

SUMMARY
Mitochondria use the electron transport chain to generate high-energy phosphate from oxidative phosphorylation, a process also regulated by the mitochondrial Ca2+ uniporter (MCU) and Ca2+ levels. Here, we show that MCUb, an inhibitor of MCU-mediated Ca2+ influx, is induced by caloric restriction, where it increases mitochondrial fatty acid utilization. To mimic the fasted state with reduced mitochondrial Ca2+ influx, we generated genetically altered mice with skeletal muscle-specific MCUb expression that showed greater fatty acid usage, less fat accumulation, and lower body weight. In contrast, mice lacking Mcub in skeletal muscle showed increased pyruvate dehydrogenase activity, increased muscle malonyl coenzyme A (CoA), reduced fatty acid utilization, glucose intolerance, and increased adiposity. Mechanistically, pyruvate dehydrogenase kinase 4 (PDK4) overexpression in muscle of Mcub-deleted mice abolished altered substrate preference. Thus, MCUb is an inducible control point in regulating skeletal muscle mitochondrial Ca2+ levels and substrate utilization that impacts total metabolic balance.
Graphical abstract
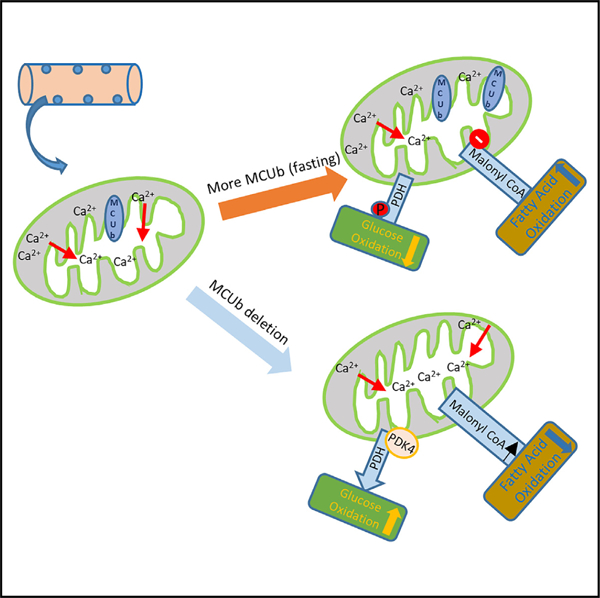
In brief
Here, we show data suggesting how the mitochondrial Ca2+ uniporter (MCU) complex has evolved the MCUb encoding gene to function as an inducible regulator of metabolism that restricts mitochondrial Ca2+ in response to environmental caloric restriction, which results in reprogramming of mitochondria toward fatty acid oxidation preference.
INTRODUCTION
Skeletal muscle can dramatically impact the overall metabolic rate and long-term energy balance of an organism. At rest or during moderate activity, aerobic metabolism within skeletal muscle serves as the major energy-producing state, while with intense exercise, skeletal muscle switches to anaerobic energy metabolism.1,2 During aerobic metabolism, both glucose and fatty acids are oxidized for energy production,3 although the utilization of either fuel source is further regulated by substrate availability and other physiological stimuli. For example, with caloric restriction, when glucose is prioritized for brain tissue, skeletal muscle preferentially utilizes fatty acids.4,5 Skeletal muscle also adapts to pathological conditions such as metabolic syndrome and type 2 diabetes, where insulin insensitivity results in metabolic inflexibility, leading to reduced fatty acid oxidation.6,7 Mitochondria are central to these disease processes and adaptation of fuel selection, but how this is regulated in skeletal muscle and other tissues is not adequately understood.
Acute mitochondrial Ca2+ elevation directly promotes aerobic metabolism by augmenting the activity of key mitochondrial metabolic enzymes, like several dehydrogenases involved in the tricarboxylic acid (TCA) cycle as well as the ATP synthase.8,9 Mitochondrial Ca2+ dynamics can also regulate substrate utilization, such as control of glucose oxidation via its rate-limiting reaction mediated by pyruvate dehydrogenase (PDH).8,10 Acute increases in mitochondrial Ca2+ stimulate PDH phosphatase 1 (PDP1), which removes the inhibitory phosphorylation of the PDH complex promoting glucose oxidation.8 However, a direct role of mitochondrial Ca2+ dynamics in stimulating or otherwise regulating fatty acid oxidation is less understood, although augmented glucose oxidation is associated with repressed fatty acid oxidation.11–13
The mitochondrial Ca2+ uniporter (MCU) complex is a primary way of importing Ca2+ into mitochondria, which can directly impact metabolism and fuel selection.14,15 We recently showed that abolishing acute mitochondrial Ca2+ uptake in skeletal muscle of genetically targeted mice for the Mcu gene impaired glucose oxidation and caused greater fatty acid oxidation, resulting in less fat accumulation with aging.16 Indeed, heart-specific deletion of Mcu in mice showed greater total work with augmented fatty acid utilization and reduced malonyl coenzyme A (CoA) levels, the rate-limiting effector of β-oxidation.17 In addition, skeletal muscle-specific Mcu-deleted mice also showed enhanced fatty acid oxidation.18
MCUb is an inhibitory subunit of the MCU complex that forms multimers with MCU, resulting in the repression of Ca2+ influx.19 In the heart, MCUb expression is induced in response to stress, such as ischemia-reperfusion (I/R) injury or ischemic preconditioning.20 Cardiac MCUb overexpression leads to repressed MCU-dependent mitochondrial Ca2+ uptake and reduced pathology following I/R injury, while loss of MCUb exaggerates I/R injury.20,21 MCUb induction has also been reported in diabetic cardiomyopathy, and inhibition of induced MCUb in these hearts resulted in augmented cardiac function and amelioration of metabolic inflexibility.22 Here we show that MCUb is selectively induced by caloric restriction in skeletal muscle, heart, liver and kidney, where it directly limits mitochondrial Ca2+ influx capacity. Mitochondria from skeletal muscle of Mcub-deleted mice no longer showed Ca2+ influx inhibition, mice lacking Mcub in skeletal muscle showed a profile of metabolic inhibition with greater fat accumulation and metabolic syndrome with aging, while mice with skeletal muscle-specific MCUb overexpression were leaner and had reduced white adipose levels with enhanced fatty acid oxidation. Thus, we show that MCUb is a fasting-induced regulator ofskeletal muscle metabolism by limiting mitochondrial Ca2+ influx and, thus, augmenting fatty acid utilization.
RESULTS
MCUb induction post fasting occurs in multiple tissues
To assess whether mitochondrial Ca2+ dynamics play a role in substrate utilization during caloric restriction, we first examined RNA expression of all MCU gene products in selected tissues following 48 h of fasting in mice (Figure 1). We observed induction in MCUb mRNA in all tissues examined except the brain, and PDH kinase 4 (PDK4) mRNA levels served as a positive control for the impact of fasting (Figures 1A–1E).23 None of the other MCU-encoding gene products were significantly altered by fasting (Figures 1A–1E). The lack of inducibility of MCUb in the brain is consistent with the fact that this tissue utilizes glucose (carbohydrates) for energy metabolism with a lack of substrate switching.24,25 Identical results were observed at the protein level in skeletal muscle, where only MCUb was induced post fasting (Figure S1A). These data suggest a correlation between MCUb induction with caloric restriction and increased fatty acid oxidation.
Figure 1. MCUb mRNA expression is induced by fasting in selected tissues.
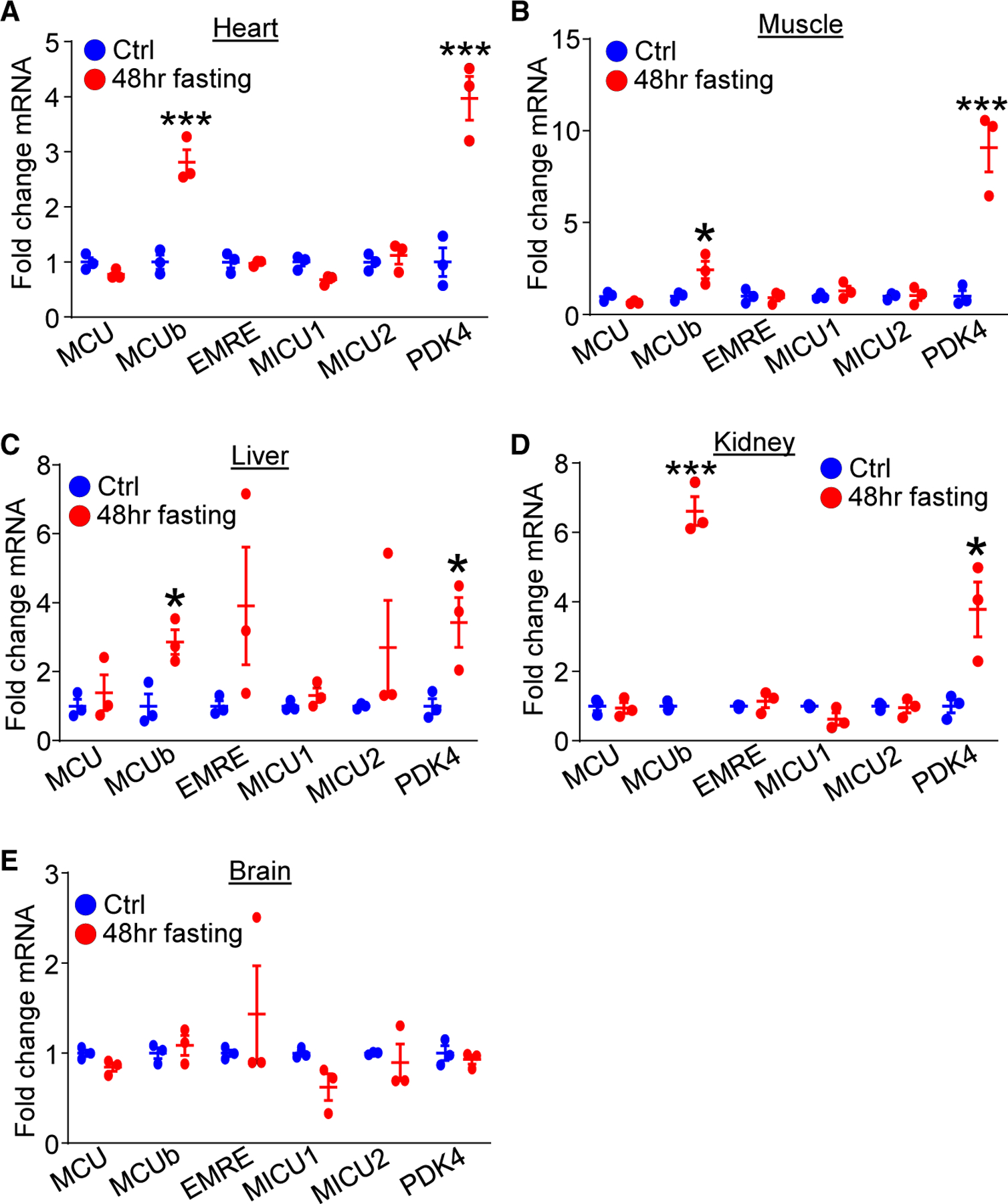
(A–E) mRNA expression of MCU gene components among (A) heart, (B) skeletal muscle, (C) liver, (D) kidney and (E) brain from control-fed mice (ctrl) and mice challenged with 48-h fasting. PDK4 was used as a control for the fasting procedure. n = 3 per group. Data are presented as mean ± SEM. Student’s t test was used for statistical analysis.
