

Abstract
One of the main components of articular cartilage is the chondrocyte’s pericellular matrix (PCM), which is critical for regulating mechanotransduction, biochemical cues, and healthy cartilage development. Here, individual primary human chondrocytes (PHC) are encapsulated and cultured in 50 µm diameter alginate microgels using drop-based microfluidics. This unique culturing method enables PCM formation and manipulation of individual cells. Over ten days, matrix formation is observed using autofluorescence imaging, and the elastic moduli of isolated cells are measured using AFM. Matrix production and elastic modulus increase are observed for the chondrons cultured in microgels. Furthermore, the elastic modulus of cells grown in microgels increases approximately 10-fold over ten days, nearly reaching the elastic modulus of in vivo PCM. The AFM data is further analyzed using a Gaussian mixture model and shows that the population of PHCs grown in microgels exhibit two distinct populations with elastic moduli averaging 9.0 kPa and 38.0 kPa. Overall, this work shows that microgels provide an excellent culture platform for the growth and isolation of PHCs, enabling PCM formation that is mechanically similar to native PCM. The microgel culture platform presented here has the potential to revolutionize cartilage regeneration procedures through the inclusion of in vitro developed PCM.
Keywords: Chondrocytes, Pericellular matrix, Atomic force microscopy, Drop-based microfluidics, Single-cell
Graphical Abstract
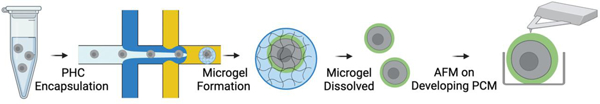
Individual primary human chondrocytes (PHCs) are encapsulated and cultured in 50 µm diameter alginate microgels using drop-based microfluidics. Chondrocytes form a mature pericellular matrix (PCM) within ten days. The alginate is removed using chemical degradation and elastic moduli of single cells are measured using atomic force microscopy (AFM). These microgels enable PCM formation that is mechanically comparable to native PCM.
Introduction:
Articular cartilage (AC) is the soft load-bearing tissue lining the interface of joints and is responsible for shock absorption and wear resistance from everyday activities such as walking, running, and jumping [1]. However, acute injuries to the AC have limited self-repair capabilities due to their avascular nature and low cell density [2]. Consequently, such injuries often lead to osteoarthritis (OA), the most common degenerative joint disease affecting over 300 million people worldwide [2–4]. Unfortunately, treatment options for acute injuries and OA are limited, and many treatments create mechanically inferior cartilage compared to native AC [2,4,5]. Matrix-assisted autologous chondrocyte transplantation (MACI), a laboratory-based tissue engineering approach to cartilage regeneration, is currently one of the most effective treatment options [2,4]. In MACI, a small portion of healthy cartilage is taken from the patient, and the chondrocytes are isolated from the cartilage and expanded without passaging using standard in vitro cell culture [2,4,6,7]. The cells are then transplanted back into the patient with a membrane typically made of porcine collagen I/III [2,4,6,7]. However, MACI has certain drawbacks: (1) During expansion on tissue culture plastic, most chondrocytes quickly dedifferentiate into osteocytes and fibroblasts, losing their ability to produce cartilage [8–13]. (2) Expanded cells lack a pericellular matrix (PCM), a 1–5 μm layer of collagens, glycoproteins, and proteoglycans that encapsulates the cells and plays a critical role in cell viability, mechanotransduction, regulating biochemical cues, and healthy cartilage development [14–18].
Culturing chondrocytes in three-dimensional (3-D) hydrogels, rather than on tissue culture plastic, has been shown to retain the chondrocyte phenotype and even reverse dedifferentiation [5,19]. Additionally, chondrocytes grown in 3-D hydrogels produce PCM and other matrix components [5,19,20]. However, the large bulk hydrogels typically used for cell culture, ranging in size from millimeters to centimeters, are not suitable for MACI [2,21]. These bulk hydrogels are often not injectable, requiring the extraction of cells from the hydrogels for compatibility with MACI [21]. An alternative to bulk hydrogel culture is the use of microscale hydrogel particles known as microgels. Microgels offer several advantages compared to bulk culture: (1) Microgels are easily injectable due to their collective shear-thinning behavior [21–23]. (2) By reducing the size of the hydrogels to the microscale, the surface area-to-volume ratio increases, facilitating the delivery of nutrients and other bioactive compounds to the encapsulated cells by reducing diffusion distances [21–23]. Our group has previously demonstrated a novel method of encapsulating single primary human chondrocytes (PHCs) in 3-D alginate microgels that are approximately 100 μm in diameter, enabling the cells to maintain their chondrocyte phenotype and form a collagen VI-rich PCM [14]. While our previous studies analyzed differences in metabolomic profiles, they did not investigate the mechanical similarity between the in vitro developed PCM in microgels and the in vivo formed PCM. Since the PCM directly transfers extracellular mechanical loads to chondrocytes, it is crucial to understand the biomechanical properties of in vitro developed PCM [14–18,24].
In this study, we encapsulate individual primary human chondrocytes (PHC) isolated from patient knees and culture them in alginate microgels created using drop-based microfluidics. This unique culturing method enables PCM formation and manipulation of individual cells [14]. Subsequently, we remove the alginate using chemical degradation and isolate the single cells into microscale wells created using photolithography for downstream atomic force microscopy (AFM). Over a period of ten days, matrix formation is observed using autofluorescence imaging and the elastic moduli of the isolated cells are measured using AFM. We compare the biochemical and biomechanical properties of cells cultured in monolayers to those cultured in microgels over ten days. We observe negligible change in both matrix formation and elastic modulus of the chondrons cultured in monolayer. In contrast, matrix production and elastic moduli increase in the chondrons cultured in microgels. Furthermore, the elastic modulus of cells grown in microgels increases approximately tenfold over ten days, nearly reaching the elastic modulus of in vivo PCM. We further analyze the AFM data using a Gaussian mixture model and find that the population of PHCs grown in microgels exhibit two heterogeneous outcomes of PCM formation. Specifically, two distinct populations were identified: the first with an average elastic modulus of 9.0 kPa, and the second with an average elastic modulus of 38.0 kPa, accounting for 54% and 46% of the population, respectively. The second population was mechanically similar to in vivo PCM, which has an elastic modulus of 25–1000 kPa [1,15–17,24–27]. Overall, our results demonstrate that the in vitro microgel culture platform enables PHCs to form PCM that is mechanically similar to native PCM.
Results:
An overview of the experimental methodology used in this work is presented in Figure 1. Briefly, we present a method for culturing individual PHCs within approximately 50 µm diameter alginate microgels. The microgels are dissolved, and the cells are measured under AFM. PHCs are harvested from donor tissue obtained from patients undergoing knee replacements and encapsulated in alginate microgels using drop-based microfluidics, as we have previously developed (Figure 1A–B)[14]. The 3-D microenvironment of the alginate microgels facilitates nutrient diffusion to the encapsulated single cells and promotes the formation of PCM surrounding PHC chondrons (Figure 1C). After 10 days in culture, the alginate surrounding the cells is dissolved (Figure 1D), and the cells are gently centrifuged onto a microscale well-plate created using photolithography (Figure 1E). The mechanical properties of the chondrons are then measured using AFM (Figure 1F).
