

Abstract
Constitutive activation of receptor tyrosine kinases (RTKs) is a frequent event in human cancer cells. Activating mutations in Fms-like tyrosine kinase 3 (FLT-3), notably, internal tandem duplications in the juxtamembrane domain (FLT-3 ITD), have been causally linked to acute myeloid leukemia. As we describe here, FLT-3 ITD exists predominantly in an immature, underglycosylated 130-kDa form, whereas wild-type FLT-3 is expressed predominantly as a mature, complex glycosylated 150-kDa molecule. Endogenous FLT-3 ITD, but little wild-type FLT-3, is detectable in the endoplasmic reticulum (ER) compartment. Conversely, cell surface expression of FLT-3 ITD is less efficient than that of wild-type FLT-3. Inhibition of FLT-3 ITD kinase by small molecules, inactivating point mutations, or coexpression with the protein-tyrosine phosphatases (PTPs) SHP-1, PTP1B, and PTP-PEST but not RPTPα promotes complex glycosylation and surface localization. However, PTP coexpression has no effect on the maturation of a surface glycoprotein of vesicular stomatitis virus. The maturation of wild-type FLT-3 is impaired by general PTP inhibition or by suppression of endogenous PTP1B. Enhanced complex formation of FLT-3 ITD with the ER-resident chaperone calnexin indicates that its retention in the ER is related to inefficient folding. The regulation of RTK maturation by tyrosine phosphorylation was observed with other RTKs as well, defines a possible role for ER-resident PTPs, and may be related to the altered signaling quality of constitutively active, transforming RTK mutants.
Cellular receptors for growth factors, hormones, cytokines, and antigens are postranslationally modified with N-linked, branched carbohydrate chains. Nascent polypeptide chains become initially glycosylated with a mannose-rich branched oligosaccharide in the endoplasmic reticulum (ER). Then, the glycoproteins are subjected to partial deglycosylation by several selective glycosidases, eventually enabling transfer to the Golgi compartment and more complex glycosylation (9). This process, designated glycoprotein maturation, is coupled to stringent quality control in the ER (4, 10). Correct folding is monitored by a complex system comprising, among other components, the chaperones calnexin and calreticulin, the oxidoreductase ERp57, and the glycosylation enzymes UDP-glucose glucosyltransferase and glucosidases I and II. Improperly folded glycoproteins are tagged by reversible glucosylation, enabling their interactions with calnexin and calreticulin and leading to their retention in the ER (4). Properly folded glycoproteins can dissociate from the chaperones and proceed to the Golgi compartment for further glycosylation.
The receptor tyrosine kinase (RTK) Fms-like tyrosine kinase 3 (FLT-3) is expressed in multiple hematopoietic lineages (21, 22). Constitutively active FLT-3 mutants, notably, versions harboring internal tandem duplications in the juxtamembrane domain (FLT-3 ITD) and versions with point mutations in the kinase activation loop, have been found in approximately 30% of acute myeloid leukemia cases (28, 38). Activated versions of FLT-3 are characterized not only by constitutive signaling but also by a different signaling quality, which is connected to their transforming capacity. Hallmarks of altered signaling are a strong activation of STAT5a and of STAT response genes, pronounced antiapoptotic effects, and the suppression of myeloid cell differentiation (25, 26, 34). The occurrence of active FLT-3 mutants is associated with a poor prognosis in patients with acute myeloid leukemia, and FLT-3 is considered a promising target for therapy (for reviews, see references 33 and 35). Tyrosine kinase inhibitors from different structural families, including AG1296 (39), SU11248 (29), PKC412 (42), and CEP-701 (17), have been shown to inhibit the signaling of activated FLT-3. Some of these compounds are presently in clinical trials.
In our analysis of FLT-3 signaling, we observed inefficient maturation of FLT-3 ITD and its reduced expression at the cell surface. The systematic investigation of these phenomena revealed that the maturation of FLT-3 ITD is impaired by its constitutive kinase activity. Entrapment by the chaperone calnexin, and therefore ER retention, indicates decreased efficiency of folding of FLT-3 ITD. This previously unrecognized mechanism appears to be generally relevant for RTKs and has several testable implications for the mechanism of transformation of constitutively active RTKs and for the cellular roles of protein-tyrosine phosphatases.
MATERIALS AND METHODS
DNA constructs.
A PCR-amplified triple-hemagglutinin (HA) tag was fused by PCR to a DNA fragment corresponding to a sequence 3′ downstream of an FLT-3 internal NdeI site. The PCR-fused fragment was subcloned with NdeI/HindIII into FLT-3-expressing pcRIITOPO (26). HA-tagged FLT-3 was subcloned with NotI/HindIII into pcDNA3.1(−). FLT-3 was PCR amplified and cloned into pEGFP-N1, introducing a six-glycine linker between the FLT-3 C terminus and enhanced green fluorescent protein (EGFP).
The Stratagene QuikChange method was used to introduce KA and YF mutations into FLT-3 cDNAs. Note that the numbering of amino acids for wild-type FLT-3 is maintained for FLT-3 ITD, despite the ITD insertion, to allow better comparison.
Primer sequences are available on request.
For the creation of a pSuper PTP1B small interfering (siRNA) expression construct, the sequence 5′-TGG AAG AAG CCC AAA GGA G-3′, corresponding to positions 221 to 239 of the human PTP1B cDNA, was used to design oligonucleotides (2) which were cloned into vector pSuper.retro.puro (Oligoengine, Seattle, Wash.). An SHP-1 siRNA expression construct in the same vector was generated in an analogous manner with the sequence 5′-GAG GTG TCC ACG GTA GCT TCC-3′, corresponding to positions 145 to 165 of the human SHP-1 cDNA (sequence kindly provided by H. Keilhack, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Mass.). All constructs were verified by DNA sequence analysis.
A eukaryotic expression construct for vesicular stomatitis virus glycoprotein (VSV-G) was kindly provided by S. Gutkind. An expression construct containing enhanced cyan fluorescent protein (ECFP) fused to an ER-targeting sequence derived from calreticulin, including a KDEL retrieval sequence, was used as an ER localization marker (pECFP-ER). ECFP fused to a Golgi compartment-targeting sequence derived from human β-1,4-galactosyltransferase was used as a Golgi compartment localization marker (pECFP-Golgi). Both constructs were purchased from BD Biosciences (Heidelberg, Germany). Expression constructs for the protein-tyrosine phosphatases (PTPs) PTP1B and PTP-PEST were kindly provided by A. Ullrich and M. Tremblay, respectively. An EGFP-SHP-1 expression construct was described earlier (37).
Cells and reagents.
HEK293 cells were grown in Dulbecco modified Eagle medium (DMEM)-F-12 medium (Gibco) containing 10% fetal calf serum (FCS) (Biochrom, Berlin, Germany). COS-7 cells were grown in DMEM (Gibco) containing 10% FCS. MV4-11 and THP-1 cells, kindly provided by S. Scholl (Department of Internal Medicine, Jena University Hospital), were grown in RPMI 1640 (Biochrom) containing 10% heat-inactivated FCS. Agarose beads linked to lectin from Galanthus nivalis, wortmannin, bis-indolylmaleimide I (BisI), neuraminidase (type V; N2876) from Clostridium perfringens, and brefeldin A were obtained from Sigma (Taufkirchen, Germany). PP1 and geldanamycin were obtained from Alexis (Grünberg, Germany). Biotinylated lectins from Dolichos biflorus (DBA), G. nivalis (GNA), Ricinus communis (RCA120), Glycine max (soybean; SBA), Sambucus nigra (SNA), and Ulex europaeus (UEA) were purchased from Vector Laboratories (Peterborough, United Kingdom). Agarose-linked streptavidin, streptavidin coupled to horseradish peroxidase (HRP), and HRP-coupled RCA120 lectin were obtained from Sigma.
AG1296 was previously described (39). SU11248 was kindly provided by Marie O'Farrell (Sugen, Inc.). MG132 and bp(V)phen were obtained from Calbiochem (Schwalbach, Germany). Redivue PRO-MIX 35S in vitro cell labeling mixture was obtained from Amersham Biosciences (Freiburg, Germany). Peptide N-glycosidase F (PNGase F) was obtained from Roche (Mannheim, Germany). Endo-β-N-acetylglucosaminidase H (endo-H) and the corresponding reaction buffers were obtained from New England Biolabs (Frankfurt, Germany).
