and copy numbers of the constituents (142.5 MDa). A possible explanation is that HAdV5 is so large that it is poorly desolvated. To test this hypothesis, measurements were made as a function of the dc voltage across the FUNPET. Lowering this voltage increases the time that the ions spend in the FUNPET, allowing more time for desolvation. The measured MW decreased by a fraction of a percent as the voltage was lowered to the point where the signal was significantly diminished. Another indication that the ions are desolvated was obtained from how their oscillation frequencies change with time. As noted earlier, the oscillation frequencies change systematically for several reasons, including the loss of small molecules.70 The average frequency shift found for AdV is similar to that found for AAV. If the AdV frequency shift is attributed entirely to the loss of small molecules, it would be equivalent to the loss of ~1 kDa (i.e., ~50 water molecules) during the 100 ms trapping time. Finally, poorly desolvated ions would be expected to yield broad peaks in the mass spectrum, while the measured peaks are relatively narrow. Taken together, these results indicate that complexes as large as adenovirus are effectively desolvated in the FUNPET, and the relative excess mass is comparable to that found for smaller ions. Thus, the large difference between the measured and expected masses for HAdV5 is not due to poor desolvation.
The copy numbers for the structural proteins in AdV are fixed by symmetry. However, an underestimate of copy numbers for the core or cement proteins could provide an explanation for the discrepancy between the expected and measured MWs. To investigate this hypothesis, CDMS measurements were performed for recombinant AdV vectors with a range of genome lengths. A plot of the vector mass against genome mass had a slope of 2.3 and an intercept of 105 MDa. The intercept is close to the expected mass of an HAdV5 empty particle with just structural and cement proteins. The slope indicates that the vector mass increases by 2.3 MDa for every 1 MDa of genome mass. The additional 1.3 MDa increase in the vector mass is attributed to core condensation proteins that are copackaged with the genome. The genome mass for HAdV5 is 22.2 MDa, so the core condensation proteins should weigh in at ~28.9 MDa, which is much greater than the 17.4 MDa determined from the published copy numbers. Thus, the copy numbers for the core condensation proteins may be underestimated. If a value of 28.9 MDa is used for the mass of the core condensation proteins, the expected mass of the mature capsid increases to 154.0 MDa, which is much closer to the measured value of 156.1 MDa.
3.7. Vaccines
Vaccines are one of the most effective means of improving public health.238 Since the emergence of the novel coronavirus in 2019, vaccination has taken center stage as the best hope for ending the COVID-19 pandemic caused by SARS-CoV-2. The vaccines that have received emergency-use authorization by the FDA use novel approaches: the Pfizer-BioNTech and Moderna vaccines are mRNA vaccines employing lipid nanoparticles for delivery. Johnson & Johnson’s Janssen vaccine is a viral vector vaccine that employs adenovirus (AdV26) to transport and deliver a DNA payload. In both cases, genetic instructions direct the production of the SARS-CoV-2 spike protein, which then induces an immune response. CDMS analysis of adenovirus vectors was described earlier.
Classical vaccination approaches fall into four main groups: whole-inactivated viruses, live-attenuated viruses, virus-like particles (VLPs), and protein subunits. The analysis of protein subunits can be challenging if they are heterogeneous, for example, the heavily glycosylated SARS-CoV-2 spike protein discussed earlier.133 The other three main groups involve virus-sized molecules that lack fast and robust analytical tools. CDMS has been used to characterize IPOL, RotaTeq, and Gardasil-9 directly from commercial sources.239 In addition to the antigen, vaccine formulations often include preservatives, adjuvants, stabilizers, and bulking agents. In the case of IPOL and RotaTeq, a simple buffer exchange into 200 mM ammonium acetate was enough to enable electrospray and CDMS measurements. For Gardasil-9 it was necessary to remove an aluminum adjuvant via a displacement reaction before buffer exchange.
IPOL is a whole-inactivated virus vaccine used to prevent poliomyelitis, which is caused by poliovirus. Poliovirus is a 30 nm nonenveloped ssRNA virus with a capsid that contains 60 copies each of four viral proteins, VP1–4, arranged in icosahedral symmetry. The poliovirus in IPOL is deactivated by formaldehyde cross-linking. Each cross-link contributes 12 Da. Figure 12a shows the CDMS mass distribution measured for IPOL. There are two peaks at around 5.94 and 8.58 MDa, which are attributed to the C-antigen and D-antigen, respectively. The vertical lines show the expected MWs (5.89 and 8.43 MDa). The C-antigen is an empty capsid, and the D-antigen has packaged a full genome. The mass of the peak attributed to the D-antigen is ~150 kDa larger than the expected MW. The excess mass is attributed mainly to the cross-links; thus, each poliovirus has ~12 500 cross-links. The number of cross-links for the empty particle (detected as a minor peak at 5.94 MDa in Figure 12a) is much less (<2 500), suggesting that the bulk of the cross-links involve RNA bases in either RNA–RNA or RNA–protein cross-links. The charge distribution for IPOL (black line in Figure 12b) is broad with well-defined maxima at around 220 and 250 e. The red line in Figure 12b shows, for comparison, the charge distribution measured for the AAV8 reference standard (discussed earlier). Poliovirus is slightly larger than AAV8 (30 versus 25 nm), and so it is expected to acquire a slightly higher charge. The charge is expected to scale as r3/2 according to the Rayleigh limit,118 so a charge of 165 e for 25 nm AAV8 would be expected to translate to 215 e for 30 nm poliovirus. This is close to the maximum at 220 e in Figure 12b, suggesting that the maximum at 250 e is due to a structure with a larger average diameter. One possibility is that this is due to the externalization of the N-terminus of VP1. This structural change is induced by receptor binding and heating in a low-salt buffer.240–242
Figure 12.
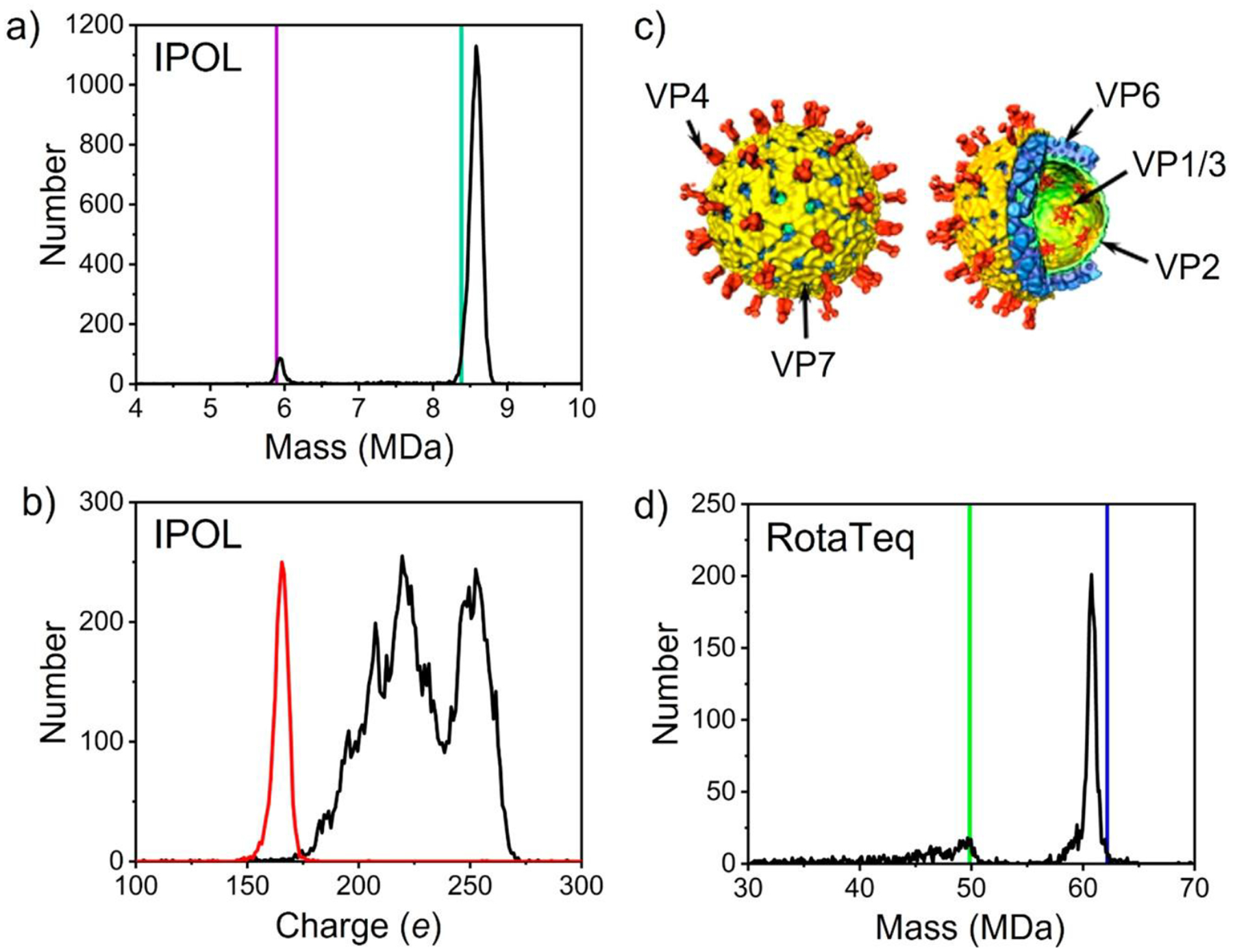
CDMS analysis of classical vaccines. (a) Mass spectrum measured by CDMS for IPOL vaccine. The expected masses of the C- and D-antigen particles (see text) are shown by the purple and green lines, respectively. The bin size is 20 kDa. (b) Charge spectra of IPOL (black) and the AAV8 reference standard (red). The bin size is 1 e. (c) Structure of the triple-layered rotavirus. (d) Mass spectrum measured by CDMS for the RotaTeq vaccine. Colored vertical lines show expected masses of the DLP with all pentonal trimers (blue) and the DLP with all pentonal trimers but missing the genome (green). The bin size is 100 kDa. (a, b, d) Adapted with permission from ref 239. Copyright 2021 American Chemical Society. (c) Adapted with permission from ref 244. Copyright 2014 Elsevier under Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License (CC By-NC-ND 3.0) https://creativecommons.org/licenses/by-nc-nd/3.0/legalcode.
RotaTeq vaccine is a live-attenuated vaccine that offers broad immunity using a reassortant combination of five human-bovine rotavirus strains. Rotavirus causes severe gastroenteritis in young children. The rotavirus is a triple-layered dsRNA virus with a segmented genome.243,244 Shedding the outer-layer proteins (VP4 and VP7) creates a double-layer particle (DLP). The middle layer (the outer layer of the DLP) is a T = 13 icosahedron with 780 copies of VP6, and the core consists of a capsid of 120 copies of VP2 surrounding the genome and 11–12 copies of VP1 (viral polymerase) and VP3 (viral capping enzyme).243–246 Taking all of the constituents into account, the expected MWs of the DLP and the triple-layer particle (TLP) are 62.16 and 106.77 MDa, respectively.239 The CDMS mass spectrum for RotaTeq is shown in Figure 12c. The TLP was not detected. The blue line in Figure 12c shows the expected MW of the DLP, and the green line shows the expected MW of the DLP minus its genome. The main peak in the spectrum is below the expected MW of the DLP (61.06 versus 62.16 MDa). This suggests that some of the components included in the summation for the expected MW are missing. The pentonal trimers have been found to dissociate from the transcriptionally active DLP in Wa strains.247,248 If 10 of the 60 pentonal trimers are lost, the expected mass would be 60.81 MDa, which is 0.4% less than the measured value. This discrepancy is within the range normally found for counterions, salt adducts, and trapped solvent. The main species detected here is the DLP, which is not infectious and has a small antigenic response. The main antigen in RotaTeq is the TLP, which has a concentration of 5–80 × 106 infectious units/mL. This is below the current limit of detection for CDMS (which is ~330 × 106 particle/mL).73 The concentration of the DLP is not specified, but it is clearly much larger than the TLP’s.
