

Abstract
The human β-like globin genes (5′-ɛ-Gγ-Aγ-δ-β-3′) are temporally expressed in sequential order from the 5′ to 3′ end of the locus, but the nonadult ɛ- and γ-globin genes are autonomously silenced in adult erythroid cells. Two cis elements have been proposed to regulate definitive erythroid γ-globin repression: the DR (direct repeat) and CCTTG elements. Since these two elements partially overlap, and since a well-characterized HPFH point mutation maps to an overlapping nucleotide, it is not clear if both or only one of the two participate in γ-globin silencing. To evaluate the contribution of these hypothetical silencers to γ-globin regulation, we generated point mutations that individually disrupted either the single DR or all four CCTTG elements. These two were separately incorporated into human β-globin yeast artificial chromosomes, which were then used to generate γ-globin mutant transgenic mice. While DR element mutation led to a dramatic increase in Aγ-globin expression only during definitive erythropoiesis, the CCTTG mutation did not affect adult stage transcription. These results demonstrate that the DR sequence element autonomously mediates definitive stage-specific γ-globin gene silencing.
The human β-globin gene locus is located on chromosome 11 and is composed of five β-like globin genes (5′-ɛ, Gγ, Aγ, δ, and β-3′). All five genes are regulated in a tissue- and developmental stage-specific fashion. The ɛ-globin gene is expressed in primitive erythroid cells of the embryonic yolk sac, while the γ- and β-globin genes are silent. During definitive erythropoiesis in the fetal liver, the two γ-globin genes are activated concomitant with ɛ-globin gene silencing. In definitive erythroid cells of the bone marrow, the δ- and β-globin genes are activated, while both the ɛ- and γ-globin genes are silenced. The developmental process that regulates stage- and organ-specific transitions from ɛ- to γ- to β-globin gene transcription, referred to as hemoglobin switching (35), is in part regulated by interactions between the locus control region (LCR) and the genes.
The expression profile of the human locus changes somewhat when the genes are examined in transgenic mice (TgM). The ɛ- and γ-globin genes are both expressed in primitive erythroid cells of the yolk sac (at 9.5 days postcoitum [dpc]). Abundant β- and diminishing γ-globin gene expression is observed in definitive erythroid cells of the fetal liver (∼14.5 dpc). The two γ-globin genes are completely silenced in adult spleen definitive erythroid cells (13, 29). Thus, in TgM the two human γ-globin genes are regulated as primitive stage-specific genes. Since the mouse lacks a true fetal stage of erythropoiesis, and the two human γ-globin genes share highest homology with the murine βh1 gene among the mouse β-like genes, and since βh1 is most abundantly expressed in the murine yolk sac, it was anticipated that the human γ-globin genes were regulated as though they were embryonic genes in the mouse (17).
Despite thousands of clinical and experimental observations (28, 35), the molecular basis for β-globin gene switching is not yet fully understood. It is generally accepted in this field that hemoglobin switching is a consequence of the interplay between developmental stage-specific transcription factors, which interact both within the promoters of the various genes and the 5′ LCR, as well as any alteration of the physical location of a gene within the locus. We and others have documented that the distance or gene order relative to the LCR is a fundamental determinant of which gene is selectively activated during the primitive stage of erythropoiesis (7, 16, 31, 39). In the definitive stage, however, the ɛ- and γ-globin genes are autonomously silenced (13, 29), regardless of their placement within the locus, or even if their expression is examined as individual transcription units (6, 32). It is therefore most probable that a sequence-specific transcription factor(s) (activators and/or repressors) regulates ɛ- and γ-globin activation and repression through nearby cis elements in a gene autonomous manner, independently from the amplifying effects of the LCR.
Erythroid cell-specific transcription factor EKLF (erythroid Krüppel-like factor) has been proposed to be one such regulator. Analysis of human adult β-globin gene expression in eklf-null mutant mice has clearly shown that EKLF is indispensable for definitive stage β-globin transcriptional activation, but additionally that it is dispensable for ɛ- and γ-globin activation (27, 44), fulfilling one anticipated requirement for a definitive stage-specific activator. EKLF binds to the proximal CACCC element of, and activates, the β-globin gene promoter (24), and a β-thalassemia point mutation that leads to the loss of EKLF binding and consequently results in a moderate reduction in β-globin transcript accumulation (10) is often found in the proximal CACCC element (21, 25, 26). However, since CACCC elements are also found in the ɛ- and γ-globin gene promoters, it becomes difficult to resolve why the eklf germ line mutation should most dramatically affect the adult gene. Although the CACCC element sequences of the β-like globins differ slightly, this difference alone cannot account for differential EKLF promoter-specific recruitment in vivo (1). Moreover, EKLF is transcribed during both primitive and definitive erythropoiesis (34) and is active during both stages (43). It therefore seems likely that preferential activation of the β-globin gene in adult definitive erythroid cells is regulated by factors in addition to EKLF.
Primitive stage-specific human and mouse globin genes harbor binding sites for putative repressors that might interfere with EKLF recruitment to their promoter CACCC sites. In a search for such elements, we and others identified two copies of direct repeat (DR) elements in the human ɛ-globin promoter, while DR elements are absent from the adult β-globin gene promoter (11, 37, 41). When we disrupted those sequences in human β-globin yeast artificial chromosome (YAC) transgenic mice (TgM), we found that the mutant ɛ-gene was activated (or failed to be repressed) during definitive erythropoiesis. Surprisingly, analysis of this ɛ-globin gene promoter mutant in a genetic background either containing or lacking EKLF demonstrated that EKLF is recruited to, and activates, the mutant promoter (41). We then identified a unique DNA binding activity that bound to the ɛ-globin gene promoter DR sequences and named this protein complex DRED (direct repeat erythroid-definitive). Based on these observations, we proposed that DRED was a definitive erythroid stage-specific repressor whose activity suppressed ɛ-globin transcription by abrogating EKLF (activator) binding to the ɛ-globin CACCC sites. The γ-globin genes, which are primitive and stage specific in the mouse, each also bear a single DR sequence element in their promoters, and we speculated that these DR elements are also involved in γ-globin gene silencing in adult definitive erythroid cells (38).
An alternative model for γ-globin gene silencing was proposed by others (22). They identified four tandem copies of CCTTG repeats within the proximal promoters of the two γ-globin genes, and the same motif was also found in the ɛ-, but not in the β-globin, gene promoter. Reporter gene transfection and PIN*POINT analyses into mouse erythroleukemia (MEL) cells revealed that this motif suppressed γ-globin transcription by interfering with the recruitment of EKLF to a neighboring CACCC site. Furthermore, an oligonucleotide containing four copies of the CCTTG motif formed a specific protein-DNA complex using nuclear extracts prepared from MEL cells.