*p < 0.05, ***p < 0.001.
Deleting MCUb in skeletal muscle shifts energy metabolism toward glucose oxidation
Given the calorie-sensitive expression characteristics of MCUb, we sought to understand its function in vivo by generating mice with skeletal muscle-specific deletion. We crossed Mcub gene-targeted mice containing loxP (fl) sites across exon 2 with skeletal muscle-specific Myod gene locus-driven Cre recombinase-containing mice (Figure 2A). We observed almost complete deletion of MCUb protein in isolated muscle mitochondria at 8 weeks of age from Mcubfl/fl-Myod-Cre mice compared with controls (Figure 2B). No other MCU components were altered with Mcub deletion in skeletal muscle (Figure S1B). Deletion of Mcub from skeletal muscle did not promote histological changes in adult muscle compared with controls (Figure 2C), nor was there a change in fiber type specificity in the quadriceps, tibialis anterior (TA), or soleus or a change in myofiber cross-sectional areas (Figures S2A–S2E). Interestingly, there was no baseline difference in mitochondrial Ca2+ content with the deletion of Mcub (Figure 2D), nor was there a change in baseline oxygen consumption rate across the different mitochondrial respiratory complexes compared with the controls (Figure 2E). However, loss of MCUb protein impaired mitochondrial Ca2+ retention capacity, suggesting that MCUb has homeostatic inhibitory activity toward MCU function in skeletal muscle mitochondria (Figure 2F). Echocardiography-magnetic resonance imaging (echoMRI) analysis of whole-body composition showed progressive fat accumulation in Mcub skeletal muscle-specific deleted mice with aging (Figure 2G). Moreover, loss of MCUb protein in skeletal muscle was associated with lipid droplet accumulation compared with the controls (Figure S2F), with no changes in lipid droplet accumulation or glycogen content in the liver (Figures S2F and S2G).
Figure 2. Skeletal muscle-specific deletion of Mcub shifts energy metabolism.
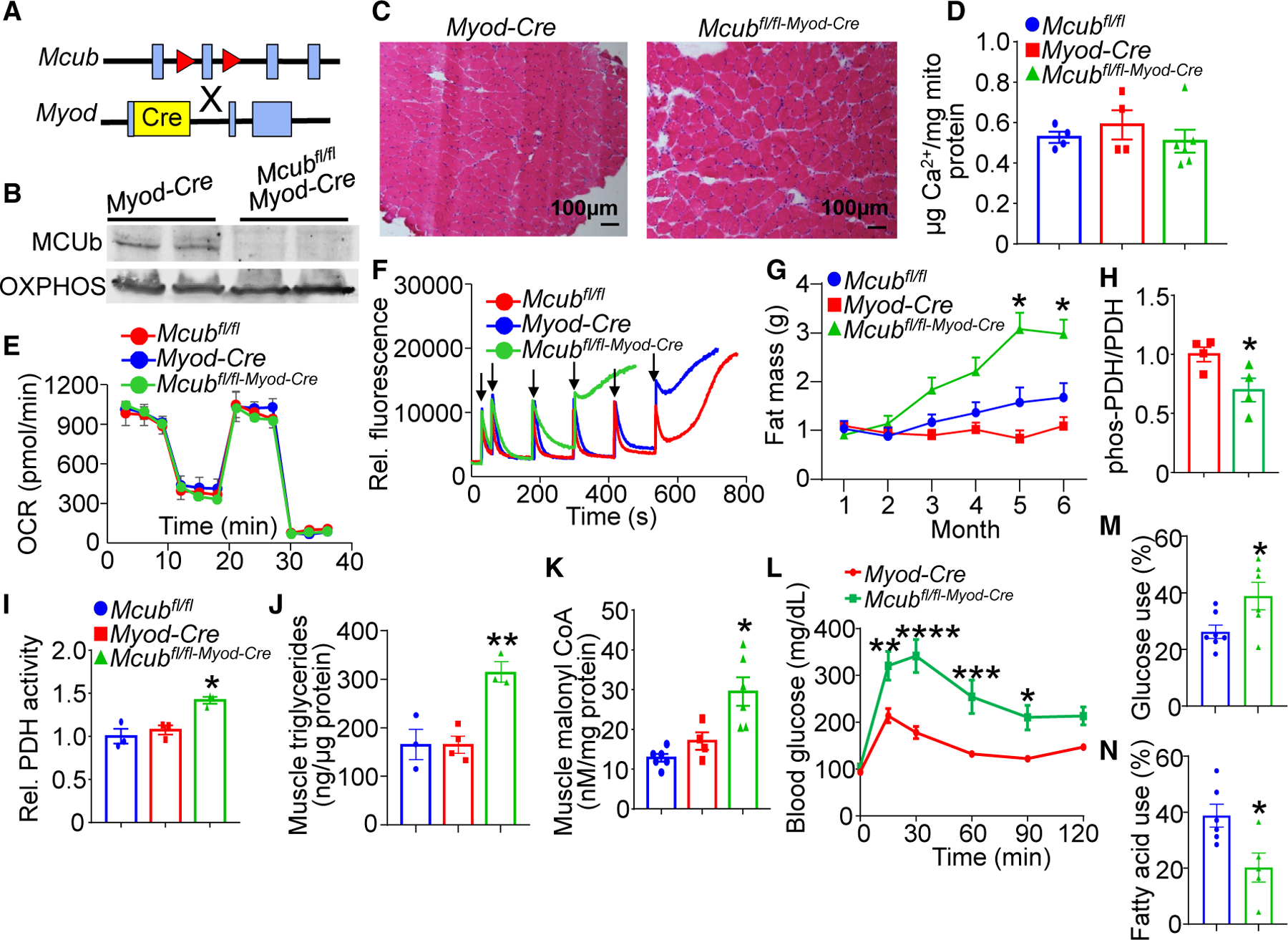
(A) Strategy to generate skeletal muscle-specific Mcub-deleted mice, with the Mcubfl/fl mouse crossed with the Myod-Cre mouse.
(B) Western blot of MCUb protein in isolated muscle mitochondria from the indicated groups of mice. Oxidative phosphorylation (OXPHOS) was used as a mitochondrial isolation and protein loading control.
(C) Representative hematoxylin and eosin (H&E)-stained soleus histological muscle sections at 100× magnification from the 2 genotypes of mice shown at 6 months of age.
(D) Quantification of baseline mitochondrial Ca2+ levels in isolated quadriceps mitochondria from the indicated groups of mice at 6 months of age. n = 4 in the Mcubfl/fl group, n = 4 in the Myod-Cre group, n = 6 in the Mcubfl/fl-Myod-Cre group. Data are presented as mean ± SEM. One-way ANOVA and Bonferroni’s multiple-comparisons test were used for statistical analysis.
(E) Oxygen consumption rate (OCR) measurement in quadriceps mitochondria from the indicated groups of mice at 4 months of age. n = 5 in the Mcubfl/fl group, n=5 in the Myod-Cre group, n = 7 in the Mcubfl/fl-Myod-Cre group.
(F) Mitochondrial Ca2+ retention capacity assay in isolated quadriceps mitochondria from the indicated groups of mice at 4 months of age. Calcium Green-5N was used as the Ca2+ indicator. The arrows indicate addition of 20 μM CaCl2 to the solution.
(G) MRI analyses of total body fat composition with aging in the indicated genotypes of mice. n = 7 in the Mcubfl/fl group, n = 8 in the Myod-Cre group, n = 8 in the Mcubfl/fl-Myod-Cre group. Data are presented as mean ± SEM. Two-way ANOVA and Bonferroni’s multiple-comparisons test were used for statistical analysis. *p < 0.05 versus either control group.
(H) Quantitation of western blotting of PDH phosphorylation over total PDH levels from the indicated genotypes of mice from quadriceps muscle at 6 months of age. n = 4 per group. Data are presented as mean ± SEM. Student’s t test was used for statistical analysis. *p < 0.05.
(I) Relative PDH enzymatic activity from quadriceps muscle from the indicated genotypes of mice at 6 months of age. n = 3 per group. Data are presented as mean ± SEM. One-way ANOVA and Bonferroni’s multiple comparison test were used for statistical analysis. *p < 0.05 versus either control group.
(J) Quadriceps triglyceride levels from the indicated mouse groups at 6 months of age. n = 3 in the Mcubfl/fl group, n = 4 in the Myod-Cre group, n = 3 in the Mcubfl/fl-Myod-Cre group. Data are presented as mean ± SEM. One-way ANOVA and Bonferroni’s multiple-comparisons test were used for statistical analysis. **p < 0.01 versus either control group.
(K) Quadriceps malonyl-CoA levels from the indicated genotypes of mice at 6 months of age. n = 6 in the Mcubfl/fl group, n = 4 in the Myod-Cre group, n = 6 in the Mcubfl/fl-Myod-Cre group. Data are presented as mean ± SEM. One-way ANOVA and Bonferroni’s multiple-comparisons test were used for statistical analysis. *p < 0.05 versus either control group.
(L) Glucose tolerance assay in the indicated groups of mice at 6 month of age. Data are presented as mean ± SEM. Two-way repeated-measures ANOVA and Bonferroni’s multiple-comparisons test were used for statistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
(M) Glucose dependency with UK5099 in isolated flexor digitorum brevis (FDB) myofibers from the indicated groups, measured as the percentage of the basal OCR (detailed in STAR Methods). N = 7 in the Mcubfl/fl group, n = 6 in the Mcubfl/fl-Myod-Cre group. Data are presented as mean ± SEM. Student’s t test was used for statistical analysis. *p < 0.05.
(N) Fatty acid dependency with etomoxir in isolated FDB myofibers from the indicated groups, presented as the percentage of the basal OCR (detailed in STAR Methods). N=6 in the Mcubfl/fl group, n=5 in the Mcubfl/fl-Myod-Cre group. Data are presented as mean ± SEM. Student’s t test was used for statistical analysis. *p < 0.05.
The increase in fat mass over time suggested a systemic metabolic shift in these mice due to deletion of Mcub in skeletal muscle. Analysis of skeletal muscle tissue showed decreased phosphorylation of PDH when normalized to total PDH levels, which would indicate elevated PDH activity and enhanced glucose oxidation with Mcub deletion (Figure 2H). Mcub-deleted mice also showed increased PDH enzymatic activity from skeletal muscle tissue extracts, supporting increased glucose oxidation (Figure 2I). Muscle triglyceride levels were significantly elevated with skeletal muscle-specific deletion of Mcub, suggesting an alteration in fatty acid metabolism (Figure 2J). To further assess the role of MCUb in fatty acid oxidation, we measured malonyl-CoA, which inhibits fatty acid oxidation by targeting enzymes controlling the rate-limiting step in β-oxidation.26 Mice with skeletal muscle-specific deletion of Mcub showed significantly enhanced muscle malonyl-CoA levels, which would impair fatty acid oxidation (Figure 2K). Consistent with these changes, glucose tolerance testing showed a defect at 6 months of age, suggesting the onset of metabolic syndrome (Figure 2L). Direct assessment of oxygen consumption in isolated myofibers showed a significant preference for glucose oxidation and a reduction in fatty acid oxidation (Figures 2M and 2N). However, these changes were balanced acutely because total indirect calorimetry showed no change in overall metabolism in skeletal muscle-specific Mcub-deleted mice, either under fed or fasted conditions (Figures S3A–S3H), although this assay is likely not sensitive enough to account for subtle changes in metabolism that could have a cumulative effect over 6 months in altering fat mass. Taken together, these results suggest prolonged impairment in fatty acid oxidation with Mcub deletion eventually leads to fat mass accumulation and secondary diabetes-like effects even though glucose oxidation is partially enhanced.
