

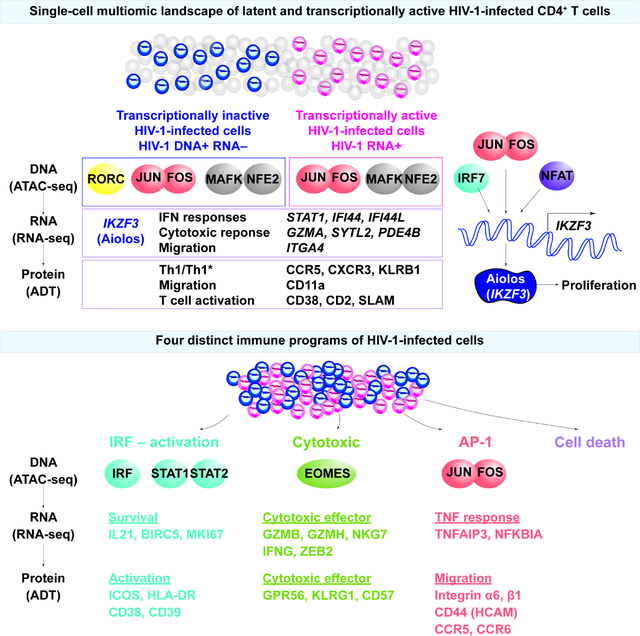
Summary
Understanding how HIV-1-infected cells proliferate and persist is key to HIV-1 eradication, but the heterogeneity and rarity of HIV-1-infected cells hampers mechanistic interrogations. Here, we used single-cell DOGMA-seq to simultaneously capture transcription factor accessibility, transcriptome, surface proteins, HIV-1 DNA, and HIV-1 RNA in memory CD4+ T cells from six people living with HIV-1 during viremia and after suppressive antiretroviral therapy. We identified increased transcription factor accessibility in latent HIV-1-infected cells (RORC) and transcriptionally active HIV-1-infected cells (IRF and AP-1). A proliferation program (IKZF3, IL21, BIRC5, and MKI67 co-expression) promoted the survival of transcriptionally active HIV-1-infected cells. Both latent and transcriptionally active HIV-1-infected cells upregulated IKZF3 (Aiolos), which correlated with proliferation of these cells. Distinct epigenetic programs drove the heterogeneous cellular states of HIV-1-infected cells: IRF:activation, Eomes:cytotoxic effector differentiation, AP-1:migration, and cell death. Our study revealed the single-cell epigenetic, transcriptional, and protein states of latent and transcriptionally active HIV-1-infected cells.
Keywords: Acute viral infection, HIV latent reservoir, single-cell ATAC-seq, single-cell RNA-seq, memory CD4+ T cells, T cell differentiation, IKZF3 (Aiolos), transcription factor, HIV persistence, HIV cure
eTOC blurb
HIV-1 latently infected cells are indistinguishable from uninfected cells, creating a barrier for cure. Using DOGMA-seq, Wei et al. identified single-cell epigenetic, transcriptional, and surface protein states of latent and transcriptionally active HIV-1-infected cells. The heterogeneous HIV-1-infected cells are driven by interferon responses, cytotoxic T cell differentiation, AP-1-driven TNF responses, and cell death. IKZF3 promotes the proliferation of HIV-1-infected cells.
Graphical Abstract
INTRODUCTION
Despite suppressive antiretroviral therapy (ART), HIV-1-infected cells persist lifelong1–3. During viremia, most HIV-1-infected cells undergo productive infection with active HIV-1 gene expression. Presumably, transcriptionally active HIV-1-infected cells should die of viral cytopathic effects or immune clearance. However, some transcriptionally active HIV-1-infected cells can survive, proliferate, and persist4. Understanding how HIV-1-infected cells proliferate and persist is key to HIV-1 eradication.
It is believed that CD4+ T activation cells support HIV-1 reactivation while resting CD4+ T cells maintain HIV-1 latency because of low expression or lack of active forms of transcription factors in the nucleus. However, CD4+ T cell differentiation, polarization, migration, and exhaustion states are heterogeneous and complex, far beyond the simplified dichotomous classification of activated versus resting states. Identification of diverse signatures for HIV-1-infected cells5–13, such as different markers for activation14, exhaustion15, migration7,16, T helper 1 (Th1) polarization12,17, cytotoxic T cell polarization11,13, or T follicular helper (Tfh) differentiation12,16, indicates the profound heterogeneity of HIV-1-infected cells and likely reflects the heterogeneous cellular states of CD4+ T cells. Understanding the heterogeneity of HIV-1-infected cells requires comprehensive genome-wide profiling to identify the epigenetic and transcriptional programs that govern CD4+ T cell differentiation and their impact on HIV-1 persistence.
The activation, proliferation, and differentiation of memory T cells is orchestrated by key transcription factors. Acute viral infection triggers type I interferon (IFN) response through IFN regulatory transcription factors (IRFs) that shape T cell differentiation18,19. Transcription factors, such as activator protein 1 (AP-1: JUN, FOS, and BATF)5,20,21, cap’n’collar (CNC: BACH1, BACH2, NFE2) family transcription factors22,23, and Maf24 orchestrate the development of adaptive immune responses. Master transcription factors guide T cell polarization and commitment, such as Tbet (TBX21) and Eomesdermin (EOMES) for Th1 and cytotoxic effector differentiation25 and RORγt (RORC) for T helper 17 (Th17) differentiation26. Understanding epigenetic and transcriptional programs that govern cell states of HIV-1-infected cells will reveal new mechanisms of HIV-1 persistence.
The development of HIV-1 cure strategies relies on identifying cellular markers that can distinguish HIV-1-infected cells from uninfected cells. In particular, latently infected cells do not express HIV-1 viral proteins and cannot be distinguished from uninfected cells. Technology advancement allows capture of rare HIV-1-infected cells (<0.1% in the CD4+ T cells in peripheral blood) for single-cell profiling5–13. Using HIV-1 RNA expression as a surrogate for transcriptionally active HIV-1-infected cells, single-cell transcriptional profiling of transcriptionally active HIV-1 RNA+ cells revealed HIV-1 persistence in cytotoxic CD4+ T cells11,13. However, single-cell transcriptome profiling of transcriptionally inactive HIV-1 RNA− infected cells remains a major challenge: no study was able to capture the single-cell transcriptional landscape of transcriptionally inactive infected cells for genome-wide examination.
Here, we examined the epigenetic, transcriptional, and surface protein expression transcriptionally inactive (HIV-1 DNA+ RNA−) and in transcriptionally active (HIV-1 RNA+) HIV-1-infected memory CD4+ T cells from six people living with HIV-1 during viremia and after suppressive ART. Using DOGMA-seq27, we identified chromatin accessible transcription factor binding regions by assay for transposase-accessible chromatin with sequencing (ATAC-seq), transcriptional programs by RNA-seq, and cell surface protein expression by antibody-derived tag (ADT) sequencing for 156 surface proteins within the same single cells. We identified transcription factor Aiolos (encoded by IKZF3) may promote proliferation of HIV-1-infected cells and found that the heterogeneous HIV-1 were driven by interferon responses, cytotoxic T cell differentiation, and AP-1-driven TNF responses. This study represents the first single-cell high-dimensional mapping of the epigenetic, transcriptional, and surface protein expression landscapes of transcriptionally inactive and active HIV-1-infected cells.
RESULTS
Single-cell DOGMA-seq captured epigenetic, transcriptional, and surface protein states of memory CD4+ T cells during HIV-1 infection
Paired memory CD4+ T cells from six people living with HIV-1 in the Sabes cohort28,29 recruited in a previous study13 during viremia [average 31 days from estimated date of detectable infection] and after one year of suppressive ART were profiled by DOGMA-seq27. Memory CD4+ T cells from four age, sex, and ethnicity-matched uninfected individuals were new to this study and served as controls (Table S1). Briefly, we used DOGMA-seq to simultaneously capture chromatin accessibility landscape and HIV-1 DNA (from ATAC-seq), cellular transcriptome and HIV-1 RNA (from RNA-seq), and 156 surface proteins (ADT sequencing) in the same single cells (Figure 1A). After removing low quality cells, we captured 93,209 single cells (25,778 in viremia, 56,771 in viral suppression, and 10,660 in uninfected controls) (Table S2). Batch effects were corrected by Harmony (for RNA-seq), reciprocal Latent Semantic Indexing (rLSI) (for ATAC-seq), and reciprocal Principal Component Analysis (rPCA) (for ADT). Cells formed distinct clusters by epigenome (ATAC-seq) (Figure S1A), transcriptome (RNA-seq) (Figure S1B), and surface protein (ADT) (Figure S1C) profiles. A weighted combination of ATAC-seq, RNA-seq, and ADT profiles allowed for an integrated view of the epigenetic regulators, transcriptional program, and surface protein expression within the same single cells on a harmonized Weighted Nearest Neighbor (WNN) Uniform Manifold Approximation and Projection for Dimension Reduction (UMAP)30 plot (Figure 1A, Figure S1D–S1E). No apparent batch effect was present after integration (Figure S1F). While ATAC-seq identified cell-type specific transcription factor accessibility (Figure S1A, S1G), RNA-seq identified cell-type specific transcriptome (Figure S1B, S1H), and ADT identified surface protein expression profiles (Figure S1C, S1I), integrating all three modalities more faithfully depicted the heterogeneous T cell differentiation states (Figure 1A). We identified 15 memory CD4+ T cell subsets (clusters) (Figure 1B, Figure S1G–I, Table S2). Per cell, we identified a median of 785 genes, 1,553 RNA unique molecular identifiers (UMIs), 62 surface proteins, 700 protein UMIs, 3,143 unique ATAC fragments, and 4,198 ATAC UMIs (Table S2).
Figure 1. Single-cell DOGMA-seq captures the heterogeneous epigenetic, transcriptional, and surface protein states of memory CD4+ T cells during HIV-1 infection.
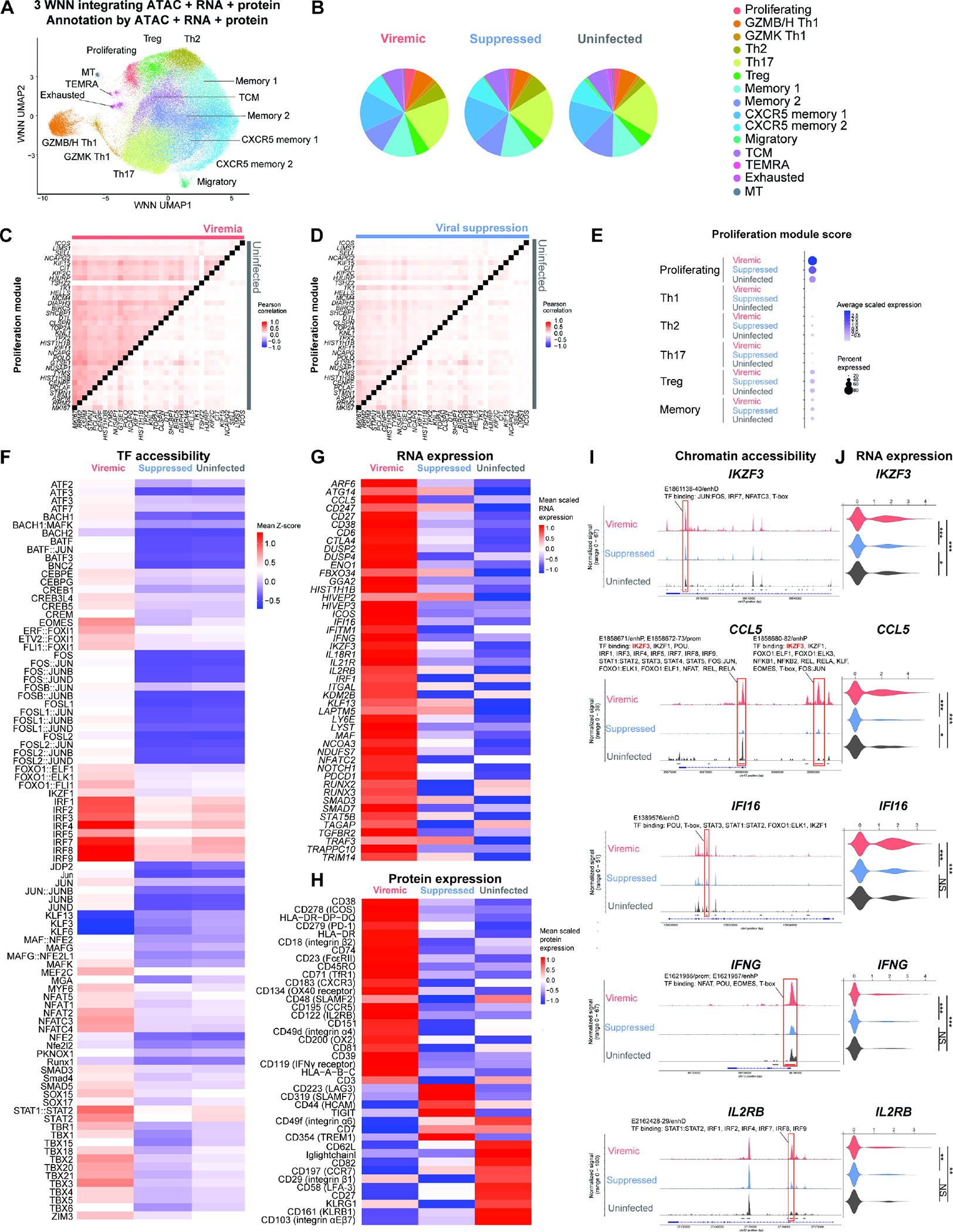
(A) Harmonized WNN UMAP projection of memory CD4+ T cells identified by cell-to-cell multimodal neighbors (weighted combination of ATAC, RNA, and protein similarities) (n = 93,209 cells, including 25,778, 56,771, 10,660 cells in the viremic, viral suppression, and uninfected conditions, respectively). (B) Pie charts indicating cell subset distributions in viremia, viral suppression, and uninfected conditions. (C–D) Heatmap comparisons of pairwise gene expression Pearson’s correlation coefficient, for the proliferation gene program identified by WGCNA. The Proliferation gene program was identified by WGCNA using top 50 genes most positively and negatively associated with the first 7 principal components in the transcriptional profiles of 25,778 cells in viremia. P < 0.05 for at least 95% Wilcoxon rank-sum tests against 10,000 random module permutations. (E) Proliferation module score per cell subset and infection condition. (F) Chromatin accessibility of transcription factor binding motifs in proliferating cluster cells grouped by conditions, represented in heatmap as comparisons of group means in ATAC-seq chromVAR bias-corrected deviations (Z-score). All motifs shown had significantly increased accessibility (P < 0.05 and mean Z-score difference ≥ 0.3). (G) Genes differentially expressed in proliferating cluster cells, represented in heatmap as comparisons of group means in normalized and scaled RNA expression. All genes passed P < 0.05 in viremic, min.pct ≥ 0.1, log2 fold change (FC) ≥ 0.25). (H) Surface proteins differentially expressed in proliferating cluster cells, represented in heatmap as comparisons of group means in DSB-normalized and scaled protein expression. All features passed P < 0.05 and log2FC ≥ 0.25. The mean expression of all protein features shown were also tested to be greater than the mean expression of their specific isotype controls in cells from the proliferating cluster (Z > 2; two-sample Z-test). For all heatmaps, n = 1,073, 1,373, 281 proliferating cells in the viremic, viral suppression, and uninfected conditions, respectively. Statistical significance for heatmaps was determined by Wilcoxon rank-sum test for comparisons between each group and all cells in the other two groups. All P values were false discovery rate (FDR)-adjusted using the Benjamini-Hochberg procedure. (I and J) Matching increase in chromatin accessibility and scaled gene expression at IKZF3, CCL5, IFI16, IFNG, and IL2RB in proliferating cells in viremia. (I) Highlighted red on the genome track corresponds to significantly differentially accessible peaks called by MACS3 (FDR-adjusted P < 0.05, min.pct ≥ 0.05, log2FC ≥ 0.15) (see Figure S2) and overlap with candidate cis-regulatory elements as predicted by ENCODE Registry of Candidate Cis-Regulatory Elements (cCREs). All transcription factors were identified using the JASPAR2022 Core Vertebrates collection, with binding confidence of P > 0.05 by PWMscan. (I) The corresponding RNA expression of genes. * P < 0.05, ** P < 0.01, *** P < 0.001, Wilcoxon rank-sum test. See also Figures S1–S4.
Acute HIV-1 infection increases IKZF3 expression in proliferating cells
We first examined how acute HIV-1 infection shaped the epigenetic, transcriptional, and protein expression states of memory CD4+ T cells (Figure S2). Chromatin accessibility of binding motifs was significantly enriched for AP-1 family transcription factors (JUN, FOS, and BATF), CNC family transcription factors (including four transcription activators (NF-E2 and NF-E2 like 1, 2, 3) and two repressors (BACH1 and BACH2)), and Maf during viremia (Figure S2A). The significantly upregulated genes were type I IFN response genes (ISG15, IFI44, IFI44L, MX2, TRIM22), PDE4B (which promotes cytokine IL-2 production)31, and SYTL2 (encoding Slp-2 for lytic granule secretion)32 during viremia (Figure S2B). The significantly upregulated surface proteins during viremia were of T cell activation (CD38, major histocompatibility complex (MHC) class II (HLA-DP, -DQ, -DR), ICOS, SLAMF2, CD74, CD134, CD2, CD3, CD101), immune checkpoint (PD-1), Th1 polarization (CXCR333, CCR534), cytotoxic T cell differentiation (KLRG1, GPR56), migration (integrin α1, α4, and β1), and anti-inflammatory Treg pathways (CD39, CD73)35 during viremia (Figure S2C).