Norovirus is the leading cause of foodborne illness in the United States. Currently there is no FDA-approved vaccine. Norovirus is a nonenveloped ssRNA virus. The capsid has T = 3 icosahedral symmetry with 90 dimers of the major capsid protein VP1. VLPs assembled from VP1 are one focus of vaccine-development efforts. While a T = 3 icosahedron is the expected VLP structure, the GII.4 variant of VP1 has been shown to adopt a T = 4 structure, and a truncated version of GI.1 VP1 has been shown to form a T = 1 VLP.249–251 CDMS mass distributions measured for GI.1 VLPs buffer-exchanged into ammonium acetate showed three main peaks at 13.7, 17.1, and 23.9 MDa, but no peak near 10.18 MDa, the expected MW of the T = 3. The peaks at 13.7 and 23.9 MDa were attributed to T = 4 and T = 7 icosahedra consisting of 120 and 210 dimers, respectively. The peak at 17.1 MDa did not match the expected MW for any icosahedron following the Caspar–Klug rules.144 It had a mass close to that expected for 150 dimers, and thus it was attributed to prolate geometry generated by elongating a T = 4 capsid along its 5-fold axis by insertion of a band of 10 hexamers, similar to the structure described earlier for woodchuck hepatitis virus.165 These results showed that the norovirus VLPs are promiscuous and can adopt many different icosahedral geometries in solution.
3.8. Antibody Binding to Viruses and VLPs
Neutralizing antibodies mediate host immunity. Quantitative information about antibody binding to individual viral particles may provide the key to understanding neutralizing efficiency and vaccine efficacy. Most methods for monitoring ligand-binding interactions such as isothermal titration calorimetry,252 surface plasmon resonance,253,254 and enzyme-linked immunosorbent assays (ELISAs)255 are ensemble measurements that provide association constants and, in some cases, average stoichiometry. They cannot provide information about the distribution of the bound ligands. The distribution, which is available from CDMS measurements, can provide valuable information about heterogeneity. For example, a situation where there are two virion subpopulations with different affinities would be revealed, under the right conditions, as two components in the mass distribution.
The binding of antigen-binding fragments (Fabs) to canine parvovirus (CPV) was investigated by measuring CDMS mass distributions for CPV–Fab complexes as a function of Fab concentration.256 This study was performed with empty CPV particles. Figure 13a shows mass distributions measured for the binding of Fab E to CPV. To provide a starting point, the top spectrum shows the mass distribution for CPV without Fab E. The peak at 4.063 MDa is attributed to the CPV capsid. The capsid, a T = 1 icosahedron, has a stochastic distribution of two capsid proteins (VP1 and VP2), and this contributes to the peak width. The middle spectrum in Figure 13 was recorded with a Fab E concentration of 1.5 μM. Fab binding caused the peak to shift to higher mass than in the top spectrum, and the peak is much broader. Peaks due to the addition of individual Fabs are not resolved because of the heterogeneity in the mass of the empty capsid. However, the large peak width reflects the broad distribution of bound Fabs. The bottom spectrum in Figure 13a shows the mass distribution measured with a Fab E concentration of 10 μM. The peak has shifted to a slightly higher mass than in the middle spectrum, and it has become narrower. Increasing the Fab E concentration further did not shift the peak to higher mass, indicating that all Fab E sites on the CPV capsid were saturated.
Figure 13.
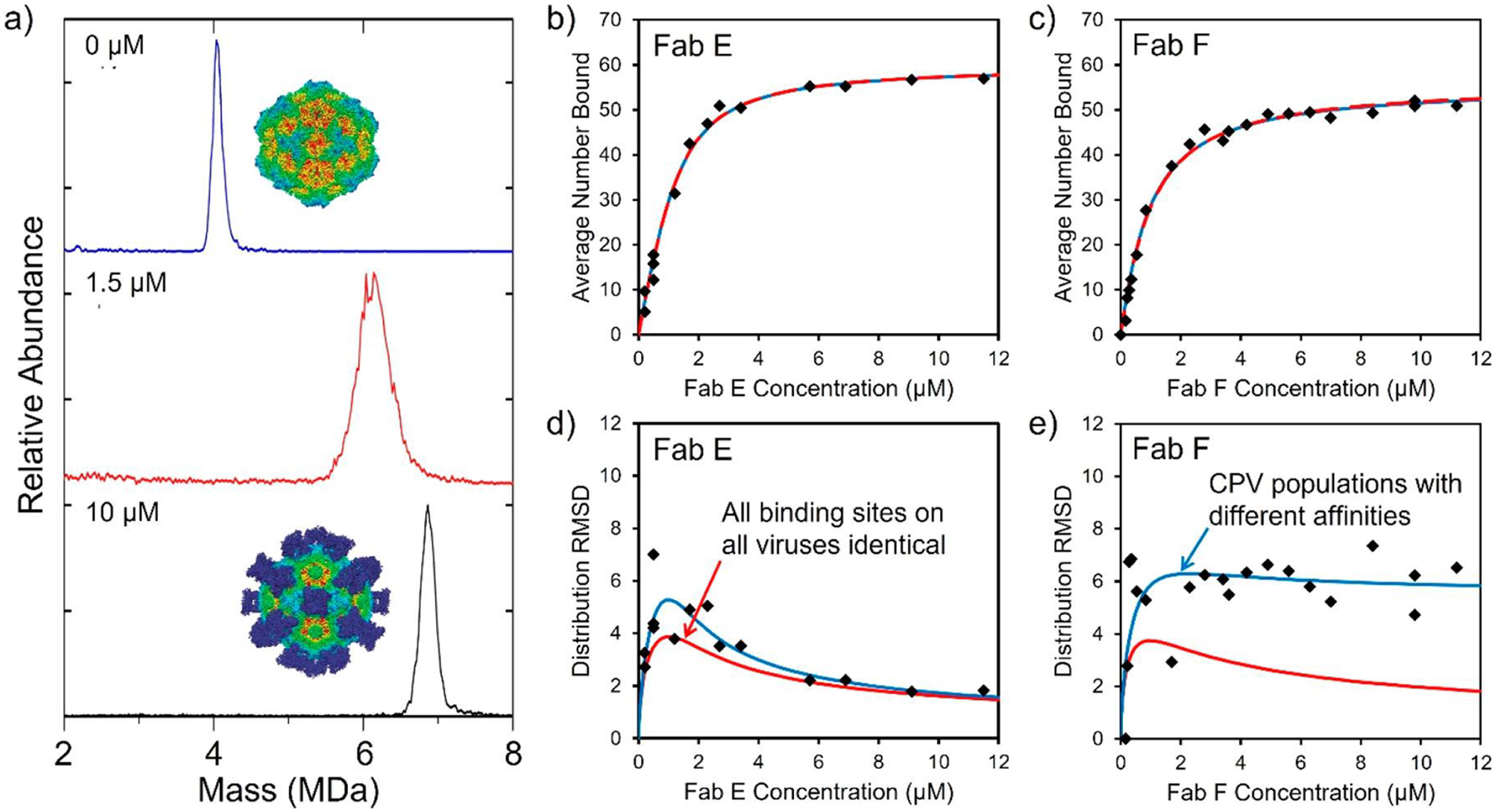
Fab binding to CPV capsids. (a) Mass distributions measured for CPV at FAB E concentrations of 0, 1.5, and 10 μM. The average mass shift provides information on the average number of Fabs bound, and the peak width provides information on the distribution of bound Fabs. The insets show cryo-EM images of CPV without Fabs (top) and saturated with 60 Fabs (bottom). (b) Plot of the average number of Fab E bound to CPV as a function of Fab E concentration. (c) Similar plot for Fab F. The black points are the measured values determined by CDMS. (d) Width of the Fab E distribution plotted against the Fab E concentration. (e) Similar plot for Fab F. The red lines show the predictions of the standard (Langmuir) model for ligand binding, where all sites have the same intrinsic affinity. The blue lines show the prediction of a model where there are CPV subpopulations with different affinities. In (b) and (c) the red and blue lines are coincident. Adapted with permission from ref 256. Copyright 2018 American Chemical Society.
Figure 13b shows the Fab E binding curve, a plot of the average number of Fab E bound as a function of the Fab E concentration. The points are the experimental measurements, and the red line is a fit of the standard (Langmuir) model for noncooperative ligand binding to n identical sites. Values for the dissociation constant (KD) and the number of sites (n) can be obtained from the fit. In this case, KD was found to be 4.20 × 10−7, and n was 60. This is consistent with cryo-EM studies, which indicate that there are 60 Fab E binding sites roughly equidistant from the 2-, 3-, and 5-fold axes of CPV.257,258 Figure 13c shows the binding curve for Fab F. The noncooperative binding model (red line) again fits the results well, yielding KD = 7.75 × 10−7. The KD values found here indicate interaction strengths at the weak end of the expected range for antibody binding. This could be partly due to the fact that antibodies have bivalent interactions versus monovalent interactions for Fabs.259
The distribution of Fabs bound at each Fab concentration can be obtained from the mass distributions. Figure 13d shows the RMSDs of the Fab E distributions plotted against Fab E concentration. The distribution initially broadens and then narrows at high Fab E concentrations where all of the sites are populated. This behavior is also evident in Figure 13a. The red line shows the prediction of the noncooperative binding model, which provides a reasonably good fit to the measurements except at the lowest concentrations. Figure 13e shows a similar plot of the distribution RMSDs for Fab F. In this case, the results are in poor agreement with the prediction of the standard model (red line). The poor fit occurs even though the standard model provides a good fit to the binding curve (Figure 13c). The blue line in Figure 13e is a good fit to the measured RMSDs. The model used to generate the blue line incorporates two CPV subpopulations with different binding affinities. The binding curves for this model cannot be distinguished from the red lines in Figure 13b and c. Thus, the heterogeneity in the affinities of the CPV particles is not revealed through the binding curve. Information on the heterogeneity can only be obtained from the distribution of bound Fabs, which is available from CDMS measurements but not available for other methods usually used to investigate ligand binding.
VLP conjugates are being developed for a variety of biomedical applications including imaging and drug delivery.260,261 As these systems become increasingly complex, their heterogeneity presents a major challenge to characterizing their composition. CDMS has been used to determine the antibody population distributions for VLP–antibody conjugates where antibodies have been covalently linked to the surface of a bacteriophage MS2 surface.262 Initial CDMS analysis of unconjugated MS2 particles suggested that they had packaged a broad distribution of extraneous genomic material. Procedures were developed to remove the unwanted genomic material, leading to a more homogeneous starting material. The most effective procedure involved disassembly, nucleic acid precipitation, and reassembly around yeast RNA, followed by high-pH treatment to remove the RNA. Using a much more homogeneous starting material led to a factor of 2 improvement in the antibody-coupling efficiency, illustrating the benefit of improved analytical characterization.
3.9. Exosomes
Exosomes are extracellular vesicles, ~30–120 nm in size, that are secreted by eukaryotic cells and appear in many bodily fluids.263 They have a lipid bilayer that surrounds a core containing proteins, RNAs, and lipids. Exosomes play a key role in intracellular communication, and they have been implicated in inflammation264 and tumor progression.265 There is a growing interest in potential clinical applications as biomarkers and therapies. CDMS analysis of exosomes derived from bovine milk shows two broad peaks: a low-mass distribution from ~2–10 MDa and a high-mass distribution from ~10–45 MDa.266 Exosomes are expected to have masses in the 10–1 200 MDa range, so the low-mass component is too small to be assigned to exosomes. However, the higher-mass peak falls within the expected range. The high-mass peak has two clearly separated low- and high-charge components in a scatter plot of charge versus mass. The exosome size distribution determined by EM imaging and by CDMS (assuming the particles were spherical) both have maxima at ~40 nm. However, the distributions determined by EM extend to larger diameters, around 70 versus 50 nm in the CDMS measurements. This could result from flattening of the particles in the EM images (leading to an overestimate of their sizes) or to discrimination against the larger particles in the CDMS measurements. Dehydration of the exosomes could also affect the size determinations.
3.10. Lipoproteins
Lipoproteins are nanometer-sized particles found in plasma. They have a broad size distribution and are challenging to characterize because of their heterogeneity. Lipoproteins are nature’s solution to the problem of transporting hydrophobic lipids through an aqueous medium. They have a micelle-like structure with a lipid core (primarily cholesterol esters and triglycerides) surrounded by a shell of phospholipid, free cholesterol, and apolipoproteins. They are usually divided into classes based on their density. The three major classes present in fasting plasma are high-density lipoproteins (HDL), which are the smallest (5–15 nm), followed by low-density lipoproteins (LDL) (18–28 nm), and finally very-low-density lipoproteins (VLDL) (30–80 nm). Each density class can be further separated into subclasses with different sizes, compositions, and metabolic properties. This is particularly opportune for HDL particles, which have many biological effects beyond lipid transport, and there is great interest in resolving and characterizing HDL subclasses.267 CDMS measurements have been performed for HDL, LDL, and VLDL.268 The CDMS spectrum for HDL (Figure 14a) shows a broad peak at ~230 kDa with a lower-intensity and poorly resolved second component at ~750 kDa. The broad peak at 230 kDa is in line with the expected HDL mass range of 50–600 kDa.