Figure 1:
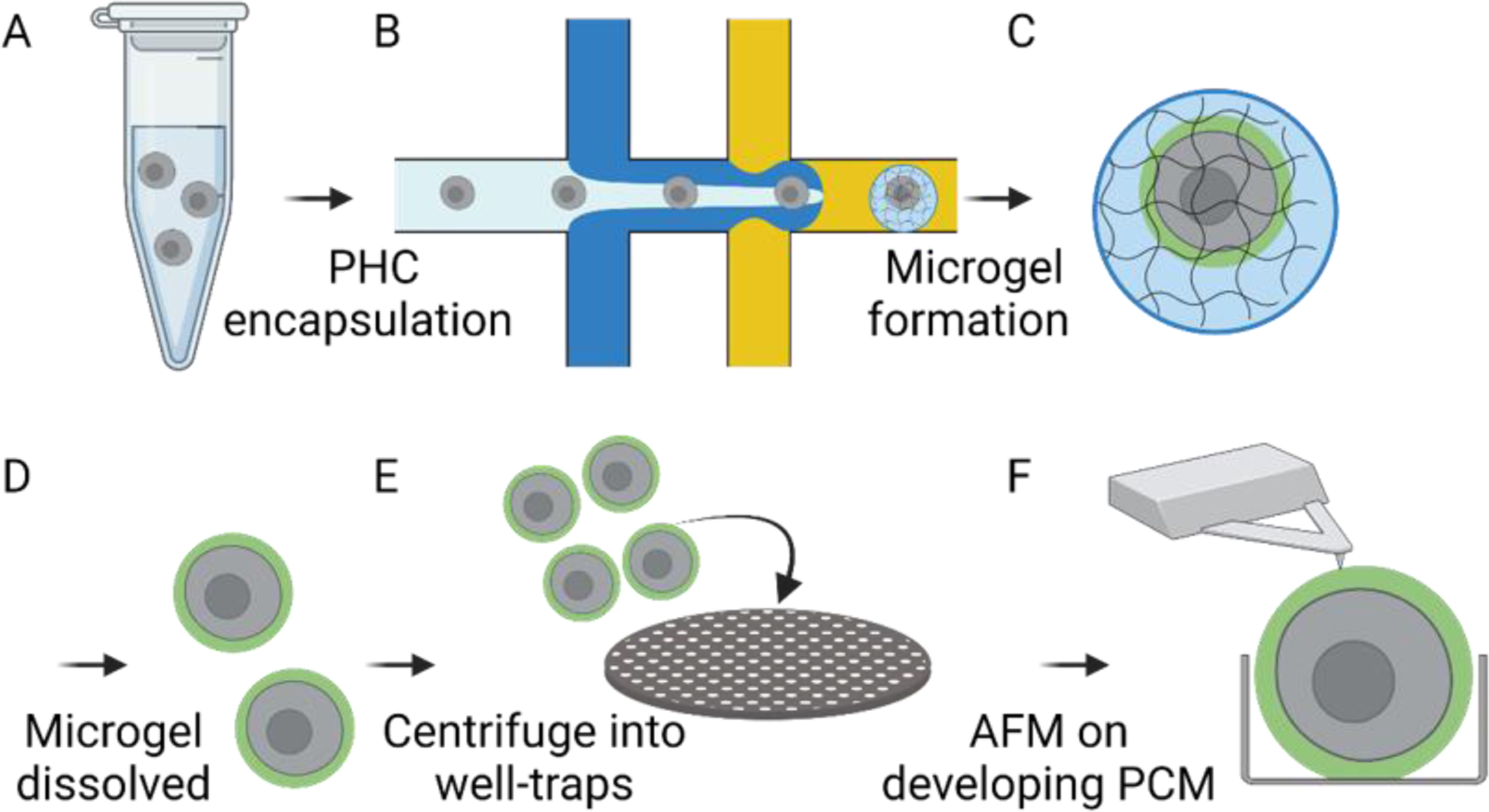
Schematic of Experimental Design: (A) Primary Human Chondrocytes (PHC) are suspended in the alginate precursor at 4 × 106 cells/mL. (B) PHCs are encapsulated in alginate microgels using drop-based microfluidics. (C) Microgels are washed and cultured in media for 10 days where chondrocytes form a collagen VI-rich PCM. (D) On days 1, 6, and 10, cells are removed from the microgels using sodium citrate and EDTA washes. (E) The released cells are centrifuged into well-traps created using photolithography. (F) The elastic moduli (E) of the cells are measured using AFM.
Analysis of PCM formation from single PHCs encapsulated within alginate microgels
Collagen VI, a biomarker for the PCM, and many other PCM components and collagens, autofluoresce when excited with wavelengths of light ranging from UV to blue (365–415 nm) [14,28–30]. In a previous study, we measured the formation of PCM in SW1353 cells, a chondrosarcoma cell line, over 10 days in microgel culture using autofluorescence imaging [14]. Results showed increased intensities surrounding the cells, indicating increased matrix protein expression of the cells cultured in alginate microgels compared to cells cultured in a monolayer [14]. This label-free approach allowed for the quantification of PCM formation over time while the cells were cultured in 3-D alginate microenvironments. In this study, we apply the same methodology to quantify PCM formation of PHC cells isolated from knee replacements of female patients aged 63 to 72 (Bozeman Deaconess Hospital, MSU).
As previously described, the isolated PHCs were cultured in both 3-D microgels and 2-D monolayers on standard tissue culture flasks. Autofluorescence imaging was performed on days 0, 1, 4, 7, and 10. PHCs grown in 2-D monolayers are known to produce minimal amounts of collagen in the PCM [19]. As a control experiment, we expanded PHCs on tissue culture flasks, and as expected, the autofluorescence images of the cells cultured in monolayers showed no observable signal on day 0 and no change in signal at any time point over the 10 days, indicating minimal matrix components (SI Figure 1).
By contrast, when PHCs were grown in microgels formed using drop-based microfluidics, autofluorescence visibly increased at each time point (Figure 2A –B). The cells grown in monolayers displayed a statistically non-significant change in fluorescence intensity over the ten days (Multiple linear regression, p = 0.85) (Figure 2B, red triangles), confirming minimal matrix component formation in monolayer cultures. However, cells grown in microgels showed a linear increase in fluorescence intensity over time (p << 0.0001). Quantitative image analysis revealed that the average fluorescence intensity across the area of each cell grown in microgels increased at a rate of 111 ± 12 a.u./day (Figure 2B, blue squares). These results indicate that PHCs grown in alginate microgels produce matrix components which remain surrounding the cell at significantly higher rates than when grown in monolayers..
Figure 2:
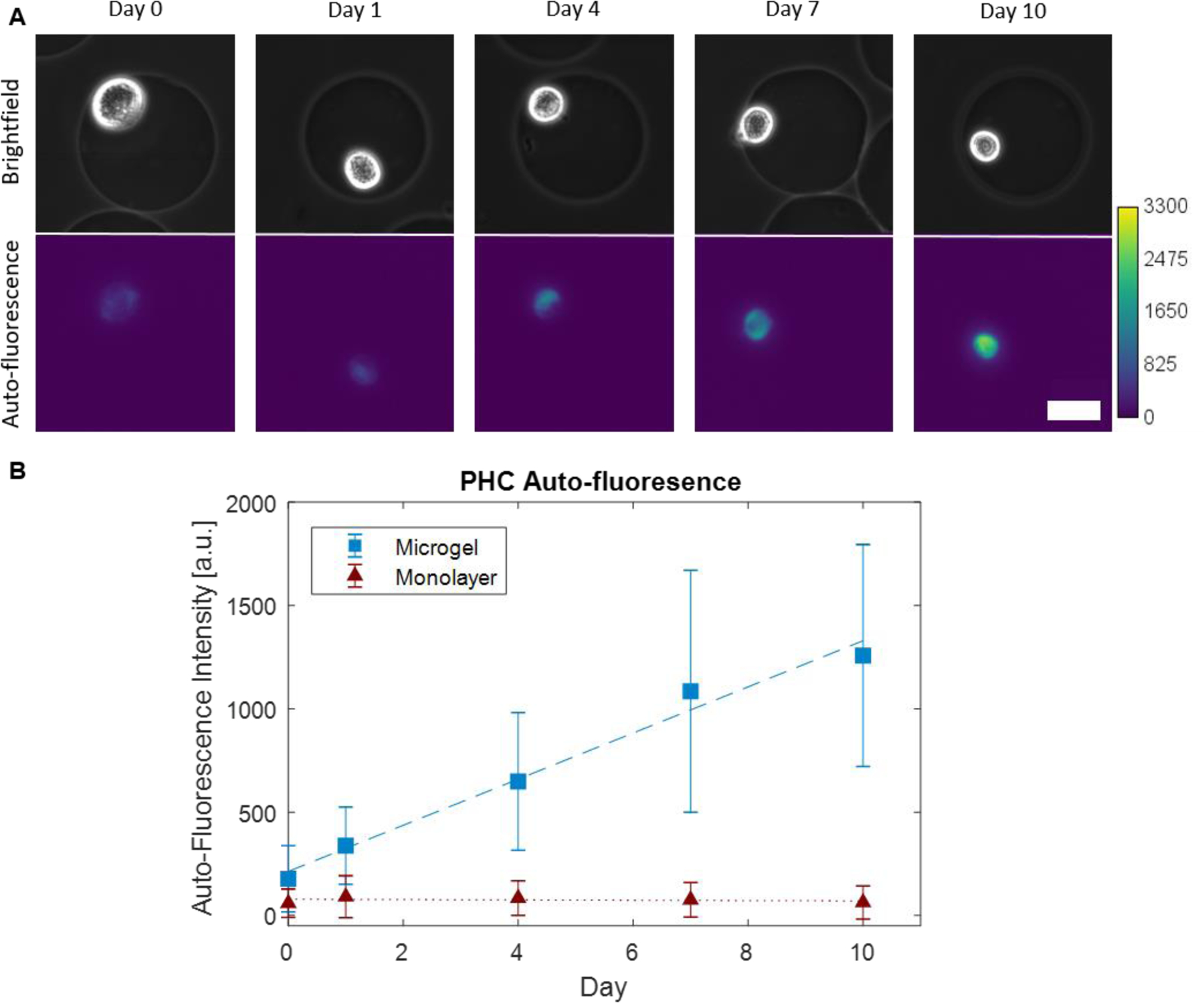
Representative images of single PHC cells cultured in microgels over 10 days using fluorescence imaging; (A) Brightfield and auto-fluorescence images (ex. 350/em. 470) of PHCs encapsulated in alginate microgels and cultured for 10 days. Scale bar is 25 μm. (B) Measured mean fluorescence intensities of imaged PHCs over time with multiple linear regressions (N =3, n = 20, p << 0.0001). PHCs encapsulated in microgels (blue squares, dashed) and PHCs grown in monolayers (red triangles, dotted).
