

Abstract
Dilated cardiomyopathy (DCM) is defined as dilation and/or reduced function of one or both ventricles and remains a common disease worldwide. An estimated 40% of cases of familial DCM have an identifiable genetic cause. Accordingly, there is a fast-growing interest in the field of molecular genetics as it pertains to DCM. Many gene mutations have been identified that contribute to phenotypically significant cardiomyopathy. DCM genes can affect a variety of cardiomyocyte functions, and particular genes whose function affects the cell–cell junction and cytoskeleton are associated with increased risk of arrhythmias and sudden cardiac death. Through advancements in next-generation sequencing and cardiac imaging, identification of genetic DCM has improved over the past couple decades, and precision medicine is now at the forefront of treatment for these patients and their families. In addition to standard treatment of heart failure and prevention of arrhythmias and sudden cardiac death, patients with genetic cardiomyopathy stand to benefit from gene mechanism–specific therapies.
Keywords: dilated cardiomyopathy, genetics, cardiac magnetic resonance imaging, epidemiology, echocardiography
EPIDEMIOLOGY AND PATHOGENESIS OF DILATED CARDIOMYOPATHY
The cardiomyopathies are defined as myocardial disorders with abnormal structure or function of the heart. Broadly, these can be subdivided into dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), arrhythmogenic cardiomyopathy (ACM), and left ventricular noncompaction cardiomyopathy. DCM is characterized by dilation and loss of function of one or both ventricles and can be due to secondary causes such as infiltrative disease, metabolic derangements, valvular disease, toxins, medication, and many more. Although ischemic cardiomyopathies are more common in the United States (59% versus 41%) (1), patients with nonischemic DCM are more likely to be women, nonwhite, and younger than those with ischemic cardiomyopathies. The true prevalence of nonischemic DCM is not fully identified but is likely underestimated. An epidemiological study performed in Olmstead county, MN, from 1975 to 1984 using autopsy data, echocardiography, and angiography found a DCM prevalence of 36.5 in 100,000 patients and a man-to-woman ratio of 3:4 (2). This differs from several other studies performed in various regions, a difference that may reflect geographical and ethnic contributions to the frequency of DCM (3–6). Up to 50% of nonischemic DCM is genetic or idiopathic (7). Familial DCM is defined as (a) the presence of two or more relatives with DCM or (b) the presence of one relative with DCM and sudden cardiac death (SCD) prior to the age of 35 years. An estimated 40% of cases of familial DCM have an identifiable genetic cause (8). As such, there is a fast-growing interest in the field of molecular genetics as it pertains to DCM. Many genes have been identified that may contribute to phenotypically significant cardiomyopathy (9).
HCM:
hypertrophic cardiomyopathy
ACM:
arrhythmogenic cardiomyopathy
A significant portion (20–38%) of DCM may have an oligogenic basis; multiple rare variants from different unlinked loci and inconstant penetrance may cause a similar phenotype of DCM. The effects of environmental insults such as myocarditis, chemotherapy, and alcohol on the phenotypic expression of DCM in patients with genetic mutations are termed gene–environment interactions (GxE) and have also been studied (10, 11). In contrast, HCM and ACM fit more classically into a Mendelian model due to few highly penetrant rare variants affecting the sarcomere or desmosome, respectively. In fact, for HCM and ACM, a single mutation may explain most genetic causes in individual families (12–14). As with HCM, variants in the sarcomere genes are a frequent cause of DCM. For example, truncating mutations in the giant sarcomeric protein Titin (TTN) are the most common cause of DCM in adults but may also cause HCM (15–18) in rare cases. Mechanistically, the distinction from HCM is that DCM variants of the sarcomere gene are generally a loss-of-function mutation resulting in impaired force generation and decreased systolic function of the ventricle. DCM-causing mutations are not limited to the sarcomere. They may affect force transmission, mechanical stress, signaling, desmosomal proteins, nuclear structure and function, ion channel activity, protein turnover, and calcium hemostasis (19).
As with adults, genetic testing has become integral to the diagnosis, prognostication, and treatment of the pediatric population. Although DCM variants that directly disturb aspects of cardiomyocyte function are also present in younger patients, many genes associated with inborn errors of metabolism cause DCM (20, 21). Interestingly, a genetic cause in a pediatric patient is identified more commonly than in an adult patient (54% versus 27%) (22, 23). The 5-year survival in pediatric DCM that is familial is 94%, but this same cohort of patients has a relatively high 5-year transplantation rate of 38% (24, 25). These trends differ in idiopathic DCM and myocarditis, emphasizing the importance of genetic testing in all pediatric patients with DCM (26).