MCUb induction post fasting enhanced fatty acid oxidation with increased mitochondrial Ca2+ uptake
To further examine the function of MCUb in skeletal muscle metabolism, Mcub muscle-specific deletion mice were fasted for 48 h, and multiple assays were performed. Protein expression of MCUb from skeletal muscle mitochondria showed strong induction of expression with fasting, which was absent in mice with Mcub deletion from skeletal muscle (Figures S4A and S4B). The phosphorylated PDH/PDH ratio was enhanced in control Mcubfl/fl mice following 48 h of fasting, and this increase was significantly reduced in Mcubfl/fl-Myod-Cre mice (Figures S4A and S4C), further suggesting that Mcub deletion in skeletal muscle enhances glucose oxidation. Indeed, PDH activity was decreased in control Mcubfl/fl mice following 48 h of fasting, while Mcub deletion restored PDH activity (Figure S4D). Moreover, Mcubfl/fl control mice showed decreased malonyl-CoA levels following 48 h of fasting, which would directly lead to greater fatty acid oxidation (Figure S4E). However, muscle malonyl-CoA levels were significantly increased in Mcubfl/fl-Myod-Cre mice with fasting, consistent with the idea that fatty acid oxidation is impaired with Mcub deletion in skeletal muscle. We also observed that 48 h of fasting increased the phosphorylated acetyl CoA carboxylase (ACC)/ACC ratio, which inhibits ACC enzymatic activity, resulting in less malonyl-CoA leading to more fatty acid oxidation (Figure S4F). However, this increase in phosphorylated ACC (p-ACC)/ACC was abolished with Mcub deletion, again suggesting impaired fatty acid oxidation.
The role of MCUb induction in mitochondrial Ca2+ dynamics during fasting was also investigated. We observed that muscle-specific Mcub deletion produced increased mitochondrial Ca2+ content compared with controls during 48 h of fasting (Figure S4G). More importantly, Mcubfl/fl control mice subjected to 48 h of fasting had increased mitochondrial Ca2+ retention capacity compared with fed controls, but this was abolished with Mcub deletion (Figure S4H). These data suggest that MCUb induction during fasting normally prevents acute mitochondrial Ca2+ loading in skeletal muscle, which then augments mitochondrial fatty acid utilization in compensation for the fasted state (see Discussion).
MCUb-dependent PDK4 regulation of metabolic dynamics in muscle
We have shown previously that skeletal muscle-specific deletion of Mcu (no uniporter complex present) exhibited a shift in energy metabolism toward fatty acid utilization,16 similar to induction of MCUb protein with fasting where MCU uniporter activity is inhibited. Mcub deletion from skeletal muscle showed the antithetic metabolic signature of Mcu-deleted mice so Mcub deletion resulted in reduced fatty acid oxidation and enhanced glucose oxidation. To further investigate the downstream molecular events associated with altered mitochondrial Ca2+, we examined global RNA gene expression changes in skeletal muscle from Mcu versus Mcub-deleted mice, which, we hypothesized, would produce opposing effects on metabolism genes. Remarkably, only 1 metabolic gene showed antithetic regulation: that of PDK4 (Figure 3A). Indeed, PDK4 mRNA and protein expression were decreased in skeletal muscle from Mcub-deleted mice compared with controls (Myod-Cre and Mcubfl/fl; Figures 3B–3D). However, 48 h of fasting increased PDK4 expression in controls, which would inhibit PDH and limit skeletal muscle glucose oxidation (Figures 3B–3D), tending to push substrate utilization away from glucose and toward fatty acid oxidation. As expected, Mcub deletion limited the fasting-induced upregulation of PDK4, undermining the shift from glucose to fatty acid oxidation of fasting muscle (Figures 3B–3D). To further investigate this regulatory relationship, we overexpressed PDK4 in Mcub skeletal muscle deletion mice. We injected 2-month-old Mcubfl/fl and Mcubfl/fl-Myod-Cre mice with MyoAAV-PDK4 or MyoAAV-luciferase as a control, and the mice were sacrificedat 4 monthsofage toexamine substrate utilization (Figure3E). Overexpressing PDK4 in skeletal muscle was observed at about the same level as the endogenous increase seen with fasting but with no changes in MCUb or MCU expression (Figures 3C and 3F). With control MyoAAV-luciferase, Mcubfl/fl-Myod-Cre fibers exhibited higher glucose utilization and reduced palmitate usage compared with the Mcubfl/fl control group (Figure 3G). However, PDK4 overexpression in skeletal muscle mitigated these differences between the Mcubfl/fl and Mcubfl/fl-Myod-Cre mice, indicating that the regulatory effect of MCUb on substrate switching can be partially attributed to PDK4 (Figure 3H).
Figure 3. MCUb induction with 48 h of fasting regulates metabolic dynamics in muscle.
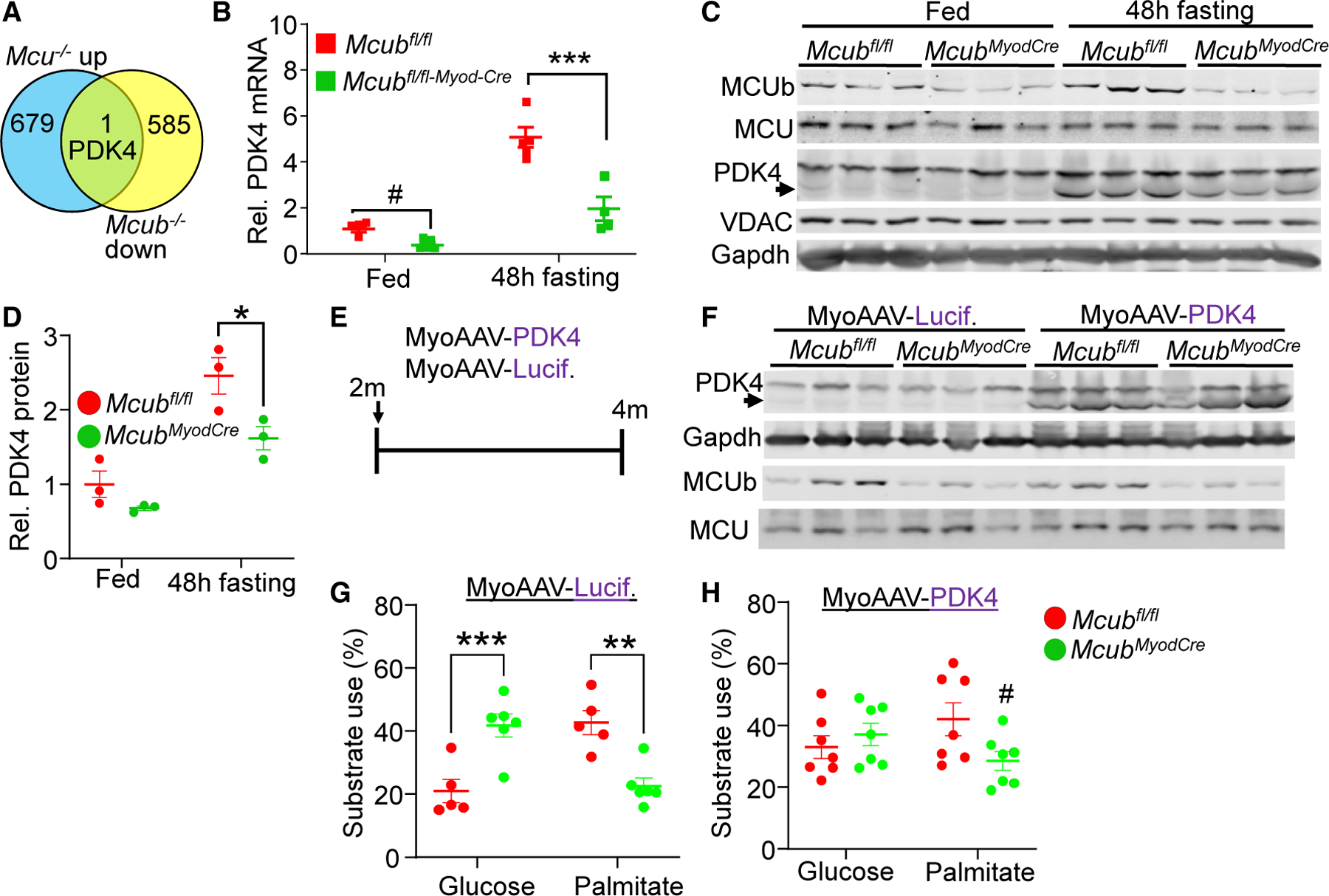
(A) Schematic identifying PDK4 as the only antithetically regulated metabolic gene from global RNA microarray analysis in muscle-specific Mcu versus Mcub-deleted mice.
(B) RNA expression of PDK4 from quadriceps at 6 months of age from the indicated groups of mice under baseline and post 48-h fasting conditions. Data are presented as mean ± SEM. ***p < 0.001 vs. the Mcubfl/fl fasting group, using two-way ANOVA and Bonferroni’s multiple-comparisons test. #p < 0.05 versus the Mcubfl/fl fed group, using Student’s t test.
(C) Representative western blot of PDK4 from quadriceps muscle from the indicated groups of mice at 6 months of age. The arrow shows the correct size to identify PDK4 from muscle. Gapdh and the voltage-dependent anion channel (VDAC) were used as controls.
(D) Quantification of PDK4 as shown in (C). n = 3 per group. Data are presented as mean ± SEM. Two-way ANOVA and Bonferroni’s multiple comparison test were used for statistical analysis. *p < 0.05.
(E) Scheme of introducing MyoAAV-PDK4 or MyoAAV-luciferase as a control at 2 months of age, followed by harvest at 4 months.
(F) Western blot of the indicated proteins from quadriceps in the indicated groups of mice. Gapdh was used as a control.
(G and H) Glucose dependency with UK5099 and fatty acid dependency with etomoxir in isolated FDB myofibers from the indicated groups, presented as the percentage of the basal OCR (detailed in STAR Methods). n = 5 in the Mcubfl/fl MyoAAV-luciferase group, n = 6 in the Mcubfl/fl-Myod-Cre MyoAAV-luciferase group, n = 7 in the Mcubfl/fl MyoAAV-PDK4 group, n = 7 in the Mcubfl/fl-Myod-Cre MyoAAV-PDK4 group. two-way ANOVA and Bonferroni’s multiple comparison test were used for statistical analysis. **p < 0.01, ***p < 0.001. #p < 0.05 vs. the Mcubfl/fl MyoAAV-PDK4-treated group using Student’s t test.
