

Abstract
Progressive hippocampal degeneration is a key component of Alzheimer’s disease (AD) progression. Therefore, identifying how hippocampal neuronal function is modulated early in AD is an important approach to eventually prevent degeneration. AD-risk factors and signaling molecules likely modulate neuronal function, including APOE genotype and angiotensin II. Compared to APOE3, APOE4 increases AD risk up to 12-fold, and high levels of angiotensin II are hypothesized to disrupt neuronal function in AD. However, the extent that APOE and angiotensin II modulates the hippocampal neuronal phenotype in AD-relevant models is unknown. To address this issue, we used electrophysiological techniques to assess the impact of APOE genotype and angiotensin II on basal synaptic transmission, presynaptic, and post-synaptic activity in mice that express human APOE3 (E3FAD) or APOE4 (E4FAD) and overproduce Aβ. We found that compared to E3FAD mice, E4FAD mice have lower synaptic activity, but higher levels of paired-pulse facilitation (PPF) and long-term potentiation (LTP) in the Schaffer Collateral Commissural Pathway (SCCP) of the hippocampus. We also found that exogenous angiotensin II has a profound inhibitory effect on hippocampal LTP in both E3FAD and E4FAD mice. Collectively, our data suggests that APOE4 and Aβ are associated with a hippocampal phenotype comprised of lower basal activity and higher responses to high-frequency stimulation, the latter of which is suppressed by angiotensin II. These novel data suggest a potential mechanistic link between hippocampal activity, APOE4 genotype, and angiotensin II in AD.
Keywords: APOE4, Hippocampus, Neuron activity, Angiotensin II
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by progressive learning and memory impairment [1, 2]. The hippocampus is one of the brain regions most affected in AD patients with high levels of Aβ plaques and neurofibrillary tangles and extensive neuronal atrophy [3–5]. Hippocampal neuronal dysfunction in AD is likely progressive, starting with altered glutamatergic activity and connectivity, culminating in cell death. Increasingly recognized is the importance of identifying how hippocampal neuronal function is modulated early in AD, to eventually prevent degeneration. A key component is understanding the impact of known AD-risk factors and signaling molecules on the hippocampal glutamatergic activity phenotype, here we focus on APOE and angiotensin II.
APOE is the greatest genetic risk factor for sporadic AD, with APOE4 increasing risk up to 12-fold compared to APOE3 [6, 7]. In AD patients, there is an increased rate of hippocampal degeneration with APOE4 [8, 9], which correlates with cognitive decline and memory deficits [10, 11]. The role of APOE in AD is complex and multifactorial, but there is evidence for an interaction with Aβ. In general, APOE4 is associated with higher Aβ levels and greater markers neuronal dysfunction in the hippocampus compared to APOE3 in vivo [12, 13]. In terms of activity, human data are mainly from younger individuals or non-AD patients and indicate higher hippocampal activity with APOE4 when assessed using fMRI [14–17]. Data on hippocampal glutamatergic activity in vivo has focused on the independent effects of APOE and human Aβ, and conflict. In fact, both higher and lower hippocampal output/glutamatergic activities have been found in familial AD (FAD) models that overproduce Aβ [18–20] and with APOE4 in vivo [11, 21–24]. Importantly, the extent that APOE modulates neuronal activity in the context of human Aβ is unclear and limited to one study that utilized acute application of Aβ to hippocampal slices [21]. Therefore, evaluating how APOE and Aβ modulate hippocampal neuronal activity is important for understanding APOE4-associated AD risk.
Angiotensin II was initially linked to AD through hypertension, which increases the risk of developing AD by ~ 35% [25, 26]. Subsequent data implied that angiotensin II may regulate hippocampal function in the absence of hypertension in AD. Higher angiotensin II [27] and angiotensin II type 1 receptor (AT1R) levels were found in brains of AD patients compared to controls [28–30]. More direct evidence was found in vivo, where inhibiting the AT1R with angiotensin receptor blockers (ARBs) resulted in improved neuronal markers and learning and memory in familial Alzheimer’s disease models [31–39]. In our own studies, we found that candesartan (an ARB) treatment of mice that express APOE4 and overproduce Aβ (E4FAD mice) altered hippocampal neuronal markers and improved short-term memory, although the magnitude of the behavioral effects were modest [36]. An important question raised by these studies is; what is the role of angiotensin II in hippocampal neuron function in the context of APOE4 and Aβ? Angiotensin II has been shown to impact neuronal activity in the hypothalamus/brainstem, supporting its potential to directly modulate activity [40–45]. Addressing this question is important for advancing our mechanistic understanding of angiotensin II in the brain and its potential role in AD.
Therefore, the goal of this study was to evaluate the role of APOE and angiotensin II on hippocampal neuron function in the context of human Aβ. To this end, we used electrophysiological techniques to evaluate changes in synaptic transmission (presynaptic versus postsynaptic activity) in mice that express human APOE3 (E3FAD) or APOE4 (E4FAD) and overproduce Aβ.
Methods
Mouse Models
All experiments were approved by the Institutional Animal Care and Use Committee at the University of Illinois at Chicago. EFAD mice were produced by crossing either APOE3- or APOE4-targeted replacement mice with mice that express 5 familial Alzheimer’s disease (5xFAD) mutations (APP K670N/M671L + I716 V + V717I and PS1 M146L + L286 V) [46]. Both female and male mice (equal numbers) were used and identified by genotyping of tail samples.
Tissue Processing
Mice were anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine (i.p) followed by transcardial perfusion using ice-cold cutting solution (in mM: 93 NMDG, 2.5 KCl, 1.2 N aH2PO4, 30 NaHCO3, 20 HEPES, 10 M gSO4, 0.5 CaCl2, 25 d-glucose, 5 sodium ascorbate, 3 sodium pyruvate). The hippocampus was dissected, sectioned (300 μm) with a vibratome (Leica VT1200), and allowed to recover in artificial cerebrospinal fluid (aCSF, in mM: 122 NaCl, 30 NaHCO3, 3 KCl, 1.25 NaH2PO4, 1 MgSO4, 10 d-glucose, and 2 CaCl2) for 30 min at 32 °C, bubbled with 95% O2–5% CO2.
Input Output Functions
Input output (I/O) response curves were generated on hippocampal slices prior to induction of high-frequency stimulation protocols. Slices were placed in a humidified interface recording chamber and continuously perfused with aCSF. A glass recording electrode (filled with aCSF) was placed over the apical dendritic layer of CA1 pyramidal neurons and Schaffer collaterals were stimulated using short current pulses delivered with a bipolar electrode roughly 300 μM apart. I/O curves were generated using stimulus intensities ranging from 0 to 200 μA in increments of 50 μA. Five fEPSPs per stimulus intensity were collected and averaged. fEPSPs for all electrophysiological experiments were recorded and analyzed using AxoGraph software.
Long-Term Potentiation
Long-term potentiation (LTP) analysis was conducted on hippocampal slices as described previously [47–49] with slight modifications. Basal synaptic transmission was recorded with single stimuli at 50% population spike threshold (ranging from 5 to 99 μA) every 15 s until stable values were obtained for 10 min. LTP was induced by a single train of high-frequency stimulation (100 Hz, 1s at test intensity) and recorded for an additional 30 min. For post-tetanic potentiation (PTP) analysis, fEPSP amplitudes recorded from 1 to 3 min after high-frequency stimulation were averaged and expressed as a percentage of the average amplitude from 10 min of pre-tetanus (baseline) recordings. For LTP analysis, fEPSP amplitudes recorded from 25 to 30 min after high-frequency stimulation were averaged and expressed as a percentage of the average amplitude from 10 min of pre-tetanus (baseline) recordings. For time course data, a bin size window of 1 min was used (i.e., mean value from 4 field responses per data point).
Paired-Pulse Facilitation
Paired-pulse facilitation (PPF) analysis was conducted on hippocampal slices as described previously, with slight modifications [50]. Two stimuli were applied to the Schaffer collaterals at an interval of 50 ms. Paired-pulse facilitation was determined by taking the ratio of the fEPSP amplitude following the second stimulus to the fEPSP amplitude following the first stimulus (referred to as the paired-pulse ratio). For between subject experiments, ten pairs of stimuli were recorded and averaged for analysis. For within subject experiments, 10 min of baseline recordings (one pair of stimuli every 15 s) were collected prior to bath application of either 10 μM angiotensin II or vehicle followed by an additional 10 min of recordings. For time course data analysis, a bin size window of 1 min was used (i.e., mean value from 4 field responses per data point).