Figure 14.
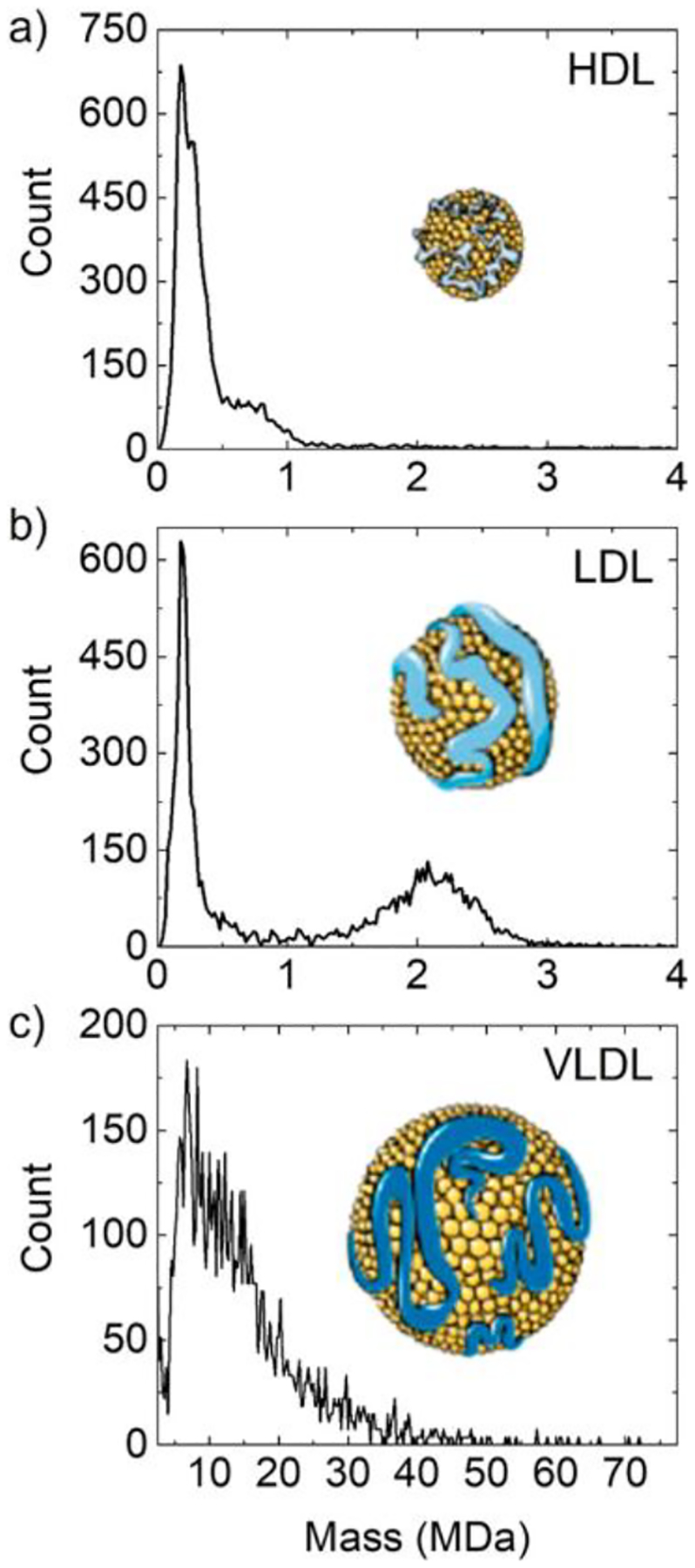
CDMS mass spectra measured for lipoproteins. (a) Mass distribution measured for HDL. (b) Mass distribution for LDL. The peak in the LDL spectrum at 230 kDa is probably an HDL impurity. (c) Mass distribution measured for VLDL. The insets show structures of the lipoprotein particles. Adapted with permission from ref 268. Copyright 2018 American Chemical Society.
For LDL, the CDMS mass spectrum (Figure 14b) shows a broad feature at 2.2 MDa and a prominent peak at 230 kDa. Apolipoprotein B100 (Apo B100) is a key structural protein for LDL and VLDL, and each particle contains one copy. Apo B100 has an MW of ~515 kDa, so the peak at 230 kDa in the LDL spectrum (Figure 14b) cannot be due to LDL. It has the same MW as the main peak in the HDL spectrum, and so it is probably an HDL impurity. The expected mass range for LDL is 2–7 MDa. The 2.2 MDa peak in the LDL spectrum falls within this range, but the peak is broad, extending from 1 to 3 MDa.
The peak at 750 kDa in the HDL spectrum (Figure 14a) lies above the expected range for HDL. A proteomics study of the HDL sample revealed the presence of Apo B100, indicating that at least some high-mass component in the HDL sample is due to an LDL impurity. However, it is difficult to imagine that the 750 kDa peak is due to LDL because Apo B100 has a mass of 515 kDa, leaving little room for any other components. Thus, this feature lacks an assignment at this point. Charge and mass scatter plots for the HDL samples revealed streaks that were attributed to HDL subgroups with different numbers of structural proteins. Finally, Figure 14c shows the CDMS spectrum measured for a VLDL sample. The mass distribution is very broad, extending from ~4 up to 40 MDa and beyond. The expected mass range (based on the 30–80 nm diameters determined by EM) is 8–160 MDa.
4. CONCLUSIONS AND OUTLOOK
CDMS opens the door to accurate MW measurements for large and heterogeneous samples that cannot be analyzed by conventional MS. While there are many powerful techniques to characterize small molecules and bulk materials, the 10–100 nm size range falls in the gap where there are fewer analytical techniques available. CDMS helps to fill this void. Currently, CDMS is only available in a handful of laboratories using homemade instruments; however, it is being commercialized. Orbitrap I2MS is expected to become available as a software upgrade. At present, Orbitrap I2MS is too new to have been employed in many applications. Table 2 provides a summary of the main performance metrics of the two types of CDMS presently in use (single-pass and ion-trap-based) and Orbitrap I2MS. Both CDMS and I2MS are still in the development phase, and further performance upgrades can be expected. In the case of ion-trap CDMS, more accurate charge measurements (due to lower noise amplifiers) and a major upgrade in the mass resolution (due to better-optimized ELIT designs) are anticipated. The coupling of CDMS to other MS-based methods (ion mobility and surface-induced dissociation, for example) will extend its reach.
Table 2.
Summary of Current Performance Metrics of Single-Pass CDMS, Ion-Trap CDMS, and Orbitrap I2MS
| single-pass CDMS | ion-trap CDMS | Orbitrap I2MS | |
|---|---|---|---|
| charge uncertainty | 50 ea | 0.2 ea | 4%b |
| lowest charge | 250 e | 1 e | 10 e |
| mass resolution | <10 | 300c | 25d |
| trapping time | 0.1–1.5 se | 2 s | |
| time to collect a spectrum | <1 min | 15–60 minf | not known |
| mass range | 1 MDa to 1 TDa | 1 kDa to 1 GDa | 10 kDa to 10 MDa |
Limited by electrical noise; almost independent of charge.
Limited mainly by ion trajectory.
Resolving power is limited by the m/z resolution.
Resolving power is limited by the accuracy of the charge measurement.
The 1.5 s trapping is required to obtain a charge RMSD of 0.2 e. With 0.1 s trapping, the RMSD is 0.8 e.
For 0.1 s single-ion trapping. It is reduced for multiple-ion trapping and increased for longer trapping times.
The applications summarized here show that CDMS can make important contributions to solving a wide range of complex problems in molecular biology and biotechnology. Several of the applications described here highlight the synergistic relationship between CDMS and cryo-EM. CDMS provides stoichiometry information and can be used to rapidly screen samples to find conditions that are optimum for cryo-EM analysis. In addition to solving research problems, there are applications for CDMS in biotechnology such as the analytical characterization of gene-therapy products and vaccines.
ACKNOWLEDGMENTS
I am happy to acknowledge the many large contributions of past and current graduate students to the CDMS project in the Jarrold lab, including Thaddeus Jones, Sarah Mabbett, Lloyd Zilch, Joshua Maze, Jonathan Smith, Indrani Bhattacharyya, Nathan Contino, Deven Shinholt, Elizabeth Pierson, David Keifer, Staci Anthony, Benjamin Draper, Carmen Dunbar, Joanna Hogan, Max Dorn, Corinne Lutomski, Kevin Bond, Nicholas Lyktey, Aaron Todd, Daniel Botamanenko, Lauren Barnes, Lohra Miller, Peyton Sayasith, Raj Parikh, and David Reitenbach. I am also happy to acknowledge our many collaborators, including Adam Zlotnick, Bogdan Dragnea, Stephen Jacobson, David Clemmer, Tuli Mukhopadhyay, Joe Wang, Colin Parrish, Matt Francis, Carol Teschke, Alan Remaley, Peter Prevelige, Charlotte Uetrecht, Thomas Powers, Brian Chait, Carol Robinson, Anthony Donsante, Aravind Asokan, and Johnson Mak. I am grateful to the National Institutes of Health for financial support under Award no. R01GM131100.
Biography
Martin Jarrold was born in the United Kingdom and earned a Ph.D. from the University of Warwick. He held a NATO postdoctoral fellowship in the Department of Chemistry at the University of California, Santa Barbara, before joining AT&T Bell Laboratories in 1985 as a member of the technical staff in the Physics Research division. In 1992 he became Professor of Chemistry at Northwestern University. He moved to Indiana University in 2002 as the Robert and Marjorie Mann Chair of Chemistry.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.chemrev.1c00377
The author declares the following competing financial interest(s): The author is a shareholder in Megadalton Solutions, a company that is engaged in commercializing CDMS.
