

Abstract
Platelets are key contributors to allergic asthma and aspirin-exacerbated respiratory disease (AERD), an asthma phenotype involving platelet activation and IL-33-dependent mast cell activation. Human platelets express the glucagon-like peptide-1 receptor (GLP-1R). GLP-1R agonists (GLP-1RA) decrease lung IL-33 release and airway hyperreactivity in mouse asthma models. We hypothesized that GLP-1RAs reduce platelet activation and downstream platelet-mediated airway inflammation in AERD. GLP-1R expression on murine platelets was assessed using flow cytometry. We tested the effect of the GLP-1RA liraglutide on lysine-aspirin (Lys-ASA)-induced changes in airway resistance, and platelet-derived mediator release in a murine AERD model. We conducted a prospective cohort study comparing the effect of pretreatment with liraglutide or vehicle on thromboxane receptor (TP) agonist-induced in vitro activation of platelets from patients with AERD and non-asthmatic controls. GLP-1R expression was higher on murine platelets than on leukocytes. A single dose of liraglutide inhibited Lys-ASA-induced increases in airway resistance and decreased markers of platelet activation and recruitment to the lung in AERD-like mice. Liraglutide attenuated TP agonist-induced activation as measured by CXCL7 release in plasma from patients with AERD and CD62P expression in platelets from both patients with AERD (N=31) and non-asthmatic, healthy controls (N=11). Liraglutide, an FDA-approved GLP-1RA for treatment of type 2 diabetes and obesity, attenuates in vivo platelet activation in an AERD murine model and in vitro activation in human platelets in patients with and without AERD. These data advance the GLP-1R axis as a new target for platelet-mediated inflammation warranting further study in asthma.
Keywords: Asthma, type 2 diabetes, obesity(1)
Introduction
Aspirin-exacerbated respiratory disease (AERD) is an eosinophilic asthma phenotype associated with nasal polyps and pathognomonic reactions to COX-1 inhibition marked by lung function decrement and incremental sinonasal congestion. Sinonasal tissues of patients with AERD contain large amounts of interleukin (IL)-33,(2) a potent type 2 cytokine that directly activates mast cells and other effector cells. Platelet-leukocyte aggregates (PLA)(3) are increased in peripheral blood and sinonasal tissues of patients with AERD where they contribute to cysteinyl leukotriene (cysLT) overproduction.(4) In a mouse model of AERD using prostaglandin E2 synthase deficient (Ptges−/−) C57BL/6 mice, antibody-mediated platelet depletion abolishes the IL-33 surge and aspirin-induced changes in airway resistance.(2, 5) More broadly, blood platelets are activated in patients with asthma at baseline,(6) and to a greater degree during exacerbations and acutely with inhaled allergen exposure.(7) Platelets are recruited to the lung during allergen challenge(1) where extravasated platelets can promote pulmonary inflammation through a variety of mechanisms.(7, 8) Platelets also accumulate in the airway wall and distal lungs of patients who die from asthma exacerbations.(1) Activated platelets release many inflammatory mediators, including CXCL7, a chemokine that attracts neutrophils and promotes inflammation and tissue remodeling.(9) Platelet activation and subsequent proinflammatory mediator release remain unaddressed by current, non-glucocorticoid,(10) asthma therapeutics.
Glucagon-like peptide-1 (GLP-1) is an incretin hormone synthesized by and secreted from enteroendocrine L cells throughout the small and large intestines (11) and by the central nervous system, predominantly in brainstem nuclei controlling metabolic, cardiovascular, and neuroprotective activities.(12) In the pancreatic islets, GLP-1 potentiates insulin secretion and suppresses glucagon secretion in a glucose-dependent fashion.(13) The GLP-1R is a group B G-protein coupled receptor that signals via stimulatory G proteins to activate adenylate cyclase, raise cAMP levels and augment intracellular calcium release to promote insulin secretion. Agonists of the GLP-1R for the treatment of type 2 diabetes and obesity are approved by the Food and Drug Administration (FDA) for age ≥10.(14, 15)
Human platelets also express the glucagon-like peptide-1 receptor.(16) GLP-1R agonists may attenuate platelet aggregation in patients with type 2 diabetes,(16, 17) contributing to their favorable cardiometabolic profile in patients with type 2 diabetes or obesity.(18) Recently, a GLP-1R agonist was shown to inhibit allergen(19–21) and viral-induced(22) airway hyperresponsiveness (AHR) in preclinical murine models. Markers of airway inflammation in these models were also decreased, including release of lung IL-33, expansion and activation of group 2 innate lymphoid cells, and generation of IL-5, IL-13, and IL-17, implicating a potential inhibitory role for the GLP-1 pathway in type 2 (T2) and non-T2 immune pathways relevant to asthma. However, these studies did not determine whether GLP-1R agonists targeted platelets as part of their mechanism of action. Subsequently, multiple clinical studies have emerged supporting a potential role for GLP-1R agonists in attenuating exacerbation risk in patients with asthma(23) and chronic obstructive pulmonary disease,(24, 25) and in improving pulmonary function in patients without diagnosed respiratory disease.(26) However, these studies have been restricted to populations with type 2 diabetes, with or without comorbid asthma that was not further phenotyped, limiting generalizability to patients without metabolic dysregulation. We recently reported that human platelets express GLP-1R, and GLP-1R agonist exposure in vivo and in vitro attenuates TP agonist-induced platelet aggregation in patients with obesity and pre-diabetes.(16)
Given the importance of platelets in AERD, our objective was to characterize the effect of liraglutide on platelet activation in AERD and non-AERD platelets. We hypothesized that GLP-1R agonist treatment would reduce platelet activation, platelet-derived mediator release, PLA formation and downstream platelet-mediated airway inflammation in AERD.
Materials and Methods
Reagents
Extract from Dermatophagoides farinae (Df) was obtained from Greer Laboratories (XPB81D3A25; Lenoir, NC). PBS was obtained from Sigma-Aldrich (St Louis, Mo). Prostaglandin (PG)D2, cysLTs, and histamine enzyme immunoassay (EIA) kits were obtained from Cayman Chemical (Ann Arbor, MI). IL-5, IL-13 and IL-33 EIA kits were purchased from R&D Systems (Minneapolis, Minn). The mucosal mast cell protease-1 (mMCP-1), CXCL7, and High-mobility box 1 (HMGB1) EIA kit were purchased from eBiosciences (San Diego, Calif), Abcam (Cambridge, Mass), and LifeSpan (Providence, RI), respectively. Preparation of the platelet stabilizing reagent has been previously described and is composed of aspirin (Sigma# A5376), apyrase (Sigma# A6635), carbaprostacyclin (Cayman Chemical #18210) and SQ 29,548 (Cayman Chemical #19025)(27, 28). Thromboxane (TXA2) analogue U46619 was from Cayman Chemical (#16450). Use of the reagent is discussed in further detail in the Human Studies section of these methods. GLP-1 peptide (Sigma, St. Louis, MO), liraglutide (Tocris, #6517) and GLP-1R antagonist Exendin-9–39 (Tocris #2081) were used for in vitro experiments. Liraglutide (Novo Nordisk, Denmark), a commercially available GLP-1R agonist, was purchased from the Brigham and Women’s Hospital (BWH) pharmacy.
Mice
For mouse platelet studies, wild-type C57BL/6 male and female mice (ages 8–12 weeks, littermates purchased from Jackson Laboratory and co-housed at VUMC for at least 1 week before harvest, 4 of each sex per treatment group) were used. Whole blood was extracted via cardiac puncture using 27G syringes in anesthetized adult mice according to the approved IACUC protocol at VUMC (M1900086), and blood was collected into tubes containing 3.8% sodium citrate. Mouse whole blood was then treated with a platelet-stabilizing agent and stained for flow cytometry.
The C57BL/6 mice lacking mPGES-1 (Ptges–/– mice) were a gift from Dr Shizuo Akira (Osaka University, Japan).(29) Six to 12-week-old male mice were used. Mice were housed at BWH’s Hale Building for Transformative Medicine (Boston, Mass). All in vivo animal studies were approved by the Animal Care and Use Committee of BWH (protocol 2016N000294; OLAW Assurance (A4752–01); AAALAC Assurance (1729); and USDA Registration (14-R-0092).