Statistical Analysis
All data are presented as mean ± S.E.M and were analyzed by Student’s t-test, Pearson’s correlation, or ANOVA using GraphPad Prism.
Results
The goals of this study were (1) to evaluate the role of APOE genotype in hippocampal neuron function and then (2) determine the effect of angiotensin II. We used EFAD mice to address these goals, as they express human APOE3 (E3FAD) or APOE4 (E4FAD) and overproduce human Aβ, through the expression of 5xFAD autosomal dominant mutations [46]. We used 6-month-old EFAD mice to focus on early/intermediate stages of changes in hippocampal function since at this age there is greater Aβ plaque accumulation with APOE4 and the beginnings of behavioral impairments.
Lower Synaptic Transmission But Higher Paired-Pulse Facilitation with APOE4 (E4FAD) Compared to APOE3 (E3FAD)
We first evaluated changes in synaptic activity, an important first step for determining the alterations in hippocampal function at the synaptic level. We therefore generated input output (I/O) functions by varying direct synaptic stimulation (input) and measuring the magnitude of the resulting synaptic responses (output) in synapses of the Schaffer Collateral Commissural Pathway (SCCP) in the Stratum Radiatum of the CA1 in E3FAD and E4FAD mice. We found that the magnitude of synaptic responses was impacted by APO genotype and stimulus intensity. The APOE genotype effect was due to lower responses in E4FAD mice compared to E3FAD mice (Fig. 1A).
Fig. 1.
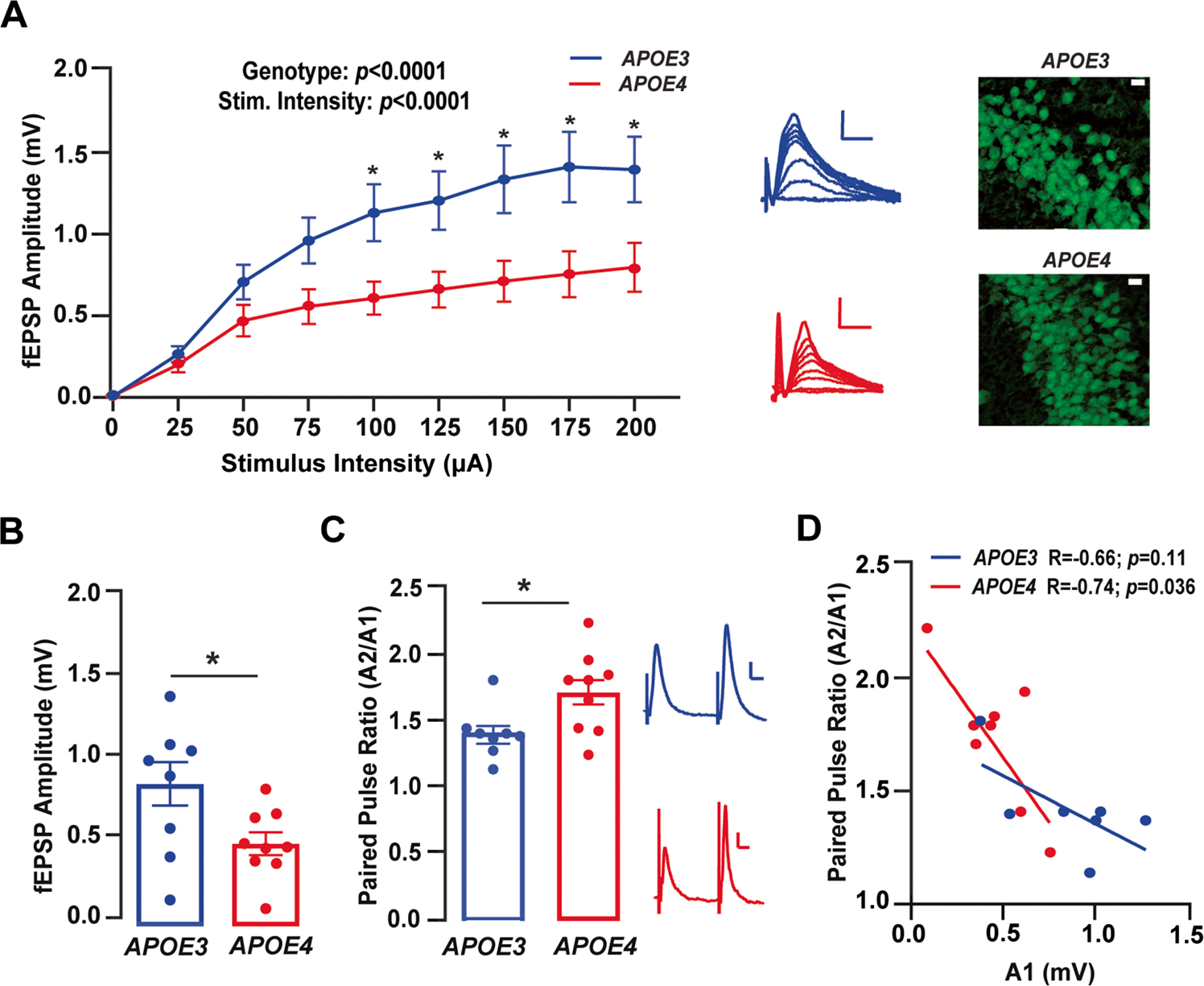
Lower synaptic transmission but higher paired-pulse facilitation with APOE4 as compared to APOE3. A Left: input output (I/O) functions in the Schaffer collaterals of E3FAD and E4FAD mice at stimulus intensities ranging from 0 to 200 μA. Evoked fEPSP amplitudes were lower in E4FAD mice compared to E3FAD when assessed by two-way ANOVA (genotype: F(1, 135) = 38.44, p < 0.0001; stimulus intensity: F(8, 135) = 15.55, p < 0.0001). Although there was no interaction between APOE genotype and stimulus intensity (interaction: F(8, 135) = 1.61, p = 0.13), we performed post hoc analysis to determine at which stimulus intensity APOE genotype-specific differences emerged. We found that E4FAD mice had lower responses at current inputs ≥ 100 μA. Data were analyzed by two-way ANOVA, *p < 0.05 by Sidek’s multiple comparisons post hoc test. n = 9 for E3FAD mice and n = 8 for E4FAD mice. Right: representative images of pyramidal neurons in the Schaffer Collateral Commissural Pathway (SCCP) from E3FAD (top) and E4FAD (bottom) mice. Scale bars = 100 μM. B Two stimuli were applied to the SCCP at 20 Hz and amplitudes of resulting fEPSPs were measured to assess paired-pulse facilitation (PPF). The amplitude of the first fEPSP (A1) was lower in E4FAD mice as compared to E3FAD mice (t(15) = 2.20, p = 0.044), C but the paired-pulse ratio (amplitude of the second fEPSP as a ratio of the amplitude of the first fEPSP – A2/A1) was higher in E4FAD as compared to E3FAD mice (t(15) = 2.45, p = 0.027). In B and C, data were analyzed by t-test, *p < 0.05. n = 8 for E3FAD mice and n = 9 for E4FAD mice. D There was also a significant correlation between A1 and PPF ratio in E4FAD (r = 0.74, p = 0.036) but not E3FAD (r = 0.66, p = 0.11) mice. For panel D, data were analyzed by Pearson’s correlation. n = 7 for E3FAD mice and n = 8 for E4FAD mice. Insets A (left) and C show representative traces from E3FAD and E4FAD mice; calibration bars = 0.2 mV, 10 ms. All data expressed as mean ± SEM (see Supplementary Table 1 for full details on n sizes and statistical comparisons)
Lower I/O responses with APOE4 compared to APOE3 could indicate that there is a lower probability of neurotransmitter release [51, 52]. To test that idea, we utilized paired-pulse facilitation (PPF), a form of short-term plasticity critical to information transfer and neural processing [52, 53]. PPF involves a transient increase in the probability of neurotransmitter release during the second of two rapidly evoked responses, an effect which can be quantified as a ratio of the second response relative to the first (A2/A1–PPF ratio) [51]. We began by assessing the magnitude of the first response (A1) at a fixed stimulus intensity of 50% of population spike threshold. In keeping with our I/O data, we found that A1 was 43% lower in E4FAD mice as compared to E3FAD mice (Fig. 1B). The PPF ratio in E4FAD mice was 22% higher than in E3FAD mice (Fig. 1C), indicating a higher degree of overall facilitation. Higher PPF ratio in E4FAD mice may be due, at least in part, to a lower baseline probability of neurotransmitter release with APOE4. In support of this idea, we found that A1 responses were negatively correlated with the PPF ratio in E4FAD, but not E3FAD, mice (Fig. 1D). Collectively, these data demonstrate that APOE genotype modulates basal synaptic transmission characterized by lower magnitude of synaptic responses in I/O curves and higher PPF ratio with APOE4 compared to APOE3. These data suggest a lower probability of neurotransmitter release in E4FAD mice in the SCCP.