REFERENCES
- (1).Macfarlane R; Torgerson D Californium-252 plasma desorption mass spectroscopy. Science 1976, 191, 920–925. [DOI] [PubMed] [Google Scholar]
- (2).Morris HR; Panico M; Barber M; Bordoli RS; Sedgwick RD; Tyler A Fast atom bombardment: a new mass spectrometric method for peptide sequence analysis. Biochem. Biophys. Res. Commun 1981, 101, 623–631. [DOI] [PubMed] [Google Scholar]
- (3).Tanaka K; Waki H; Ido Y; Akita S; Yoshida Y; Yoshida T; Matsuo T Protein and polymer analyses up to m/z 100 000 by laser ionization time-of flight mass spectrometry. Rapid Commun. Mass Spectrom 1988, 2, 151–153. [Google Scholar]
- (4).Karas M; Hillenkamp F Laser desorption ionization of proteins with molecular masses exceeding 10,000 Da. Anal. Chem 1988, 60, 2299–2301. [DOI] [PubMed] [Google Scholar]
- (5).Fenn JB; Mann M; Meng CK; Wong SF; Whitehouse CM Electrospray ionization-principles and practice. Mass Spectrom. Rev 1990, 9, 37–70. [Google Scholar]
- (6).Chowdhury SK; Katta V; Chait BT Probing conformational changes in proteins by mass spectrometry. J. Am. Chem. Soc 1990, 112, 9012–9013. [Google Scholar]
- (7).Katta V; Chait BT Observation of the heme-globin complex in native myoglobin by electrospray-ionization mass spectrometry. J. Am. Chem. Soc 1991, 113, 8534–8535. [Google Scholar]
- (8).Ganem B; Li YT; Henion JD Detection of noncovalent receptor-ligand complexes by mass spectrometry. J. Am. Chem. Soc 1991, 113, 6294–6296. [Google Scholar]
- (9).Baca M; Kent SBH Direct observation of a ternary complex between the dimeric enzyme HIV-1 protease and a substrate-based inhibitor. J. Am. Chem. Soc 1992, 114, 3992–3993. [Google Scholar]
- (10).Loo J Electrospray ionization mass spectrometry: a technology for studying noncovalent macromolecular complexes. Int. J. Mass Spectrom 2000, 200, 175–186. [Google Scholar]
- (11).van den Heuvel RHH; Heck AJR Native protein mass spectrometry: from intact oligomers to functional machineries. Curr. Opin. Chem. Biol 2004, 8, 519–526. [DOI] [PubMed] [Google Scholar]
- (12).Mehmood S; Allison TM; Robinson CV Mass spectrometry of protein complexes: from origins to applications. Annu. Rev. Phys. Chem 2015, 66, 453–474. [DOI] [PubMed] [Google Scholar]
- (13).Rostom AA; Robinson CV Detection of the intact GroEL chaperonin assembly by mass spectrometry. J. Am. Chem. Soc 1999, 121, 4718–4719. [Google Scholar]
- (14).Hernández H; Robinson CV Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nat. Protoc 2007, 2, 715–26. [DOI] [PubMed] [Google Scholar]
- (15).Mann M; Meng CK; Fenn JB Interpreting mass spectra of multiply charged ions. Anal. Chem 1989, 61, 1702–1708. [Google Scholar]
- (16).Tito MA; Tars K; Valegard K; Hajdu J; Robinson CV Electrospray time-of-flight mass spectrometry of the intact MS2 virus capsid. J. Am. Chem. Soc 2000, 122, 3550–3551. [Google Scholar]
- (17).Stockley PG; Rolfsson O; Thompson GS; Basnak G; Francese S; Stonehouse NJ; Homans SW; Ashcroft AE A simple, RNA-mediated allosteric switch controls the pathway to formation of a T = 3 viral capsid. J. Mol. Biol 2007, 369, 541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Uetrecht C; Versluis C; Watts NR; Roos WH; Wuite GJL; Wingfield PT; Steven AC; Heck AJR High-resolution mass spectrometry of viral assemblies: molecular composition and stability of dimorphic hepatitis B virus capsids. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 9216–9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Snijder J; Rose RJ; Veesler D; Johnson JE; Heck AJR Studying 18 MDa virus assemblies with native mass spectrometry. Angew. Chem., Int. Ed 2013, 52, 4020–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Snijder J; van de Waterbeemd M; Damoc E; Denisov E; Grinfeld D; Bennett A; Agbandje-McKenna M; Makarov A; Heck AJR Defining the stoichiometry and cargo load of viral and bacterial nanoparticles by Orbitrap mass spectrometry. J. Am. Chem. Soc 2014, 136, 7295–7299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).van de Waterbeemd M; Snijder J; Tsvetkova IB; Dragnea BG; Cornelissen JJ; Heck AJR Examining the heterogeneous genome content of multipartite viruses BMV and CCMV by native mass spectrometry. J. Am. Soc. Mass Spectrom 2016, 27, 1000–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Philip MA; Gelbard F; Arnold S An absolute method for aerosol particle mass and charge measurement. J. Colloid Interface Sci 1983, 91, 507–515. [Google Scholar]
- (23).Peng W-P; Yang Y-C; Kang M-W; Lee YT; Chang H-C Measuring masses of single bacterial whole cells with a quadrupole ion trap. J. Am. Chem. Soc 2004, 126, 11766–11767. [DOI] [PubMed] [Google Scholar]
- (24).Wuerker RF; Shelton H; Langmuir RV Electrodynamic containment of charged particles. J. Appl. Phys 1959, 30, 342–349. [Google Scholar]
- (25).Hars G; Tass Z Application of quadrupole ion trap for the accurate mass determination of submicron size charged particles. J. Appl. Phys 1995, 77, 4245–4250. [Google Scholar]
- (26).Nie Z; Tzeng Y-K; Chang H-C; Chiu C-C; Chang C-Y; Chang C-M; Tao M-H Microscopy-based mass measurement of a single whole virus in a cylindrical ion trap. Angew. Chem., Int. Ed 2006, 45, 8131–8134. [DOI] [PubMed] [Google Scholar]
- (27).Cai Y; Peng WP; Kuo SJ; Sabu S; Han CC; Chang HC Optical detection and charge-state analysis of MALDI-generated particles with molecular masses larger than 5 MDa. Anal. Chem 2002, 74, 4434–4440. [DOI] [PubMed] [Google Scholar]
- (28).Cai Y; Peng WP; Chang HC Ion trap mass spectrometry of fluorescently labeled nanoparticles. Anal. Chem 2003, 75, 1805–1811. [DOI] [PubMed] [Google Scholar]
- (29).Bell DM; Howder CR; Johnson RC; Anderson SL Single CdSe/ZnS nanocrystals in an ion trap: charge and mass determination and photophysics evolution with changing mass, charge, and temperature. ACS Nano 2014, 8, 2387–2398. [DOI] [PubMed] [Google Scholar]
- (30).Howder CR; Long BA; Bell DM; Anderson SL Thermally brightened CdSe/ZnS quantum dots as noncontact probes for surface chemistry studies of dark nanoparticles trapped in the gas phase. J. Phys. Chem. C 2015, 119, 14561–14570. [Google Scholar]
- (31).Peng W-P; Yang Y-C; Lin C-W; Chang H-C Molar mass and molar mass distribution of polystyrene particle size standards. Anal. Chem 2005, 77, 7084–7089. [DOI] [PubMed] [Google Scholar]
- (32).Peng W-P; Yang Y-C; Kang M-W; Tzeng Y-K; Nie Z; Chang H-C; Chang W; Chen C-H Laser-induced acoustic desorption mass spectrometry of single bioparticles. Angew. Chem., Int. Ed 2006, 45, 1423–1426. [DOI] [PubMed] [Google Scholar]
- (33).Zhang N; Zhan L; Xiong C; Nie Z Review of particle ion trap mass spectrometry. Zhongguo Kexue: Huaxue 2014, 44, 801–806. [Google Scholar]
- (34).Schlemmer S; Illemann J; Wellert S; Gerlich D Nondestructive high-resolution and absolute mass determination of single charged particles in a three-dimensional quadrupole trap. J. Appl. Phys 2001, 90, 5410–5418. [Google Scholar]
- (35).Grimm M; Langer B; Schlemmer S; Lischke T; Becker U; Widdra W; Gerlich D; Flesch R; Rühl E Charging mechanisms of trapped element-selectively excited nanoparticles exposed to soft X rays. Phys. Rev. Lett 2006, 96, 066801. [DOI] [PubMed] [Google Scholar]
- (36).Smith RD; Cheng X; Brace JE; Hofstadler SA; Anderson GA Trapping, detection and reaction of very large single molecular ions by mass spectrometry. Nature 1994, 369, 137–139. [Google Scholar]
- (37).Bruce JE; Cheng X; Bakhtiar R; Wu Q; Hofstadler SA; Anderson GA; Smith RD Trapping, detection, and mass measurement of individual ions in a Fourier transform ion cyclotron resonance mass spectrometer. J. Am. Chem. Soc 1994, 116, 7839–7847. [Google Scholar]
- (38).Cheng X; Camp DG; Wu Q; Bakhtiar R; Springer DL; Morris BJ; Bruce JE; Anderson GA; Edmonds CG; Smith RD Molecular weight determination of plasmid DNA using electrospray ionization mass spectrometry. Nucleic Acids Res 1996, 24, 2183–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Abdillahi AM; Lee KW; McLuckey SA Mass Analysis of macromolecular analytes via multiply charged ion attachment. Anal. Chem 2020, 92, 16301–16306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).van de Waterbeemd M; Fort KL; Boll D; Reinhardt-Szyba M; Routh A; Makarov A; Heck AJR High-fidelity mass analysis unveils heterogeneity in intact ribosomal particles. Nat. Methods 2017, 14, 283–286. [DOI] [PubMed] [Google Scholar]
- (41).Gilmore IS; Seah MP Ion detection efficiency in SIMS: dependencies on energy, mass and composition for microchannel plates used in mass spectrometry. Int. J. Mass Spectrom 2000, 202, 217–229. [Google Scholar]
- (42).Twerenbold D; Vuilleumier J-L; Gerber D; Tadsen A; van den Brandt B; Gillevet PM Detection of single macromolecules using a cryogenic particle detector coupled to a biopolymer mass spectrometer. Appl. Phys. Lett 1996, 68, 3503–3505. [Google Scholar]
- (43).Frank M; Mears CA; Labov SE; Benner WH; Horn D; Jaklevic JM; Barfknecht AT High efficiency detection of 66,000 Da protein molecules using a cryogenic detector in a matrix-assisted laser desorption/ionization time-of-flight mass spectrometer. Rapid Commun. Mass Spectrom 1996, 10, 1946–1950. [Google Scholar]
- (44).Frank M; Labov SE; Westmacott G; Benner WH Energy-sensitive cryogenic detectors for high-mass biomolecule mass spectrometry. Mass Spectrom. Rev 1999, 18, 155–186. [DOI] [PubMed] [Google Scholar]
- (45).Rabin MW; Hilton GC; Martinis JM Application of microcalorimeter energy measurement to biopolymer mass spectrometry. IEEE Trans. Appl. Supercond 2001, 11, 242–247. [Google Scholar]
- (46).Wenzel RJ; Matter U; Schultheis L; Zenobi R Analysis of megadalton ions using cryodetection MALDI time-of-flight mass spectrometry. Anal. Chem 2005, 77, 4329–4337. [DOI] [PubMed] [Google Scholar]
- (47).Aksenov AA; Bier ME The analysis of polystyrene and polystyrene aggregates into the mega dalton mass range by cryodetection MALDI TOF MS. J. Am. Soc. Mass Spectrom 2008, 19, 219–230. [DOI] [PubMed] [Google Scholar]
- (48).Sipe DM; Plath LD; Aksenov AA; Feldman JS; Bier ME Characterization of mega-dalton-sized nanoparticles by super-conducting tunnel junction cryodetection mass spectrometry. ACS Nano 2018, 12, 2591–2602. [DOI] [PubMed] [Google Scholar]
- (49).Shockley W Currents to conductors induced by a moving point charge. J. Appl. Phys 1938, 9, 635–636. [Google Scholar]
- (50).Weinheimer AJ The charge induced on a conducting cylinder by a point charge and its application to the measurement of charge on precipitation. J. Atmos. Oceanic Technol 1988, 5, 298–304. [Google Scholar]
- (51).Shelton H; Hendricks CD; Wuerker RF Electrostatic acceleration of microparticles to hypervelocities. J. Appl. Phys 1960, 31, 1243–1246. [Google Scholar]
- (52).Fuerstenau SD; Benner WH Molecular weight determination of megadalton DNA electrospray ions using charge detection time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom 1995, 9, 1528–1538. [DOI] [PubMed] [Google Scholar]
- (53).Schultz JC; Hack CA; Benner WH Mass determination of megadalton-DNA electrospray ions using charge detection mass spectrometry. J. Am. Soc. Mass Spectrom 1998, 9, 305–313. [DOI] [PubMed] [Google Scholar]
- (54).Schultz JC; Hack CA; Benner WH Polymerase chain reaction products analyzed by charge detection mass spectrometry. Rapid Commun. Mass Spectrom 1999, 13, 15–20. [Google Scholar]
- (55).Fuerstenau SD; Benner WH; Thomas JJ; Brugidou C; Bothner B; Siuzdak G Mass spectrometry of an intact virus. Angew. Chem., Int. Ed 2001, 40, 541–544. [DOI] [PubMed] [Google Scholar]
- (56).Dahan M; Fishman R; Heber O; Rappaport M; Altstein N; Zajfman D; van der Zande WJ A new type of electrostatic ion trap for storage of fast ion beams. Rev. Sci. Instrum 1998, 69, 76–83. [Google Scholar]