We also compared the growth of PHCs and SW1353 cells in monolayers versus microgels as a control experiment. The autofluorescence signal from PHCs grown in microgels increased at a rate of approximately 70 a.u./day, which was about 5 times faster than SW1353 cells in microgels. The final pixel intensity reached 1258 ± 537 a.u. for PHCs and 347 ± 73 a.u. for SW1353 cells (SI Figure 2). As expected, these results confirm that PHCs produce more matrix components than SW1535 cells, likely due to the higher expression levels of anabolic genes involved in matrix production [31].
Our prior work using immunofluorescence has demonstrated that PHCs cultured in alginate microgels produce a PCM-like structure composed of collagen VI [14]. However, in these experiments, the PHCs were not released from the microgels. While recent advancements have allowed for imaging of cell organelles using AFM force curves without breaching the cell wall, quantifying the elastic modulus of subsurface structures at unknown distances beneath the imaging surface of the probe tip remains infeasible [32]. To directly measure the elastic modulus of the PCM-like structure, we first dissolved the microgels using sodium citrate and EDTA, which are typical reagents for alginate dissolution, before conducting AFM characterization of the elastic modulus. This approach enables direct actuation of the PHCs by the AFM probe tip without the presence of alginate. To assess whether the dissolution process affected the structure of the developing PCM, PHCs on days 1, 6, and 10 were fixed and stained for collagen VI using antibodies before and after microgel dissolution. We compared the fluorescence of the cells before and after microgel dissolution on these specific days. Representative cell images of intact microgels before and after dissolution can be seen in Figure 3.
Figure 3:
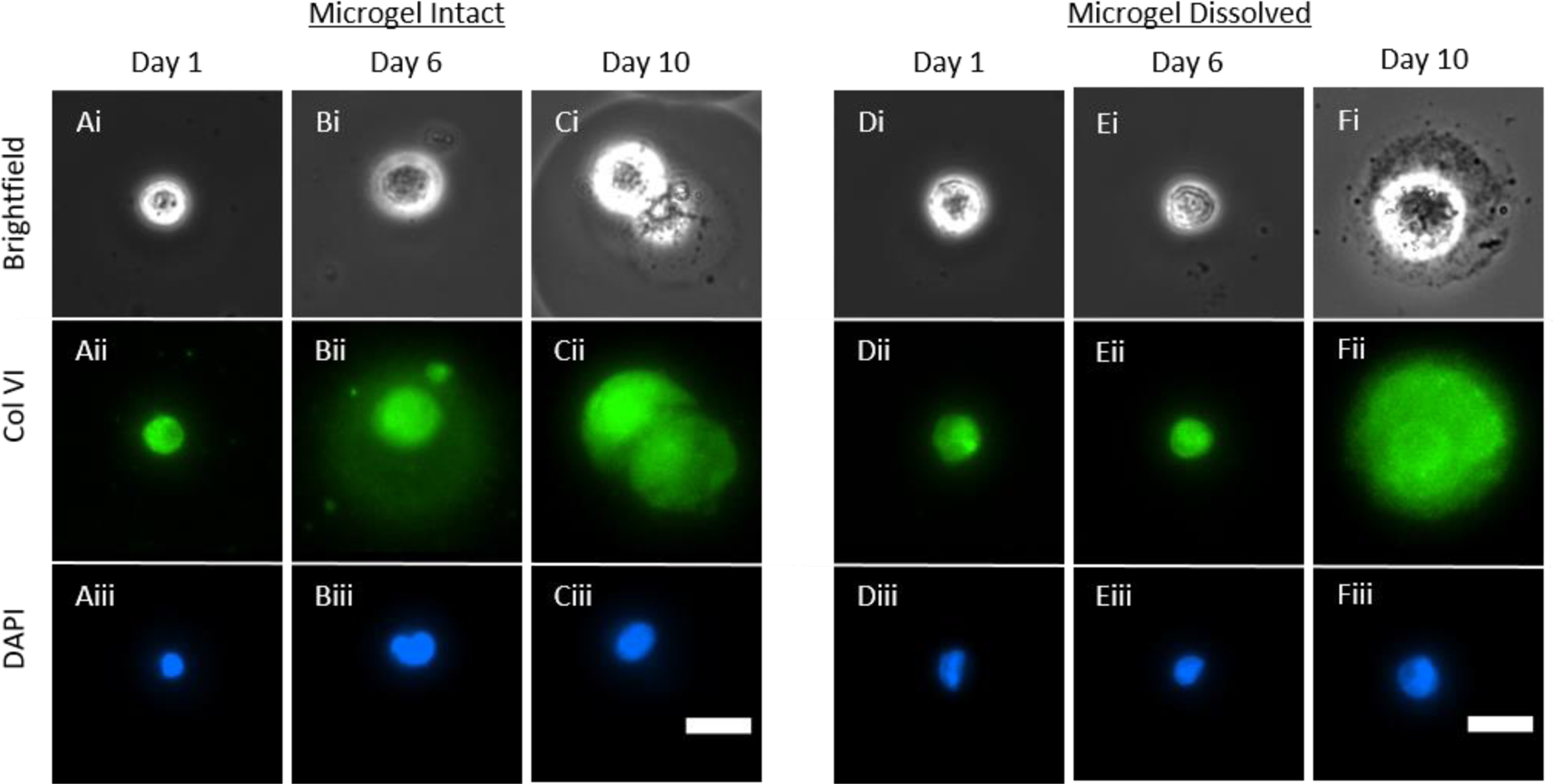
Collagen VI immunofluorescence; Representative Human Primary Chondrocyte (HPC) cultured in 1.5 % w/w alginate microgels with the microgels intact (A-C) and after dissolving the microgels using sodium citrate and EDTA (D-F). Cells were fixed and stained for collagen VI (green) and nuclei (DAPI blue). Cells were imaged on days 1, 6, and 10 to match with AFM time points. Scale bars are 25 μm.
Prior to dissolution, a thin layer of collagen VI can be observed surrounding the cell on day 1 (Figure 3A ii), which becomes thicker by day 6 (Figure 3Bii). By day 10, the entire region surrounding the cell shows a significant amount of collagen VI, resembling the PCM typically observed in vivo (Figure 3Cii). Following microgel dissolution on day 1, there appeared to be no difference in collagen VI expression between cells in microgels and cells after dissolution, with a thin layer of collagen VI remaining immediately surrounding each cell (Figure 3Aii and 3Dii). Before microgel dissolution on day 6, more diffuse signal is observed radiating from the cell (Figure 3Bii). However, after microgel dissolution on day 6, the diffuse signal observed further from the cell in intact microgels is no longer present (Figure 3Bii vs. 3Eii). In fact, only a thin layer of collagen VI immediately surrounding the cell remained (Figure 3Eii), similar to cells in microgels on day 1 (Figure 3Aii). After microgel dissolution on day 10, the region surrounding the cells exhibits a significant amount of collagen VI immediately encasing the cell, which extends into the surrounding area (Figure 3Cii vs. 3Fii). The PCM is also visible in brightfield once released from the microgel (Figure 3Fi). By day 10, the matrix is stable and strong enough that the dissolution of the microgel does not visibly affect the PCM produced by the PHC. These results show that on day 6, the loss of the alginate disrupts the developing PCM, whereas on day 10, the PCM is well-formed enough to remain intact after dissolution of the alginate. It is possible that the EDTA dissolution of the alginate microgels could disrupt some disulfide bonds in PCM crosslinking. However, based on the presence of collagen VI on day 10, we hypothesize that the loss of collagen VI on day 6 is due to the loss of the alginate support, rather than the chemical effects of the EDTA [33].
Elastic moduli (E) of individual PHCs released from alginate microgels and measured using AFM
Understanding the mechanical properties, primarily the elastic modulus (E), of PCM development in vitro is critical to understanding how the chondrons will respond to mechanical forces upon transplantation into patients. Here, AFM was used to measure the elastic moduli of individual PHCs at different time points after their development in alginate microgels. First, the microgels were dissolved to isolate the growing chondrons on days 1, 6, and 10. The isolated chondrons were then suspended in PBS and gently centrifuged into microscale wells (30 × 30 × 10 μm) on a silicon wafer created using photolithography. These wells allowed for the immobilization of single cells for subsequent AFM measurements. As a control, PHCs grown in monolayers were also removed from 2-D tissue culture using sodium citrate and EDTA and centrifuged into microscale wells.