Larger Magnitude of Response to High-Frequency Stimulation with APOE4 as Compared to APOE3
We next looked at more persistent forms of synaptic plasticity. Long-term potentiation (LTP), thought to be a cellular basis of learning and memory [54, 55], is a form of synaptic strengthening that occurs following a train of high-frequency stimulation. In general, synaptic responses following high-frequency stimulation can be separated into two components: post-tetanic potentiation (PTP) or short-term potentiation [56, 57], which is principally mediated by presynaptic mechanisms, and long-term potentiation (LTP), which is mediated by alterations in glutamate receptors at the postsynaptic site [58, 59]. Therefore, we analyzed PTP and LTP separately to gain a more complete understanding of the synaptic phenotype of E3FAD and E4FAD mice (Fig. 2A). We found that the magnitude of the PTP response was 30% higher in E4FAD as compared to E3FAD mice (Fig. 2B). Similarly, we found that levels of the LTP component were 20% higher in E4FAD mice as compared to E3FAD mice (Fig. 2C). Together, these data suggest a change in basal release probability and a higher magnitude of presynaptic and postsynaptic response to high-frequency stimulation with APOE4 as compared to APOE3.
Fig. 2.
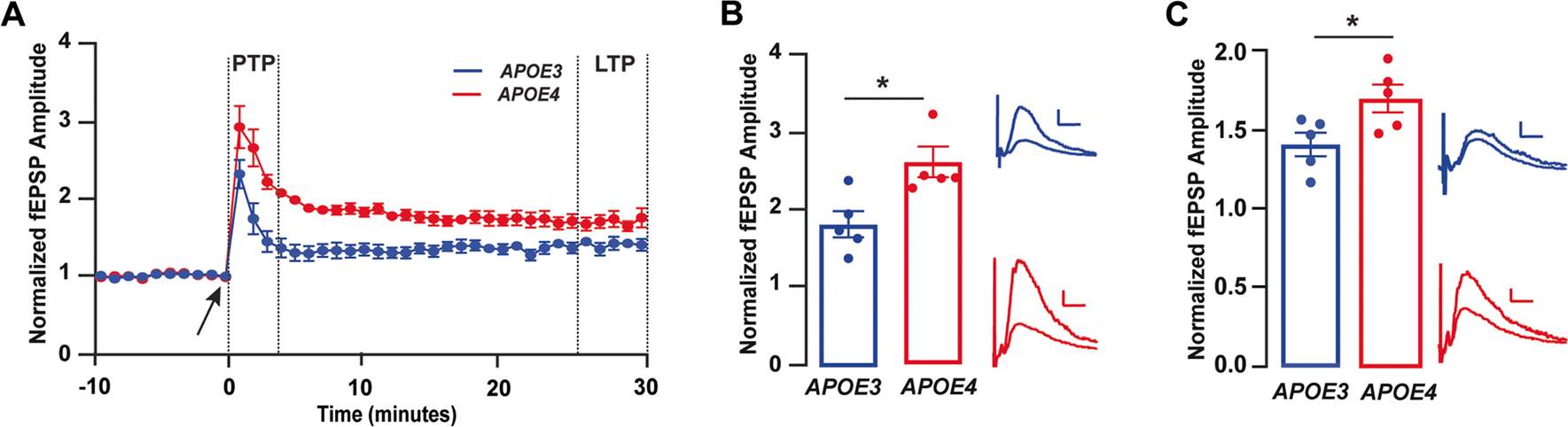
Larger magnitude of response to high-frequency stimulation with APOE4 compared to APOE3. A Time course data depicting the effect of high-frequency stimulation (HFS) on the amplitude of fEPSPs in E3FAD and E4FAD mice. The arrow at 0 min indicates induction of HFS protocol. Dashed lines between minutes 1–3 indicate post-tetanic potentiation (PTP) period and dashed lines between minutes 25–30 indicate long-term potentiation (LTP) period. Amplitude of fEPSPs was higher in E4FAD as compared to E3FAD mice during B PTP (t(8) = 2.95, p = 0.018) and C LTP (t(8) = 2.77, p = 0.024) time periods. Insets B and C show representative traces from E3FAD and E4FAD mice during the PTP and LTP time periods, respectively; calibration bars = 0.1 mV, 10 ms. All data expressed as mean ± SEM. *p < 0.05 by t-test. n = 5 for E3FAD mice and n = 5 for E4FAD mice (see Supplementary Table 1 for full details on n sizes and statistical comparisons)
No Effect of Angiotensin II on Synaptic Transmission or Synaptic Facilitation with APOE3 or APOE4
We next evaluated the impact of angiotensin II on the APOE modulated synaptic response. We started by examining the effect of bath applied angiotensin II on synaptic transmission and PPF in E3FAD and E4FAD mice (Fig. 3). As we found before (Fig. 1), APOE genotype modulates the magnitude of the first evoked fEPSP A1 response (lower in E4FAD mice, Fig. 3B) and the PPF ratio (higher in E4FAD mice, Fig. 3D). However, we found no effect of angiotensin II treatment on either A1 (Fig. 3B) or the PPF ratio (Fig. 3D). These data imply that exogenous angiotensin II does not modulate synaptic transmission or paired-pulse facilitation in EFAD mice.
Fig. 3.
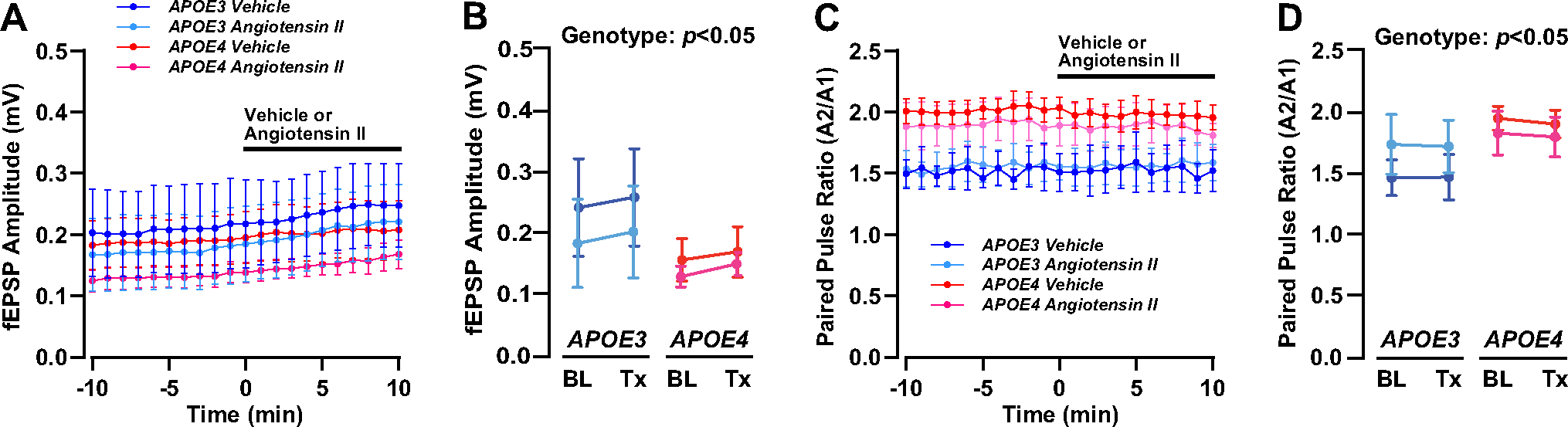
No effect of angiotensin II on basal synaptic transmission or synaptic facilitation with APOE3 or APOE4. A Paired stimuli were applied to the SCCP at 20 Hz every 15 s for 20 min to assess basal synaptic transmission and paired-pulse facilitation (PPF). Time course data of the amplitude of the first response (mV) is depicted. Time 0 indicates bath application of either 10 μM of angiotensin II or vehicle treatment. B There was an APOE genotype effect on the amplitude of the first evoked fEPSP A1 response (A1), which was higher in E4FAD mice than E3FAD mice (genotype: F(1, 28) = 4.51, p = 0.043). However, there were no differences between angiotensin II and vehicle treatment on A1 (treatment: F(1, 28) = 1.78, p = 0.29). C Time course data of the paired-pulse facilitation ratio (A2/A1) is depicted. D The PPF ratio was higher in E4FAD mice than E3FAD mice (genotype: F(1, 28) = 4.35, p = 0.046). There were differences between angiotensin II and vehicle treatment on the PPF ratio (treatment: F(1, 28) = 0.77, p = 0.39). All data expressed as mean ± SEM. p > 0.05 by three-way ANOVA. n = 4 for E3FAD vehicle, n = 5 for E3FAD angiotensin II, n = 4 for E4FAD vehicle, and n = 5 for E4FAD angiotensin II (see Supplementary Table 1 for full details on n sizes and statistical comparisons)
Angiotensin II Suppresses the Magnitude of Response to High-Frequency Stimulation with APOE3 and APOE4
Finally, we evaluated the effects of angiotensin II on the magnitude of synaptic responses to high frequency stimulation in E3FAD and E4FAD mice (Fig. 4A). We found that APOE genotype and treatment impacted PTP and LTP responses. As we found in Fig. 2, LTP and PTP were higher with APOE4 compared to APOE3. Angiotensin II treatment resulted in ~ 20% lower magnitude of PTP and LTP responses (Fig. 4B and C). Taken together, this data suggests that angiotensin II impacts longer-term forms of plasticity. Effects on PTP suggest a presynaptic effect while those on LTP imply a potential postsynaptic mechanism of action.