Immunization and challenge
Airway inflammation was induced by repetitive intransal administration of Df (1 or 3 μg) to sedated Ptges−/− mice as described. Liraglutide was administered intraperitoneally (1.5mg/kg) 24 hours prior to challenge with aerosolized Lysine-aspirin (Lys-ASA). This dose was chosen based on the quantity required to inhibit OVA-induced airway inflammation(21).
Measurement of airway resistance
Airway resistance (RL) in response to Lys-ASA was assessed with an Invasive Pulmonary Function Device (Buxco®, Sharon, Conn). Briefly, the mice were anesthetized 24 hours after GLP-1R agonist administration, and tracheotomy was performed. After allowing for RL to reach a stable baseline, Lys-ASA (20 μL of 100 mg/mL) was delivered to the lung via a nebulizer, and RL was recorded for 45 minutes. The results were expressed as the percentage of change in RL from baseline.
Immunohistochemistry staining for CD41
Lung tissue was fixed in 4% PFA (wt/ vol) for 24 hours. Tissue was embedded in FFPE and tissue blocks were cut into 5 um sections and mounted on positively charged slides. After deparaffinization and rehydration, antigen retrieval was performed in citrate buffer at PH 9.0 (Dako.com) with a steamer. Endogenous peroxidase was inhibited in 0.03% H2O2 in methanol. Slides were incubated with rabbit Anti mouse CD41 mAb (2.5 ug/ml; Abcam ab225896) overnight at 4°C. The slides were counterstained with anti-rabbit HRP-polymer conjugated secondary antibody (BioCare Medical), using AEC (Abcam) as the chromogen. Sections were counterstained with Gill’s hematoxylin #2 and mounted with VectaMount AQ.
ImageJ quantification
Images were taken on a Leica DM LB2 microscope (Leica 10x objective) fitted with a Leica digital camera (DMC6200). Images were acquired using Leica LAS X software. We used ImageJ Fiji software to Semi- quantify CD41 expression. Briefly, ImageJ Fiji software was used to deconvolute and separate the staining of the IHC image. ImageJ separates the IHC image into AEC staining (red staining) for the CD41 and Hematoxylin for the nucleus. The AEC-stained image was analyzed at a maximum threshold value at 45, Filters with Median Radius 2.0 Pixels. Total CD41+ area and area fraction were acquired from three randomly selected fields in each tissue section.
Human studies
Inclusion criteria for all participants specified ≥18 years of age, no history of type 2 diabetes, and no history of GLP-1R agonist use. AERD was defined as physician-diagnosed asthma, chronic rhinosinusitis with nasal polyposis and either a respiratory reaction during a formal aspirin challenge or a history of two or more respiratory reactions to a cyclooxygenase-1 inhibitor. Participants were prospectively recruited from BWH and Vanderbilt University Medical Center (VUMC) Allergy/Clinical Immunology clinical sites. Control participants were recruited from VUMC; a history of physician-diagnosed or self-identified asthma were exclusion criteria. Demographics and concurrent medication use (e.g., aspirin, biologics) were recorded. Peripheral blood sample collection was optimized for platelet assays. Primary analyses compared platelet activation in liraglutide versus vehicle treated samples. An exploratory subgroup analysis was conducted based on concurrent AERD treatment. For all human studies, corresponding protocols and informed consent forms were reviewed and approved by the respective Institutional Review Board (BWH#2018P000488, BWH#2003P002088; VUMC#200203).
Ex vivo human platelet assays
Human peripheral blood was treated with liraglutide (50nM), the circulating concentration of liraglutide in vivo under treatment conditions,(30) or vehicle for 30 minutes then activated by the TXA2 analogue U46619 (500nM) or vehicle control for 30 minutes and treated with a platelet-stabilizing agent.(27) Samples were then shipped to VUMC on icepack overnight or stored at 4°C for analyses of PLA (CD45+CD41+) and the platelet activation marker p-selection (CD62P) by flow cytometry on whole blood the following day. After preparing whole blood samples for flowcytometry, tubes were centrifuged at 3200 rpm, at room temperature, without brakes, in a swing rotor bucket centrifuge to prepare platelet-poor plasma (PPP). PPP samples were stored at −80C until time of analysis.
To permit delayed analyses, we used an established platelet-stabilizing agent that has been previously shown to preserve existing platelet activation state and inhibit further in vitro platelet activation that may occur during manipulation of blood samples ex vivo.(29) Prior to using the platelet stabilizing reagent for delayed evaluation of platelet activation in our study, we first confirmed that the addition of the platelet stabilizing reagent had no impact on platelet activation status on platelets from participants with AERD. We compared whole blood samples from subjects with AERD at VUMC, treated with and without the platelet-stabilizing reagent before activation with U46619 (500nM) or vehicle control and analyzed at time 0 and 24 hours. The platelet stabilizing reagent prevented subsequent platelet activation that occurs over time without altering the readout of the initial stimulation (Supplemental Fig.1a–c)
Flow Cytometry
Whole blood samples from human subjects were stained with human anti-CD45, anti-CD41, anti-P-selectin (CD62P), and anti-PAC-1 (BD Bioscience #557748, #555467, #550888, #340507) monoclonal antibodies.
GLP-1R expression on whole blood from C57BL/6 mice was evaluated using a far-red fluorescent GLP-1R antagonist peptide label (LUXendin645) for flow cytometry.(31) After incubating the labeled peptide for an hour at room temp, whole blood was stained with mouse anti-CD45, anti-CD41, and anti-CD62P (BD Bioscience #557748, #555467, #550888) monoclonal antibodies to identify platelets (CD45−CD41+), leukocytes (CD45+CD41−), and PLA (CD45+CD41+) and analyzed by flow cytometry as previously described.(16)
EIA
Mouse BAL fluid was obtained 45 minutes after Lys-ASA challenge by 2 repeated lavages with 1.0 ml of Ca2+- and Mg2+-free HBSS with 0.5 mM EDTA. Lungs were homogenized with a Tissue-Tearor homogenizer (Biospec Products) in 1 ml of T-PER Tissue Protein Extraction Reagent (ThermoFisher) containing protease inhibitor (Roche). Total IL-5, −13, and −33 content in lungs was measured with a commercial EIA (R&D Systems) and corrected for the protein content of each sample. Concentrations of PGD2, cysLTs, Histamine, mMCP1, CXCL7, and HMGB1 in BAL fluids were assayed by commercially available EIA kits according to the instructions of manufacturers Cayman Chemical (PGD2, cysLTs, and histamine), eBiosciences (mMCP-1), Abcam and R&D (CXCL7), and LifeSpan (HMGB1).
The concentration of CXCL7 in human PPP samples were assayed by commercially available EIA kits according to the manufacturer’s instructions (Human NAP-2/CXCL7 ELISA kit# RAB0135, Sigma)
Statistical analysis
Data are expressed as means plus or minus SEMs from at least 4 mice from each sex, except where otherwise indicated. Human sample sizes are indicated in each figure. Descriptive analyses of human cohorts are presented. Quantitative analyses in murine and human samples were performed with Prism software (GraphPad Software version 9.5.0, La Jolla, Calif). Group means were compared and differences between 2 treatment groups were assessed by using the Student t- test or Mann-Whitney test, and differences among multiple groups were assessed by using 1-way ANOVA after checking for normality with post-hoc Fisher’s least squares difference group tests unless otherwise indicated in the Supplementary figure legend text. Statistical significance was defined by a two-sided P-value of ≤0.05.