Once cells were isolated into the microscale wells, AFM measurements were conducted on individual cells. Force versus displacement curves were acquired for 20 cells from each donor (N = 3) on days 1, 6, and 10. Additionally, an 8 × 8 force map covering a 1 µm2 area was acquired for each cell and every other data point was removed from analysis to minimize bias from neighboring indentations. Representative force curves are depicted in Figure 4A and 4B. The indentation depth to reach 2 nN for PHCs cultured in monolayer was compared to that for PHCs cultured in microgels. Cells cultured in monolayer reached an indentation force of 2 nN at average indentation depths ranging from 1.33 to 1.91 µm on days 1, 6, and 10 (Figure 4A). For chondrons cultured in microgels on day 1, the indentation depths were similar to those of cells cultured in monolayers, with an average indentation depth of 0.90 µm (Figure 4B, Day 1). However, on days 6 and 10, significant decreases in the indentation depths were observed, with the average indentation depths of 0.51 and 0.33 µm, respectively (Figure 4B, Day 6 and Day 10). This substantial decrease in the indentation depth suggests that the chondrons grown in microgels are undergoing matrix production and stiffening.
Figure 4:
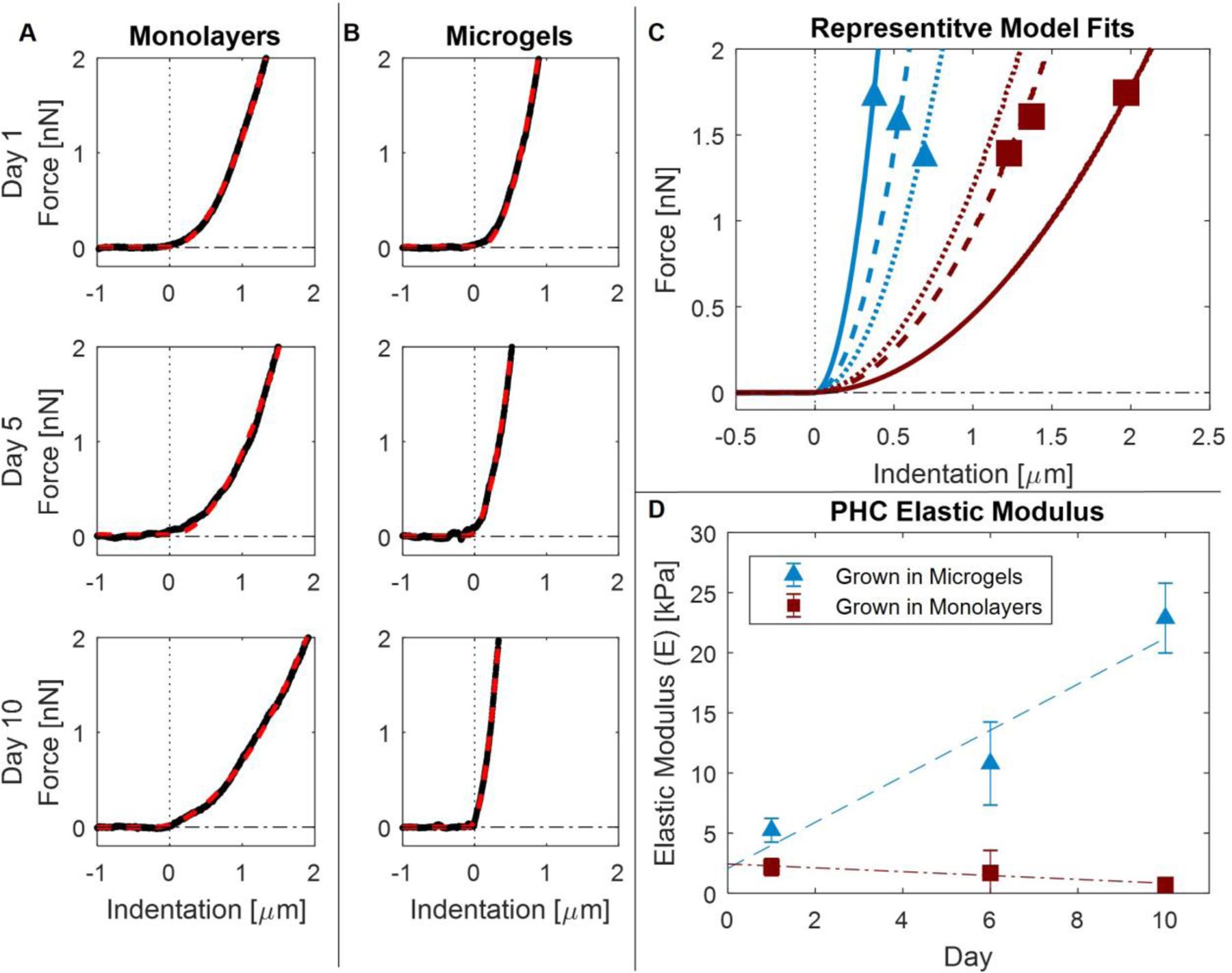
Atomic Force Microscopy Measurements; (A) Representative force vs. indentation curves of PHCs grown in monolayer on days 1, 6, and 10. Modeled Hertz fits are overlaid (red dashed). (B) Representative force vs. indentation curves of PHCs grown in microgels on days 1, 6, and 10. Modeled Hertz fits are overlaid (red dashed). (C) Average Hertz models for cells cultured in microgels (red squares) and cells cultured in monolayers (blue triangles) on days 1 (dotted), 6 (dashed), and 10 (solid). (D) Average elastic modulus and the standard deviation plotted for cells cultured in microgels (blue triangles, dashed) and monolayer (red squares, dotted dashes). At each time point, 20 cells were measured for each donor and culture condition (N = 3). When compared with a linear mixed effects model, modeled slopes were significantly different from each other (p < 0.0001) while the modeled intercepts were not (p = 0.50) Error bars represent the standard deviation between donors.
To calculate E of the cells, the indentation force curves were fit using a Hertzian model for a cone with a half angle of 36 degrees [32], and representative model fits are shown in Figure 4C. The representative model fits for cells grown in monolayers on days 1, 6, and 10 exhibit a slight softening with force versus indentation curves becoming flatter as time progresses (Figure 4C, red squares). Conversely, the representative model fits for cells grown in microgels on days 1, 6, and 10 demonstrate substantial stiffening of the chondrons with force versus indentation curves becoming steeper as time increases (Figure 4C, blue triangles). Cells initially cultured in monolayer displayed an average elastic modulus of 2.1 ± 0.7 kPa on day 1, and a minor decrease in stiffness with an average elastic modulus of 0.7 ± 0.3 kPa on day 10 (Figure 4D, red squares). By contrast, chondrons cultured in microgels exhibited an average elastic modulus of 5.2 ± 1.0 kPa on day 1, and a significant increase in stiffness with an average elastic modulus of 22.9 ± 2.9 kPa by day 10 (Figure 4D, blue triangles). The data were modeled using a linear mixed effects model to quantify trends in the elastic moduli data. The linear mixed effects model revealed that over the course of 10 days, the slopes of the two lines were significantly different from each other (p < 1.0 × 10−30); cells grown in monolayers decreased in stiffness at a rate of 0.16 ± 0.1 kPa/day, while cells grown in microgels increased in stiffness at a rate of 1.91 ± 0.2 kPa/day. Interestingly, cells grown in both monolayers and microgels showed no significant difference in the modeled day 0 elastic modulus (p = 0.50), suggesting that the observed difference in elastic moduli by day 1 was due to culturing over 24 h. These results indicate that microgel culture promotes rapid matrix production within 24 h.
Heterogeneous outcomes of PCM formation in microgels
Chondrocytes exhibit spatial heterogeneity, with phenotypic differences depending on their location within the cartilage [11,19,34,35]. The E of cells grown in microgels was further analyzed, and when comparing the three donors, there was no difference in mean E between donors on days 1, 6, and 10 (p > 0.46). Since there was no donor-related difference at any time point, the E data from cells grown in microgels were pooled together. On day 10, cells grown in microgels displayed a wide distribution of E ranging from 3 to 119 kPa (Figure 5A Day 10). The histograms of individual E on day 1 showed a normal distribution with no skewness (S = 0.05), and the mean and median were approximately equal at 5.2 and 5.0 kPa, respectively (Figure 5A, Day 1). However, on day 10, the distribution becomes highly skewed (S = 0.43), and significant differences in the mean and median were observed at 22.8 and 3.2 kPa, respectively. These results indicate that on day 10, the mean E is highly dominated by the skewed population.