Antibodies to the following were used: FLT-3* (clone 4G8; recognizes the extracellular domain of FLT-3), pan-Erk, and PTP1B (monoclonal) (BD Biosciences); pY591 (3461S; recognizes phosphorylated Y591 in activated FLT-3) (Cell Signaling, Frankfurt, Germany); HA and vinculin (V248) (Upstate, Biomol, Hamburg, Germany); FLT-3 (sc-480), c-Src (sc-18), SHP-1 (sc-287), calnexin (sc-6465), c-Kit (sc-168), and GFP (sc-9996) (Santa Cruz, Santa Cruz, Calif.); and β-actin, mouse immunoglobulin G2a (IgG2a), mouse IgG1(κ), and VSV-G (ascitic fluid) (Sigma). Dichlorotriazenyl amino fluorescein (DTAF)-coupled anti-mouse IgG, Cy3-coupled anti-mouse IgG, and anti-rabbit IgG were obtained from Molecular Probes (Karlsruhe, Germany).
FACS analysis.
MV4-11 and THP-1 cells were indirectly labeled with FLT-3* antibody or mouse IgG1(κ) as an isotype control and with DTAF-coupled goat anti-mouse IgG according to standard protocols. Fluorescence-activated cell sorting (FACS) analysis was performed with a FACSCalibur (Becton Dickinson) instrument. Subsequent data analysis was performed with WinMDI software.
Immunoprecipitation and Western blotting.
Vesicle immunoprecipitation was performed as described by Waugh et al. (41). MV4-11 and THP-1 cells were suspended in 4.3 M glycerol in 10 mM Tris-HCl (pH 7.4) for 10 min. Cells then were resuspended in homogenization buffer (0.25 M sucrose, 10 mM Tris-HCl [pH 7.4], 1 mM EGTA, 0.5 mM EDTA, protease inhibitors) and homogenized. ER vesicles were isolated by adding 0.5 μg of anticalnexin antibody and 60 μl of protein G-Sepharose and incubating the mixture at 4°C overnight. Beads were washed three times with washing buffer (0.25 M sucrose, 10 mM Tris-HCl [pH 7.4], 150 mM NaCl) and boiled in 30 μl of 2× sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) sample buffer. Samples were subjected to SDS-PAGE and immunoblotting for FLT-3 and calnexin.
HEK293 cells were transfected by the calcium phosphate method, and COS-7 cells were transfected with Lipofectamine 2000 (Invitrogen). Cell lysis with lysis buffer containing 0.5% Nonidet P-40 and immunoblotting were performed as previously described (15). For FLT-3-calnexin coimmunoprecipitation, cell lysates were incubated overnight with the corresponding antibody and 1 h with protein A/G-Sepharose beads, and the beads were sedimented and washed three times with 20 mM HEPES (pH 7.5)-150 mM NaCl-10% glycerol-0.1% Triton X-100 (HNGT buffer). Isolation of high-mannose FLT-3 was performed by incubation of cell lysates with 15 μl of immobilized GNA lectin for 2 h and three washes with HNGT buffer. Treatment with PNGase F was performed as previously described (14). For digestion with endo-H, immunoprecipitates from 5 × 106 cells with endogenous FLT-3 expression or 2.4 × 105 transfected cells were subjected to denaturation with 25 μl of denaturation buffer (supplied with the enzyme) at 95°C for 10 min. Then, 25 μl of 2× reaction buffer (supplied with the enzyme) and 0.5 μl of endo-H were added, and the mixture was incubated at 37°C overnight. The reaction was terminated with SDS-PAGE sample buffer, and the reaction products were analyzed by immunoblotting.
Neuraminidase treatment of immunoprecipitates was done after denaturation with SDS-PAGE sample buffer. SDS was diluted to 0.5% with sodium phosphate buffer (pH 7.4; final concentration, 50 mM), and 0.5 μl of neuraminidase was added to a 60-μl sample. The mixture was incubated at 37°C for 2 h.
For lectin pull-down assays, THP-1 cells or FLT-3 KA-overexpressing COS-7 cells were lysed with lysis buffer containing 1% Nonidet P-40. The lysate of 2 × 107 to 2.5 × 107 THP-1 cells or 1.2 × 105 transfected COS-7 cells was mixed with 40 μg of biotinylated lectin and incubated with end-over-end rotation at 4°C overnight. Then, 35 μl of a 1:1 suspension of agarose-coupled streptavidin was added, and incubation was continued for 1 h. The beads were washed three times with HNGT buffer and subsequently extracted with SDS-PAGE sample buffer at 37°C for 30 min. The samples were separated by SDS-7.5% PAGE and subjected to immunoblotting with anti-FLT-3 antibodies. For lectin blotting, FLT-3 was immunoprecipitated from lysates of THP-1 cells or transfected COS-7 cells. Immunoprecipitates corresponding to 1 × 107 to 2 × 107 THP-1 cells or 2.5 × 105 transfected COS-7 cells per lane were resolved by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked by incubation with 2% Tween 20 in Tris-buffered saline (TBS) at room temperature for 20 min, washed three times with TBS, and incubated with biotinylated lectin (5 to 20 μg/ml) or HRP-coupled lectin (1 to 5 μg/ml) in TBS containing 1 mM MnCl2, 1 mM MgCl2, and 1 mM CaCl2 at room temperature overnight. The blots were either developed directly (HRP-coupled lectin) or incubated with HRP-conjugated streptavidin (0.2 μg/ml) at room temperature for 1 h and then developed with enhanced chemiluminescence detection.
PTP suppression by siRNA.
HEK293 cells were transiently transfected by the calcium phosphate method with 0.4 μg of an expression construct for HA-tagged wild-type FLT-3 plus 4 μg of the corresponding PTP1B siRNA construct per well of a six-well plate. Cells were allowed to express PTP1B siRNA for 4 days and then were lysed. For suppression of SHP-1 expression, THP-1 cells were infected with retroviruses containing the SHP-1 siRNA expression contruct pSuper.retro.puro, produced in the Phoenix Amphopack packaging cell line (kindly provided by G. Nolan, Stanford University). Cells were selected for 10 days with 2 μg of puromycin/ml and expanded. For suppression of PTP1B, THP-1 cells were transfected with siGENOME SMARTpool siRNAs (NM_002827; Dharmacon) by using Nucleofector kit V and optimized protocol V-01 according to the instructions of the supplier (Amaxa GmbH, Cologne, Germany). Three days after transfection, cells were lysed and FLT-3 was immunoprecipitated. PTP1B expression, FLT-3 maturation, and loading (β-actin or vinculin) were analyzed by immunoblotting.
Inhibitor treatments.
Cells were starved overnight in medium containing 0.5% FCS and then treated with kinase or signal transduction inhibitors at the following concentrations: 10 μM AG1296, 1 μM SU11248, 5 μM PP1, 100 nM wortmannin, and 500 nM BisI. For treatment with geldanamycin and brefeldin A, transfected COS-7 cells were allowed to express FLT-3 constructs for 2 days. Cells then were treated for 6 h with either 3 μM geldanamycin or 5 μg of brefeldin A/ml before lysis. For the inhibition of total PTP activity, transfected COS-7 cells were left untreated or were treated with 1 mM bp(V)phen for up to 3 h. In some experiments, cells were preincubated with 25 μM MG132 for 4 h before PTP inhibition.
Pulse-chase labeling.
Pulse-chase analysis was carried out as previously described (21). Briefly, MV4-11 cells or transfected COS-7 cells were labeled for 20 min with 200 μCi of Redivue/ml in cysteine- and methionine-free DMEM, with subsequent chasing into DMEM containing 0.1 mM methionine and 0.2 mM cysteine. Immunoprecipitated FLT-3 was analyzed by SDS-PAGE and fluorography.
Immunohistochemistry studies.
COS-7 cells were grown on collagen-coated coverslips and transiently transfected with Lipofectamine 2000 (Invitrogen). The cells were allowed to express HA-tagged FLT-3 constructs for 1 day before fixation with 4% paraformaldehyde. After blocking was done with 10% normal goat serum in phosphate-buffered saline (PBS)-1.5% bovine serum albumin (BSA), cells were incubated with FLT-3* antibody (1:100) in PBS-1% BSA overnight, washed, and stained with Cy3-coupled anti-mouse IgG. Coexpression of EGFP-tagged FLT-3 or HA-tagged FLT-3 in COS-7 cells was performed with a number of other proteins. Cells were fixed with methanol at −20°C, quenched with 0.1% NaBH4 at room temperature, blocked as described above, and incubated with pY591 antibody (1:100) in TBS-1% BSA overnight and then with Cy3-coupled anti-rabbit IgG (1:600). For SHP-1 immunostaining, cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100 in PBS, blocked as described above, and incubated with SHP-1 antibody at a dilution of 1:100 and then with Cy3-coupled anti-rabbit IgG as described above. Microscopy was done with a Zeiss LSM 510 confocal laser scanning microscope.