- (57).Schmidt HT; Cederquist H; Jensen J; Fardi A Conetrap: a compact electrostatic ion trap. Nucl. Instrum. Methods Phys. Res., Sect. B 2001, 173, 523–527. [Google Scholar]
- (58).Benner WH A gated electrostatic ion trap to repetitiously measure the charge and m/z of large electrospray ions. Anal. Chem 1997, 69, 4162–4168. [Google Scholar]
- (59).Contino NC; Jarrold MF Charge detection mass spectrometry for single ions with a limit of detection of 30 charges. Int. J. Mass Spectrom 2013, 345–347, 153–159. [Google Scholar]
- (60).Gamero-Castaño M Induction charge detector with multiple sensing stages. Rev. Sci. Instrum 2007, 78, 043301. [DOI] [PubMed] [Google Scholar]
- (61).Gamero-Castaño M Retarding potential and induction charge detectors in tandem for measuring the charge and mass of nanodroplets. Rev. Sci. Instrum 2009, 80, 053301. [DOI] [PubMed] [Google Scholar]
- (62).Mabbett SR; Zilch LW; Maze JT; Smith JW; Jarrold MF Pulsed acceleration charge detection mass spectrometry: application to weighing electrosprayed droplets. Anal. Chem 2007, 79, 8431–8439. [DOI] [PubMed] [Google Scholar]
- (63).Smith JW; Siegel EE; Maze JT; Jarrold MF Image charge detection mass spectrometry: pushing the envelope with sensitivity and accuracy. Anal. Chem 2011, 83, 950–956. [DOI] [PubMed] [Google Scholar]
- (64).Barney BL; Daly RT; Austin DE A multi-stage image charge detector made from printed circuit boards. Rev. Sci. Instrum 2013, 84, 114101. [DOI] [PubMed] [Google Scholar]
- (65).Draper BE; Anthony SN; Jarrold MF The FUNPET- a new hybrid ion funnel-ion carpet atmospheric pressure interface for the simultaneous transmission of a broad mass range. J. Am. Soc. Mass Spectrom 2018, 29, 2160–2172. [DOI] [PubMed] [Google Scholar]
- (66).Schmidt HT; Cederquist H; Jensen J; Fardi A Conetrap: a compact electrostatic ion trap. Nucl. Instrum. Methods Phys. Res., Sect. B 2001, 173, 523–527. [Google Scholar]
- (67).Hogan JA; Jarrold MF Optimized electrostatic linear ion trap for charge detection mass spectrometry. J. Am. Soc. Mass Spectrom 2018, 29, 2086–2095. [DOI] [PubMed] [Google Scholar]
- (68).Todd AR; Jarrold MF Dynamic calibration enables high accuracy charge measurements on individual ions for charge detection mass spectrometry. J. Am. Soc. Mass Spectrom 2020, 31, 1241–1248. [DOI] [PubMed] [Google Scholar]
- (69).Draper BE; Jarrold MF Real time analysis and signal optimization for charge detection mass spectrometry. J. Am. Soc. Mass Spectrom 2019, 30, 898–904. [DOI] [PubMed] [Google Scholar]
- (70).Keifer DZ; Alexander AW; Jarrold MF Spontaneous mass and charge losses from single multi-megadalton ions studied by charge detection mass spectrometry. J. Am. Soc. Mass Spectrom 2017, 28, 498–506. [DOI] [PubMed] [Google Scholar]
- (71).Elliott AG; Harper CC; Lin H-W; Williams ER Mass, mobility and MSn measurements of single ions using charge detection mass spectrometry. Analyst 2017, 142, 2760–2769. [DOI] [PubMed] [Google Scholar]
- (72).Todd AR; Alexander AW; Jarrold MF Implementation of a charge sensitive amplifier without a feedback resistor for charge detection mass spectrometry reduces noise and enables detection of individual ions carrying a single charge. J. Am. Soc. Mass Spectrom 2020, 31, 146–154. [DOI] [PubMed] [Google Scholar]
- (73).Todd AR; Jarrold MF Dramatic improvement in sensitivity with pulsed mode charge detection mass spectrometry. Anal. Chem 2019, 91, 14002–14008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Keifer DZ; Shinholt DL; Jarrold MF Charge detection mass spectrometry with almost perfect charge accuracy. Anal. Chem 2015, 87, 10330–10337. [DOI] [PubMed] [Google Scholar]
- (75).Pierson EE; Contino NC; Keifer DZ; Jarrold MF Charge detection mass spectrometry for single ions with an uncertainty in the charge measurement of 0.65 e. J. Am. Soc. Mass Spectrom 2015, 26, 1213–1220. [DOI] [PubMed] [Google Scholar]
- (76).Contino NC; Pierson EE; Keifer DZ; Jarrold MF Charge detection mass spectrometry with resolved charge states. J. Am. Soc. Mass Spectrom 2013, 24, 101–108. [DOI] [PubMed] [Google Scholar]
- (77).Bertuccio G; Rehak P; Xi DM A novel charge sensitive preamplifier without the feedback resistor. Nucl. Instrum. Methods Phys. Res., Sect. A 1993, 326, 71–76. [Google Scholar]
- (78).Todd AR; Barnes LF; Young K; Zlotnick A; Jarrold MF Higher resolution charge detection mass spectrometry. Anal. Chem 2020, 92, 11357–11364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Harper CC; Elliott AG; Oltrogge LM; Savage DF; Williams ER Multiplexed charge detection mass spectrometry for high-throughput single ion analysis of large molecules. Anal. Chem 2019, 91, 7458–7465. [DOI] [PubMed] [Google Scholar]
- (80).Harper CC; Williams ER Enhanced multiplexing in Fourier transform charge detection mass spectrometry by decoupling ion frequency from mass to charge ratio. J. Am. Soc. Mass Spectrom 2019, 30, 2637–2645. [DOI] [PubMed] [Google Scholar]
- (81).Elliott AG; Harper CC; Lin H-W; Williams ER Effects of individual ion energies on charge measurements in Fourier transform charge detection mass spectrometry. J. Am. Soc. Mass Spectrom 2019, 30, 946–955. [DOI] [PubMed] [Google Scholar]
- (82).Botamanenko DY; Jarrold MF Ion-ion interactions in charge detection mass spectrometry. J. Am. Soc. Mass Spectrom 2019, 30, 2741–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).McKay AR; Ruotolo BT; Ilag LL; Robinson CV Mass measurements of increased accuracy resolve heterogeneous populations of intact ribosomes. J. Am. Chem. Soc 2006, 128, 11433–11442. [DOI] [PubMed] [Google Scholar]
- (84).Lössl P; Snijder J; Heck AJR Boundaries of mass resolution in native mass spectrometry. J. Am. Soc. Mass Spectrom 2014, 25, 906–917. [DOI] [PubMed] [Google Scholar]
- (85).Larsson DSD; Liljas L; van der Spoel D Virus capsid dissolution studied by microsecond molecular dynamics simulations. PLoS Comput. Biol 2012, 8, No. e1002502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Andoh Y; Yoshii N; Yamada A; Fujimoto K; Kojima H; Mizutani K; Nakagawa A; Nomoto A; Okazaki S All-atom molecular dynamics calculation study of entire poliovirus empty capsids in solution. J. Chem. Phys 2014, 141, 165101. [DOI] [PubMed] [Google Scholar]
- (87).El-Baba TJ; Raab SA; Brown CJ; Lutomski CA; Henderson LW; Woodall DW; Shen J; Trinidad JC; Niu H; Jarrold MF; et al. Thermal analysis of a mixture of ribosomal proteins by vT-ESI-MS: towards a new parallel approach for characterizing the stabilityome. Anal. Chem 2021, 93, 8484–8492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Chen R; Wu Q; Mitchell DW; Hofstadler SA; Rockwood AL; Smith RD Direct charge number and molecular weight determination of large individual ions by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem 1994, 66, 3964–3969. [Google Scholar]
- (89).Chen R; Cheng X; Mitchell DW; Hofstadler SA; Wu Q; Rockwood AL; Sherman MG; Smith RD Trapping, detection, and mass determination of coliphage T4 DNA ions by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem 1995, 67, 1159–1163. [Google Scholar]
- (90).Eliuk S; Makarov A Evolution of Orbitrap mass spectrometry instrumentation. Annu. Rev. Anal. Chem 2015, 8, 61–80. [DOI] [PubMed] [Google Scholar]
- (91).Makarov A; Denisov E Dynamics of ions of intact proteins in the Orbitrap mass analyzer. J. Am. Soc. Mass Spectrom 2009, 20, 1486–1495. [DOI] [PubMed] [Google Scholar]
- (92).Kafader JO; Melani RD; Senko MW; Makarov AA; Kelleher NL; Compton PD Measurement of individual ions sharply increases the resolution of Orbitrap mass spectra of proteins. Anal. Chem 2019, 91, 2776–2783. [DOI] [PubMed] [Google Scholar]
- (93).McGee JP; Melani RD; Yip PF; Senko MW; Compton PD; Kafader JO; Kelleher NL Isotopic resolution of protein complexes up to 466 kDa using individual ion mass spectrometry. Anal. Chem 2021, 93, 2723–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Bruce JE; Anderson GA; Udseth HR; Smith RD Large molecule characterization based upon individual ion detection with electrospray ionization-FTICR mass spectrometry. Anal. Chem 1998, 70, 519–525. [DOI] [PubMed] [Google Scholar]
- (95).Kafader JO; Melani RD; Durbin KR; Ikwuagwu B; Early BP; Fellers RT; Beu SC; Zabrouskov V; Makarov AA; Maze JT; et al. Multiplexed mass spectrometry of individual ions improves measurement of proteoforms and their complexes. Nat. Methods 2020, 17, 391–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Wörner TP; Snijder J; Bennett A; Agbandje-McKenna M; Makarov AA; Heck AJR Resolving heterogeneous macromolecular assemblies by Orbitrap-based single-particle charge detection mass spectrometry. Nat. Methods 2020, 17, 395–398. [DOI] [PubMed] [Google Scholar]
- (97).Loumaigne M; Midelet C; Doussineau T; Dugourd P; Antoine R; Stamboul M; Débarre A; Werts MHV Optical extinction and scattering cross sections of plasmonic nanoparticle dimers in aqueous suspension. Nanoscale 2016, 8, 6555–6570. [DOI] [PubMed] [Google Scholar]
- (98).Faucon A; Benhelli-Mokrani H; Fleury F; Dubreil L; Hulin P; Nedellec S; Doussineau T; Antoine R; Orlando T; Lascialfari A; et al. Tuning the architectural integrity of high-performance magneto-fluorescent core-shell nanoassemblies in cancer cells. J. Colloid Interface Sci 2016, 479, 139–0841149. [DOI] [PubMed] [Google Scholar]
- (99).Doussineau T; Kerleroux M; Dagany X; Clavier C; Barbaire M; Maurelli J; Antoine R; Dugourd P Charging megadalton poly(ethylene oxide)s by electrospray ionization. A charge detection mass spectrometry study. Rapid Commun. Mass Spectrom 2011, 25, 617–623. [DOI] [PubMed] [Google Scholar]
- (100).Doussineau T; Bao CY; Antoine R; Dugourd P; Zhang W; D’Agosto F; Charleux B Direct molar mass determination of self-assembled amphiphilic block copolymer nanoobjects using electrospray-charge detection mass spectrometry. ACS Macro Lett 2012, 1, 414–417. [DOI] [PubMed] [Google Scholar]
- (101).Doussineau T; Santacreu M; Antoine R; Dugourd P; Zhang W; Chaduc I; Lansalot M; D’Agosto F; Charleux B The charging of micellar nanoparticles in electrospray ionization. Chem-PhysChem 2013, 14, 603–609. [DOI] [PubMed] [Google Scholar]
- (102).Ouadah N; Doussineau T; Hamada T; Dugourd P; Bordes C; Antoine R Correlation between the charge of polymer particles in solution and in the gas phase investigated by zeta-potential measurements and electrospray ionization mass spectrometry. Langmuir 2013, 29, 14074–14081. [DOI] [PubMed] [Google Scholar]
- (103).Chaduc I; Parvole J; Doussineau T; Antoine R; Désert A; Dugas P-Y; Ravaine S; Duguet E; Bourgeat-Lami E; Lansalot M Towards a one-step method for preparing silica/polymer heterodimers and dimpled polymer particles. Polymer 2015, 70, 118–126. [Google Scholar]
- (104).Doussineau T; Désert A; Lambert O; Taveau J-C; Lansalot M; Dugourd P; Bourgeat-Lami E; Ravaine S; Duguet E; Antoine R Charge detection mass spectrometry for the characterization of mass and surface area of composite nanoparticles. J. Phys. Chem. C 2015, 119, 10844–10849. [Google Scholar]
- (105).Warren NJ; Mykhaylyk OO; Ryan AJ; Williams M; Doussineau T; Dugourd P; Antoine R; Portale G; Armes SP Testing the vesicular morphology to destruction: birth and death of diblock copolymer vesicles prepared via polymerization-induced self-assembly. J. Am. Chem. Soc 2015, 137, 1929–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Viodé A; Dagany X; Kerleroux M; Dugourd P; Doussineau T; Charles L; Antoine R Coupling of size-exclusion chromatography with electrospray ionization charge-detection mass spectrometry for the characterization of synthetic polymers of ultra-high molar mass. Rapid Commun. Mass Spectrom 2016, 30, 132–136. [DOI] [PubMed] [Google Scholar]