Figure 5: Intercell and Intracell Elastic Moduli Heterogeneity.
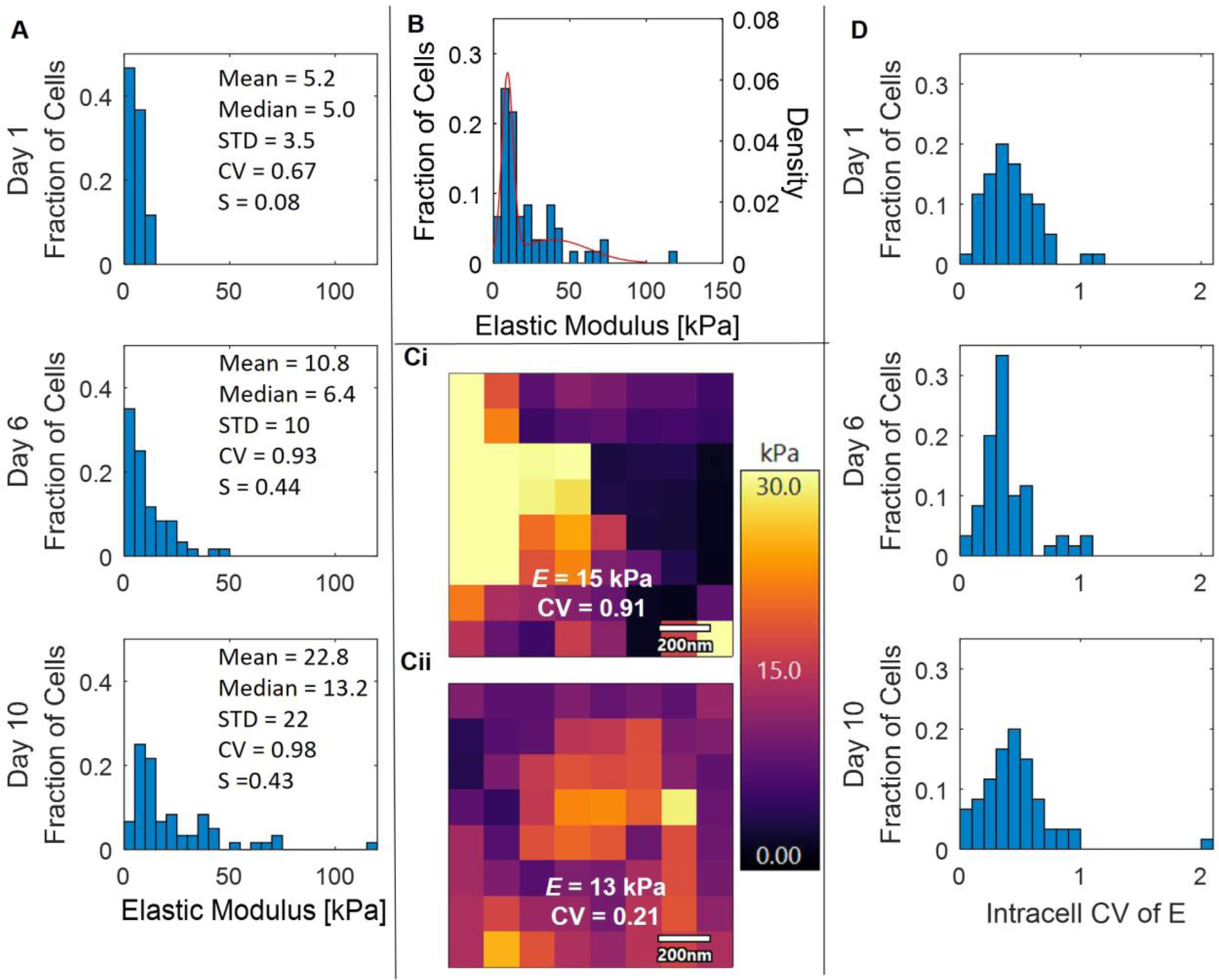
(A) Histograms of individual E from PHCs grown in microgels on days 1, 6, and 10. (B) Histogram of E for cells grown in microgels on day 10 plotted with the probability distribution function of the Gaussian mixture model with k =2. (C) Representative force maps of PHCs cultured in microgels on day 10 with similar E (15 and 13 kPa) but different coefficients of variation (CV = 0.91 and 0.21). Every other point shown was removed for statistical analysis to minimize bias from neighboring indentations. Scale bar is 200 nm. (D) Histograms of the intracell E CV from each cell’s force map on days 1, 6, and 10. Cells were cultured in microgels.
Therefore, a Gaussian mixture model (GMM) was applied to understand if the skew in E on day 10 was due to a subpopulation of cells. After applying the GMM for up to three populations (k =1 AIC = 536, k=2 AIC = 485, k=3 AIC = 490), the model found that two distinct distributions provided the best fit. The first population had a mean of 9.1 ± 3.6 kPa, accounting for 54.0% of the population, while the second population had a mean of 39 ± 7.9 kPa, accounting for 46.0% of the population (Figure 5B). These results indicate that one subpopulation, accounting for 54.0% of the population, exhibited only a slight increase in stiffness compared to day 1, with a difference in E of approximately 4 kPa. The second subpopulation, representing 46.0% of the population, became significantly stiffer with an E approximately 35 kPa higher than on day 1. The second subpopulation falls within the range of native PCM stiffness (25–1000 kPa, found through AFM, micropipette aspiration, and other methods) [1,15–17,24–27].
In addition to studying the variations in PCM outcomes between cells, AFM enables the study of intracellular E differences within individual cells, or the heterogeneity in elastic moduli at different points on individual cells. During AFM force curve acquisition, 8 × 8 force maps covering a 1 µm2 area were acquired for each cell grown in microgels and every other data point was removed from analysis to minimize bias from neighboring indentations. The force maps were further analyzed, and the coefficient of variation (CV) was quantified for each cell, revealing whether the produced matrix exhibited homogeneous or heterogeneous E over the 1 × 1 μm probed region. For example, representative force maps for two cells grown in microgels on day 10 are displayed in Figure 5Ci–ii with similar E values(15 and 13 kPa). However, the cells displayed significantly different CVs, indicating highly heterogeneous matrix in the probed regions of one cell (Figure 5Ci, CV = 0.91), while the other cell exhibited a more homogenous matrix (Figure 5Cii, CV = 0.21). Histograms of the intracellular heterogeneity for N = 60 cells on days 1, 6, and 10 exhibited a wide range of CV values, ranging from 0.06 to 2.0 (Figure 5D). Interestingly, the distributions of intracellular E CV did not change over time for the PHCs cultured in microgels (p > 0.16). To determine if the CV correlated with E, linear regressions were performed on cells grown in microgels on days 1, 6, and 10. The linear regression analysis revealed that on day 1, there was no relationship between the average E and the CV of the cell (p = 0.84) (SI Figure 3 Day 1). However, on days 6 and 10, a positive correlation was found between E and the CV of the cell (p = 0.011 and 0.000011), indicating that cells producing a higher E were more likely to exhibit heterogeneous matrix compared to cells that did not produce a stiff matrix (SI Figure 3, Day 6 and Day 10).
Discussion:
In this work, we introduce a novel methodology for culturing single chondrocytes isolated from human knees within alginate microgels created using drop-based microfluidics. This approach allows for the formation of PCM around each cell over a 10 day incubation period. After dissolving the alginate surrounding the cells, the biochemical and mechanical properties of the cells were analyzed using autofluorescence, immunofluorescence, and AFM.
To monitor and quantify matrix formation in microgels, we employed autofluorescence imaging. We observed a significant increase in the autofluorescence signal of PHCs cultured in microgels, compared to PHCs cultured in monolayer with negligible change in signal. This difference in autofluorescence corresponds to the expected levels of collagen production, as PHCs produce less collagen I, II, IV, and VI when cultured in monolayer and have potential to de-differentiate compared to when cultured in 3-D. Measuring cellular autofluorescence allows for non-invasive monitoring of matrix production over time without disturbing cells, unlike other assays such as immunofluorescence staining. Additionally, this method enables quantitative comparisons between different cell types. We discovered that the autofluorescence signal of PHCs increases nearly 5 times faster than SW1353 cells, providing further evidence to support that even osteoarthritic PHCs from patients undergoing knee replacements produce significantly more collagen than SW1353 cells. PCM from patients with osteoarthritis have been shown to have 30–90% lower elastic moduli compared to normal cartilage [36]. Further studies with PHCs from younger patients would likely demonstrate higher elastic moduli.