Results
Murine platelets express GLP-1R
To establish if murine platelets expressed GLP-1R and provide rationale for investigating the impact of liraglutide in a platelet-dependent murine model of AERD, whole blood from C57BL/6 mice was treated with a platelet-stabilizing reagent(27) and stained with CD41, CD45, and a far-red fluorescent GLP1R antagonist peptide label (LUXendin645).(31) CD45−CD41+ platelets demonstrated stronger expression of GLP-1R than CD45+CD41+ PLA and CD45+CD41− leukocytes by flow cytometry (Figure 1). In a competitive inhibition assay, the selective GLP-1R antagonist exendin-9–39 (EX9) out competed LUXendin645 staining on CD45−CD41+ platelets, CD45+CD41− leukocytes, and CD45+CD41+ PLA and at a concentration consistent with the known biology of EX9 competition at GLP-1R(31) (Figure 1B). LUXendin645 staining decreased with EX9 pretreatment on platelets and PLA (Figure 1C and 1D, respectively) from male and female C57BL/6 mice, confirming GLP-1R expression on murine platelets. A modest but significant decrease in LUXendin expression was noted with EX9 pretreatment on CD45+ cells without adherent platelets (Figure 1E). GLP-1R expression did not differ by sex in this cohort (Supplemental Fig. 2).
Figure 1: The glucagon-like peptide (GLP)-1 receptor is expressed on murine platelets.
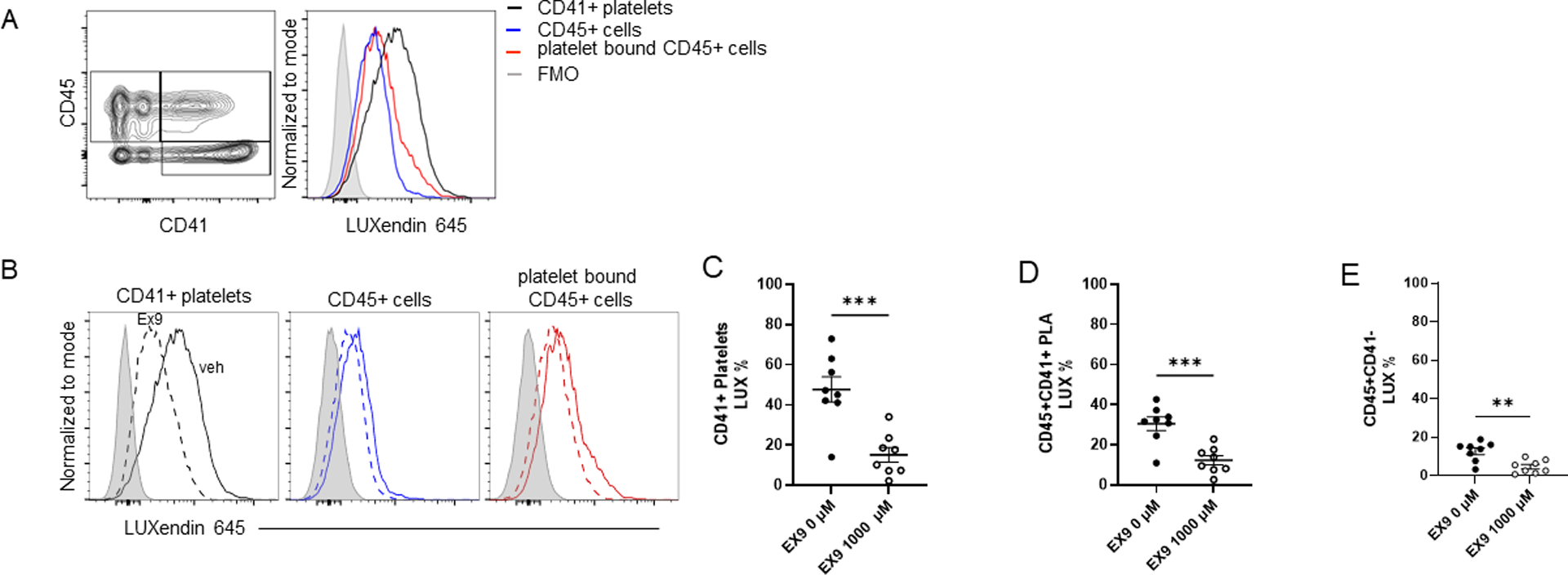
In wild-type C57B6 mice, whole blood was treated with a platelet stabilizing reagent and stained with CD41, CD45, and a far-red fluorescent GLP-1 receptor antagonistic peptide label (LUXendin645[4], 50 nM) to denote GLP-1R expression for flowcytometry. A) Representative gating strategy for CD45−CD41+ platelets, CD45+CD41− leukocytes, and CD45+CD41+ platelet-leukocyte aggregates (PLA) and corresponding LUXendin645 signal. CD45−CD41+ platelets (black line) exhibit greater GLP-1R expression as compared with leukocytes (blue line), PLA (red line), and the LUXendin645 (gray) FMO. B) In a competitive inhibition assay, pretreatment with the selective GLP-1R antagonist exendin-9–39 (EX9) for 5 minutes prior to LUXendin645 incubation (dashed lines) attenuated LUXendin645 signal (solid lines). C) LUXendin645 signal following EX9 pretreatment on CD41+ platelets, D) CD45+CD41+ PLA and E) CD45+ leukocytes. 8 adult wild-type C57B6 mice, 4 male and 4 female, per treatment group were used in all experiments. Mean and SE plotted. ** = p<0.01, **** = p<0.0001
GLP-1R agonist inhibits ASA-induced changes in airway resistance in an AERD model
In AERD-like Df challenged Ptges−/− mice, a single dose of GLP-1R agonist inhibited Lys-ASA-induced increases in airway resistance (Figure 2A–B; Supplemental Fig. 3). Downstream features of the model were also blunted by GLP-1R agonist (Supplemental Fig. 3).
Figure 2: Glucagon-like peptide (GLP)-1 receptor agonists attenuate airway resistance, and platelet presence following aspirin challenge in the Ptges−/− AERD mouse model, and human platelet-dependent mediator release.