DIAGNOSIS AND IMAGING
When evaluating for genetic DCM, it is essential to perform a detailed medical history. In addition, clinicians should obtain baseline hematologic and metabolic data, as well as a baseline electrocardiogram, which may abnormal in HCM, ACM, or DCM. Furthermore, patients should undergo ambulatory cardiac monitoring to evaluate arrythmia burden for further risk stratification. Building a detailed three-generation family history is crucial for determining risk factors for disease progression and SCD as well as classifying cases as familial or sporadic DCM (27). Echocardiography should be included in the initial work-up and screening for patients with suspected DCM. Cardiac magnetic resonance imaging (CMRI) can also be useful in characterization of cardiomyopathy and is frequently utilized in the diagnosis of genetic DCM.
The imaging diagnosis of DCM on echocardiogram is defined as the presence of fractional shortening of <25%, left ventricular ejection fraction (LVEF) of <45%, and left ventricular end diastolic diameter of >2.7 cm/m2 or >117% predicted when corrected for age and body surface area (>2 standard deviations from the upper limits of normal values) (28). Any known secondary cause of myocardial disease must be excluded prior to making a diagnosis of genetic or idiopathic DCM (29). Phenotype-negative individuals who have confirmed variant mutations for DCM may undergo screening by echocardiography every 1–5 years.
Echocardiography
On 2D echocardiography, the LVEF is assessed using the biplane Simpson method with the use of contrast agents to opacify the left ventricular endocardium in the case of poor image quality (30). Linear measurements for estimation of LVEF (Teichholz method) should be avoided due to inaccuracy. Also useful in the estimation of LVEF is 3D echocardiography, which has shown greater reproducibility, accuracy, and inter- and intra-reader consistency when compared to conventional 2D echocardiography techniques (31, 32). This improvement is likely due to several factors, including geometric assumptions with 2D echocardiography in which the heart is modeled as an ellipsoid shape, multiple acquisitions with 2D that can introduce more error, and foreshortening that can affect 2D calculation. Compared to CMRI, a 3D echocardiogram underestimates left ventricular volumes due to overall lower spatial resolution.
Aside from the imaging definition of DCM, other associated findings may be present on a DCM patient’s echocardiogram. As DCM progresses, the left ventricle continues to remodel, becoming more spherical. This change is directly measured by an increased ratio of the short axis to the long axis (33). Functional mitral regurgitation results from dilation of the mitral valve annulus and consequent tethering of the mitral valve leaflets. Careful attention should be given to evaluate for left ventricular thrombus, pulmonary hypertension, and right ventricular dysfunction or dilation.
Cardiac Magnetic Resonance Imaging
CMRI is currently the gold standard for assessment of biventricular volumes and function (34). In addition, the main strength of CMRI is the enhanced ability for tissue characterization and scar pattern analysis (35–38). It is a useful additional test in conjunction with echocardiography to further characterize the phenotype of DCM, as well as to rule out secondary causes of DCM (39). For instance, postischemic DCM with severe myocardial hypoperfusion or scarring may have a similar phenotype of ventricular dilation with reduced function but have distinctive findings on CMRI compared with nonischemic DCM, associated with different prognosis and treatment (40).
Late gadolinium enhancement (LGE) imaging comprises a standard sequence with CMRI to evaluate for focal myocardial fibrosis. A majority (58%) of DCM patients have no LGE, while up to 28% may have a characteristic midmyocardial pattern of enhancement (41, 42). The presence or absence of LGE has large prognostic implications. A study following 472 patients with DCM for 5.3 years noted that those with midmyocardial LGE had a threefold increase in all-cause mortality and fivefold increase in a composite end point of SCD and aborted SCD. A smaller study with 65 patients also showed worse prognosis for DCM patients with midmyocardial LGE, reporting an eightfold increase in heart failure, appropriate internal defibrillator (ICD) firing, and cardiac death (43).
ICD:
internal defibrillator
There is overlap between ACM and DCM. Specific genes such as LMNA, SCN5A, FLNC, RBM20, PLN, DSP, DES, and TMEM43 can cause an arrhythmogenic DCM and may require ICD outside of the traditional primary prevention criteria (44–49). CMRI is also used clinically in DCM patients for additional SCD risk stratification. For example DSP and FLNC mutations may have a characteristic extensive ring-shaped LGE, which is a midmyocardial or subepicardial LGE pattern in three contiguous walls on the short axis view (50). Other forms of DCM do not typically present with this ring-shaped enhancement and can have heterogenous scar patterns (Figure 1).
Figure 1.