Muscle-specific MCUb expression showed reduced fat accumulation with enhanced PDK4
To further examine the role of MCUb in energy metabolism in skeletal muscle, we generated skeletal muscle-specific MCUb overexpression mice by crossing Col1a1Mcub and Myod-Cre mice (Figure 4A). The Mcub cDNA was inserted into exon 51 of the Col1a1 gene in the opposite orientation with inverted loxP sites so that Cre-mediated recombination would give cDNA expression for the first time, downstream of the promoter cassette (Figure 4A). With Myod-Cre, a significant increase in MCUb protein in muscle was observed in Col1a1Mcub-Myod-Cre mice compared with the controls (Figure 4B), and the total level of protein overexpression was similar to endogenous MCUb protein induction in muscle with caloric restriction (Figure S4A). MCUb induction in muscle showed an increase in PDK4 protein expression, further suggesting the importance of a MCUb-PDK4 circuit in regulating energy metabolism (Figures 4B and 4C). Importantly, MCUb overexpression in muscle increased mitochondrial Ca2+ retention capacity, like the increase observed with 48 h of fasting when endogenous MCUb is induced (Figure 4D). Importantly, mice with skeletal muscle-specific MCUb overexpression showed significantly reduced body weight and fat (Figures 4E and 4F). No differences were observed in interscapular brown adipose tissue mass with MCUb overexpression compared with the controls (Figure 4G). However, MCUb overexpression showed less intramuscular white adipose tissue content compared with the controls, further supporting the concept that MCUb induction shifts energy metabolism toward fatty acid utilization in skeletal muscle, leading to less adiposity (Figure 4H). Indeed, direct assessment of oxygen consumption in isolated myofibers from these mice showed a significant preference for fatty acid oxidation and a reduction in glucose oxidation with MCUb induction in muscle (Figures 4I and 4J). Taken together, these results indicate that MCUb serves as a critical control point of mitochondrial Ca2+ levels to affect substrate usage and total metabolic activity.
Figure 4. Muscle-specific MCUb induction showed reduced fat accumulation with enhanced PDK4 levels.
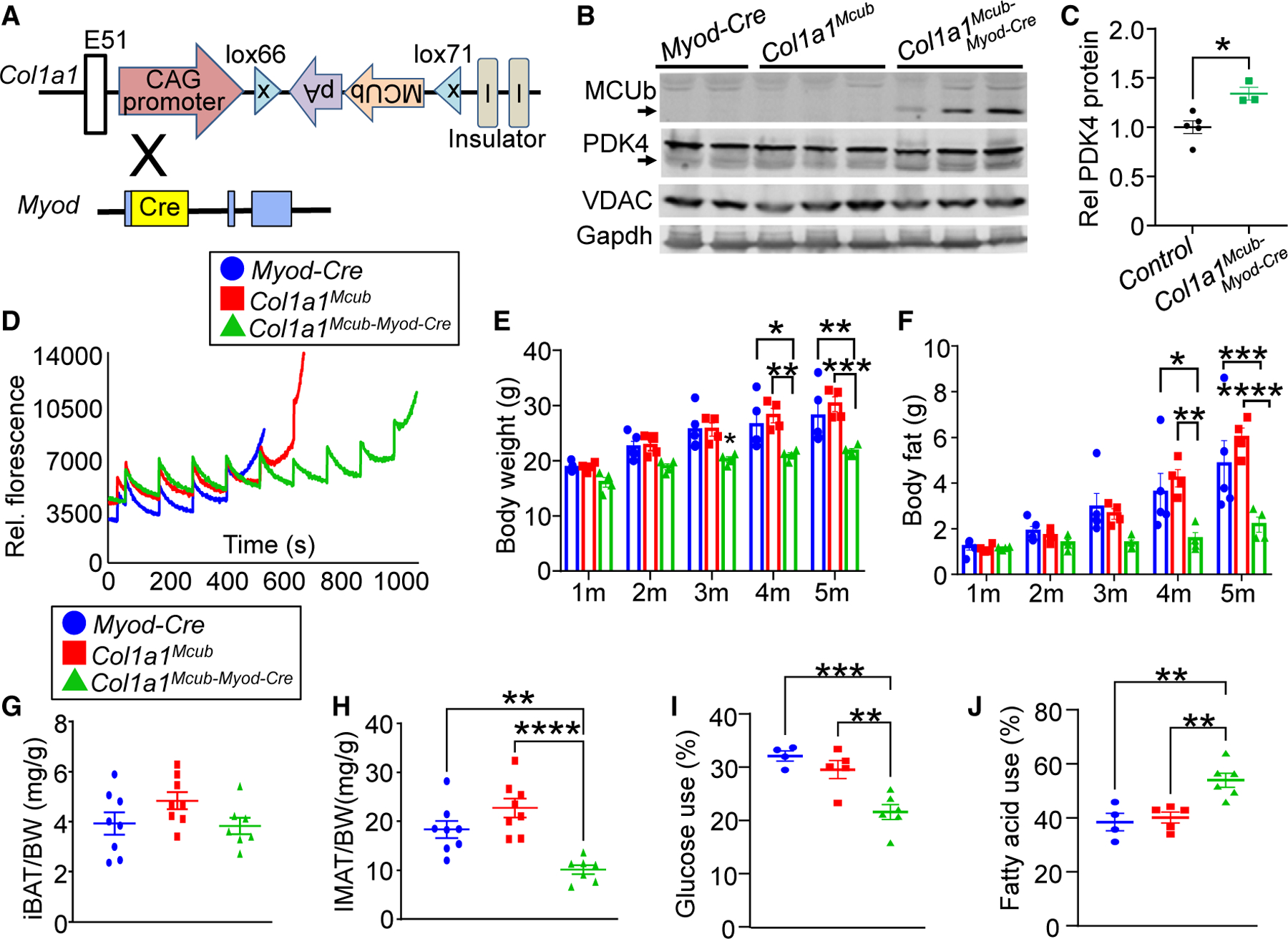
(A) Strategy to generate skeletal muscle-specific Mcub overexpression in mice using exon 51 insertion of the mouse MCUb cDNA into the Col1a1 locus in the opposite orientation with inverted loxP sites and a ubiquitous promoter. Col1a1-Mcub mice were crossed with Myod-Cre-expressing mice to generate recombination of the MCUb cDNA and constitutive expression in skeletal muscle.
(B) Western blot of the indicated proteins from quadriceps in the indicated groups of mice. Gapdh and VDAC were used as controls.
(C) Quantification of PDK4 protein expression as shown in (B). Student’s t test was used for statistical analysis. *p < 0.05.
(D) Ca2+ retention capacity assay in isolated quadriceps mitochondria from the indicated groups of mice at 3–4 months of age. Calcium Green-5N was used as the Ca2+ indicator, and Ca2+ additions were given at each pulse point.
(E) MRI analyses of total body mass with aging in the indicated genotypes of mice. n = 4 per group. Data are presented as mean ± SEM. Two-way ANOVA and Bonferroni’s multiple comparison test were used for statistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001.
(F) MRI analyses of total body fat composition with aging in the indicated genotypes of mice. n = 4 per group. Data presented as mean ± SEM. Two-way ANOVA and Bonferroni’s multiple-comparisons test were used for statistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
(G) Interscapular brown adipose tissue (iBAT) weight-to-body weight ratio in the indicated genotypes of mice. n = 8 in the Myod-Cre group, n = 8 in the Col1a1Mcub group, n = 7 in the Col1a1Mcub-Myod-Cre group. Data are presented as mean ± SEM. One-way ANOVA and Bonferroni’s multiple comparison test were used for statistical analysis.
(H) Intermuscular adipose tissue (IMAT) weight-to-body weight ratio in the indicated genotypes of mice. n = 8 in the Myod-Cre group, n = 8 in the Col1a1Mcub group, n = 7 in the Col1a1Mcub-Myod-Cre group. Data are presented as mean ± SEM. One-way ANOVA and Bonferroni’s multiple comparison test were used for statistical analysis. **p < 0.01, ****p < 0.0001.
(I) Glucose dependency with UK5099 in isolated FDB myofibers from the indicated groups of mice, measured as percentage of basal OCR (detailed in STAR Methods). n = 4 in the Myod-Cre group, n = 5 in the Col1a1Mcub group, n = 6 in the Col1a1Mcub-Myod-Cre group. Data are presented as mean ± SEM. One-way ANOVA and Bonferroni’s multiple comparison test were used for statistical analysis. **p < 0.01, ***p < 0.001.
(J) Fatty acid dependency with etomoxir in isolated FDB myofibers from the indicated groups of mice, measured as the percentage of basal OCR (detailed in STAR Methods). n = 4 in the Myod-Cre group, n = 5 in the Col1a1Mcub group, n = 6 in the Col1a1Mcub-Myod-Cre group. Data are presented as mean ± SEM. One-way ANOVA and Bonferroni’s multiple comparison test were used for statistical analysis. **p < 0.01.
DISCUSSION
Mitochondria directly sample intracellular Ca2+ oscillations to adapt energy output to the physiological state. Acute augmentation in mitochondrial matrix Ca2+ activates energy production by stimulating several dehydrogenases involved in the TCA cycle as well as by stimulating the maximal enzymatic rate of the ATP synthase.8,27 One key mitochondrial Ca2+-regulated enzyme is PDH, catalyzing the irreversible conversion of pyruvate to acetyl CoA, which is the rate-limiting step in glucose oxidation. PDH activity is directly regulated by the Ca2+-activated PDPs, especially PDP isoform 1, which dephosphorylates and reinstates PDH complex activity.11–13 While mitochondrial Ca2+ regulates glucose metabolism via its rate-limiting reaction, the direct role of mitochondrial Ca2+ in fatty acid metabolism is not fully understood. Here we identified a role of mitochondrial Ca2+ dynamics in fatty acid oxidation via malonyl-CoA, a highly regulated metabolite that serves as the rate-limiting step in β-oxidation, and this metabolite plays a critical role in balancing between states of glucose verses fatty acid usage.28,29 Thus, changes in mitochondrial matrix Ca2+ levels can impact fuel selection and overall rates of glucose and fatty acid oxidation through malonyl-CoA levels in skeletal muscle.26
The mitochondrial Ca2+ uniporter, which underlies acute alterations of Ca2+ in the matrix of the mitochondria, has been implicated as a regulator of energy metabolism. Remarkably, mice lacking the Mcu gene, which show no acute Ca2+ influx in isolated mitochondria, were partially viable as adults with enhanced PDH phosphorylation, decreased PDH activity, as well as increased lactate levels in skeletal muscle, suggesting impaired glucose oxidation.15 Similarly, heart-specific Mcu deletion in mice resulted in increased PDH phosphorylation, decreased PDH activity, and an increase in the nicotinamide adenine dinucleotide (NAD+)/nicotinamide adenine dinucleotide H (NADH) ratio.30,31 Mcu gene deletion in cardiomyocytes showed impaired acute energy production after β-adrenergic stimulation,30,31 and mice with heart-specific Mcu deletion were deficient in rapidly adapting to maximal treadmill performance, indicating that mitochondrial Ca2+ influx is critical for acute “fight-or-flight” increases in energy production.31 However, in the long term, Altamimi et al.17 observed enhancement in both glucose and fatty acid oxidation in heart-specific Mcu-deleted mice at baseline or with isoproterenol stimulation. Mechanistically, they provided evidence that MCU-regulated mitochondrial Ca2+ levels impacted fatty acid oxidation via malonyl-CoA with acetylation of key enzymes involved in fatty acid oxidation.
Mice with skeletal muscle-specific Mcu deletion also showed altered substrate utilization with increased PDH phosphorylation as well as impaired PDH activity.16 Skeletal muscle-specific Mcu-deleted mice showed reduced malonyl-CoA levels following exercise, and these mice showed less fat accumulation with aging and enhanced fatty acid oxidation.16 Gherardi et al.18 also reported enhanced fatty acid oxidation in muscle-specific Mcu-deleted mice,18 with greater fatty acid dependency, less PDH activity, and impaired exercise performance.