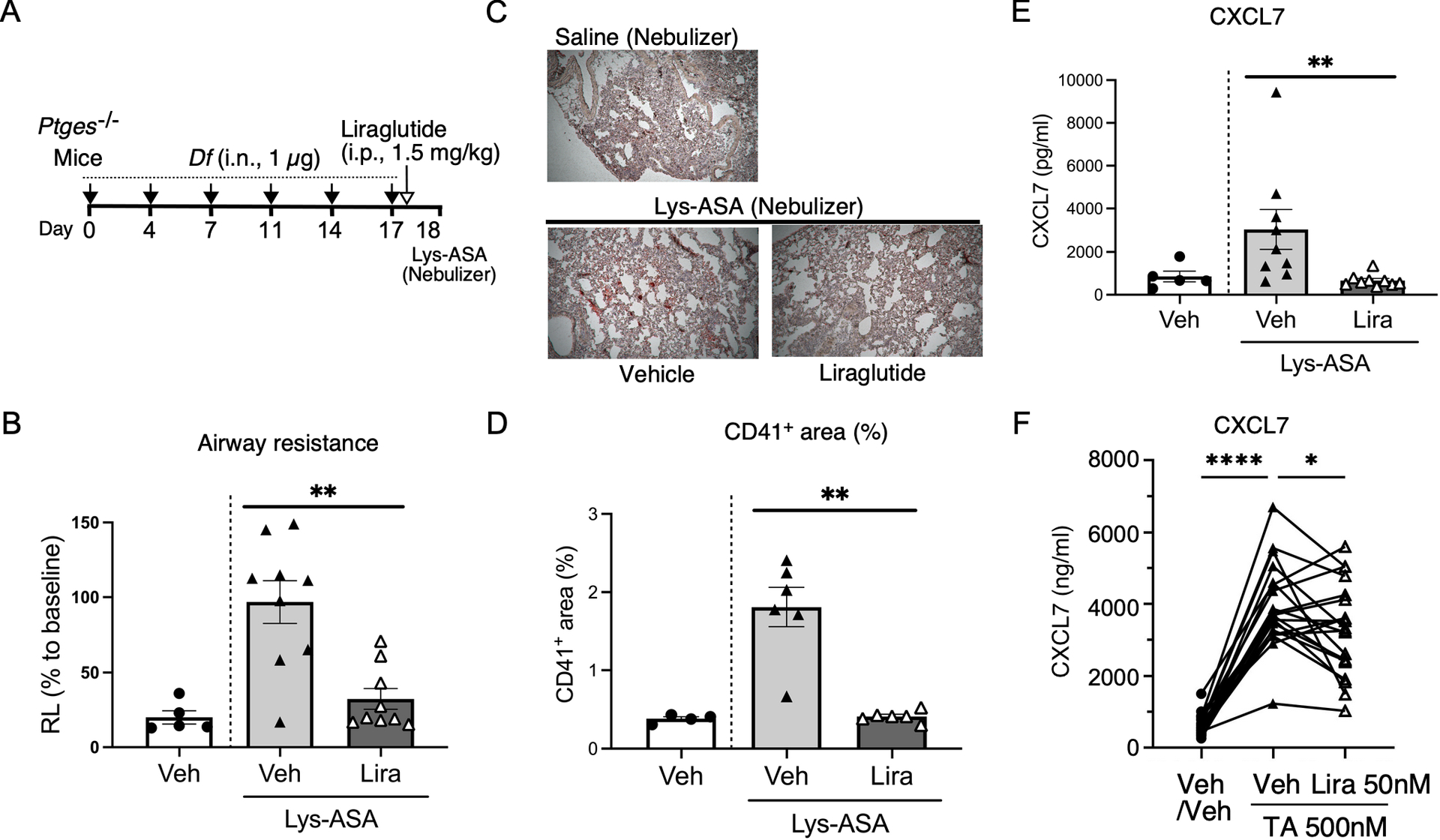
A) Protocol for intranasal administration of Df (1 μg) followed by Lysine-aspirin (Lys-ASA) challenge. Liraglutide or PBS was administered 24 hours prior to Lys-ASA challenge. B) Airway resistance (RL) in response to Lys-ASA was assessed with an Invasive Pulmonary Function Device and compared across treatment groups. (PBS/Veh: n =5, Veh/Lys-ASA: n=9, Liraglutide/Lys-ASA: n=9 mice from 3 experiments). Values are mean ± SEMs. **=p<0.01, by Mann Whitney test. C) Representative lung tissue sections from Vehicle- or Liraglutide-treated AERD model mice stained with platelet marker anti-CD41 antibody. D) The quantification of CD41+ area (%) was calculated by comparing it to the lung area in each image by ImageJ. Three random fields were selected in each lung tissue section from 2 independent experiments. (PBS/Veh: n =4, Veh/Lys-ASA: n=6, Liraglutide/Lys-ASA: n=6 mice from 2 experiments). Values are mean ± SEMs. **=p<0.01, by Mann Whitney test. E) Platelet-derived mediator release measured in murine BAL fluids (CXCL7: 45 minutes following aspirin challenge in the Ptges−/− AERD mouse model. (PBS/Veh: n =5, Veh/Lys-ASA: n=9, Liraglutide/Lys-ASA: n=9 mice from three independent experiments). Values were compared across treatment conditions. Values are mean ± SEMs. **=p<0.01, by Mann Whitney test F) GLP-1R agonists attenuate TA-induced CXCL7 release in humans. Plasma from whole blood samples from patients with the aspirin-exacerbated respiratory disease (AERD) phenotype of asthma pre-treated with Liraglutide (Lira, 50nM) or vehicle (Veh) for 30 minutes followed by TX-analogue (TA, 500nM) or veh activation for 30 minutes (as in Fig. 3). (n=20, p<0.0001, one-way ANOVA with post-hoc paired T-test reported, * P<0.05, **** P<0.0001).
GLP-1R agonist significantly attenuates platelet activation and platelet recruitment in the lungs of AERD-like mice
To better define the impact of GLP-1R activation on platelet function in vivo, we performed additional experiments in Ptges−/− mice, using a lower Df dose (1 μg) to optimize the platelet-dependent events (Fig. 2A). We focused on platelet activation and recruitment that characteristically occurs in this model and is necessary for the changes in airway physiology. Liraglutide treatment markedly inhibited the Lys-ASA induced increase in airway resistance (Fig. 2B). ImageJ-based analysis confirmed a marked inhibition of platelet recruitment following GLP-1R agonist treatment in the murine lung tissue (P=0.02; Figure 2C). Lys-ASA challenges induced platelet recruitment to the lung tissue, as evidenced by immunohistology for CD41, which was markedly attenuated by liraglutide (Fig. 2D). Lys-ASA-induced increases in BAL fluid levels of platelet derived chemokine CXCL7 were also significantly suppressed by liraglutide treatment (P=0.02; Figure 2E).
AERD cohort demographics and clinical features
Forty-four study participants were recruited, 11 controls and 33 AERD patients (Table I). Mean age in the AERD cohort was 49.2(12.3) years, 58% were female and 85% were White, consistent with prior demographic reports of AERD patients from these clinical sites.(32, 33) Eight patients were on high-dose (≥650mg) daily aspirin and 11 were on dupilumab 300 mg every 2 weeks; all patients on these therapies had used them for ≥90 days. Among AERD patients on dupilumab, 18.2% had concurrent aspirin use (Table I). All control participants and 30 AERD patients had available samples for analysis. The three missing samples did not meet study procedure criteria due to shipping delays during the COVID-19 pandemic.
Table I.
Patient Characteristics
| Characteristic | No Asthma (N=11) |
AERD (N = 33*) |
|---|---|---|
| Age, years (SD) | 36.1 (17.0) | 49.2 (12.3) |
| Sex, female, n (%) | 4 (36) | 19 (58) |
| Race, White, n (%) | 9 (82) | 28 (85) |
| Ethnicity, Hispanic, n (%) | 0 (0) | 2 (6) |
| Aspirin use,† n (%) | 0 (0) | 8 (24) |
| Dupilumab use,‡ n (%) | 0 (0) | 11 (33) |
| Concurrent aspirin use | -- | 2 (6) |
| Nasal steroid use, n (%) | 0 (0) | 27 (81.8) |
| Inhaled steroid use, n (%) | 0 (0) | 13 (39.4) |
| VUMC enrolled, n (%) | 11 (100) | 11 (33) |
VUMC=Vanderbilt University Medical Center; remainder recruited at Brigham and Women’s Hospital
30 patients with samples available for analysis.
At a dose equivalent to ≥650mg/day
Treated for a minimum of 90 days
Ex vivo GLP-1R agonist treatment decreases platelet activation, including platelets from patients with AERD
To identify the effect of GLP-1R agonists on human platelets, we assessed TP agonist-induced platelet activation in whole blood samples from the AERD and healthy non-asthmatic populations. Stimulation of samples from both subject groups with the TP agonist U46619 induced platelet activation as measured by an increase in CD62P expression on CD41+ platelets alone (Figure 3A–B). Pretreatment with GLP-1R agonist liraglutide at doses comparable to the serum concentrations achieved in vivo during active clinical treatment (50nM) attenuated TP-induced CD62P expression on CD41+ platelets in both groups (Figure 3A–B). In the subset of participants with AERD with available PPP samples (n=20), liraglutide treatment similarly attenuated the TP-induced platelet-derived mediator CXCL7 (Figure 2F). Samples from the subsets of participants with AERD receiving dupilumab (n=10; no dupilumab, n=20), or high-dose aspirin therapy (n=8; no aspirin, n=15) displayed TP-induced CD62P expression and liraglutide-induced suppression of CD62P expression at levels that were similar to those in samples from patients not on these treatments (Supplemental Fig. 4a–b, dupilumab treatment analysis; c-d aspirin treatment analysis). TP stimulation significantly increased in vitro PLA formation in subjects with AERD but not non-AERD controls. Liraglutide pretreatment did not significantly reduce TP-induced PLA formation in either patient population (Figure 3C–D) and did not significantly attenuate CD62P expression on PLAs (Figure 3E–F).
Figure 3: Glucagon-like peptide (GLP)-1 receptor agonists attenuate thromboxane-induced platelet activation in subjects with and without AERD.