To identify distinct gene programs that were induced during viremia de novo, we performed Weighted Gene Correlation Network Analysis (WGCNA)36 to identify genes that are highly co-expressed in cells in viremia but not in viral suppression and uninfected conditions. WGCNA discovers sets of coregulated gene that may be co-regulated as they are determined to be pairwise correlated37. Among five statistically significant gene modules identified by WGCNA in viremia (Table S2), the proliferation gene program revealed co-expression of the proliferation gene MKI67, cell survival gene BIRC5 (Survivin)38, and T cell activation gene ICOS. To contrast the degree that RNA expression was correlated among genes identified in the proliferation gene program, the Pearson’s correlation coefficients for gene pair RNA expression were visualized and compared in heatmap (Figure 1C–1D). Indeed, genes in the proliferation program had higher RNA co-expression in viremia in comparison to viral suppression and uninfected conditions. To verify that the proliferation gene program was most enriched in the proliferating cluster during viremia, the normalized per-cell averaged RNA expression for proliferation gene program (module score, see Methods) was measured as a score of gene program enrichment. Indeed, the proliferation gene program scores were overall higher in viremia than in viral suppression and uninfected conditions in the proliferating cluster (Figure 1E, S3A, S3B).
We next focused on the proliferating cluster to determine the cellular states of proliferating T cells during in HIV-1 infection versus uninfected condition (Figure 1F–1H). We first identified transcription factors having binding motifs that were enriched in accessible chromatin. During viremia, accessibility of transcription factors of type I IFN responses (IRF, STAT1/STAT2), T cell activation [AP-1 (JUN, FOS, BATF), NFAT], effector differentiation (TBX21 and EOMES), and memory development (FOXO1) were all enriched (Figure 1G). Comparing proliferating versus all other clusters, accessibility of IFN regulatory transcription factors (IRF, STAT1/STAT2) were the most prominently enriched (Figure S3C, S3D).
To determine which genes were epigenetically regulated by transcription factors that had enriched binding motif chromatin accessibility during viremia in proliferating cells, we identified gene regions that had significantly increased chromatin accessibility (ATAC peaks) and overlapped with The Encyclopedia of DNA Elements (ENCODE) candidate cis-regulatory elements (cCREs) (Figure 1I, S3E, Table S2) and increased RNA expression (Figure 1G, 1J) in the proliferating cluster in viremia, and the corresponding transcription factor binding motifs that were present in these regions of increased accessibility (Figure 1I). Examples were T cell differentiation genes (IKZF3, CCL5, IFNG), type I IFN response gene (IFI16), and T cell proliferation gene (IL2RB). For instance, IKZF3 (encoding Aiolos, an Ikaros zinc finger family transcription factor that regulates lymphocyte proliferation and differentiation)39 accessibility was increased in viremia at a putative regulatory element (identified as an open chromatin ATAC peak) (Figure 1I). Motifs found in this highly accessible footprint bind to IRF7, JUN:FOS, NFAT, and T-box transcription factors, all of which had enriched binding motif accessibility in the proliferating cluster in viremia (Figure 1F). Consistent with increased chromatin accessibility, IKZF3 had increased RNA expression in the proliferating cluster (Figure 1J). To provide a more direct demonstration of the relationships shown between chromatin accessibility at the highlighted cCRE (Figure 1I) and RNA expression (1J), we found that IKZF3 RNA+ cells had increased chromatin accessibility at the IKZF3 gene than IKZF3 RNA− cells in the Proliferating cluster (Figure S4A). Similarly, CCL5 RNA+ cells had increased chromatin accessibility at the CCL5 gene, IFI16 RNA+ cells had increased chromatin accessibility at the IFI16 gene, IFNG RNA+ cells had increased chromatin accessibility at the IFNG gene, and IL2RB RNA+ cells had increased chromatin accessibility at the IL2RB gene. Importantly, ATAC peaks that had increased chromatin accessibility (highlighted in red in Figure S4A–S4E) shared the same gene coordinates as significantly differentially accessible peaks identified in Figure 1I. Together, these results suggest that type I IFN response, T cell differentiation, and proliferation may be epigenetically upregulated during viremia in proliferating cells.
We identified a relationship between Aiolos (IKZF3) binding at CCL5 locus and CCL5 expression in proliferating cells. First, we found that Aiolos binding motif in the CCL5 cCREs had increased chromatin accessibility in viremia (Figure 1I, peaks highlighted red). Second, the same CCL5 cCREs had increased chromatin accessibility in proliferating cells that were enriched in Aiolos binding motif accessibility (IKZF3 Chromatin Variation Across Regions (chromVAR) z-score > 0) in comparison to proliferating cells that were low in Aiolos binding motif accessibility (IKZF3 chromVAR z-score < 0) (Figure S4F). Third, proliferating cells that were enriched in Aiolos binding motif accessibility had upregulated CCL5 expression in comparison to proliferating cells that were low in Aiolos binding motif accessibility (Figure S4G). Finally, a differential transcription factor binding motif accessibility (chromVAR) analysis between CCL5+ proliferating cells and CCL5− proliferating cells revealed that IKZF3 had significantly increased global accessibility in cells that were CCL5+. We noted that, in addition to Aiolos, AP-1, IRF, STAT1:STAT2, and T-box family transcription factors may also bind at CCL5 cCREs (Figure 1I). In addition to CCL5, we also identified Aiolos binding motif in significantly differentially accessible ATAC peaks in several gene loci (See Table S2 for annotated cCREs and binding transcription factors for differentially accessible ATAC peaks shown in Figure S3E, and the corresponding gene expression and protein expression fold changes).
To identify immune pathways that were induced during viremia in proliferating cells, we identified significantly upregulated genes in viremia in the proliferating cluster (Figure 1G). We found increased expression of type I IFN response gene (IFI16), T cell differentiation genes (IKZF3, CCL5, IFNG), and T cell proliferation gene (IL2RB) during viremia but not in viral suppression, in agreement with Gene Ontology analysis (Figure S3F, S3G). Using significantly downregulated genes, Gene Ontology revealed downregulated response to virus and type I IFN response in viral suppression in the proliferating cluster (Table S2). In agreement with above, Gene Set Enrichment Analysis (GSEA) revealed enrichment of type I IFN responses (Figure S3H) and Th1 polarization (Figure S3I; including SERPINB9, a granzyme B inhibitor) during viremia in proliferating cells.
Finally, differential surface protein expression analysis revealed significant upregulation of markers of activation, Th1 polarization [CXCR3, CCR5, CD119 (IFNγ receptor)], and T cell migration (integrin α4, integrin β2) (Figure 1H) during viremia in proliferating CD4+ T cells. Of note, IL2RB had consistently higher chromatin accessibility (Figure 1I, S3E), higher RNA expression (Figure 1G, 1J), and higher surface protein expression (Figure 1H) in proliferating cells during viremia versus viral suppression and uninfected conditions.
Single-cell DOGMA-seq identified the cellular states of transcriptionally inactive and transcriptionally active HIV-1-infected cells
To examine the epigenetic, transcriptional, and protein expression differences between HIV-1-infected cells versus uninfected cells, we mapped ATAC-seq reads and RNA-seq reads to HXB2 HIV-1 reference genome and to autologous virus (Figure 2) to identify HIV-1 DNA+ cells and HIV-1 RNA+ cells, respectively. Of note, HIV-1 DNA+ cells can be transcriptionally silent or have low HIV-1 RNA expression below the detection limit.
Figure 2. HIV-1 proviral and viral genome landscape captured by DOGMA-seq.
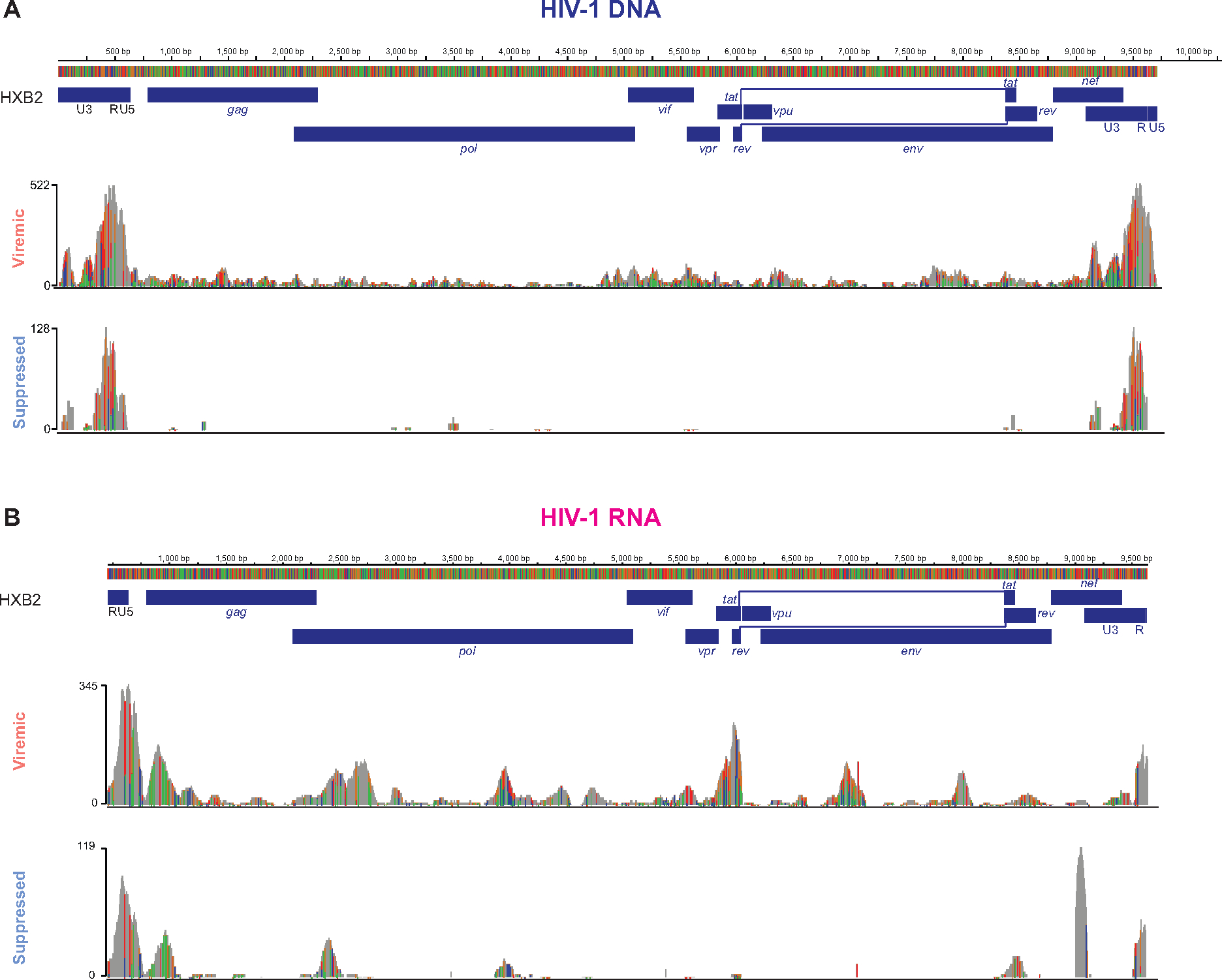
Integrative genomics viewer (IGV) plots of HIV-1 DNA+ reads (A) and of HIV-1 RNA+ reads (B) mapped to HXB2 reference genome and autologous HIV-1 sequences. See also Figures S5.
To define the sensitivity of HIV-1 DNA detection, we mapped HIV-1 DNA from a separate DOGMA-seq dataset that we recently described40–42. Of 14,780 cells from HIV-1-infected, stably integrated Jurkat cell lines, each have known HIV-1 integration sites10,41,42, we identified 5,220 HIV-1 DNA+ cells (35.32% sensitivity) at 1 read per cell threshold and 4,197 HIV-1 DNA+ cells (28.40% sensitivity) at 2 reads per cell threshold. Despite that these HIV-1-infected Jurkat cells had active HIV-1 expression and high chromatin accessibility at the HIV-1 provirus40, the detection of HIV-1 DNA remained low (28.4%). We anticipated that using ATAC-seq to detect HIV-1 DNA in HIV-1-infected cells in the clinical samples may be even lower. Given the small proportion of the ~9,719 bp HIV-1 genome among the 6×109 bp for the diploid human genome, an integrated HIV-1 proviral genome only accounted for 0.00016% of the human genome in one infected cell. Given that each infected cell has only one copy of HIV-1 proviral DNA but many copies of HIV-1 RNA, we expected to identify HIV-1 RNA+ cells with no detectable HIV-1 DNA (not captured by ATAC-seq). In addition, HIV-1 may integrate into low accessible heterochromatin which would not be captured by ATAC-seq. To define the sensitivity of HIV-1 RNA detection using DOGMA-seq, a threshold of 2 HIV-1 RNA reads among a mean of 48,223 total RNA reads captured per cell from the HIV-1+ participants indicated that if 0.004% of cellular RNA reads were HIV-1 reads then the cell would be detected as HIV-1+.
To increase the specificity of HIV-1 genome mapping and to guard against potential sequencing artifacts, HIV-1+ cells were defined by the presence of ≥2 reads of HIV-1 RNA or HIV-1 DNA or both per cell (Table S3). This threshold yielded no HIV-1 RNA+ cells or HIV-1 DNA+ cells in samples from four uninfected participants (n = 10,660) or from uninfected Jurkat cells (n = 9,560). Given the rarity of HIV-1-infected cells (~879/106, 0.09%) during viremia43, the false negative rate (HIV-1 DNA− and HIV-1 RNA− cells actually being an HIV-1-infected cell) was <0.1%.
Per participant, we identified 1.19%–3.07% of memory CD4+ T cells in viremia (Figure S5A, Table S3) and 0.02%–0.06% in viral suppression were HIV-1-infected (Figure S5B, Table S3). No HIV-1-infected cells were detected in uninfected controls (Figure S5C, Table S3). We defined HIV-1 DNA+ RNA− cells as HIV-1 DNA+ cells that had undetectable HIV-1 expression (transcriptionally inactive HIV-1-infected cells). We combined HIV-1 DNA− RNA+ cells with HIV-1 DNA+ RNA+ cells and defined them as HIV-1 RNA+ cells (transcriptionally active HIV-1-infected cells). In total, we identified 233 HIV-1 DNA+ RNA− cells (0.90%) and 256 HIV-1 RNA+ cells (0.99%) [including 218 HIV-1 DNA− RNA+ cells (0.85%) and 38 HIV-1 DNA+ RNA+ cells (0.15%)] in viremia (Figure 3A), and 19 HIV-1 DNA+ (0.03%) and 14 HIV-1 RNA+ cells (0.02%) [including 11 HIV-1 DNA− RNA+ cells (0.02%) and 3 HIV-1 DNA+ RNA+ RNA− cells (0.005%)] in viral suppression (Figure 3B). Pooled HIV-1 DNA+ cells and HIV-1 RNA+ cells were distributed across different memory CD4+ T cell subsets without clear bias in cell-type compositions when compared with HIV-1− cells (Figure 3C). In addition, no significant bias was observed in the numbers of HIV-1 RNA reads recovered in HIV-1 RNA+ cells per cell-type (Figure S5D, S5E). Of note, we sequenced on average 50,131 ATAC read pairs and 48,223 RNA read pairs per cell and reached sequencing saturation across 10x runs (Figure S5F). Most HIV-1 DNA reads were captured within the HIV-1 LTR promoter (Figure 2) and reflected increased chromatin accessibility at HIV-1 promoter than protein coding regions40. Between participants, no notable differences were observed in the HIV-1 genome mapping (Figure S5G–S5J).
Figure 3. Single-cell DOGMA-seq captures transcriptionally inactive HIV-1-infected cells (HIV-1 DNA+ HIV-1 RNA−) and transcriptionally active HIV-1-infected cells (HIV-1 RNA+) and their respective epigenetic landscape.