We also investigated the mechanical properties of chondrons grown in microgels over a 10 day period. We observed a rapid increase in E of cells cultured in microgels, reaching nearly 10 times higher values compared to day 1, and approaching the range of native PCM stiffness seen in vivo (25 – 1000 kPa) [1,15–17,24–27]. Interestingly, the increase in E was evident as early as 24h in culture. In contrast, PHCs cultured in monolayers exhibited negligible change in E over the same 10 day period. These findings suggest that microgels provide an excellent growth environment for single-cell PHCs, allowing them to form PCM that closely resembles native PCM. Notably, the distribution of E from PHCs grown in microgels on day 10 was heterogeneous, indicating the presence of subpopulations with varying PCM production capacities. By applying a Gaussian mixture model to the E data, we identified two distinct subpopulations with mean values centered at 9.1 ± 3.6 and 39 ± 7.9 kPa, accounting for approximately half of the cells each. This suggests that half of the PHCs contributed significantly more to matrix formation on our study. One possible explanation is that the chondrocytes in this study were extracted from the entirety of full-thickness knee cartilage, which has diverse chondrocyte populations with chondrocytes in the deep zone experiencing higher compressive forces compared to chondrocytes in the superficial zone [1,11,19,34,35]. The subpopulations observed in our experiments could be due to the heterogeneity of cells within the cartilage, but additional studies targeting particular locations within the cartilage would be needed to confirm this. Future work comparing isolated populations from different cartilage zones (e.g. superficial versus deep) cultured in microgels could provide insights into the ability to isolate more advantageous cells for cartilage repair. Additionally, future experiments comparing the elastic moduli of PHCs cultured in bulk alginate gels compared to alginate microgel culture could provide interesting insights into PHC PCM formation.
Our results demonstrate that microgel culture offers a superior alternative to traditional methods for in vitro growth and expansion of chondrocytes that can be applied to innovating regenerative medicine techniques. The microgels allow chondrocytes to rapidly form a mature PCM in as short as 10 days. In existing techniques such as MACI, cells are cultured for 3–6 weeks, but by incorporating the microgel method for the final two weeks of culture, cells could potentially be implanted with a more developed PCM without altering the overall culturing time. Moreover, techniques like MACI rely on cell adherence to a membrane scaffold to prevent leakage and improve cell viability. In contrast, microgels are easily injectable and would reduce the reliance on cell adherence to membranes, as the microgels are less likely to leak due to their size [2,4,21,23]. Additionally, by utilizing non-invasive autofluorescence measurements, chondrocytes that produce the most matrix components can potentially be isolated and expanded, accelerating recovery times in regenerative cartilage procedures. Overall, our study demonstrates that microgels provide an excellent culture platform for the growth and isolation of PHCs, enabling PCM formation that is mechanically similar to native PCM. The microgel culture platform presented here has the potential to revolutionize cartilage regeneration procedures by enhancing PCM growth around individual chondrocytes.
Methods:
Primary human chondrocyte harvest and cell culture
Primary Human Chondrocytes (PHCs) were obtained from female Stage-IV osteoarthritis patients aged 63–73 undergoing total joint replacement under IRB approval using established methods (N = 3/experiment) [37,38]. Isolated chondrocytes were seeded onto 60 × 15 mm tissue culture dishes and maintained in Dulbecco’s Modified Eagle’s medium (DMEM) (Gibco, Waltham, MA, USA) supplemented with Fetal Bovine Serum (FBS) (10% v/v) (Bio-Techne, Minneapolis, MN, USA), penicillin (10,000 I.U./mL), and streptomycin (10,000 μg/mL) in 5% CO2 at 37 °C. PHCs were passaged at 90% confluence and used in all experiments between passages 1–4. For autofluorescence imaging and AFM experiments, cells were cultured with DMEM supplemented with FBS (10% v/v), penicillin (10,000 I.U./mL), streptomycin (10,000 μg/mL), and 50 μg/mL sodium ascorbate in 5% CO2 at 37 °C.
SW1353 cell culture
SW1353 chondrocytes, a cell line initiated from a human chondrosarcoma and obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA), were cultured using standard tissue culture techniques [14]. SW1353 cells were seeded onto 60 × 15 mm tissue culture dishes and were maintained in DMEM supplemented with FBS (10% v/v), penicillin (10,000 I.U./mL), and streptomycin (10,000 μg/mL) in 5% CO2 at 37 °C. SW1353 cells were passaged at 90% confluence and used in all experiments between passages 5–20. For autofluorescence imaging experiments, cells were cultured with DMEM supplemented with FBS (10% v/v), penicillin (10,000 I.U./mL), streptomycin (10,000 μg/mL), and 50 μg/mL sodium ascorbate in 5% CO2 at 37 °C.
PDMS microfluidic device fabrication
Negative master molds for the microfluidic devices were prepared using standard photolithography techniques [14]. Negative master molds were made with Nano SU-8-100 photoresist (Microchem, Round Rock, TX, USA) on 3” silicon wafers (University Wafer Inc., Boston, MA, USA University Wafer ID: 447). The microgel drop maker was fabricated to be 50 µm in height. Devices were treated with (tridecafluoro-1,1,2,2-tetrahydrooctyl) trichlorosilane (1 v/v%) (Gelest) in NOVEC HFE 7500 fluorinated oil (3M, Saint Paul, MN, USA) and left for solvent evaporation overnight at 55 °C.
AFM well-trap fabrication
The well-traps were fabricated with Nano SU-8-100 photoresist (Microchem, Round Rock, TX, USA) on 3” silicon wafers (University Wafer Inc., Boston, MA, USA University Wafer ID: 447) using standard photolithography techniques [14]. To fit on the AFM, wafers were cut into ~ 1 × 1 cm pieces using a diamond-tipped scribe. Wells were designed as 30 × 30 × 10 μm rectangular prisms with heights of 10 µm. Developed well-traps were rinsed with phosphate buffered saline (PBS) prior to use.
Preparation of microfluidic microgel reagents
To make the continuous/oil phase, the surfactant Krytox 157 FSH (4% w/w) (Miller-Stephenson, Danbury, CT, USA) was added to NOVEC HFE 7500 fluorinated oil (3M, Saint Paul, MN, USA) [14]. The solution was filtered through a 0.2 µm hydrophobic polytetrafluoroethylene (PTFE) syringe filter to remove dust and other contaminants.
Two dispersed phase alginate precursor solutions were made as previously described [14,39]. The first (Alginate - precursor A) by mixing CaCl2 (80 mM), Na2EDTA (Ethylenediaminetetraacetic acid) (80 mM) (Fisher Chemical, Hampton, NH, USA), and 3-(Morpholin-4-yl) propane-1-sulfonic acid (MOPS) (40 mM) (VWR, Randor, PA, USA) with ultra-pure water (18.2 MΩ●cm). The second (Alginate - precursor B) was made by mixing Zn (CH3CO2)2 (80 mM), EDDA (Ethylenediaminediacetic acid) (80 mM) (TCI Chemicals, Tokyo, Japan), and MOPS (40 mM) with ultra-pure water (50 mL) (18.2 MΩ●cm). The pH of both solutions was adjusted to 7.2 using NaOH (10 M). Sodium alginate (1.5% w/w) (Sigma, St. Louis, MO, USA, 9005-38-3) was mixed with each solution, then each was filtered through a 0.2 µm PES syringe filter (Argos Technologies).
Cell encapsulation and culture in microgels
For cell encapsulation and culture in microgels, cells were prepared by removing them from monolayers using Trypsin-EDTA and then suspending them in DMEM supplemented with FBS (10% v/v), penicillin (10,000 I.U./mL), and streptomycin (10,000 μg/mL))to inactivate the trypsin [14]. Cells were centrifuged at 500 × g for 5 min and washed 3× with PBS. The cells were then suspended in alginate-precursor A at 4 × 106 cells/mL and loaded into 1 mL syringes. Cells were encapsulated in 50 µm diameter alginate microgels following previous protocols [14,39]. The continuous flow rate used was QOil = 2000 µL/h and the dispersed phase flow rate used was QAlgA = QAlgB = 250 µL/h. After encapsulation and gelation, the microgels were washed with equal volumes of 1H,1H,2H,2H-Perfluoro-octanal (PFO) (647-42-7, VWR) in HFE7500 (20% v/v) and PBS to drops. The microgels were collected from the PBS and suspended in DMEM supplemented with FBS (10% v/v), penicillin (10,000 I.U./mL), streptomycin (10,000 μg/mL)), and 50 µg/mL sodium ascorbate. Microgels were added to 6 well plates at 1 × 106 cells/well and the volume was adjusted to 2 mL with media. Sodium ascorbate was added every 3 days to supplement the media. Cells were incubated in 5% CO2 at 37 °C.