Fig. 4.
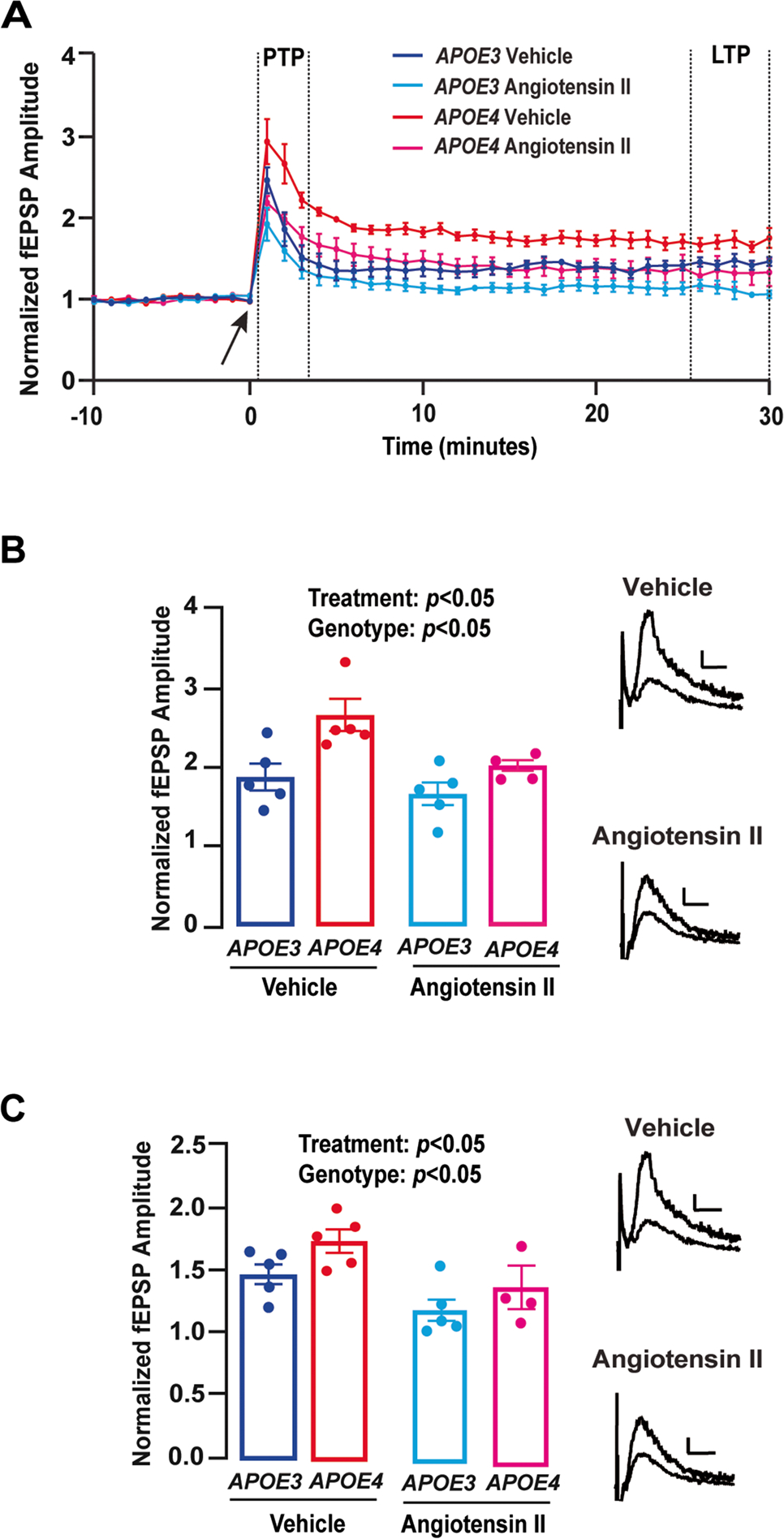
Angiotensin II suppresses the magnitude of response to high frequency stimulation with APOE3 and APOE4. A Time course data depicting the effect of high-frequency stimulation (HFS) on the amplitude of fEPSPs in E3FAD and E4FAD mice. The arrow at 0 min indicates induction of HFS protocol. Dashed lines between minutes 1 and 3 indicate post-tetanic potentiation (PTP) period and dashed lines between minutes 25 and 30 indicate long-term potentiation (LTP) period. B During the PTP period, amplitude of the fEPSPs was lower with the addition of 10 μM angiotensin II (treatment: F(1, 15) = 6.87, p = 0.019) and amplitudes were higher overall in E4FAD than E3FAD mice (genotype: F(1, 15) = 12.36, p = 0.0031). C During the LTP period, amplitude of the fEPSPs were also lower with the addition of 10 μM angiotensin II (treatment: F(1, 15) = 9.40, p = 0.008) and amplitudes were higher overall in E4FAD than E3FAD mice (genotype: F(1, 15) = 6.61, p = 0.021). Insets B and C show representative control and angiotensin II traces for both the PTP and LTP periods. Calibration bars = 0.1 mV, 10 ms. All data expressed as mean ± SEM. *p < 0.05 by two-way ANOVA. n =5 for E3FAD vehicle, n =5 for E3FAD angiotensin II, n = 5 for E4FAD vehicle, and n = 4 for E4FAD angiotensin II (see Supplementary Table 1 for full details on n sizes and statistical comparisons)
Discussion
APOE4 and Neuron Function
Identifying how known AD-risk factors impact neuronal activity is important for our understanding of the disease, and here we found APOE4 and high Aβ levels are associated with lower synaptic transmission and greater responses to high-frequency stimulation in the hippocampus. This phenotype is in partial agreement with previous human and in vivo studies. In non-AD context, compared to APOE3, higher hippocampal activity has been found with APOE4 in several studies using fMRI [14–17]. However, age and AD status may impact the extent that APOE4 differs from APOE3. For example, it has been suggested that higher hippocampal activity represents a feature of cognitive impairment for all APOE genotypes [60] or alternatively that hippocampal activity is lower with age with APOE3 but not APOE4 carriers [61]. Somewhat related is the higher association of APOE4 with seizures and epilepsy that implies network hyperexcitability is a general feature with APOE4 in humans [62–65]. Taken together these human studies broadly imply higher hippocampal activity with APOE4 as compared to APOE3, with the caveat that age, disease severity and region are important considerations. Data from mouse models highlights a complex interaction with APOE genotype and hippocampal activity. In the dentate gyrus/medial perforant pathway [11, 21], there is lower LTP induction and maintenance with APOE4 compared to APOE3 in young mice. However, there is no difference between APOE genotypes in old mice in the same circuit, data that implies the early changes are negated due to age-related impairments in hippocampal plasticity [66, 67]. In layers II/III of the entorhinal cortex, higher spontaneous glutamatergic neuronal transmission has been found with APOE4 compared to APOE3 that with age lead to lower potentiation following high-frequency stimulation [68]. In the CA1/SCCP and similar to our findings, LTP responses are generally higher with APOE4 [22–24] in young mice, although a few have also reported lower responses [69, 70]. Thus, in the absence of high Aβ, the impact of APOE appears to be dependent on brain circuit and age. Future studies could focus on clarifying the overall impact of APOE4, brain region and age on hippocampal activity. In general, however, there is some consensus that for the CA1 APOE4 is associated with hippocampal hyperactivity, however whether this is impacted by high levels of Aβ is unknown.