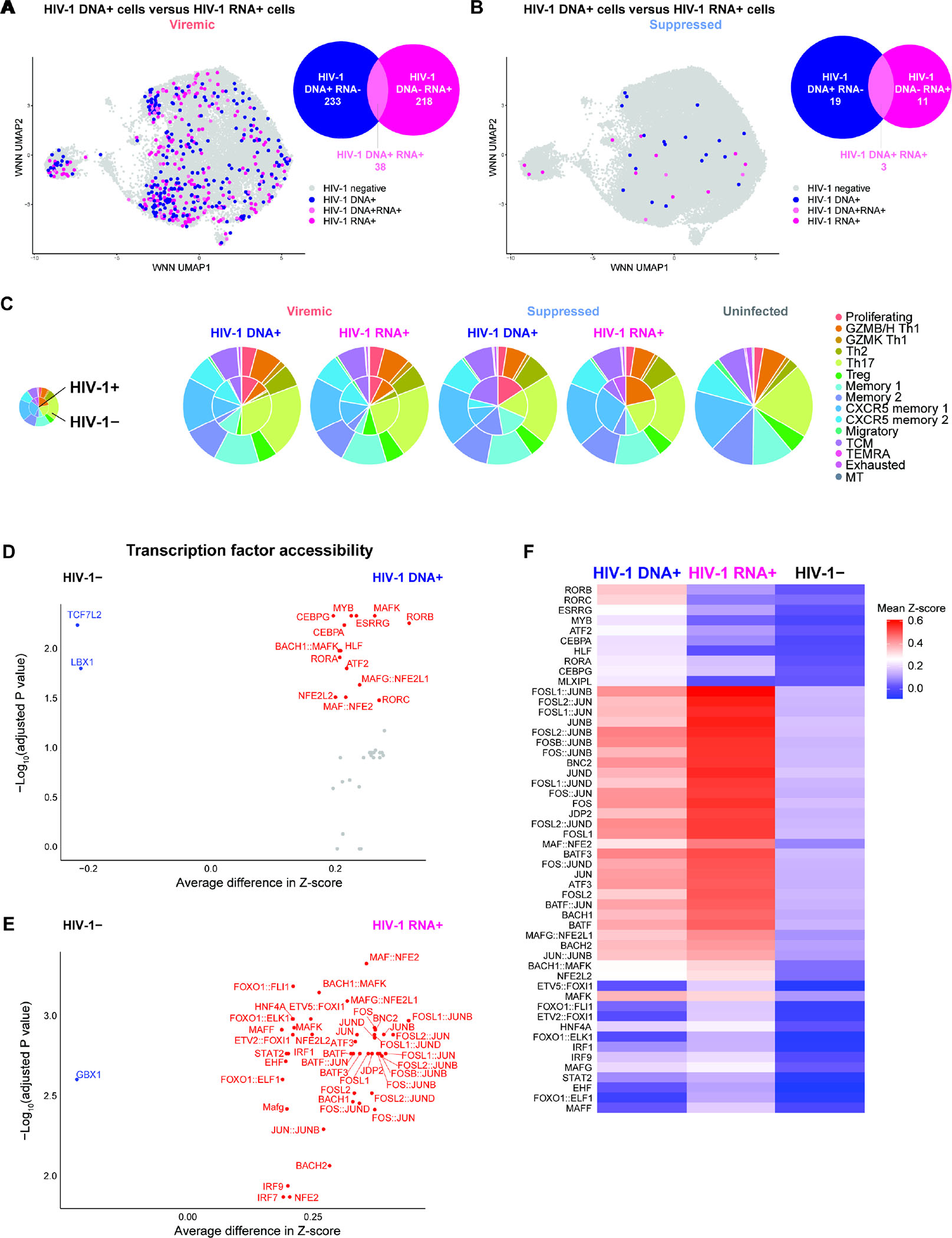
(A–B) HIV-1 DNA+ cells and HIV-1 RNA+ cells were identified by mapping ATAC-seq and RNA-seq reads, respectively, to autologous HIV-1 and HXB2 reference sequences. An HIV-1-infected cell was defined as having at least 2 HIV-1 reads per barcode to guard against index hopping and sequencing artifacts. The Venn diagrams were drawn to scale with intersection depicting the subset of HIV-1-infected cells that are HIV-1 DNA+ RNA+ (light pink). For all downstream figures, HIV-1+ DNA+ RNA− cells were termed HIV-1 DNA+ cells (blue) while HIV-1 DNA− RNA+ cells and HIV-1 DNA+ RNA+ cells were merged and termed HIV-1 RNA+ cells (pink). (C) Pie charts indicating cell subset distributions. (D–E) Volcano plots indicating transcription factors binding motifs having increased global chromatin accessibility (measured by chromVAR bias-corrected deviations) between HIV-1 DNA+ cells (n = 233) and HIV-1− cells (n = 2,238) in viremia (D) and between HIV-1 RNA+ cells (n = 256) and HIV-1− cells (n = 2,238) in viremia (E). (F) Global chromatin accessibility of transcription factor binding motifs in HIV-1 DNA+ cells and in HIV-1 RNA+ cells, with respect to each other and to HIV-1− cells, in viremia, represented in heatmap as comparisons of group means in chromVAR bias-corrected deviations (Z-score). All motifs shown had significantly increased accessibility (P <0.05 and mean Z-score difference ≥ 0.2). All volcano plots and heatmap showed comparisons for viremia (n = 233, 256, 2,238 cells in HIV-1 DNA+, HIV-1 RNA+, and HIV-1−). Statistical significance for heatmaps was determined by Wilcoxon rank-sum test for comparisons between each group and all cells in the other two groups. For fair comparisons between groups in volcano plots and heatmap, HIV-1− cells were downsized to match the same number of cells in HIV-1+ groups with 1,000 bootstrap replicates. The 1,000 P values were transformed to follow a standard Cauchy distribution and a combined P value was calculated as the weighted sum of Cauchy transformed P values, followed by correction for multiple comparisons using the Benjamini-Hochberg (FDR) procedure. See also Figure S7.
RORC, BACH2, and Maf transcription factors shaped transcriptionally inactive HIV-1-infected cells while AP-1, IRF, BACH2, and Maf shaped transcriptionally active HIV-1-infected cells
We first examined the epigenetic landscape of HIV-1-infected cells (Figure 3D–3F). During viremia, binding motifs for retinoid acid-related (RAR) orphan receptors (RORB and RORC, encoding RORβ and RORγt) had significantly increased chromatin accessibility in HIV-1 DNA+ cells (Figure 3D, 3F). Notably, RORγt (encoded by RORC) is a master transcription factor that drives Th17 differentiation. We then compared the transcription factor binding potential of HIV-1 RNA+ cells versus uninfected cells. We found that the binding motifs of AP-1 (JUN, FOS, BATF), the CNC family (NFE2L2, BACH1, BACH2), Maf, and IRF transcription factors were prominently enriched in accessible chromatin of HIV-1 RNA+ cells (Figure 3E, 3F). Similarly, HIV-1 DNA+ cells also had enriched AP-1, CNC, and Maf transcription factor binding motif accessibility but to a lesser extent than HIV-1 RNA+ cells (Figure 3F). Of note, these were the same transcription factors that had enriched chromatin accessibility in memory CD4+ T cells during viremia (Figure S2A). Overall, binding motifs of AP-1, CNC, Maf, and IRF family transcription factors had enriched accessibility in HIV-1-infected cells, while accessibility of RORβ and RORγt transcription factor binding motifs were additionally enriched in HIV-1 DNA+ cells.
Transcription factor IKZF3 was associated with the survival and proliferation of transcriptionally active HIV-1-infected cells
To identify the transcriptional landscape of HIV-1-infected cells, we identified significantly upregulated genes in these cells versus HIV-1− cells during viremia (Figure 4A–4C). We identified 19 genes that were significantly upregulated in both HIV-1 DNA+ cells and HIV-1 RNA+ cells (Table S2). Among them, eight genes were closely associated with T cell differentiation: a lymphocyte differentiation gene (IKZF3, encoding transcription factor Aiolos), three type I IFN response genes (STAT1, IFI44, IFI44L), two cytotoxic T cell effector genes [GZMA, SYTL2 (mediating lytic granule secretion)44], one T cell activation gene (PDE4B mediating IL-2 production)45, one T cell migration gene (ITGA4)(Figure 4A, 4B). Of note, IKZF3 was also upregulated in the proliferating cells (Figure 1G).
Figure 4. Transcriptionally inactive HIV-1-infected cells upregulate IKZF3, type I IFN, cytotoxic T cell response, and homing RNA expression..

(A–B) Volcano plots indicating differentially expressed genes in viremia between HIV-1 DNA+ cells (n = 233) or HIV-1 RNA+ cells (n = 256) versus HIV-1− cells (n = 2,238). P < 0.05, min.pct ≥ 0.15, log2FC ≥ 0.25. (C) Genes highly expressed in viremia in HIV-1 RNA+ cells and in HIV-1 DNA+ cells, represented in heatmap as comparisons of group means in normalized and scaled RNA expression. All genes passed P < 0.05, min.pct ≥ 0.15, log2FC ≥ 0.25. Statistical significance for heatmaps was determined by Wilcoxon rank-sum test for comparisons between each group and all cells in the other two groups. For fair comparisons between groups in volcano plots and heatmap, HIV-1− cells were downsized to match the same number of cells in HIV-1+ groups with 1,000 bootstrap replicates. The 1,000 P values were transformed to follow a standard Cauchy distribution and a combined P value is calculated as the weighted sum of Cauchy transformed P values, followed by correction for multiple comparisons using the Benjamini-Hochberg (FDR) procedure. (D–E) Dot plot of average normalized and scaled RNA expression for significantly upregulated genes identified in HIV-1 DNA+ cells in viremia as shown in (A) and in HIV-1 RNA+ cells in viremia as shown in (B) that were also upregulated in HIV-infected cells during viral suppression. Log2FC > 0 between HIV-1+ cells and HIV-1− cells in viral suppression. P values did not reach statistical significance in viral suppression because of the low number of HIV+ cells captured. n = 19, 14, 56,738 HIV-1 DNA+ cells, HIV-1 RNA+ cells, and uninfected cells in viral suppression, respectively. (F–G) Heatmap comparisons of pairwise gene expression Pearson’s correlation coefficients, for the proliferation (MKI67)-driven gene program identified by WGCNA, between HIV-1 DNA-1+ cells versus HIV-1− cells in viremia (F) and between HIV-1 RNA-1+ cells versus HIV-1− cells in viremia (G). This module was identified in HIV-1 RNA+ cells in viremia by considering top 50 genes most positively and negatively associated in the first 8 principal components. Module met significance with P <0.05 for at least 95% Wilcoxon rank-sum tests against 10,000 random module permutations. (H) Enriched Gene Ontology biological processes determined using genes in WGCNA-identified proliferation gene program as query. (I–J) To identify immune pathways, all 36,601 genes are ranked by log2 fold change in normalized gene expression between HIV-1+ and HIV-1− cell groups in viremia. Significantly enriched pathways were identified using GSEA. Top 10 representative leading-edge genes were shown. See also Figures S6–S7.
We noted that many HIV-1+ cells were recovered in the Th17 cluster during viremia (Figure 3C, S5A). To examine whether the significantly upregulated genes identified in HIV-1+ cells versus HIV-1− cells were driven by HIV-1 enrichment in specific cell subsets such as Th17, we examined the RNA expression for the eight key genes described above (IKZF3, STAT1, IFI44, IFI44L, GZMA, SYTL2, PDE4B, and ITGA4) in the 15 cell clusters in viremia (Figure S6A). We found that the RNA expression of these genes were not significantly higher in Th17 in comparison with other clusters, albeit Th17 cells harbored the largest number of HIV-1− and HIV-1+ cells (Figure S6B, S6C, Table S3), suggesting that Th17 cellular RNA profile was not the dominate contributor of the transcriptional profile of HIV-1+ cells.
In HIV-1 DNA+ cells, the upregulated genes were involved in cytotoxic T cell differentiation (GZMA, GZMK, KLRB1, CCL5, LTB) and T cell migration (ITGA4, T cell homing integrin α4) (Figure 4A), consistent with previous finding of enrichment of HIV-1-infected cells in cytotoxic CD4+ T cells11,13 and migratory cells7,16,46. In HIV-1 RNA+ cells, the upregulated genes included cytotoxic T cell differentiation [GZMA, CTSC (encoding cathepsin C)], T cell migration [ITGA4 and ITGB7 (integrin β7)], type I IFN responses (STAT1, STAT2, MX1), T cell differentiation transcription factors (FOSL2, REL, MAF, STAT5B), and a cell survival gene (CFLAR, encoding c-FLIP that prevents T cell death) (Figure 4B). Gene Ontology analysis of significantly upregulated genes showed that HIV-1 DNA+ cells were enriched of integrin pathways (Figure S6D–S6E) while HIV-1 RNA+ cells had heightened type I IFN and viral responses (Figure S6F–S6G). In contrast, Gene Ontology analysis of significantly downregulated genes showed significant downregulation of autophagy regulation in HIV-1 DNA+ cells and significant downregulation of negative regulations of cell growth and differentiation in HIV-1 RNA+ cells (Table S2).
We wanted to identify the transcriptional landscape of HIV-1-infected cells during viral suppression. However, we did not feel confident using the low number of cells captured to identify cellular markers. Instead, we asked whether cellular genes upregulated during viremia remained upregulated during viral suppression. In HIV-1 DNA+ cells, we found that cytotoxic markers GZMA, GZMK, KLRB1, and LTB were upregulated both in viremia and after viral suppression (Figure 4D). In HIV-1 RNA+ cells, we found that cytotoxic T cell markers (CTSC, SYTL2), type I IFN response genes (IFI44, STAT1, STAT2), survival gene CFLAR, and T cell differentiation transcription factor IKZF3 were upregulated both in viremia and after viral suppression (Figure 4E).
To distinguish the transcriptional programs in HIV-1-infected cells, we identified co-expressed genes de novo using WGCNA. We found a proliferation program that distinguished co-expressed genes between HIV-1 DNA+ cells, HIV-1 RNA+ cells, and HIV-1− cells (Figure 4F, 4G). These co-expressed genes (such as IKZF3, IL21 (a key Tfh cytokine), BIRC5 (Survivin), and MKI67) were involved in cell division and differentiation in Gene Ontology analysis (Figure 4H) and may support the proliferation of HIV-1 RNA+ cells despite active HIV-1 RNA expression. Of note, BIRC538 and IL-216 are cellular factors known to promote HIV-1 persistence. We next identified distinct immune pathways that shaped the transcriptional landscape of HIV-1-infected during viremia. Using GSEA, we found significant enrichment of an integrin pathway in HIV-1 DNA+ cells (P < 0.05, Figure 4I) and IL-6/JAK/STAT3 signaling in HIV-1 RNA+ cells (P < 0.05, Figure 4J).
Transcriptionally inactive HIV-1-infected cells expressed Th1/Th17, T cell activation, and migratory markers
Increased expression of cellular markers have been reported in HIV-1-infected cells, such as Th1 polarization (CXCR3 and CCR5)5,12, activation markers (HLA-DR14, CD247, ICOS12, SLAM5), cytotoxic T cell markers [KLRB148 and Granzyme B13], exhaustion markers (PD-1, CTLA-4, and TIGIT)15, Tfh polarization (CXCR5 and PD-1)12, and homing marker VLA-4 (integrin α4β1)5,7,16. Furthermore, targeted HIV-1 proviral genome sequencing identified protein signatures enriched in CD4+ T cells harboring intact HIV-1 proviruses, such as PD-1, TIGIT, KLRG1, HVEM, CD49d (integrin α4), and CD95 (Fas receptor)6. Using DOGMA-seq, we identified 7 surface proteins that were significantly upregulated in both HIV-1 DNA+ cells and HIV-1 RNA+ cells, including Th1 polarization markers (CCR5, CXCR3, and KLRB1), T cell migration marker (ITGAL) and T cell activation markers [CD38, LFA-2, and SLAM] (Figure 5A, 5B, 5C), consistent with previous studies5,6. A total of 18 proteins were upregulated in HIV-1 RNA+ cells, including markers of Th1 and Th1* (Th1/Th17) polarization (CCR5, CCR6, CXCR3, KLRB1, and the IFNγ receptor CD119), migration (Integrin α4, Integrin αL, and HCAM), and T cell activation (Figure 5B). Notably, CD81 degrades SAM Domain And HD Domain 1 (SAMHD1) and enhances HIV-1 reverse transcription49. Given that the low number of HIV-1 DNA+ cells and HIV-1 RNA+ cells captured during viral suppression were not sufficient to perform robust statistical analysis, we asked whether protein markers upregulated during viremia were also upregulated during viral suppression. We found seven protein markers that were significantly upregulated in HIV-1 RNA+ cells in viremia were also upregulated in HIV-1 RNA+ cells in viral suppression (Figure 5D, 5E). Of note, SLAM and CCR5 protein expression were upregulated in both HIV-1 DNA+ cells and HIV-1 RNA+ cell in viremia and viral suppression. Overall, we found that surface protein markers of Th1 polarization and T cell activation were upregulated in HIV-1 DNA+ cells versus HIV-1− cells, whereas surface protein markers of Th1 and Th1/Th17 polarization (Figure 5F), migration (Figure 5G), and T cell activation (Figure 5H) were upregulated in HIV-1 RNA+ cells versus HIV-1− cells.
Figure 5. Transcriptionally inactive HIV-1-infected cells upregulate Th1/Th17, T cell activation, and homing protein expression. .