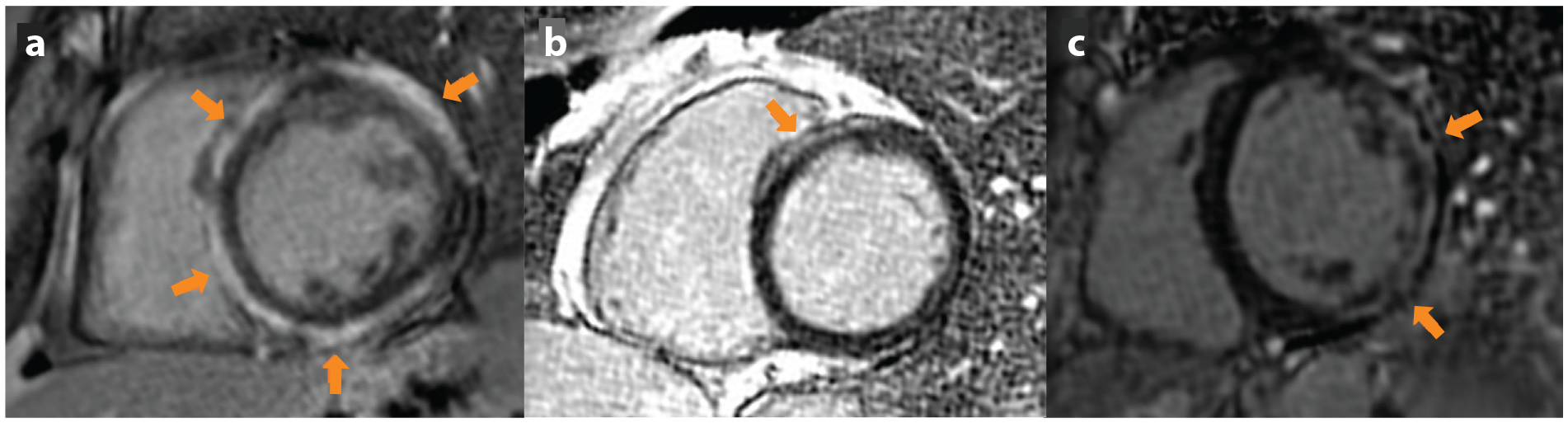
Phase-sensitive inversion recovery MRI sequences showing examples of nonischemic patterns of LGE in DCM (arrows).
(a) Characteristic ring-like subepicardial LGE in patient with DCM and DSP mutation. (b) Anteroseptal midmyocardial LGE in a patient with idiopathic DCM. (c) Patient with Duchenne’s muscular dystrophy demonstrating subepicardial LGE of the anterolateral and inferolateral walls. Abbreviations: DCM, dilated cardiomyopathy; LGE, late gadolinium enhancement; MRI, magnetic resonance imaging.
Genetics
There have been remarkable achievements in the field of genetics due to advancements in next-generation sequencing, and now a person’s entire genome can be evaluated with a single test. Many gene variants have been identified in the pathogenesis of DCM (Table 1). Most of the DCM genes are inherited in an autosomal dominant pattern and have variable penetrance (51). Autosomal recessive, X-linked, mitochondrial inheritance patterns and de novo mutations also occur (52). As discussed, DCM genes encode a wide variety of cellular functions.
Table 1.
Genes causing dilated cardiomyopathy
| Gene | Gene symbol | Mode of inheritance | Classificationa |
|---|---|---|---|
| ATP Binding Cassette Subfamily C Member 9 | ABCC9 | AD | Limited |
| Actin Alpha Cardiac Muscle 1 | ACTCI | AD | Moderate |
| Ankyrin Repeat Domain 1 | ANKRDI | AD | Limited |
| BAG Cochaperone 3 | BAG3 | AD | Definitive |
| Cysteine And Glycine Rich Protein 3 | CSRP3 | AD | Limited |
| Cardiotrophin 1 | CTFI | AD | Limited |
| Desmin | DES | AD | Definitive |
| Desmoglein 2 | DSG2 | AD | Limited |
| Desmoplakin | DSP | AD | Definitiveb |
| Dystrobrevin Alpha | DTNA | AD | Limited |
| EYA Transcriptional Coactivator And Phosphatase 4 | EYA4 | AD | Limited |
| Filamin C | FLNC | AD | Definitive |
| GATA Zinc Finger Domain Containing 1 | GATADI | AR | Limited |
| Integrin Linked Kinase | ILK | AD | Limited |
| Junctophilin 2 | JPH2 | AR | Moderate |
| Laminin Subunit Alpha 4 | LAMA4 | AD | Limited |
| LIM Domain Binding 3 | LDB3 | AD | Limited |
| Lamin A/C | LMNA | AD | Definitive |
| Leucine Rich Repeat Containing 10 | LRRC10 | AR | NKDR/AMO |
| MIB E3 Ubiquitin Protein Ligase 1 | MIB1 | AD | NKDR/AMO |
| Myosin Binding Protein C 3 | MYBPC3 | AD | Limited |
| Myosin Heavy Chain 6 | MYH6 | AD | Limited |
| Myosin Heavy Chain 7 | MYH7 | AD | Definitive |
| Myosin Light Chain 2 | MYL2 | AD | Limited |
| Myosin Light Chain 3 | MYL3 | AD | Disputed |
| Myopalladin | MYPN | AD | Limited |
| Nebulette | NEBL | AD | Limited |
| Nexilin F-Actin Binding Protein | NEXN | AD | Moderate |
| NK2 Homeobox 5 | NKX2–5 | AD | Limited |
| Natriuretic Peptide A | NPPA | AR | NKDR |
| Obscurin | OBSCN | AD | Limited |
| PDZ And LIM Domain 3 | PDLIM3 | AD | Disputed |
| Phospholamban | PLN | AD | Moderateb |
| Plakophillin 2 | PKP2 | AD | Disputed |
| Pleckstrin Homology And RUN Domain Containing M2 | PLEKHM2 | AR | Limited |
| PR/SET Domain 16 | PRDM16 | AD | Limited |
| Presenilin 1 | PSEN1 | AD | Disputed |