Here, we observed that deletion of Mcub in skeletal muscle had the antithetic effect to Mcu deletion. Mcub-null muscle showed augmented mitochondrial Ca2+ uptake with increased glucose oxidation and impaired fatty acid oxidation, while MCUb-overexpressing mice had a Ca2+ retention capacity profile suggesting lower mitochondrial Ca2+ and a molecular profile suggesting augmented fatty acid utilization. Skeletal muscle-specific Mcub-deleted mice also showed greater fat mass with aging, less fatty acid oxidation through malonyl-CoA inhibition, and features of secondary metabolic syndrome, while the skeletal muscle-specific MCUb-overexpressing mice had reduced body weight and reduced white adipose levels. Mcub deletion in skeletal muscle also resulted in decreased PDH phosphorylation and enhanced PDH activity, suggesting greater glucose oxidation. Moreover, Mcub null mice showed decreased PDK4 levels, which contributes to the preferential glucose utilization in muscle, while MCUb overexpressors had increased PDK4 levels.
Because MCUb is the only starvation-inducible gene component of the MCU complex, it suggests a critical role of MCUb in adapting mitochondrial Ca2+ to altered caloric states. Indeed, fasting induces MCUb expression in skeletal muscle in 1–2 days, and then either extinguishes a previous flight-or-flight mechanism of rapid Ca2+ influx or it reduces baseline mitochondrial Ca2+ influx, thereby ensuring that muscle relies more heavily on fatty acid oxidation, preserving glucose for total systemic usage. Indeed, genetically altered mice with forced MCUb protein expression in skeletal muscle showed decreased body weight and white fat levels in muscle due to augmented fatty acid oxidation. Hence, MCUb serves as a critical regulator of total systemic metabolism by fine-tuning mitochondrial Ca2+ homeostasis, explaining why skeletal muscle specific Mcub-deleted mice add fat mass with aging and why Mcu-null mice have reduced fat mass like the muscle-specific MCUb-overexpressing mice described here. Thus, our observations provide insight into how the MCU complex has evolved MCUb to function as a dynamic regulator of metabolism by restricting mitochondrial Ca2+ in response to environmental caloric conditions. Our results also suggest that development of a non-toxic and cell-permeable MCU inhibitor could have an anti-obesity effect.
Limitations of the study
Menezes-Filho et al.32 have shown impaired acute mitochondrial Ca2+ uptake and decreased mitochondrial Ca2+ retention capacity in liver mitochondria following overnight fasting, results that are consistent with MCUb induction in liver with fasting, which would inhibit mitochondrial Ca2+ uptake. However, Paillard et al.33 have shown distinct MCU-dependent mitochondrial Ca2+ uptake profiles and oxidative metabolism among heart, muscle, and liver.33 Unlike the heart, where fatty acid is the predominant fuel for energy metabolism, there is less evidence suggesting whether glucose or fatty acid predominates in skeletal muscle as the baseline energy substrate.34,35 Our data show that increased acute mitochondrial Ca2+ uptake with Mcub deletion enhances glucose oxidation, which also resulted in impaired fatty acid oxidation through malonyl-CoA and the Randle cycle.36 However, for our observations to reflect the known accumulation of fat in Mcub muscle-specific null mice with aging, fatty acid utilization must play a more important role in skeletal muscle than currently appreciated, which would override a potentially less central role of augmented glucose oxidation that we observed with Mcub deletion. Indeed, earlier studies have demonstrated that the concentration of malonyl-CoA in skeletal muscle is diminished by 80% after 48 h of fasting,37 which supports the dramatic need for fatty acid oxidation in the fasting state of muscle. Human studies have shown a significant increase in fat oxidation with a reduction in glucose oxidation with prolonged fasting, suggesting again that the skeletal muscle metabolism can alter total body caloric balance and propensity for obesity by burning fatty acids.38 However, a limitation of this hypothesis and the current study is the lack of understanding how forcing skeletal muscle to use fatty acid fuel sources over glucose leads to weight loss and an augmented systemic metabolic profile.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Jeffery D, Molkentin (jeff.molkentin@cchmc.org).
Materials availability
All mice generated in the laboratory of the corresponding investigator will be provided upon request, as well as the plasmids encoding the AAV vectors that were used. All original microarray data generated from skeletal muscle of control versus Mcufl/fl-Myod-Cre versus Mcubfl/fl-Myod-Cre mice are deposited with NCBI’s Gene Expression Omnibus database, with release upon acceptance of the manuscript (GSE205193). Any information required to reanalyze the data reported in this paper is available from the lead contact.
Data and code availability
This paper does not report any original code.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animals
Mice were housed and cared for in an AAALAC-accredited animal care facility at Cincinnati Children’s Hospital Medical Center, which meets or exceeds the requirements of the Office of Laboratory Animal Welfare. Mice were inspected daily by veterinary technicians to ensure health. All animal experiments were approved by the Institutional Animal Care and Use Committee of the Cincinnati Children’s Hospital Medical Center (Protocol# IACUC 2021–0047). The number of mice used in this study reflects the minimum number needed to achieve statistical significance based on previous power analysis and our experience with such assays. All mice used in this study were in the C57BL/6 genetic background and both sexes of mice were used in equal ratios and all animals were housed at 21–22°C, 40–60% humidity, 12-h light/12-h dark cycle, and unless specified differently they had free access to food and water ad libitum. A maximum duration of 48 h of fasting was used for some experiments. Forty-eight hours of fasting was also selected to ensure that caloric restriction was maximally achieved apart from unknown residual nutrition from the digestive system. Mice were randomly assigned to groups and experiments were conducted with blinding when possible. Ages of the mice and numbers used are described in the figure legends or results section for each experiment.
Skeletal muscle-specific Mcub deleted mice (Mcubfl/fl-Myod-Cre) were generated by crossing Mcubfl/fl mice with mice containing a Cre cDNA inserted into the Myod genetic locus (Myod-Cre).20,39 We also utilized Mcufl/fl-Myod Cre mice as previously published,16 which here we used to generate muscle RNA for comparison analysis with Mcub deleted muscle.
To generate tissue-specific MCUb overexpressing genetically targeted mice, a Col1a1-MCUb cDNA containing targeting vector was constructed extending 2.5 kb upstream and 3 kb downstream of the sgRNA target sequence (GGGAGGAAACCTGCCCTTGG) just downstream of the 3′-untranslated region (UTR) of the Col1a1 gene at the exon 51, which does not impact expression of the Col1a1 gene.40,41 The constructed DNA targeting vector also contain a CAG promoter, lox66, an inverted SV40 poly(A)-addition signal, lox71, and two copies of insulators.42 An inverted cDNA coding for mouse Mcub was amplified from BC049571 (Horizon Discovery) and was inserted into the targeting vector using a Mlu I restriction site following the inverted SV40 poly(A) signal. Col1a1 Locus targeted mice were generated via pronuclear injection of newly fertilized C57BL/6 embryos at the 1-cell stage with Cas9 protein (IDT, Catalog #1081061), synthetic sgRNA (IDT), and the targeting vector at a concentration of 40 ng/μL, 27 ng/μL, and 4 ng/μL, respectively. The injected embryos were transferred on the same day into the oviductal ampulla of pseudopregnant CD-1 females (Charles River Laboratories, Strain code 022) at approximately 25 embryos per recipient. The pups were born and genotyped by long-range PCR and Sanger sequencing. Muscle-specific MCUb overexpression mice were generated by crossing Col1a1-Mcub and Myod-Cre mice. Both alleles were maintained in the heterozygous state.
AAV virus generation and delivery
Pdk4 cDNA was cloned from genomic cDNA generated from mouse heart tissue using CloneAmp HiFi polymerase (Clontech Laboratories). The cDNA was then cloned into the pAAV-MCS expression vector (Cell Biolabs Inc.) utilizing the NEBuilder HiFi DNA Assembly polymerase (New England BioLabs) (for primers, see key resources table). MyoAAV-PDK4 virus was produced in-house using the MyoAAV 1A capsid as previously described.43,44 Briefly, AAV Pro 293T cells (Takara) were plated in 15 cm dishes at a density of 2 ×107 cells/dish. The next day, each plate was transfected with 10.5 μg helper plasmid, 5.25 μg of the Rep/Cap plasmid, and 5.25 μg of the PDK4 MyoAAV-1A plasmid. Recombinant virus was harvested from the cells and media, and purified by ultracentrifuge using an iodixanol gradient.43 AAV titers were quantified by Taqman-based qPCR with C1000 Touch Thermal Cycler (Bio-Rad Laboratories). MyoAAV-luciferase was used as the control virus. For delivery, virus was delivered 4xE1013 vg/Kg of mice via retro-orbital injection at 2-month of age.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| MCUb (rabbit polyclonal against | custom-made and affinity purified | N/A |
| mouse MCUb) | from YenZym Antibodies | |
| MCU | Cell Signaling Technology | RRID:AB_2721812 |
| CBARA1/MICU1 (D4P8Q) | Cell Signaling Technology | RRID:AB_2797943 |
| MICU2 | Bethyl laboratories | Catalog# A300-BL19212 |
| PDK4 | Novus Biologicals | RRID:AB_1625832 |
| phospho-PDHE1α Ser293 | Novus Biologicals | Catalog#NB110-93479; RRID:AB_1237282 |
| PDHE1α | Abcam | Catalog#Ab110330; RRID:AB_10858459 |
| phospho-Acetyl-CoA Carboxylase Ser79 | Cell Signaling Technology | Catalog#3661S; RRID:AB_330337 |
| Acetyl-CoA Carboxylase | Cell Signaling Technology | Catalog#3662S; RRID:AB_2219400 |
| OXPHOS cocktail | Abcam | Catalog#: ab110413; RRID:AB_2629281 |