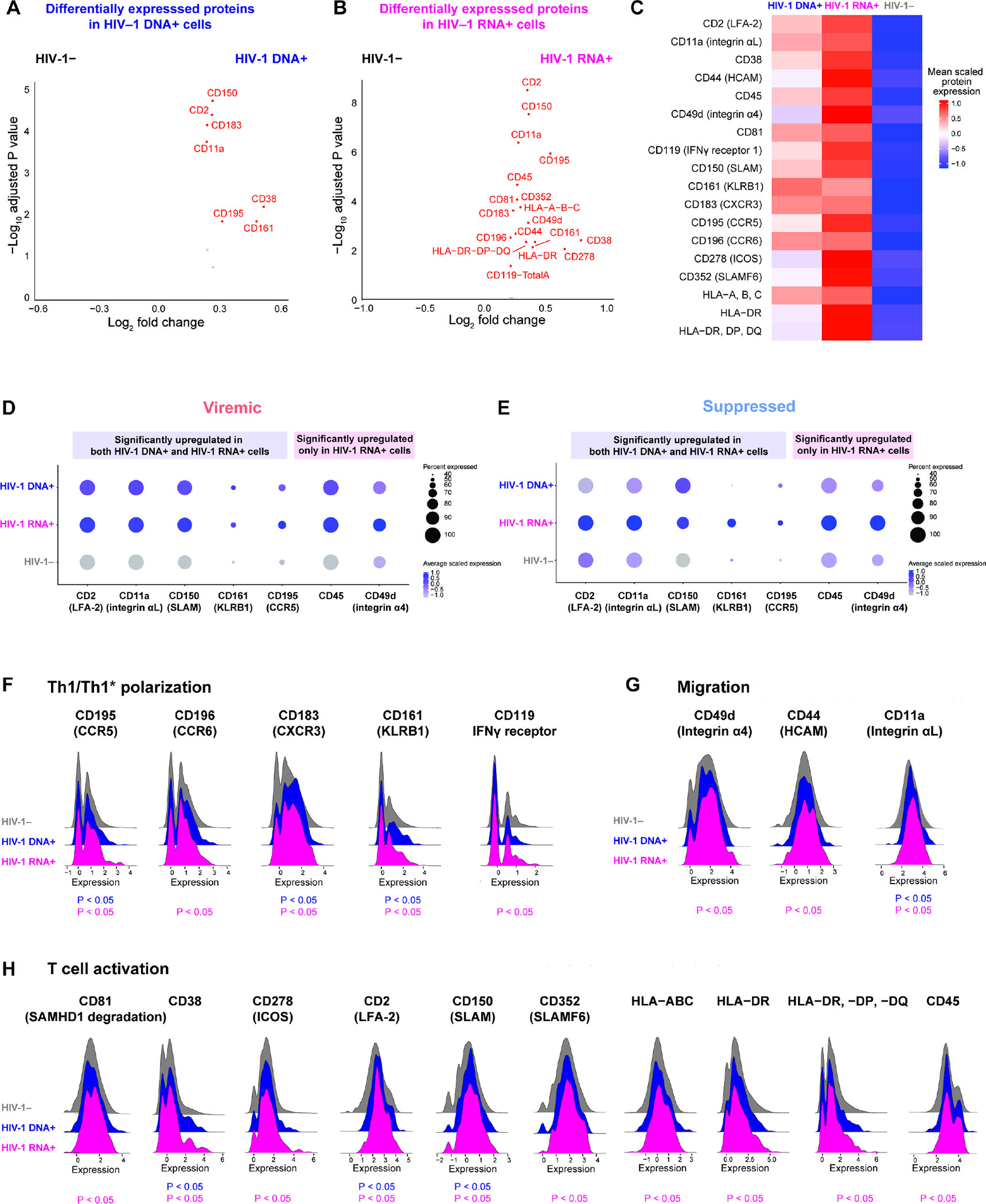
(A–B) Volcano plots indicating differentially expressed surface proteins in viremia between HIV-1 DNA+ cells (n = 233) or HIV-1 RNA+ cells (n = 256) versus HIV-1− cells (n = 2,238). P < 0.05 and log2FC ≥ 0.25. (C) Surface proteins highly expressed in viremia in HIV-1 DNA+ cells versus HIV-1 RNA+ cells versus HIV-1− cells, represented in heatmap as comparisons of group means in DSB-normalized and scaled protein expression. All proteins passed P < 0.05 and log2FC ≥ 0.3. The mean expression of all protein features shown were also tested to be greater than the mean expression of their specific isotype controls (Z > 2; two-sample Z-test) in HIV-1+ cells. Statistical significance for heatmaps was determined by Wilcoxon rank-sum test for comparisons between each group and all cells in the other two groups. For fair comparisons between groups, HIV-1− cells were downsized to match the same number of cells in HIV-1+ groups with 1,000 bootstrap replicates. The 1,000 P values were transformed to follow a standard Cauchy distribution and a combined P value is calculated as the weighted sum of transformed P values, followed by correction for multiple comparisons using the Benjamini-Hochberg (FDR) procedure. (D–E) Significantly upregulated proteins identified in HIV-1 DNA+ cells and in HIV-1 RNA+ cells (A–B) in viremia (D) that were also upregulated in HIV-1-infected cells in viral suppression (log2FC > 0 between HIV-1+ cells and HIV-1− cells in viral suppression) (E). Protein expression is visualized in dots plot of average CLR-normalized and scaled expression. P values did not reach statistical significance in viral suppression because of the low number of HIV-1+ cells captured. n = 19, 14, 56,738 HIV-1 DNA+ cells, HIV-1 RNA+ cells, and uninfected cells in viral suppression, respectively. (F–H) Ridge plot of DSB-normalized and scaled protein expression distributions for significantly highly expressed surface proteins identified in (A–B). See also Figures S6–S7.
The heterogeneous HIV-1-infected cells comprised four distinct cell states: IRF, cytotoxic, AP-1, and cell death
We identified epigenetic, transcriptional, and protein signatures in HIV-1+ memory CD4+ T cells. While differences existed between participants, these programs were not driven by specific participants (Figure S7). We asked whether we could dissect the heterogeneity of HIV-1-infected CD4+ T cell reservoir. First, we collected all identified HIV-1-infected cells to determine distinct cell clusters by their transcriptional profiles (Figure 6A). We found that clustering of HIV-1+ cells did not result from batch effects by infection conditions, HIV-1 RNA expression (Figure 6B), or from study participants who differed in peak viral loads (Figure 6C, Table S1). Each cluster had distinct transcriptional profiles (Figure 6D), distinctly enriched transcription factor accessibility (Figure 6E), and distinct surface protein expression profiles (Figure 6E). Indeed, four phenotypically distinct populations of HIV-1+ cells were determined across six HIV-1+ study participants.
Figure 6. The heterogeneous HIV-1-infected T cell reservoir comprised four distinct cell states: IRF, cytotoxic, AP-1, and cell death.
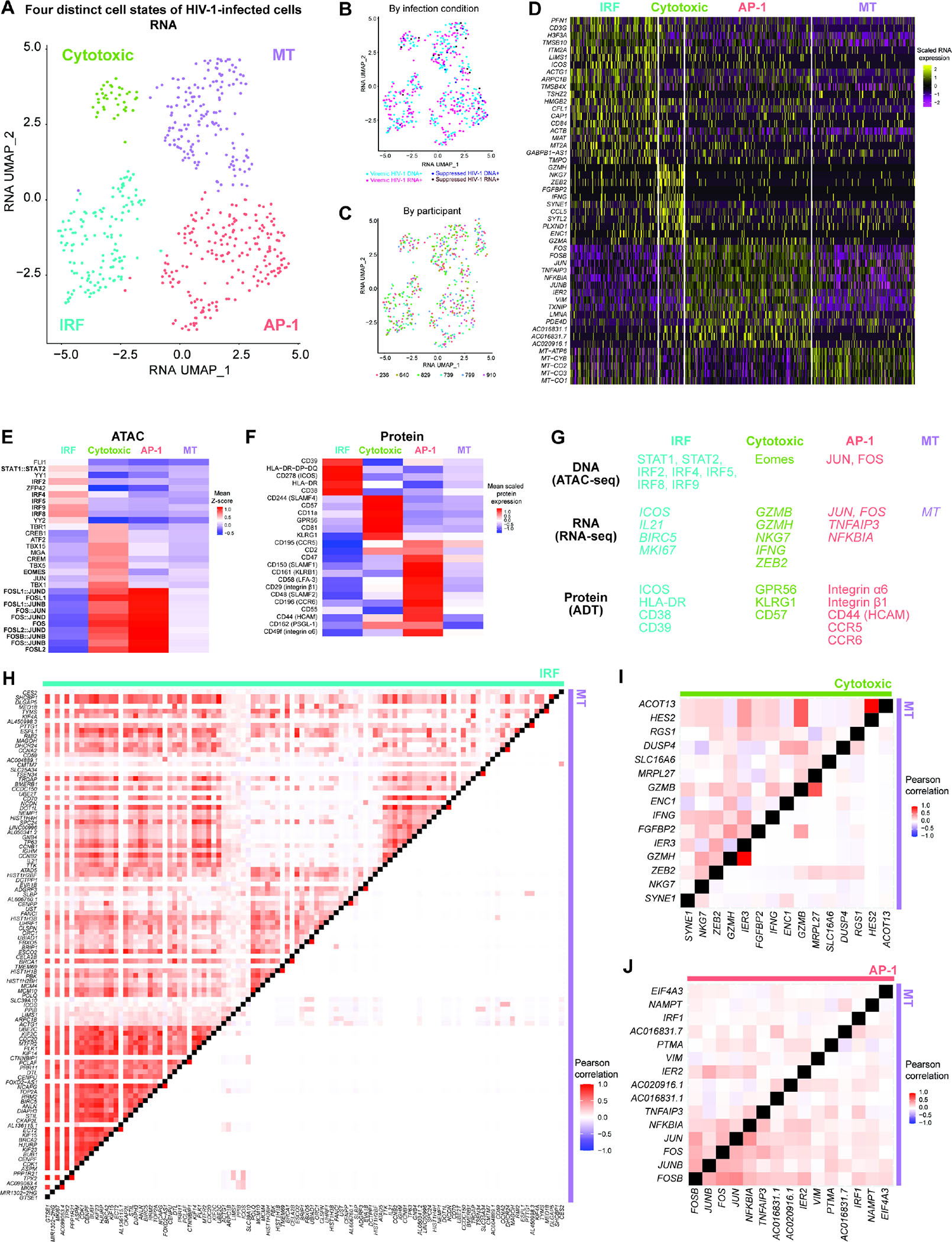
(A–C) RNA UMAP projection of all HIV-1+ cells (n = 233, 256, 19, 14 for viremic HIV-1 DNA+, viremic HIV-1 RNA+, suppressed HIV-1 DNA+, and suppressed HIV-1 RNA+, respectively) defined by transcriptional profile. Four phenotypically distinct clusters–IRF, Cytotoxic, AP-1, and MT clusters–were annotated (A). No apparent batch effects were observed by infection conditions or by HIV-1 RNA expression (B) or by study participants (C). Batch effects between participants were corrected by Harmony. (D) Heatmap of normalized and scaled RNA expression for most highly differentially expressed genes (by log2FC) in each cluster of cells. P < 0.05, min.pct ≥ 0.25, log2FC ≥ 0.25. (E) Differential accessibility for transcription factor binding motifs, represented in heatmap as comparisons of means of ATAC-seq chromVAR bias-corrected deviations (Z-score) between group. All motifs shown had significantly increased accessibility, by P < 0.05 and mean difference ≥ 0.3. (F) Proteins highly expressed per cell group, represented in heatmap as comparisons of group means in DSB-normalized and scaled protein expression. All features passed P < 0.05, and log2FC ≥ 0.25. The mean expression of all protein features shown were also tested to be greater than the mean expression of their specific isotype controls in HIV-1-infected cells (Z > 2; two-sample Z-test). Statistical significance for all heatmaps was determined by Wilcoxon rank-sum test for comparisons between each group and all cells in the other three groups. (G) Transcription factors with enriched binding motif accessibility and upregulated genes and proteins in each of the four phenotypically distinct clusters of HIV-1+ cells. (H–J) Heatmap comparisons of pairwise gene expression Pearson’s correlation coefficients for modules identified by WGCNA (using with top 50 genes most positively and negatively associated with the first 8 principal components in the transcriptional profiles of all 522 HIV-1+ cells). See also Figures S6.
We annotated each cluster of HIV-1-infected cells based on their distinct cellular profiles (transcription factor accessibility, transcriptional programs, and surface protein expression): (a) the IRF cluster had increased IRF and STAT1/STAT2 transcription factor accessibility (Figure 6E), ‘activation/proliferation’ gene co-expression program (Figure 6H: co-expression of ICOS, BIRC5, IL21, and MKI67), and activation surface proteins (Figure 6F: HLA-DR, ICOS, and CD38); (b) the Cytotoxic cluster had increased Eomes transcription factor accessibility (Figure 6E), upregulated cytotoxic effector T cell gene markers (Figure 6D: GZMA, GZMH, NKG7, CCL5, IFNG, ZEB2), ‘cytotoxic’ gene co-expression program (Figure 6I: co-expression of GZMB, GZMH, NKG7, IFNG, and ZEB2), and cytotoxic surface proteins (Figure 6F: GPR56 and KLRG1); (c) the AP-1 cluster had increase AP-1 transcription factor accessibility (Figure 6E), increased JUN and FOS RNA expression (Figure 6D), ‘TNF’ gene co-expression program (Figure 6J: co-expression of JUN, FOS, TNFAIP3, and NFKBIA), and migratory surface proteins (Figure 6F: HCAM, PSGL-1, integrin α6 and β1); (d) the MT (mitochondrial gene) cluster had upregulated mitochondrial gene expression (Figure 6D), likely reflecting cell death.
Both transcriptionally inactive and active HIV-1-infected cells upregulate IKZF3 expression
Our findings all pointed to the plausibility that IKZF3 promoted the proliferation and survival of HIV-1-infected cells, particularly during acute HIV-1 infection. We showed that AP-1, IRF7, NFATC3, and Tbox transcription factors may bind to an accessible cis-regulatory element in IKZF3 (Figure 1I) to regulate its expression (Figure 1J). In addition, IKZF3 was co-expressed with cellular proliferation and survival genes such as IL21, MKI67, and BIRC5 (Figure 4G) in HIV-1 RNA+ cells. We next compared IKZF3 gene accessibility and gene expression by infection conditions (Figure 7A) and between HIV-1+ and HIV-1− cells in viremia (Figure 7B) across memory CD4+ T cell subsets. IKZF3 gene accessibility (Figure 7A) and RNA expression (Figure 7A) were increased during viremia in proliferating cells. Comparing between HIV-1+ and HIV-1− proliferating cells, IKZF3 gene accessibility was significantly higher in HIV-1 DNA+ cells (Figure 7B–7D) and IKZF3 gene expression was significantly higher in both HIV-1 DNA+ cells and HIV-1 RNA+ cells (Figure 7B, 7E). Overall, IKZF3 had increased gene accessibility and gene expression in proliferating cells under viremia, especially HIV-1-infected cells.
Figure 7. Both transcriptionally inactive and active HIV-1-infected cells upregulate IKZF3 expression.
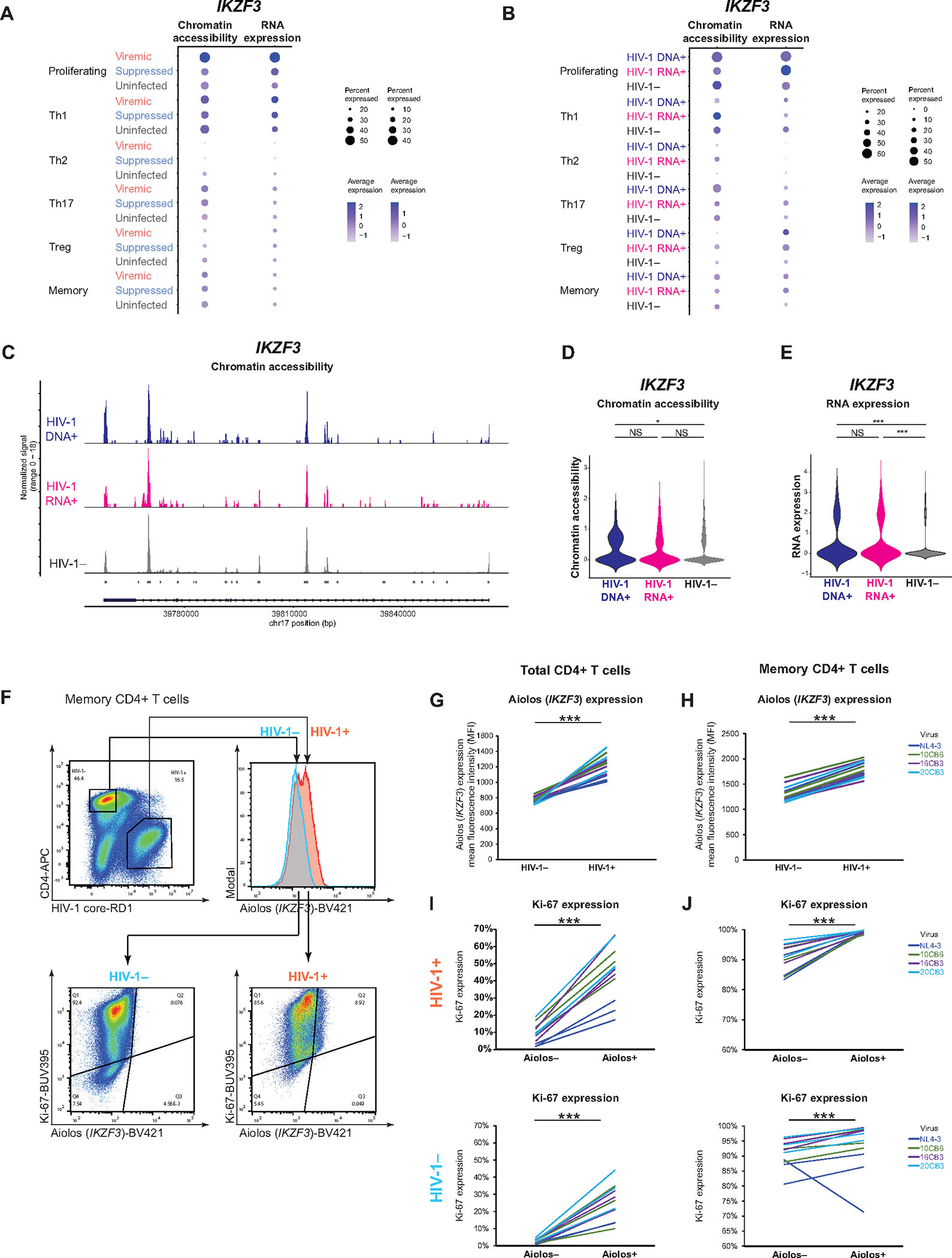
(A–B) Dot plots showing mean normalized and scaled chromatin accessibility (left) and RNA expression (right) of IKZF3 in six main cell subsets. (C) Chromatin accessibility at IKZF3. (D–E) Violin plots showing differences in IKZF3 chromatin accessibility (D) and RNA expression (E). * P < 0.05, *** P <0.001, Wilcoxon rank-sum tests. (F) Representative flow cytometry plot showing Aiolos protein expression in HIV-1-infected versus uninfected cells. Activated primary CD4+ T cells were infected with the replication-competent NL4–3 reference strain and three R5-tropic clinical isolates 10CB6, 16CB3, and 20CB350. After lymphocyte gating, doublet discrimination, and gating for cell viability, HIV-1-infected cells were determined based on HIV-1 core-RD1 expression and CD4 downregulation. (G, H) Aiolos protein expression in HIV-1-infected and uninfected cells. Each line represents one biological replicate from one uninfected donor. Total CD4+ T cells (G) and memory CD4+ T cells (H) from 3 uninfected donors were tested for each virus. (I–J) Proportion of Ki-67 expression among Aiolos+ HIV-1+ cells versus Aiolos− HIV-1+ cells in total CD4+ T cells (I) and memory CD4+ T cells (J). *** P < 0.001, paired two-tail Student’s t-test (G–J).