| Presenilin 2 | PSEN2 | AD | Limited |
| RNA Binding Motif Protein 20 | RBM20 | AD | Definitive |
| Sodium Voltage-Gated Channel Alpha Subunit 5 | SCN5A | AD | Definitive |
| Sarcoglycan Delta | SGCD | AD | Limited |
| T-Box Transcription Factor 20 | TBX20 | AD | Limited |
| Titin-Cap | TCAP | AD | Limited |
| Transmembrane Protein 43 | TMEM43 | AD | Definitiveb |
| Troponin C1, Slow Skeletal And Cardiac Type | TNNC1 | AD | Definitive |
| Troponin I3, Cardiac Type | TNNI3 | AD | Moderate |
| TNNI3 Interacting Kinase | TNNI3K | AD | Limited |
| Troponin T2, Cardiac Type | TNNT2 | AD | Definitive |
| Tropomyosin 1 | TPM1 | AD | Moderate |
| Titin | TTN | AD | Definitive |
| Vinculin | VCL | AD | Moderate |
In arrhythmogenic right ventricular cardiomyopathy.
Abbreviations: AD, autosomal dominant; AMO, animal model only; AR, autosomal recessive; NKDR, no known disease relationship.
TTN
The giant protein TTN forms the “elastic” filament of the sarcomere, essential for the mechanical compliance of the heart muscle (53). TTN is the largest macromolecule in the human body, composed of 27,000–33,000 amino acids, and is encoded by the gene TTN, which contains 363 exons. It is heavily expressed in striated muscle tissue including cardiac myocytes (54). Modifications at the genetic, transcriptional, and post-translational levels can lead to loss or gain of function and present as HCM, ACM, or DCM. Truncating TTN variants (TTNtv) are the most common causes of genetic DCM, accounting for 20–25% of all cases. Interestingly, at least a subset of peripartum cardiomyopathy cases are attributable to TTNtv (55, 56). Greater than 60,000 missense variants of TTN have been identified, but the clinical significance of missense variations in TTN remains unknown (9).
LMNA
LMNA missense and truncating mutations account for up to 8% of genetic DCM cases. Proteins Lamin A and Lamin C are encoded by the LMNA gene via differential splicing. Mutations in LMNA lead to phenotypic expressions including premature aging, myopathies, and DCM (57). LMNA mutations also lead to conduction abnormalities, atrial and ventricular arrythmias, and SCD, which usually precede DCM and have nearly complete penetrance by the seventh decade of life (58, 59).
PLN
The gene PLN encodes phospholamban, a small, 52–amino acid transmembrane protein that inhibits sarcoplasmic reticulum Ca2+ATPase in its unphosphorylated form. The R14del mutation of PLN is a founder mutation in the Netherlands and Germany, associated with ACM and DCM. Although, as with other DCM genes, mutations in PLN have variable penetrance, lethal arrythmias have been described (60, 61).
RBM20
The gene RBM20 encodes the RNA binding motif 20 protein, a 1,227–amino acid protein that is expressed in both the atria and the ventricles. Mutations in RBM20 are responsible for 1–5% of genetic DCM (62). RBM20 function regulates cardiac splicing including the splicing of TTN. Thus, given the downstream consequences of RBM20 mutations, their presentation may be similar to those of TTNtv.