| VDAC | Abcam | Catalog#: ab14734; RRID:AB_443084 |
| GAPDH | Fitzgerald | Catalog#: 10R-G109A; RRID:AB_1285808 |
| Myh-7 | Developmental Studies Hybridoma Bank | RRID:AB_10572253 |
| Myh-2 | Developmental Studies Hybridoma Bank | RRID:AB_2147165 |
| Myh-4 | Developmental Studies Hybridoma Bank | RRID:AB_2266724 |
| Laminin | Sigma-Aldrich | Catalog#: L9393; RRID:AB_477163 |
| IRDye® 800CW Goat anti-Rabbit IgG Secondary Antibody | LI-COR Biosciences | Catalog#: 926-32211; RRID:AB_621843 |
| IRDye® 680RD Goat anti-Mouse IgG Secondary Antibody | LI-COR Biosciences | Catalog#: 925-68070; RRID:AB_2651128 |
| Goat anti Mouse IgG2b Secondary Antibody, Alexa Fluor 568 | Thermo Fisher Scientific | Catalog#: A21144; RRID:AB_2535780 |
| Goat anti Mouse IgG2b Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Catalog#: A-21141; RRID:AB_2535778 |
| Goat anti Mouse IgG1 Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific | Catalog#: A21240; RRID:AB_2535809 |
| Goat anti-Mouse IgG Secondary Antibody, Alexa Fluor 405 | Thermo Fisher Scientific | Catalog#: A-31553; RRID:AB_221604 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| MyoAAV-PDK4 | This paper | N/A |
| MyoAAV-luciferase | This paper | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| RIPA buffer | Sigma-Aldrich | Catalog#: R0278 |
| cOmplete™, Mini Protease Inhibitor Cocktail | Millipore Sigma | Catalog#: 4693124001 |
| PhosSTOP Phosphatase Inhibitor Cocktail Tablets, EASYpack | Roche | Catalog#: 04906837001 |
| TRIzol™ Reagent | Thermo Fisher Scientific | Catalog#: 15596018 |
| SuperScript® III First-Strand Synthesis SuperMix | Thermo Fisher Scientific | Catalog#: 18080400 |
| SsoAdvanced™ Universal SYBR® Green Supermix | Bio-Rad Laboratories | Catalog#: 172-5274 |
| D-Mannitol, ≥98% | Sigma-Aldrich | Catalog#: M4125-100G |
| Sucrose | Sigma-Aldrich | Catalog#: S9378 |
| HEPES | Sigma-Aldrich | Catalog#: H-3375 |
| EGTA | Millipore Sigma | Catalog#: 324626 |
| Trypsin | Worthington Biochemical Corporation | Catalog#: LS003703 |
| Bovine Serum Albumin | Sigma-Aldrich | Catalog#: A6003-25G |
| Potassium Chloride | Sigma-Aldrich | Catalog#: P5405-250G |
| Magnesium Chloride | Fluka | Catalog#: 63065 |
| Potassium Phosphate monobasic | Thermo Fisher Scientific | Catalog#: BP362-500 |
| Seahorse XF DMEM medium | Agilent Technologies | Catalog#: 103575-100 |
| Sodium Pyruvate solution | Cytiva | Catalog#: SH30239.01 |
| L-Malic Acid | Sigma-Aldrich | Catalog#: M-1125 |
| Seahorse XF Palmitate-BSA FAO Substrate | Agilent Technologies | Catalog#: 102720-100 |
| Seahorse XF Glucose Solution | Agilent Technologies | Catalog#: 103577-100 |
| Calcium Green™−5N | Thermo Fisher Scientific | Catalog#: C-3737 |
| Calcium Chloride | Thermo Fisher Scientific | Catalog#: C79-500 |
| Adenosine 5′-diphosphate sodium salt | Sigma-Aldrich | Catalog#: A2754-500MG |
| Corning Laminin, Mouse | Thermo Fisher Scientific | Catalog#: CB-40232 |
| Sodium Chloride | Thermo Fisher Scientific | Catalog#: BP358-10 |
| 10% Buffered Formalin Phosphate | Thermo Fisher Scientific | Catalog#: SF100-4 |
| Igepal CA-630 (NP-40) | Sigma-Aldrich | Catalog#: I8896-100mL |
| Paraformaldehyde (32%) | Electron Microscopy Sciences | Catalog#: 15714 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Calcium Detection Kit | Abcam | Catalog#: ab102505 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent Technologies | Catalog#: 103015-100 |
| Seahorse XF Mito Fuel Flex Test Kit | Agilent Technologies | Catalog#: 103260-100 |
| Pyruvate dehydrogenase (PDH) Enzyme Activity Microplate Assay Kit | Abcam | Catalog#: ab109902 |
| Malonyl coenzyme A ELISA Kit | MyBioSource | Catalog#: MBS705127 |
| Glycogen Assay Kit | Millipore Sigma | Catalog#: MAK016 |
| Triglyceride Assay Kit - Quantification | Abcam | Catalog#: ab65336 |
|
| ||
| Deposited data | ||
|
| ||
| Expression Omnibus database | In house (Affymetrix, | GEO: GSE205193 |
| (GSE205193). Wt TA muscle versus Mcufl/fl-Myod-Cre and Mcubfl/fl-Myod-Cre | Clariom S Mouse) | |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: Myod-Cre, C57BL/6 | Previously generated | https://doi.org/10.1016/j.ydbio.2009.05.554 |
| Mouse: Mcubfl/fl, C57BL/6 | Previously generated | https://doi.org/10.1161/CIRCRESAHA.119.316369 |
| Mouse: Mcufl/fl—Myod-Cre, C57BL/6 | Previously generated | https://doi.org/10.1172/jci.insight.121689 |
| Mouse: Col1a1MCUb, C57BL/6 | This paper | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| MCU forward: GTGCCCTCTGATGACGTGACGG | Thermo Fisher Scientific | N/A |
| MCU reverse: ATGACAAGCTTAAAGTCATG | Thermo Fisher Scientific | N/A |
| MCUb forward: GAAGAGCCAAGTGGAGAGCA | Thermo Fisher Scientific | N/A |
| MCUb reverse: TTCCGACCGGGCTTCTATTG | Thermo Fisher Scientific | N/A |
| MICU1 forward: ACACCCTCAAGTCTGGCTTAT | Thermo Fisher Scientific | N/A |
| MICU1 reverse: TTCCCATCTTTGAAGTGCTTCTT | Thermo Fisher Scientific | N/A |
| MICU2 forward: TCGGCGCAGAAAAATTATTTGG | Thermo Fisher Scientific | N/A |
| MICU2 reverse: GTGTCATGTAATACTCTCCGTCG | Thermo Fisher Scientific | N/A |
| EMRE forward: TCTACACCGTACCGGGCAG | Thermo Fisher Scientific | N/A |
| EMRE reverse: AGTGTCCCGACATAGAGAAAGG | Thermo Fisher Scientific | N/A |
| PDK4 forward: AGGGAGGTCGAGCTGTTCTC | Thermo Fisher Scientific | N/A |
| PDK4 reverse: GGAGTGTTCACTAAGCGGTCA | Thermo Fisher Scientific | N/A |
| RPL7 forward: GAAGCTCATCTATGAGAAGGC | Thermo Fisher Scientific | N/A |
| RPL7 reverse: AAGACGAAGGAGCTGCAGAAC | Thermo Fisher Scientific | N/A |
| PDK4 forward (cloning): ATGAAGGCAGCCCGC | Thermo Fisher Scientific | N/A |
| PDK4 reverse (cloning): TCACACTGCCAGCTTCT | Thermo Fisher Scientific | N/A |
| PDK4 forward (infusion): TGGGATTCGAACATCGATATGAAGGCAGCCCGC | Thermo Fisher Scientific | N/A |
| PDK4 reverse (infusion): ACCCGTAGATCTCTCGAGTCACACTGCCAGCTTCT | Thermo Fisher Scientific | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Odyssey CLx Imaging System | LI-COR Biosciences | https://www.licor.com/bio/odyssey-dlx/ |
| GraphPad Prism 9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | N/A | https://ImageJ.nih.gov/ij/ |
| NIS Elements | Nikon Instruments Inc. | RRID:SCR_014329 |
| PTI FelixGX | Horiba Scientific | https://www.horiba.com/usa/scientific/products/detail/action/show/Product/pti-felixgx-1652/ |
| Transcriptome Analysis Console | Applied Biosystems, Thermofisher | Software version 4.0.2 |
| iPathwayGuide | Advaita Bioinformatics | Software Version. 1.4.0 |
|
| ||
| Other | ||
|
| ||
| Omni Tissue Master 125 Handheld Homogenizer | Omni International, Inc | https://pr.vwr.com/store/product/12377007/omni-tissue-master-125-handheld-homogenizer-omni-international-inc |
| Seahorse XF Pro Analyzer | Agilent Technologies, Inc. | https://www.agilent.com/en/product/cell-analysis/real-time-cell-metabolic-analysis/xf-analyzers/seahorse-xf-pro-analyzer-1980223 |
| Seahorse XFe24 Analyzer | Agilent Technologies, Inc. | https://www.agilent.com/en/product/cell-analysis/real-time-cell-metabolic-analysis/xf-analyzers/seahorse-xfe24-analyzer-740878 |
| PTI QuantaMaster™ 800 | Horiba Scientific | https://www.horiba.com/fileadmin/uploads/Scientific/Fluorescence/Downloads/QM-800.pdf |
| Nikon A1 confocal Laser Microscope | Nikon Instruments Inc. | RRID:SCR_020318 |
| Olympus BX60 Fluorescent microscope | Olympus Life Science | https://www.olympus-lifescience.com/en/technology/museum/micro/1993_02/ |
| Bio-Rad CFX96 Real-Time PCR Detection System | Bio-Rad | RRID:SCR_018064 |
| Direct Detect® Spectrometer | Millipore Sigma | Catalog#: C134681 |
| Direct Detect® assay-free cards | Millipore Sigma | Catalog#: DDAC00010-8P |
| Contour Next EZ Blood Glucose Meter | Ascensia Diabetes Care | https://www.ascensiadiabetes.com/products/contour-next-ez/?utm_source=google&utm_medium=cpc&utm_campaign=brand |
| Bioruptor™ Standard | Diagenode Inc. | Catalog#: UCD-200 TM (1.5 mL) |
METHOD DETAILS
Western blotting
Skeletal muscle tissue or isolated muscle mitochondria were lysed in RIPA buffer (Sigma-Aldrich) on ice containing protease/phosphatase inhibitor cocktail (Millipore Sigma). Muscle tissues were minced on ice and homogenized using Omni Tissue Master 125 Handheld Homogenizer (Omni International, Inc). After sitting on ice for 10 min, muscle tissues were then sonicated using a Biorupter UCD-200 sonicator (Diagenode Inc.) at 4°C for 5 min followed by 14,000 rpm centrifugation at 4°C for 10 min to get clean protein lysate. Muscle mitochondria were prepared as described in the section of “Mitochondrial isolation and analyses” below. After lysis in RIPA buffer, isolated muscle mitochondria were sonicated at 4°C for 5 min followed by 14,000 xg centrifugation at 4°C for 10 min. Protein concentration was measured using Direct Detect Spectrometer (Millipore Sigma). Protein lysates were loaded onto a 6–12% SDS-PAGE gel and electrophoresed at 100 V and transferred onto a 0.2 μm nitrocellulose membrane (Bio-Rad Laboratories) at 100 V for 1 h on ice. Western blots were performed using primary antibodies (listed in the key resources table) at 4°C overnight. Fluorescence-based secondary antibodies were used and incubated at room temperature for 1 h (see STAR Methods listing of antibodies). Membranes were analyzed using Odyssey CLx Imaging System (LI-COR Biosciences).
Real-time qPCR RNA measurements
RNA extraction from different tissues (heart, quadriceps muscle, liver, kidney, and brain) of mice was performed as previously described.20 Briefly, RNA was extracted from indicated tissues using TrizolTM Reagent and cDNA was synthesized using SuperScript III First-Strand Synthesis SuperMix. RT-qPCR was performed with C1000 Touch Thermal Cycler (Bio-Rad Laboratories) using SsoAdvanced Universal SYBR Green Supermix. RT-qPCR cycling conditions were: 95°C for 30 s, [95°C for 15 s, 60°C for 30 s, followed by fluorescence reads] (40 cycles), 65°C–95°C melting curve with 0.5°C inclement every 5 s, followed by fluorescence reads. The cycle threshold values were normalized to the values obtained from the RPL7 internal loading control. Data were presented as fold change relative to controls. Sequences of primers used in RT-PCR are listed in the key resources table.