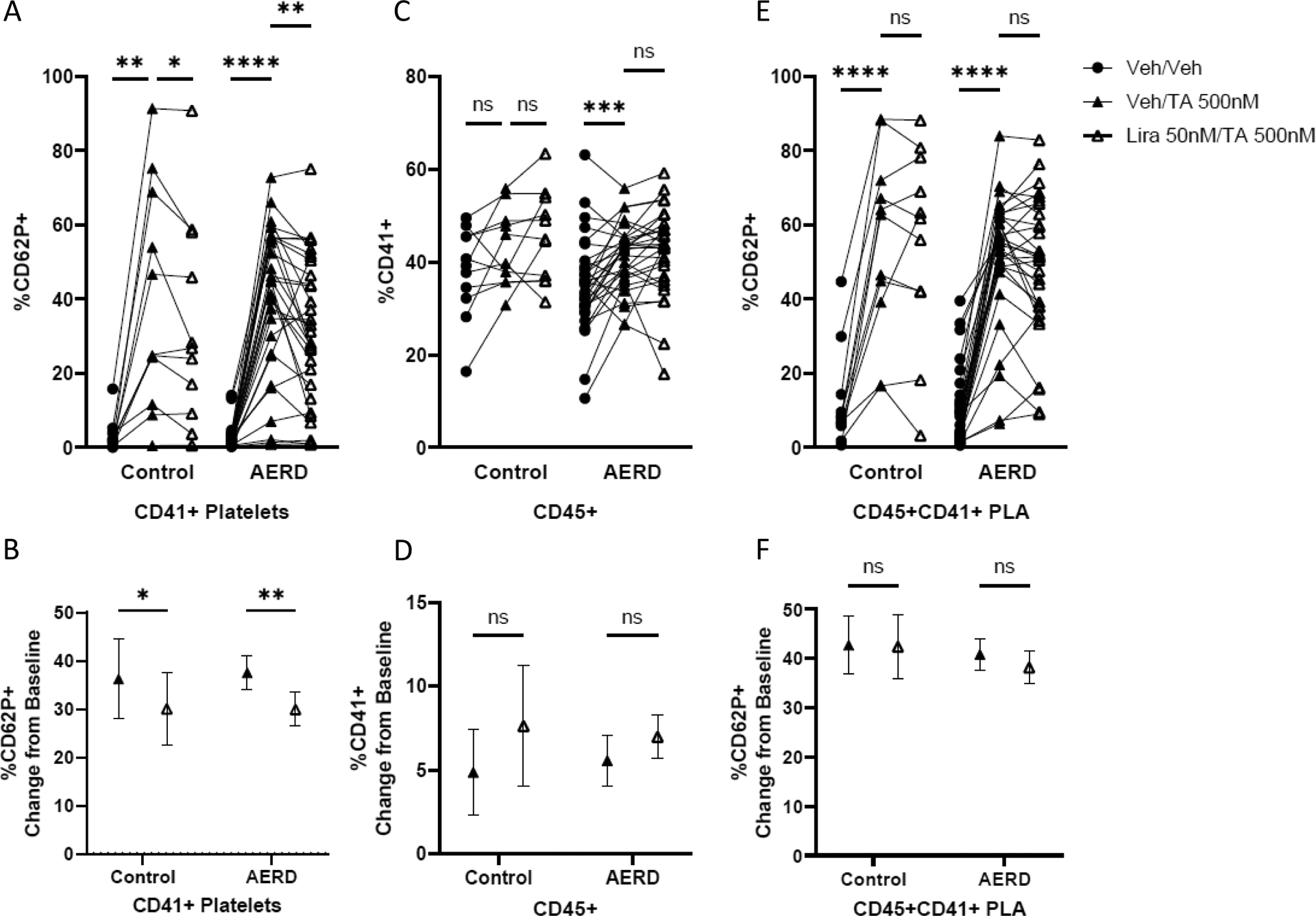
In healthy controls (n=11) and subjects with the platelet-dependent phenotype of asthma, aspirin-exacerbated respiratory disease (AERD, n=31), whole blood samples were treated with the GLP-1R agonist liraglutide (Lira 50 nM) or vehicle (Veh) for 30 minutes then stimulated with the thromboxane receptor agonist, U46619 (TA 500 nM) for 30 minutes and stained for flow cytometry. Effect of pre-treatment with liraglutide or vehicle on TA-induced A) CD41+ platelet CD62P expression, B) CD45+CD41+ PLA formation and C) CD45+CD41+ PLA CD62P expression. Two-way ANOVA confirmed outcomes were not different between clinical phenotypes. One-way ANOVA with post-hoc Fisher’s Least Significant Difference group comparisons reported. Group mean with standard error of the mean of the percent change from baseline in D) CD41+ platelet CD62P expression, E) CD45+CD41+ PLA formation and F) CD45+CD41+ PLA CD62P expression. Paired T-test reported. *=p<0.05, ** = p<0.01, ***=p<0.001, **** = p<0.0001
Discussion
In this study, we demonstrate that a single dose of a GLP-1R agonist decreases platelet-derived mediator release and markedly attenuates platelet recruitment to the lungs of a mouse model of AERD.(2) In patients with and without AERD, GLP-1R agonists similarly decrease in vitro platelet activation and proinflammatory platelet-derived mediator release, but not in vitro PLA formation. These effects are consistent among patients on a range of concurrent AERD therapy. In conducting these proof-of-concept studies we also confirmed the presence of the GLP-1R on murine platelets using a validated peptide probe (31), providing the opportunity to address prior knowledge gaps in the GLP-1R literature.(34, 35) Murine GLP-1R expression did not differ by sex; sex differences in the clinical response to GLP-1R agonists in humans remains an active area of research though no difference in human receptor expression has been reported.(36) Together, these results characterize platelets as a target of GLP-1R agonists in an AERD-like mouse model and in humans with and without AERD.
In prior studies, Lys-ASA challenges of Df-sensitized AERD-like Ptges−/− mice induced rapid recruitment of platelets to the lung tissue, accompanied by increases in BAL fluid levels of CXCL7, a highly abundant platelet-derived chemokine. Depletion of platelets(37) and inhibition of platelet activation with a TP receptor antagonist markedly attenuated Lys-ASA-induced changes in lung function in the AERD-like Ptges−/− model mice.(2) Use of this model in our study permitted assessment of airway resistance as well as platelet-focused measurements in murine lungs and BAL fluid. While our study focused on characterizing the involvement of platelets as targets for GLP-1R agonists, we were also able to explore the effect of GLP-1R agonists on previously demonstrated downstream features of the models including cysLTs production and mast cell activation. (Supplemental Fig. 3). It is likely that GLP-1R agonism on platelets impacts several of these downstream features, leading to its marked effect on changes in airway resistance (Fig. 2B, Supplemental Fig. 3b). Additionally, the inhibition of CXCL7 release could impact the incremental recruitment of blood born granulocytes to the lungs following challenges.