To validate whether Aiolos (encoded by IKZF3) protein expression was indeed increased during acute HIV-1 infection, we infected activated primary CD4+ T cells (both total CD4+ T cells and memory CD4+ T cells) from uninfected individuals with replication-competent NL4–3 reference strain and three R5-tropic clinical isolates as previously reported50 and measured Aiolos protein expression using flow cytometry. HIV-1 infection was measured by HIV-1 p24 expression and CD4 downregulation. We found that HIV-1-infected cells had higher Aiolos protein expression as measured by mean fluorescent intensity (MFI), both in total CD4+ T cells and in memory CD4+ T cells (Figure 7F, 7G). We then asked whether Aiolos protein expression correlated with Ki-67 protein expression, as we saw in transcriptome analysis (Figure 4F). Indeed, Aiolos+ cells had higher Ki-67 (encoded by MKI67) protein expression, which was more prominent in HIV-1+ cells than HIV-1− cells (Figure 7I–7J), indicating that Aiolos expression may promote the proliferation of HIV-1-infected cells. Overall, both single-cell DOGMA-seq and in vitro validation results showed that IKZF3 expression was increased in HIV-1-infected cells and correlated with proliferation marker Ki-67 expression.
DISCUSSION
Our study examined the single-cell epigenetic, transcriptional, and protein expression profiles of a total of 93,209 memory CD4+ T cells, including 25,778 cells from viremia and 56,771 cells from viral suppression from six people living with HIV-1 and 10,660 cells from four uninfected individuals. Among them, we identified 489 HIV-1-infected cells (233 HIV-1 DNA+ cells and 256 HIV-1 RNA+ cells) in viremia and 33 HIV-1-infected cells (19 HIV-1 DNA+ cells and 14 HIV-1 RNA+ cells) in viral suppression.
Our study revealed the single-cell trimodal cellular states of HIV-1-infected cells, particularly the latent HIV-1-infected cells. We identified transcription factors that governed cellular programs of HIV-1-infected cells across the central dogma of molecular biology. Specifically, we found four distinct populations HIV-1-infected cells: IRF, cytotoxic, AP-1, and MT clusters. Through a genome wide understanding of the cellular programs of HIV-1-infected cells, we identified T cell immune programs that shaped the cell states of HIV-1-infected cells (Figure 6): some cells may die of viral cytopathic effects (MT cluster), others may survive and proliferate by taking advantage of T cell differentiation programs. During acute infection, type I IFN responses shaped a subset of HIV-1-infected cells that had heightened IRF transcription factor binding motif accessibility and drove T cell activation and proliferation programs (characterized by upregulation of survival and proliferation genes IL21, BIRC5, and MKI67 and surface proteins of activation ICOS, HLA-DR, CD38) (IRF cluster). Some cells differentiated toward cytotoxic CD4+ T cells, driven by terminal effector transcription factors (EOMES) and characterized by markers of cytotoxic T cell effectors (upregulated GZMB, GZMH, NKG7, IFNG and surface proteins GRP56 and KLRG1) (Cytotoxic cluster). Other infected cells were driven by AP-1 transcription factors and were upregulated in migratory proteins (integrin β1, HCAM, PSGL-1, CCR5, CCR6) (AP-1 cluster). Overall, we identified distinct immune programs among the heterogeneous HIV-1-infected CD4+ T cells.
We postulated that upregulation of specific cellular markers in HIV-1-infected cells could be caused by preferential infection of cells that expressed these markers, or preferential proliferation of these cells despite HIV-1-infection, or both. For example, HIV-1 may preferentially infect activated Th1 cells because they expressed higher co-receptor CCR5 protein expression (Figure 5F). Alternatively, HIV-1 infection can be stochastic, but cells that had strong activation and proliferation programs (IL21, BIRC538, MKI67) (Figure 6G) or cytotoxic T cells (GZMB, GZMH, IFNG, NKG713) (Figure 6H) could more favorably proliferate and persist over time.
Taking advantage of the genome wide epigenetic and transcriptional profiling, we identified the transcription factor Aiolos (encoded by IKZF3) and its impact on the proliferation of HIV-1-infected cells, which could not be identified by existing multi-omic methods. Aiolos had enriched chromatin accessibility and RNA expression in proliferating cells (Figure 1G, 1J, 7A) and in HIV-1-infected cells (Figure 4A, 4B, 7B, 7D, 7E). Enrichment of Aiolos protein expression in HIV-1-infected cells was validated during in vitro productive infection (Figure 7F–7J) and ex vivo in activated CD4+ T cells from virally suppressed individual in an independent study9. Transcription factors that may activate IKZF3 gene expression were IRF7, AP-1, and NFAT (Figure 1I), which were also the major transcription factors that govern T cell proliferation in viremia (Figure 1F). Aiolos expression correlated with proliferation maker Ki-67 expression in HIV-1-infected cells, both at RNA expression (Figure 4G) and at protein expression (Figure 7G–7J). Given that Aiolos drives Bcl-2 expression39 and NF-κB signaling51 and promotes cellular survival and proliferation, Aiolos may be a transcription factor that drives the proliferation of HIV-1-infected cells. Testing whether Aiolos inhibitors (such as lenalidomide, an FDA-approved drug for multiple myeloma52) can halt the proliferation of HIV-1-infected cells without damaging normal immune responses can be a strategy for HIV eradication interventions.
While no cellular markers can serve as the sole marker specific for HIV-1-infected cells for therapeutic targeting, understanding epigenetic and transcriptional regulation of HIV-1-infected cells in the contexts of T cell differentiation53,54 and T cell clonal expansion dynamics55 guide the development of therapeutic strategies. Our study advances our understanding of HIV-1 reservoir to a big picture understanding of T cell proliferation and the phenotypic complexity of HIV-1-infected cells.
Limitations of the study
The major limitations of the study were the low number of HIV-1-infected cells identified during viral suppression which inherently limited the conclusions that could be drawn from the suppressed time point, the inability to infer proviral genome intactness (DOGMA-seq recovers fragmented DNA and RNA reads), the limited HIV-1 DNA detection by ATAC-seq, and that HIV-1 DNA+ RNA− cells having undetectable HIV-1 RNA may not necessarily be transcriptionally silent because of the detection limit of HIV-1 RNA by single-cell RNA-seq-based approaches. While plate-based methods56 may enhance HIV-1+ read recovery, the low-throughput is not sufficient for clinical samples. An additional limitation of the study is that all participants were young Hispanic males, so the generalizability of our conclusions to people of different age, sex, and ethnicity remains to be determined. Given that a significant proportion of our findings are consistent with previous single-cell profiling of HIV-1-infected cells, such as AP-1 in ATAC-seq5, T cell phenotypes in RNA-seq and surface protein profiling5–7,12, and identification of cytotoxic T cells as the most clonal population13, at least part of our results have been confirmed by other studies.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ya-Chi Ho (ya-chi.ho@yale.edu)
Materials availability
This study did not generate new reagents.
Data and code availability
Single-cell DOGMA-seq sequencing data have been deposited at Gene Expression Omnibus and is publicly available from the date of publication. Accession numbers are listed in the Key Resources Table.
All code used to generate results have been deposited on Github and is publicly available from the date of publication. DOI of the analysis scripts is listed in the Software and Algorithms section of the Key Resources Table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TotalSeq-A Human Universal Cocktail V1.0 | BioLegend | CAT # 399907; RRID: AB_2888692 |
| TotalSeq-A CD30, clone BY88 | BioLegend | CAT # 333913; RRID: AB_2749966 |
| TotalSeq-A CD197, clone G043H7 | BioLegend | CAT # 353247; RRID: AB_2750357 |
| TotalSeq-A anti-human Hashtag 1 | BioLegend | CAT # 394601; RRID: AB_2750015 |
| TotalSeq-A anti-human Hashtag 2 | BioLegend | CAT # 394603; RRID: AB_2750016 |
| TotalSeq-A anti-human Hashtag 3 | BioLegend | CAT # 394605; RRID: AB_2750017 |
| TotalSeq-A anti-human Hashtag 4 | BioLegend | CAT # 394607; RRID: AB_2750018 |
| TotalSeq-A anti-human Hashtag 5 | BioLegend | CAT # 394609; RRID: AB_2750019 |
| TotalSeq-A anti-human Hashtag 6 | BioLegend | CAT # 394611; RRID: AB_2750020 |
| CD4-APC, clone OKT4 | BioLegend | CAT # 317416; RRID: AB_571945 |
| HIV-1 core antigen-RD1, clone KC57 | Beckman Coulter | CAT # 6604667 |
| IKZF3-BV421, clone 14C4C97 | BioLegend | CAT # 371010; RRID: AB_2616875 |
| Ki67-BUV395, clone 56 | BD Biosciences | CAT # 564071 |
| Bacterial and virus strains | ||
| NL4–3 | NIH HIV Reagents Program | CAT # ARP-114 |
| 10CB6 | Ho et al.50 | N/A |
| 16CB3 | Ho et al.50 | N/A |
| 20CB3 | Ho et al.50 | N/A |
| Biological samples | ||
| Blood samples from Sabes Cohort (Table S1) | This study and Collora et al.40 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Digitonin 5% | ThermoFisher | CAT # BN2006 |
| xGen Lockdown Reagents | IDT | CAT # 1072281 |
| Protector RNase Inhibitor | Sigma-Aldrich | PN-3335399001 |
| Human Cot-1 DNA | Invitrogen | CAT # 15279011 |
| Dynabeads M-270 Streptavidin | Invitrogen | CAT # 65306 |
| Human TruStain FcX | BioLegend | CAT # 422302; RRID: AB_2818986 |
| LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit | Thermo Fisher | CAT # L10119 |
| Human BD Fc Block | BD Biosciences | CAT # 564219 |
| eBioscience Foxp3 / Transcription Factor Staining Buffer | Thermo Fisher | CAT # 00–5523-00 |
| BD Pharmingen Purified Mouse Anti-Human CD3, Clone UCHT1 | BD Biosciences | CAT # 555330 |
| BD Pharmingen™ Purified Mouse Anti-Human CD28, Clone CD28.2 | BD Biosciences | CAT # 556620 |
| Recombinant Human Interleukin-2 | Conn Stem | CAT#C1002 |
| Critical commercial assays | ||
| Chromium Next GEM Single Cell Multiome ATAC + Gene Expression Reagent Bundle | 10x Genomics | PN-1000283 |
| Chromium Nuclei Isolation Kit with RNase Inhibitor | 10x Genomics | PN-1000494 |
| Chromium Next GEM Chip J Single Cell | 10x Genomics | PN-1000230 |
| Dual Index Kit TT Set A | 10x Genomics | PN-1000215 |
| Single Index Kit N Set A | 10x Genomics | PN-1000212 |
| 3’ Feature Barcode Kit | 10x Genomics | PN-1000262 |
| EasySeq Dead Cell Removal Annexin V kit | STEMCELL | CAT # 17899 |
| EasySep Human CD4+ T Cell Isolation Kit | STEMCELL | CAT # 17952 |
| Memory CD4+ T cell Isolation Kit | Miltenyi Biotec | CAT # 130–091-893 |
| Kapa Hifi Hotstart Readymix | Kapa Biosystems | CAT # KK2602 |
| SPRIselect 5 mL reagent kit | Beckman Coulter | CAT # B23317 |
| LIVE/DEAD Fixable Near-IR-Dead Cell Stain Kit | Thermo Fisher | L34975 |
| Deposited data | ||
| DOGMAseq memory CD4+ T cells | This study | GEO: GSE239916 |
| Oligonucleotides | ||
| ADT: * indicates phosphorothioate: CCTTGGCACCCGAGAATT*C*C | Mimitou et al.27 | DOGMAseq |
| HTO: * indicates phosphorothioate: GTGACTGGAGTTCAGACGTGTGC*T*C | Mimitou et al.27 | DOGMAseq |
| SIPCR: Dual index common primer for ADT/HTO | This study, see Methods S1 | DOGMAseq |
| RPX: Dual index common primer for ADT | This study, see Methods S1 | DOGMAseq |
| D7X: Dual index common primer for HTO | This study, see Methods S1 | DOGMAseq |
| Software and algorithms | ||
| CellRanger-Arc v2.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-multiome-atac-gex/software/pipelines/latest/installation#download |
| CellRanger v5.0.1 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| STAR v2.7 | Dobin et al.57 | https://github.com/alexdobin/STAR |
| Bowtie 2 v2.4.2 | Langmead and Salzberg58 | https://github.com/BenLangmead/bowtie2 |
| Cutadapt v4.2 | Martin59 | https://cutadapt.readthedocs.io/en/stable/installation.html |
| Seqtk v1.3 | https://github.com/lh3/seqtk | https://anaconda.org/bioconda/seqtk |
| SAMtools v1.16.1 | Li et al.60 | https://anaconda.org/bioconda/samtools |
| IGV v2.16 | Robinson et al.61 | https://software.broadinstitute.org/software/igv/download |
| Seurat v4.3.0 | Hao et al.62 | https://cran.r-project.org/web/packages/Seurat/index.html |
| Signac v1.7 | Stuart et al.63 | https://cran.r-project.org/web/packages/Signac/index.html |
| Harmony v3.8 | Korsunsky et al.64 | https://portals.broadinstitute.org/harmony/articles/quickstart.html |
| Clustree v0.5.0 | Zappia and Oshlack65 | https://cran.r-project.org/web/packages/clustree/index.html |
| MACS3 v3.0.0 | Zhang et al.66 | https://github.com/macs3-project/MACS |
| EnsDb.Hsapiens.v86 v3.16 | Rainer67 | https://bioconductor.org/packages/release/data/annotation/html/EnsDb.Hsapiens.v86.html |
| BSgenome.Hsapiens.UCSC.hg38 v1.4.5 | Team TBD68 | https://bioconductor.org/packages/release/data/annotation/html/BSgenome.Hsapiens.UCSC.hg38.html |
| org.Hs.eg.db v3.8.2 | Carlson et al.69 | https://bioconductor.org/packages/release/data/annotation/html/org.Hs.eg.db.html |
| HOMER v4.11 | Heinz et al.70 | http://homer.ucsd.edu/homer/introduction/install.htsml |
| TFBSTools v3.16 | Tan and Lenhard71 | https://bioconductor.org/packages/release/bioc/html/TFBSTools.html |
| chromVAR v1.18 | Schep et al.72 | http://bioconductor.org/packages/release/bioc/html/chromVAR.html |
| JASPAR2022 | Castro-Mondragon et al.73 | https://bioconductor.org/packages/release/data/annotation/html/JASPAR2022.html |
| DSB v1.0.3 | Mulè et al.74 | https://cran.r-project.org/web/packages/dsb/index.html |
| ACAT v0.91 | Liu et al.75 | https://github.com/yaowuliu/ACAT |
| WGCNA v1.70 | Langfelder and Horvath37 | https://cran.r-project.org/web/packages/WGCNA/index.html |
| topGO v2.48 | Alexa76 | https://bioconductor.org/packages/release/bioc/html/topGO.html |
| fGSEA v1.22 | Korotkevich et al.77 | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| msigdbr v7.5.1 | Dolgalev78 | https://cran.r-project.org/web/packages/msigdbr/vignettes/msigdbr-intro.html |
| R version 4.0.5 | R Core Team79 | https://www.r-project.org/ |
| FlowJo V10.8.1 | FlowJo | https://www.flowjo.com/solutions/flowjo/downloads |
| Analysis scripts | This study | https://doi.org/10.5281/zenodo.8351067 |
EXPERIMENTAL MODELS AND STUDY PARTICIPANT DETAILS
Participant details
The demographics of six people living with HIV-1 and four age, sex, and ethnicity-matched uninfected study participants from the Sabes study28,29 are detailed in Table S1. The Sabes study was reviewed and approved by the Institutional Review Board (IRB) of the Fred Hutchinson Cancer Research Center, the non-government organization Associatión Civil Impacta Salud y Educación, Lima, Peru (Impacta), and the ethics committee of Impacta and the Peruvian National Institute of Health. All participants provided informed consent, including consent for storage and future use of specimens.
From the six participants living with HIV-1, we obtained paired blood samples during viremia and after viral suppression under the Sabes study protocol28,29. Of note, the six participants living with HIV-1 were the same participants participated in our previous study13. Briefly, uninfected study participants were tested monthly with third-generation HIV-1 immunoassays. Seronegative samples were tested for HIV-1 RNA by pooled nucleic acid amplification (NAAT) assays. After specimens were collected during viremia, participants were randomly assigned to immediate and deferred ART (either EFV/FTC/TDF or EGV/COBI/TFC/TDF) initiation arms. In the immediate ART arm, ART was initiated at the baseline visit (< 2 months of estimated date of infection). In the deferred arm, ART was initiated 24 weeks after the baseline visit (6 to 8 months after estimated date of infection). The virally suppressed specimens were taken after one year of suppressive ART (plasma viral load < 200 copies/mL at 6 months prior to blood sampling). We obtained blood samples from 3 participants from the immediate ART arm (SI236, SI640, SI829) and 3 participants from the deferred arm (SD739, SD799, SD910) to ensure that our results are generalizable to participants who started ART early (<6 months) versus late (>6 months). Uninfected study participants were identified at the initial HIV-1 screening (seronegative by whole blood HIV-1 antibody immunoassay followed by NAAT test for HIV-1 RNA) and re-tested monthly for a 2-year period.
Experimental models
For in vitro models, we established four HIV-1-infected and stably integrated Jurkat cell lines (using single-round HIV-1 reporter virus HIV-1-d6-GFP), each having known HIV-1 integration sites in actively transcribed genes as described previously10,41,42. For flow cytometry, primary CD4+ T cells and primary memory CD4+ T cells from de-identified uninfected individuals (New York Blood Center) were infected with replication-competent NL4–3 reference strain and three R5-tropic reconstructed clinical isolates 10CB6, 16CB3, and 20CB3 as previously reported50 for 3 days.