Mitochondrial isolation and analyses
Muscle mitochondria were isolated by differential centrifugation as described previously.15,16 Briefly, skeletal muscles were harvested from mice and minced in 5 mL MS-EGTA buffer (225 mmol/L mannitol, 75 mmol/L sucrose, 5 mmol/L HEPES and 1 mmol/L EGTA, pH 7.4) containing 0.001 g Trypsin (220 unit/mg protein) for 2 min. The digestion was stopped by adding 5 mL MS-EGTA buffer containing 0.2% bovine serum albumin. Three milliliters of the solution was transferred for homogenization with a Potter-Elvehjem tissue homogenizer (VWR International, LLC). The homogenates underwent 1,000 xg centrifugation for 5 min, and the supernatants were transferred for a second spin at 10,000 xg for 10 min to generate mitochondrial pellets. The mitochondrial pellets were re-suspended in selected buffers depending on analyses (see below).
For mitochondrial Ca2+ content measurements, isolated muscle mitochondria were re-suspended in de-ionized H2O and sonicated for 5 min. Samples were centrifuged at 12,000 xg for 5 min at 4°C and supernatants were used to measure baseline mitochondrial Ca2+ content with a Calcium Detection Kit (Abcam). One hundred micrograms of mitochondria were added as directed by the vendor’s protocol.
For the mitochondrial Ca2+ retention capacity assay, which shows Ca2+ uptake until the mitochondrial permeability pore opens, isolated muscle mitochondria were re-suspended in KCl buffer (125 mmol/L KCl, 20 mmol/L HEPES, 2 mmol/L MgCl2, 2 mmol/L KH2PO4, and 40 μmol/L EGTA, pH 7.2). Mitochondrial Ca2+ retention capacity was measured using a PTI QuantaMaster Spectrofluorometer (HORIBA), with excitation as 506 nm and emission as 533 nm. One milligram of isolated muscle mitochondria was added to the KCl buffer containing 1 μmol/L malate, 7 μmol/L pyruvate, 250 nmol/L of Calcium Green 5-N (Thermo Fisher Scientific). Malate and pyruvate were substrates to activate mitochondrial function. Calcium Green 5-N was used as Ca2+ indicator in the solution. Twenty micromolar CaCl2 solution with 2 μL aliquots were given sequentially until mitochondria swelled and the pore opened, hence no longer taking up Ca2+.
For oxygen consumption rate measurements, isolated muscle mitochondria were re-suspended in KCl buffer containing 25 mmol/L pyruvate, 10 mmol/L malate and 5 mmol/L ADP. Oxygen consumption rates were determined with the Mito Stress Test Kit using the standard protocol provided by Agilent Technologies with a Seahorse Extracellular Flux (XF) Analyzer (Agilent Technologies) as described previously.20,45 Briefly, after basal respiration was measured, the mitochondria were treated sequentially with 2 μmol/L oligomycin, 5 μmol/L carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP), and 0.5 μmol/L Rotenone/antimycin A to generate the 4 characteristic plateaus of the traces shown.
Mito fuel selection assay in isolated myofibers
The Mito fuel examination assay was performed in isolated mouse flexor digitorum brevis (FDB) myofibers as previously described.16,18 Briefly, FDB muscle myofibers were isolated and plated on laminin-precoated cell culture 96-well plates formulated for oxygen consumption rate (OCR) analysis using the Seahorse system (Agilent Technologies). Following a 2-h incubation, assay media containing 5 mmol/L glucose and/or 100 μmol/L palmitate-bovine serum albumin (BSA) were replaced and cultured in non-CO2 chambers for 45 min before the direct assessment in the Seahorse instrument. OCR was determined using the Mito Fuel Selection Test Kit (Agilent Technologies) using the modified protocol provided by Agilent Technologies with a Seahorse Extracellular Flux (XF) Analyzer (Agilent Technologies). After basal respiration measurement, UK5099 or Etomoxir (25 μmol/L) were used to assess OCR based on glucose utilization versus fatty acid. Then, oligomycin (2 μmol/L), FCCP (0.6 μmol/L) and Rotenone/antimycin A (1 μmol/L) were added sequentially to assess the full scope of mitochondrial activity across the mitochondrial oxidative complexes. Thus, one complete OCR trace consists of five conditions with triplicate measurements: baseline, treatment, oligomycin, FCCP and Rotenone/antimycin A. The data points shown (Figures 2M, 2N, 4G, and 4H) are from individual mice, which were collected and analyzed in 4 separate wells with approximately 5–20 myofibers in each well. Data were normalized to myofiber numbers and analyzed as (Averagebaseline-Averagetreatment)/(Averagebaseline-AverageRotenone/antimycin A) X 100% for glucose dependency/fatty acid dependency.
Histological analyses
Muscles were harvested and fixed in 10% formalin overnight processed and embedded in paraffin. Five micrometer thick histological sections were cut using an HM 355S Automatic Microtome (Thermo Fisher Scientific). Hematoxylin and eosin-staining (H&E) was performed and images were obtained using Olympus BX60 microscope with 10× objective. Myofiber cross sectional area was measured using National Institutes of Health (NIH) ImageJ software from images of histological sections. For immunofluorescence, slides were stained overnight at 4°C with 1X PBS containing a combination of anti-Myh7 (BA-F8, 1:50) primary antibody, anti-Myh2 primary antibody (SC-71, 1:20), anti-Myh4 primary antibody (BF-F3, 1:10) to show fiber-type specificity, and with anti-laminin antibody (L9393, 1:200) to delineate myofiber outlines present in a muscle histological section. Primary antibodies were visualized using Alexa 568 goat anti-mouse IgG2b, Alexa 488 goat anti-mouse IgG2b, Alexa 674 anti-mouse IgG1 and Alexa 405 goat anti-rabbit IgG secondary antibodies, diluted 1:500 in 1X PBS. Immunofluorescence images were captured using a Nikon Eclipse Ti microscope (Nikon Instruments Inc.).
Oil Red O staining was performed using Cincinnati Children’s Pathology Research Core service (RRID: SCR_022637). Briefly, muscles and livers were harvested and fixed in 4% paraformaldehyde (PFA) overnight and rinsed in 30% sucrose before Optimal Cutting Temperature compound (OCT) embedding. Eight micrometer thick histological sections were cut using a Leica CM1860 Cryostat (Leica Biosystems). Air-dried slides were placed in 100% propylene glycol for 5 min and then transferred to Oil Red O solution in a 60°C oven for 10 min. Slides were then placed in 85% propylene glycol for 1 min. Slides were counterstained in Hematoxylin 560 MX for 15 s. Images were obtained using Olympus BX60 microscope with 10× objective.
Glucose tolerance test
For glucose tolerance testing, mice were fasted overnight, and blood samples were taken from the tail as time 0 using Contour Next EZ Blood Glucose Meter (Ascensia Diabetes Care). Then 1.5 g/kg glucose was administered into awake mice via intraperitoneal injection according to the body weight of the mice. Blood samples were then withdrawn at 15, 30, 60, 90 and 120 min following injection for measurement of plasma glucose concentrations.
Measurement of IMAT and iBAT
Interscapular brown adipose tissue (iBAT) and Intermuscular adipose tissue (IMAT) were collected from indicated mice as previously described.46,47
PDH activity assay
PDH activity was measured using a pyruvate dehydrogenase combo microplate assay kit (Abcam). Quadriceps were collected from mice at 6-month of age. Samples were homogenized in ice-cold 1X PBS protease/phosphatase inhibitor cocktail (Millipore Sigma) was added and the PDH activity assay, and determination was conducted according to vendor’s protocol (Abcam).
Muscle malonyl CoA measurement
Quadriceps from 6 month-old mice were collected at baseline and post 48-h fasting. Muscles were homogenized in ice-cold 1X PBS followed by two freeze-thaw cycles. Lysates were obtained by centrifugation at 5,000 xg for 5 min at 4°C. Malonyl-CoA levels were measured using a malonyl-CoA Elisa Kit (MyBioSource) following the vendor’s protocol.
Triglyceride level measurements
Triglyceride measurements from tissue were performed with Triglyceride Quantification Assay Kit (Abcam). One hundred milligrams of tissue was harvested and homogenized in 1 mL of 5% NP-40/ddH2O solution using a Dounce homogenizer with 10–15 passes. Samples were heated slowly to 80°C–100°C in a water bath for 2–5 min, then cooled to room temperature (repeated twice). Samples were then centrifuged at 14,000 xg for 2 min. Supernatants were used for triglycerides measurement using vendor’s protocol (Abcam).
Glycogen measurement
Glycogen measurement was performed using Glycogen Assay Kit (Millipore Sigma). Ten milligrams of tissue was homogenized in 100 μL of ddH2O on ice. Homogenates were boiled at 100°C for 5 min to inactivate enzymes. Samples were centrifuged at 13,000 xg for 5 min and supernatants were used for glycogen measurement following vendor’s protocol.
Magnetic resonance imaging (MRI) body composition analysis
Fat mass and lean mass of mice were measured with EchoMRI body composition analyzer (EchoMRI LLC) as described previously.16 Briefly, the mouse was placed in a special plastic holder with restricted movement, and the holder was inserted in the magnet for MRI measurements. Measurements were performed as duplicates for each mouse. Mice were examined every month starting at 1 month of age, until 6 months of age.
RNA microarray analyses
Tibialis anterior were collected from mice at 3–4 months of age for RNA microarray analyses. Briefly, total RNA was isolated with TrizolTM Reagent (Thermo Fisher Scientific), and RNA quality assessed using an Agilent 2100 Bioanalyzer. Microarray analysis was carried out at Cincinnati Children’s Hospital Medical Center Gene Expression Core Facility using the Affymetrix Clariom S platform. Differential gene expression between samples was determined via bioinformatics analyses of resultant data (CHP files) using Transcriptome Analysis Console (Applied Biosystems), the Clariom_S_Mouse TAC Configuration file, and the iPathwayGuide (Advaita Bioinformatics). All original microarray data generated from skeletal muscle of Mcufl/fl-Myod-Cre and Mcubfl/fl-Myod-Cre mice are deposited with NCBI’s Gene Expression Omnibus database, with release upon acceptance of the manuscript (GSE205193).
Indirect calorimetry measurement
Mice at 6-month of age were transferred to the Mouse Metabolic Phenotyping Center at University of Cincinnati. Energy expenditure measurements were performed using the Columbus Instruments Oxymax Equal Flow System (https://med.uc.edu/research/core/Index/50/Facility/).48 After acclimation, mice were monitored for 48-h with free access to food and water followed by 18-h fasting with no food provided. Total food intake was measured during the 48-h fed condition. Total water intake was measured during each course. Other metabolic parameters (VO2, VCO2, Respiratory quotient, and heat production) were reported as average values during each investigative course.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were presented as mean ± SEM. Group sizes were determined by experience with similar studies performed within the Molkentin laboratory, and by an a priori power analysis for a two-tailed, two-sample t test with an α of 0.05 and power of 0.8, in order to detect a 10% difference in signal.
All statistical analysis was conducted using GraphPad Prism 9 software. The normality of all data was ascertained using the Shapiro-Wilk normality test. For statistical analysis comparing only two groups (Figures 1A–1E, 2H, 2M, 2N, 4C, S2B–S2D, and S2G), student’s t-test was used and a p value of <0.05 was considered significant. For statistical analysis comparing multiple groups with one variable (Figures 2D, 2I–2K, 4G–4J, S2E, S3A–S3H, and S4B–S4G), a one-way ANOVA was used to analyze sample variance. If the one-way ANOVA confirmed a significant difference among groups, a post hoc Bonferroni test for multiple comparisons was performed and an adjusted p value of <0.05 was considered significant. For statistical analysis with two variables (Figures 2G, 2L, 3B, 3D, 3G, 3H, 4E, and 4F), a two-way ANOVA was used to determine possible interactions between the variables. If the two-way ANOVA confirmed a significant interaction between variations, a post hoc Bonferroni’s multiple comparisons test was performed and an adjusted p value of <0.05 was considered significant.