We recently reported in vivo exposure to liraglutide attenuates platelet activation after 2 weeks of treatment in a cohort of adults with prediabetes and obesity.(16) Current limitations of FDA approval for GLP-1RAs (to type 2 diabetes and obesity treatment) preclude direct in vivo assessment of the effects on platelets and airway inflammation in patients with asthma without these comorbid conditions. Thus, it was necessary to examine the effect of in vitro GLP-1R agonist treatment of whole blood on markers of platelet activation in these two AERD cohorts, as well as samples from healthy controls. Platelet activation leads to α-granule secretion, which we assessed by CD62P expression and plasma CXCL7 levels at baseline and following activation with a TP receptor agonist. TP receptor agonists mimic the effect of TXA2, an inflammatory mediator elevated in patients with AERD.(38) We focused on CD62P in this proof-of-principle study because of previous reports demonstrating that CD62P and CD63 correlate strongly with one another upon platelet activation,(39) with platelet-adherent eosinophils, and with urinary LTE4 levels at baseline (a marker of disease severity) in patients with AERD.(40)
The significant attenuation of platelet activation after brief exposure to a GLP-1R agonist in whole blood from patients with and without AERD supports anti-platelet actions of GLP-1R agonists independent of inflammatory disease state. This extends recent findings that in vivo exposure to GLP-1R agonists in patients with prediabetes and obesity attenuates TXA2-induced platelet aggregation, a measure of platelet binding to each other as in processes leading to homeostasis.(16) Future studies on the effect of in vivo GLP-1R agonist exposure on platelet aggregation and activation in the context of asthma are needed. While we were not powered to look at differential response to GLP-1R agonist treatment by concurrent AERD therapies, post-hoc analysis demonstrated GLP-1R agonist pretreatment reduced platelet activation across three real-world standard treatment conditions: no aspirin/biologics, aspirin, or dupilumab. Other clinical studies have demonstrated treatment with dupilumab and high-dose aspirin therapy do not alter PLA formation or CD62P expression in platelets from patients with AERD.(41, 42) While high-dose aspirin can inhibit endogenous platelet TXA2 production critical for platelet activation amplification and platelet recruitment to the site of vascular injury, use of an exogenous TP receptor analogue can overcome this inhibition and allow for the continued assessment of platelet response to activating stimuli.
Adherence of platelets to leukocytes is necessary for transcellular platelet-based conversion of LTA4 to LTC4, a process that is greatly increased in AERD and contributes to the elevated levels of LTC4 production observed in this disease. Adherent platelets account for the majority of LTC4S activity of blood granulocyte fractions from AERD patients(4). Both free platelets and PLA are recruited to the lungs of Ptges−/− mice,(37) as well as in other models of allergen induced pulmonary inflammation,(43) and our histological analysis does not distinguish between recruited PLA and free platelets. In contrast to attenuation of CD62P expression, GLP-1R agonists did not consistently demonstrate significant reduction of TP agonist-induced PLA formation or PLA-associated platelet activation in vitro. PLA response to GLP-1R agonists was heterogenous and attenuation was only observed in a subgroup of patients with AERD. PLA formation can result from CD40/CD40L and other cell surface protein interactions which may occur independently of CD62P expression, explaining the observed discordance between the significant reduction in the % of CD62P+ platelets and PLA formation.(44, 45) Additionally, leukocytes forming PLAs may preferentially take up platelets with the strongest CD62 expression which may or may not be equally susceptible to agonist, and may rapidly clear the adherent platelets through phagocytosis.(46) PLA formation in CD45+ cell subsets was not pursued in this pilot human study on platelet activation due to the lack of effect on total PLA formation and the unknown impact of the stabilizing reagent on leukocyte subsets after 24 hours of exposure. Given that platelets from subjects with AERD displayed no differences in responsiveness to agonists of either TP or GLP-1R, it is likely that any platelet-associated features of AERD reflect endogenous factors that influence platelets, rather than differences inherent to platelets per se. In vivo studies involving longer exposure to GLP-1R agonists are needed to assess the true impact of GLP-1R agonists on PLA formation, including PLA subsets, and augmentation of leukotriene generation from peripheral blood granulocytes including leukotriene generation from peripheral blood granulocytes. It will also be important to determine whether GLP-1R agonists impact indices of airway inflammation and clinical reactions to NSAIDs in human subjects with AERD.
While studies in mice that utilize depletion strategies have demonstrated a prominent role for platelets in airway inflammation, few clinical trials have targeted platelet-associated inflammation in asthma. Clinical trials evaluating the purinergic receptor P2Y12 antagonist, prasugrel, in allergic asthma(47) and AERD(48) have been largely negative. Notably, P2Y12 receptor blockade does not alter CD62P expression or PLA percentage in the blood of subjects with AERD, indicating that other targets are needed to attenuate platelet activation in human inflammatory disease(48). Recent studies demonstrate that platelets accumulate in airway walls and distal lung tissues of patients with asthma, with especially high levels observed in asthma fatalities, suggesting a potential role in asthma exacerbations.(1) It is tempting to speculate that the impact of GLP-1R on asthma exacerbations in patients with concomitant type 2 diabetes could reflect the impact of these drugs on platelets.(23) Our findings support that GLP-1R agonist may address platelet mediated inflammation not addressed by current asthma therapies.
GLP-1R agonists attenuate ex vivo human platelet activation including proinflammatory mediator release, and target platelets and platelet -dependent airway inflammation in an AERD murine model. These findings support the GLP-1R axis as a therapeutic target for platelet-mediated inflammation, an area particularly relevant to the AERD asthma phenotype. Over a decade of FDA-approved, commercially available GLP-1R agonist drugs in broad use for type 2 diabetes and obesity and safety in euglycemic states highlights the translational potential for these drugs in common human pulmonary diseases such as asthma. Prospective clinical study of the impact of GLP-1R agonists in AERD and other asthma phenotypes is warranted.
Supplementary Material
Key points:
GLP-1RAs attenuate ex vivo human platelet activation and proinflammatory mediators
GLP-1RAs target platelets/platelet-dependent airway inflammation in an AERD model
The GLP-1R axis may regulate platelet-mediated inflammation in asthma
Acknowledgements
Drs. Cahill and Boyce are co-senior authors who contributed equally to this work. The authors gratefully acknowledge Johannas Broichhagen, PhD and David J. Hodson, PhD for their contribution of the LUXendin reagent; R. Stokes Peebles, MD for facilitating the studies performed at VUMC; David K. Flaherty and Brittany K. Matlock for their technical expertise; Jonathan Hacker for assistance in sample processing; BWH’s AERD Center providers; and our patients at BWH and VUMC.
Grant support:
National Institutes of Health grants: NIH R01AI078908, R01HL117945, R37AI052353, R01AI136041, U19AI095219, U01AI155299, K23AI118804, K23HL161332, UL1 TR002243; Brigham and Women’s Hospital Research Institute Pilot Award; and by generous contributions from the Vinik Family and the Kaye Family.
Conflict of Interest Statement:
D.F., T.A., W.E.S., A.M., J.N, Y.T., C.F., T.L., D.C.N., A.L., H.H., J.Y., K.D.N. have no relevant conflicts of interest to disclose. T.M.L. has served on scientific advisory boards for GlaxoSmithKline and Sanofi-Genzyme, Novartis, and Regeneron. K.N.C. has served on scientific advisory boards for Novartis, GlaxoSmithKline, Regeneron, and Sanofi and reports personal fees from Third Harmonic Bio, Ribon Therapeutics, Verantos, Elsevier ClinicalKey, and UpToDate, research funding from the National Institutes of Health (U01AI155299), and research support in the form of investigational product from Novo Nordisk outside the submitted work. J.A.B. has served as a scientific advisory board member for Sanofi-Regeneron, Third Harmonic Bio, and Siolta Therapeutics, unrelated to GLP-1R agonists.