SCN5A
The gene SCN5A encodes the alpha unit of the main cardiac sodium channel, Nav1.5 (63). Mutations in this gene have been associated with ACM syndromes such as Brugada and long QT syndrome. Missense mutations in SCN5A have also been identified in DCM and carry a higher risk for arrythmias (46, 47).
Cytoskeletal Genes
Various genes encode cardiac cytoskeletal proteins and are associated with DCM. Mutations in the dystrophin gene can lead to DCM in Duchenne’s muscular dystrophy, which has an X-linked inheritance pattern (64). Mutations in the sarcoglycan genes can also produce cardiomyopathy associated with muscular dystrophy from sarcolemmal instability. The gene FLNC encodes filamin C, and mutations have been described in both ACM and DCM. Filamin C has a critical function in cardiomyocytes, interacting with actin, Z-disk, the desmosome, and the dystrophin complex (65). Truncation mutations in FLNC cause DCM that is associated with a high rate of arrythmias and SCD (66).
MANAGEMENT AND EMERGING THERAPIES
The current standard treatment of DCM with reduced LVEF (<40%) is directed toward heart failure, aiming to promote reverse remodeling, improve left ventricular dilation, and improve cardiac function. The current guidelines include a combination of four evidence-based therapies (67): (a) angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, or angiotensin receptor/neprilysin inhibitor; (b) evidence-based beta-blockers; (c) aldosterone antagonists; and (d) SGLT2 (sodium-glucose transport protein 2) inhibitors. Reassessment of LVEF is typically performed after 3 months of uninterrupted medical therapy. Patients with persistently low LVEF (<35%) are at high risk for SCD and benefit from ICD (67), while those with a wide QRS (>150 ms), left bundle branch block, and NYHA (New York Heart Association) class II benefit from cardiac resynchronization therapy. Finally, patients who have refractory heart failure should be referred for advanced therapies including left ventricular assist device and transplant.
However, the heart failure guidelines concept of “one size fits all” does not fully apply to DCM. Patients who are carriers of ACM gene mutations, such as mutations of FLNC, DSP, LMNA, or PLN, may require ICD based on arrhythmic risk factors (68) rather than based on severe left ventricular dysfunction. Also, the understanding of the genetic cause provides novel treatment opportunities. Emerging treatments for DCM include gene therapy for gene replacement (as in regenerative medicine advanced therapy trials) or direct genome editing by CRISPR/Cas-9 technology (currently being tested in vitro and in vivo), signaling pathway modifiers [REALM-DCM trial (NCT03439514)], and modifiers of myofilament function 9 (65, 69, 70).
CONCLUSIONS
DCM is defined as dilation or loss of one or both ventricles and remains a common disease process worldwide. Through advancements in next-generation sequencing and cardiac imaging, identification of genetic DCM has improved over the past couple decades, and precision medicine is now at the forefront of treatment for these patients and their families. In addition to standard treatment of heart failure and prevention of SCD, patients with genetic cardiomyopathy stand to benefit from gene mechanism–specific therapies.
DISCLOSURE STATEMENT
L.M. and M.G.R.T. receive grant support from Tenaya Therapeutics, Pfizer, Owkin, Greenstone Bioscience, and Bristol-Meyers-Squibb. L.M. is member of the scientific advisory board of Tenaya Therapeutics. L.M. and M.R.G.T. receive grant support from the National Institutes of Health (X01 HL139403, R01HL153325, R01HL164634).