Autofluorescence imaging and analysis
Cells were imaged using an epi-fluorescence microscope (Nikon Ti2 Eclipse) with a 20× objective over 10 days to capture bright field and autofluorescence (ex. 375nm/ em. 415nm). Cells were imaged on days 0, 1, 4, 7, and 10. To image cells in microgels, 5 µL of microgels and media were pipetted onto a PTFE-printed microscope slide. To image cells in monolayers, cells were grown on 35 mm glass-bottom dishes (MatTek). Approximately 20 cells from each donor (N = 3) were imaged per condition for each time point. The background intensity was subtracted from each image using Fiji ImageJ and the fluorescence intensity of each cell was measured using the Fiji ImageJ distribution of TrackMate [40,41]. Autofluorescence curves were analyzed using a multiple linear regression model in Matlab.
Alginate dissolution and chondron isolation
To isolate chondrons from the alginate microgels, microgels were collected from the media via centrifugation for 1 min at 200 × g and vortexed for 2 min with sodium citrate (50mM) and EDTA (80mM) in PBS. The chondrons were then collected via centrifugation for 5 min at 500 × g and washed 3× in PBS.
Immunofluorescence staining and imaging
After days 1, 6, and 10 in culture, PHCs grown in microgels were collected in microcentrifuge tubes and washed with PBS. The sample was split with half the cells remaining in microgels and half the cells being removed from microgels by dissolving the alginate. Cells were fixed with paraformaldehyde (PFA) (4% v/v) in PBS for 15 min at room temperature and washed 3× with PBS. Cells were blocked Bovine Serum Albumin (BSA) (2% w/v) in PBS at room temperature for 1 h and then washed 3× with PBS. Cells were permeabilized with Triton-X (0.1% w/v) and BSA (0.125% w/v) in PBS and then washed 3× with BSA (0.125% w/v) in PBS. The microgels were then incubated with the primary antibody to collagen VI (5 µg/mL) (Rabbit Polyclonal Anti-collagen VI antibody, ab6588 from Abcam, Boston, MA, USA), Triton-X (0.1% w/v), and BSA (0.125% w/v) in PBS, incubated at room temperature overnight and then washed 3× with BSA (0.125% w/v) in PBS. A 1mL mixture of secondary antibody (Donkey anti-Rabbit IgG H&L Alexa FluorR488) (5µg/mL) (Abcam, Boston, MA, USA), Triton-X (0.1% w/v), and BSA (0.125% w/v) in PBS, and Hoechst 33342 Solution (20 µg/mL) (ThermoFisher Scientific, USA)/PBS were added for 1 h at 37 °C and then washed 3× with 0.125% BSA-PBS.
Cells and microgels were loaded onto glass slides and imaged on a custom-built Nikon Ti2 epi-fluorescent microscope. Cells were imaged in Phase Contrast/DAPI/GFP. A total of 3–10 cells were imaged per condition.
Force curve acquisition and analysis
Cells grown in microgels were prepared for AFM by first collecting the microgels on days 1, 6, and 10, and dissolving the alginate microgels. Cells were then washed with PBS and placed into the well-traps by pipetting the cell suspension onto the trap and centrifuging at 2000 × g for 1 min.
Cells grown in monolayer were prepared for AFM by collecting the cells using a cell scraper and sodium-citrate (50mM)/EDTA (80mM) in PBS. Cells were then washed with PBS and placed into the well-traps by pipetting the cell suspension onto the traps and centrifuging at 2000 × g for 1 min.
The cell-laden traps were placed in an AFM (Cypher S, Asylum Research) outfitted with a droplet holding stage and qp-BioAC AFM probe (Nanosensors). A cantilever with 0.1 N/m spring constant was selected for force curve collection and calibrated using the Sader method [42]. Force curves were performed using extension and retraction speeds of 1 µm/s and a maximum force of 2 nN was reached. A Hertzian contact model was applied to the force-distance data to calculate the average quasi-static contact modulus of the substrate volume actuated under the probe tip (referred to as elastic modulus in this work). The Hertzian model applied used a tip conical tip geometry with a half angle of 36 degrees [32]. The contact point was identified when the applied force was larger than the noise floor of the cantilever with no contact and was identified by the AFM software package (Asylum Research Igor PRO 6.3B). The 2nN max force was selected to minimize indentation depth and confine the actuated volume, consistent with AFM cell mechanics literature [43]. Accordingly, indentation depths did not exceed 10% of the cell diameter. Cells were located in brightfield, and the tip was placed approximately above the middle of the cell (SI Figure 4). Cell slippage was not observed. 8 × 8 force maps were acquired for each cell, probing over a 1 × 1 µm area. Every other data point was removed from analysis to minimize bias from neighboring indentations. Image surfaces were smooth at tip-relevant length scales and no topography-related artifacts were noted. Surface curvature was an order of magnitude greater than the indentation depth and periodic, nanometer scale features were not present in the topographic scans. Twenty cells were imaged for each of cells grown in microgels and monolayers on days 1, 6, and 10 for each donor (N = 3). Force curves were analyzed using a Hertz equation to calculate E of each cell The Hertzian model applied used a tip conical tip geometry with a half angle of 36 degrees [32].
Statistical differences in E between cells grown in monolayer and microgels (Figure 4D) were assessed using a linear mixed effects model. Statistical differences between microgels from each donor were tested with two-sample T-tests. Differences in E of microgels on day 10 were examined using a Gaussian mixture model. Differences in CV of microgels were assessed using through two-sample T-tests and trends between E and CV were tested using linear regressions. All statistics were performed in Matlab (Mathworks, USA).
Representative force curves and maps were generated in the Igor Pro software package and Matlab.
Statistical Analysis:
For auto-fluoresence experiments, approximately 20 cells from each donor (N = 3) were imaged per condition for each time point. The background intensity was subtracted from each image using Fiji ImageJ and the fluorescence intensity of each cell was measured using the Fiji ImageJ distribution of TrackMate [40,41]. Autofluorescence curves were analyzed using a multiple linear regression model in Matlab.
For AFM experiments, where force maps were acquired, every other data point was removed from analysis to minimize bias from neighboring indentations. Twenty cells were imaged for each of cells grown in microgels and monolayers on days 1, 6, and 10 for each donor (N = 3). Force curves were analyzed using a Hertz equation to calculate E of each cell The Hertzian model applied used a tip conical tip geometry with a half angle of 36 degrees [32].
Statistical differences in E between cells grown in monolayer and microgels (Figure 4D) were assessed using a linear mixed effects model. Statistical differences between microgels from each donor were tested with two-sample T-tests. Differences in E of microgels on day 10 were examined using a Gaussian mixture model. Differences in CV of microgels were assessed using through two-sample T-tests and trends between E and CV were tested using linear regressions. All statistics were performed in Matlab (Mathworks, USA).
Representative force curves and maps were generated in the Igor Pro software package and Matlab.
Supplementary Material
Acknowledgments
This work was performed in part at the Montana Nanotechnology Facility, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (Grant# ECCS-2025391).
Funding
This research was funded by the National Institutes of Health [R01AR073964 to RKJ] and National Science Foundation [CAREER 1753352 to CBC, 1554708 to RKJ]. The funding sources had no role in the design or execution of the study.
Footnotes
Informed Consent Statement
The Montana State University Institutional Review Board (IRB) defined these studies as exempt (exemption 4B) on 13 October 2011. Patient consent was provided using an IRB approved consent form.
Conflicts of Interest
June owns stock in Beartooth Biotech, which was not involved in this study.
Data Availability
All raw data is available upon request from the corresponding author.