Abbreviations:
- AHR
Airway hyper-responsiveness
- AERD
Aspirin-exacerbated respiratory disease
- BAL
Bronchoalveolar lavage
- CD41
glycoprotein IIb/IIIa integrin
- CXCL7
Chemokine (C-X-C motif) ligand 7
- CysLT
Cysteinyl leukotriene
- Df
Extract from Dermatophagoides farinae
- IL
Interleukin
- ILC2
Group 2 innate lymphoid cell
- GLP-1R
Glucagon-like peptide-1 receptor
- HMGB1
High-mobility box 1
- LO
5-Lipoxygenase
- LT
Leukotriene
- LTC4S
Leukotriene C4 synthase
- Lys-ASA
Lysine-aspirin
- mMCP-1
Mucosal mast cell protease-1
- PG
Prostaglandin
- P2Y12 inhibitor
Purinergic P2Y12 receptor antagonist
- PLA
Platelet-leukocyte aggregates
- PRP
Platelet-rich plasma
- PPP
Platelet-poor plasma
- PTGES
Prostaglandin E2 synthase
- RAGE
Receptor for advanced glycation end products
- RL
Lung resistance
- TXA2
Thromboxane A2
- T2
Type 2
- TP
Thromboxane receptor
References
- 1.Shah SA, Kanabar V, Riffo-Vasquez Y, Mohamed Z, Cleary SJ, Corrigan C, James AL, Elliot JG, Shute JK, Page CP, and Pitchford SC. 2021. Platelets independently Recruit into asthmatic lungs and models of allergic inflammation via CCR3. Am J Respir Cell Mol Biol. 64(5):557–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu T, Kanaoka Y, Barrett NA, Feng C, Garofalo D, Lai J, Buchheit K, Bhattacharya N, Laidlaw TM, Katz HR, and Boyce JA. 2015. Aspirin-exacerbated respiratory disease involves a cysteinyl leukotriene-driven IL-33-mediated mast cell activation pathway. J Immunol. 195(8):3537–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finsterbusch M, Schrottmaier WC, Kral-Pointner JB, Salzmann M, and Assinger A. 2018. Measuring and interpreting platelet-leukocyte aggregates. Platelets. 29(7):677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laidlaw TM, Kidder MS, Bhattacharyya N, Xing W, Shen S, Milne GL, Castells MC, Chhay H, and Boyce JA. 2012. Cysteinyl leukotriene overproduction in aspirin-exacerbated respiratory disease is driven by platelet-adherent leukocytes. Blood. 119(16):3790–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu T, Barrett NA, Kanaoka Y, Buchheit K, Laidlaw TM, Garofalo D, Lai J, Katz HR, Feng C, and Boyce JA. 2019. Cysteinyl leukotriene receptor 2 drives lung immunopathology through a platelet and high mobility box 1-dependent mechanism. Mucosal Immunol. 12(3):679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Idzko M, Pitchford S, and Page C. 2015. Role of platelets in allergic airway inflammation. J Allergy Clin Immunol. 135(6):1416–1423. [DOI] [PubMed] [Google Scholar]
- 7.Knauer KA, Lichtenstein LM, Adkinson NF Jr., and Fish JE. 1981. Platelet activation during antigen-induced airway reactions in asthmatic subjects. N Engl J Med. 304(23):1404–1407. [DOI] [PubMed] [Google Scholar]
- 8.Takeda T, Morita H, Saito H, Matsumoto K, and Matsuda A. 2018. Recent advances in understanding the roles of blood platelets in the pathogenesis of allergic inflammation and bronchial asthma. Allergol Int. 67(3):326–333. [DOI] [PubMed] [Google Scholar]
- 9.Pitchford S, Cleary S, Arkless K, and Amison R. 2019. Pharmacological strategies for targeting platelet activation in asthma. Curr Opin Pharmacol. 46:55–64. [DOI] [PubMed] [Google Scholar]
- 10.Bhoria P, Sharma S, Varma N, Malhotra P, Varma S, and Luthra-Guptasarma M. 2015. Effect of steroids on the activation status of platelets in patients with Immune thrombocytopenia (ITP). Platelets. 26(2):119–126. [DOI] [PubMed] [Google Scholar]
- 11.Habib AM, Richards P, Cairns LS, Rogers GJ, Bannon CA, Parker HE, Morley TC, Yeo GS, Reimann F, and Gribble FM. 2012. Overlap of endocrine hormone expression in the mouse intestine revealed by transcriptional profiling and flow cytometry. Endocrinology. 153(7):3054–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marchetti P, Lupi R, Bugliani M, Kirkpatrick CL, Sebastiani G, Grieco FA, Del Guerra S, D’Aleo V, Piro S, Marselli L, Boggi U, Filipponi F, Tinti L, Salvini L, Wollheim CB, Purrello F, and Dotta F. 2012. A local glucagon-like peptide 1 (GLP-1) system in human pancreatic islets. Diabetologia. 55(12):3262–3272. [DOI] [PubMed] [Google Scholar]
- 13.Drucker DJ 2006. The biology of incretin hormones. Cell Metab. 3(3):153–165. [DOI] [PubMed] [Google Scholar]
- 14.Campbell JE, and Drucker DJ. 2013. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 17(6):819–837. [DOI] [PubMed] [Google Scholar]
- 15.Sarafidis P, Ferro CJ, Morales E, Ortiz A, Malyszko J, Hojs R, Khazim K, Ekart R, Valdivielso J, Fouque D, London GM, Massy Z, Ruggenenti P, Porrini E, Wiecek A, Zoccali C, Mallamaci F, and Hornum M. 2019. SGLT-2 inhibitors and GLP-1 receptor agonists for nephroprotection and cardioprotection in patients with diabetes mellitus and chronic kidney disease. A consensus statement by the EURECA-m and the DIABESITY working groups of the ERA-EDTA. Nephrol Dial Transplant. 34(2):208–230. [DOI] [PubMed] [Google Scholar]
- 16.Cahill KN, Amin T, Boutaud O, Printz R, Newcomb DC, Foer D, Hodson DJ, Broichhagen J, Beckman JA, Yu C, Nian H, Mashayekhi M, Silver HJ, Luther JM, Brown NJ, Peebles RS Jr., and Niswender K. 2022. Glucagon-like peptide-1 receptor regulates thromboxane-induced human platelet activation. JACC Basic Transl Sci. 7(7):713–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loganathan J, Cohen AC, Kaloupis GM, Harris C, Chronopoulos A, James V, Hamilton J, Green S, Wallis A, Morgan S, Dauer R, Gilfillan C, and Dear AE. 2022. A pilot clinical study to evaluate liraglutide-mediated anti-platelet activity in patients with type-2 diabetes (ELAID study). J Diabetes Complications. 36(5):108188. [DOI] [PubMed] [Google Scholar]
- 18.Bethel MA, Patel RA, Merrill P, Lokhnygina Y, Buse JB, Mentz RJ, Pagidipati NJ, Chan JC, Gustavson SM, Iqbal N, Maggioni AP, Öhman P, Poulter NR, Ramachandran A, Zinman B, Hernandez AF, and Holman RR. 2018. Cardiovascular outcomes with glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes: a meta-analysis. Lancet Diabetes Endocrinol. 6(2):105–113. [DOI] [PubMed] [Google Scholar]
- 19.Toki S, Goleniewska K, Reiss S, Zhang J, Bloodworth MH, Stier MT, Zhou W, Newcomb DC, Ware LB, Stanwood GD, Galli A, Boyd KL, Niswender KD, and Peebles RS Jr. 2018. Glucagon-like peptide 1 signaling inhibits allergen-induced lung IL-33 release and reduces group 2 innate lymphoid cell cytokine production in vivo. J Allergy Clin Immunol. 142(5):1515–1528e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toki S, Newcomb DC, Printz RL, Cahill KN, Boyd KL, Niswender KD, and Peebles RS Jr. 2021. Glucagon-like peptide-1 receptor agonist inhibits aeroallergen-induced activation of ILC2 and neutrophilic airway inflammation in obese mice. Allergy. 76(11):3433–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hur J, Kang JY, Kim YK, Lee SY, and Lee HY. 2021. Glucagon-like peptide 1 receptor (GLP-1R) agonist relieved asthmatic airway inflammation via suppression of NLRP3 inflammasome activation in obese asthma mice model. Pulm Pharmacol Ther. 67:102003. [DOI] [PubMed] [Google Scholar]