Supplementary Material
Highlights.
MCUb deletion in skeletal muscle causes fat accumulation with less fatty acid oxidation
MCUb induction mediated by fasting stimulates fatty acid oxidation in muscle
MCUb overexpression in skeletal muscle stimulates fatty acid oxidation and fat loss
MCUb regulation of mitochondrial substrate utilization is PDK4 dependent
ACKNOWLEDGMENTS
We would like to thank Dr. Gary Lopaschuk (University of Alberta, Canada) for critical evaluation of the manuscript and helpful suggestions. This work was supported by a grant from National Institutes of Health, National Heart, Lung, and Blood Institute to J.D.M. and D.M.B. (2R01HL132831).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113465.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
REFERENCES
- 1.Hargreaves M, and Spriet LL (2020). Skeletal muscle energy metabolism during exercise. Nat. Metab 2, 817–828. [DOI] [PubMed] [Google Scholar]
- 2.Spriet LL (1992). Anaerobic metabolism in human skeletal muscle during short-term, intense activity. Can. J. Physiol. Pharmacol 70, 157–165. [DOI] [PubMed] [Google Scholar]
- 3.Kelley D, Mitrakou A, Marsh H, Schwenk F, Benn J, Sonnenberg G, Arcangeli M, Aoki T, Sorensen J, Berger M, et al. (1988). Skeletal muscle glycolysis, oxidation, and storage of an oral glucose load. J. Clin. Invest 81, 1563–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelley DE, Goodpaster B, Wing RR, and Simoneau JA (1999). Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am. J. Physiol 277, E1130–E1141. [DOI] [PubMed] [Google Scholar]
- 5.Smith RL, Soeters MR, Wüst RCI, and Houtkooper RH (2018). Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr. Rev 39, 489–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelley DE, and Mandarino LJ (2000). Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes 49, 677–683. [DOI] [PubMed] [Google Scholar]
- 7.Battaglia GM, Zheng D, Hickner RC, and Houmard JA (2012). Effect of exercise training on metabolic flexibility in response to a high-fat diet in obese individuals. Am. J. Physiol. Endocrinol. Metab 303, E1440–E1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denton RM (2009). Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 1787, 1309–1316. [DOI] [PubMed] [Google Scholar]
- 9.Gherardi G, Monticelli H, Rizzuto R, and Mammucari C (2020). The Mitochondrial Ca(2+) Uptake and the Fine-Tuning of Aerobic Metabolism. Front. Physiol 11, 554904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wescott AP, Kao JPY, Lederer WJ, and Boyman L (2019). Voltage-energized Calcium-sensitive ATP Production by Mitochondria. Nat. Metab 1, 975–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vassylyev DG, and Symersky J (2007). Crystal structure of pyruvate dehydrogenase phosphatase 1 and its functional implications. J. Mol. Biol 370, 417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turkan A, Hiromasa Y, and Roche TE (2004). Formation of a complex of the catalytic subunit of pyruvate dehydrogenase phosphatase isoform 1 (PDP1c) and the L2 domain forms a Ca2+ binding site and captures PDP1c as a monomer. Biochemistry 43, 15073–15085. [DOI] [PubMed] [Google Scholar]
- 13.Glancy B, Willis WT, Chess DJ, and Balaban RS (2013). Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 52, 2793–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chaudhuri D, Sancak Y, Mootha VK, and Clapham DE (2013). MCU encodes the pore conducting mitochondrial calcium currents. Elife 2, e00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, et al. (2013). The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol 15, 1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwong JQ, Huo J, Bround MJ, Boyer JG, Schwanekamp JA, Ghazal N, Maxwell JT, Jang YC, Khuchua Z, Shi K, et al. (2018). The mitochondrial calcium uniporter underlies metabolic fuel preference in skeletal muscle. JCI Insight 3, e121689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altamimi TR, Karwi QG, Uddin GM, Fukushima A, Kwong JQ, Molkentin JD, and Lopaschuk GD (2019). Cardiac-specific deficiency of the mitochondrial calcium uniporter augments fatty acid oxidation and functional reserve. J. Mol. Cell. Cardiol 127, 223–231. [DOI] [PubMed] [Google Scholar]
- 18.Gherardi G, Nogara L, Ciciliot S, Fadini GP, Blaauw B, Braghetta P, Bonaldo P, De Stefani D, Rizzuto R, and Mammucari C (2019). Loss of mitochondrial calcium uniporter rewires skeletal muscle metabolism and substrate preference. Cell Death Differ 26, 362–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabò I, and Rizzuto R (2013). The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J 32, 2362–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huo J, Lu S, Kwong JQ, Bround MJ, Grimes KM, Sargent MA, Brown ME, Davis ME, Bers DM, and Molkentin JD (2020). MCUb Induction Protects the Heart from Post-Ischemic Remodeling. Circ. Res 127, 379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lambert JP, Luongo TS, Tomar D, Jadiya P, Gao E, Zhang X, Lucchese AM, Kolmetzky DW, Shah NS, and Elrod JW (2019). MCUB Regulates the Molecular Composition of the Mitochondrial Calcium Uniporter Channel to Limit Mitochondrial Calcium Overload During Stress. Circulation 140, 1720–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cividini F, Scott BT, Suarez J, Casteel DE, Heinz S, Dai A, Diemer T, Suarez JA, Benner CW, Ghassemian M, and Dillmann WH (2021). Ncor2/PPARalpha-Dependent Upregulation of MCUb in the Type 2 Diabetic Heart Impacts Cardiac Metabolic Flexibility and Function. Diabetes 70, 665–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang S, Hulver MW, McMillan RP, Cline MA, and Gilbert ER (2014). The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr. Metab 11, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dienel GA (2019). Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev 99, 949–1045. [DOI] [PubMed] [Google Scholar]
- 25.Schönfeld P, and Reiser G (2017). Brain energy metabolism spurns fatty acids as fuel due to their inherent mitotoxicity and potential capacity to unleash neurodegeneration. Neurochem. Int 109, 68–77. [DOI] [PubMed] [Google Scholar]
- 26.Ruderman NB, Saha AK, Vavvas D, and Witters LA (1999). Malonyl-CoA, fuel sensing, and insulin resistance. Am. J. Physiol 276, E1–E18. [DOI] [PubMed] [Google Scholar]
- 27.Hubbard MJ, and McHugh NJ (1996). Mitochondrial ATP synthase F1-beta-subunit is a calcium-binding protein. FEBS Lett 391, 323–329. [DOI] [PubMed] [Google Scholar]
- 28.Balu D, Ouyang J, Parakhia RA, Pitake S, and Ochs RS (2016). Ca(2+) effects on glucose transport and fatty acid oxidation in L6 skeletal muscle cell cultures. Biochem. Biophys. Rep 5, 365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spriet LL (2014). New insights into the interaction of carbohydrate and fat metabolism during exercise. Sports Med 44 (Suppl 1), S87–S96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, et al. (2015). The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep 12, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, and Molkentin JD (2015). The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep 12, 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Menezes-Filho SL, Amigo I, Luévano-Martínez LA, and Kowaltowski AJ (2019). Fasting promotes functional changes in liver mitochondria. Biochim. Biophys. Acta. Bioenerg 1860, 129–135. [DOI] [PubMed] [Google Scholar]
- 33.Paillard M, Csordás G, Szanda G, Golenár T, Debattisti V, Bartok A, Wang N, Moffat C, Seifert EL, Spät A, and Hajnóczky G (2017). Tissue-Specific Mitochondrial Decoding of Cytoplasmic Ca(2+) Signals Is Controlled by the Stoichiometry of MICU1/2 and MCU. Cell Rep 18, 2291–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lopaschuk GD, Ussher JR, Folmes CDL, Jaswal JS, and Stanley WC (2010). Myocardial fatty acid metabolism in health and disease. Physiol. Rev 90, 207–258. [DOI] [PubMed] [Google Scholar]
- 35.van der Vusse GJ, Glatz JF, Stam HC, and Reneman RS (1992). Fatty acid homeostasis in the normoxic and ischemic heart. Physiol. Rev 72, 881–940. [DOI] [PubMed] [Google Scholar]
- 36.Hue L, and Taegtmeyer H (2009). The Randle cycle revisited: a new head for an old hat. Am. J. Physiol. Endocrinol. Metab 297, E578–E591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGarry JD, Mills SE, Long CS, and Foster DW (1983). Observations on the affinity for carnitine, and malonyl-CoA sensitivity, of carnitine palmitoyltransferase I in animal and human tissues. Demonstration of the presence of malonyl-CoA in non-hepatic tissues of the rat. Biochem. J 214, 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horton TJ, and Hill JO (2001). Prolonged fasting significantly changes nutrient oxidation and glucose tolerance after a normal mixed meal. J. Appl. Physiol 90, 155–163. [DOI] [PubMed] [Google Scholar]
- 39.Kanisicak O, Mendez JJ, Yamamoto S, Yamamoto M, and Goldhamer DJ (2009). Progenitors of skeletal muscle satellite cells express the muscle determination gene, MyoD. Dev. Biol 332, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huijbers IJ, Del Bravo J, Bin Ali R, Pritchard C, Braumuller TM, van Miltenburg MH, Henneman L, Michalak EM, Berns A, and Jonkers J (2015). Using the GEMM-ESC strategy to study gene function in mouse models. Nat. Protoc 10, 1755–1785. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Z, and Lutz B (2002). Cre recombinase-mediated inversion using lox66 and lox71: method to introduce conditional point mutations into the CREB-binding protein. Nucleic Acids Res 30, e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chung JH, Bell AC, and Felsenfeld G (1997). Characterization of the chicken beta-globin insulator. Proc. Natl. Acad. Sci. USA 94, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Challis RC, Kumar SR, Chan KY, Challis C, Jang MJ, Rajendran PS, Tompkins JD, Shivkumar K, Deverman BE, and Gradinaru V (2018). Widespread and Targeted Gene Expression by Systemic AAV Vectors: Production, Purification, and administration. Preprint at bioRxiv
- 44.Tabebordbar M, Lagerborg KA, Stanton A, King EM, Ye S, Tellez L, Krunnfusz A, Tavakoli S, Widrick JJ, Messemer KA, et al. (2021). Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell 184, 4919–4938.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karch J, Bround MJ, Khalil H, Sargent MA, Latchman N, Terada N, Peixoto PM, and Molkentin JD (2019). Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci. Adv 5, eaaw4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bagchi DP, and MacDougald OA (2019). Identification and Dissection of Diverse Mouse Adipose Depots. J. Vis. Exp 10.3791/59499. [DOI] [PMC free article] [PubMed]
- 47.Mann A, Thompson A, Robbins N, and Blomkalns AL (2014). Localization, identification, and excision of murine adipose depots. J. Vis. Exp 10.3791/52174. [DOI] [PMC free article] [PubMed]
- 48.Weng J, Lou D, Benoit SC, Coschigano N, Woods SC, Tso P, and Lo CC (2017). Energy homeostasis in apolipoprotein AIV and cholecystokinin-deficient mice. Am. J. Physiol. Regul. Integr. Comp. Physiol 313, R535–R548. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper does not report any original code.