METHOD DETAILS
Memory CD4+ T cell isolation, cell hashing for pooling, and cell staining with barcoded antibodies
Aliquots of 20 million viably frozen peripheral blood mononuclear cells (PBMC) were thawed. Dead cells were removed by immunomagnetic depletion using EasySep Dead Cell Removal Annexin V kit (STEMCELL Technologies, catalog no. 17899) following vendor-recommended protocols. Then, memory CD4+ T cells were purified by immunomagnetic negative selection using Memory CD4+ T cell Isolation Kit removing CD45RA+ cells and non-CD4+ T cells (Miltenyi Biotec, catalog no. 130-091-893).
To pool samples, purified memory CD4+ T cells from participants were each stained with uniquely barcoded TotalSeq-A hashing antibodies (BioLegend) and incubated at 4°C for 30 minutes. Cells were washed with 1 mL wash media and pelleted (500 g for 5 minutes at 4°C) for a total of 3 washes. Cells were then resuspended in 1 mL wash media and pooled (6 viremic samples, 6 virally suppressed samples, and 4 uninfected samples were pooled separately).
The pooled samples were stained with a panel of 154 cell surface proteins and 9 isotype controls included in TotalSeq-A Human Universal Cocktail (V1.0, BioLegend, catalog no. 399907). We additionally stained for CD30 (clone BY88, BioLegend, catalog no. 333913) and CD197 (CCR7, clone G043H7, BioLegend, catalog no. 353247) that were not included in the Universal Cocktail (for a total of 156 surface proteins). All barcoded antibodies were prepared following recommended protocols. The pooled samples (~1.5 × 106 cells in each viremic and virally suppressed pools) were resuspended in 61.5 μL staining buffer and incubated with 7.5 μL Human TruStain FcX (BioLegend, catalog no. 422302) at 4°C for 10 minutes. Next, 3 μL of CD197 was added to each sample followed by incubation at 37°C for 10 minutes then chilled on ice for 5 minutes. Next, 3 μL of CD30 and 75 μL of TotalSeq-A Human Universal Cocktail (3 tests) were added to each sample at 4°C for 30 minutes. Cells were washed with 1 mL wash media and pelleted (500 g for 5 minutes at 4°C) for a total of 3 washes. The pooled uninfected sample (~0.5 × 106 cells) were stained with the same protocols at one third volumes.
Cell permeabilization
After antibody staining, cells were permeabilized without fixation with 0.01% digitonin solution according to the DOGMA-seq protocol27. The digitonin lysis buffer (0.01% DIG, 20 mM Tris-HCl pH 7.4, 150 mM NaCl, 3 mM MgCl2 and 2 U/μL RNase inhibitor) and digitonin wash buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 3 mM MgCl2 and 1 U/μL RNAse inhibitor) were prepared and chilled on ice. Antibody-stained cells were resuspended in 100 μL digitonin lysis buffer on ice for 5 minutes then washed with 1 mL digitonin wash buffer and pelleted (500 g for 5 minutes at 4°C). Cells were then resuspended in 150 μL digitonin wash buffer and counted using Trypan blue staining to verify permeabilization.
DOGMA-seq library preparation and sequencing
Cells were processed according to the Chromium Next GEM Single Cell Multiome ATAC + Gene Expression protocol (10x documentation GC000338 Rev A) with modifications described in the original DOGMA-seq protocol27. We chose the digitonin (DIG) protocol instead of the low-loss lysis (LLL) protocol for its lower mitochondrial DNA capture (0.04% versus 25.3%) and to increase ATAC-seq-based HIV-1 DNA capture. The digitonin protocol showed superior performance in a separate benchmarking study80. See Document S1 for specific protocol modifications and all primers used (ADT and HTO additive primers, SI-PCR and RPX primers, and D7X primers). Pooled samples were sequenced in multiple 10x runs (n = 6, 3, 1 for virally suppressed, viremic, and uninfected samples, respectively). Per run, 41,000 cells were loaded into the 10x Genomics Chromium Controller with a target barcode recovery of 24,000 and singlet recovery of 19,400. Libraries were sequenced on NovaSeq 6000 with a target of ~50,000 ATAC read pairs (500 million per run), ~40,000 RNA read pairs (400 million per run), and ~10,000 ADT/HTO barcode reads (100 million per run) for a total of 100,000 reads per cell. Because our goal was to capture rare HIV-1 reads, our sequencing specifications were two times the recommended sequencing requirements for Single Cell Multiome (10x Genomics, 25,000 ATAC read pairs/cell and 20,000 RNA read pairs/cell) and for CITE-seq (10x Genomics, 20,000 RNA read pairs/cell and 5,000 ADT reads/cell) to ensure good sequencing saturation (Table S2) and to increase sensitivity of HIV-1 read detection. We recovered on average 13,229 cells that passed CellRanger knee call, and an average of 50,131 ATAC read pairs, 48,223 RNA read pairs, and 1,049 usable Antibody barcodes per cell (Table S2). The final libraries were quantified using a Qubit dsDNA HS Assay Kit (Invitrogen) and a High Sensitivity D1000 DNA kit on Agilent 2200 TapeStation system.
Single-cell analyses
DOGMA-seq data pre-processing
Raw sequence files from Chromium Single Cell Multiome ATAC + Gene Expression sequencing (ATAC & RNA) were demultiplexed with CellRanger-Arc v2 (10x Genomics) mkfastq and reads were aligned to the hg38 reference genome using CellRanger-Arc count. Raw sequence files from Single Cell Gene Expression with Feature Barcoding (RNA & ADT/HTO) were demultiplexed with CellRanger v5 (10x Genomics) mkfastq and reads were aligned to the hg38 reference genome using CellRanger count. Cell barcodes that passed knee call by CellRanger and CellRanger-Arc were used for downstream analyses.
Removal of low-quality cells and demultiplexing
Cells that passed knee call in filtered count matrices were used to initialize Seurat Objects 62,81. The objects were filtered by RNA to remove cells with ≥ 25% mitochondrial gene content, ≤ 200 genes, and RNA UMI counts ≤ 500 or ≥ 10,000. Of note, mitochondrial DNA and RNA are inevitably captured in the DOGMA-seq method. Therefore, we used a higher mitochondrial gene cutoff than RNA-based capture such as ECCITE-seq. The objects were then filtered by ATAC to remove cells with ≤ 500 unique ATAC fragments, nucleosome signal strength ≥ 1, transcription start site (TSS) enrichment score ≤ 2, and ATAC UMI counts ≤ 500 or ≥ 100,000.
To remove doublet and negative cells by hashtag oligos (HTOs), the HTO data was normalized by centered log-ratio (CLR) transformation. Cells from individual sequencing runs were demultiplexed based on HTO enrichment using MULTIseqDemux82 implemented in Seurat v481, with automated threshold finding set to TRUE, range of quantile values from 0.1 to 0.999, and maximum number of iterations set to 10. Hashtag-defined doublets and barcodes without hashtag assignment were discarded. ATAC & RNA Seurat objects from individual sequencing runs were then merged by unified set of ATAC peaks across datasets using Signac v1.763. To create a Seurat object containing all three assays (ATAC, RNA, and protein), the ATAC & RNA object was merged with the RNA & protein object by shared GEX barcode and 10x run.
Data normalization, batch effect correction by integration, and data visualization
We normalized and corrected for batch effect for each ATAC, RNA, and protein data to account for technical variation from separate 10x runs. The ATAC data was normalized by latent semantic indexing [LSI: term frequency-inverse document frequency (TF-IDF) followed by singular value decomposition (SVD)] in Signac v1.763 to correct for differences in cellular sequencing depth and across peaks to assign higher values to rare peaks. To correct for batch effects between ATAC datasets, we performed integration by identifying pairwise anchors (pairwise correspondences between single cells across datasets) using FindIntegrationAnchors in Seurat v4 with reciprocal LSI reduction method. The RNA data were normalized by LogNormalize (feature expression measurement divided by the total expression in each cell multiplied by a scale factor of 10,000 then natural-log transformed) in Seurat v4. To correct for batch effects between RNA datasets, we performed PCA before integration by Harmony64. Finally, the ADT (protein) data was normalized across cells using the centered log ratio (CLR) method in Seurat v4. To correct for batch effects between ADT datasets, we performed integration by identifying pairwise anchors with reciprocal PCA reduction method.
We next clustered the cells by shared nearest neighbor space and visualized them in low dimensional projections. To visualize the integrated ATAC data in low dimensional space, the LSI coordinates were integrated across the dataset using IntegrateEmbeddings (Signac v1.7) with ATAC integration anchors and visualized using UMAP. To visualize the integrated RNA and the integrated protein data, the batch corrected data were scaled and centered followed by PCA dimensionality reduction, and cells were visualized in UMAP. To cluster the cells, we simultaneously considered all 3 (ATAC, RNA, and protein) modalities that were independently preprocessed by performing weighted-nearest neighbor (WNN) analysis62. Twenty nearest neighbors for each cell were calculated based on the weighted combination of ATAC, RNA, and protein similarities using FindMultiModalNeighbors in Seurat v4 with integrated ATAC LSI, Harmony, and integrated protein PCA components (2:30, 1:13, and 1:10 numbers of components, respectively). Weighted nearest neighbors were used in cluster determination in Seurat v4 with the smart local moving (SLM) algorithm and an optimized resolution parameter of 0.6 as determined by Clustree evaluation65 and visualized by UMAP.
Downstream data processing
Gene accessibility was measured in Signac v1.7 (determined as number of fragments mapping to each gene coordinate extended by 2kb upstream region to include promoter regions). Gene accessibility data were normalized by LogNormalize in Seurat v4 to adjust for gene accessibility measurements by the total accessibility in each cell multiplied by median count as scale factor.
For differentially accessible ATAC peak analyses, ATAC peaks celled by CellRanger-Arc were recalled using Model-based Analysis of ChIP-Seq 3 (MACS3) v3.0.066 with default parameters in Signac v1.7. Peaks were assigned to genes by distance to nearest TSS and the detailed genomic annotations of the regions occupied by center of peaks were determined by annotatePeaks.pl with hg38 reference genome using HOMER v4.1170. In-peak candidate cis-regulatory elements were predicted by ENCODE Registry of candidate cis-Regulatory Elements (cCREs)83 All predicted TF-binding motifs found in accessible peaks were obtained using genome.ucsc.edu with JASPAR Transcription Factors Track Settings. Transcription factor binding motifs were retrieved from JASPAR CORE 202273 collection and all predicted transcription factor binding sites found in accessible regions met prediction confidence of P < 0.05 as determined by PWMScan and visualized in UCSC Genome Browser84. Transcription factor accessibility deviations (measured in Z-scores) were computed using chromVAR v1.1872 with human genome reference hg38 and transcription factor binding motif references from the JASPAR2022 Core Vertebrates database73.
For protein expression datasets, we normalized the ADT data and performed two steps of corrections for protein expression composition biases. For all analyses of differentially expressed surface proteins, ADT data was normalized and denoised using DSB v1.074 to correct for ambient unbound protein noise by considering protein expression in empty droplets and to correct for cell-to-cell technical noise by shared variance between isotype controls and background protein counts (denoise.counts = TRUE, use.isotype.control = TRUE). For all visualizations of protein expression trends (in dot plot), we normalized the data across cells (margin = 2) using NormalizeData in Seurat v4 with centered-log-ratio (CLR) transformation method as this approach maintains authentic values for fractions of cells expressing the protein feature in dot plots (versus DSB transformation which reflects the standard deviation scores against ambient capture noise). Both DSB and CLR are standard normalization approaches that account for composition biases in single-cell ADT datasets85, with DSB values additionally correct for background noise74 for a more statistically robust differential expression analysis (for volcano plots) but do not reflect true 0 count data (in dot plot). In the second steps, for all features used in comparisons between populations (or cells grouped by different conditions), the group mean expression of each surface proteins was compared to that of its specific isotype controls by Two-Sample Z-test. A protein feature is kept for population comparisons if Z > 2 (2 standard deviation difference).
Cell subset identification
15 clusters of memory CD4+ T cells were distinguished by weighted nearest neighbor analysis, and cell subsets were manually annotated by accessibility of key transcriptional factors in the epigenomic landscape, then by RNA expression of marker genes in the transcriptional profile, then by differential surface protein expression.
HIV-1+ cell identification
To identify HIV-1 RNA+ cells, HIV-1 transcripts were identified per cell barcode by mapping RNA reads to HXB2 reference sequence and participant autologous HIV-1 sequences (Table S3, mapping to autologous sequences was previously shown to increase mapping rate by approximately 20% and increase spliced HIV-1 RNA capture for these same participant samples13) using STAR v2.757 with a pipeline that we have previously established for ECCITE-seq13. Briefly, STAR was run in two pass modes: first pass to identify and annotate splice sites in input HIV-1 reference genomes, and second pass to realign reads to annotated HXB2 genome. The maximum number of multiple alignments allowed was set to 100. Reads that mapped to multiple HIV-1 references or sites (e.g., LTR) were deduplicated. Barcodes and UMIs from associated Read 1 file were extracted and matched to mapped reads. To guard against index hopping and sequencing artifacts, cells with a minimum of 2 HIV-1 reads per barcode were considered positive. No false positive HIV-1 RNA+ cells were identified in uninfected participant samples (n = 10,660 cells).
To identify HIV-1 DNA+ cells, HIV-1 DNA fragments were identified per cell barcode by mapping adapter-trimmed mate-pair ATAC reads to participant autologous HIV-1 sequence and to HXB2 reference sequence using Bowtie 2 v2.4.258 in local alignment and -a (search for and report all alignments) modes. To account for in vivo hypermutation events and HIV-1 sequence diversity, we allowed for 1 mismatch (-N 1) per seed length of 10 (-L 10) during multi-seed alignment. All other parameters were kept at --very-sensitive pre-set (-D 20; -R 3; -i S,1,0.50). Reads that mapped to multiple HIV-1 references or sites (e.g., LTR) were deduplicated. Barcodes and UMIs from associated Read 2 file were then extracted and matched to mapped reads. To guard against false positive detection, all HIV-1-mapped reads were verified by matching to the Los Alamos HIV Sequence Database (https://www.hiv.lanl.gov/). Testing on 14,780 HIV-1-infected primary CD4+ T cells, our HIV-1 DNA alignment approach recovered 5,220 HIV-1 DNA+ cells (35.32% sensitivity) at 1 HIV-1 read per cell threshold and 4,197 HIV-1 DNA+ cells (28.40% sensitivity) at 2 HIV-1 reads per cell threshold. To guard against index hopping and sequencing artifacts, cells with a minimum of 2 HIV-1 reads per barcode were considered positive. No false positive HIV-1 DNA+ cells were identified in uninfected ATAC datasets (n = 10,660 and 9,560 cells in uninfected participant samples and uninfected Jurkat samples). We additionally performed alignment by BWA-MEM v0.7.1786, which was less sensitive than Bowtie2 for our datasets (capturing 90% – 95% HIV-1 reads identified by Bowtie2) and yielded no additional HIV-1 DNA reads.
Analyses of differential gene and peak, and transcription factor accessibility, RNA and surface protein expression
Differential gene and peak and transcription factor accessibility analyses and differential RNA and protein expression were performed using FindMarkers (comparisons of 2 groups) or FindAllMarkers (comparisons of > 2 groups) in Seurat v4, with the following specifications: 1) pseudocount.use = 1, 2) minimum percent of cells expressing the feature (min.pct) and 3) log2 fold change (logfc.threshold) feature selection filter cutoffs as specified in figure legends. As intended by Seurat V4, only features that met ‘min.pct’ and ‘logfc.threshold’ cutoffs were retained, so that poorly expressed genes or genes having extremely low differential expression do not skew the differential expression results (i.e., the FDR-adjusted P value). Of note, no min.pct values were set for chromVAR deviation scores or DSB-normalized protein expression since they were Z-scores. For differential gene accessibility analyses, accessibility expression were normalized with LogNormalize method using a scale factor of median UMI count in each cell. The Wilcoxon Rank-sum test was used to determine fold change significance. For differential peak accessibility analyses, we added as latent variable the total number of in-peak UMIs to account for the effect of differential sequencing depth on the result. Logistic regression was used to determine fold change significance. For differential transcription factor accessibility analyses, the Wilcoxon Rank-sum test was used to determine significant differences of mean deviation scores. For differential gene expression analyses, RNA expression was normalized with LogNormalize method using a scale factor of 10,000. The Wilcoxon Rank-sum test was used to determine fold change significance. For differential surface protein expression analyses, protein expression was normalized by DSB. A protein feature was kept for comparison if its group mean expression is greater than the group mean expression of its isotype control with Z > 2 (2 standard deviation difference) by Two-Sample Z-test. The Wilcoxon Rank-sum test was used to determine fold change significance. To adjust for sample size for a fair comparison between groups when HIV-1+ cells were compared to HIV-1− cells, HIV-1− cell groups were downsized to match the same number of cells in HIV-1+ cell groups with 1,000 bootstrap replicates. The 1,000 P values were transformed to follow a standard Cauchy distribution and a combined P value is calculated as the weighted sum of Cauchy transformed P values75. For all comparisons, P values were corrected for multiple comparisons using the Benjamini-Hochberg (FDR) procedure87.
The averaged expression for all significantly differentially expressed features shown in heatmaps and dot plots were calculated using feature-level scaled values (using ScaleData in Seurat v4: distribution of each feature is centered to mean of 0 and per-cell feature expression is the standard deviation from the mean) using all cells that belong to the cell clusters or conditions under comparison, so that all features can be shown on the same scale (all feature expression share the same scale). This scaling is applied for all features shown in heatmaps and dot plots (gene, protein, peak) except for transcription factor accessibility (which is in chromVAR Z-score). For all dot plot comparisons between HIV-1+ cells versus HIV-1− cells, all features that were significantly upregulated in the HIV-1-infected cell groups during viremia were retained if they also had ‘percent expressed’ in significantly upregulated group ≥ ‘precent expressed’ in HIV-1-negative cell group in viremia, where ‘percent expressed’ is the percentage of cells in the group that express the feature (percentage of cells with feature count > 0).