High Aβ levels are a major pathological hallmark of AD and therefore may interact with APOE to modulate hippocampal activity. In models of high Aβ caused by overexpression of familial AD (FAD) mutations, data are conflicted as to the effects of chronic exposure to Aβ on hippocampal electrophysiology. For example, there are reports of age-dependent reductions in LTP in the CA1/SCCP [71–74] and in the dentate gyrus/perforant pathway [75–78] of various FAD mouse models including 5xFAD [74]. Conversely, there is also evidence indicating transient enhancements in hippocampal activity in the CA/SCCP [79–82] and in the dentate gyrus [83]. In terms of LTP responses with Aβ and APOE, there is only one report on the role of exogenously added oligomeric Aβ in young mice that express the human APOE gene. Those data demonstrate an isoform-specific inhibitory effect on hippocampal neuronal activity in the medial perforant pathway following the order APOE4 > APOE3 > APOE2 [21]. However, in our model system, we show that chronic high levels of Aβ with APOE4 is associated with enhanced levels of LTP in the CA1/SCCP. The differences between data may be related to the circuit (perforant vs CA1/SCCP), age, and/or model (chronic vs acute). Future studies could address how these factors affect the interaction of APOE4 and Aβ on neuron activity.
Our data raise the important question of what higher hippocampal neuron activity in the CA1/SCCP may mean in the context of AD. One possibility is that higher activity is a general property associated with APOE4 across the lifespan and has no impact on neural circuit disruption of cognitive dysfunction in AD. The other extreme is that hyperactivity is a detrimental or maladaptive response due to higher Aβ levels and/or the response of APOE4 to Aβ. There are also several alternatives to these extremes. For example, as Aβ accumulation can have inhibitory effects on hippocampal activity (discussed above), heightened activity with APOE4 may be an important compensatory mechanism early on in disease progression to preserve neural output. Conversely, high levels of hippocampal activity with APOE4 may represent an example of antagonistic pleiotropy, a function that is beneficial early in life, but detrimental later. Evidence for the APOE4 antagonistic pleiotropy hypothesis comes from studies conducted in young APOE4-carriers that outperform non-carriers on memory and neurocognitive tasks early in life, potentially due to greater involvement of executive processes [84, 85]. The idea is that due to continuous higher activity, the circuit is predisposed to dysfunction in AD; or, to compensate for declines in older age, this same recruitment mechanism leads to detrimental hippocampal hyperactivity, ultimately contributing to accelerated cognitive decline. Consistent with this, is the idea that lowering hippocampal excitability levels with APOE4 may be beneficial in AD. Indeed, preventing hyperexcitability has been documented to improve memory performances in AD transgenic mice [86, 87] most likely by enhancing responsiveness to GABAergic interneuron inputs [68, 88]. Future studies will ultimately reveal to what extent APOE4-driven hyperactivity may be a contributing factor to increased AD risk.
Our data also raise the question of what potential mechanisms may underlie the altered hippocampal activity with APOE4 and Aβ. In general, the question of how APOE impacts neuronal function is considered pleiotropic including modulating neuronal function indirectly and directly. As broad examples, APOE4 is associated with greater neurovascular dysfunction, metabolic dysfunction, neuroinflammation, and peripheral inflammation, processes that independently can all disrupt neuronal activity [7, 89]. There are also specific neuronal mechanisms that are disrupted with APOE4 including inhibitory network function within the hippocampus (reviewed in [88]). For example, in APOE-targeted replacement mice, compared to APOE3, with APOE4, there are lower levels of GABAergic somatostatin-positive interneurons in the hippocampus, an effect that appears driven by apoE production in neurons [88]. Thus, the loss of GABAergic interneurons could contribute to network hyperexcitability and higher levels of pyramidal cell firing [68]. Another possibility is the idea that apoE4 derived specifically from astrocytes enhances neuronal excitability [90], potentially due to lysosome dysregulation, altered membrane lipidomes, and/or Ca2+-induced hyperactivity [91]. Collectively, all these factors could contribute to the phenotype we found in hippocampal neurons of lower basal synaptic transmission combined with enhanced PPF and LTP.
At the cellular level in glutamatergic neurons, our data suggests that the impact of APOE4, either due to the mechanisms described above or others, causes changes in both the presynapse and postsynapse. In the presynapse, we found lower magnitudes of evoked fEPSPs combined with enhanced PPF ratios with APOE4 compared to APOE3. This phenotype could be caused by dysregulation in presynaptic calcium homeostasis with APOE4 [92, 93]. In AD, neurons tend to have higher levels of resting calcium which has been attributed to enhanced calcium entry and/or enhanced calcium leakage from intracellular stores [94]. If there are higher neuronal calcium levels with APOE4 due to calcium leakage and/or buffering, it would mean that baseline neuronal activity would be lower because it would interfere with membrane depolarization and thus the probability of firing action potentials. In addition, repeated stimulation (i.e., tetanus) would trigger the release of abnormally high levels of intracellular calcium from organelles such as the mitochondria and endoplasmic reticulum with APOE4. This would, in turn, result in a higher number of neurotransmitter-containing vesicles to fuse with the plasma membrane, thereby increasing presynaptic glutamate release resulting in higher levels of responses to high-frequency stimulation, in agreement with our data. Relatedly, it has been proposed that APOE modulates the glutamate-glutamine cycle, in that with APOE4 there is lower glutamate production and ultimately less efficient vesicular loading [24, 95]. Consistent with this, our PPF data support a lower probability of neurotransmitter release as part of the APOE4 phenotype which could be explained by lower glutamate production, less efficient loading of glutamate into synaptic vesicles, and/or dysfunctions in the presynaptic vesicular fusion/release mechanisms (potentially due to calcium buffering deficits). Due to any combination of these factors, higher levels of presynaptic input may be required with APOE4 to elicit the same postsynaptic responses as APOE3 under basal conditions. We have also provided direct evidence that APOE4 modulates post-synaptic neuronal signaling mechanisms. In keeping with other reports [23], we observed a substantially larger magnitude of response to high-frequency stimulation with APOE4 as compared to APOE3. Higher post-synaptic activity can be caused by changes in AMPA and NMDA composition, levels, and signaling. In terms of APOE, most data on postsynaptic mechanisms are related to receptor signaling. ApoE4 is thought to enhance ERK1/2 activation through interactions with the LRP1 receptor which promotes induction of LTP to a greater extent than apoE3 [23, 96]. It has also been reported that APOE4 suppresses LTP induced by reelin due to modulating glutamate receptor phosphorylation and/or sequestration [24, 95]. The lower response to reelin in vivo could cause a compensatory upregulation response with APOE4 and Aβ. Therefore, with APOE4, there could be changes at the postsynapse in signaling, receptor levels, or calcium responses [79] that result in greater LTP responses following tetanic stimulation. A final explanation for our data is lower overall GABAergic inputs to CA1 neurons, resulting in a heightened response to repeated glutamatergic inputs manifesting in aberrantly increased hippocampal activation with APOE4 [88]. Future mechanistic studies could inform how alterations in presynaptic and postsynaptic signaling with APOE genotype modulate hippocampal circuitry in AD.