LITERATURE CITED
- 1.Shore S, Grau-Sepulveda MV, Bhatt DL, et al. 2015. Characteristics, treatments, and outcomes of hospitalized heart failure patients stratified by etiologies of cardiomyopathy. JACC Heart Failure 3:906–16 [DOI] [PubMed] [Google Scholar]
- 2.Codd MB, Sugrue DD, Gersh BJ, Melton L Jr. 1989. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975–1984. Circulation 80:564–72 [DOI] [PubMed] [Google Scholar]
- 3.Torp A 1978. Incidence of congestive cardiomyopathy. Postgrad. Med. J 54(633):435–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manolio TA, Baughman KL, Rodeheffer R, et al. 1992. Prevalence and etiology of idiopathic dilated cardiomyopathy (summary of a National Heart, Lung, and Blood Institute workshop). Am. J. Cardiol 69(17):1458–66 [DOI] [PubMed] [Google Scholar]
- 5.Miura K, Nakagawa H, Morikawa Y, et al. 2002. Epidemiology of idiopathic cardiomyopathy in Japan: results from a nationwide survey. Heart 87(2):126–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amoah AG, Kallen C. 2000. Aetiology of heart failure as seen from a National Cardiac Referral Centre in Africa. Cardiology 93(1–2):11–18 [DOI] [PubMed] [Google Scholar]
- 7.McNally EM, Mestroni L. 2017. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ. Res 121:731–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ganesh SK, Arnett DK, Assimes TL, et al. 2013. Genetics and genomics for the prevention and treatment of cardiovascular disease: update: a scientific statement from the American Heart Association. Circulation 128(25):2813–51 [DOI] [PubMed] [Google Scholar]
- 9.Gigli M, Begay RL, Morea G, et al. 2016. A review of the giant protein titin in clinical molecular diagnostics of cardiomyopathies. Front. Cardiovasc. Med 3:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas DC. 2004. Statistical Methods in Genetic Epidemiology New York: Oxford Univ. Press [Google Scholar]
- 11.Hazebroek MR, Moors S, Dennert R, et al. 2015. Prognostic relevance of gene-environment interactions in patients with dilated cardiomyopathy: applying the MOGE (S) Classification. J. Am. Coll. Cardiol 66(12):1313–23 [DOI] [PubMed] [Google Scholar]
- 12.Morales A, Kinnamon DD, Jordan E, et al. 2020. Variant interpretation for dilated cardiomyopathy: refinement of the American College of Medical Genetics and Genomics/ClinGen Guidelines for the DCM Precision Medicine Study. Circ. Genom. Precis. Med 13(2):e002480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haas J, Frese K, Peil B, et al. 2015. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J 36(18):1123–35 [DOI] [PubMed] [Google Scholar]
- 14.Cowan JR, Kinnamon DD, Morales A, et al. 2018. Multigenic disease and bilineal inheritance in dilated cardiomyopathy is illustrated in nonsegregating LMNA pedigrees. Circ. Genom. Precis. Med 11(7):e002038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akhtar MM, Lorenzini M, Cicerchia M, et al. 2020. Clinical phenotypes and prognosis of dilated cardiomyopathy caused by truncating variants in the TTN gene. Circ. Heart Fail 13:e006832. [DOI] [PubMed] [Google Scholar]
- 16.Roberts AM, Ware JS, Herman DS, et al. 2015. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci. Transl. Med 7:270ra6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fatkin D, Calkins H, Elliott P, et al. 2021. Contemporary and future approaches to precision medicine in inherited cardiomyopathies: JACC focus seminar 3/5. J. Am. Coll. Cardiol 7(20):2551–72 [DOI] [PubMed] [Google Scholar]
- 18.Eldemire R, Taylor MRG, Mestroni L. 2020. Understanding the role of titin in dilated cardiomyopathy. Int. J. Cardiol 316:186–87 [DOI] [PubMed] [Google Scholar]
- 19.Fatkin D, Seidman CE, Seidman JG. 2014. Genetics and disease of ventricular muscle. Cold Spring Harb. Perspect. Med 4(1):a021063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harakalova M, Kummeling G, Sammani A, et al. 2015. A systematic analysis of genetic dilated cardiomyopathy reveals numerous ubiquitously expressed and muscle-specific. Eur. J. Heart Fail 17(5):484–93 [DOI] [PubMed] [Google Scholar]
- 21.Posafalvi A, Herkert JC, Sinke RJ, et al. 2013. Clinical utility gene card for: dilated cardiomyopathy (CMD). Eur. J. Hum. Genet 21(10). 10.1038/ejhg.2012.276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Towbin JA. 2020. Pediatric primary dilated cardiomyopathy gene testing and variant reclassification: Does it matter? J. Am. Heart Assoc 9(11):e016910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Long PA, Evans JM, Olson TM. 2017. Diagnostic yield of whole exome sequencing in pediatric dilated cardiomyopathy. J. Cardiovasc. Dev. Dis 4(3):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Towbin JA, Lowe AM, Colan SD, et al. 2006. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 296(15):1867–76 [DOI] [PubMed] [Google Scholar]