Quantitative analysis.
Densitometric analysis of immunoblots was performed with NIH Image 1.61 software. The ratio of the amount of the complex glycosylated (150-kDa) form to the amount of the high-mannose (130-kDa) form was taken as a measure of maturation and is given in arbitrary units. Results are expressed as the mean ± standard error of the mean (SEM). Data were analyzed by using paired two-tailed Student's t tests. A P value of <0.05 was considered significant.
RESULTS
Tyrosine phosphorylation of FLT-3 is related to inefficient maturation.
When comparing the signaling mechanisms of wild-type FLT-3 with those of the transforming version FLT-3 ITD, we noted that FLT-3 ITD was usually detectable in smaller amounts at the cell surface than was the wild-type receptor. This finding is exemplified here for the cell lines THP-1, harboring wild-type FLT-3, and MV4-11, expressing FLT-3 ITD (Fig. 1A, left panel). Lower surface levels of FLT-3 seem to be caused not by different expression levels but rather by different cellular distributions. FLT-3 ITD was detectable in membrane vesicles enriched in ER membranes, obtained by immunoprecipitating membrane vesicles containing calnexin, an ER-resident molecule. Wild-type FLT-3 was not recovered in detectable amounts in this vesicle fraction (Fig. 1A, right panel).
FIG. 1.


Impaired maturation of the constitutively active FLT-3 ITD RTK. (A) (Left panel) Cell surface-localized FLT-3 was detected by FACS analysis of MV4-11 cells expressing FLT-3 ITD and of THP-1 cells expressing wild-type FLT-3. (Right panel) ER membrane vesicles from both cell lines were immunoprecipitated (vesIP) with anticalnexin antibodies (anti-CNX) and analyzed for the presence of FLT-3 by immunoblotting. Equal levels of expression of calnexin in both cell lines were detected by immunoblotting of cell lysate aliquots. (B) FLT-3 ITD or wild-type FLT-3 was immunoprecipitated (IP) from MV4-11 cells or THP-1 cells, respectively, and analyzed by immunoblotting. (C) Cell lysates of HEK293 cells expressing either wild-type (WT) FLT-3 or FLT-3 ITD (HA tagged) either were subjected directly to immunoblotting or were first subjected to pull-down assays with immobilized GNA lectin or to immunoprecipitation and then digested with PNGase F and analyzed by immunoblotting. CG, complex glycosylated species; HM, high-mannose form; DG, deglycosylated form. (D) Wild-type FLT-3 or FLT-3 ITD was immunoprecipitated from THP-1 cells or MV4-11 cells, respectively, and either mock treated or digested with endo-H or PNGase F. The digestion products were analyzed by immunoblotting with anti-FLT-3 antibodies. (E) Glycosylated proteins were affinity purified from lysates of FLT-3 K644A-expressing COS-7 cells (upper panel) or THP-1 cells (lower panel) with the indicated biotinylated lectins and agarose-linked streptavidin. For lectin selectivity and abbreviations, see the text and the legend to Fig. 2. The presence of FLT-3 in the glycoprotein pull-down assay was detected by immunoblotting with anti-FLT-3 antibodies. (F) FLT-3 was immunoprecipitated from THP-1 cells and either mock treated or digested with neuraminidase (N′ase). DS, desialylated species. (G) (Upper panels) MV4-11 cells were treated with AG1296 or SU11248 for 3 h. Endogenous FLT-3 ITD was immunoprecipitated and detected by immunoblotting. (Lower panel) MV4-11 cells were metabolically labeled with [35S]methionine-[35S]cysteine for 20 min. An excess of unlabeled methionine-cysteine was added to the medium (chase) either in the presence or in the absence of 1 μM SU11248. FLT-3 ITD was immunoprecipitated at the indicated times and analyzed by SDS-PAGE and fluorography. (H) Wild-type FLT-3, FLT-3 ITD, or their kinase-inactive K644A counterparts were expressed in COS-7 cells. The corresponding cell lysates were subjected to immunoblotting.
It was previously reported that two species of FLT-3 can be observed in intact cells: a 150-kDa species representing a complex glycosylated, mature form and a 130-kDa species representing an underglycosylated, immature form which contains mannose-rich structures (21). We noted that in cells expressing FLT-3 ITD, predominantly a 130-kDa species was detectable, whereas for wild-type FLT-3, proportionally more of the 150-kDa species was detectable (Fig. 1B and Fig. 1C, left panel). The different proportions were not caused by solubility differences, since they were equally apparent when cells were solubilized directly with SDS-PAGE sample buffer. To confirm that the 130- and 150-kDa species differ in size because of differences in their glycosylation, we transiently overexpressed wild-type FLT-3 and FLT-3 ITD and treated the corresponding immunoprecipitates with PNGase F. Both receptor isoforms could be completely converted into an approximately 110-kDa molecule, proving that the observed size differences were caused by different modifications with N-linked carbohydrates. Since the material from both glycosylated receptor forms adds up in this band, its intensity is higher than those of the individual 150- and 130-kDa bands in the mock-treated controls. Furthermore, GNA lectin, which is selective for mannose-rich structures (36), interacted specifically with the 130-kDa species of both FLT-3 and FLT-3 ITD, further supporting its identity as a high-mannose form (Fig. 1C, right panel). Finally, the 130-kDa form was selectively deglycosylated upon digestion with endo-H, an enzyme which is highly specific for immature mannose-rich carbohydrates (Fig. 1D). Since mannose-rich glycoproteins are exclusively present in the ER, these biochemical observations provided further evidence that the FLT-3 130-kDa species resides in an ER compartment.
The glycosylation state of the 150-kDa form was analyzed by testing its reactivity with a set of lectins in both pull-down (Fig. 1E) and blotting (Fig. 2) assays. The 150-kDa form selectively interacted with SNA, RCA120, and SBA lectins, indicating the presence of terminal residues of sialic acid, galactose, and N-acetylgalactosamine, respectively, which are absent in the 130-kDa form. Selective reactivity of the 150-kDa form with lectins specific for these sugars was also derived from blotting experiments (Fig. 2). SNA lectin recognizes sialic acid preferentially in a form which is α-2,6-linked to subsequent galactose. Thus, SNA lectin reactivity indicates the presence of this terminal disaccharide. As shown before, GNA lectin in both pull-down and blotting assays reacted preferentially with the 130-kDa form. Interaction assays with UEA lectin revealed little reactivity, suggesting that neither of the FLT-3 variants contains significant amounts of fucose. DBA lectin is specific for N-acetylgalactosamine in an α-glycosidic linkage. The absence of reactivity with DBA lectin but the reactivity with SBA lectin therefore indicates that the terminal N-acetylgalactosamine residues are engaged in a β-glycosidic linkage. Treatment of FLT-3 immunoprecipitates from THP-1 cells with neuraminidase resulted in a clear size reduction of the 150-kDa form, further supporting the presence of significant amounts of sialic acid (Fig. 1F). Thus, the carbohydrate part of the 150-kDa form has features which are typical of complex glycosylated glycoproteins (9) and similar to those of oligosaccharide chains in the cell surface epidermal growth factor receptor (45). A tentative structure is indicated in Fig. 2. Taken together, these results indicate that FLT-3 ITD exists in a much higher proportion than wild-type FLT-3 as an ER-bound, immature form containing mannose-rich carbohydrates. Conversely, wild-type FLT-3 exists predominantly as a 150-kDa form containing a larger proportion of carbohydrates with features typical of complex glycosylation.
FIG. 2.

Reactivity of FLT-3 with lectins in blotting experiments. Lectin reactivity is given in a semiquantitative scale ranging from − (no reactivity) to +++ (very strong reactivity). In the left panel, structural elements for which there is direct evidence from blotting and pull-down assays are shown in black type, and other elements are shown in gray type, in analogy with literature data.