- 22.Bloodworth MH, Rusznak M, Pfister CC, Zhang J, Bastarache L, Calvillo SA, Chappell JD, Boyd KL, Toki S, Newcomb DC, Stier MT, Zhou W, Goleniewska K, Moore ML, Hartert TV, Niswender KD, and Peebles RS Jr. 2018. Glucagon-like peptide 1 receptor signaling attenuates respiratory syncytial virus-induced type 2 responses and immunopathology. J Allergy Clin Immunol. 142(2):683–687e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foer D, Beeler PE, Cui J, Karlson EW, Bates DW, and Cahill KN. 2021. Asthma exacerbations in patients with type 2 diabetes and asthma on glucagon-like peptide-1 receptor agonists. Am J Respir Crit Care Med. 203(7):831–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Albogami Y, Cusi K, Daniels MJ, Wei YJ, and Winterstein AG. 2021. Glucagon-like peptide 1 receptor agonists and chronic lower respiratory disease exacerbations among patients with type 2 diabetes. Diabetes Care. 44(6):1344–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pradhan R, Lu S, Yin H, Yu OHY, Ernst P, Suissa S, and Azoulay L. 2022. Novel antihyperglycaemic drugs and prevention of chronic obstructive pulmonary disease exacerbations among patients with type 2 diabetes: population based cohort study. BMJ. 379:e071380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.López-Cano C, Ciudin A, Sánchez E, Tinahones FJ, Barbé F, Dalmases M, García-Ramírez M, Soto A, Gaeta AM, Pellitero S, Martí R, Hernández C, Simó R, and Lecube A. 2022. Liraglutide improves forced vital capacity in individuals with type 2 diabetes: data from the randomized crossover LIRALUNG study. Diabetes. 71(2):315–320. [DOI] [PubMed] [Google Scholar]
- 27.Zagol-Ikapite I, Sosa IR, Oram D, Judd A, Amarnath K, Amarnath V, Stec D, Oates JA, and Boutaud O. 2015. Modification of platelet proteins by malondialdehyde: prevention by dicarbonyl scavengers. J Lipid Res. 56(11):2196–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oates J, Boutaud O, and Zagol-Ikapitte I. Methods of preventing platelet activation. Patent number: 10166248. Date of patent issue: Jaunary 1, 2019. [Google Scholar]
- 29.Uematsu S, Matsumoto M, Takeda K, and Akira S. 2002. Lipopolysaccharide-dependent prostaglandin E(2) production is regulated by the glutathione-dependent prostaglandin E(2) synthase gene induced by the Toll-like receptor 4/MyD88/NF-IL6 pathway. J Immunol. 168(11):5811–5816. [DOI] [PubMed] [Google Scholar]
- 30.Knudsen LB, and Lau J. 2019. The discovery and development of liraglutide and semaglutide. Front Endocrinol (Lausanne). 10:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ast J, Arvaniti A, Fine NHF, Nasteska D, Ashford FB, Stamataki Z, Koszegi Z, Bacon A, Jones BJ, Lucey MA, Sasaki S, Brierley DI, Hastoy B, Tomas A, D’Agostino G, Reimann F, Lynn FC, Reissaus CA, Linnemann AK, D’Este E, Calebiro D, Trapp S, Johnsson K, Podewin T, Broichhagen J, and Hodson DJ. 2020. Super-resolution microscopy compatible fluorescent probes reveal endogenous glucagon-like peptide-1 receptor distribution and dynamics. Nat Commun. 11(1):467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cahill KN, Wu P, Milne GL, Amin T, Singer J, Murphy K, Lewis E, Gapko D, Boyce JA, and Laidlaw TM. 2022. Mediator production and severity of aspirin-induced respiratory reactions: Impact of sampling site and body mass index. J Allergy Clin Immunol. 150(1):170–177e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mullur J, Steger CM, Gakpo D, Bensko JC, Maurer R, Laidlaw TM, and Buchheit KM. 2022. Aspirin desensitization and biologics in aspirin-exacerbated respiratory disease: Efficacy, tolerability, and patient experience. Ann Allergy Asthma Immunol. 128(5):575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barale C, Buracco S, Cavalot F, Frascaroli C, Guerrasio A, and Russo I. 2017. Glucagon-like peptide 1-related peptides increase nitric oxide effects to reduce platelet activation. Thromb Haemost. 117(6):1115–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cameron-Vendrig A, Reheman A, Siraj MA, Xu XR, Wang Y, Lei X, Afroze T, Shikatani E, El-Mounayri O, Noyan H, Weissleder R, Ni H, and Husain M. 2016. Glucagon-like peptide 1 receptor activation attenuates platelet aggregation and thrombosis. Diabetes. 65:1714–1723. [DOI] [PubMed] [Google Scholar]
- 36.Rentzeperi E, Pegiou S, Koufakis T, Grammatiki M, and Kotsa K. 2022. Sex differences in response to treatment with glucagon-like peptide 1 receptor agonists: opportunities for a tailored approach to diabetes and obesity care. J Pers Med. 12(3):454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu T, Laidlaw TM, Katz HR, and Boyce JA. 2013. Prostaglandin E2 deficiency causes a phenotype of aspirin sensitivity that depends on platelets and cysteinyl leukotrienes. Proc Natl Acad Sci U S A. 110(42):16987–16992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cahill KN, Bensko JC, Boyce JA, and Laidlaw TM. 2015. Prostaglandin D₂: a dominant mediator of aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 135(1):245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blair TA, Michelson AD, and Frelinger AL. 2018. Mass cytometry reveals distinct platelet subtypes in healthy subjects and novel alterations in surface glycoproteins in glanzmann thrombasthenia. Sci Rep. 8(1):10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mitsui C, Kajiwara K, Hayashi H, Ito J, Mita H, Ono E, Higashi N, Fukutomi Y, Sekiya K, Tsuburai T, Akiyama K, Yamamoto K, and Taniguchi M. 2016. Platelet activation markers overexpressed specifically in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 137(2):400–411. [DOI] [PubMed] [Google Scholar]
- 41.Cahill KN, Cui J, Kothari P, Murphy K, Raby BA, Singer J, Israel E, Boyce JA, and Laidlaw TM. 2019. Unique effect of aspirin therapy on biomarkers in aspirin-exacerbated respiratory disease: a prospective trial. Am J Respir Crit Care Med. 200(6):704–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buchheit KM, Sohail A, Hacker J, Maurer R, Gakpo D, Bensko JC, Taliaferro F, Ordovas-Montanes J, and Laidlaw TM. 2022. Rapid and sustained effect of dupilumab on clinical and mechanistic outcomes in aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 150(2):415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pitchford SC, Momi S, Baglioni S, Casali L, Giannini S, Rossi R, Page CP, and Gresele P. 2008. Allergen induces the migration of platelets to lung tissue in allergic asthma. Am J Respir Crit Care Med. 177(6):604–612. [DOI] [PubMed] [Google Scholar]
- 44.Lievens D, Zernecke A, Seijkens T, Soehnlein O, Beckers L, Munnix IC, Wijnands E, Goossens P, van Kruchten R,Thevissen L, Boon L, Flavell RA, Noelle RJ, Gerdes N, Biessen EA, Daemen MJ, Heemskerk JW, Weber C, and Lutgens E. 2010. Platelet CD40L mediates thrombotic and inflammatory processes in atherosclerosis. Blood. 116(20):4317–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schrottmaier WC, Kral JB, Badrnya S, and Assinger A. 2015. Aspirin and P2Y12 Inhibitors in platelet-mediated activation of neutrophils and monocytes. Thromb Haemost. 114(3):478–489. [DOI] [PubMed] [Google Scholar]
- 46.Maugeri N, Rovere-Querini P, Evangelista V, Covino C, Capobianco A, Bertilaccio MT, Piccoli A, Totani L, Cianflone D, Maseri A, and Manfredi AA. 2009. Neutrophils phagocytose activated platelets in vivo: a phosphatidylserine, P-selectin, and {beta}2 integrin-dependent cell clearance program. Blood. 113(21):5254–5265. [DOI] [PubMed] [Google Scholar]
- 47.Lussana F, Di Marco F, Terraneo S, Parati M, Razzari C, Scavone M, Femia EA, Moro A, Centanni S, and Cattaneo M. 2015. Effect of prasugrel in patients with asthma: results of PRINA, a randomized, double-blind, placebo-controlled, cross-over study. J Thromb Haemost. 13(1):136–141. [DOI] [PubMed] [Google Scholar]
- 48.Laidlaw TM, Cahill KN, Cardet JC, Murphy K, Cui J, Dioneda B, Kothari P, Raby BA, Israel E, and Boyce JA. 2019. A trial of type 12 purinergic (P2Y12) receptor inhibition with prasugrel identifies a potentially distinct endotype of patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 143(1):316–324 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.