- (107).Halim MA; Clavier C; Dagany X; Kerleroux M; Dugourd P; Dunbar RC; Antoine R Infrared laser dissociation of single megadalton polymer ions in a gated electrostatic ion trap: the added value of statistical analysis of individual events. Phys. Chem. Chem. Phys 2018, 20, 11959–11966. [DOI] [PubMed] [Google Scholar]
- (108).Antoine R Weighing synthetic polymers of ultra-high molar mass and polymeric nanomaterials: what can we learn from charge detection mass spectrometry? Rapid Commun. Mass Spectrom 2020, 34, No. e8539. [DOI] [PubMed] [Google Scholar]
- (109).Hendricks CD Charged droplet experiments. J. Colloid Sci 1962, 17, 249–259. [Google Scholar]
- (110).Maze JT; Jones TC; Jarrold MF Negative droplets from positive electrospray. J. Phys. Chem. A 2006, 110, 12607–12612. [DOI] [PubMed] [Google Scholar]
- (111).Zilch LW; Maze JT; Smith JW; Ewing GE; Jarrold MF Charge separation in the aerodynamic break-up of micrometer-sized water droplets. J. Phys. Chem. A 2008, 112, 13352–13363. [DOI] [PubMed] [Google Scholar]
- (112).Zilch LW; Maze JT; Smith JW; Jarrold MF Freezing, fragmentation, and charge separation in sonic sprayed water droplets. Int. J. Mass Spectrom 2009, 283, 191–199. [Google Scholar]
- (113).Bhattacharyya I; Maze JT; Ewing GE; Jarrold MF Charge separation from the bursting of bubbles on water. J. Phys. Chem. A 2011, 115, 5723–5728. [DOI] [PubMed] [Google Scholar]
- (114).Gao J; Austin DE Mechanistic investigation of charge separation in electrospray ionization using microparticles to record droplet charge state. J. Am. Soc. Mass Spectrom 2020, 31, 2044–2052. [DOI] [PubMed] [Google Scholar]
- (115).Harper CC; Brauer DD; Francis MB; Williams ER Direct observation of ion emission from charged aqueous droplets: effects on gaseous macromolecular charging. Chem. Sci 2021, 12, 5185–5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Fernandez de la Mora J Electrospray ionization of large multiply charged species proceeds via Dole’s charged residue mechanism. Anal. Chim. Acta 2000, 406, 93–104. [Google Scholar]
- (117).Konermann L; Ahadi E; Rodriguez AD; Vahidi S Unraveling the mechanism of electrospray ionization. Anal. Chem 2013, 85, 2–9. [DOI] [PubMed] [Google Scholar]
- (118).Rayleigh FRS On the equilibrium of liquid conducting masses charged with electricity. Philos. Mag. Series 5 1882, 14, 184–186. [Google Scholar]
- (119).Doussineau T; Yu Bao C; Clavier C; Dagany X; Kerleroux M; Antoine R; Dugourd P Infrared multiphoton dissociation tandem charge detection-mass spectrometry of single megadalton electrosprayed ions. Rev. Sci. Instrum 2011, 82, 084104. [DOI] [PubMed] [Google Scholar]
- (120).Doussineau T; Antoine R; Santacreu M; Dugourd P Pushing the limit of infrared multiphoton dissociation to megadalton-size DNA ions. J. Phys. Chem. Lett 2012, 3, 2141–2145. [DOI] [PubMed] [Google Scholar]
- (121).Doussineau T; Paletto P; Dugourd P; Antoine R Multiphoton dissociation of electrosprayed megadalton-sized DNA ions in a charge-detection mass spectrometer. J. Am. Soc. Mass Spectrom 2015, 26, 7–13. [DOI] [PubMed] [Google Scholar]
- (122).Halim MA; Bertorelle F; Doussineau T; Antoine R Direct determination of molecular weight distribution of calf-thymus DNAs and study of their fragmentation under ultrasonic and low-energy infrared irradiations. A charge detection mass spectrometry investigation. Rapid Commun. Mass Spectrom 2019, 33, 35–39. [DOI] [PubMed] [Google Scholar]
- (123).Dong X; Huang Y; Cho BG; Zhong J; Gautam S; Peng W; Williamson SD; Banazadeh A; Torres-Ulloa KY; Mechref Y Advances in mass spectrometry-based glycomics. Electrophoresis 2018, 39, 3063–3081. [DOI] [PubMed] [Google Scholar]
- (124).Huang C; Wang Y; Li X; Ren L; Zhao J; Hu Y; Zhang L; Fan G; Xu J; Gu X; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).WHO Coronavirus Disease (COVID-19) Dashboard https://covid19.who.int/ (accessed October 10, 2020).
- (126).Yang X; Yu Y; Xu J; Shu H; Xia J; Liu H; Wu Y; Zhang L; Yu Z; Fang M; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir. Med 2020, 8, 475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Zhou P; Yang X-L; Wang X-G; Hu B; Zhang L; Zhang W; Si H-R; Zhu Y; Li B; Huang C-L; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Wu A; Peng Y; Huang B; Ding X; Wang X; Niu P; Meng J; Zhu Z; Zhang Z; Wang J; et al. Genome composition and divergence of the novel coronavirus (2019-NCoV) originating in China. Cell Host Microbe 2020, 27, 325–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Amanat F; Krammer F SARS-CoV-2 vaccines: status report. Immunity 2020, 52, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Tai W; He L; Zhang X; Pu J; Voronin D; Jiang S; Zhou Y; Du L Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol 2020, 17, 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Watanabe Y; Allen JD; Wrapp D; McLellan JS; Crispin M Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Shajahan A; Supekar NT; Gleinich AS; Azadi P Deducing the N- and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 2020, 30, 981–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Miller LM; Barnes LF; Raab SA; Draper BE; El-Baba TJ; Lutomski CA; Robinson CV; Clemmer DE; Jarrold MF Heterogeneity of glycan processing on trimeric SARS-CoV-2 spike protein revealed by charge detection mass spectrometry. J. Am. Chem. Soc 2021, 143, 3959–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Nal B; Chan C; Kien F; Siu L; Tse J; Chu K; Staropoli I; Crescenzo-Chaigne B; Escriou N; van der Werf S; et al. Differential maturation and subcellular localization of severe acute respiratory syndrome coronavirus surface proteins S, M, and E. J. Gen. Virol 2005, 86, 1423–1434. [DOI] [PubMed] [Google Scholar]
- (135).Pierson EE; Keifer DZ; Contino NC; Jarrold MF Probing higher order multimers of pyruvate kinase with charge detection mass spectrometry. Int. J. Mass Spectrom 2013, 337, 50–56. [Google Scholar]
- (136).Kim SJ; Fernandez-Martinez J; Nudelman I; Shi Y; Zhang W; Raveh B; Herricks T; Slaughter BD; Hogan JA; Upla P; et al. Structure and functional anatomy of the nuclear pore complex. Nature 2018, 555, 475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Chiti F; Dobson C Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu. Rev. Biochem 2017, 86, 27–68. [DOI] [PubMed] [Google Scholar]
- (138).Bernstein SL; Dupuis NF; Lazo ND; Wyttenbach T; Condron MM; Bitan G; Teplow DB; Shea J-E; Ruotolo BT; Robinson CV; et al. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat. Chem 2009, 1, 326–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Bleiholder C; Dupuis NF; Wyttenbach T; Bowers MT Ion mobility-mass spectrometry reveals a conformational conversion from random assembly to β-sheet in amyloid fibril formation. Nat. Chem 2011, 3, 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Pujol-Pina R; Vilaprinyó-Pascual S; Mazzucato R; Arcella A; Vilaseca M; Orozco M; Carulla N SDS-PAGE analysis of Aβ oligomers is disserving research into Alzheimerś disease: appealing for ESI-IM-MS. Sci. Rep 2015, 5, 14809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Doussineau T; Mathevon C; Altamura L; Vendrely C; Dugourd P; Forge V; Antoine R Mass determination of entire amyloid fibrils by using mass spectrometry. Angew. Chem 2016, 128, 2386–2390. [DOI] [PubMed] [Google Scholar]
- (142).Bascetin R; Admane K; Agniel R; Boudou T; Doussineau T; Antoine T; Gallet O; Leroy-Dudal J; Vendrely C Amyloid-like aggregates formation by blood plasma fibronectin. Int. J. Biol. Macromol 2017, 97, 733–743. [DOI] [PubMed] [Google Scholar]
- (143).Pansieri J; Halim MA; Vendrely C; Dumoulin M; Legrand F; Sallanon MM; Chierici S; Denti S; Dagany X; Dugourd P; et al. Mass and charge distributions of amyloid fibers involved in neurodegenerative diseases: mapping heterogeneity and polymorphism. Chem. Sci 2018, 9, 2791–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Caspar DL; Klug A Physical principles in the construction of regular viruses. Cold Spring Harbor Symp. Quant. Biol 1962, 27, 1–24. [DOI] [PubMed] [Google Scholar]
- (145).Kang S; Uchida M; O’Neil A; Li R; Prevelige PE; Douglas T Implementation of P22 viral capsids as nanoplatforms. Biomacromolecules 2010, 11, 2804–2809. [DOI] [PubMed] [Google Scholar]
- (146).Wen AM; Steinmetz NF Design of virus-based nanomaterials for medicine. biotechnology, and energy. Chem. Soc. Rev 2016, 45, 4074–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Zlotnick A; Ceres P; Singh S; Johnson JM A small molecule inhibits and misdirects assembly of hepatitis B virus capsids. J. Virol 2002, 76, 4848–4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Stray SJ; Bourne CR; Punna S; Lewis WG; Finn MG; Zlotnick A A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 8138–8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Billioud G; Pichoud C; Puerstinger G; Neyts J; Zoulim F The main hepatitis B virus (HBV) mutants resistant to nucleoside analogs are susceptible in vitro to non-nucleoside inhibitors of HBV replication. Antiviral Res 2011, 92, 271–276. [DOI] [PubMed] [Google Scholar]
- (150).Katen SP; Chirapu SR; Finn MG; Zlotnick A Trapping of hepatitis B virus capsid assembly intermediates by phenylpropenamide assembly accelerators. ACS Chem. Biol 2010, 5, 1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Ruan L; Hadden JA; Zlotnick A Assembly properties of hepatitis B virus core protein mutants correlate with their resistance to assembly-directed antivirals. J. Virol 2018, 92, No. e01082–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Crowther RA; Kiselev NA; Böttcher B; Berriman JA; Borisova GP; Ose V; Pumpens P Three-dimensional structure of hepatitis B virus core particles determined by electron cryomicroscopy. Cell 1994, 77, 943–950. [DOI] [PubMed] [Google Scholar]
- (153).Wingfield PT; Stahl SJ; Williams RW; Steven AC Hepatitis core antigen produced in Escherichia coli: subunit composition, conformation analysis, and in vitro capsid assembly. Biochemistry 1995, 34, 4919–4932. [DOI] [PubMed] [Google Scholar]
- (154).Ceres P; Zlotnick A Weak protein-protein interactions are sufficient to drive assembly of hepatitis B virus capsids. Biochemistry 2002, 41, 11525–11531. [DOI] [PubMed] [Google Scholar]
- (155).Endres D; Zlotnick A Model based analysis of assembly kinetics for virus capsids or other spherical polymers. Biophys. J 2002, 83, 1217–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Zlotnick A To build a virus capsid. An equilibrium model of the self-assembly of polyhedral protein complexes. J. Mol. Biol 1994, 241, 59–67. [DOI] [PubMed] [Google Scholar]
- (157).Jack RL; Hagan MF; Chandler D Fluctuation-dissipation ratios in the dynamics of self-assembly. Phys. Rev. E 2007, 76, 021119. [DOI] [PubMed] [Google Scholar]
- (158).Pierson EE; Keifer DZ; Selzer L; Lee LS; Contino NC; Wang JC-Y; Zlotnick A; Jarrold MF Detection of late intermediates in virus capsid assembly by charge detection mass spectrometry. J. Am. Chem. Soc 2014, 136, 3536–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Lutomski CA; Lyktey NA; Zhao Z; Pierson EE; Zlotnick A; Jarrold MF HBV capsid completion occurs through error correction. J. Am. Chem. Soc 2017, 139, 16932–16938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Lutomski CA; Lyktey NA; Pierson EE; Zhao Z; Zlotnick A; Jarrold MF Multiple pathways in capsid assembly. J. Am. Chem. Soc 2018, 140, 5784–5790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Luque A; Reguera D; Morozov A; Rudnick J; Bruinsma R Physics of shell assembly: line tension, hole implosion, and closure catastrophe. J. Chem. Phys 2012, 136, 184507. [DOI] [PubMed] [Google Scholar]
- (162).Hagan MF Modeling viral capsid assembly. Adv. Chem. Phys 2014, 155, 1–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Nguyen HD; Reddy VS; Brooks CL Deciphering the kinetic mechanism of spontaneous self-assembly of icosahedral capsids. Nano Lett 2007, 7, 338–344. [DOI] [PubMed] [Google Scholar]