To our surprise, treatment of cells with FLT-3 tyrosine kinase inhibitors, such as AG1296 or SU11248, resulted in the formation of larger amounts of the mature, 150-kDa form of FLT-3 ITD (Fig. 1G, upper panel). Similar data can be found in the literature (17, 34, 43) but have not received attention. Consistent with the effects of kinase inhibitors, the kinase-inactive FLT-3 K644A and FLT-3 ITD K644A mutants were present in the mature form in significantly larger amounts than their active counterparts (Fig. 1H). These findings indicated that FLT-3 kinase activity may impair efficient maturation. To directly test the effect of kinase activity on FLT-3 maturation, we performed pulse-chase experiments, comparing transiently expressed active FLT-3 ITD and wild-type FLT-3 with catalytically inactive FLT-3 ITD K644A. As shown in Fig. 3A, interconversion of the immature, 130-kDa species into the mature, 150-kDa species occurred rapidly for FLT-3 ITD K644A, whereas FLT-3 ITD very inefficiently matured within the observation time of 3 h. Wild-type FLT-3 matured better than FLT-3 ITD but clearly less efficiently than FLT-3 ITD K644A. Treatment of FLT-3 ITD-expressing MV4-11 cells with the FLT-3 kinase inhibitor SU11248 also led to much more efficient maturation in this assay (Fig. 1G, lower panel). Thus, kinase inactivation strongly promoted the maturation of FLT-3 ITD. Confirming previous observations for wild-type FLT-3 (21), these data also show that the 130-kDa species gives rise to the 150-kDa species and exclude the possibility that the 130-kDa species is a degradation product of the 150-kDa species. Further, they exclude the possibility that the low cellular levels of the 150-kDa species in cells expressing active FLT-3 ITD are the consequence of correct formation but subsequent rapid degradation. Consistent with improved maturation, both FLT-3 K644A and FLT-3 ITD K644A were localized to a much larger extent at the surface of transfected cells than were the kinase-active receptors (Fig. 3B).
FIG.3.

Tyrosine phosphorylation regulates FLT-3 maturation. (A) Proteins in COS-7 cells expressing wild-type (WT) FLT-3, FLT-3 ITD, or FLT-3 ITD K644A were metabolically labeled with [35S]methionine-[35S]cysteine for 20 min. An excess of unlabeled methionine-cysteine was added to the medium (chase). FLT-3 was immunoprecipitated (IP) at the indicated times and analyzed by SDS-PAGE and fluorography. The ratio of 150-kDa signals to 130-kDa signals was calculated in arbitrary units (a.u.). The graphic was compiled from four experiments with consistent results. (B) COS-7 cells expressing wild-type FLT-3, FLT-3 ITD, FLT-3 K644A, or FLT-3 ITD K644A were fixed and subjected to immunostaining with anti-FLT-3* antibody and Cy3-labeled secondary antibody. Images were recorded with a confocal laser scanning microscope. White arrowheads indicate membrane localization; the red arrowhead indicates perinuclear staining. (C) FLT-3 variants were expressed alone or coexpressed with pp60c-src in HEK293 cells. Lysate aliquots were analyzed by immunoblotting. Apparent differences in endogenous c-Src levels reflect loading differences and were not the result of FLT-3 expression.
Src family kinases cooperate with a number of RTKs in signal generation and have the capacity to phosphorylate RTKs with a selectivity overlapping that of autophosphorylation (8, 40). We therefore wondered whether the phosphorylation of kinase-inactive FLT-3 K644A by a Src family kinase would occur and would have an effect on maturation. Interestingly, the coexpression of pp60c-src resulted in FLT-3 tyrosine phosphorylation (data not shown) and strongly impaired the maturation of the kinase-inactive KA mutants of wild-type FLT-3 and FLT-3 ITD (Fig. 3C). Thus, tyrosine phosphorylation of the kinase-deficient receptors themselves or of other cellular substrates by a heterologous kinase could complement defective FLT-3 kinase activity and still result in a maturation block.
PTPs promote and enable FLT-3 maturation.
To further establish a relationship of FLT-3 maturation with tyrosine phosphorylation, we explored the possible effects of PTPs. The overexpression of SHP-1, a PTP that is expressed in all hematopoietic lineages (44), resulted in the dephosphorylation of FLT-3 ITD (Fig. 4A and B) and in elevated levels of mature, 150-kDa FLT-3 ITD (Fig. 4A). Both effects were dependent on PTP activity and were not observed with catalytically inactive SHP-1 C455S (Fig. 4A). The 150-kDa form was less efficiently dephosphorylated in these experiments, probably indicating that this form has a localization different from that of SHP-1. The promoting effect of SHP-1 on FLT-3 ITD maturation could also be observed in pulse-chase experiments (Fig. 4C). To explore whether PTP expression has a general effect on cellular glycoprotein maturation, we analyzed the effect of SHP-1 expression on the maturation of VSV-G. VSV-G was previously used to study glycoprotein maturation (23). Since the size difference between mature VSV-G and immature VSV-G is very small, the presence of the immature form can be better assessed by analyzing endo-H sensitivity. In this type of analysis, neither the coexpression of SHP-1 nor that of FLT-3 ITD affected the maturation of VSV-G (Fig. 4D), indicating that both have no general influence on the maturation machinery.
FIG. 4.



PTPs promote FLT-3 maturation. (A) FLT-3 ITD was transiently coexpressed with various amounts of wild-type (WT) PTP SHP-1 or the catalytically inactive SHP-1 CS variant. Lysate aliquots were analyzed by immunoblotting. (B) COS-7 cells were transfected with expression constructs for FLT-3 ITD alone or together with EGFP-tagged SHP-1. Phosphorylated FLT-3 was detected by immunostaining with anti-pY591 antibody and Cy3-labeled secondary antibody. The expression of SHP-1 was monitored by EGFP fluorescence. (C) FLT-3 ITD was expressed in the absence or in the presence of SHP-1 in COS-7 cells, and the cells were metabolically labeled with [35S]methionine-[35S]cysteine. Maturation of FLT-3 was analyzed in a pulse-chase experiment as described in the legend to Fig. 3A. IP, immunoprecipitation. (D) VSV-G was expressed in HEK293 cells either alone or in the presence of FLT-3 ITD, FLT-3 ITD KA, or SHP-1. VSV-G was immunoprecipitated and either mock treated or digested with endo-H. Digestion products were analyzed by SDS-PAGE and immunoblotting with anti-VSV antibodies. Immature, ER-localized VSV-G species are indicated by the appearance of a lower-molecular-weight band after endo-H digestion (EndoH sens.). Mature VSV-G remains resistant to endo-H (EndoH resist.). (E) EGFP-tagged FLT-3 ITD was coexpressed either with an ECFP-tagged Golgi compartment marker or with an ECFP-tagged ER marker alone or in the presence of SHP-1 in COS-7 cells. Localization of FLT-3 ITD was detected by EGFP fluorescence and compared to the ECFP fluorescence of the indicated localization markers. The expression of SHP-1 was monitored by immunostaining with anti-SHP-1 antibody and Cy3-labeled secondary antibody. Images were recorded with a confocal laser scanning microscope. White arrowheads indicate membrane localization; red arrowheads indicate perinuclear staining. (F) FLT-3 ITD was transiently coexpressed with various amounts of PTP1B or PTP-PEST in HEK293 cells. Lysate aliquots were analyzed by immunoblotting. (G) COS-7 cells were transfected with wild-type FLT-3 and preincubated either with proteasome inhibitor MG132 (lower panels) or with the corresponding solvent (upper panels). The broad-range PTP inhibitor bp(V)phen was added, and incubation was continued for the indicated times. Lysate aliquots were analyzed with the indicated antibodies. (H) THP-1 cells expressing wild-type FLT-3 endogenously were subjected to treatment with bp(V)phen as described for panel G. FLT-3 was immunoprecipitated, and expression was analyzed by immunoblotting. (I) COS-7 cells were transfected with an expression construct for wild-type FLT-3 or FLT-3 ITD and treated with brefeldin A for 6 h. Lysate aliquotes were analyzed by immunoblotting. (J) (Left panels) HEK293 cells were transfected with wild-type FLT-3 and cotransfected with a pSuper PTP1B siRNA expression construct or the corresponding vector. Immunoblots of lysate aliquots are shown. (Right panels) THP-1 cells stably transduced either with empty pSuper.retro plasmid or with a pSuper.retro.puro SHP-1 siRNA expression construct were transfected with PTP1B siRNAs by using Amaxa electroporation. FLT-3 was immunoprecipitated, and expression was analyzed by immunoblotting. Lysate aliquots were analyzed by immunoblotting for the suppression of PTP1B and SHP-1. Relative expression levels were determined by densitometric analysis and are indicated below the blots (control, 1.0).