In humans, expression of the γ-globin genes is approximately 1% of the total β-like globin protein in adult erythrocytes. However, patients with hereditary persistence of fetal hemoglobin (HPFH) have increased basal levels of γ-globin, comprising some 5 to 20% of total (35). Among the score of well-characterized HPFH alleles, the Greek syndrome is the result of a point mutation in the Aγ-globin proximal promoter (−117: G→A) (5, 14). The two putative repressor elements discussed above (the DR and CCTTG motifs) overlap by two nucleotides, and the Greek HPFH point mutation could affect any factor binding to both elements (Fig. 1A). Therefore, this mutation could elicit a phenotype in humans as a consequence of disrupting either the DR or CCTTG motif.
FIG. 1.

Human Aγ-globin gene promoter mutagenesis. (A) Schematic structure of the human β-globin locus used for creating transgenic mice. The LCR and β-like globin genes are shown by the open and solid boxes, respectively (top). An enlarged map of Aγ-globin gene promoter is shown (middle) with two putative repressor sequences, a DR (shaded arrow) motif and four CCTTG motifs (open rectangles). The two motifs partially overlap, and the Greek-type HPFH mutation (Aγ-117, indicated as a vertical line) maps in this overlapping segment. Detailed sequences of the Aγ promoter are shown (bottom) with a CAC and distal CAAT boxes (bracketed). The DR motif is underlined with CCTTG motifs italicized. (B) Sequence alignment of wild-type (WT) and mutant Aγ-globin promoters (mutDR and mutCCTTG) used for YAC mutagenesis as well as the ɛ- and β-globin promoters (Epsi and Beta, respectively). These are aligned with DR motifs underlined, except for Beta, which has no DR motif (corresponding portions are still underlined). The CCTTG motifs are italicized and mutated nucleotides are indicated by periods. (C) Competitive EMSA analysis of DRED binding to the β-like globin gene promoters. Increasing amounts of MEL cell nuclear extract (NE) were incubated with [γ-32P]-labeled Epsi probe and analyzed by polyacrylamide gel electrophoresis. An unlabeled 50- or 200-fold molar excess of competitor oligonucleotides listed in panel B were included in the reaction. The relative abundance of DRED EMSA product is shown at the bottom of each lane (the signal intensity with no added competitor was set at 100%). −, no added MEL cell NE.
To clarify which sequence motif, either DR or CCTTG (or possibly both), function as the cis-element(s) responsible for adult erythroid γ-globin gene silencing, we created two groups of point mutations: one that would solely disrupt the DR motif and another that would modify all four CCTTG repeat sequences without affecting the DR element. These two Aγ-globin promoter mutants were individually incorporated into a human β-globin YAC and used to generate TgM. Expression analysis of all the human β-like globin genes revealed that the DR mutation led to a dramatic increase in Aγ-globin mRNA accumulation only in definitive erythroid cells. The CCTTG mutations, in contrast, exhibited only a modest suppression of Aγ-globin expression, and only during the fetal liver, but not the adult, definitive stage of erythropoiesis. These results therefore demonstrate that the DR sequence is a potent definitive stage-specific γ-globin gene silencer element in vivo.
MATERIALS AND METHODS
EMSA.
Electrophoretic mobility shift assay (EMSA) was performed as described previously (41) with only slight modifications. Nuclear extracts were prepared from MEL cells. Sequences of oligonucleotides used as probes or competitors are listed in Fig. 1B (only sense strands are shown). Double-stranded probe DNAs were end-labeled with [γ-32P]ATP, column-purified, and used for binding reactions. The binding buffer used was 11.1 mM Tris-HCl (pH 7.6), 55.5 mM NaCl, 1.1 mM EDTA, 2.2 mM MgCl2, 1.1 mM dithiothreitol, and 11% glycerol.
YAC mutagenesis and TgM.
The targeting vector for mutating the DR motif in the human Aγ-globin promoter was constructed as follows. PCR-directed mutagenesis was performed with the following oligonucleotides using the human β-globin YAC (A201F4.3 [39]) as a template. Artificially introduced restriction enzyme sites are underlined and shown in parentheses: HAGP1-5S, 5′-CTGGAATGACTGGATCCGAACAAGGC-3′ (BamHI); HAGP5-3A, 5′-TTGCCTTGTCAAGGAGATTGGACAAGGC-3′; HAGP6-5S, 5′-CCTTGACAAGGCAAGCTTGACCAATAG-3′ (HindIII); and HAGP2-3A, 5′-GTCCATGTCTAGACAACCAGGAGCCTGTGA-3′ (XbaI).
PCRs were first initiated with the HAGP1-5S/HAGP5-3A and HAGP6-5S/HAGP2-3A primer sets, respectively. The PCR products from the first of synthesis were combined, and a second round of PCR was performed with the HAGP1-5S/HAGP2-3A primer set. The resultant mutant DNA fragment, corresponding to the human Aγ-globin gene promoter (nucleotides [nt] 39143 to 39704; HUMHBB; GenBank), was then digested with BamHI and XbaI and subcloned into BamHI/XbaI-digested pRS306 yeast-targeting vector. The mutation was verified by DNA sequencing. The yeast targeting plasmid DNA was digested with MscI (at nucleotide position 39281) prior to transformation of yeast bearing the wild-type human β-globin YAC to initiate homologous recombination (3).
Essentially the same protocol was followed for mutating the four tandem copies of the γ-globin CCTTG motifs. Oligonucleotides used for PCR-directed mutagenesis were the HAGP1-5S, HAGP2-3A (above) and the following two sequences: HAGP4-5S, 5′-TGACCAATAGCCTGCAGTCGGCAAACTTGAC-3′ (PstI), and HAGP3-3A,5′-AGGCTATTGGTCACTGCTCGGCTGGCCAACC-3′.
The targeting vector DNA was linearized with PflMI (at nucleotide position 39273) and used for targeted recombination of the wild-type human β-globin YAC. Generation and structural analysis of human β-globin YAC transgenic mice was performed as described earlier (41).
Semiquantitative RT-PCR analysis.
Total RNA from a minimum of two animals from each of the transgenic lines was extracted from the yolk sac (9.5 dpc), fetal liver (14.5 dpc), adult spleen (made anemic by phenylhydrazine injection; 1 month old), or peripheral blood (from nonanemic mice) using ISOGEN (Nippon Gene). First-strand cDNA was synthesized with Moloney murine reverse transcriptase (ReverTra Ace; TOYOBO) using 2.5 μg of total RNA in a 20-μl reaction volume. PCR was performed with 1 μl of cDNA in a 10-μl reaction volume (50 μM each deoxynucleoside triphosphates, 50 ng of each gene-specific primer set, 0.2 units of Taq polymerase [Invitrogen], 0.5 μCi of [α-32P]dCTP, 1× PCR buffer [2.5 mM MgCl2]) under the following conditions: 94°C for 30 s, 58°C for 1 min, and 72°C for 1 min. PCR cycles were as followed: 12 cycles for β/α and 21 cycles for γ (adult spleen); 12 cycles for β/α and 18 cycles for γ (fetal liver); 12 cycles for γ/α and 18 cycles for ɛ (yolk sac). Parallel control reactions showed that these cycle numbers were within the exponential amplification range. A sample of each PCR was electrophoresed on 8% polyacrylamide gels, dried, and subjected to PhosphorImager (Typhoon, Amersham) quantification and autoradiography. The PCR primers used were previously described (3, 39).