- (164).Kukreja AA; Wang JC-Y; Pierson E; Keifer DZ; Selzer L; Tan Z; Dragnea B; Jarrold MF; Zlotnick A Structurally similar woodchuck and human hepadnavirus core proteins have distinctly different temperature dependences of assembly. J. Virol 2014, 88, 14105–14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Pierson EE; Keifer DZ; Kukreja AA; Wang JC-Y; Zlotnick A; Jarrold MF Charge detection mass spectrometry identifies preferred non-icosahedral polymorphs in the self-assembly of woodchuck hepatitis virus capsids. J. Mol. Biol 2016, 428, 292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Moody MF The shape of the T-even bacteriophage head. Virology 1965, 26, 567–576. [DOI] [PubMed] [Google Scholar]
- (167).Moody MF Geometry of phage head construction. J. Mol. Biol 1999, 293, 401–433. [DOI] [PubMed] [Google Scholar]
- (168).Fokine A; Chipman PR; Leiman PG; Mesyanzhinov VV; Rao VB; Rossmann MG Molecular architecture of the prolate head of bacteriophage T4. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 6003–6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Hull R Molecular biology of rice tungro viruses. Annu. Rev. Phytopathol 1996, 34, 275–297. [DOI] [PubMed] [Google Scholar]
- (170).Hull R; Hills GJ; Markham R Studies on alfalfa mosaic virus. II. The structure of the virus components. Virology 1969, 37, 416–428. [DOI] [PubMed] [Google Scholar]
- (171).Heijtink RA; Houwing CJ; Jaspars EMJ Molecular weights of particles and RNAs of alfalfa mosaic virus. Number of subunits in protein capsids. Biochemistry 1977, 16, 4684–4693. [DOI] [PubMed] [Google Scholar]
- (172).Luque A; Reguera D The structure of elongated viral capsids. Biophys. J 2010, 98, 2993–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Kann M; Schmitz A; Rabe B Intracellular transport of hepatitis B virus. World J. Gastroenterol 2007, 13, 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Chen C; Wang JC-Y; Pierson EE; Keifer DZ; Delaleau M; Gallucci L; Cazenave CM; Kann M; Jarrold MF; Zlotnick A Importin β can bind hepatitis B virus core protein and empty core-like particles and induce structural changes. PLoS Pathog 2016, 12, No. e1005802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Dykeman EC; Stockley PG; Twarock R Solving a Levinthal’s paradox for virus assembly identifies a unique antiviral strategy. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 5361–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Twarock R; Stockley PG RNA-mediated virus assembly: mechanisms and consequences for viral evolution and therapy. Annu. Rev. Biophys 2019, 48, 495–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Aniagyei SE; DuFort C; Kao C; Dragnea B Self-assembly approaches to nanomaterial encapsulation in viral protein cages. J. Mater. Chem 2008, 18, 3763–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Kao CC; Sivakumaran K Brome Mosaic Virus, Good for an RNA Virologist’s Basic Needs. Mol. Plant Pathol 2000, 1, 91–97. [DOI] [PubMed] [Google Scholar]
- (179).Bond K; Tsvetkova I; Wang JC-Y; Jarrold MF; Dragnea B Virus assembly pathways: straying away but not too far. Small 2020, 16, 2004475. [DOI] [PubMed] [Google Scholar]
- (180).Rao A; Grantham GL Molecular studies on bromovirus capsid protein. II. Functional analysis of the amino-terminal arginine-rich motif and its role in encapsidation, movement, and pathology. Virology 1996, 226, 294–305. [DOI] [PubMed] [Google Scholar]
- (181).Ni P; Wang Z; Ma X; Das NC; Sokol P; Chiu W; Dragnea B; Hagan M; Kao CC An examination of the electrostatic interactions between the N-Terminal tail of the brome mosaic virus coat protein and encapsidated RNAs. J. Mol. Biol 2012, 419, 284–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (182).Belyi VA; Muthukumar M Electrostatic origin of the genome packing in viruses. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 17174–17178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Krol MA; Olson NH; Tate J; Johnson JE; Baker TS; Ahlquist P RNA-controlled polymorphism in the in vivo assembly of 180-subunit and 120-subunit virions from a single capsid protein. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 13650–13655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Maassen SJ; de Ruiter MV; Lindhoud S; Cornelissen JJLM Oligonucleotide length-dependent formation of virus-like particles. Chem. - Eur. J 2018, 24, 7456–7463. [DOI] [PubMed] [Google Scholar]
- (185).Thurm AR; Beren C; Duran-Meza AL; Knobler CM; Gelbart WM RNA homopolymers form higher-curvature virus-like particles than do normal-composition RNAs. Biophys. J 2019, 117, 1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (186).Bond KM; Lyktey NA; Tsvetkova IB; Dragnea B; Jarrold MF Disassembly intermediates of brome mosaic virus identified by charge detection mass spectrometry. J. Phys. Chem. B 2020, 124, 2124–2131. [DOI] [PubMed] [Google Scholar]
- (187).Chakravarty A; Reddy VS; Rao ALN Unravelling the stability and capsid dynamics of the three virions of brome mosaic virus assembled autonomously in vivo. J. Virol 2020, 94, No. e01794–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Brisco M; Hull R; Wilson TMA Swelling of isometric and of bacilliform plant virus nucleocapsids is required for virus-specific protein synthesis in vitro. Virology 1986, 148, 210–217. [DOI] [PubMed] [Google Scholar]
- (189).Shields SA; Brisco MJ; Wilson TMA; Hull R Southern bean mosaic virus RNA remains associated with swollen virions during translation in wheat germ cell-free extracts. Virology 1989, 171, 602–606. [DOI] [PubMed] [Google Scholar]
- (190).Prevelige PE; Thomas D; King J Scaffolding protein regulates the polymerization of P22 coat subunits into icosahedral shells in vitro. J. Mol. Biol 1988, 202, 743–757. [DOI] [PubMed] [Google Scholar]
- (191).Keifer DZ; Pierson EE; Hogan JA; Bedwell G; Prevelige PE; Jarrold MF Charge detection mass spectrometry of bacteriophage P22 procapsid distributions above 20 MDa. Rapid Commun. Mass Spectrom 2014, 28, 483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (192).Thuman-Commike PA; Greene B; Malinski JA; King J; Chiu W Role of the scaffolding protein in P22 procapsid size determination suggested by T = 4 and T = 7 procapsid structures. Biophys. J 1998, 74, 559–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Zlotnick A; Suhanovsky MM; Teschke CM The energetic contributions of scaffolding and coat proteins to the assembly of bacteriophage procapsids. Virology 2012, 428, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Suhanovsky MM; Teschke CM Bacteriophage P22 capsid size determination: roles for the coat protein telokin-like domain and the scaffolding protein amino-terminus. Virology 2011, 417, 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Keifer DZ; Motwani T; Teschke CM; Jarrold MF Acquiring structural information on virus particles via charge detection mass spectrometry. J. Am. Soc. Mass Spectrom 2016, 27, 1028–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Motwani T; Lokareddy RK; Dunbar CA; Cortines JR; Jarrold MF; Cingolani G; Teschke CM A viral scaffolding protein triggers portal ring oligomerization and incorporation during procapsid assembly. Sci. Adv 2017, 3, No. e1700423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Tang J; Lander GC; Olia A; Li R; Casjens S; Prevelige P; Cingolani G; Baker TS; Johnson JE Peering down the barrel of a bacteriophage portal: the genome packaging and release valve in P22. Structure 2011, 19, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Keifer DZ; Motwani T; Teschke CM; Jarrold MF Measurement of the accurate mass of a 50 MDa infectious virus. Rapid Commun. Mass Spectrom 2016, 30, 1957–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Chang J; Weigele P; King J; Chiu W; Jiang W Cryo-EM asymmetric reconstruction of bacteriophage P22 reveals organization of its DNA packaging and infecting machinery. Structure 2006, 14, 1073–1082. [DOI] [PubMed] [Google Scholar]
- (200).Pedulla ML; Ford ME; Karthikeyan T; Houtz JM; Hendrix RW; Hatfull GF; Poteete AR; Gilcrease EB; Winn-Stapley DA; Casjens SR Corrected sequence of the bacteriophage P22 genome. J. Bacteriol 2003, 185, 1475–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Casjens S; Hayden M Analysis in vivo of the bacteriophage P22 headful nuclease. J. Mol. Biol 1988, 199, 467–474. [DOI] [PubMed] [Google Scholar]
- (202).Strauss JH; Strauss EG The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev 1994, 58, 491–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Jose J; Snyder JE; Kuhn RJ A structural and functional perspective of alphavirus replication and assembly. Future Microbiol 2009, 4, 837–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Sokoloski KJ; Hayes CA; Dunn MP; Balke JL; Hardy RW; Mukhopadhyay S Sindbis virus infectivity improves during the course of infection in both mammalian and mosquito cells. Virus Res 2012, 167, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Hernandez R; Sinodis C; Brown DT Sindbis virus: propagation, quantification, and storage. Curr. Protoc. Microbiol 2005, 00, 15B.1.1–15B.1.34. [DOI] [PubMed] [Google Scholar]
- (206).Dunbar CA; Rayaprolu V; Wang C-Y; Brown CJ; Leishman E; Jones-Burrage S; Trinidad JC; Bradshaw HB; Clemmer DE; Mukhopadhyay S; et al. Dissecting the components of sindbis virus from arthropod and vertebrate hosts: implications for infectivity differences. ACS Infect. Dis 2019, 5, 892–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Wang D; Tai PWL; Gao G Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discovery 2019, 18, 358–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Keeler AM; Flotte TR Recombinant adeno-associated virus gene therapy in light of Luxterna (and Zolgensma and Glybera): where are we, and how did we get here? Annu. Rev. Virol 2019, 6, 601–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Atchison RW; Casto BC; Hammon WM Electron microscopy of adeno-associated virus (AAV) in cell cultures. Virology 1966, 29, 353–357. [DOI] [PubMed] [Google Scholar]
- (210).Hoggan MD; Blacklow NR; Rowe WP Studies of small DNA viruses found in various adenovirus preparations: physical, biological, and immunological characteristics. Proc. Natl. Acad. Sci. U. S. A 1966, 55, 1467–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Srivastava A; Lusby EW; Berns KI Nucleotide sequence and organization of the adeno-associated virus 2 genome. J. Virol 1983, 45, 555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (212).Kronenberg S; Kleinschmidt JA; Böttcher B Electron cryomicroscopy and image reconstruction of adeno-associated virus type 2 empty capsids. EMBO Rep 2001, 2, 997–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (213).Kaludov N; Padron E; Govindasamy L; McKenna R; Chiorini JA; Agbandje-McKenna M Production, purification, and preliminary X-ray crystallographic studies of adeno-associated virus serotype 4. Virology 2003, 306, 1–6. [DOI] [PubMed] [Google Scholar]
- (214).Nam H; Lane M; Padron E; Gurda B; McKenna R; Kohlbrenner E; Aslanidi G; Byrne B; Muzyczka N; Zolotukhin S; et al. Structure of adeno-associated virus serotype 8, a gene therapy vector. J. Virol 2007, 81, 12260–12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (215).Rose JA; Maizel JV; Inman JK; Shatkin AJ Structural proteins of adenovirus-associated viruses. J. Virol 1971, 8, 766–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (216).Johnson FB; Ozer HL; Hoggan MD Structural proteins of adenovirus associated virus type 3. J. Virol 1971, 8, 860–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Buller RML; Rose JA Characterization of adenovirus associated virus induced polypeptides in KB cells. J. Virol 1978, 25, 331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Samulski JR; Muzyczka N AAV-Mediated gene therapy for research and therapeutic purposes. Annu. Rev. Virol 2014, 1, 427–451. [DOI] [PubMed] [Google Scholar]