We also analyzed the effect of SHP-1 coexpression on the cellular localization of FLT-3 ITD. In the absence of SHP-1, EGFP-tagged FLT-3 ITD showed a perinuclear localization, largely overlapping the localization of a coexpressed ECFP-tagged ER marker but only partially overlapping the localization of a corresponding Golgi compartment marker. Coexpression of SHP-1 led to a change in FLT-3 ITD localization, with reduced amounts in the perinuclear compartment and elevated amounts in the cell periphery, including the plasma membrane (Fig. 4E). To evaluate the specificity of the SHP-1 effect, we tested additional PTPs for their influence on FLT-3 ITD maturation. PTP1B and, to a lesser extent, PTP-PEST dephosphorylated FLT-3 ITD and promoted its maturation, whereas RPTPα neither dephosphorylated the receptor nor had an effect on its maturation (Fig. 4F). Thus, not all PTPs can promote maturation, and the effect correlates with the capacity of a PTP to dephosphorylate FLT-3. Conversely, the general inhibition of cellular PTPs by the cell-permeating PTP inhibitor bp(V)phen strongly compromised the maturation of wild-type FLT-3 (Fig. 4G and H). At longer treatment times, FLT-3 protein was lost. In the presence of the proteasome inhibitor MG132, the bp(V)phen-induced loss of FLT-3 was attenuated to some extent (Fig. 4G, lower panel), suggesting that the massive block of FLT-3 maturation upon general PTP inhibition eventually triggered proteasome-mediated degradation. Thus, general PTP inhibition had an effect on FLT-3 maturation similar to the general blockade of transport from the ER to the Golgi compartment by brefeldin A (Fig. 4I).
To address more specifically the role of PTPs in maturation, we chose to perform siRNA-mediated suppression of two PTPs. PTP1B is anchored to ER membranes via a C-terminal targeting sequence and may control the phosphorylation status of newly synthesized RTKs (5). Transfection of HEK293 cells with an expression vector directing the synthesis of an siRNA which specifically targets human PTP1B mRNA reduced the expression of endogenous PTP1B by at least 80% (Fig. 4J, left panel). Transiently expressed wild-type FLT-3 clearly matured less efficiently in these cells than in the corresponding control cells. Thus, endogenous PTP1B was limiting for efficient FLT-3 maturation in this setting. Interestingly, the suppression of PTP1B was also associated with hyperphosphorylation of the immature, 130-kDa form of FLT-3 (Fig. 4J, left panel). In an attempt to suppress PTP1B expression in THP-1 cells, we succeeded only in a mild reduction in protein levels (by 35%) compared to the results in mock-treated cells. Still, less efficient maturation of endogenous wild-type FLT-3 could clearly be detected (Fig. 4J, right panel). On the other hand, efficient reduction of endogenous SHP-1 expression had only a rather weak effect on maturation (Fig. 4J, right panel); however, this finding was consistently seen in many experiments. This analysis, which needs to be extended to additional PTPs, indicates the differential importance of endogenous PTPs for FLT-3 maturation.
Reduced folding efficiency causes ER retention of FLT-3 ITD.
To explore the mechanism leading to the retarded maturation of FLT-3 ITD, we first considered that its constitutive signaling activity could inhibit components that are important for ER-Golgi compartment translocation and/or complex glycosylation. If this were the case, then it should be possible to assign the maturation block to a selective downstream signaling event. We first tested several inhibitors of signaling pathways which are known to be activated by FLT-3 ITD (for a review, see reference 35). Among them, the protein kinase C (PKC) family inhibitor BisI and the Src family kinase inhibitor PP1 reproducibly but only weakly promoted maturation, whereas the phosphatidylinositol 3-kinase inhibitor wortmannin had no effect (Fig. 5A and B).
FIG. 5.
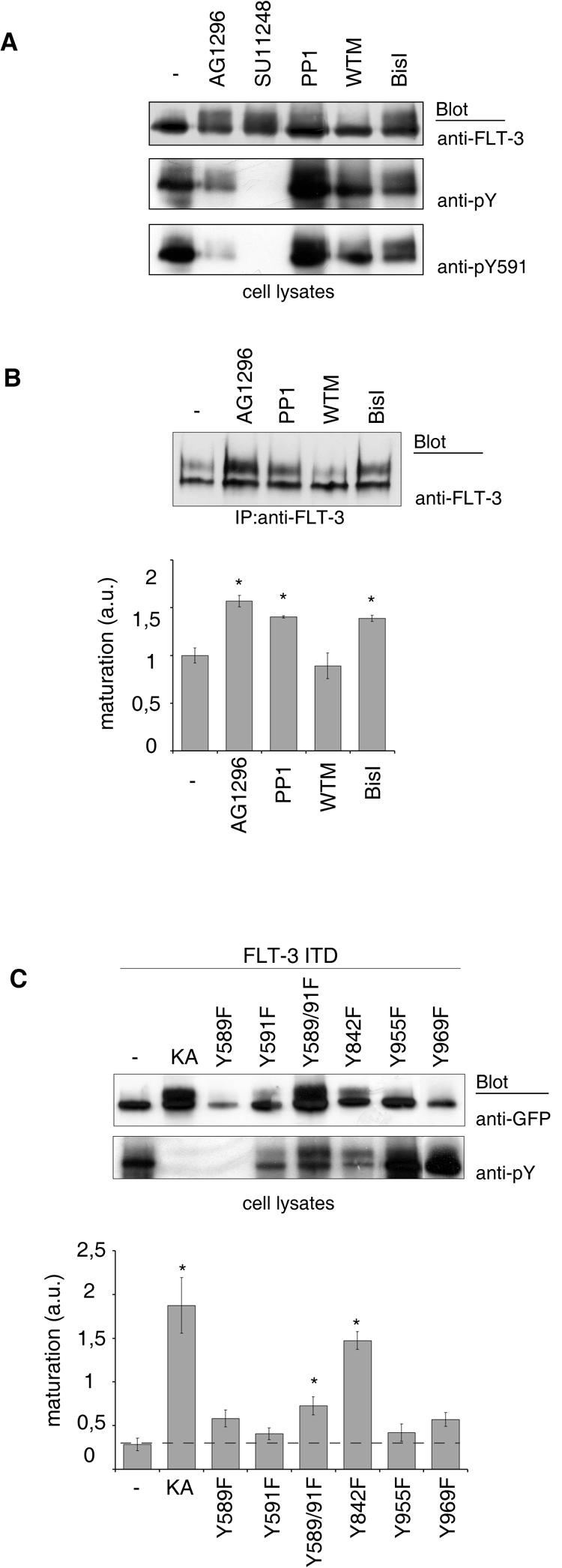
Roles of signal transduction and of individual phosphorylation sites in FLT-3 maturation. FLT-3 ITD-expressing COS-7 cells (A) or MV4-11 cells (B) were treated with the FLT-3 tyrosine kinase inhibitor AG1296 or SU11248, the Src family kinase inhibitor PP1, the phosphatidylinositol 3-kinase inhibitor wortmannin (WTM), or the PKC family inhibitor BisI for 3 h. FLT-3 expression was analyzed by immunoblotting of lysate aliquots (A) or of FLT-3 immunoprecipitates (IP) (B, upper panel). The results of three experiments with MV4-11 cells were quantitated and are shown in the lower panel (mean ± SEM; the asterisk indicates a P value of <0.05). a.u., arbitrary units. (C) The indicated mutants of EGFP-tagged FLT-3 ITD were expressed in COS-7 cells. Expression and phosphorylation were evaluated by immunoblotting of lysate aliquots; representative results are shown in the upper panels. The results of three or four independent experiments were quantitated by densitometric scanning, and maturation was calculated as the ratio of 150-kDa signals to 130-kDa signals; the results are shown in the lower panel (mean ± SEM; the asterisk indicates a P value of <0.05). The broken horizontal line indicates the control level of maturation, for better comparison.