RESULTS
Aγ-globin promoter mutagenesis.
As described in the introduction, two putative repressor elements have been proposed to regulate the human Aγ-globin promoter from positions neighboring the CAAT box (Fig. 1A). The single DR motif and one of the four tandem copies of CCTTG motifs overlap by two base pairs, including the nucleotide that is mutated in the Greek-type HPFH (Aγ −117: G→A) (5, 14). In order to discriminate which element is responsible for the HPFH phenotype, we designed two human Aγ-globin promoter mutations. In the first mutant, we changed four nucleotides within and around the DR motif, while all four CCTTG motifs were left unchanged (mutDR, Fig. 1B). In the second mutant, nine nucleotide changes were introduced to alter all four CCTTG elements, leaving the single DR motif intact (mutCCTTG, Fig. 1B). Nucleotide −117 that is mutated in the Greek HPFH was not altered in either of the mutants.
We previously showed that the DR motif in the ɛ- and γ-globin promoters form a unique complex with a protein recovered from MEL cell nuclear extracts, and we designated that complex DRED (41). On the basis of both biochemical and genetic evidence, we proposed that DRED might be the factor responsible for silencing of the ɛ- and γ-globin genes in adult definitive stage erythroid cells. In order to determine if the DR mutation (mutDR) successfully disrupted DRED binding to the γ-globin promoter, we conducted EMSA using MEL cell nuclear extracts (Fig. 1C). Since we had demonstrated that DRED bound with high affinity to the wild-type ɛ-globin promoter fragment that contains two DR sequences (Epsi in Fig. 1B) (41), we used the ɛ-globin promoter fragment as a probe and all other sequences as competitors in the EMSA reactions. Radiolabeled Epsi probe and increasing amounts of MEL cell nuclear extract formed the anticipated complex (labeled DRED in Fig. 1C). This complex was disrupted by including unlabeled Epsi fragment in the reaction. Wild-type (WT) and mutCCTTG Aγ promoter fragments efficiently competed with the labeled probe for DRED binding, indicating that functional DRED binding sites are present in these oligonucleotides. In obvious contrast, the mutDR as well as the adult β-globin promoter (Beta) fragments failed to disrupt DRED complex formation. Since the Beta fragment does not bear any DR sites, we conclude that the mutDR promoter lost significant affinity for DRED, while the mutCCTTG oligonucleotide retains undiminished DRED binding activity.
Lee et al. reported detection of a binding activity to the CCTTG-repeat sequences in MEL cell nuclear extract using EMSA (22). We therefore attempted to determine if the mutCCTTG sequence lost its affinity for a putative CCTTG-binding factor. Although we used an identical probe (WTX4) and the identical binding buffer as reported in the previous work (22) in attempting to reproduce those data, we were unable to detect a binding activity in MEL cell nuclear extracts (data not shown) under those or any modified binding conditions, the reasons for which are currently unresolved.
Generation of YAC transgenic mice.
The two Aγ-globin promoter mutations, mutDR and mutCCTTG, were introduced separately into a 150-kb human β-globin YAC (A201F4.3) (40) by homologous recombination in yeast (Fig. 2A). Successful mutagenesis was confirmed by Southern blot analysis of the yeast DNA (data not shown). The YAC DNA was purified by standard procedures (3) and was injected into fertilized oocytes to generate transgenic mice. Tail DNA from offspring (F0) was screened by PCR using human β-globin gene-specific primers followed by detailed Southern blot analysis (Fig. 2B and data not shown) to confirm the integrity and organization of the integrated YAC transgenes.
FIG. 2.

Structural analysis of human β-globin YAC TgM. (A) Schematic representation of the human β-globin YAC. The positions of the β-like globin genes are shown relative to the LCR. SfiI restriction enzyme sites are indicated as vertical lines. Probes (hatched boxes) used for long-range structural analysis and anticipated restriction enzyme fragments after SfiI digestion are shown with their sizes (solid thick lines). (B) Long-range transgene analysis of mutDR (left panel) and mutCCTTG (right panel) YAC transgenic mice. The whole β-globin locus is contained within two SfiI fragments (10 and 100 kb, as in panel A). DNA from thymus cells of transgenic mice was digested with SfiI in agarose plugs, separated by pulsed-field gel electrophoresis, and hybridized separately to probes (indicated on the top of each panel) from the β-globin locus or the right YAC vector arm. The sizes of the expected bands are shown on the left.
To verify the integrity of the integrated YAC DNAs in the mouse genome, high-molecular-weight DNA was prepared from thymi embedded in agarose plugs followed by SfiI enzyme digestion, pulsed-field gel electrophoresis, and Southern blot hybridization (Fig. 2B). DNA fragments from the LCR (HS4-3′), the ɛ-globin gene (E3I), the β-globin gene (B3O), and inside of the SfiI site in the YAC right vector arm (R arm) were used as probes; all four probes detected hybridization bands of the expected sizes (10 and 100 kb; Fig. 2B), demonstrating that each TgM line bore intact, unfragmented copies of YAC transgene(s). We identified two independent transgenic lines for each mutant YAC construct (lines 74 and 75 for mutDR and lines 251 and 428 for mutCCTTG). Further end-fragment and copy number analyses (using a fragment from the endogenous angiotensinogen locus as an internal control) (4) revealed that lines 74 and 251 carried a single YAC, while the other two (lines 75 and 428) bore two tandemly repeated YAC copies (data not shown).
Expression of the mutDR YACs in definitive and primitive erythroid cells.
Two animals (1 month old) from each line of wild-type (single-copy lines 31 and 60, equal to the 60-loxP [39, 42]) or mutDR (single-copy line 74 and two-copy line 75) TgM were treated with phenylhydrazine. After five days, anemic spleen samples were collected for RNA extraction and semiquantitative RT-PCR analysis (Fig. 3A). Human β- and γ-globin gene expression was compared between wild-type and mutant TgM, where endogenous mouse α-globin gene expression was used as the internal control. γ-Globin expression dramatically increased in the mutDR TgM compared with that in the wild-type control (Fig. 3A, top), while β-globin gene expression did not differ significantly (Fig. 3A, bottom). Essentially the same result was obtained with the RNA obtained from the peripheral blood of nonanemic TgM (Fig. 3B).
FIG. 3.