Angiotensin II and Neuron Function
We found that angiotensin II suppresses neuronal activity, which raises the important question of the significance of this finding in the context of AD and APOE4. In general, higher angiotensin II levels and/or receptor signaling are considered detrimental in AD [29, 38, 97, 98]. This proposal is based on data that in the medial frontal cortex of AD patients there are up 40% higher angiotensin II levels [27] as well as higher ACE and AT1R levels in the hippocampus and prefrontal cortex as compared to age matched controls [28–30]. Further, AT1R levels are 2.5× higher in the hippocampus [99] and 3× higher in the cortex [100] of APPJ20 mice as compared to wild type controls. In support that enhanced levels of angiotensin II is detrimental for brain function are findings that blocking the AT1R is beneficial in FAD mouse models [31–39]. Specific to APOE4, we found a slight improvement in behavior in EFAD mice after ARB treatment [36]. However, caution may be warranted in assigning a beneficial vs. detrimental impact of angiotensin II to brain function, including with APOE4. Angiotensin II binds receptors on multiple cell types including glia and endothelial cells to exert pleotropic mechanisms of action. In fact, in many in vivo studies, including ours in E4FAD mice, the strongest effect of ARB treatment appears to be preventing enhanced glial activation and modulating neuroinflammatory markers due to high Aβ levels. However, despite a strong effect on glia in our previous study, the corresponding change in behavior was relatively modest in E4FAD mice. This raises the possibility that if higher hippocampal output is detrimental for APOE4 carriers, then angiotensin II-dependent suppression may be beneficial, and therefore, blocking the AT1R globally is not optimal. Alternatively, if higher LTP is a beneficial compensatory mechanism, then preventing the angiotensin II-dependent suppression of LTP is optimal. Therefore, there may also be a balance, whereby neither too low nor too high levels of LTP are optimal for both APOE3 and APOE4, and therefore, maintaining a certain moderate level of LTP is more important. Interestingly, while use of angiotensin system blockers was associated with slower global Aβ accumulation over time and a lower incidence of AD in APOE4 non-carriers, this effect was not seen in APOE4 carriers [101, 102]. Ultimately, understanding how the fundamental cell-type-specific functions of angiotensin II/AT1R collectively contribute in vivo to behavior is important for a deeper mechanistic and therapeutic understanding of the angiotensin system in AD. Recognizing the complexity of AD, the relative contribution of AT1R on each cell type to disease progression may depend on the stage of AD and the relative contribution of inflammation, vascular dysfunction, and neuron hyperactivity to cognitive impairment in each patient.
Mechanistically, our data supports that while exogenous angiotensin II does not impact synaptic transmission or neural facilitation, it does have a profound inhibitory effect on hippocampal LTP in mice that express human APOE. In general, data are mixed on the role of angiotensin II on neuronal excitability with reports of both excitatory and inhibitory effects at the single cell level depending on the brain region and neuronal subpopulation [42, 103, 104]. However, our LTP result is in agreement with other studies demonstrating the inhibitory effects of angiotensin II on synaptic plasticity including in the medial perforant pathway [105] and the lateral nucleus of the amygdala [106]. The majority of the functions associated with angiotensin II signaling in neurons is mediated through AT1R signaling pathways. The AT1R is a G-protein-coupled receptor of the Gαq subtype. Gαq receptors activate protein kinase C (PKC), which regulates calcium-dependent inactivation of NMDA receptors [107, 108]. Lower levels of NMDA receptor activation at the post-synapse would lead to a lower responsiveness to glutamate and therefore suppression of LTP, consistent with our data. Taken together, this suggests that high levels of AT1R activation with angiotensin II interferes with NMDA receptor-dependent synaptic plasticity in the SCCP.
In addition to the unresolved mechanistic questions, our study design limits the extent that we can conclude how APOE regulates the hippocampal neuronal phenotype. An important question alluded to above is how age, sex and APOE genotype interact to modulate hippocampal activity. In general, the interaction between female sex and APOE4 results in greater AD risk and/or progression. In our initial analysis, we did not find an effect of sex (see Supplementary Figure 1A&B) on PPF in statistical analysis, and we therefore designed our study to compare APOE genotype rather than the interactions between sex and APOE. In addition, although we lack power to conduct statistical analysis, visually there is no apparent interaction of APOE and sex on LTP and/or responses to angiotensin II (Supplementary Figure 1C&D). These data are somewhat surprising given the association with sex and APOE genotype in AD. There are several potential explanations including, but not limited to, that in slices the contribution of sex effects is negated because soluble factors that are regulated by sex are absent, that sex impacts the number of “healthy” neurons, that sex impacts other neuronal sub-types and/or brain regions, and/or that the effects of sex on neuron function occurs at later or earlier ages. Future studies could provide more in-depth evaluation of how sex interacts with APOE genotype to impact neuronal function and circuitry in relation to behavior.
Conclusions
Collectively, our data suggests that APOE4 and Aβ are associated with a hippocampal phenotype comprised of lower activity and higher stimulus evoked responses, the latter of which is suppressed by angiotensin II. These novel data suggest a potential mechanistic link between hippocampal activity, APOE4 genotype, and angiotensin II in AD.
Supplementary Material
Funding
This work was supported by National Institutes of Health grants R01AG061114 (LMT), R61NS114353 (LMT), R01MH086507 (KYT), and University of Illinois at Chicago Institutional funds (LMT & KYT).
Abbreviations
- AD
Alzheimer’s disease
- ARBs
angiotensin receptor blockers
- AT1R
angiotensin type 1 receptor
- aCSF
artificial cerebrospinal fluid
- EFAD mice
mice that express human APOE3- or APOE4- and 5 FAD mutations APP K670N/M671L + I716 V + V717I and PS1 M146L + L286 V
- FAD
familial AD models
- fEPSPs
field excitatory post-synaptic potential
- HFS
high-frequency stimulation
- LTP
long-term potentiation
- PPF
paired-pulse facilitation
- PTP
post-tetanic potentiation
- SCCP
Schaffer Collateral Commissural Pathway
Footnotes
Conflict of Interest The authors declare no competing interests.
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s12035-023-03556-9.
Research Involving Human Participants and/or Animals This article does not contain any studies with human participants performed by any of the authors.
Ethical Approval All protocols follow the UIC Institutional Animal Care and Use Committee protocols.
Consent to Participate Not applicable.
Consent for Publication Not applicable.
Data Availability
The datasets used and/or analyzed during the current study are provided as a supplementary file and are available from the corresponding author on reasonable request.
References
- 1.Alzheimer’s disease facts and figures (2022) Alzheimers Dement. 18(4):700–789 [DOI] [PubMed] [Google Scholar]
- 2.Collaborators GBDDF (2022) Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet. Public Health 7(2):e105–e125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mufson EJ et al. (2015) Hippocampal plasticity during the progression of Alzheimer’s disease. Neuroscience 309:51–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balestrieri JVL et al. (1992) Structural volume of hippocampus and Alzheimer’s disease. Rev Assoc Med Bras 66(4):512–515 [DOI] [PubMed] [Google Scholar]
- 5.Scaduto P et al. (2023) Functional excitatory to inhibitory synaptic imbalance and loss of cognitive performance in people with Alzheimer’s disease neuropathologic change. Acta Neuropathol 145(3):303–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu CC et al. (2013) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9(2):106–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liao F, Yoon H, Kim J (2017) Apolipoprotein E metabolism and functions in brain and its role in Alzheimer’s disease. Curr Opin Lipidol 28(1):60–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kerchner GA et al. (2014) APOE epsilon4 worsens hippocampal CA1 apical neuropil atrophy and episodic memory. Neurology 82(8):691–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mueller SG, Weiner MW (2009) Selective effect of age, Apo e4, and Alzheimer’s disease on hippocampal subfields. Hippocampus 19(6):558–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolk DA, Dickerson BC, I. Alzheimer’s Disease Neuroimaging (2010) Apolipoprotein E (APOE) genotype has dissociable effects on memory and attentional-executive network function in Alzheimer’s disease. Proc Natl Acad Sci U S A 107(22):10256–10261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trommer BL et al. (2004) ApoE isoform affects LTP in human targeted replacement mice. Neuroreport 15(17):2655–2658 [DOI] [PubMed] [Google Scholar]