- 25.Singh RK, Canter CE, Shi L, et al. 2017. Survival without cardiac transplantation among children with dilated cardiomyopathy. J. Am. Coll. Cardiol 70(21):2663–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van der Meulen MH, Herkert JC, den Boer SL, et al. 2022. Genetic evaluation of a nation-wide Dutch pediatric DCM cohort: the use of genetic testing in risk stratification. Circ. Genom. Precis. Med 15(5):e002981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hershberger RE, Givertz MM, Ho CY, et al. 2018. Genetic evaluation of cardiomyopathy—a Heart Failure Society of America practice guideline. J. Cardiac Fail 24:281–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinamonti B, Abate E, De Luca A, et al. 2019. Role of cardiac imaging: echocardiography. In Dilated Cardiomyopathy: From Genetics to Clinical Management, ed. Sinagra G, Merlo M, Pinamonti B, et al. , ch. 7. Cham, Switz.: Springer. https://www.ncbi.nlm.nih.gov/books/NBK553855/ [Google Scholar]
- 29.Mestroni L, Maisch B, McKenna WJ, et al. 1999. Guidelines for the study of familial dilated cardiomyopathies. Eur. Heart J 20(2):93–102 [DOI] [PubMed] [Google Scholar]
- 30.Hoffmann R, Barletta G, von Bardeleben S, et al. 2104. Analysis of left ventricular volumes and function: a multicenter comparison of cardiac magnetic resonance imaging, cine ventriculography, and unenhanced and contrast-enhanced two-dimensional and three-dimensional echocardiography. J. Am. Soc. Echocardiogr 27(3):292–301 [DOI] [PubMed] [Google Scholar]
- 31.Gutiérrez-Chico JL, Zamorano JL, Pérez de Isla L, et al. 2005. Comparison of left ventricular volumes and ejection fractions measured by three-dimensional echocardiography versus by two-dimensional echocardiography and cardiac magnetic resonance in patients with various cardiomyopathies. Am. J. Cardiol 95(6):809–13 [DOI] [PubMed] [Google Scholar]
- 32.Shiota T, McCarthy PM, White RD, et al. 1999. Initial clinical experience of real-time three-dimensional echocardiography in patients with ischemic and idiopathic dilated cardiomyopathy. Am. J. Cardiol 84(9):1068–73 [DOI] [PubMed] [Google Scholar]
- 33.Douglas PS, Morrow R, Loli A, Reichek N. 1989. Left ventricular shape, afterload and survival in idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol 13(2):311–15 [DOI] [PubMed] [Google Scholar]
- 34.Strohm O, Schulz-Menger J, Pilz B, et al. 2001. Measurement of left ventricular dimensions and function in patients with dilated cardiomyopathy. J. Magn. Reson. Imaging 13(3):367–71 [DOI] [PubMed] [Google Scholar]
- 35.Schulz-Menger J, Friedrich MG. 2000. Magnetic resonance imaging in patients with cardiomyopathies: when and why. Herz 25(4):384–91 [DOI] [PubMed] [Google Scholar]
- 36.Francone M, Carbone I, Agati L, et al. 2011. Utility of T2-weighted short-tau inversion recovery (STIR) sequences in cardiac MRI: an overview of clinical applications in ischaemic and non-ischaemic heart disease. Radiol. Med 116(1):32–46 [DOI] [PubMed] [Google Scholar]
- 37.Eitel I, Behrendt F, Schindler K, et al. 2008. Differential diagnosis of suspected apical ballooning syndrome using contrast-enhanced magnetic resonance imaging. Eur. Heart J 29(21):2651–59 [DOI] [PubMed] [Google Scholar]
- 38.Bruder O, Wagner A, Jensen CJ, et al. 2010. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol 56(11):875–87 [DOI] [PubMed] [Google Scholar]
- 39.Francone M 2014. Role of cardiac magnetic resonance in the evaluation of dilated cardiomyopathy: diagnostic contribution and prognostic significance. ISRN Radiol 2014:365404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gulati A, Jabbour A, Ismail TF, et al. 2013. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA 309(9):896–908 [DOI] [PubMed] [Google Scholar]
- 41.McCrohon JA, Moon JC, Prasad SK, et al. 2003. Differentiation of heart failure related to dilated cardiomyopathy and coronary artery disease using gadolinium-enhanced cardiovascular magnetic resonance. Circulation 108(1):54–59 [DOI] [PubMed] [Google Scholar]
- 42.Kramer CM. 2015. Role of cardiac MR imaging in cardiomyopathies. J. Nucl. Med 56(Suppl. 4):39S–45S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu KC, Weiss RG, Thiemann DR, et al. 2008. Late gadolinium enhancement by cardiovascular magnetic resonance heralds an adverse prognosis in nonischemic cardiomyopathy. J. Am. Coll. Cardiol 51(25):2414–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fatkin D, MacRae C, Sasaki T, et al. 1999. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med 341(23):1715–24 [DOI] [PubMed] [Google Scholar]
- 45.Cadrin-Tourigny J, Bosman LP, Nozza A, et al. 2019. A new prediction model for ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J 40(23):1850–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mann SA, Castro ML, Ohanian M, et al. 2012. R222Q SCN5A mutation is associated with reversible ventricular ectopy and dilated cardiomyopathy. J. Am. Coll. Cardiol 60(16):1566–73 [DOI] [PubMed] [Google Scholar]