Taking an alternative approach, we generated point mutations in individual FLT-3 autophosphorylation sites and tested the mutants for maturation efficiency. If selective signaling events were important, then maturation should be enhanced by mutation of the corresponding binding sites of respective signaling molecules. This analysis revealed differential effects of the YF point mutations (Fig. 5C). Single mutations of tyrosines 589, 591, 955, and 969 had little promoting effect on maturation. The Y589F mutation strongly reduced expression for unknown reasons. Clear promotion of maturation occurred with mutation of tyrosine 842, which resides in the FLT-3 kinase activation loop. By analogy with related RTKs, phosphorylation of this site should be important for kinase activation (11). Indeed, the FLT-3 ITD Y842F molecule exhibited reduced general phosphorylation. Thus, this mutation did not selectively affect signaling but rather impaired global phosphorylation. The promoted maturation of FLT-3 ITD Y842F was therefore consistent with the effects of kinase inhibitors, K644A mutations, or dephosphorylation by PTPs described above. The combined mutation of tyrosines 589 and 591 had no significant effect on kinase activity, global phosphorylation, or expression level but still reproducibly enhanced maturation weakly, suggesting that these mutations may inhibit a signaling activity of FLT-3 ITD which negatively regulates maturation. By analogy with homologous platelet-derived growth factor (PDGF) receptors and the RTK c-Kit (19, 27), one candidate signaling molecule for binding to these sites is pp60c-src, consistent with the maturation-promoting effect of a Src family kinase inhibitor. Thus, selective signaling events downstream of active FLT-3, notably, Src family kinases, may play some role in the attenuation of the maturation process. However, global FLT-3 phosphorylation appeared to be the dominant factor for retarded maturation. These findings also suggest that it is not the kinase activity of FLT-3 ITD toward other substrates but rather the constitutive phosphorylation of the molecule itself that may primarily cause impaired maturation.
As an obvious possible reason for the inefficient maturation of FLT-3 ITD independent from signaling processes, we considered the inefficient folding of the autophosphorylated kinase. A reduced folding capacity should lead to an enhanced interaction with ER-resident chaperones in the process of ER quality control. The observed ER retention of FLT-3 ITD would be consistent with such a model. An enhanced chaperone dependence of FLT-3 ITD was previously suggested. FLT-3 ITD but not wild-type FLT-3 was shown to associate with heat shock protein 90 (Hsp90), and its activity and expression were dependent on this association (24). In support of this observation, we found that geldanamycin, an inhibitor of Hsp90 function, potently reduced the level of expression of FLT-3 ITD, whereas wild-type FLT-3 was affected to a much lesser extent (Fig. 6A). While this article was in preparation, similar findings were reported by others (7). We therefore tested whether FLT-3 ITD would exhibit an enhanced interaction with the ER chaperone calnexin, a key component of the ER quality control mechanism for glycoproteins (4). FLT-3 ITD indeed associated with calnexin, whereas wild-type FLT-3 and the kinase-inactive FLT-3 ITD K644A mutant interacted with calnexin much less efficiently (Fig. 6B and C). Thus, constitutive phosphorylation of FLT-3 ITD appears to result in inefficient folding, indicated by chaperone dependence and enhanced recognition by the ER quality control system.
FIG. 6.

Impaired stability of FLT-3 ITD, enhanced association with the ER chaperone calnexin, and generality of phosphorylation-dependent RTK maturation. (A) COS-7 cells were transfected with expression constructs for wild-type (WT) FLT-3 or FLT-3 ITD and treated with the Hsp90 inhibitor geldanamycin for 6 h. The expression of FLT-3 was detected by immunoblotting of lysate aliquots. (B) FLT-3 ITD or its kinase-inactive FLT-3 ITD K644A counterpart was expressed in COS-7 cells. FLT-3 was immunoprecipitated (IP), and the immunoprecipitates were probed for associated calnexin. Lysate aliquots were analyzed by immunoblotting for equal levels of expressionof calnexin. (C) MV4-11 or THP-1 cells were used to analyze calnexin (CNX) association with endogenous FLT-3 ITD or wild-type FLT-3, respectively. Either FLT-3 was detected in calnexin immunoprecipitates (left panels) or FLT-3 was immunoprecipitated and probed for associated calnexin (right panels). CG, complex glycosylated species; HM, high-mannose form. (D) The following proteins were expressed in HEK293 cells: PDGFβ receptor (PDGFβR), its kinase-inactive KA version (alone or in the presence of pp60c-src kinase), wild-type c-Kit, KitΔGNNK, or Trk-Ros-ECFP (a chimeric receptor containing the extracellular domain of human TrkA and the intracellular domain of mouse c-Ros and tagged with ECFP) (alone or in combination with wild-type [WT] SHP-1 or its CS variant). Corresponding cell lysates were analyzed by immunoblotting.
Inverse relationship of maturation efficiency and tyrosine phosphorylation for other RTKs.
To explore whether the inverse relationship of RTK phosphorylation and maturation obeys a more common mechanism, we analyzed additional members of the RTK family. For the PDGFβ receptor, expression of the kinase-inactive K634A mutant led to the formation of more mature receptor than expression of the wild type (Fig. 6D). However, coexpression of pp60c-src with the kinase-inactive receptor resulted in the accumulation of an immature receptor, similar to what was observed before for FLT-3. In a comparison of wild-type c-Kit with its constitutively active counterpart c-KitΔGNNK (30), it was evident that wild-type c-Kit matured properly, while c-KitΔGNNK existed to a considerable extent in an immature, underglycosylated form (Fig. 6D). Finally, the structurally less related RTK c-Ros also yielded mature and underglycosylated forms upon overexpression. Coexpression with the potent PTP SHP-1 (15) strongly promoted maturation (Fig. 6D). Thus, the regulation of RTK maturation by tyrosine phosphorylation appears to be a more general phenomenon and is not restricted to FLT-3.
DISCUSSION
Mutations in membrane molecules which impair expression at the cell surface have been found in different diseases (32). Examples are loss-of-function mutations in the extracellular domain of the RTK RET, which are associated with Hirschsprung disease. In this condition, immature forms of mutant RET protein accumulate intracellularly and are prevented from interaction with the coreceptor GFRα1 and the ligand GDNF (16). Similar observations have been reported for the insulin receptor in certain rare cases of insulin-resistant diabetes (31). As a common principle, these disease-causing mutations impair the folding efficiency of the kinases, leading to entrapment by the chaperones that are components of the ER quality control. Another class of mutations which lead to proteins with pronounced chaperone interactions are gain-of-function mutations in oncogenic kinases. Such proteins, for example, BCR-Abl, v-Src, and mutated Kit, are dependent on continuous interactions with the chaperone Hsp90, as indicated by their rapid degradation in the presence of the Hsp90-depleting agent geldanamycin or its analogs (12).
As we describe here, the transforming RTK FLT-3 ITD interacts with both classes of chaperones, indicating inefficient folding and stability. An enhanced interaction with calnexin in the ER is associated with partial ER retention and reduced surface expression. As a previously unrecognized phenomenon, ER retention of FTL-3 ITD is linked to its constitutive kinase activity and can be overcome by kinase-inactivating mutations, treatment with kinase inhibitors, or coexpression of PTPs. A negative regulatory function of tyrosine phosphorylation for RTK maturation is most likely a more general mechanism, since we observed it also for three other tested members of the RTK family—c-Kit, the PDGFβ receptor, and Ros. While this article was in preparation, Lievens et al. (18) reported that highly activated mutant versions of fibroblast growth factor receptor 3 (FGFR-3), which cause a severe form of dwarfism in men, mature very inefficiently. Kinase inactivation, however, restores maturation and surface localization. These findings are consistent with the observations that we report here and support the generality of an inhibitory role of tyrosine phosphorylation in the maturation of RTKs.