Expression of human β-like globin genes in the adult spleens of mutDR YAC TgM. (A) Semiquantitative RT-PCR analysis of β-like globin gene expression. Total RNA from two individuals from each line of TgM was prepared from the spleens of 1-month-old anemic mice. Expression of human γ (hγ)- and human β (hβ)-globin in comparison to the endogenous mouse α (mα)-globin genes was analyzed separately by semiquantitative RT-PCR. The signals for hγ-globin at 21 cycles and hβ/mα-globin at 12 cycles were quantified by PhosphorImager, and the ratios of hγ/mα (top) and hβ/mα (bottom) were calculated (the mouse α signal at 12 cycles was set at 100%, and the values are normalized by transgene copy numbers) and statistically analyzed (n = 3). The average and standard deviation are graphically depicted. Representative results are shown below each panel. (B) Total RNA from the peripheral blood of TgM was analyzed as in panel A. The PCR cycle numbers used were 20 and 11 for hγ-globin and hβ/mα-globin, respectively. (C to E) RT-PCR analysis of human Gγ/Aγ transcription ratios in the adult spleen. (C) Schematic representation of RT-PCR products amplified with primer sets specific for human γ- (common to both human Gγ- and Aγ-) and mouse α-globin genes. The positions of the primers (solid box), the total length of each RT-PCR product (hatched box for human Gγ, solid box for human Aγ, and open box for mouse α; sizes in base pairs are indicated in parentheses), and the positions of PstI restriction sites with the fragment sizes produced after enzyme digestion are shown in parentheses. (D) PstI digestion of RT-PCR products. The PCR products were digested with PstI and separated on an 8% polyacrylamide gel. A PstI site in the Aγ but not in the Gγ gene enabled separation and quantification of products derived from the two individual γ-globin genes. To internally control for complete PstI digestion, a PstI site was artificially introduced into the mouse α-globin gene PCR primer. The sizes of the expected bands are shown (on the left for undigested PCR products, and on the right for those digested with PstI; sizes in base pairs are indicated in parentheses). (E) The contribution of the Gγ and Aγ genes relative to total γ-globin synthesis are quantified from panel D and plotted as hatched and solid bars, respectively. Average values from two independent experiments are shown.
Since two types of γ-globin genes (Gγ and Aγ) are present in the human locus and the γ gene-specific primer set simultaneously amplifies both gene products, we next determined which of the two γ-globin genes are ectopically activated in the mutDR TgM. Since the Gγ- and Aγ-globin genes diverged from a common ancestral gene quite recently during molecular evolution, their sequences are very similar (8, 33). However, polymorphisms in restriction enzyme recognition sites can be found, notably here that a PstI site present in the Aγ-globin gene is absent from the Gγ gene (Fig. 3C). Therefore, RT-PCR amplification of the γ-globin, as well as endogenous α-globin, mRNA in the exponential amplification range followed by PstI digestion of the PCR products allowed a distinction between Aγ- and Gγ-globin transcripts (Fig. 3D). Quantification of the bands shown in Fig. 3D after executing the discriminating PstI digests revealed that the expression of the Aγ-globin gene in the mutDR TgM increased in comparison with the wild-type, while expression from the (unmodified) Gγ-globin gene in mutDR TgM did not differ from wild-type (Fig. 3E). Thus, the promoter mutDR disruption autonomously affects Aγ-, but not Gγ-, globin gene transcription.
We next analyzed β-like globin gene expression in fetal liver definitive erythroid cells of the same lines of TgM (14.5 dpc). Elevated expression (two- to approximately threefold, compared with the wild-type control) of γ-globin transcript was also observed in mutDR TgM at this stage, while β-globin expression was not statistically affected (Fig. 4A). We again separately analyzed the expression of each γ-globin gene (Fig. 4B and 4C) and found that mutant Aγ-globin gene expression increased by three- to fivefold in comparison with the wild-type control. Curiously, expression of the (wild-type) Gγ-globin gene in the mutDR TgM was also significantly higher (twofold) than that in the wild-type β-globin YAC TgM, when single-copy TgM lines were compared (lines 31, 60, and 74).
FIG. 4.

Expression of human β-like globin genes in the fetal liver of mutDR TgM. (A) Semiquantitative RT-PCR analysis of β-like globin gene expression. Total RNA was prepared from the livers of two fetuses (14.5 dpc) from two independent litters for each line, derived from the intercross of male transgenic and female wild-type animals. Expression of human γ (hγ)- and human β (hβ)-globin to endogenous mouse α (mα)-globin genes was analyzed separately by semiquantitative RT-PCR. The signals for hγ-globin at 18 cycles and hβ/mα-globin at 12 cycles were quantified by PhosphorImager, and the ratios of hγ/mα (top) and hβ/mα (bottom) were calculated (the mouse α signal at 12 cycles was set at 100% and the values are normalized by transgene copy numbers). The averages ± standard deviations from at least three independent experiments were calculated and are graphically depicted. Representative results are shown below each panel. (B and C) RT-PCR analysis of human Gγ/Aγ transcription ratios in the fetal liver. (B) PstI digestion of RT-PCR products. See the legend to Fig. 3 for details. Undigested PCR products are included (uncut) as a reference. (C) The contribution of the Gγ and Aγ genes relative to total γ-globin synthesis are quantified from panel B and plotted as hatched and solid bars, respectively. Average values obtained from two independent experiments are shown.
We finally analyzed the expression of the human ɛ- and γ-globin genes in the yolk sac (9.5 dpc). No significant difference was observed in the expression of these genes between the wild-type (line 31 and 60) (42) and the mutDR TgM lines (Fig. 5). These results clearly demonstrate that mutation of the DR motif in the Aγ-globin gene promoter affected its expression only in the definitive stage of erythropoiesis, which is consistent with our hypothesis that the DR elements in the ɛ- and γ-globin promoters are the target motifs for a definitive stage-specific repressor.
FIG. 5.
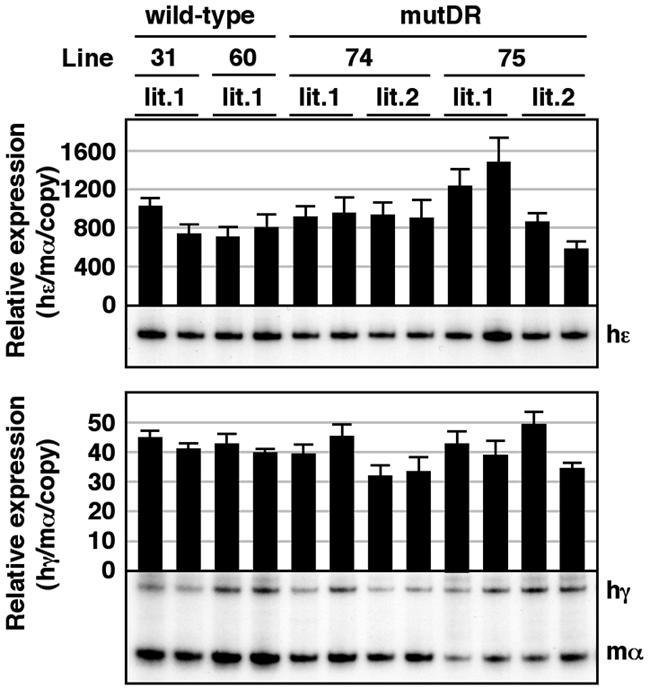
Expression of human β-like globin genes in the yolk sac of mutDR TgM. Semiquantitative RT-PCR analysis of β-like globin gene expression. Total RNA was prepared from the yolk sac of two embryos (9.5 dpc) derived from the intercross of male transgenic and female wild-type animals. Samples were collected from two independent litters from each mutant line. Expression of human ɛ (hɛ)- and human γ (hγ)-globin compared to endogenous mouse α (mα)-globin genes was analyzed separately by semiquantitative RT-PCR. The signals for hɛ-globin at 18 cycles and hγ/mα-globin at 12 cycles were quantified by PhosphorImager, and the ratios of hɛ/mα (top) and hγ/mα (bottom) were calculated (the mouse α signal at 12 cycles was set at 100% and the values are normalized by transgene copy numbers). The average ± standard deviation from at least three independent experiments was calculated and graphically depicted. Representative results are shown below each panel.