- 12.Tai LM et al. (2017) EFAD transgenic mice as a human APOE relevant preclinical model of Alzheimer’s disease. J Lipid Res 58(9):1733–1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu DS et al. (2015) APOE4 enhances age-dependent decline in cognitive function by down-regulating an NMDA receptor pathway in EFAD-Tg mice. Mol Neurodegener 10:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trivedi MA et al. (2008) fMRI activation during episodic encoding and metacognitive appraisal across the lifespan: risk factors for Alzheimer’s disease. Neuropsychologia 46(6):1667–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bookheimer SY et al. (2000) Patterns of brain activation in people at risk for Alzheimer’s disease. N Engl J Med 343(7):450–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Filippini N et al. (2009) Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc Natl Acad Sci U S A 106(17):7209–7214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kunz L et al. (2015) Reduced grid-cell-like representations in adults at genetic risk for Alzheimer’s disease. Science 350(6259):430–433 [DOI] [PubMed] [Google Scholar]
- 18.Busche MA et al. (2012) Critical role of soluble amyloid-beta for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 109(22):8740–8745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Billings LM et al. (2005) Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 45(5):675–688 [DOI] [PubMed] [Google Scholar]
- 20.Vyas Y, Montgomery JM, Cheyne JE (2020) Hippocampal deficits in amyloid-beta-related rodent models of Alzheimer’s disease. Front Neurosci 14:266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trommer BL et al. (2005) ApoE isoform-specific effects on LTP: blockade by oligomeric amyloid-beta1–42. Neurobiol Dis 18(1):75–82 [DOI] [PubMed] [Google Scholar]
- 22.Kitamura HW et al. (2004) Age-dependent enhancement of hippocampal long-term potentiation in knock-in mice expressing human apolipoprotein E4 instead of mouse apolipoprotein E. Neurosci Lett 369(3):173–178 [DOI] [PubMed] [Google Scholar]
- 23.Korwek KM et al. (2009) ApoE isoform-dependent changes in hippocampal synaptic function. Mol Neurodegener 4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y et al. (2010) ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci U S A 107(26):12011–12016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lennon MJ et al. (2019) Midlife hypertension and Alzheimer’s disease: a systematic review and meta-analysis. J Alzheimers Dis 71(1):307–316 [DOI] [PubMed] [Google Scholar]
- 26.de Gasparo M et al. (2000) International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev 52(3):415–472 [PubMed] [Google Scholar]
- 27.Kehoe PG et al. (2016) Angiotensin-converting enzyme 2 is reduced in Alzheimer’s disease in association with increasing amyloid-beta and tau pathology. Alzheimers Res Ther 8(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cosarderelioglu C et al. (2022) Higher angiotensin II type 1 receptor levels and activity in the postmortem brains of older persons with Alzheimer’s dementia. J Gerontol A Biol Sci Med Sci 77(4):664–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ismael S et al. (2021) Renin-angiotensin system alterations in the human Alzheimer’s disease brain. J Alzheimers Dis 84(4):1473–1484 [DOI] [PubMed] [Google Scholar]
- 30.Savaskan E et al. (2001) Cortical alterations of angiotensin converting enzyme, angiotensin II and AT1 receptor in Alzheimer’s dementia. Neurobiol Aging 22(4):541–546 [DOI] [PubMed] [Google Scholar]
- 31.Mogi M et al. (2008) Telmisartan prevented cognitive decline partly due to PPAR-gamma activation. Biochem Biophys Res Commun 375(3):446–449 [DOI] [PubMed] [Google Scholar]
- 32.Ongali B et al. (2014) Angiotensin II type 1 receptor blocker losartan prevents and rescues cerebrovascular, neuropathological and cognitive deficits in an Alzheimer’s disease model. Neurobiol Dis 68:126–136 [DOI] [PubMed] [Google Scholar]
- 33.Torika N et al. (2017) Intranasal telmisartan ameliorates brain pathology in five familial Alzheimer’s disease mice. Brain Behav Immun 64:80–90 [DOI] [PubMed] [Google Scholar]
- 34.Saavedra JM (2017) Beneficial effects of angiotensin II receptor blockers in brain disorders. Pharmacol Res 125(Pt A):91–103 [DOI] [PubMed] [Google Scholar]
- 35.Saavedra JM (2012) Angiotensin II AT(1) receptor blockers ameliorate inflammatory stress: a beneficial effect for the treatment of brain disorders. Cell Mol Neurobiol 32(5):667–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scheinman SB et al. (2021) Systemic candesartan treatment modulates behavior, synaptic protein levels, and neuroinflammation in female mice that express human APOE4. Front Neurosci 15:628403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davies NM et al. (2011) Associations of anti-hypertensive treatments with Alzheimer’s disease, vascular dementia, and other dementias. J Alzheimers Dis 26(4):699–708 [DOI] [PubMed] [Google Scholar]
- 38.Saavedra JM (2016) Evidence to consider angiotensin II receptor blockers for the treatment of early Alzheimer’s disease. Cell Mol Neurobiol 36(2):259–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loera-Valencia R et al. (2021) Brain renin-angiotensin system as novel and potential therapeutic target for Alzheimer’s disease. Int J Mol Sci 22(18) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh MR, Vigh J, Amberg GC (2021) Angiotensin-II modulates GABAergic neurotransmission in the mouse substantia nigra. eNeuro 8(2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Richards EM et al. (1999) Angiotensin II type 1 receptor-modulated signaling pathways in neurons. Mol Neurobiol 19(1):25–41 [DOI] [PubMed] [Google Scholar]
- 42.Pan HL (2004) Brain angiotensin II and synaptic transmission. Neuroscientist 10(5):422–431 [DOI] [PubMed] [Google Scholar]
- 43.Mooney RD, Zhang Y, Rhoades RW (1994) Effects of angiotensin II on visual neurons in the superficial laminae of the hamster’s superior colliculus. Vis Neurosci 11(6):1163–1173 [DOI] [PubMed] [Google Scholar]
- 44.Li DP, Pan HL (2005) Angiotensin II attenuates synaptic GABA release and excites paraventricular-rostral ventrolateral medulla output neurons. J Pharmacol Exp Ther 313(3):1035–1045 [DOI] [PubMed] [Google Scholar]
- 45.Li DP, Chen SR, Pan HL (2003) Angiotensin II stimulates spinally projecting paraventricular neurons through presynaptic disinhibition. J Neurosci 23(12):5041–5049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Youmans KL et al. (2012) APOE4-specific changes in Abeta accumulation in a new transgenic mouse model of Alzheimer disease. J Biol Chem 287(50):41774–41786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Church E et al. (2022) Synaptic integration of subquantal neurotransmission by colocalized G protein-coupled receptors in presynaptic terminals. J Neurosci 42(6):980–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zaldua S et al. (2020) Epidermal growth factor treatment of female mice that express APOE4 at an age of advanced pathology mitigates behavioral and cerebrovascular dysfunction. Heliyon 6(5):e03919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bashir ZI et al. (1991) Long-term potentiation of NMDA receptor-mediated synaptic transmission in the hippocampus. Nature 349(6305):156–158 [DOI] [PubMed] [Google Scholar]
- 50.Cochilla AJ, Alford S (1998) Metabotropic glutamate receptor-mediated control of neurotransmitter release. Neuron 20(5):1007–1016 [DOI] [PubMed] [Google Scholar]
- 51.Leung LS, Fu XW (1994) Factors affecting paired-pulse facilitation in hippocampal CA1 neurons in vitro. Brain Res 650(1):75–84 [DOI] [PubMed] [Google Scholar]
- 52.Santschi LA, Stanton PK (2003) A paired-pulse facilitation analysis of long-term synaptic depression at excitatory synapses in rat hippocampal CA1 and CA3 regions. Brain Res 962(1–2):78–91 [DOI] [PubMed] [Google Scholar]
- 53.Jackman SL, Regehr WG (2017) The mechanisms and functions of synaptic facilitation. Neuron 94(3):447–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Opitz B (2014) Memory function and the hippocampus. Front Neurol Neurosci 34:51–59 [DOI] [PubMed] [Google Scholar]
- 55.Takeuchi T, Duszkiewicz AJ, Morris RG (2014) The syn-aptic plasticity and memory hypothesis: encoding, storage and persistence. Philos Trans R Soc Lond B Biol Sci 369(1633):20130288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bao JX, Kandel ER, Hawkins RD (1997) Involvement of pre- and postsynaptic mechanisms in posttetanic potentiation at Aplysia synapses. Science 275(5302):969–973 [DOI] [PubMed] [Google Scholar]
- 57.Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361(6407):31–39 [DOI] [PubMed] [Google Scholar]
- 58.Langdon RB, Johnson JW, Barrionuevo G (1995) Posttetanic potentiation and presynaptically induced long-term potentiation at the mossy fiber synapse in rat hippocampus. J Neurobiol 26(3):370–385 [DOI] [PubMed] [Google Scholar]
- 59.Sastry BR, Goh JW, Auyeung A (1986) Associative induction of posttetanic and long-term potentiation in CA1 neurons of rat hippocampus. Science 232(4753):988–990 [DOI] [PubMed] [Google Scholar]