Gene module identification by WGCNA, Gene Ontology (GO) analysis, and pathway enrichment by GSEA
Co-expressed gene modules were discovered through Weighted Gene Co-expression Network Analysis (WGCNA)36 as previously described88. Briefly, Seurat object was split by condition of interest and the expression matrix of 50 genes most positively and negatively associated with the first < 10 principal components were used as input for WGCNA v1.7037. A soft power was selected as recommended where possible88: the first power with a scale-free topology > 0.8, but this soft power was reduced if fewer than 3 modules with a minimum gene size of 10 were identified. The vectorized topological overlap matrix (TOM) dissimilarity matrix was compared with 10,000 randomly generated and vectorized TOM dissimilarity matrices from the same gene list used for module discovery. Module was determined as significant if FDR-adjusted P < 0.05 (one-sided Wilcoxon rank-sum test) in at least 95% of 10,000 tests. Similar modules were merged using a dissimilarity threshold of 0.5 (MEDissThres = 0.5). See Table S2 for all WGCNA modules identified. To visualize gene pair co-expression, 36601 × 36601 gene pair Pearson’s correlation coefficients were determined using the corSparse function in qlcMatrix R package. For list of genes identified in WGCNA gene modules, we made a custom script to generate a pairwise gene expression Pearson’s correlation coefficient heatmap for cells in each condition. Condition-specific heatmaps were merged along the diagonal split (highlighted in black) to provide a visual contrast of the degree of gene pair co-expression in each of the two conditions (no correlations were calculated for cells between conditions).
For GSEA, all 36,601 genes were ranked by log2 fold change in gene expression comparisons between groups. Gene ranks were used to determine significantly enriched gene sets and build enrichment plot using fGSEA v1.22 with default settings77. Queried gene sets were retrieved from MSigDB v2022.189,90 using msigdbr v7.5.178(R package version 7.5.1). Specifically, a total of 111,635 reference gene sets from the H (hallmark), C2 (curated), and C7 (immunologic signature) collections were retrieved from MSigDB. Enrichment score for each gene set was measured by the weighted Kolmogorov–Smirnov statistic. Significance of enrichment scores was estimated by the nominal P value. A gene set was considered significantly enriched when P < 0.05.
For Gene Ontology analysis, we used all significantly differentially expressed genes to test for significant Gene Ontology terms using topGO v2.4876(R package version 2.48.0.) with Biology Process (GO:BP) subontology91 and org.Hs.eg.db v3.8.2 (human genome wide annotation primarily based on mapping using Entrez Gene identifiers69, R package version 3.8.2). Gene Ontology terms were considered significant when P < 0.01 using Fisher’s exact test. For Figure S6, Gene Ontology analysis was performed using all genes detected in WGCNA modules to test for significant GO terms in the H (hallmark), C2 (curated), GO (Gene Ontology), and C7 (immunologic signature) MSigDB v2022.1 collections as test references. The hypergeometric P value was calculated as the probability of randomly drawing k number of observed overlapping genes between query gene list and reference gene set from a total of 36,601 genes. Gene Ontology terms were considered significant when FDR-adjusted P < 0.05.
Enrichment of WGCNA gene module was scored as previously described92 using AddModuleScore in Seurat v4, which calculates the per-cell average RNA expression of all genes in module subtracted by the aggregated expression of a set of 100 randomly selected genes in the same cells. Note that module score considers the average expression of all genes in a gene set where the weight of each gene expression is the same. This may lead to some cells having high module scores scattered across the UMAP albeit having poor gene co-expression in those cells, since the scores may be skewed by genes that are highly expressed across clusters. Significance of module score differences between cell groups were tested using Wilcoxon rank-sum test.
Flow cytometry
Primary CD4+ T cells or primary memory CD4+ T cells were isolated by magnetic negative depletion (by STEMCELL catalog no. 17952 or Miltenyi catalog no. 130-091-893) from de-identified uninfected individuals (New York Blood Center) and activated with anti-CD3/CD28 antibodies for 3 days in the presence of IL-2 at 30 U/ml. Cells were infected with replication-competent NL4–3 reference strain and three R5-tropic reconstructed clinical isolates 10CB6, 16CB3, and 20CB3 as previously reported50 for 3 days. Cells were stained with LIVE/DEAD Fixable Near-IR–Dead Cell Stain Kit (Thermo Fisher catalog no. L34975), stained with Human BD Fc Block (BD Biosciences catalog no. 564219), stained with surface protein markers (CD4-APC, clone OKT4, BioLegend catalog no. 317416), permeabilized with eBioscience Foxp3 / Transcription Factor Staining Buffer (Thermo Fisher catalog no. 00-5523-00), and stained intracellular proteins HIV-1 core antigen-RD1 (clone KC57, Beckman Coulter catalog no. 6604667), Aiolos (BV421, clone 14C4C97, BioLegend catalog no. 371010) and Ki67 (BUV395, clone 56, BD Biosciences catalog no. 564071). Note that Aiolos antibody clone 16D9C97 resulted in nonspecific staining and was not used. Flow cytometry was performed at Beckman CytoFLEX and analyzed using FlowJo v10.8.1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise stated, statistical analysis was performed using R (version 4.0.5). For all analyses, the statistical tests used, sample size n, mean, median, and standard deviation are described in the figure legends when applicable. For additional details of the statistical tests used for each type of analysis (e.g., WGCNA, Gene Ontology, down-sampling and bootstrapping for comparisons between HIV-1+ vs HIV-1− cells), see also relevant Methods sections. Unless otherwise stated, P values were corrected for multiple comparisons using the Benjamini-Hochberg (FDR) procedure87 when applicable. For all statistical analyses, significance was defined as FDR-adjusted P < 0.05 or P < 0.05. Poor quality cells were excluded from analysis (see Methods for quality control metrics). No data or subjects were excluded from analysis.
Supplementary Material
Highlights.
Single-cell multiomics captures in vivo states of HIV-1 DNA+ and HIV-1 RNA+ cells
HIV-1 latently infected cells are distinct at the DNA, RNA, and protein levels
HIV-1-infected cells have four distinct states: IRF, cytotoxic, AP-1, and cell death
IKZF3 (Aiolos) promotes the proliferation of HIV-1-infected cells
ACKNOWLEDGEMENTS
We thank all study participants. We thank Guilin Wang and Yale Center for Genome Analysis. This work is supported by Yale Top Scholar, NIH R01 AI141009, NIH R01 AI174863, NIH R61/R33 DA047037, NIH P01 AI169768, NIH R37 AI147868, NIH R01 DA051906, NIH R01AI145164, NIH UM1 DA051410, NIH U01 DA053628, NIH CHEETAH P50 AI150464, NIH REACH Martin Delaney Collaboratory UM1 AI164565, NIH BEAT-HIV Martin Delaney Collaboratory UM1 AI164570, Natural Sciences and Engineering Research Council of Canada (NSERC) Postdoctoral Fellowship (PDF557234) (Y.W.), AIDS and Cancer Specimen Resource Pilot Grant (Y.W.), Yale Gruber Fellowship (J.A.C. and K.H.M.), NIH T32 AI055403 (J.A.C.). The Sabes and MERLIN studies were supported by NIH R01 DA032106 and NIH R01 DA040532 (A.D.). We thank ART drug donation from Merck & Co. and Gilead Sciences Inc for the Sabes and Merlin cohorts.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, Lloyd AL, Nowak MA, and Fauci AS (1997). Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A 94, 13193–13197. 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, et al. (1997). Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278, 1295–1300. 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 3.Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, and Richman DD (1997). Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278, 1291–1295. 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- 4.Einkauf KB, Osborn MR, Gao C, Sun W, Sun X, Lian X, Parsons EM, Gladkov GT, Seiger KW, Blackmer JE, et al. (2022). Parallel analysis of transcription, integration, and sequence of single HIV-1 proviruses. Cell. 10.1016/j.cell.2021.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu VH, Nordin JML, Nguyen S, Joy J, Mampe F, Del Rio Estrada PM, Torres-Ruiz F, González-Navarro M, Luna-Villalobos YA, Ávila-Ríos S, et al. (2023). Profound phenotypic and epigenetic heterogeneity of the HIV-1-infected CD4(+) T cell reservoir. Nat Immunol 24, 359–370. 10.1038/s41590-022-01371-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun W, Gao C, Hartana CA, Osborn MR, Einkauf KB, Lian X, Bone B, Bonheur N, Chun TW, Rosenberg ES, et al. (2023). Phenotypic signatures of immune selection in HIV-1 reservoir cells. Nature 614, 309–317. 10.1038/s41586-022-05538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dufour C, Richard C, Pardons M, Massanella M, Ackaoui A, Murrell B, Routy B, Thomas R, Routy JP, Fromentin R, and Chomont N (2023). Phenotypic characterization of single CD4+ T cells harboring genetically intact and inducible HIV genomes. Nat Commun 14, 1115. 10.1038/s41467-023-36772-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clark IC, Mudvari P, Thaploo S, Smith S, Abu-Laban M, Hamouda M, Theberge M, Shah S, Ko SH, Pérez L, et al. (2023). HIV silencing and cell survival signatures in infected T cell reservoirs. Nature 614, 318–325. 10.1038/s41586-022-05556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohn LB, da Silva IT, Valieris R, Huang AS, Lorenzi JCC, Cohen YZ, Pai JA, Butler AL, Caskey M, Jankovic M, and Nussenzweig MC (2018). Clonal CD4(+) T cells in the HIV-1 latent reservoir display a distinct gene profile upon reactivation. Nat Med 24, 604–609. 10.1038/s41591-018-0017-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu R, Yeh YJ, Varabyou A, Collora JA, Sherrill-Mix S, Talbot CC Jr., Mehta S, Albrecht K, Hao H, Zhang H, et al. (2020). Single-cell transcriptional landscapes reveal HIV-1-driven aberrant host gene transcription as a potential therapeutic target. Sci Transl Med 12. 10.1126/scitranslmed.aaz0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weymar GHJ, Bar-On Y, Oliveira TY, Gaebler C, Ramos V, Hartweger H, Breton G, Caskey M, Cohn LB, Jankovic M, and Nussenzweig MC (2022). Distinct gene expression by expanded clones of quiescent memory CD4(+) T cells harboring intact latent HIV-1 proviruses. Cell Rep 40, 111311. 10.1016/j.celrep.2022.111311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gantner P, Buranapraditkun S, Pagliuzza A, Dufour C, Pardons M, Mitchell JL, Kroon E, Sacdalan C, Tulmethakaan N, Pinyakorn S, et al. (2023). HIV rapidly targets a diverse pool of CD4(+) T cells to establish productive and latent infections. Immunity 56, 653–668 e655. 10.1016/j.immuni.2023.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collora JA, Liu R, Pinto-Santini D, Ravindra N, Ganoza C, Lama JR, Alfaro R, Chiarella J, Spudich S, Mounzer K, et al. (2022). Single-cell multiomics reveals persistence of HIV-1 in expanded cytotoxic T cell clones. Immunity 55, 1013–1031 e1017. 10.1016/j.immuni.2022.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee E, Bacchetti P, Milush J, Shao W, Boritz E, Douek D, Fromentin R, Liegler T, Hoh R, Deeks SG, et al. (2019). Memory CD4 + T-Cells Expressing HLA-DR Contribute to HIV Persistence During Prolonged Antiretroviral Therapy. Front Microbiol 10, 2214. 10.3389/fmicb.2019.02214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fromentin R, Bakeman W, Lawani MB, Khoury G, Hartogensis W, DaFonseca S, Killian M, Epling L, Hoh R, Sinclair E, et al. (2016). CD4+ T Cells Expressing PD-1, TIGIT and LAG-3 Contribute to HIV Persistence during ART. PLoS Pathog 12, e1005761. 10.1371/journal.ppat.1005761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pardons M, Baxter AE, Massanella M, Pagliuzza A, Fromentin R, Dufour C, Leyre L, Routy JP, Kaufmann DE, and Chomont N (2019). Single-cell characterization and quantification of translation-competent viral reservoirs in treated and untreated HIV infection. PLoS Pathog 15, e1007619. 10.1371/journal.ppat.1007619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee GQ, Orlova-Fink N, Einkauf K, Chowdhury FZ, Sun X, Harrington S, Kuo HH, Hua S, Chen HR, Ouyang Z, et al. (2017). Clonal expansion of genome-intact HIV-1 in functionally polarized Th1 CD4+ T cells. J Clin Invest 127, 2689–2696. 10.1172/JCI93289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurachi M, Barnitz RA, Yosef N, Odorizzi PM, DiIorio MA, Lemieux ME, Yates K, Godec J, Klatt MG, Regev A, Wherry EJ, and Haining WN (2014). The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat Immunol 15, 373–383. 10.1038/ni.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kolumam GA, Thomas S, Thompson LJ, Sprent J, and Murali-Krishna K (2005). Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med 202, 637–650. 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shaulian E, and Karin M (2002). AP-1 as a regulator of cell life and death. Nature Cell Biology 4, E131–E136. 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 21.Wherry EJ, Ha S-J, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, and Ahmed R (2007). Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 27, 670–684. 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 22.Roychoudhuri R, Clever D, Li P, Wakabayashi Y, Quinn KM, Klebanoff CA, Ji Y, Sukumar M, Eil RL, Yu Z, et al. (2016). BACH2 regulates CD8+ T cell differentiation by controlling access of AP-1 factors to enhancers. Nature Immunology 17, 851–860. 10.1038/ni.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao C, Lou G, Sun H-W, Zhu Z, Sun Y, Chen Z, Chauss D, Moseman EA, Cheng J, D’Antonio MA, et al. (2021). BACH2 enforces the transcriptional and epigenetic programs of stem-like CD8+ T cells. Nature Immunology 22, 370–380. 10.1038/s41590-021-00868-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zuberbuehler MK, Parker ME, Wheaton JD, Espinosa JR, Salzler HR, Park E, and Ciofani M (2019). The transcription factor c-Maf is essential for the commitment of IL-17-producing γδ T cells. Nat Immunol 20, 73–85. 10.1038/s41590-018-0274-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, Mullen AC, Gasink CR, Kaech SM, Miller JD, et al. (2005). Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nature Immunology 6, 1236–1244. 10.1038/ni1268. [DOI] [PubMed] [Google Scholar]
- 26.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, and Littman DR (2006). The Orphan Nuclear Receptor RORγt Directs the Differentiation Program of Proinflammatory IL-17+ T Helper Cells. Cell 126, 1121–1133. 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 27.Mimitou EP, Lareau CA, Chen KY, Zorzetto-Fernandes AL, Hao Y, Takeshima Y, Luo W, Huang T-S, Yeung BZ, Papalexi E, et al. (2021). Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nature Biotechnology 39, 1246–1258. 10.1038/s41587-021-00927-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lama JR, Brezak A, Dobbins JG, Sanchez H, Cabello R, Rios J, Bain C, Ulrich A, De la Grecca R, Sanchez J, and Duerr A (2018). Design Strategy of the Sabes Study: Diagnosis and Treatment of Early HIV Infection Among Men Who Have Sex With Men and Transgender Women in Lima, Peru, 2013–2017. Am J Epidemiol 187, 1577–1585. 10.1093/aje/kwy030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lama JR, Ignacio RAB, Alfaro R, Rios J, Cartagena JG, Valdez R, Bain C, Barbarán KS, Villaran MV, Pilcher CD, et al. (2020). Clinical and Immunologic Outcomes after Immediate or Deferred Antiretroviral Therapy Initiation during Primary HIV Infection: The Sabes Randomized Clinical Study. Clin Infect Dis. 10.1093/cid/ciaa167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McInnes L, Healy J, and Melville J (2018). UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXivLabs, arXiv:1802.03426. [Google Scholar]
- 31.Arp J, Kirchhof MG, Baroja ML, Nazarian SH, Chau TA, Strathdee CA, Ball EH, and Madrenas J (2003). Regulation of T-Cell Activation by Phosphodiesterase 4B2 Requires Its Dynamic Redistribution during Immunological Synapse Formation. Molecular and Cellular Biology 23, 8042–8057. doi: 10.1128/MCB.23.22.8042-8057.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ménasché G, Ménager MM, Lefebvre JM, Deutsch E, Athman R, Lambert N, Mahlaoui N, Court M, Garin J, Fischer A, and de Saint Basile G (2008). A newly identified isoform of Slp2a associates with Rab27a in cytotoxic T cells and participates to cytotoxic granule secretion. Blood 112, 5052–5062. 10.1182/blood-2008-02-141069. [DOI] [PubMed] [Google Scholar]
- 33.Becattini S, Latorre D, Mele F, Foglierini M, De Gregorio C, Cassotta A, Fernandez B, Kelderman S, Schumacher TN, Corti D, Lanzavecchia A, and Sallusto F (2015). T cell immunity. Functional heterogeneity of human memory CD4(+) T cell clones primed by pathogens or vaccines. Science 347, 400–406. 10.1126/science.1260668. [DOI] [PubMed] [Google Scholar]
- 34.Loetscher P, Uguccioni M, Bordoli L, Baggiolini M, Moser B, Chizzolini C, and Dayer J-M (1998). CCR5 is characteristic of Th1 lymphocytes. Nature 391, 344–345. 10.1038/34814. [DOI] [PubMed] [Google Scholar]