For the mechanism of maturation arrest, we considered the possibility that signaling downstream of constitutively active FLT-3 ITD negatively regulates components that participate in the maturation process, for example, molecules of the quality control mechanism or molecules with importance for ER-Golgi vesicle transport. We observed, however, no clear effects of FLT-3 ITD expression on the maturation of VSV-G. Thus, signaling of FLT-3 ITD was obviously without prominent effects on the general glycoprotein maturation machinery. Attempts to link the constitutive signaling activity of FLT-3 ITD with ER retention by testing a panel of signal transduction inhibitors and a panel of YF mutants likewise revealed no clue for a critical signaling event. We observed some weak maturation-promoting effects of the PKC inhibitor BisI, the Src family inhibitor PP1, and a Y589/591F double mutation, which may also be related to impaired interactions with Src family kinases. These potential pathways for the regulation of ER quality control or further steps in FLT-3 maturation deserve further investigation. However, complete inactivity or global dephosphorylation of FLT-3 ITD showed the best correlation with maturation, suggesting that it may be phosphorylation of the RTK itself which reduces folding efficiency and triggers the calnexin interaction. Again, this activity could not be linked to phosphorylation of single sites in our mutational analysis. Instead, the physicochemical effect of phosphorylation on multiple sites may impair folding and potentially also protein stability. Answering these questions will require folding experiments at the level of recombinant FLT-3 protein.
Our findings suggest a novel function for ER-associated PTPs. General PTP inhibition drastically impaired the maturation of wild-type FLT-3 to the point that degradation was initiated. Based on our results, we propose that suppression of the basal activities of newly synthesized RTKs is essential for their release from ER quality control and further processing. Analysis of individual PTPs for their roles in this process is highly warranted. It should be noted that several PTPs have ER localization domains (PTP1B and T-cell PTP) (5, 20) or are enriched in a perinuclear compartment (SHP-1) (37). A role for PTP1B in suppressing the activities of ER-bound RTKs was proposed earlier but has not been linked to RTK maturation (6). A function of PTP1B in this process is supported by our finding of impaired FLT-3 maturation upon siRNA-mediated suppression of endogenous PTP1B expression in HEK293 cells transiently expressing wild-type FLT-3. Also, siRNA-mediated partial reduction of endogenous PTP1B levels in THP-1 cells led to less efficient maturation of endogenous wild-type FLT-3. However, the suppression of SHP-1 had only a weak effect on endogenous FLT-3 maturation, suggesting differential importance of the two PTPs in this process. Further experiments are required to firmly establish the role of PTP1B and to evaluate the role of other PTP family members.
Another interesting implication from our study is the possibility that ER retention of constitutively active RTKs changes the quality of signal transduction by providing access to substrates which are not accessible to surface-bound RTKs. This may be an important aspect of the transforming activities of constitutively active RTKs. Indeed, there are strong qualitative differences in the signaling of FLT-3 ITD compared with ligand-stimulated wild-type FLT-3 which presently cannot be explained (26). ER-retained, constitutively active versions of FGFR-3 are capable of activating the Janus kinase (Jak)/STAT pathway by directly recruiting Jak1, whereas wild-type FGFR-3 cannot activate STAT1 (18). For the RTK Ros, it has been shown that aberrant Golgi compartment localization in glioblastoma cells, conferred by fusion with the Golgi compartment-targeted partner FIG, is transforming (3). Intracellular activation of PDGF receptors by the v-Sis protein has been related to sis-mediated transformation (1, 13). Thus, ER or Golgi compartment retention may emerge as a more general pathway for RTK-mediated pathological signaling and transformation.
Acknowledgments
We are grateful to S. Scholl for providing THP-1 and MV4-11 cells as well as for help with FACS analysis. We thank W. Birchmeier, L. Claesson-Welsh, D. Fujita, S. Gutkind, C. H. Heldin, L. Rönnstrand, M. Tremblay, and A. Ullrich for providing various cDNAs and H. Keilhack, M. O'Farrel, and G. Nolan for providing other reagents and tools.
This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB604, A1, and Bo 1043/4-3 to F.-D.B. and Se600/2-3 to H.S.), from the Deutsche Krebshilfe (10-2100-Do2 to F.-D.B.), and from the IZKF Münster (to H.S.).
REFERENCES
- 1.Bejcek, B. E., D. Y. Li, and T. F. Deuel. 1989. Transformation by v-sis occurs by an internal autoactivation mechanism. Science 245:1496-1499. [DOI] [PubMed] [Google Scholar]
- 2.Brummelkamp, T. R., R. Bernards, and R. Agami. 2002. A system for stable expression of short interfering RNAs in mammalian cells. Science 296:550-553. [DOI] [PubMed] [Google Scholar]
- 3.Charest, A., V. Kheifets, J. Park, K. Lane, K. McMahon, C. L. Nutt, and D. Housman. 2003. Oncogenic targeting of an activated tyrosine kinase to the Golgi apparatus in a glioblastoma. Proc. Natl. Acad. Sci. USA 100:916-921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ellgaard, L., and A. Helenius. 2003. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 4:181-191. [DOI] [PubMed] [Google Scholar]
- 5.Frangioni, J. V., P. H. Beahm, V. Shifrin, C. A. Jost, and B. G. Neel. 1992. The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid C-terminal sequence. Cell 68:545-560. [DOI] [PubMed] [Google Scholar]
- 6.Frangioni, J. V., A. Oda, M. Smith, E. W. Salzman, and B. G. Neel. 1993. Calpain-catalyzed cleavage and subcellular relocation of protein phosphotyrosine phosphatase 1B (PTP-1B) in human platelets. EMBO J. 12:4843-4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.George, P., P. Bali, P. Cohen, J. Tao, F. Guo, C. Sigua, A. Vishvanath, W. Fiskus, A. Scuto, S. Annavarapu, L. Moscinski, and K. Bhalla. 2004. Cotreatment with 17-allylamino-demethoxygeldanamycin and FLT-3 kinase inhibitor PKC412 is highly effective against human acute myelogenous leukemia cells with mutant FLT-3. Cancer Res. 64:3645-3652. [DOI] [PubMed] [Google Scholar]
- 8.Hansen, K., M. Johnell, A. Siegbahn, C. Rorsman, U. Engström, C. Wernstedt, C. H. Heldin, and L. Rönnstrand. 1996. Mutation of a Src phosphorylation site in the PDGF beta-receptor leads to increased PDGF-stimulated chemotaxis but decreased mitogenesis. EMBO J. 15:5299-5313. [PMC free article] [PubMed] [Google Scholar]
- 9.Helenius, A., and M. Aebi. 2001. Intracellular functions of N-linked glycans. Science 291:2364-2369. [DOI] [PubMed] [Google Scholar]
- 10.Helenius, A., and M. Aebi. 2004. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73:1019-1049. [DOI] [PubMed] [Google Scholar]
- 11.Hubbard, S. R. 2002. Autoinhibitory mechanisms in receptor tyrosine kinases. Front. Biosci. 7:d330-d340. [DOI] [PubMed] [Google Scholar]
- 12.Kamal, A., M. F. Boehm, and F. J. Burrows. 2004. Therapeutic and diagnostic implications of Hsp90 activation. Trends Mol. Med. 10:283-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keating, M. T., and L. T. Williams. 1988. Autocrine stimulation of intracellular PDGF receptors in v-sis-transformed cells. Science 239:914-916. [DOI] [PubMed] [Google Scholar]
- 14.Keilhack, H., U. Hellman, J. van Hengel, F. van Roy, J. Godovac-Zimmermann, and F. D. Böhmer. 2000. The protein-tyrosine phosphatase SHP-1 binds to and dephosphorylates p120 catenin. J. Biol. Chem. 275:26376-26384. [DOI] [PubMed] [Google Scholar]