- 60.Tran TT et al. (2017) Increased hippocampal activation in ApoE-4 carriers and non-carriers with amnestic mild cognitive impairment. Neuroimage Clin 13:237–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nichols LM et al. (2012) Interactive effect of apolipoprotein e genotype and age on hippocampal activation during memory processing in healthy adults. Arch Gen Psychiatry 69(8):804–813 [DOI] [PubMed] [Google Scholar]
- 62.Diaz-Arrastia R et al. (2003) Increased risk of late posttraumatic seizures associated with inheritance of APOE epsilon4 allele. Arch Neurol 60(6):818–822 [DOI] [PubMed] [Google Scholar]
- 63.Liang Y et al. (2019) Association of apolipoprotein E genotypes with epilepsy risk: a systematic review and meta-analysis. Epilepsy Behav 98(Pt A):27–35 [DOI] [PubMed] [Google Scholar]
- 64.Gouras GK et al. (1997) Increased apolipoprotein E epsilon 4 in epilepsy with senile plaques. Ann Neurol 41(3):402–404 [DOI] [PubMed] [Google Scholar]
- 65.Lamoureux L et al. (2021) APOE4 promotes tonic-clonic seizures, an effect modified by familial Alzheimer’s disease mutations. Front Cell Dev Biol 9:656521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lynch MA (1998) Analysis of the mechanisms underlying the age-related impairment in long-term potentiation in the rat. Rev Neurosci 9(3):169–201 [DOI] [PubMed] [Google Scholar]
- 67.Barnes CA (2003) Long-term potentiation and the ageing brain. Philos Trans R Soc Lond B Biol Sci 358(1432):765–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nuriel T et al. (2017) Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nat Commun 8(1):1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sun GZ et al. (2017) Hippocampal synaptic and neural network deficits in young mice carrying the human APOE4 gene. CNS Neurosci Ther 23(9):748–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Qiao F et al. (2014) Apolipoprotein E4 impairs in vivo hippocampal long-term synaptic plasticity by reducing the phosphorylation of CaMKIIalpha and CREB. J Alzheimers Dis 41(4):1165–1176 [DOI] [PubMed] [Google Scholar]
- 71.Balducci C et al. (2011) The gamma-secretase modulator CHF5074 restores memory and hippocampal synaptic plasticity in plaque-free Tg2576 mice. J Alzheimers Dis 24(4):799–816 [DOI] [PubMed] [Google Scholar]
- 72.Gong B et al. (2004) Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest 114(11):1624–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chang EH et al. (2006) AMPA receptor downscaling at the onset of Alzheimer’s disease pathology in double knockin mice. Proc Natl Acad Sci U S A 103(9):3410–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kimura R, Ohno M (2009) Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol Dis 33(2):229–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.D’Amelio M et al. (2011) Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat Neurosci 14(1):69–76 [DOI] [PubMed] [Google Scholar]
- 76.Richards JG et al. (2003) PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J Neurosci 23(26):8989–9003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Palop JJ et al. (2007) Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55(5):697–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mango D et al. (2019) Targeting synaptic plasticity in experimental models of Alzheimer’s disease. Front Pharmacol 10:778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zaman SH et al. (2000) Enhanced synaptic potentiation in transgenic mice expressing presenilin 1 familial Alzheimer’s disease mutation is normalized with a benzodiazepine. Neurobiol Dis 7(1):54–63 [DOI] [PubMed] [Google Scholar]
- 80.Parent A et al. (1999) Synaptic transmission and hippocampal long-term potentiation in transgenic mice expressing FAD-linked presenilin 1. Neurobiol Dis 6(1):56–62 [DOI] [PubMed] [Google Scholar]
- 81.Oddo S et al. (2003) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39(3):409–421 [DOI] [PubMed] [Google Scholar]
- 82.Auffret A et al. (2009) Age-dependent impairment of spine morphology and synaptic plasticity in hippocampal CA1 neurons of a presenilin 1 transgenic mouse model of Alzheimer’s disease. J Neurosci 29(32):10144–10152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Davis KE, Fox S, Gigg J (2014) Increased hippocampal excitability in the 3xTgAD mouse model for Alzheimer’s disease in vivo. PLoS One 9(3):e91203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tuminello ER, Han SD (2011) The apolipoprotein e antagonistic pleiotropy hypothesis: review and recommendations. Int J Alzheimers Dis 2011:726197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Han SD, Bondi MW (2008) Revision of the apolipoprotein E compensatory mechanism recruitment hypothesis. Alzheimers Dement 4(4):251–254 [DOI] [PubMed] [Google Scholar]
- 86.Kazim SF et al. (2017) Early-onset network hyperexcitability in presymptomatic Alzheimer’s disease transgenic mice is suppressed by passive immunization with anti-human APP/Abeta antibody and by mGluR5 blockade. Front Aging Neurosci 9:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hijazi S et al. (2020) Early restoration of parvalbumin interneuron activity prevents memory loss and network hyperexcitability in a mouse model of Alzheimer’s disease. Mol Psychiatry 25(12):3380–3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Najm R, Jones EA, Huang Y (2019) Apolipoprotein E4, inhibitory network dysfunction, and Alzheimer’s disease. Mol Neurodegener 14(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Flowers SA, Rebeck GW (2020) APOE in the normal brain. Neurobiol Dis 136:104724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Konings SC et al. (2021) Astrocytic and neuronal apolipoprotein E isoforms differentially affect neuronal excitability. Front Neurosci 15:734001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Larramona-Arcas R et al. (2020) Sex-dependent calcium hyperactivity due to lysosomal-related dysfunction in astrocytes from APOE4 versus APOE3 gene targeted replacement mice. Mol Neurodegener 15(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ramakrishna S et al. (2021) APOE4 affects basal and NMDAR-mediated protein synthesis in neurons by perturbing calcium homeostasis. J Neurosci 41(42):8686–8709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Veinbergs I et al. (2002) Neurotoxic effects of apolipoprotein E4 are mediated via dysregulation of calcium homeostasis. J Neurosci Res 67(3):379–387 [DOI] [PubMed] [Google Scholar]
- 94.Berridge MJ (2011) Calcium signalling and Alzheimer’s disease. Neurochem Res 36(7):1149–1156 [DOI] [PubMed] [Google Scholar]
- 95.Dumanis SB et al. (2013) APOE genotype affects the pre-synaptic compartment of glutamatergic nerve terminals. J Neurochem 124(1):4–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Weeber EJ et al. (2002) Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J Biol Chem 277(42):39944–39952 [DOI] [PubMed] [Google Scholar]
- 97.Wright JW, Harding JW (2019) Contributions by the brain renin-angiotensin system to memory, cognition, and Alzheimer’s disease. J Alzheimers Dis 67(2):469–480 [DOI] [PubMed] [Google Scholar]
- 98.Jackson L et al. (2018) Within the brain: the renin angiotensin system. Int J Mol Sci 19(3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Trigiani LJ et al. (2018) Pleiotropic benefits of the angiotensin receptor blocker candesartan in a mouse model of Alzheimer disease. Hypertension 72(5):1217–1226 [DOI] [PubMed] [Google Scholar]
- 100.Royea J et al. (2020) AT2R’s (angiotensin II type 2 receptor’s) role in cognitive and cerebrovascular deficits in a mouse model of Alzheimer disease. Hypertension 75(6):1464–1474 [DOI] [PubMed] [Google Scholar]
- 101.Ouk M et al. (2021) The use of angiotensin-converting enzyme inhibitors vs. angiotensin receptor blockers and cognitive decline in Alzheimer’s disease: the importance of blood-brain barrier penetration and APOE epsilon4 carrier status. Alzheimers Res Ther 13(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Qiu WQ et al. (2013) Angiotensin converting enzyme inhibitors and the reduced risk of Alzheimer’s disease in the absence of apolipoprotein E4 allele. J Alzheimers Dis 37(2):421–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Barnes KL, DeWeese DM, Andresen MC (2003) Angiotensin potentiates excitatory sensory synaptic transmission to medial solitary tract nucleus neurons. Am J Physiol Regul Integr Comp Physiol 284(5):R1340–R1353 [DOI] [PubMed] [Google Scholar]
- 104.Xiong H, Marshall KC (1994) Angiotensin II depresses glutamate depolarizations and excitatory postsynaptic potentials in locus coeruleus through angiotensin II subtype 2 receptors. Neuroscience 62(1):163–175 [DOI] [PubMed] [Google Scholar]
- 105.Denny JB et al. (1991) Angiotensin II blocks hippocampal long-term potentiation. Brain Res 567(2):321–324 [DOI] [PubMed] [Google Scholar]
- 106.von Bohlenund Halbach O, Albrecht D (1998) Angiotensin II inhibits long-term potentiation within the lateral nucleus of the amygdala through AT1 receptors. Peptides 19(6):1031–1036 [DOI] [PubMed] [Google Scholar]
- 107.Lu WY et al. (2000) In CA1 pyramidal neurons of the hippocampus protein kinase C regulates calcium-dependent inactivation of NMDA receptors. J Neurosci 20(12):4452–4461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xiong ZG et al. (1998) Regulation of N-methyl-D-aspartate receptor function by constitutively active protein kinase C. Mol Pharmacol 54(6):1055–1063 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are provided as a supplementary file and are available from the corresponding author on reasonable request.