Expression of mutCCTTG YACs in definitive and primitive erythroid cells.
Finally, we investigated the in vivo role of the CCTTG repeat motifs in γ-globin gene silencing. In the adult spleen, the level of γ-globin transcription was unaltered in the mutCCTTG TgM, while that in the mutDR TgM dramatically increased (Fig. 6, top). Among the wild-type, mutDR, and mutCCTTG TgM, the level of adult β-globin transcript accumulation did not significantly differ (Fig. 6, bottom). Moreover, the level of either γ-globin gene in the mutCCTTG transgenic mice was unaltered (data not shown).
FIG. 6.
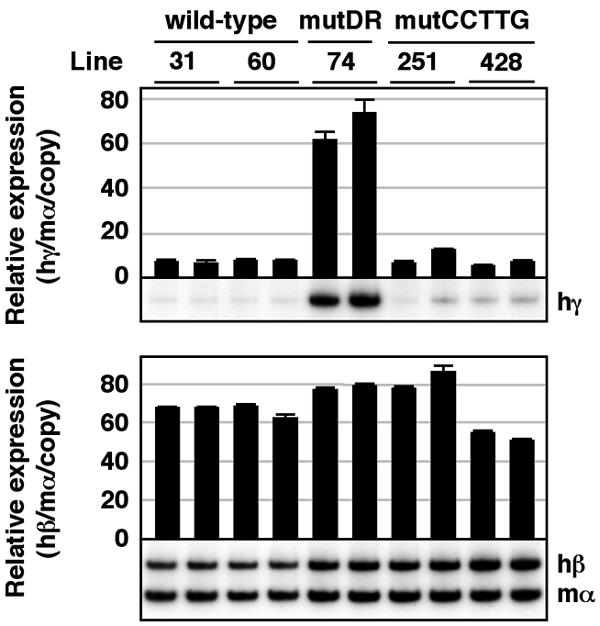
Expression of human β-like globin genes in the adult spleens of mutCCTTG TgM. Semiquantitative RT-PCR analysis of β-like globin gene expression. Total RNA from two individuals for each line was prepared from the spleens of anemic mice (1 month old). The average and standard deviation from three independent experiments are graphically depicted. Representative results are shown below each panel. See the legend to Fig. 3 for details.
In the fetal liver, however, expression of the γ-globin gene in mutCCTTG TgM was slightly but significantly elevated (1.5-fold) when compared to wild-type TgM (Fig. 7A, top), and this phenotype was mostly attributable to mutant Aγ-gene expression, which increased ca. twofold when compared to wild type (Fig. 7B and C). Adult β-globin transcript levels were invariant comparing wild-type and mutCCTTG TgM (Fig. 7A, bottom). In the yolk sac of mutCCTTG TgM, the level of the ɛ- or the γ-globin gene was unaffected when compared to wild-type TgM (Fig. 8). These results demonstrate that the CCTTG motif may represent a weak repressor element that is active only in fetal liver erythroid cells, but it cannot represent a major silencing element for Aγ-globin in adult definitive erythroid cells.
FIG. 7.

Expression of human β-like globin genes in the fetal liver of mutCCTTG TgM. (A) Semiquantitative RT-PCR analysis of β-like globin gene expression. Total RNA was prepared from the livers of two fetuses (14.5 dpc) in two independent litters (lit.1 and lit.2) for each line. The average and standard deviation from at least three independent experiments are graphically depicted. Representative results are shown below each panel. See the legend to Fig. 4 for details. (B and C) RT-PCR analysis of human Gγ/Aγ transcription ratios in the fetal liver. (B) PstI digestion of RT-PCR products. See the legend to Fig. 3 for details. (C) The contribution of the Gγ and Aγ genes relative to total γ-globin synthesis are quantified from panel B and plotted as hatched and solid bars, respectively. Average values obtained from two independent experiments are shown.
FIG. 8.
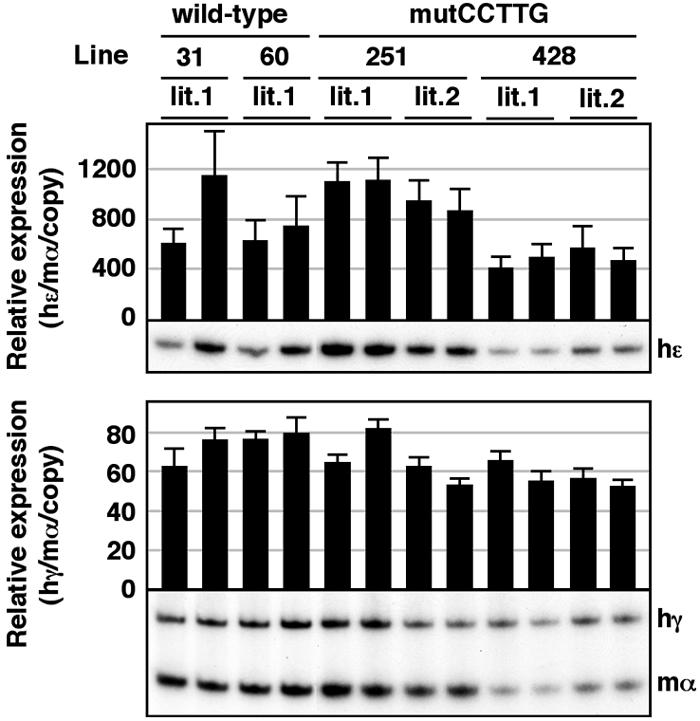
Expression of human β-like globin genes in the yolk sac of mutCCTTG TgM. Semiquantitative RT-PCR analysis of β-like globin gene expression. Total RNA was prepared from the yolk sac of two embryos (9.5 dpc) derived from the intercross of male transgenic and female wild-type animals. Samples were collected from two independent litters (lit.1 and lit.2) for each mutant line. The average ± standard deviation was calculated (n = 3) and is graphically depicted. Representative results are shown below each panel. See the legend to Fig. 5 for details.