- (219).Sommer JM; Smith PH; Parthasarathy S; Isaacs J; Vijay S; Kieran J; Powell SK; McClelland A; Wright JF Quantification of adeno-associated virus particles and empty capsids by optical density measurement. Mol. Ther 2003, 7, 122–128. [DOI] [PubMed] [Google Scholar]
- (220).Hauswirth WW; Berns KI Adeno-associated virus DNA replication: non-unit length molecules. Virology 1979, 93, 57–68. [DOI] [PubMed] [Google Scholar]
- (221).Laughlin CA; Myers MW; Risin DL; Carter BJ Defective-interfering particles of the human parvovirus adeno-associated virus. Virology 1979, 94, 162–174. [DOI] [PubMed] [Google Scholar]
- (222).Pierson EE; Keifer DZ; Asokan A; Jarrold MF Resolving adeno-associated viral particle diversity with charge detection mass spectrometry. Anal. Chem 2016, 88, 6718–6725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (223).Smith PH; Wright JF; Qu G; Patarroyo-White S; Parker A; Sommer JM Packaging of host cell and plasmid DNA into recombinant adeno-associated virus particles produced by triple transfection. Mol. Ther 2003, 7, S348. [Google Scholar]
- (224).Chadeuf G; Ciron C; Moullier P; Salvetti A Evidence for encapsidation of prokaryotic sequences during recombinant adeno-associated virus production and their in vivo persistence after vector delivery. Mol. Ther 2005, 12, 744–753. [DOI] [PubMed] [Google Scholar]
- (225).Wright JF; Le T; Prado J; Bahr-Davidson J; Smith PH; Zhen Z; Sommer JM; Pierce GF; Qu G Identification of factors that contribute to recombinant AAV2 particle aggregation and methods to prevent its occurrence during vector purification and formulation. Mol. Ther 2005, 12, 171–178. [DOI] [PubMed] [Google Scholar]
- (226).Werle A; Powers T; Zobel J; Wappelhorst C; Jarrold M; Lyktey N; Sloan C; Wolf A; Adams-Hall S; Baldus P; et al. Comparison of analytical techniques to quantitate the capsid content of adeno-associated viral vectors. Mol. Ther.-Methods Clin. Dev 2021. DOI: 10.1016/j.omtm.2021.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Barnes LF; Draper BE; Chen Y-T; Powers TW; Jarrold MF Quantitative analysis of genome packaging in recombinant AAV vectors by charge detection mass spectrometry. Mol. Ther.-Methods Clin. Dev 2021, 23, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Garmann RF; Comas-Garcia M; Koay MST; Cornelissen JJLM; Knobler CM; Gelbart WM Role of electrostatics in the assembly pathway of a single-stranded RNA virus. J. Virol 2014, 88, 10472–10479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Nam H-J; Gurda BL; McKenna R; Potter M; Byrne B; Salganik M; Muzyczka N; Agbandje-McKenna M Structural studies of adeno-associated virus serotype 8 capsid transitions associated with endosomal trafficking. J. Virol 2011, 85, 11791–11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Dong J; Fan P; Frizzell R Quantitative analysis of packaging capacity of recombinant adeno-associated virus. Hum. Gene Ther 1996, 7, 2101–2112. [DOI] [PubMed] [Google Scholar]
- (231).Hermonat PL; Quirk GJ; Bishop BM; Han L The packaging capacity of adeno-associated virus (AAV) and the potential for wild-type-plus AAV gene therapy vector. FEBS Lett 1997, 407, 78–84. [DOI] [PubMed] [Google Scholar]
- (232).Reddy VS; Nemerow GR Structures and organization of adenovirus cement proteins provide insights into the role of capsid maturation in virus entry and infection. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 11715–11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (233).Ahi YS; Mittal SK Components of adenovirus genome packaging. Front. Microbiol 2016, 7, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (234).Kulanayake S; Tikoo SK Adenovirus core proteins: Structure and function. Viruses 2021, 13, 388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (235).Barnes LF; Draper BE; Jarrold MF Analysis of recombinant adenovirus vectors by ion trap charge detection mass spectrometry: accurate molecular weight measurements beyond 150 MDa. Anal. Chem (under review) 2021 [DOI] [PubMed] [Google Scholar]
- (236).Takahashi E; Cohen SL; Tsai PK; Sweeney JA Quantitation of adenovirus type 5 empty capsids. Anal. Biochem 2006, 349, 208–217. [DOI] [PubMed] [Google Scholar]
- (237).Condezo GN; Marabini R; Ayora S; Carazo JM; Alba R; Chillón M; San Martín C Structures of Adenovirus Incomplete Particles Clarify Capsid Architecture and Show Maturation Changes of Packaging Protein L1 52/55k. J. Virol 2015, 89, 9653–9664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (238).Rappuoli R; Hanon E Sustainable vaccine development: a vaccine manufacturer’s perspective. Curr. Opin. Immunol 2018, 53, 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Miller LM; Bond KM; Draper BE; Jarrold MF Characterization of classical vaccines by charge detection mass spectrometry. Anal. Chem 2021, 93, 11965–11972. [DOI] [PubMed] [Google Scholar]
- (240).Curry S; Chow M; Hogle JM The poliovirus 135S particle is infectious. J. Virol 1996, 70, 7125–7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (241).Bubeck D; Filman DJ; Cheng N; Steven AC; Hogle JM; Belnap DM The structure of the poliovirus 135S cell entry intermediate at 10-Angstrom resolution reveals the location of an externalized polypeptide that binds to membranes. J. Virol 2005, 79, 7745–7755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Wilton T; Dunn G; Eastwood D; Minor PD; Martin J Effect of formaldehyde inactivation on poliovirus. J. Virol 2014, 88, 11955–11964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Pesavento JB; Crawford SE; Estes MK; Venkataram Prasad BV Rotavirus proteins: structure and assembly. Curr. Top. Microbiol. Immunol 2006, 309, 189–219. [DOI] [PubMed] [Google Scholar]
- (244).Desselberger U Rotaviruses. Virus Res 2014, 190, 75–96. [DOI] [PubMed] [Google Scholar]
- (245).McClain B; Settembre E; Temple BRS; Bellamy AR; Harrison SC X-ray crystal structure of the rotavirus inner capsid particle at 3.8 Å resolution. J. Mol. Biol 2010, 397, 587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (246).Matthijnssens J; Joelsson DB; Warakomski DJ; Zhou T; Mathis PK; van Maanen M-H; Ranheim TS; Ciarlet M Molecular and biological characterization of the 5 human-bovine rotavirus (WC3)-based reassortant strains of the pentavalent rotavirus vaccine, RotaTeq®. Virology 2010, 403, 111–127. [DOI] [PubMed] [Google Scholar]
- (247).McClain B; Settembre E; Temple BRS; Bellamy AR; Harrison SC X-ray crystal structure of the rotavirus inner capsid particle at 3.8 Å resolution. J. Mol. Biol 2010, 397, 587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (248).Greig SL; Berriman JA; O’Brien JA; Taylor JA; Bellamy AR; Yeager MJ; Mitra AK Structural determinants of rotavirus subgroup specificity mapped by cryo-electron microscopy. J. Mol. Biol 2006, 356, 209–221. [DOI] [PubMed] [Google Scholar]
- (249).Devant JM; Hofhaus G; Bhella D; Hansman GS Heterologous expression of human norovirus GII.4 VP1 leads to assembly of T = 4 virus-like particles. Antiviral Res 2019, 168, 175–182. [DOI] [PubMed] [Google Scholar]
- (250).Jung J; Grant T; Thomas DR; Diehnelt CW; Grigorieff N; Joshua-Tor L High-resolution cryo-EM structures of outbreak strain human norovirus shells reveal size variations. Proc. Natl. Acad. Sci.U. S. A 2019, 116, 12828–12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Pogan R; Weiss VU; Bond K; Dülfer J; Krisp C; Lyktey N; Müller-Guhl J; Zoratto S; Allmaier G; Jarrold MF; et al. N-terminal VP1 truncations favor T = 1 norovirus-like particles. Vaccines 2021, 9, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (252).Damian L Isothermal titration calorimetry for studying protein-ligand interactions. Methods Mol. Biol 2013, 1008, 103–118. [DOI] [PubMed] [Google Scholar]
- (253).Pattnaik P Surface plasmon resonance applications in understanding receptor-ligand interaction. Appl. Biochem. Biotechnol 2005, 126, 79–92. [DOI] [PubMed] [Google Scholar]
- (254).Dubs M-C; Altschuh D; Van Regenmortel MHV Interaction between viruses and monoclonal antibodies studied by surface plasmon resonance. Immunol. Lett 1992, 31, 59–64. [DOI] [PubMed] [Google Scholar]
- (255).Nelson CDS; Palermo LM; Hafenstein SL; Parrish CR Different mechanisms of antibody-mediated neutralization revealed using the Fab fragment of monoclonal antibodies. Virology 2007, 361, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (256).Dunbar CA; Callaway HM; Parrish CR; Jarrold MF Probing antibody binding to canine parvovirus with charge detection mass spectrometry. J. Am. Chem. Soc 2018, 140, 15701–15711. [DOI] [PubMed] [Google Scholar]
- (257).Hafenstein S; Bowman VD; Sun T; Nelson CDS; Palermo LM; Chipman PR; Battisti AJ; Parrish CR; Rossmann MG Structural comparison of different antibodies interaction with parvovirus capsids. J. Virol 2009, 83, 5556–5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Organtini LJ; Lee H; Iketani S; Huang K; Ashley RE; Makhov AM; Conway JF; Parrish CR; Hafenstein S Near-atomic resolution structure of a highly neutralizing fab bound to canine parvovirus. J. Virol 2016, 90, 9733–9742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (259).Yuan W; Parrish CR Comparison of two single-chain antibodies that neutralize canine parvovirus: analysis of an antibody-combining site and mechanisms of neutralization. Virology 2000, 269, 471–480. [DOI] [PubMed] [Google Scholar]
- (260).Wang Q; Kaltgrad E; Lin T; Johnson JE; Finn MG Natural supramolecular building blocks: wild-type cowpea mosaic virus. Chem. Biol 2002, 9, 805–811. [DOI] [PubMed] [Google Scholar]
- (261).Kovacs EW; Hooker JM; Romanini DW; Holder PG; Berry KE; Francis MB Dual-surface-modified bacteriophage MS2 as an ideal scaffold for a viral capsid-based drug delivery system. Bioconjugate Chem 2007, 18, 1140–1147. [DOI] [PubMed] [Google Scholar]
- (262).Bond KM; Aanei IL; Francis MB; Jarrold MF Determination of antibody population distributions for virus-antibody conjugates by charge detection mass spectrometry. Anal. Chem 2020, 92, 1285–1291. [DOI] [PubMed] [Google Scholar]
- (263).Schorey JS; Cheng Y; Singh PP; Smith VL Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep 2015, 16, 24–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Słomka A; Urban SK; Lukacs-Kornek V; Zekanowska E; Kornek M Large extracellular vesicles: have we found the holy grail of inflammation? Front. Immunol 2018, 9, 2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Fang T; Lv H; Lv G; Li T; Wang C; Han Q; Yu L; Su B; Guo L; Huang S; et al. Tumor-derived exosomal miR-1247–3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat. Commun 2018, 9, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (266).Brown BA; Zeng X; Todd AR; Barnes LF; Winstone JMA; Trinidad JC; Novotny MV; Jarrold MF; Clemmer DE Charge detection mass spectrometry measurements of exosomes and other extracellular particles enriched from bovine milk. Anal. Chem 2020, 92, 3285–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (267).Rosenson RS; Brewer HB; Chapman MJ; Fazio S; Hussain MM; Kontush A; Krauss RM; Otvos JD; Remaley AT; Schafer EJ HDL measures, particle heterogeneity, proposed nomenclature, and relation to atherosclerotic cardiovascular events. Clin. Chem 2011, 57, 392–410. [DOI] [PubMed] [Google Scholar]
- (268).Lutomski CA; Gordon SM; Remaley AT; Jarrold MF Resolution of lipoprotein subclasses by charge detection mass spectrometry. Anal. Chem 2018, 90, 6353–6356. [DOI] [PubMed] [Google Scholar]