- 35.Antonioli L, Pacher P, Vizi ES, and Haskó G (2013). CD39 and CD73 in immunity and inflammation. Trends in Molecular Medicine 19, 355–367. 10.1016/j.molmed.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang B, and Horvath S (2005). A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4, Article17. 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 37.Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuo HH, Ahmad R, Lee GQ, Gao C, Chen HR, Ouyang Z, Szucs MJ, Kim D, Tsibris A, Chun TW, et al. (2018). Anti-apoptotic Protein BIRC5 Maintains Survival of HIV-1-Infected CD4(+) T Cells. Immunity 48, 1183–1194 e1185. 10.1016/j.immuni.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Romero F, Martínez AC, Camonis J, and Rebollo A (1999). Aiolos transcription factor controls cell death in T cells by regulating Bcl-2 expression and its cellular localization. EMBO J 18, 3419–3430. 10.1093/emboj/18.12.3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Collora JA, and Ho YC (2023). Integration site-dependent HIV-1 promoter activity shapes host chromatin conformation. Genome Res. 10.1101/gr.277698.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pedersen SF, Collora JA, Kim RN, Yang K, Razmi A, Catalano AA, Yeh YJ, Mounzer K, Tebas P, Montaner LJ, and Ho YC (2022). Inhibition of a Chromatin and Transcription Modulator, SLTM, Increases HIV-1 Reactivation Identified by a CRISPR Inhibition Screen. J Virol 96, e0057722. 10.1128/jvi.00577-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yeh YJ, Jenike KM, Calvi RM, Chiarella J, Hoh R, Deeks SG, and Ho YC (2020). Filgotinib suppresses HIV-1-driven gene transcription by inhibiting HIV-1 splicing and T cell activation. J Clin Invest 130, 4969–4984. 10.1172/jci137371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chun T-W, Justement JS, Lempicki RA, Yang J, Dennis G, Hallahan CW, Sanford C, Pandya P, Liu S, McLaughlin M, et al. (2003). Gene expression and viral prodution in latently infected, resting CD4+ T cells in viremic versus aviremic HIV-infected individuals. Proceedings of the National Academy of Sciences 100, 1908–1913. doi: 10.1073/pnas.0437640100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holt O, Kanno E, Bossi G, Booth S, Daniele T, Santoro A, Arico M, Saegusa C, Fukuda M, and Griffiths GM (2008). Slp1 and Slp2-a localize to the plasma membrane of CTL and contribute to secretion from the immunological synapse. Traffic 9, 446–457. 10.1111/j.1600-0854.2008.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arp J, Kirchhof MG, Baroja ML, Nazarian SH, Chau TA, Strathdee CA, Ball EH, and Madrenas J (2003). Regulation of T-cell activation by phosphodiesterase 4B2 requires its dynamic redistribution during immunological synapse formation. Mol Cell Biol 23, 8042–8057. 10.1128/mcb.23.22.8042-8057.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tokarev A, McKinnon LR, Pagliuzza A, Sivro A, Omole TE, Kroon E, Chomchey N, Phanuphak N, Schuetz A, Robb ML, et al. (2020). Preferential Infection of α4β7+ Memory CD4+ T Cells During Early Acute Human Immunodeficiency Virus Type 1 Infection. Clin Infect Dis 71, e735–e743. 10.1093/cid/ciaa497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iglesias-Ussel M, Vandergeeten C, Marchionni L, Chomont N, and Romerio F (2013). High levels of CD2 expression identify HIV-1 latently infected resting memory CD4+ T cells in virally suppressed subjects. J Virol 87, 9148–9158. 10.1128/jvi.01297-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li X, Liu Z, Li Q, Hu R, Zhao L, Yang Y, Zhao J, Huang Z, Gao H, Li L, Cai W, and Deng K (2019). CD161(+) CD4(+) T Cells Harbor Clonally Expanded Replication-Competent HIV-1 in Antiretroviral Therapy-Suppressed Individuals. mBio 10. 10.1128/mBio.02121-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rocha-Perugini V, Suárez H, Álvarez S, López-Martín S, Lenzi GM, Vences-Catalán F, Levy S, Kim B, Muñoz-Fernández MA, Sánchez-Madrid F, and Yáñez-Mó M (2017). CD81 association with SAMHD1 enhances HIV-1 reverse transcription by increasing dNTP levels. Nat Microbiol 2, 1513–1522. 10.1038/s41564-017-0019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, Lai J, Blankson JN, Siliciano JD, and Siliciano RF (2013). Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 155, 540–551. 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lazarian G, Yin S, Ten Hacken E, Sewastianik T, Uduman M, Font-Tello A, Gohil SH, Li S, Kim E, Joyal H, et al. (2021). A hotspot mutation in transcription factor IKZF3 drives B cell neoplasia via transcriptional dysregulation. Cancer Cell 39, 380–393 e388. 10.1016/j.ccell.2021.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, et al. (2014). Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305. 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, Olsen BN, Mumbach MR, Pierce SE, Corces MR, et al. (2019). Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat Biotechnol 37, 925–936. 10.1038/s41587-019-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Giles JR, Manne S, Freilich E, Oldridge DA, Baxter AE, George S, Chen Z, Huang H, Chilukuri L, Carberry M, et al. (2022). Human epigenetic and transcriptional T cell differentiation atlas for identifying functional T cell-specific enhancers. Immunity 55, 557–574 e557. 10.1016/j.immuni.2022.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA, et al. (2019). Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med 25, 1251–1259. 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng H, Pui H. p., Lentini A, Kolbeinsdóttir S, Andrews N, Pei Y, Reinius B, Deng Q, and Enge M (2021). Smart3-ATAC: a highly sensitive method for joint accessibility and full-length transcriptome analysis in single cells. bioRxiv, 2021.2012.2002.470912. 10.1101/2021.12.02.470912. [DOI] [Google Scholar]
- 57.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 17, 3. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 60.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stuart T, Srivastava A, Madad S, Lareau CA, and Satija R (2021). Single-cell chromatin state analysis with Signac. Nat Methods 18, 1333–1341. 10.1038/s41592-021-01282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296. 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zappia L, and Oshlack A (2018). Clustering trees: a visualization for evaluating clusterings at multiple resolutions. Gigascience 7. 10.1093/gigascience/giy083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rainer J (2017). EnsDb.Hsapiens.v86: Ensembl based annotation package.
- 68.TEAM TBD R (2023). BSgenome.Hsapiens.UCSC.hg38: Full genomic sequences for Homo sapiens (UCSC genome hg38). .
- 69.Carlson M, Falcon S, Pages H, and Li N (2019). org. Hs. eg. db: Genome wide annotation for Human. Bioconductor. [Google Scholar]
- 70.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tan G, and Lenhard B (2016). TFBSTools: an R/bioconductor package for transcription factor binding site analysis. Bioinformatics 32, 1555–1556. 10.1093/bioinformatics/btw024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nature Methods 14, 975–978. 10.1038/nmeth.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, Lemma RB, Turchi L, Blanc-Mathieu R, Lucas J, Boddie P, Khan A, Manosalva Pérez N, et al. (2022). JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res 50, D165–D173. 10.1093/nar/gkab1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mulè MP, Martins AJ, and Tsang JS (2022). Normalizing and denoising protein expression data from droplet-based single cell profiling. Nat Commun 13, 2099. 10.1038/s41467-022-29356-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu Y, and Xie J (2020). Cauchy combination test: a powerful test with analytic p-value calculation under arbitrary dependency structures. J Am Stat Assoc 115, 393–402. 10.1080/01621459.2018.1554485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Alexa AR, Jorg (2022). topGO: Enrichment Analysis for Gene Ontology. Bioconductor. [Google Scholar]
- 77.Korotkevich G, Sukhov V, and Sergushichev A (2019). Fast gene set enrichment analysis. bioRxiv, 060012. 10.1101/060012. [DOI] [Google Scholar]
- 78.Dolgalev I (2022). msigdbr: MSigDB Gene Sets for Multiple Organisms in a Tidy Data Format. GitHub. [Google Scholar]
- 79.R Core Team R (2021). R: A language and environment for statistical computing
- 80.Xu Z, Heidrich-O’Hare E, Chen W, and Duerr RH (2022). Comprehensive benchmarking of CITE-seq versus DOGMA-seq single cell multimodal omics. Genome Biology 23, 135. 10.1186/s13059-022-02698-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e1821. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McGinnis CS, Patterson DM, Winkler J, Conrad DN, Hein MY, Srivastava V, Hu JL, Murrow LM, Weissman JS, Werb Z, Chow ED, and Gartner ZJ (2019). MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nat Methods 16, 619–626. 10.1038/s41592-019-0433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, Adrian J, Kawli T, Davis CA, Dobin A, Kaul R, et al. (2020). Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710. 10.1038/s41586-020-2493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nassar LR, Barber GP, Benet-Pagès A, Casper J, Clawson H, Diekhans M, Fischer C, Gonzalez JN, Hinrichs AS, Lee BT, et al. (2023). The UCSC Genome Browser database: 2023 update. Nucleic Acids Res 51, D1188–d1195. 10.1093/nar/gkac1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Heumos L, Schaar AC, Lance C, Litinetskaya A, Drost F, Zappia L, Lücken MD, Strobl DC, Henao J, Curion F, Schiller HB, and Theis FJ (2023). Best practices for single-cell analysis across modalities. Nat Rev Genet, 1–23. 10.1038/s41576-023-00586-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J R Stat Soc B 57, 289–300. [Google Scholar]
- 88.Kazer SW, Aicher TP, Muema DM, Carroll SL, Ordovas-Montanes J, Miao VN, Tu AA, Ziegler CGK, Nyquist SK, Wong EB, et al. (2020). Integrated single-cell analysis of multicellular immune dynamics during hyperacute HIV-1 infection. Nat Med 26, 511–518. 10.1038/s41591-020-0799-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chagoyen M, and Pazos F (2010). Quantifying the biological significance of gene ontology biological processes--implications for the analysis of systems-wide data. Bioinformatics 26, 378–384. 10.1093/bioinformatics/btp663. [DOI] [PubMed] [Google Scholar]
- 92.Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell DOGMA-seq sequencing data have been deposited at Gene Expression Omnibus and is publicly available from the date of publication. Accession numbers are listed in the Key Resources Table.
All code used to generate results have been deposited on Github and is publicly available from the date of publication. DOI of the analysis scripts is listed in the Software and Algorithms section of the Key Resources Table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TotalSeq-A Human Universal Cocktail V1.0 | BioLegend | CAT # 399907; RRID: AB_2888692 |
| TotalSeq-A CD30, clone BY88 | BioLegend | CAT # 333913; RRID: AB_2749966 |
| TotalSeq-A CD197, clone G043H7 | BioLegend | CAT # 353247; RRID: AB_2750357 |
| TotalSeq-A anti-human Hashtag 1 | BioLegend | CAT # 394601; RRID: AB_2750015 |
| TotalSeq-A anti-human Hashtag 2 | BioLegend | CAT # 394603; RRID: AB_2750016 |
| TotalSeq-A anti-human Hashtag 3 | BioLegend | CAT # 394605; RRID: AB_2750017 |
| TotalSeq-A anti-human Hashtag 4 | BioLegend | CAT # 394607; RRID: AB_2750018 |
| TotalSeq-A anti-human Hashtag 5 | BioLegend | CAT # 394609; RRID: AB_2750019 |
| TotalSeq-A anti-human Hashtag 6 | BioLegend | CAT # 394611; RRID: AB_2750020 |
| CD4-APC, clone OKT4 | BioLegend | CAT # 317416; RRID: AB_571945 |
| HIV-1 core antigen-RD1, clone KC57 | Beckman Coulter | CAT # 6604667 |
| IKZF3-BV421, clone 14C4C97 | BioLegend | CAT # 371010; RRID: AB_2616875 |
| Ki67-BUV395, clone 56 | BD Biosciences | CAT # 564071 |
| Bacterial and virus strains | ||
| NL4–3 | NIH HIV Reagents Program | CAT # ARP-114 |
| 10CB6 | Ho et al.50 | N/A |
| 16CB3 | Ho et al.50 | N/A |
| 20CB3 | Ho et al.50 | N/A |
| Biological samples | ||
| Blood samples from Sabes Cohort (Table S1) | This study and Collora et al.40 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Digitonin 5% | ThermoFisher | CAT # BN2006 |
| xGen Lockdown Reagents | IDT | CAT # 1072281 |
| Protector RNase Inhibitor | Sigma-Aldrich | PN-3335399001 |
| Human Cot-1 DNA | Invitrogen | CAT # 15279011 |
| Dynabeads M-270 Streptavidin | Invitrogen | CAT # 65306 |
| Human TruStain FcX | BioLegend | CAT # 422302; RRID: AB_2818986 |
| LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit | Thermo Fisher | CAT # L10119 |
| Human BD Fc Block | BD Biosciences | CAT # 564219 |
| eBioscience Foxp3 / Transcription Factor Staining Buffer | Thermo Fisher | CAT # 00–5523-00 |
| BD Pharmingen Purified Mouse Anti-Human CD3, Clone UCHT1 | BD Biosciences | CAT # 555330 |
| BD Pharmingen™ Purified Mouse Anti-Human CD28, Clone CD28.2 | BD Biosciences | CAT # 556620 |
| Recombinant Human Interleukin-2 | Conn Stem | CAT#C1002 |
| Critical commercial assays | ||
| Chromium Next GEM Single Cell Multiome ATAC + Gene Expression Reagent Bundle | 10x Genomics | PN-1000283 |
| Chromium Nuclei Isolation Kit with RNase Inhibitor | 10x Genomics | PN-1000494 |
| Chromium Next GEM Chip J Single Cell | 10x Genomics | PN-1000230 |
| Dual Index Kit TT Set A | 10x Genomics | PN-1000215 |
| Single Index Kit N Set A | 10x Genomics | PN-1000212 |
| 3’ Feature Barcode Kit | 10x Genomics | PN-1000262 |
| EasySeq Dead Cell Removal Annexin V kit | STEMCELL | CAT # 17899 |
| EasySep Human CD4+ T Cell Isolation Kit | STEMCELL | CAT # 17952 |
| Memory CD4+ T cell Isolation Kit | Miltenyi Biotec | CAT # 130–091-893 |
| Kapa Hifi Hotstart Readymix | Kapa Biosystems | CAT # KK2602 |
| SPRIselect 5 mL reagent kit | Beckman Coulter | CAT # B23317 |
| LIVE/DEAD Fixable Near-IR-Dead Cell Stain Kit | Thermo Fisher | L34975 |
| Deposited data | ||
| DOGMAseq memory CD4+ T cells | This study | GEO: GSE239916 |
| Oligonucleotides | ||
| ADT: * indicates phosphorothioate: CCTTGGCACCCGAGAATT*C*C | Mimitou et al.27 | DOGMAseq |
| HTO: * indicates phosphorothioate: GTGACTGGAGTTCAGACGTGTGC*T*C | Mimitou et al.27 | DOGMAseq |
| SIPCR: Dual index common primer for ADT/HTO | This study, see Methods S1 | DOGMAseq |
| RPX: Dual index common primer for ADT | This study, see Methods S1 | DOGMAseq |
| D7X: Dual index common primer for HTO | This study, see Methods S1 | DOGMAseq |
| Software and algorithms | ||
| CellRanger-Arc v2.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-multiome-atac-gex/software/pipelines/latest/installation#download |
| CellRanger v5.0.1 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| STAR v2.7 | Dobin et al.57 | https://github.com/alexdobin/STAR |
| Bowtie 2 v2.4.2 | Langmead and Salzberg58 | https://github.com/BenLangmead/bowtie2 |
| Cutadapt v4.2 | Martin59 | https://cutadapt.readthedocs.io/en/stable/installation.html |
| Seqtk v1.3 | https://github.com/lh3/seqtk | https://anaconda.org/bioconda/seqtk |
| SAMtools v1.16.1 | Li et al.60 | https://anaconda.org/bioconda/samtools |
| IGV v2.16 | Robinson et al.61 | https://software.broadinstitute.org/software/igv/download |
| Seurat v4.3.0 | Hao et al.62 | https://cran.r-project.org/web/packages/Seurat/index.html |
| Signac v1.7 | Stuart et al.63 | https://cran.r-project.org/web/packages/Signac/index.html |
| Harmony v3.8 | Korsunsky et al.64 | https://portals.broadinstitute.org/harmony/articles/quickstart.html |
| Clustree v0.5.0 | Zappia and Oshlack65 | https://cran.r-project.org/web/packages/clustree/index.html |
| MACS3 v3.0.0 | Zhang et al.66 | https://github.com/macs3-project/MACS |
| EnsDb.Hsapiens.v86 v3.16 | Rainer67 | https://bioconductor.org/packages/release/data/annotation/html/EnsDb.Hsapiens.v86.html |
| BSgenome.Hsapiens.UCSC.hg38 v1.4.5 | Team TBD68 | https://bioconductor.org/packages/release/data/annotation/html/BSgenome.Hsapiens.UCSC.hg38.html |
| org.Hs.eg.db v3.8.2 | Carlson et al.69 | https://bioconductor.org/packages/release/data/annotation/html/org.Hs.eg.db.html |
| HOMER v4.11 | Heinz et al.70 | http://homer.ucsd.edu/homer/introduction/install.htsml |
| TFBSTools v3.16 | Tan and Lenhard71 | https://bioconductor.org/packages/release/bioc/html/TFBSTools.html |
| chromVAR v1.18 | Schep et al.72 | http://bioconductor.org/packages/release/bioc/html/chromVAR.html |
| JASPAR2022 | Castro-Mondragon et al.73 | https://bioconductor.org/packages/release/data/annotation/html/JASPAR2022.html |
| DSB v1.0.3 | Mulè et al.74 | https://cran.r-project.org/web/packages/dsb/index.html |
| ACAT v0.91 | Liu et al.75 | https://github.com/yaowuliu/ACAT |
| WGCNA v1.70 | Langfelder and Horvath37 | https://cran.r-project.org/web/packages/WGCNA/index.html |
| topGO v2.48 | Alexa76 | https://bioconductor.org/packages/release/bioc/html/topGO.html |
| fGSEA v1.22 | Korotkevich et al.77 | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| msigdbr v7.5.1 | Dolgalev78 | https://cran.r-project.org/web/packages/msigdbr/vignettes/msigdbr-intro.html |
| R version 4.0.5 | R Core Team79 | https://www.r-project.org/ |
| FlowJo V10.8.1 | FlowJo | https://www.flowjo.com/solutions/flowjo/downloads |
| Analysis scripts | This study | https://doi.org/10.5281/zenodo.8351067 |