DISCUSSION
Globin gene switching has long been a central model for analyzing the molecular events controlling the temporal regulation of tissue-specific gene expression. In spite of intensive studies, we do not yet completely understand how β-globin gene switching is controlled. Human β-globin locus TgM have been generated using large cosmid (36), YAC (13, 29), and bacterial artificial chromosome (18, 19) DNAs for analysis of this issue, since tissue- and developmental stage-specific globin gene switching can be properly achieved only by using these technically challenging experimental tools. The Greek-type HPFH mutation has been introduced into a human β-globin YAC and then examined in TgM (30). In this model, human γ- to β-globin gene switching was delayed in comparison with TgM bearing the wild-type locus. This result thus ensured the validity of an approach attempting to investigate the mechanisms that regulate switching in human β-globin YAC TgM.
Two cis DNA motifs, the DR and CCTTG sequences, have been proposed as silencing elements for definitive stage-specific human embryonic and fetal globin gene repression (22, 41). To complicate matters, these overlapping motifs in the Aγ-globin promoter contain the nucleotide mutated in the Greek HPFH syndrome therefore obscuring which motif might be responsible for the HPFH phenotype. Hence, we evaluated the roles of these two putative silencing motifs in γ-globin gene repression by generating separately mutated Aγ-globin gene promoters in human β-globin YAC TgM.
RT-PCR analysis of YAC TgM bearing mutDR clearly demonstrated that this motif functions as a definitive stage-specific repressor cis-element and is required for γ-globin gene silencing (Fig. 3, 4, and 5). In the adult spleen, Aγ-globin mRNA levels were almost 10-fold higher in mutDR TgM in comparison to wild-type controls (Fig. 3E), while Gγ- as well as β-globin gene expression levels were not altered (Fig. 3A and E). One might presume that a significant increase in γ-globin gene expression would lead to a reciprocal decrease in β-gene expression, since these genes compete for the LCR enhancer activity within the locus in the competition model. Even in human patients with HPFH, however, increased γ-globin expression is approximately 5 to 20% of total β-like globin protein in adult erythrocytes (35). Consistent with this clinical observation, TgM carrying a human β-globin YAC with the Greek-type HPFH mutation in the Aγ-globin promoter displayed ∼10% of γ-globin expression (at day 17 after birth [30]). Therefore, it is unlikely that the level of γ-gene expression in our mutDR TgM exceeds 10% of total β-like globin expression in adult erythrocytes, which may not be enough to competitively suppress β-globin gene transcription.
Curiously, expression of the human Gγ-globin gene in the mutDR TgM was slightly elevated in fetal liver erythroid cells (Fig. 4C). Since this phenotype was observed in only one of the lines (single-copy TgM line 74), it may simply be a position effect difference between the lines. However, this phenotype could also be genuine, since a similar increase was not observed in yolk sac erythroid cells (line 74 in Fig. 5C) and that “expression per copy” value tends to be lower in Tg lines bearing multiple transgene copies. If this is a meaningful phenotype, it must be attributable to the DR motif mutation in the Aγ-globin promoter, since the remainder of the β-globin locus is otherwise unaltered. Since the human Gγ- and Aγ-globin genes are separated by approximately 5 kb, we feel that it is unlikely that transcription factors associated with the Aγ-globin promoter directly influence Gγ-globin transcription. Recently, an attractive model has been proposed for human β-globin gene switching (2, 9, 15). According to this model, the β-globin locus is divided into three subdomains, including either the LCR, the ɛ- and γ-globin genes, or the δ- and β-globin genes, and the model postulates that the “open-closed” status of the subdomains is developmentally regulated (12, 20). Since the Gγ- and Aγ-globin genes would be in the same subdomain, the phenotype in mutDR could be explained as follows: when Aγ-globin expression is activated by factors recruited to the promoter region of this gene, the chromatin structure of the entire ɛ/γ subdomain is in an “open” configuration, and thus the Gγ-globin promoter region becomes more readily accessible to activating transcription factors.
In primitive erythroid cells, no significant difference in Aγ gene expression was observed when comparing wild-type and mutDR TgM. This was expected, since we hypothesized that the repressor that bound to the DR motif was likely to be definitive stage specific. In summary, the data shown here demonstrate that the DR motif in the Aγ-globin gene promoter significantly contributes to the mechanism controlling globin gene switching.
We also examined the role of the CCTTG repeat motifs in Aγ-globin transcriptional regulation. These analyses showed that γ-globin transcription in mutCCTTG TgM was slightly elevated, and only during the fetal liver stage, in comparison to wild-type controls (Fig. 6, 7, and 8), and further that this increase was mostly attributable to elevated Aγ-globin transcript accumulation (Fig. 7B and C). Since we were unable to detect a CCTTG-specific binding activity in MEL cell nuclear extracts, we were unable to confirm if this mutation completely disrupted the binding of any CCTTG-associated proteins (22). To ascertain a loss of binding activity as best we could, we introduced point mutations into all four CCTTG motifs in the Aγ-globin promoter by referring to the reported oligonucleotide (MUTX4) that showed no binding activity in the previous report (22). Although these genetic results suggest that the CCTTG repeats might collectively function as weak fetal liver stage-specific silencer cis elements, their effects are neither as pronounced nor as specific as those exerted by the DR motif.
Alternatively, it is possible that the small increase in the γ-globin gene expression in the fetal liver of mutCCTTG TgM may still be due to the effect on the DR motif and its interacting protein (presumably DRED), since these two motifs are in very close proximity (Fig. 1B). A small change in affinity (of DRED) may not have been detected in gel shift assay (Fig. 1C) in this case.
There have been two protein complexes that are reported to bind to DR sequences in definitive erythroid cells. One is the stage-specifically expressed nuclear receptor COUP-TFII (11), and the other is DRED (41); indeed, both factors displayed reduced affinity to the DR target sequence when a Greek-type HPFH point mutation was introduced (23, 38). Thus, it is important to determine the stage-specific activity and binding affinity of these factors to the wild-type and mutant Aγ-globin gene promoters in order to conclude which protein is participating in γ-globin gene silencing. Furthermore, it is probably mandatory to also analyze EKLF recruitment to the wild-type versus mutant promoters in vivo to more clearly elaborate the molecular mechanisms controlling γ-globin gene activation and silencing. Such experiments should be feasible once antibodies against DRED and EKLF become available.