Citations:
- 1.Muir H The chondrocyte, architect of cartilage. Biomechanics, structure, function and molecular biology of cartilage matrix macromolecules. Bioessays 1995;17: 1039–1048. [DOI] [PubMed] [Google Scholar]
- 2.Huang BJ, Hu JC, Athanasiou KA. Cell-based tissue engineering strategies used in the clinical repair of articular cartilage. Biomaterials 2016;98: 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Safiri S, Kolahi A-A, Smith E, Hill C, Bettampadi D, Mansournia MA, et al. Global, regional and national burden of osteoarthritis 1990–2017: a systematic analysis of the Global Burden of Disease Study 2017. Ann Rheum Dis 2020;79: 819–828. [DOI] [PubMed] [Google Scholar]
- 4.Jacobi M, Villa V, Magnussen RA, Neyret P. MACI - a new era? Sports Med Arthrosc Rehabil Ther Technol 2011;3: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amr M, Mallah A, Yasmeen S, Van Wie B, Gozen A, Mendenhall J, et al. From Chondrocytes to Chondrons, Maintenance of Phenotype and Matrix Production in a Composite 3D Hydrogel Scaffold. Gels 2022;8. doi: 10.3390/gels8020090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minas T, Peterson L. Advanced techniques in autologous chondrocyte transplantation. Clin Sports Med 1999;18: 13–44, v–vi. [DOI] [PubMed] [Google Scholar]
- 7.Batty L, Dance S, Bajaj S, Cole BJ. Autologous chondrocyte implantation: an overview of technique and outcomes. ANZ J Surg 2011;81: 18–25. [DOI] [PubMed] [Google Scholar]
- 8.Benya PD, Padilla SR, Nimni ME. Independent regulation of collagen types by chondrocytes during the loss of differentiated function in culture. Cell 1978;15: 1313–1321. [DOI] [PubMed] [Google Scholar]
- 9.Mayne R, Vail MS, Mayne PM, Miller EJ. Changes in type of collagen synthesized as clones of chick chondrocytes grow and eventually lose division capacity. Proc Natl Acad Sci U S A 1976;73: 1674–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.von der Mark K, Gauss V, von der Mark H, Müller P. Relationship between cell shape and type of collagen synthesised as chondrocytes lose their cartilage phenotype in culture. Nature 1977;267: 531–532. [DOI] [PubMed] [Google Scholar]
- 11.Darling EM, Athanasiou KA. Rapid phenotypic changes in passaged articular chondrocyte subpopulations. J Orthop Res 2005;23: 425–432. [DOI] [PubMed] [Google Scholar]
- 12.Costa E, González-García C, Gómez Ribelles JL, Salmerón-Sánchez M. Maintenance of chondrocyte phenotype during expansion on PLLA microtopographies. J Tissue Eng 2018;9: 2041731418789829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh S, Scott AK, Seelbinder B, Barthold JE, Martin BMS, Kaonis S, et al. Dedifferentiation alters chondrocyte nuclear mechanics during in vitro culture and expansion. Biophys J 2022;121: 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fredrikson JP, Brahmachary PP, Erdoğan AE, Archambault ZK, Wilking JN, June RK, et al. Metabolomic Profiling and Mechanotransduction of Single Chondrocytes Encapsulated in Alginate Microgels. Cells 2022;11. doi: 10.3390/cells11050900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexopoulos LG, Haider MA, Vail TP, Guilak F. Alterations in the mechanical properties of the human chondrocyte pericellular matrix with osteoarthritis. J Biomech Eng 2003;125: 323–333. [DOI] [PubMed] [Google Scholar]
- 16.Wilusz RE, Sanchez-Adams J, Guilak F. The structure and function of the pericellular matrix of articular cartilage. Matrix Biol 2014;39: 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chondrons Zhang Z. and the pericellular matrix of chondrocytes. Tissue Eng Part B Rev 2015;21: 267–277. [DOI] [PubMed] [Google Scholar]
- 18.Zelenski NA, Leddy HA, Sanchez-Adams J, Zhang J, Bonaldo P, Liedtke W, et al. Type VI Collagen Regulates Pericellular Matrix Properties, Chondrocyte Swelling, and Mechanotransduction in Mouse Articular Cartilage. Arthritis Rheumatol 2015;67: 1286–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeon JE, Schrobback K, Meinert C, Sramek V, Hutmacher DW, Klein TJ. Effect of preculture and loading on expression of matrix molecules, matrix metalloproteinases, and cytokines by expanded osteoarthritic chondrocytes. Arthritis Rheum 2013;65: 2356–2367. [DOI] [PubMed] [Google Scholar]
- 20.Ng L, Hung H-H, Sprunt A, Chubinskaya S, Ortiz C, Grodzinsky A. Nanomechanical properties of individual chondrocytes and their developing growth factor-stimulated pericellular matrix. J Biomech 2007;40: 1011–1023. [DOI] [PubMed] [Google Scholar]
- 21.Caldwell AS, Aguado BA, Anseth KS. Designing Microgels for Cell Culture and Controlled Assembly of Tissue Microenvironments. Adv Funct Mater 2020;30. doi: 10.1002/adfm.201907670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caliari SR, Burdick JA. A practical guide to hydrogels for cell culture. Nat Methods 2016;13: 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Y, Zhu H, Denduluri A, Ou Y, Erkamp NA, Qi R, et al. Recent Advances in Microgels: From Biomolecules to Functionality. Small 2022;18: e2200180. [DOI] [PubMed] [Google Scholar]
- 24.Danalache M, Kleinert R, Schneider J, Erler AL, Schwitalle M, Riester R, et al. Changes in stiffness and biochemical composition of the pericellular matrix as a function of spatial chondrocyte organisation in osteoarthritic cartilage. Osteoarthritis Cartilage 2019;27: 823–832. [DOI] [PubMed] [Google Scholar]
- 25.Arnold KM, Sicard D, Tschumperlin DJ, Westendorf JJ. Atomic Force Microscopy Micro-Indentation Methods for Determining the Elastic Modulus of Murine Articular Cartilage. Sensors 2023;23. doi: 10.3390/s23041835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chery DR, Han B, Li Q, Zhou Y, Heo S-J, Kwok B, et al. Early changes in cartilage pericellular matrix micromechanobiology portend the onset of post-traumatic osteoarthritis. Acta Biomater 2020;111: 267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chery DR, Han B, Zhou Y, Wang C, Adams SM, Chandrasekaran P, et al. Decorin regulates cartilage pericellular matrix micromechanobiology. Matrix Biol 2021;96: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rigacci L, Alterini R, Bernabei PA, Ferrini PR, Agati G, Fusi F, et al. Multispectral imaging autofluorescence microscopy for the analysis of lymph-node tissues. Photochem Photobiol 2007;71: 737–742. [DOI] [PubMed] [Google Scholar]
- 29.Hoell T, Huschak G, Beier A, Hüttmann G, Minkus Y, Holzhausen HJ, et al. Auto fluorescence of intervertebral disc tissue: a new diagnostic tool. Eur Spine J 2006;15 Suppl 3: S345–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andersson-Engels S, Johansson J, Svanberg K, Svanberg S. Fluorescence imaging and point measurements of tissue: applications to the demarcation of malignant tumors and atherosclerotic lesions from normal tissue. Photochem Photobiol 1991;53: 807–814. [DOI] [PubMed] [Google Scholar]
- 31.Gebauer M, Saas J, Sohler F, Haag J, Söder S, Pieper M, et al. Comparison of the chondrosarcoma cell line SW1353 with primary human adult articular chondrocytes with regard to their gene expression profile and reactivity to IL-1beta. Osteoarthritis Cartilage 2005;13: 697–708. [DOI] [PubMed] [Google Scholar]
- 32.Guerrero CR, Garcia PD, Garcia R. Subsurface Imaging of Cell Organelles by Force Microscopy. ACS Nano 2019;13: 9629–9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trivedi MV, Laurence JS, Siahaan TJ. The Role of Thiols and Disulfides in Protein Chemical and Physical Stability. Current Protein & Peptide Science [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee DA, Noguchi T, Knight MM, O’Donnell L, Bentley G, Bader DL. Response of chondrocyte subpopulations cultured within unloaded and loaded agarose. J Orthop Res 1998;16: 726–733. [DOI] [PubMed] [Google Scholar]
- 35.McLeod MA, Wilusz RE, Guilak F. Depth-dependent anisotropy of the micromechanical properties of the extracellular and pericellular matrices of articular cartilage evaluated via atomic force microscopy. J Biomech 2013;46: 586–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilusz RE, Zauscher S, Guilak F. Micromechanical mapping of early osteoarthritic changes in the pericellular matrix of human articular cartilage. Osteoarthritis Cartilage 2013;21: 1895–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zignego DL, Hilmer JK, June RK. Mechanotransduction in primary human osteoarthritic chondrocytes is mediated by metabolism of energy, lipids, and amino acids. J Biomech 2015;48: 4253–4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zignego DL, Hilmer JK, Bothner B, Schell WJ, June RK. Primary human chondrocytes respond to compression with phosphoproteomic signatures that include microtubule activation. J Biomech 2019;97: 109367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Håti AG, Bassett DC, Ribe JM, Sikorski P, Weitz DA, Stokke BT. Versatile, cell and chip friendly method to gel alginate in microfluidic devices. Lab Chip 2016;16: 3718–3727. [DOI] [PubMed] [Google Scholar]
- 40.Tinevez J-Y, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, et al. TrackMate: An open and extensible platform for single-particle tracking. Methods 2017;115: 80–90. [DOI] [PubMed] [Google Scholar]
- 41.Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, et al. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 2017;18: 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sader JE, Chon JWM, Mulvaney P. Calibration of rectangular atomic force microscope cantilevers. Rev Sci Instrum 1999;70: 3967–3969. [Google Scholar]
- 43.Gavara N A beginner’s guide to atomic force microscopy probing for cell mechanics. Microsc Res Tech 2017;80: 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data is available upon request from the corresponding author.