- 47.McNair WP, Sinagra G, Taylor MR, et al. 2011. SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism. J. Am. Coll. Cardiol 57(21):2160–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith ED, Lakdawala NK, Papoutsidakis N, et al. 2020. Desmoplakin cardiomyopathy, a fibrotic and inflammatory form of cardiomyopathy distinct from typical dilated or arrhythmogenic right ventricular cardiomyopathy. Circulation 141(23):1872–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ortiz-Genga MF, Cuenca S, Dal Ferro M, et al. 2016. Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J. Am. Coll. Cardiol 68(22):2440–51 [DOI] [PubMed] [Google Scholar]
- 50.Augusto JB, Eiros R, Nakou E, et al. 2020. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: a comprehensive genotype-imaging phenotype study. Eur. Heart J. Cardiovasc. Imaging 21(3):326–36 [DOI] [PubMed] [Google Scholar]
- 51.Hershberger RE, Hedges DJ, Morales A. 2013. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat. Rev. Cardiol 10:531–47 [DOI] [PubMed] [Google Scholar]
- 52.McNally EM, Golbus JR, Puckelwartz MJ. 2013. Genetic mutations and mechanisms in dilated cardiomyopathy. J. Clin. Investig 123(1):19–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eldemire R, Tharp CA, Taylor MRG, et al. 2021. The sarcomeric spring protein titin: biophysical properties, molecular mechanisms, and genetic mutations associated with heart failure and cardiomyopathy. Curr. Cardiol. Rep 23(9):121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maruyama K, Kimura S, Yoshidomi H, et al. 1984. Molecular size and shape of beta-connectin, an elastic protein of striated muscle. J. Biochem 95:1423–33 [DOI] [PubMed] [Google Scholar]
- 55.van Spaendonck-Zwarts KY, Posafalvi A, van den Berg MP, et al. 2014. Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. Eur. Heart J. Cardiovasc. Imaging 35(32):2165–73 [DOI] [PubMed] [Google Scholar]
- 56.Ware JS, Li J, Mazaika E, et al. 2016. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N. Engl. J. Med 374:233–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu JT, Muchir A, Nagy PL, Worman HJ. 2011. LMNA cardiomyopathy: cell biology and genetics meet clinical medicine. Dis. Model. Mech 4(5):562–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kumar S, Baldinger SH, Gandjbakhch E, et al. 2016. Long-term arrhythmic and nonarrhythmic outcomes of lamin A/C mutation carriers. J. Am. Coll. Cardiol 68(21):2299–307 [DOI] [PubMed] [Google Scholar]
- 59.Pasotti M, Klersy C, Pilotto A, et al. 2008. Long-term outcome and risk stratification in dilated cardiolaminopathies. J. Am. Coll. Cardiol 52(15):1250–60 [DOI] [PubMed] [Google Scholar]
- 60.Haghighi K, Kolokathis F, Gramolini AO, et al. 2006. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. PNAS 103(5):1388–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu GS, Morales A, Vafiadaki E, et al. 2015. A novel human R25C-phospholamban mutation is associated with super-inhibition of calcium cycling and ventricular arrhythmia. Cardiovasc. Res 107(1):164–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stillitano F, Turnbull IC, Karakikes I, et al. 2016. Genomic correction of familial cardiomyopathy in human engineered cardiac tissues. Eur. Heart J 37:3282–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Veerman CC, Wilde AA, Lodder EM. 2015. The cardiac sodium channel gene SCN5A and its gene product NaV1.5: role in physiology and pathophysiology. Gene 573(2):177–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adorisio R, Mencarelli E, Cantarutti N, et al. 2020. Duchenne dilated cardiomyopathy: cardiac management from prevention to advanced cardiovascular therapies. J. Clin. Med 9(10):3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen SN, Lam CK, Wan YW, et al. 2022. Activation of PDGFRA signaling contributes to filamin C-related arrhythmogenic cardiomyopathy. Sci. Adv 8(8):eabk0052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gigli M, Stolfo D, Graw SL, et al. 2021. Phenotypic expression, natural history, and risk stratification of cardiomyopathy caused by filamin C truncating variants. Circulation 144(20):1600–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022. AHA/ACC/HFSA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 45(18):e895–1032 [DOI] [PubMed] [Google Scholar]
- 68.Towbin JA, McKenna WJ, Abrams DJ, et al. 2019. HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 16(11):e301–72 [DOI] [PubMed] [Google Scholar]
- 69.Helms AS, Thompson AD, Day SM. 2021. Translation of new and emerging therapies for genetic cardiomyopathies. JACC Basic Transl. Sci 7(1):70–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Teerlink JR, Diaz R, Felker GM, et al. 2021. Cardiac myosin activation with omecamtiv mecarbil in systolic heart failure. N. Engl. J. Med 384:105–16 [DOI] [PubMed] [Google Scholar]