The first molecularly characterized human hemoglobinopathy was sickle cell anemia (SCD). A single amino acid substitution in the β-globin polypeptide causes production of an abnormal hemoglobin (hemoglobin S; HbS), and condensation of these abnormal hemoglobins into long polymers result in a debilitating, life-threatening disease. Hemoglobin F (HbF), containing γ-globin chains, is known to inhibit polymerization of HbS, and HPFH mutations increase the synthesis of HbF, thus ameliorating SCD phenotypes and severity. Therefore, increasing HbF in patients with SCD or β-thalassemia by therapeutic means could reduce disease severity (35). In this study, we showed that the destruction of the DR motif in the Aγ-globin gene promoter recapitulates the HPFH phenotype in mice and therefore we expect that the DR sequence, or proteins that bind to it, could serve as targets for future SCD therapeutic development.
Acknowledgments
We thank Y. Tanimoto for technical assistance in generating transgenic mice, K.-C. Lim for creative efforts, and our laboratory members for helpful discussions and encouragement.
This work was supported by research grants from the NIH (HL24415 and HL73465 to J.D.E. and O.T.), the Cooley's Anemia Foundation (K.T. and O.T.), the Inamori Foundation (K.T.), the Naito Foundation (K.T.), the 21st Century COE Program (A.F.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT), and Grants-in-Aid for Scientific Research (A, MEXT; A.F.), Scientific Research on Priority Areas (MEXT; K.T.), and Young Scientists (A, MEXT; K.T.)
REFERENCES
- 1.Asano, H., and G. Stamatoyannopoulos. 1998. Activation of beta-globin promoter by erythroid Kruppel-like factor. Mol. Cell. Biol. 18:102-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashe, H. L., J. Monks, M. Wijgerde, P. Fraser, and N. J. Proudfoot. 1997. Intergenic transcription and transinduction of the human beta-globin locus. Genes Dev. 11:2494-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bungert, J., U. Dave, K. C. Lim, K. H. Lieuw, J. A. Shavit, Q. Liu, and J. D. Engel. 1995. Synergistic regulation of human beta-globin gene switching by locus control region elements HS3 and HS4. Genes Dev. 9:3083-3096. [DOI] [PubMed] [Google Scholar]
- 4.Clouston, W. M., B. A. Evans, J. Haralambidis, and R. I. Richards. 1988. Molecular cloning of the mouse angiotensinogen gene. Genomics 2:240-248. [DOI] [PubMed] [Google Scholar]
- 5.Collins, F. S., J. E. Metherall, M. Yamakawa, J. Pan, S. M. Weissman, and B. G. Forget. 1985. A point mutation in the A gamma-globin gene promoter in Greek hereditary persistence of fetal haemoglobin. Nature 313:325-326. [DOI] [PubMed] [Google Scholar]
- 6.Dillon, N., and F. Grosveld. 1991. Human gamma-globin genes silenced independently of other genes in the beta-globin locus. Nature 350:252-254. [DOI] [PubMed] [Google Scholar]
- 7.Dillon, N., T. Trimborn, J. Strouboulis, P. Fraser, and F. Grosveld. 1997. The effect of distance on long-range chromatin interactions. Mol. Cell 1:131-139. [DOI] [PubMed] [Google Scholar]
- 8.Efstratiadis, A., J. W. Posakony, T. Maniatis, R. M. Lawn, C. O′Connell, R. A. Spritz, J. K. DeRiel, B. G. Forget, S. M. Weissman, J. L. Slightom, A. E. Blechl, O. Smithies, F. E. Baralle, C. C. Shoulders, and N. J. Proudfoot. 1980. The structure and evolution of the human beta-globin gene family. Cell 21:653-668. [DOI] [PubMed] [Google Scholar]
- 9.Engel, J. D., and K. Tanimoto. 2000. Looping, linking, and chromatin activity: new insights into beta-globin locus regulation. Cell 100:499-502. [DOI] [PubMed] [Google Scholar]
- 10.Feng, W. C., C. M. Southwood, and J. J. Bieker. 1994. Analyses of beta-thalassemia mutant DNA interactions with erythroid Kruppel-like factor (EKLF), an erythroid cell-specific transcription factor. J. Biol. Chem. 269:1493-1500. [PubMed] [Google Scholar]
- 11.Filipe, A., Q. Li, S. Deveaux, I. Godin, P. H. Romeo, G. Stamatoyannopoulos, and V. Mignotte. 1999. Regulation of embryonic/fetal globin genes by nuclear hormone receptors: a novel perspective on hemoglobin switching. EMBO J. 18:687-697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forsberg, E. C., K. M. Downs, H. M. Christensen, H. Im, P. A. Nuzzi, and E. H. Bresnick. 2000. Developmentally dynamic histone acetylation pattern of a tissue-specific chromatin domain. Proc. Natl. Acad. Sci. USA 97:14494-14499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaensler, K. M., M. Kitamura, and Y. W. Kan. 1993. Germ-line transmission and developmental regulation of a 150-kb yeast artificial chromosome containing the human beta-globin locus in transgenic mice. Proc. Natl. Acad. Sci. USA 90:11381-11385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gelinas, R., B. Endlich, C. Pfeiffer, M. Yagi, and G. Stamatoyannopoulos. 1985. G to A substitution in the distal CCAAT box of the A gamma-globin gene in Greek hereditary persistence of fetal haemoglobin. Nature 313:323-325. [DOI] [PubMed] [Google Scholar]
- 15.Gribnau, J., K. Diderich, S. Pruzina, R. Calzolari, and P. Fraser. 2000. Intergenic transcription and developmental remodeling of chromatin subdomains in the human beta-globin locus. Mol. Cell 5:377-386. [DOI] [PubMed] [Google Scholar]
- 16.Hanscombe, O., D. Whyatt, P. Fraser, N. Yannoutsos, D. Greaves, N. Dillon, and F. Grosveld. 1991. Importance of globin gene order for correct developmental expression. Genes Dev. 5:1387-1394. [DOI] [PubMed] [Google Scholar]
- 17.Hardison, R., and W. Miller. 1993. Use of long sequence alignments to study the evolution and regulation of mammalian globin gene clusters. Mol. Biol. Evol. 10:73-102. [DOI] [PubMed] [Google Scholar]
- 18.Huang, Y., D. P. Liu, L. Wu, T. C. Li, M. Wu, D. X. Feng, and C. C. Liang. 2000. Proper developmental control of human globin genes reproduced by transgenic mice containing a 160-kb BAC carrying the human beta-globin locus. Blood Cells Mol. Dis. 26:598-610. [DOI] [PubMed] [Google Scholar]
- 19.Kaufman, R. M., C. T. Pham, and T. J. Ley. 1999. Transgenic analysis of a 100-kb human beta-globin cluster-containing DNA fragment propagated as a bacterial artificial chromosome. Blood 94:3178-3184. [PubMed] [Google Scholar]
- 20.Kim, A., and A. Dean. 2004. Developmental stage differences in chromatin subdomains of the beta-globin locus. Proc. Natl. Acad. Sci. USA 101:7028-7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kulozik, A. E., A. Bellan-Koch, S. Bail, E. Kohne, and E. Kleihauer. 1991. Thalassemia intermedia: moderate reduction of beta globin gene transcriptional activity by a novel mutation of the proximal CACCC promoter element. Blood 77:2054-2058. [PubMed] [Google Scholar]