- 15.Keilhack, H., M. Müller, S. A. Böhmer, C. Frank, K. M. Weidner, W. Birchmeier, T. Ligensa, A. Berndt, H. Kosmehl, B. Günther, T. Müller, C. Birchmeier, and F. D. Böhmer. 2001. Negative regulation of Ros receptor tyrosine kinase signaling. An epithelial function of the SH2 domain protein tyrosine phosphatase SHP-1. J. Cell Biol. 152:325-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kjaer, S., and C. F. Ibanez. 2003. Intrinsic susceptibility to misfolding of a hot-spot for Hirschsprung disease mutations in the ectodomain of RET. Hum. Mol. Genet. 12:2133-2144. [DOI] [PubMed] [Google Scholar]
- 17.Levis, M., J. Allebach, K. F. Tse, R. Zheng, B. R. Baldwin, B. D. Smith, S. Jones-Bolin, B. Ruggeri, C. Dionne, and D. Small. 2002. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood 99:3885-3891. [DOI] [PubMed] [Google Scholar]
- 18.Lievens, P. M., C. Mutinelli, D. Baynes, and E. Liboi. 2004. The kinase activity of fibroblast growth factor receptor 3 with activation loop mutations affects receptor trafficking and signaling. J. Biol. Chem. 279:43254-43260. [DOI] [PubMed] [Google Scholar]
- 19.Linnekin, D., C. S. DeBerry, and S. Mou. 1997. Lyn associates with the juxtamembrane region of c-Kit and is activated by stem cell factor in hematopoietic cell lines and normal progenitor cells. J. Biol. Chem. 272:27450-27455. [DOI] [PubMed] [Google Scholar]
- 20.Lorenzen, J. A., C. Y. Dadabay, and E. H. Fischer. 1995. COOH-terminal sequence motifs target the T cell protein tyrosine phosphatase to the ER and nucleus. J. Cell Biol. 131:631-643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lyman, S. D., L. James, J. Zappone, P. R. Sleath, M. P. Beckmann, and T. Bird. 1993. Characterization of the protein encoded by the flt3 (flk2) receptor-like tyrosine kinase gene. Oncogene 8:815-822. [PubMed] [Google Scholar]
- 22.Matthews, W., C. T. Jordan, G. W. Wiegand, D. Pardoll, and I. R. Lemischka. 1991. A receptor tyrosine kinase specific to hematopoietic stem and progenitor cell-enriched populations. Cell 65:1143-1152. [DOI] [PubMed] [Google Scholar]
- 23.Mezzacasa, A., and A. Helenius. 2002. The transitional ER defines a boundary for quality control in the secretion of tsO45 VSV glycoprotein. Traffic 3:833-849. [DOI] [PubMed] [Google Scholar]
- 24.Minami, Y., H. Kiyoi, Y. Yamamoto, K. Yamamoto, R. Ueda, H. Saito, and T. Naoe. 2002. Selective apoptosis of tandemly duplicated FLT3-transformed leukemia cells by Hsp90 inhibitors. Leukemia 16:1535-1540. [DOI] [PubMed] [Google Scholar]
- 25.Mizuki, M., R. Fenski, H. Halfter, I. Matsumura, R. Schmidt, C. Müller, W. Gruning, K. Kratz-Albers, S. Serve, C. Steur, T. Buchner, J. Kienast, Y. Kanakura, W. E. Berdel, and H. Serve. 2000. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood 96:3907-3914. [PubMed] [Google Scholar]
- 26.Mizuki, M., J. Schwable, C. Steur, C. Choudhary, S. Agrawal, B. Sargin, B. Steffen, I. Matsumura, Y. Kanakura, F. D. Böhmer, C. Muller-Tidow, W. E. Berdel, and H. Serve. 2003. Suppression of myeloid transcription factors and induction of STAT response genes by AML-specific Flt3 mutations. Blood 101:3164-3173. [DOI] [PubMed] [Google Scholar]
- 27.Mori, S., L. Rönnstrand, K. Yokote, A. Engstrom, S. A. Courtneidge, L. Claesson-Welsh, and C. H. Heldin. 1993. Identification of two juxtamembrane autophosphorylation sites in the PDGF beta-receptor; involvement in the interaction with Src family tyrosine kinases. EMBO J. 12:2257-2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakao, M., S. Yokota, T. Iwai, H. Kaneko, S. Horiike, K. Kashima, Y. Sonoda, T. Fujimoto, and S. Misawa. 1996. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 10:1911-1918. [PubMed] [Google Scholar]
- 29.O'Farrell, A. M., T. J. Abrams, H. A. Yuen, T. J. Ngai, S. G. Louie, K. W. Yee, L. M. Wong, W. Hong, L. B. Lee, A. Town, B. D. Smolich, W. C. Manning, L. J. Murray, M. C. Heinrich, and J. M. Cherrington. 2003. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood 101:3597-3605. [DOI] [PubMed] [Google Scholar]
- 30.Reith, A. D., C. Ellis, S. D. Lyman, D. M. Anderson, D. E. Williams, A. Bernstein, and T. Pawson. 1991. Signal transduction by normal isoforms and W mutant variants of the Kit receptor tyrosine kinase. EMBO J. 10:2451-2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rouard, M., J. Bass, F. Grigorescu, T. P. Garrett, C. W. Ward, G. Lipkind, C. Jaffiole, D. F. Steiner, and G. I. Bell. 1999. Congenital insulin resistance associated with a conformational alteration in a conserved beta-sheet in the insulin receptor L1 domain. J. Biol. Chem. 274:18487-18491. [DOI] [PubMed] [Google Scholar]
- 32.Sanders, C. R., and J. K. Myers. 2004. Disease-related misassembly of membrane proteins. Annu. Rev. Biophys. Biomol. Struct. 33:25-51. [DOI] [PubMed] [Google Scholar]
- 33.Sawyers, C. L. 2002. Finding the next Gleevec: FLT3 targeted kinase inhibitor therapy for acute myeloid leukemia. Cancer Cell. 1:413-415. [DOI] [PubMed] [Google Scholar]
- 34.Scheijen, B., H. T. Ngo, H. Kang, and J. D. Griffin. 2004. FLT3 receptors with internal tandem duplications promote cell viability and proliferation by signaling through Foxo proteins. Oncogene 23:3338-3349. [DOI] [PubMed] [Google Scholar]
- 35.Schmidt-Arras, D., J. Schwäble, F. D. Böhmer, and H. Serve. 2004. Flt3 receptor tyrosine kinase as a drug target in leukemia. Curr. Pharm. Des. 10:1867-1883. [DOI] [PubMed] [Google Scholar]
- 36.Shibuya, N., I. J. Goldstein, E. J. Van Damme, and W. J. Peumans. 1988. Binding properties of a mannose-specific lectin from the snowdrop (Galanthus nivalis) bulb. J. Biol. Chem. 263:728-734. [PubMed] [Google Scholar]
- 37.Tenev, T., S. A. Böhmer, R. Kaufmann, S. Frese, T. Bittorf, T. Beckers, and F. D. Böhmer. 2000. Perinuclear localization of the protein-tyrosine phosphatase SHP-1 and inhibition of epidermal growth factor-stimulated STAT1/3 activation in A431 cells. Eur. J. Cell Biol. 79:261-271. [DOI] [PubMed] [Google Scholar]
- 38.Thiede, C., C. Steudel, B. Mohr, M. Schaich, U. Schakel, U. Platzbecker, M. Wermke, M. Bornhauser, M. Ritter, A. Neubauer, G. Ehninger, and T. Illmer. 2002. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 99:4326-4335. [DOI] [PubMed] [Google Scholar]
- 39.Tse, K. F., E. Novelli, C. I. Civin, F. D. Böhmer, and D. Small. 2001. Inhibition of FLT3-mediated transformation by use of a tyrosine kinase inhibitor. Leukemia 15:1001-1010. [DOI] [PubMed] [Google Scholar]
- 40.Wasilenko, W. J., D. M. Payne, D. L. Fitzgerald, and M. J. Weber. 1991. Phosphorylation and activation of epidermal growth factor receptors in cells transformed by the src oncogene. Mol. Cell. Biol. 11:309-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waugh, M. G., D. Lawson, and J. J. Hsuan. 1999. Epidermal growth factor receptor activation is localized within low-buoyant density, non-caveolar membrane domains. Biochem. J. 337:591-597. [PMC free article] [PubMed] [Google Scholar]
- 42.Weisberg, E., C. Boulton, L. M. Kelly, P. Manley, D. Fabbro, T. Meyer, D. G. Gilliland, and J. D. Griffin. 2002. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell 1:433-443. [DOI] [PubMed] [Google Scholar]
- 43.Yee, K. W., A. M. O'Farrell, B. D. Smolich, J. M. Cherrington, G. McMahon, C. L. Wait, L. S. McGreevey, D. J. Griffith, and M. C. Heinrich. 2002. SU5416 and SU5614 inhibit kinase activity of wild-type and mutant FLT3 receptor tyrosine kinase. Blood 100:2941-2949. [DOI] [PubMed] [Google Scholar]
- 44.Yi, T. L., J. L. Cleveland, and J. N. Ihle. 1992. Protein tyrosine phosphatase containing SH2 domains: characterization, preferential expression in hematopoietic cells, and localization to human chromosome 12p12-p13. Mol. Cell. Biol. 12:836-846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhen, Y., R. M. Caprioli, and J. V. Staros. 2003. Characterization of glycosylation sites of the epidermal growth factor receptor. Biochemistry 42:5478-5492. [DOI] [PMC free article] [PubMed] [Google Scholar]