- 22.Lee, J. S., H. Ngo, D. Kim, and J. H. Chung. 2000. Erythroid Kruppel-like factor is recruited to the CACCC box in the beta-globin promoter but not to the CACCC box in the gamma-globin promoter: the role of the neighboring promoter elements. Proc. Natl. Acad. Sci. USA 97:2468-2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liberati, C., M. R. Cera, P. Secco, C. Santoro, R. Mantovani, S. Ottolenghi, and A. Ronchi. 2001. Cooperation and competition between the binding of COUP-TFII and NF-Y on human epsilon- and gamma-globin gene promoters. J. Biol. Chem. 276:41700-41709. [DOI] [PubMed] [Google Scholar]
- 24.Miller, I. J., and J. J. Bieker. 1993. A novel, erythroid cell-specific murine transcription factor that binds to the CACCC element and is related to the Kruppel family of nuclear proteins. Mol. Cell. Biol. 13:2776-2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Orkin, S. H., S. E. Antonarakis, and H. H. Kazazian, Jr. 1984. Base substitution at position −88 in a beta-thalassemic globin gene. Further evidence for the role of distal promoter element ACACCC. J. Biol. Chem. 259:8679-8681. [PubMed] [Google Scholar]
- 26.Orkin, S. H., H. H. Kazazian, Jr., S. E. Antonarakis, S. C. Goff, C. D. Boehm, J. P. Sexton, P. G. Waber, and P. J. Giardina. 1982. Linkage of beta-thalassaemia mutations and beta-globin gene polymorphisms with DNA polymorphisms in human beta-globin gene cluster. Nature 296:627-631. [DOI] [PubMed] [Google Scholar]
- 27.Perkins, A. C., K. M. Gaensler, and S. H. Orkin. 1996. Silencing of human fetal globin expression is impaired in the absence of the adult beta-globin gene activator protein EKLF. Proc. Natl. Acad. Sci. USA 93:12267-12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peterson, K. R. 2003. Hemoglobin switching: new insights. Curr. Opin. Hematol. 10:123-129. [DOI] [PubMed] [Google Scholar]
- 29.Peterson, K. R., C. H. Clegg, C. Huxley, B. M. Josephson, H. S. Haugen, T. Furukawa, and G. Stamatoyannopoulos. 1993. Transgenic mice containing a 248-kb yeast artificial chromosome carrying the human beta-globin locus display proper developmental control of human globin genes. Proc. Natl. Acad. Sci. USA 90:7593-7597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peterson, K. R., Q. L. Li, C. H. Clegg, T. Furukawa, P. A. Navas, E. J. Norton, T. G. Kimbrough, and G. Stamatoyannopoulos. 1995. Use of yeast artificial chromosomes (YACs) in studies of mammalian development: production of beta-globin locus YAC mice carrying human globin developmental mutants. Proc. Natl. Acad. Sci. USA 92:5655-5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peterson, K. R., and G. Stamatoyannopoulos. 1993. Role of gene order in developmental control of human gamma- and beta-globin gene expression. Mol. Cell. Biol. 13:4836-4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raich, N., T. Enver, B. Nakamoto, B. Josephson, T. Papayannopoulou, and G. Stamatoyannopoulos. 1990. Autonomous developmental control of human embryonic globin gene switching in transgenic mice. Science 250:1147-1149. [DOI] [PubMed] [Google Scholar]
- 33.Slightom, J. L., A. E. Blechl, and O. Smithies. 1980. Human fetal G gamma- and A gamma-globin genes: complete nucleotide sequences suggest that DNA can be exchanged between these duplicated genes. Cell 21:627-638. [DOI] [PubMed] [Google Scholar]
- 34.Southwood, C. M., K. M. Downs, and J. J. Bieker. 1996. Erythroid Kruppel-like factor exhibits an early and sequentially localized pattern of expression during mammalian erythroid ontogeny. Dev. Dyn. 206:248-259. [DOI] [PubMed] [Google Scholar]
- 35.Stamatoyannopoulos, G., and A. W. Neinhuis. 1994. Hemoglobin switching, p. 107-155. In G. Stamatoyannopoulos, A. W. Neinhuis, P. Majerus, and H. Varmus (ed.), The molecular basis of blood diseases, 2nd ed. W. B. Saunders Co., New York, N.Y.
- 36.Strouboulis, J., N. Dillon, and F. Grosveld. 1992. Developmental regulation of a complete 70-kb human beta-globin locus in transgenic mice. Genes Dev. 6:1857-1864. [DOI] [PubMed] [Google Scholar]
- 37.Stuve, L. L., and R. M. Myers. 1993. Identification and characterization of a beta-globin promoter-binding factor from murine erythroleukemia cells. Mol. Cell. Biol. 13:4311-4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanabe, O., F. Katsuoka, A. D. Campbell, W. Song, M. Yamamoto, K. Tanimoto, and J. D. Engel. 2002. An embryonic/fetal beta-type globin gene repressor contains a nuclear receptor TR2/TR4 heterodimer. EMBO J. 21:3434-3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanimoto, K., Q. Liu, J. Bungert, and J. D. Engel. 1999. Effects of altered gene order or orientation of the locus control region on human beta-globin gene expression in mice. Nature 398:344-348. [DOI] [PubMed] [Google Scholar]
- 40.Tanimoto, K., Q. Liu, J. Bungert, and J. D. Engel. 1999. The polyoma virus enhancer cannot substitute for DNase I core hypersensitive sites 2-4 in the human beta-globin LCR. Nucleic Acids Res. 27:3130-3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanimoto, K., Q. Liu, F. Grosveld, J. Bungert, and J. D. Engel. 2000. Context-dependent EKLF responsiveness defines the developmental specificity of the human epsilon-globin gene in erythroid cells of YAC transgenic mice. Genes Dev. 14:2778-2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tanimoto, K., A. Sugiura, A. Omori, G. Felsenfeld, J. D. Engel, and A. Fukamizu. 2003. Human beta-globin locus control region HS5 contains CTCF- and developmental stage-dependent enhancer-blocking activity in erythroid cells. Mol. Cell. Biol. 23:8946-8952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tewari, R., N. Gillemans, M. Wijgerde, B. Nuez, M. von Lindern, F. Grosveld, and S. Philipsen. 1998. Erythroid Kruppel-like factor (EKLF) is active in primitive and definitive erythroid cells and is required for the function of 5′HS3 of the beta-globin locus control region. EMBO J. 17:2334-2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wijgerde, M., J. Gribnau, T. Trimborn, B. Nuez, S. Philipsen, F. Grosveld, and P. Fraser. 1996. The role of EKLF in human beta-globin gene competition. Genes Dev. 10:2894-2902. [DOI] [PubMed] [Google Scholar]